

Abstract
The Institute of Medicine recently convened a workshop to review the state of the various domains of nutritional genomics research and policy and to provide guidance for further development and translation of this knowledge into nutrition practice and policy. Nutritional genomics holds the promise to revolutionize both clinical and public health nutrition practice and facilitate the establishment of (a) genome-informed nutrient and food-based dietary guidelines for disease prevention and healthful aging, (b) individualized medical nutrition therapy for disease management, and (c) better targeted public health nutrition interventions (including micronutrient fortification and supplementation) that maximize benefit and minimize adverse outcomes within genetically diverse human populations. As the field of nutritional genomics matures, which will include filling fundamental gaps in knowledge of nutrient–genome interactions in health and disease and demonstrating the potential benefits of customizing nutrition prescriptions based on genetics, registered dietitians will be faced with the opportunity of making genetically driven dietary recommendations aimed at improving human health.
Public health nutrition continues to be challenged by increasing expectations from the food supply on one hand, and fundamental gaps in nutrition knowledge on the other, which can constrain the development and implementation of nutrition and food policy (1). Current demands on the food supply are no longer limited to ensuring general safety and preventing micronutrient deficiencies. Increasingly, there is interest in engineering medicinal qualities into the food supply to enable diets that promote health and “nurture” a sense of well-being that transcends the mere absence of disease by improving biological functions and even increasing lifespans.
Unquestionably, nutrition is one of the primary environmental exposures that determines health. Common human chronic diseases, including type 2 diabetes, metabolic syndrome, cardiovascular and neurological disease, and many cancers are initiated and/or accelerated by nutrient/food exposures. However, it is also recognized that chronic diseases are complex in their etiology and include a substantial genetic component; individuals respond differently to foods and even individual nutrients. Investigation in this new field of nutrition research, often referred to as nutritional genomics, focuses on deciphering the biological mechanisms that underlie both the acute and persistent genome-nutrient interactions that influence health.
Nutritional genomics, while centered on the biology of individuals, distinguishes itself from other “omics” fields by its unique focus on disease prevention and healthy aging through the manipulation of gene–diet interactions. Nutritional genomics promises to revolutionize both clinical and public health nutrition practice and facilitate the establishment of (a) genome-informed nutrient and food-based dietary guidelines for disease prevention and healthful aging, (b) individualized medical nutrition therapy for disease management, and (c) better targeted public health nutrition interventions, including micronutrient fortification and supplementation, that maximize benefit and minimize adverse outcomes within genetically diverse human populations. Research dietitians are among the leading scientists pioneering this field, and food and nutrition professionals will be primarily responsible for its implementation.
In 2006, the Institute of Medicine convened a workshop to review the state of the various domains of nutritional genomics research and policy and to provide guidance for further development and translation of this knowledge into nutrition practice and policy (2). Three scientific domains of nutritional genomics were discussed: (a) nutritional genetics or nutrigenetics, which involves the identification, classification, and characterization of human genetic variation that modifies nutrient metabolism/ utilization and food tolerances (Figure 1); (b) nutritional epigenetics, which refers to the effect of nutrients on deoxyribonucleic acid (DNA)/chromatin (and hence gene expression), which programs or reprograms biological networks with multigenerational consequences; and (c) systems biology and nutritional engineering, which is the application of nutrigenomics information to manipulate biological pathways and networks for benefit through nutrition, including the use of food-based diets, dietary restriction, or nutritional supplements to affect genome expression, stability, and/or direct dietary compensation for metabolic deficiencies (2). This Institute of Medicine report provides background for this review, which is restricted in scope to highlighting the interactions of the B vitamin folate with the human genome and the current gaps in knowledge that must be overcome to achieve genomically driven nutrition practice and policies.
Figure 1.
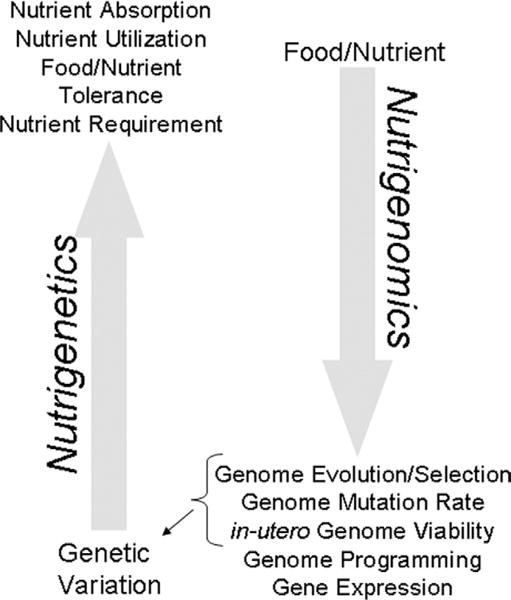
Nutrient-genome interactions. Nutritional genomics encompasses both nutrigenetics, the influence of genetic variation on nutrient utilization/metabolism, food tolerances, and nutrient requirements; and nutrigenomics, the modulatory role of nutrients on genome evolution, mutation rate, in-utero viability, programming, and expression. In turn, several of the nutrigenomic outcomes (ie, genome evolution) contribute to the genetic variation observed within genetically diverse human populations. NOTE: This figure is available online at www.adajournal.org as part of a PowerPoint presentation.
ORIGIN OF GENE–NUTRIENT INTERACTIONS
The human genome, and the genetic variation present within human populations, is in part a product of adaptive evolution to an often scant and unpredictable food supply (3,4). Single nucleotide polymorphisms (SNPs), which are common, single base-pair differences in DNA sequence, represent a primary form of human genetic variation. Of the approximately 10 million SNPs in the human genome, many are believed to have functional consequences (eg, alter the activity/function of the protein product) (5,6). SNPs arise through the sequential process of DNA mutation and subsequent expansion of the mutation within a population. Food and nutrient exposures affect both of these processes. For example, B-vitamin deficiencies impair DNA synthesis/stability and increase DNA mutation rates (germ line and somatic DNA mutations) as do excesses of prooxidants, including iron. Likewise, the nutritional environment can accelerate the expansion of fortuitous germ line DNA mutations within a population such that they accumulate and contribute to human genetic variation.
Indeed, many SNPs that affect nutrient utilization display genomic “signatures” of such positive selection (3). For example, a SNP located near the gene that encodes lactase enables carriers of this SNP to produce this enzyme throughout adulthood and thus continue to tolerate milk (7). This SNP penetrated populations whose ancestors came from places where dairy herds could be raised safely and economically, such as in Europe (8,9). However, several gene variants that arose through positive selection are modern-day candidates for disease alleles; gene variants that permit adaptation to one environment can be deleterious when the environmental conditions change (eg, the nature and abundance of the food supply). For example, the HFE gene variant that is associated with risk for hemochromatosis may have conferred advantage in iron-poor regions but confers risk for iron overload in iron-rich environments (10,11). Expansion of a SNP also requires that the associated changes in biochemistry permit embryonic survival in the interuterine environment. Both malnutrition and some gene variants that impair nutrient metabolism and/or utilization are risk factors for spontaneous abortion (12).
