

Abstract
Class I hyaluronan synthases (HAS) assemble [GlcNAc(β1,4)GlcUA(β1,3)]n-UDP at the reducing end and also make chitin. Streptococcus equisimilis HAS (SeHAS) also synthesizes chitin-UDP oligosaccharides, (GlcNAc-β1,4)n-GlcNAc(α1→)UDP (Weigel et al. 2015). Here we determined if HAS uses chitin-UDPs as primers to initiate HA synthesis, leaving the non-HA primer at the nonreducing (NR) end. HA made by SeHAS membranes was purified, digested with streptomyces lyase, and hydrophobic oligomers were enriched by solid phase extraction and analyzed by MALDI-TOF MS. Jack bean hexosaminidase (JBH) and MS/MS were used to analyze 19 m/z species of possible GnHn ions with clustered GlcNAc (G) residues attached to disaccharide units (H): (GlcNAcβ1,4)2–5[GlcUA(β1,3)GlcNAc]2–6. JBH digestion sequentially removed GlcNAc from the NR-end of GnHn oligomers, producing successively smaller GnH2–3 series members. Since lyase releases dehydro-oligos (dHn; M−18), only the unique NR-end oligo lacks dehydro-GlcUA. Hn oligomers were undetectable in lyase digests, whereas JBH treatment created new H2–6m/z peaks (i.e. HA tetra- through dodeca-oligomers). MS/MS of larger GnHn species produced chitin (2–5 GlcNAcs), HA oligomers and multiple smaller series members with fewer GlcNAcs. All NR-ends (97%) started with GlcNAc, as a chitin trimer (three GlcNAcs), indicating that GlcNAc(β1,4)2GlcNAc(α1→)-UDP may be optimal for initiation of HA synthesis. Also, HA made by live S. pyogenes cells had G4Hn chitin-oligo NR-ends. We conclude that chitin-UDP functions in vitro and in live cells as a primer to initiate synthesis of all HA chains and these primers remain at the NR-ends of HA chains as residual chitin caps [(GlcNAc-β1,4)3–4].
Keywords: chitin oligosaccharide, Class I hyaluronan synthase, polymer synthesis, reducing end elongation, UDP-activated
Introduction
Class I hyaluronan (HA) synthases (HASs) are CAZy GT2 family members (Campbell et al. 1997; Spicer and McDonald 1998; Itano and Kimata 2002; Weigel 2002; Weigel and DeAngelis 2007) that are lipid-dependent integral membrane proteins (Tlapak-Simmons et al. 1999; Weigel et al. 2006; Sakr et al. 2008; Kultti et al. 2010; Ontong et al. 2014). HA synthesis requires no exogenous primer, since purified HAS from streptococcus (Tlapak-Simmons et al. 1999) or mouse (Yoshida et al. 2000) only needs GlcUA-UDP, GlcNAc-UDP and Mg+2 ions to synthesize HA. Class I HASs are novel glycosyltransferases (GTases) because sugar addition is at the reducing end, not the nonreducing end (NR-end), so that UDP-sugars are acceptors that receive the growing HA chain from an HA-UDP donor (Prehm 1983; Asplund et al. 1998; Bodevin-Authelet et al. 2005; Tlapak-Simmons et al. 2005; Prehm 2006; Weigel et al. 2015). Multiple studies confirmed this mechanism for streptococcal, mouse and human HAS, including showing the presence of UDP at the reducing end of growing chains (Prehm 1983; Asplund et al. 1998; Bodevin-Authelet et al. 2005; Tlapak-Simmons et al. 2005; Prehm 2006). If the reducing end UDP is cleaved to generate free UDP and an HA chain, then further elongation cannot proceed and the final HA size becomes fixed. HAS is also unusual in that the enzyme continuously translocates the growing HA-UDP chain across the cell membrane through an intraprotein pore to the exterior (Tlapak-Simmons et al. 1999; Hubbard et al. 2012).
Interestingly, Class I HAS from frog and mouse synthesize (GlcNAc-β1,4)n, chitin oligosaccharides (oligos), in the presence of only GlcNAc-UDP (Semino et al. 1996; Yoshida et al. 2000). We recently found that Streptococcus equisimilis HAS (SeHAS), the smallest member of the Class I HAS family (417 aa), also makes chitin, confirming this function as a general Class I HAS characteristic (Weigel et al. 2015). We also discovered that HAS synthesizes novel products, not previously observed and consistent with its reducing end mechanism of sugar addition: (GlcNAc-β1,4)nGlcNAc(α1→)UDP oligos. Free chitin oligos arise from cleavage of these labile activated-UDP linkages.