Nutrients and other bioactive food components can also regulate gene expression (Figure 1). All organisms have acquired the ability to sense and adapt to their nutrient environment by altering the expression of proteins that function in metabolic and signaling pathways. Salient examples include, but are not limited to, the regulation of gene transcription by vitamin A or vitamin D through interaction with their respective nuclear receptors. This ability of nutrients to communicate with the genome is an essential feature of organismal evolution. Nutrients can elicit transient alterations in gene expression and/or influence more permanent whole genome reprogramming events that can be inherited (ie, passed onto offspring). The term epigenetics refers to the inheritance of traits through mechanisms that are independent of DNA primary sequence and includes the inheritance of gene expression patterns and/or expression levels that contribute to phenotypic differences among individuals, including monozygotic twins (13).
The embryo seems to be especially susceptible to nutrient-induced adaptations in gene expression, a phenomenon referred to as metabolic imprinting or metabolic programming (14). These adaptations occur within critical windows during embryonic development and can persist into adulthood. The associated changes in metabolism resulting from these reprogramming events are believed to enable in utero survival in suboptimal nutrient environments, but predispose the individual to metabolic disease in adulthood (14). This relationship among maternal nutrition, fetal epigenetic programming, and adult-onset chronic disease is the basis of the fetal origins of adult disease hypothesis, which proposes that nutrition acts very early in life to program risk for adverse outcomes in adult life (Figure 2) (15). This theory, which was originally supported only by epidemiological associations, has now been validated in whole-animal studies. These studies demonstrate that early nutrition exposures increased risk in adulthood for obesity, hypertension, and insulin resistance, which are the antecedents of adult chronic disease including cardiovascular disease and diabetes (15). The genome, in turn, can constrain diet (Figure 1). Genetic variation and/or variations in epigenetic programming can affect nutrient absorption and utilization (eg, hemochromatosis) and thereby confer differences in food/nutrient tolerances (eg, iron) and may contribute to the variation in human nutrient requirements (3).
Figure 2.
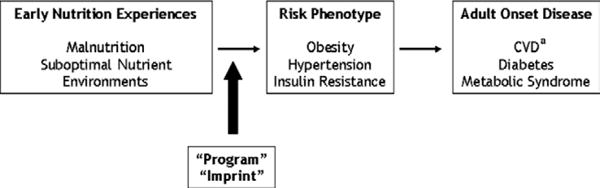
The fetal origins of disease hypothesis. In utero environmental exposures, including nutrition, act early in life to program risk for adult health outcomes. aCVD=cardiovascular disease. NOTE: Information from this figure is available online at www.adajournal.org as part of a PowerPoint presentation.
FOLATE METABOLISM: A CONDUIT BETWEEN THE NUTRIENT ENVIRONMENT AND THE GENOME
Folate is a B vitamin that functions as a metabolic cofactor by carrying and chemically activating single carbons for the biosynthesis of purine nucleotides and thymidylate and for the remethylation of homocysteine to methionine, a metabolic network known as folate-mediated one-carbon metabolism (Figure 3). Methionine in turn can be adenosylated to form S-adenosylmethionine (SAM), which is a co-substrate for numerous cellular methylation reactions (16). Purines and thymidylate are required for DNA synthesis, and SAM is required for genome methylation. The human genome is methylated on cytosine bases present within DNA at specific CpG sequences; lysine residues on histone proteins bound to DNA are also targets for SAM–dependent methylation (16,17). Methylation of chromatin (a complex of DNA and histones) regulates the expression of numerous genes throughout the genome. Methylation is associated with inactivation of an X chromosome in females and with silencing genetically imprinted genes where only the maternal or paternal allele is expressed and the other is silenced by methylation (13). Methionine and thymidylate biosynthesis are the most sensitive pathways within the folate metabolic network, and their impairments compromise the rate and fidelity of DNA synthesis and repair, DNA stability, and the efficiency of cellular methylation reactions (16,18–21).
Figure 3.
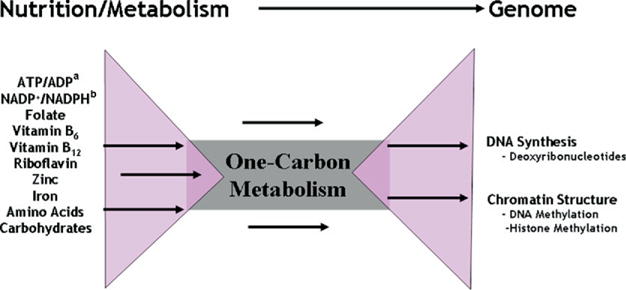
Folate-mediated one-carbon metabolism is a conduit between the cellular nutrient environment and genome synthesis and methylation. aATP/ADP=adenosine triphosphate/adenosine diphosphate. bNADP+/NADPH=Nicotinamide adenine dinucleotide phosphate. NOTE: This figure is available online at www.adajournal.org as part of a PowerPoint presentation.
The folate-dependent biosynthesis of nucleotide precursors for DNA synthesis and of SAM for genome methylation is dependent on the availability of many vitamins including B-12, B-6, niacin, riboflavin and minerals (zinc, cobalt) and is subject to regulation by other nutrients not directly involved in DNA or SAM biosynthesis (iron, vitamin A). Therefore, folate-mediated one-carbon metabolism is a conduit that enables communication between the cellular nutrient environment and regulation of the genome (Figure 3). Impairments in one-carbon metabolism, and the SAM cycle in particular, induced by nutritional deficiencies and/or SNPs in genes that encode folate-dependent enzymes, alter genome methylation patterns and gene expression levels (16,17,22), including the expression of tumor suppressor genes (23,24). For example, severe impairments in the homocysteine remethylation pathway, as seen in those with hyperhomocysteinemia, decreases DNA methylation, which results in a homocysteine-dependent shift from monoallelic to biallelic expression of genetically imprinted genes. Folate supplementation reverses DNA hypomethylation and restores monoallelic expression of imprinted genes in these individuals (25). Therefore, folate is key for genome synthesis, stability, and expression. Alterations in DNA and histone methylation are the likely epigenetic signatures that are metastable, heritable, and may enable adaptation to suboptimal nutrient environments through genome programming (Figure 2).
Disruptions in folate metabolism are common and increase risk for pathologies that include certain cancers, cardiovascular disease, neurological disorders and developmental anomalies such as spina bifida, cleft palate, and spontaneous abortion, among others (16). Folate supplementation can reduce the risk of these disorders developing, with genetically susceptible individuals/populations accruing the most benefit; this suggests that folate nutrition can compensate for genetically induced impairments in folate metabolism (26). However, the biochemical mechanisms underlying folate-related pathologies have remained elusive despite intensive investigation.
Gene variants that encode folate-dependent enzymes and alter the efficiency of nucleotide and SAM biosynthesis can confer both protection and risk for specific pathologies and developmental anomalies (16). A common SNP in the methylenetetrahydrofolate reductase gene (MTHFR), 677C>T, and methylenetetrahydrofolate dehydrogenase gene (MTHFD1), 1958G>A, which encode folate-dependent enzymes, increase risk for neural tube defects (NTDs) (27,28). However, the MTHFR 677C>T SNP is also protective against colon cancer in folate-replete individuals (29). The prevalence of the MTHFR 677C>T SNP varies markedly among human populations; the allelic frequency is approximately 20% in some Hispanic populations, but it is rare in African populations (30), potentially indicating that this gene variant has emerged through positive selection (31). The functional role of these polymorphisms in the etiology of neural tube defects and cancer is unknown but is likely related to DNA synthesis, repair, and/or methylation.
NUTRITION PRACTICE AND POLICY: APPLICATION OF GENETIC AND EPIGENETIC INFORMATION
The incorporation of genomics into nutrition practice and policy offers the potential to “personalize” nutrition and aid in the prevention of chronic disease by targeting the molecular antecedents of disease that are responsive to dietary components (32). Classic monogenic disorders, including phenylketonuria and other inborn errors of metabolism, illustrate the severe consequences that can result from genetic disruptions, but more importantly demonstrate that genetic diseases can be managed and/or alleviated through diet. Likewise, nutrients, like pharmaceuticals, are powerful effectors of genome expression and stability, and gene–nutrient interactions can be optimized for disease prevention.
Folate and Reproduction
Folic Acid Fortification and the Prevention of Birth Defects
Micronutrient food fortifications have been successful approaches in eliminating micronutrient deficiencies and their associated pathologies, including iodine to prevent cretinism (33) and iron to prevent anemia (34). The more recent folic acid fortification policy in the United States was unique from previous fortification initiatives in that it did not seek to remedy a nutrient deficiency in the general population. Rather, it targeted a distinct group, women of childbearing age with genetic susceptibility to bear a child with an NTD, to prevent neural tube effects and other common developmental anomalies (35). Notwithstanding many years of research and changes in public health policy, the mechanism(s) by which alterations in folate status and/or metabolism affect neural tube closure remain poorly understood, although it is assumed that folic acid is acting to overcome metabolic impairments whose etiologies are genetic. In this regard, genetic variants of two genes that encode folate-dependent enzymes, MTHFR 677C>T and MTHFD1 1958G>A, have been identified as risk alleles for folate-responsive developmental anomalies, including NTDs (28). The success of the folic acid fortification policy is marked by the reduction in NTD rates (35,36). Recent concerns have been expressed regarding the potential adverse impact on colon cancer incidence (37) and cognitive functions in the elderly (38).
Folate and the Prevention of Miscarriage
Maternal environments as well as maternal and fetal genotypes that do not support basic processes during embryonic development are usually eliminated by spontaneous abortion (39–42). Humans exhibit high rates of fetal loss (43), with approximately 75% of human conceptions lost spontaneously before term, half of which occur before the first 3 weeks of gestation and generally are unnoticed because many embryos fail to implant (40–42,44). Maternal folate status, impaired folate metabolism as evidenced by elevated plasma homocysteine, and SNPs in folate-related genes that are associated with risk for birth defects have also been associated with risk for miscarriage (45–51). In a recent study, increased maternal folate status also increased the likelihood of twin births after in vitro fertilization (52). The shared risk for both birth defects and spontaneous abortion conferred by MTHFR polymorphic alleles, and evidence that folic acid reduces risk for neural tube defects, has led to the suggestion that women with recurrent miscarriage might benefit from folate and/or vitamin B-12 supplementation (47). The notion that maternal nutritional status can rescue embryos that carry genomes that increase risk for miscarriage, or as stated, “good diet hides genetic mutations” (53), has been demonstrated by numerous examples of nutritional rescue of gene disruptions in mice (53–56). Research is needed to establish the mechanisms and levels of maternal nutrient intakes that prevent spontaneous miscarriage, and to determine whether “rescued” fetal genomes confer dependencies on elevated intakes of these nutrients for the fetus that persist into adulthood.
Very Early Nutrition; Assisted Reproduction
Children conceived by assisted reproductive techniques may be at higher risk for genetic imprinting disorders (57). In some but not all studies, elevated rates of spontaneous abortion have been associated with human in vitro fertilization pregnancies compared with natural conceptions (39). There is increasing evidence that technical procedures, including early harvest and manipulations of eggs, the use of spermatogenesis, impaired gametes, and/or exposure of embryos to culture medium may result in the development of embryos with altered methylation pathways and/or reprogramming of genomic imprints and physiological pathways (39,57). Imprinting disorders and in vitro fertilization are also associated with human inter uterine growth retardation, which increases risk for late-onset chronic disease (58,59). To date, studies of assisted reproduction in humans have not conclusively established a connection between assisted reproduction and subtle effects on genome imprinting. However, studies in animal models have shown that embryo culture medium formulation affects the expression and methylation status of imprinted genes in mammalian embryos, resulting in imprinting-related disorders, including large offspring syndrome (59–63). These and other studies suggest that even the earliest nutritional exposures may affect genomic reprogramming processes, indicating the need for long-term and large-scale monitoring of children conceived through assisted reproduction and their risk for late-onset metabolic diseases.
Maternal Nutrition and Genome Programming
Human epidemiological studies as well as an increasing number of animal models demonstrate that maternal nutrition can “program” gene expression patterns in the embryo that persist into adulthood and contribute to metabolic disease (58). The most celebrated animal model that demonstrates this phenomenon is the yellow agouti (Avy) mouse (64). The agouti gene influences coat color as well as the propensity to become obese. In this model, dietary folate- and choline-mediated alterations in genome methylation can be set irreversibly or “imprinted” during early development such that maternal diet determines the density of cytosine methylation at the agouti locus and hence the level of agouti gene transcription, coat color, and propensity for obesity (65). The methylation patterns and subsequent effects on coat color and, presumably, associated metabolic characteristics are maintained throughout the lifetime of experimental animals (64,65).