Chitin-UDP oligos made by HAS or free chitin derived by hydrolysis might have several functions. Since chitin oligos are signaling regulators in lower vertebrate development (Semino et al. 1996; Van der Holst et al. 2001), they might have similar, as yet unrecognized, functions in mammalian development. If so, these chitin oligos would be made by HAS1-3 rather than chitin synthase, which is not in the human genome. A second important possible function of chitin-UDP oligos made by HAS is that they could serve as self-made primers for HAS to initiate HA synthesis (Weigel et al. 2015). Although addition of an exogenous primer is not required to initiate HA chain assembly, a kinetic lag is often detected before steady-state synthesis begins (Baggenstoss and Weigel 2006), indicating that initiation of HA assembly is the rate-limiting step in overall synthesis.
None of the previous findings about HA biosynthesis exclude the possibility that an endogenous primer, made by HAS itself, is required to start HA synthesis. Consistent with an obligatory role for chitin-UDPs to prime HA synthesis, we detected these products only when HA synthesis could not occur (i.e. the absence of UDP-GlcUA), indicating that the syntheses of HA-UDP and chitin-UDP are not independent; they are dependent and operate in a serial, rather than parallel, way.
Here we tested the hypothesis that chitin-UDP oligos made by HAS serve as primers for initiating HA synthesis. If this is true, then growing and newly released HA-UDP (or free HA) chains would contain this residual chitin primer as a unique and non-HA structure at their NR-ends. Purified HA made by SeHAS was digested with HA lyase and product oligos were analyzed by MALDI-TOF MS. The presence of (GlcNAc-β1,4)n chitin oligos at the NR-end of HA was confirmed by MS/MS analysis of candidate m/z ions corresponding to novel hybrid chitin-HA oligos. Treatment with Jack bean N-acetylhexosaminidase (JBH) decreased these species and produced smaller chitin-HA oligos and nondehydro-HA oligos, which are not eliminase products. The results confirm a proposed 12th function of HAS (Weigel et al. 2015), which is needed to create the first HA disaccharide, GlcNAc(β1,4)GlcUA(β1,3), at the NR-end, attached to the chitin primer.
Results
Development of a protocol for purification of hybrid (GlcNAcn)-HA oligomers
Based on the discovery that HAS assembles UDP-activated chitin oligos and our hypothesis that these novel activated species are self-made primers for starting HA synthesis, we established a protocol to identify the predicted chitin-HA end cap products by enriching for HA oligos, in lyase digests, that are more hydrophobic than normal HA oligos (i.e. >50% GlcNAc). We also limited the size of newly made HA. Since HA size is very sensitive to the HAS:substrate ratio and decreases as this ratio increases (Baggenstoss and Weigel 2006), we used a high HAS-membrane protein concentration and limited the time of synthesis, by quickly heat-inactivating the enzyme (Tlapak-Simmons et al. 2004), to obtain HA product sizes of ~50 kDa. In an extensive HA lyase digest of 4 MDa HA (>21,100 sugars) that yields ~3015 oligo products per HA chain (seven sugars average size), it might be difficult to detect one novel chitin-HA oligo (0.033% of total products). However, starting with 50 kDa HA increases the end-cap oligo frequency 80-fold, making detection easier (about 2.6% of total products).
Freshly made HA was purified to remove lipids, protein and small molecules by organic solvent extraction (Folch et al. 1957), ethanol precipitation and size exclusion chromatography (SEC). The two latter steps removed small molecule contaminants ≤3 kDa, the size range for the oligos of interest. HA was well separated by SEC (Figure 1A) from the many metabolites with significant A280, and the exogenous UDP-sugars used for synthesis. The starting HA (triangles) eluted between fractions #14–22, with A210 peaking at #17–18 and no A280, as expected, whereas both A210 and A280 increased in the included volume starting at fraction #26. The HA from fractions #15–21 was pooled, lyophilized, dissolved and subjected to SEC again (Figure 1A, circles). The HA peak showed strong A210 and no A280, but the large A210 and A280 peaks at fractions #26–35 were absent, confirming that small molecules had been removed from the final purified HA. Sodium was eliminated from buffers during HA purification and lyase digestion in order to decrease the fraction of parent ions present as multiple sodiated species.
Fig. 1.