Dietary Recommendations
Dietary Recommendations for Populations
Nutrient-based dietary guidelines help individuals and populations achieve adequate dietary patterns to maintain health. These numeric values are quantitatively derived when possible and are based on a nutrient intake level that prevents a clinical and/or biochemical outcome that signifies a particular nutrient deficiency (66,67). Age, sex, life stage, and other variables contribute to the variation in nutrient requirements among individuals within a population; often requirements are derived separately for these population subgroups. Although genetic variation confers tolerance/intolerance for certain foods (8,68), the genetic contribution to nutrient requirements within and among human populations remains to be evaluated rigorously.
Recommended Dietary Allowance (RDA) and Tolerable Upper Intake Level
The RDA is the level of dietary intake that is sufficient to meet the requirement of 97% of healthy individuals in a particular life stage and sex group (Figure 4). Although homozygosity for the MTHFR 677C>T genotype is associated with higher folate requirements, the folate RDA of 400 g dietary folate equivalent/day achieves/maintains normal folate status in premenopausal women differing in MTHFR 677C>T genotype (69). However, a higher level of folate intake is required by men with the MTHFR 677TT genotype (70). The Tolerable Upper Intake Level (UL) is the highest level of nutrient intake that can be achieved without incurring risk of adverse health effects within the general population. To date, no common allelic variant has been identified and shown to warrant genotype-specific numeric standards for ULs.
Figure 4.
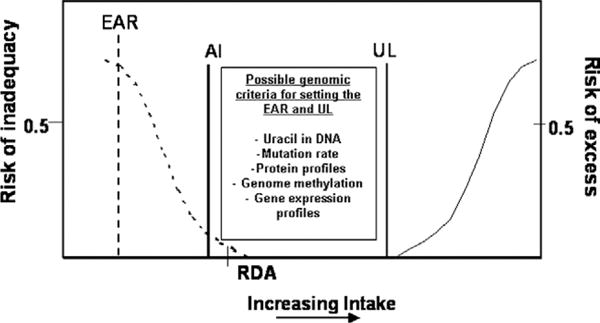
Possible genomic criteria for establishing dietary requirements. Estimated Average Requirement (EAR) represents the intake at which the risk of inadequacy is 0.5 (50%) to an individual in a population. Recommended Dietary Allowance (RDA) represents a level of nutrient intake at which the risk of inadequacy is less than 3%. Adequate Intake (AI) is not based on an EAR but is assumed to be near or above the RDA if one could be calculated. The Tolerable Upper Intake Level (UL) represents risk of excess that is set close to 0. NOTE: This figure is available online at www.adajournal.org as part of a PowerPoint presentation.
A priori, genetic variation is not expected to confer extreme variation to nutrient requirements among individuals and populations because genomes that are not compatible with the in utero nutrient environment are eliminated by spontaneous abortion. Genetic variation that confers more subtle differences in nutrient requirements or food tolerances may be enriched in subgroups or populations and contribute to metabolic disease in certain environmental contexts. Much genetic variation remains to be characterized, especially in geographically and culturally isolated populations that have existed in unique or challenging nutritional environments for many generations where adaptive alleles may have expanded. Certain populations have been shown to be particularly vulnerable to adverse health consequences when dietary patterns change rapidly, especially energy intake (71).
In general, genotype-specific recommendations may only be required if the level of nutrient intake that represents minimal nutrient adequacy for one genetic subgroup exceeds the UL for another group, assuming that avoidance of nutrient deficiency and harm/toxicity are the primary criteria for setting numeric values. For example, although it is generally appreciated that optimal folate intakes differ among identifiable genetic subgroups, the magnitude of the genetic contribution to overall population variation may not warrant genotype-specific recommendations, especially considering that folic acid intakes up to 1 mg/day are not associated with known toxicities, although more study is needed.
Iron may also warrant consideration of genotype-specific recommendations (34,72–74). Although more than 90% of patients with hemochromatosis are homozygous for the HFE C282Y polymorphism, identification of such homozygotes in the general population has revealed that phenotypic penetrance may be as little as 1%. Low penetrance may reflect differences in lifestyle, including dietary intake, which provides another example of a nutrition-by-genomics interaction. However, low penetrance may also be the result of genetic and biochemical redundancy that prevents penetrance of a mutation unless there are multiple functional mutations carried by the same person. Metabolic and biochemical pathways that are genetically and physiologically redundant are less likely to impact dietary requirements. For these and other examples, it will be important to evaluate rigorously both the penetrance of the gene variant (contribution of the individual allele to variation in nutrient requirements) and the prevalence of the gene variant within the population.
Genomic Criteria for Setting Requirements and Toxicities
Genomic technologies that quantify the molecular antecedents of disease promise to provide new criteria for establishing numeric standards for nutrient requirements and toxicities (Figure 4). For example, somatic cell DNA mutation rates or methylation state can be quantified in controlled experimental settings and may predict the risk for late-onset degenerative diseases and cancers that may be modifiable by nutrient intakes (75). Intake levels of nutrients required for DNA synthesis, including folate, vitamin B-12, niacin, and zinc, may influence uracil content in DNA and other indicators of genome stability in humans (76). Antioxidants, including carotenoids, vitamin C, and vitamin E, may prevent DNA damage due to oxidative stress. A recent randomized, double-blind, placebo-controlled folic acid intervention indicated that healthy men and women who consumed a supplement containing 1.2 mg/day folic acid exhibited significantly lower uracil content in lymphocyte DNA compared with the placebo group (76). Further validation of such protective effects on genomic outcomes in controlled human trials may indicate benefit in increasing the recommended intake levels, potentially to levels that are not normally achievable from a natural food-based diet. Likewise, the use of other genomic technologies, including expression profiling and proteomics, may provide a comprehensive set of quantitative and physiologically relevant biomarkers to assess the relationships among nutrient intakes and “metabolic health” (77).
Personalized Nutrition
The prescription of nutrient intakes to individual genetic background, including the use of single nutrients or nutrient restriction to “engineer” physiological responses for health benefit or to manage chronic disease, is a hallmark of nutritional genomics. High-dose vitamin therapy has been advocated to compensate for impaired metabolism resulting from gene mutations and SNPs that impair enzyme function (78). Vitamin E and n-3 fatty acid supplements may promote healthy aging and potentially longevity by modulating the inflammatory response by altering gene transcription (79). Achieving full benefit of such approaches may require intervention during embryogenesis. For example, choline (80) or docosahexaenoic acid (81) supplementation during prenatal and/or early postnatal period may improve central nervous system function and cognitive performance throughout adulthood. Continued progress in these areas requires a greater understanding of genome-nutrient interactions that achieve the desired outcome and the effectiveness, the associated critical windows, and validation of the safety of the nutritional interventions.
However, caution is warranted when dietary approaches aim to optimize functional abilities rather than achieve the more traditional goal of preventing nutrient deficiencies. Human physiology is not adapted to nutrient intake exposures that exceed what can or has been achieved in historical and healthful food-based diets. Therefore, new risks and toxicities should be anticipated in population subgroups when nutrients are administered at pharmacological levels, as illustrated by the unintended consequences of increased consumption of naturally occurring food substances when they are used as food additives (eg, fructose) (82). Rigorous hazard identification is essential (83).
CONCLUSIONS
The variation in the human genome that emerged as a consequence of adaptation to local food environments is a present-day determinant of food and nutrient tolerances/intolerances, risk for metabolic disease, and human nutritional requirements. The more recent and unprecedented ability to manipulate the food supply and optimize dietary practices offers the opportunity to manage environmental exposures and potentially tailor them to individual genetics.
As the field of nutritional genomics matures and the potential benefits of customizing nutritional prescriptions based on genetics are demonstrated, registered dietitians will be faced with the opportunity of making genetically driven dietary recommendations aimed at improving human health. This capacity may also present potential dilemmas because genetic subgroups may respond differently to nutrient exposures, with some groups benefiting while others accrue risk. Registered dietitians can best prepare to meet these challenges by pursuing graduate degrees with genetic and/or molecular biology components, attending conferences/seminars featuring presentations on nutritional genomics, and/or engaging themselves in the growing body of literature aimed at elucidating relationships between diet and genes. In doing so, registered dietitians will be uniquely equipped with the knowledge and skills essential for the management and possible exploitation of genome-nutrient interactions aimed at improving human health through personalized nutrition. Equally important, registered dietitians will need to increase the public’s awareness of the false and exaggerated product claims that threaten to undermine the acceptance and credibility of approaches based on genomic information. Lastly, major advancements in the social behavioral sciences are essential to apply knowledge gained from nutritional genomics.
The promise of nutritional engineering for optimal health through diet remains a research endeavor and a public expectation, as evidenced by the common use of dietary supplements. Clearly, addressing current gaps in knowledge is essential for assuring safety as we proceed in engineering genome function for optimal health. Among the immediate priorities are the need to better understand the role of maternal nutrition in programming and reprogramming biological pathways, the associated mechanisms, and long-term health consequences for the embryo. Similarly, identifying and classifying human genetic variation that impacts nutrient utilization and physiological function will be critical for identifying genetic subgroups for whom generalized nutrient requirements do not apply. In the meantime, the Institutes of Medicine’s Dietary Reference Intakes (84) represent the best translation of current nutrition knowledge into practice.
References
- 1.Garza C, Stover PJ. The role of science in identifying common ground in the GMO debate. Trend Food Tech. 2003;14:182–190. [Google Scholar]
- 2.IOM. Nutrigenomics and beyond: Informing the future. Washington, DC: The National Academies Press; 2007. [Google Scholar]
- 3.Stover PJ. Human nutrition and genetic variation. Food Nutr Bull. 2007;28(Suppl International 1):S101–S115. doi: 10.1177/15648265070281S109. [DOI] [PubMed] [Google Scholar]
- 4.Tishkoff SA, Verrelli BC. Role of evolutionary history on haplotype block structure in the human genome: Implications for disease mapping. Curr Opin Genet Dev. 2003;13:569–575. doi: 10.1016/j.gde.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 5.Cargill M, Altshuler D, Ireland J, Sklar P, Ardlie K, Patil N, Shaw N, Lane CR, Lim EP, Kalyanaraman N, Nemesh J, Ziaugra L, Friedland L, Rolfe A, Warrington J, Lipshutz R, Daley GQ, Lander ES. Characterization of single-nucleotide polymorphisms in coding regions of human genes. Nat Genet. 1999;22:231–238. doi: 10.1038/10290. [DOI] [PubMed] [Google Scholar]
- 6.Tishkoff SA, Kidd KK. Implications of biogeography of human populations for ’race’ and medicine. Nat Genet. 2004;36(Suppl 11):S21–S27. doi: 10.1038/ng1438. [DOI] [PubMed] [Google Scholar]
- 7.Enattah NS, Sahi T, Savilahti E, Terwilliger JD, Peltonen L, Jarvela I. Identification of a variant associated with adult-type hypolactasia. Nat Genet. 2002;30:233–237. doi: 10.1038/ng826. [DOI] [PubMed] [Google Scholar]
- 8.Bersaglieri T, Sabeti PC, Patterson N, Vanderploeg T, Schaffner SF, Drake JA, Rhodes M, Reich DE, Hirschhorn JN. Genetic signatures of strong recent positive selection at the lactase gene. Am J Hum Genet. 2004;74:1111–1120. doi: 10.1086/421051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bloom G, Sherman PW. Dairying barriers and the distribution of lactose malabsorption. Evolution and Human Behavior. 2005;26:301–312. [Google Scholar]
- 10.Toomajian C, Ajioka RS, Jorde LB, Kushner JP, Kreitman M. A method for detecting recent selection in the human genome from allele age estimates. Genetics. 2003;165:287–297. doi: 10.1093/genetics/165.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toomajian C, Kreitman M. Sequence variation and haplotype structure at the human HFE locus. Genetics. 2002;161:1609–1623. doi: 10.1093/genetics/161.4.1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stover PJ, Garza C. Bringing individuality to public health recommendations. J Nutr. 2002;132(8 Suppl):S2476–S2480. doi: 10.1093/jn/132.8.2476S. [DOI] [PubMed] [Google Scholar]
- 13.Dennis C. Epigenetics and disease: Altered states. Nature. 2003;421:686–688. doi: 10.1038/421686a. [DOI] [PubMed] [Google Scholar]
- 14.Waterland RA, Garza C. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr. 1999;69:179–197. doi: 10.1093/ajcn/69.2.179. [DOI] [PubMed] [Google Scholar]
- 15.Barker DJ. Intrauterine programming of coronary heart disease and stroke. Acta Paediatr Suppl. 1997;423:178–182. doi: 10.1111/j.1651-2227.1997.tb18408.x. discussion 183. [DOI] [PubMed] [Google Scholar]
- 16.Stover PJ. Physiology of folate and vitamin B12 in health and disease. Nutr Rev. 2004;62(6 Pt 2):S3–S12. doi: 10.1111/j.1753-4887.2004.tb00070.x. discussion S13. [DOI] [PubMed] [Google Scholar]
- 17.Henikoff S, McKittrick E, Ahmad K. Epigenetics, histone H3 variants, and the inheritance of chromatin states. Cold Spring Harb Symp Quant Biol. 2004;69:235–243. doi: 10.1101/sqb.2004.69.235. [DOI] [PubMed] [Google Scholar]
- 18.Blount BC, Mack MM, Wehr CM, MacGregor JT, Hiatt RA, Wang G, Wickramasinghe SN, Everson RB, Ames BN. Folate deficiency causes uracil misincorporation into human DNA and chromosome breakage: Implications for cancer and neuronal damage. Proc Natl Acad Sci U S A. 1997;94:3290–3295. doi: 10.1073/pnas.94.7.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finkelstein JD. Homocysteine: A history in progress. Nutr Rev. 2000;58:193–204. doi: 10.1111/j.1753-4887.2000.tb01862.x. [DOI] [PubMed] [Google Scholar]
- 20.Finkelstein JD. Pathways and regulation of homocysteine metabolism in mammals. Semin Thromb Hemost. 2000;26:219–225. doi: 10.1055/s-2000-8466. [DOI] [PubMed] [Google Scholar]
- 21.Clarke S, Banfield K. S-adenosylmethionine-dependent methyltransferases. In: Carmel R, Jacobson DW, editors. Homocysteine in Health and Disease. Cambridge: Cambridge Press; 2001. pp. 63–78. [Google Scholar]
- 22.Kimmins S, Sassone-Corsi P. Chromatin remodelling and epigenetic features of germ cells. Nature. 2005;434:583–589. doi: 10.1038/nature03368. [DOI] [PubMed] [Google Scholar]
- 23.Zingg JM, Jones PA. Genetic and epigenetic aspects of DNA methylation on genome expression, evolution, mutation and carcinogenesis. Carcinogenesis. 1997;18:869–882. doi: 10.1093/carcin/18.5.869. [DOI] [PubMed] [Google Scholar]
- 24.Song J, Sohn KJ, Medline A, Ash C, Gallinger S, Kim YI. Chemopreventive effects of dietary folate on intestinal polyps in Apc+/−Msh2−/−mice. Cancer Res. 2000;60:3191–3199. [PubMed] [Google Scholar]
- 25.Ingrosso D, Cimmino A, Perna AF, Masella L, De Santo NG, De Bonis ML, Vacca M, D’Esposito M, D’Urso M, Galletti P, Zappia V. Folate treatment and unbalanced methylation and changes of allelic expression induced by hyperhomocysteinaemia in patients with uraemia. Lancet. 2003;361:1693–1699. doi: 10.1016/S0140-6736(03)13372-7. [DOI] [PubMed] [Google Scholar]
- 26.De Marco P, Moroni A, Merello E, de Franchis R, Andreussi L, Finnell RH, Barber RC, Cama A, Capra V. Folate pathway gene alterations in patients with neural tube defects. Am J Med Genet. 2000;95:216–223. doi: 10.1002/1096-8628(20001127)95:3<216::aid-ajmg6>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 27.Christensen B, Arbour L, Tran P, Leclerc D, Sabbaghian N, Platt R, Gilfix BM, Rosenblatt DS, Gravel RA, Forbes P, Rozen R. Genetic polymorphisms in methylenetetrahydrofolate reductase and methionine synthase, folate levels in red blood cells, and risk of neural tube defects. Am J Med Genet. 1999;84:151–157. doi: 10.1002/(sici)1096-8628(19990521)84:2<151::aid-ajmg12>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 28.Brody LC, Conley M, Cox C, Kirke PN, McKeever MP, Mills JL, Molloy AM, O’Leary VB, Parle-McDermott A, Scott JM, Swanson DA. A polymorphism, R653Q, in the trifunctional enzyme methylenetetrahydrofolate dehydrogenase/methenyltetrahydrofolate cyclohydrolase/formyltetrahydrofolate synthetase is a maternal genetic risk factor for neural tube defects: Report of the birth defects research group. Am J Hum Genet. 2002;71:1207–1215. doi: 10.1086/344213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ma J, Stampfer MJ, Giovannucci E, Artigas C, Hunter DJ, Fuchs C, Willett WC, Selhub J, Hennekens CH, Rozen R. Methylenetetrahydrofolate reductase polymorphism, dietary interactions, and risk of colorectal cancer. Cancer Res. 1997;57:1098–1102. [PubMed] [Google Scholar]
- 30.Esfahani ST, Cogger EA, Caudill MA. Heterogeneity in the prevalence of methylenetetrahydrofolate reductase gene polymorphisms in women of different ethnic groups. J Am Diet Assoc. 2003;103:200–207. doi: 10.1053/jada.2003.50030. [DOI] [PubMed] [Google Scholar]
- 31.Tishkoff SA, Williams SM. Genetic analysis of African populations: Human evolution and complex disease. Nat Rev Genet. 2002;3:611–621. doi: 10.1038/nrg865. [DOI] [PubMed] [Google Scholar]
- 32.Stover PJ. Nutritional genomics. Physiol Genomics. 2004;16:161–165. doi: 10.1152/physiolgenomics.00204.2003. [DOI] [PubMed] [Google Scholar]
- 33.Delange FM. Control of iodine deficiency in Western and Central Europe. Cent Eur J Public Health. 2003;11:120–123. [PubMed] [Google Scholar]
- 34.Swanson CA. Iron intake and regulation: Implications for iron deficiency and iron overload. Alcohol. 2003;30:99–102. doi: 10.1016/s0741-8329(03)00103-4. [DOI] [PubMed] [Google Scholar]
- 35.Honein MA, Paulozzi LJ, Mathews TJ, Erickson JD, Wong LY. Impact of folic acid fortification of the US food supply on the occurrence of neural tube defects. JAMA. 2001;285:2981–2986. doi: 10.1001/jama.285.23.2981. [DOI] [PubMed] [Google Scholar]
- 36.De Wals P, Tairou F, Van Allen MI, Uh SH, Lowry RB, Sibbald B, Evans JA, Van den Hof MC, Zimmer P, Crowley M, Fernandez B, Lee NS, Niyonsenga T. Reduction in neural-tube defects after folic acid fortification in Canada. N Engl J Med. 2007;357:135–142. doi: 10.1056/NEJMoa067103. [DOI] [PubMed] [Google Scholar]
- 37.Cole BF, Baron JA, Sandler RS, Haile RW, Ahnen DJ, Bresalier RS, McKeown-Eyssen G, Summers RW, Rothstein RI, Burke CA, Snover DC, Church TR, Allen JI, Robertson DJ, Beck GJ, Bond JH, Byers T, Mandel JS, Mott LA, Pearson LH, Barry EL, Rees JR, Marcon N, Saibil F, Ueland PM, Greenberg ER. Folic acid for the prevention of colorectal adenomas: A randomized clinical trial. JAMA. 2007;297:2351–2359. doi: 10.1001/jama.297.21.2351. [DOI] [PubMed] [Google Scholar]
- 38.Morris MS, Jacques PF, Rosenberg IH, Selhub J. Folate and vitamin B-12 status in relation to anemia, macrocytosis, and cognitive impairment in older Americans in the age of folic acid fortification. Am J Clin Nutr. 2007;85:193–200. doi: 10.1093/ajcn/85.1.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bulletti C, Flamigni C, Giacomucci E. Reproductive failure due to spontaneous abortion and recurrent miscarriage. Hum Reprod Update. 1996;2:118–136. doi: 10.1093/humupd/2.2.118. [DOI] [PubMed] [Google Scholar]
- 40.Brent RL, Beckman DA. The contribution of environmental teratogens to embryonic and fetal loss. Clin Obstet Gynecol. 1994;37:646–670. doi: 10.1097/00003081-199409000-00018. [DOI] [PubMed] [Google Scholar]
- 41.Edmonds DK, Lindsay KS, Miller JF, Williamson E, Wood PJ. Early embryonic mortality in women. Fertil Steril. 1982;38:447–453. [PubMed] [Google Scholar]
- 42.Edwards RG. Recent scientific and medical advances in assisted human conception. Int J Dev Biol. 1997;41:255–262. [PubMed] [Google Scholar]
- 43.Delhanty JD. Preimplantation genetics: An explanation for poor human fertility? Ann Hum Genet. 2001;65:331–338. doi: 10.1017/S0003480001008739. [DOI] [PubMed] [Google Scholar]
- 44.Wilcox AJ, Weinberg CR, O’Connor JF, Baird DD, Schlatterer JP, Canfield RE, Armstrong EG, Nisula BC. Incidence of early loss of pregnancy. N Engl J Med. 1988;319:189–194. doi: 10.1056/NEJM198807283190401. [DOI] [PubMed] [Google Scholar]
- 45.Zetterberg H. Methylenetetrahydrofolate reductase and transcobalamin genetic polymorphisms in human spontaneous abortion: biological and clinical implications. Reprod Biol Endocrinol. 2004;2:7. doi: 10.1186/1477-7827-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zetterberg H, Regland B, Palmer M, Ricksten A, Palmqvist L, Rymo L, Arvanitis DA, Spandidos DA, Blennow K. Increased frequency of combined methylenetetrahydrofolate reductase C677T and A1298C mutated alleles in spontaneously aborted embryos. Eur J Hum Genet. 2002;10:113–118. doi: 10.1038/sj.ejhg.5200767. [DOI] [PubMed] [Google Scholar]
- 47.Zetterberg H, Regland B, Palmer M, Rymo L, Zafiropoulos A, Arvanitis DA, Spandidos DA, Blennow K. The transcobalamin codon 259 polymorphism influences the risk of human spontaneous abortion. Hum Reprod. 2002;17:3033–3036. doi: 10.1093/humrep/17.12.3033. [DOI] [PubMed] [Google Scholar]
- 48.Zetterberg H, Zafiropoulos A, Spandidos DA, Rymo L, Blennow K. Gene-gene interaction between fetal MTHFR 677C>T and transcobalamin 776C>G polymorphisms in human spontaneous abortion. Hum Reprod. 2003;18:1948–1950. doi: 10.1093/humrep/deg375. [DOI] [PubMed] [Google Scholar]
- 49.Nelen WL, Blom HJ, Steegers EA, den Heijer M, Eskes TK. Hyperhomocysteinemia and recurrent early pregnancy loss: A meta-analysis. Fertil Steril. 2000;74:1196–1199. doi: 10.1016/s0015-0282(00)01595-8. [DOI] [PubMed] [Google Scholar]
- 50.Nelen WL. Hyperhomocysteinaemia and human reproduction. Clin Chem Lab Med. 2001;39:758–763. doi: 10.1515/CCLM.2001.126. [DOI] [PubMed] [Google Scholar]
- 51.Isotalo PA, Wells GA, Donnelly JG. Neonatal and fetal methylenetetrahydrofolate reductase genetic polymorphisms: an examination of C677T and A1298C mutations. Am J Hum Genet. 2000;67:986–990. doi: 10.1086/303082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haggarty P, McCallum H, McBain H, Andrews K, Duthie S, McNeill G, Templeton A, Haites N, Campbell D, Bhattacharya S. Effect of B vitamins and genetics on success of in-vitro fertilisation: prospective cohort study. Lancet. 2006;367:1513–1519. doi: 10.1016/S0140-6736(06)68651-0. [DOI] [PubMed] [Google Scholar]
- 53.Pennisi E. Evolution of developmental diversity. Evodevo devotees eye ocular origins and more. Science. 2002;296:1010–1011. doi: 10.1126/science.296.5570.1010. [DOI] [PubMed] [Google Scholar]
- 54.Pasqualetti M, Neun R, Davenne M, Rijli FM. Retinoic acid rescues inner ear defects in Hoxa1 deficient mice. Nat Genet. 2001;29:34–39. doi: 10.1038/ng702. [DOI] [PubMed] [Google Scholar]
- 55.Zhao R, Russell RG, Wang Y, Liu L, Gao F, Kneitz B, Edelmann W, Goldman ID. Rescue of embryonic lethality in reduced folate carrier-deficient mice by maternal folic acid supplementation reveals early neonatal failure of hematopoietic organs. J Biol Chem. 2001;276:10224–10228. doi: 10.1074/jbc.c000905200. [DOI] [PubMed] [Google Scholar]
- 56.Eudy JD, Spiegelstein O, Barber RC, Wlodarczyk BJ, Talbot J, Finnell RH. Identification and characterization of the human and mouse SLC19A3 gene: A novel member of the reduced folate family of micronutrient transporter genes. Mol Genet Metab. 2000;71:581–590. doi: 10.1006/mgme.2000.3112. [DOI] [PubMed] [Google Scholar]
- 57.Paoloni-Giacobino A. Epigenetics in reproductive medicine. Pediatr Res. 2007;61(5 Pt 2):R51–R57. doi: 10.1203/pdr.0b013e318039d978. [DOI] [PubMed] [Google Scholar]
- 58.McMillen IC, Robinson JS. Developmental origins of the metabolic syndrome: Prediction, plasticity, and programming. Physiol Rev. 2005;85:571–633. doi: 10.1152/physrev.00053.2003. [DOI] [PubMed] [Google Scholar]
- 59.De Rycke M, Liebaers I, Van Steirteghem A. Epigenetic risks related to assisted reproductive technologies: Risk analysis and epigenetic inheritance. Hum Reprod. 2002;17:2487–2494. doi: 10.1093/humrep/17.10.2487. [DOI] [PubMed] [Google Scholar]
- 60.Sinclair KD, Dunne LD, Maxfield EK, Maltin CA, Young LE, Wilmut I, Robinson JJ, Broadbent PJ. Fetal growth and development following temporary exposure of day 3 ovine embryos to an advanced uterine environment. Reprod Fertil Dev. 1998;10:263–269. doi: 10.1071/r98021. [DOI] [PubMed] [Google Scholar]
- 61.Khosla S, Dean W, Reik W, Feil R. Culture of preimplantation embryos and its long-term effects on gene expression and phenotype. Hum Reprod Update. 2001;7:419–427. doi: 10.1093/humupd/7.4.419. [DOI] [PubMed] [Google Scholar]
- 62.Young LE, Fernandes K, McEvoy TG, Butterwith SC, Gutierrez CG, Carolan C, Broadbent PJ, Robinson JJ, Wilmut I, Sinclair KD. Epigenetic change in IGF2R is associated with fetal overgrowth after sheep embryo culture. Nat Genet. 2001;27:153–154. doi: 10.1038/84769. [DOI] [PubMed] [Google Scholar]
- 63.Gao S, Latham KE. Maternal and environmental factors in early cloned embryo development. Cytogenet Genome Res. 2004;105:279–284. doi: 10.1159/000078199. [DOI] [PubMed] [Google Scholar]
- 64.Waterland RA, Jirtle RL. Early nutrition, epigenetic changes at transposons and imprinted genes, and enhanced susceptibility to adult chronic diseases. Nutrition. 2004;20:63–68. doi: 10.1016/j.nut.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 65.Morgan HD, Sutherland HG, Martin DI, Whitelaw E. Epigenetic inheritance at the agouti locus in the mouse. Nat Genet. 1999;23:314–318. doi: 10.1038/15490. [DOI] [PubMed] [Google Scholar]
- 66.Schneeman BO, Mendelson R. Dietary guidelines: Past experience and new approaches. J Am Diet Assoc. 2002;102:1498–1500. doi: 10.1016/s0002-8223(02)90332-9. [DOI] [PubMed] [Google Scholar]
- 67.Schneeman BO. Evolution of dietary guidelines. J Am Diet Assoc. 2003;103(suppl 2):S5–S9. doi: 10.1016/j.jada.2003.09.030. [DOI] [PubMed] [Google Scholar]
- 68.Poulter M, Hollox E, Harvey CB, Mulcare C, Peuhkuri K, Kajander K, Sarner M, Korpela R, Swallow DM. The causal element for the lactase persistence/non-persistence polymorphism is located in a 1 Mb region of linkage disequilibrium in Europeans. Ann Hum Genet. 2003;67:298–311. doi: 10.1046/j.1469-1809.2003.00048.x. [DOI] [PubMed] [Google Scholar]
- 69.Guinotte CL, Burns MG, Axume JA, Hata H, Urrutia TF, Alamilla A, McCabe D, Singgih A, Cogger EA, Caudill MA. Methylenetetrahydro-folate reductase 677C–>T variant modulates folate status response to controlled folate intakes in young women. J Nutr. 2003;133:1272–1280. doi: 10.1093/jn/133.5.1272. [DOI] [PubMed] [Google Scholar]
- 70.Solis CS, Veenema K, Ivanov AA, Tran S, Li R, Wang W, Moriarty DJ, Maletz CV, Caudill MA. Folate intake at RDA levels is inadequate for Mexican American men with the methylenetetrahydrofolate reductase 677TT genotype. J Nutr. 2008;138:1–6. doi: 10.1093/jn/138.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Baier LJ, Hanson RL. Genetic studies of the etiology of type 2 diabetes in Pima Indians: Hunting for pieces to a complicated puzzle. Diabetes. 2004;53:1181–1186. doi: 10.2337/diabetes.53.5.1181. [DOI] [PubMed] [Google Scholar]
- 72.Moirand R, Guyader D, Mendler MH, Jouanolle AM, Le Gall JY, David V, Brissot P, Deugnier Y. HFE based re-evaluation of heterozygous hemochromatosis. Am J Med Genet. 2002;111:356–361. doi: 10.1002/ajmg.10547. [DOI] [PubMed] [Google Scholar]
- 73.Beutler E. Iron absorption in carriers of the C282Y hemochromatosis mutation. Am J Clin Nutr. 2004;80:799–800. doi: 10.1093/ajcn/80.4.799. [DOI] [PubMed] [Google Scholar]
- 74.Hunt JR, Zeng H. Iron absorption by heterozygous carriers of the HFE C282Y mutation associated with hemochromatosis. Am J Clin Nutr. 2004;80:924–931. doi: 10.1093/ajcn/80.4.924. [DOI] [PubMed] [Google Scholar]
- 75.Fenech M. Genome health nutrigenomics: Nutrition and the science of optimal genome maintenance. Asia Pac J Clin Nutr. 2004;13:S15. [Google Scholar]
- 76.Basten GP, Duthie SJ, Pirie L, Vaughan N, Hill MH, Powers HJ. Sensitivity of markers of DNA stability and DNA repair activity to folate supplementation in healthy volunteers. Br J Cancer. 2006;94:1942–1947. doi: 10.1038/sj.bjc.6603197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Csete M, Doyle J. Bow ties, metabolism and disease. Trends Biotechnol. 2004;22:446–450. doi: 10.1016/j.tibtech.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 78.Ames BN, Elson-Schwab I, Silver EA. High-dose vitamin therapy stimulates variant enzymes with decreased coenzyme binding affinity (increased K(m)): Relevance to genetic disease and polymorphisms. Am J Clin Nutr. 2002;75:616–658. doi: 10.1093/ajcn/75.4.616. [DOI] [PubMed] [Google Scholar]
- 79.Kornman KS, Martha PM, Duff GW. Genetic variations and inflammation: A practical nutrigenomics opportunity. Nutrition. 2004;20:44–49. doi: 10.1016/j.nut.2003.09.008. [DOI] [PubMed] [Google Scholar]
- 80.Mellott TJ, Williams CL, Meck WH, Blusztajn JK. Prenatal choline supplementation advances hippocampal development and enhances MAPK and CREB activation. Faseb J. 2004;18:545–547. doi: 10.1096/fj.03-0877fje. [DOI] [PubMed] [Google Scholar]
- 81.Heird WC, Lapillonne A. The role of essential fatty acids in development. Annu Rev Nutr. 2004 doi: 10.1146/annurev.nutr.24.012003.132254. [DOI] [PubMed] [Google Scholar]
- 82.Cox TM. The genetic consequences of our sweet tooth. Nat Rev Genet. 2002;3:481–487. doi: 10.1038/nrg815. [DOI] [PubMed] [Google Scholar]
- 83.State-of-the-Science-Panel N. National Institutes of Health State-of-the-Science-Conference Statement: Multivitamin/Mineral Supplements and Chronic Disease Prevention. Annals of Internal Medicine. 2006;145:364–371. doi: 10.7326/0003-4819-145-5-200609050-00136. [DOI] [PubMed] [Google Scholar]
- 84.Otten JJ, Pitzi Hellwig J, Meyers LD, editors. IOM. Dietary reference intakes: The essential guide to nutrient requirements. The National Academies Press; 2006. [Google Scholar]