Purification of SeHAS HA and the strategy to identify HA oligos derived from NR-ends. (A) SEC purification. HA synthesized by SeHAS membranes was collected and purified as described in Methods. The HA was loaded onto a Sephacryl HR 300 SEC column and eluted with 50 mM ammonium acetate 20% ethanol. Fractions (2.4 mL) from this first SEC run (▲, ∆) were lyophilized, dissolved in water, and analyzed for A210 (▲) and A280 (∆). HA-containing fractions were identified near the void-volume by agarose gel electrophoreses (insert above graph) with high A210 values and no A280. Included volume fractions showed both high A280 and A210 values. Fractions 15–20 were pooled, lyophilized and run over the SEC column again (●, ○). The HA was recovered at the same position with strong A210 (●) and no A280 (○), but little or no A210 or A280 in the included region, indicating that the first SEC fractionation successfully removed smaller contaminants. (B) Scheme for lyase digestion of a putative (GlcNAcβ1,4)4-HA chain synthesized by HAS. A portion of the NR-end of a proposed HA chain (G4Hn) is shown with a sequence of four GlcNAc(β1,4) residues (G, blue squares) followed by a stretch of n repeating HA disaccharides (H) with alternating GlcNAc(β1,4) residues and GlcUA(β1,3) residues (blue/white diamonds). Extensive digestion with HA lyase produces HA oligo fragments, which all contain dehydro-GlcUA at the NR-end (asterisks, *). The only fragment in the lyase digest that is different, in lacking a dehydro NR-end, is the original NR-end of the starting HA chain. This is also the only oligo in the lyase digest that is a substrate for JBH, which is specific for hexNAc(β1) residues at the NR-end of a glycan. JBH sequentially removes the four G residues to liberate an H2 oligo (HA tetrasaccharide; boldface font) that was the NR-end of the intact HA. C. The scheme shows the products generated by ovine testicular hyaluronidase during digestion of the (GlcNAcβ1,4)4-HA chain in B. The mechanism of this enzyme, which is a hydrolase, cleaves GlcNAc(β1,4)GlcUA bonds to give A at the NR-end and G at the reducing end; so all released fragments, except the NR-end, are Hn oligos with the repeating sequence A–G-(A–G)n.
An important aspect of the strategy to identify NR-end oligos (Figure 1B) is that the purified intact HA is digested with streptomyces HA lyase to give dehydro-oligos (d-oligos) for all the internal HA oligos in the digest; these contain GlcUA with a double bond at the NR-end and GlcNAc at the reducing end (Shimada and Matsumura 1980). The only lyase digestion fragment derived from each HA chain that does not have a double bond, due to the cleavage mechanism of the HA eliminase, is the oligo derived from, and thus containing, the NR-end. In contrast, ovine testicular hyaluronidase (OTH) gives normal HA oligos with GlcUA at the NR-end and GlcNAc at the reducing (Figure 1C). Lyase d-oligo digests were passed over SepPak (C18) cartridges, washed three times and more tightly bound species were eluted with ACN. Figure 2 shows MS profiles, in negative mode, for the 700–2600 m/z range of a typical starting HA d-oligo digest (Figure 2A) and the Wash 1, Wash 2, Wash 3 and 10% ACN fractions (Figure 2B–E). Solid phase extraction removed the majority of expected HA d-oligos (with equal G and A content and identical charge:mass ratios) by the failure of these species to bind strongly. The predominant species in the starting digest (Figure 2A) were d-oligos (Table I) representing tetra-, hexa-, octa- deca- and dodeca-oligos (dH2, dH3, dH4, dH5, and dH6, respectively; where Hn is the number of HA disaccharides). The major digest product detected was dH3, the hexamer, followed by dH2 and dH4 with comparable signals. The largest digestion product detected was dH7 (a tetra-deca oligo). The dHn oligos were also major species in Wash 1 (Figure 2B) and Wash 2 and dH3 could also be detected in the Wash 3 and ACN fractions (Figure 2D and E).
Fig. 2.

Identification of candidate chitin-HA oligos in fractionated lyase digests of SeHAS HA. HA was synthesized, purified, treated with HA lyase and the digest was then fractionated by solid phase extraction over Sep-Pak C-18 cartridges as described in Methods. The starting digest (A) and the Wash 1 (B), Wash 2 (C), Wash 3 (D) and ACN (E) fractions were examined by MALDI-TOF MS in negative mode. Labels indicate m/z peaks corresponding to M−1 ions for many of the candidate dHn and GnHn species listed in Tables I and II. Panel E is at a different scale than panels (A–D). Note that signals for some minor species such as G5H2,3 (at m/z 1790 and 2169) are not easily discerned at the scale used to show the more abundant species.
Table I.
Monoisotopic m/z values of normal and dehydro-HA oligos. Observed species values are the mean for the number of times each was observed (in parentheses) in untreated lyase digests or after treatment with JBH. ND, not detected
| HA Oligo | [M]−Predicted | [M]−Observed (lyase digest) | [M]−Observed (JBH treated) | |
|---|---|---|---|---|
| dHn | Hn | |||
| H2 | 775.2262 | ND | 775.2046 (44) | |
| H3 | 1154.3377 | ND | 1154.2942 (39) | |
| H4 | 1533.4491 | ND | 1533.3603 (24) | |
| H5 | 1912.5606 | ND | 1912.4254 (16) | |
| H6 | 2291.6721 | ND | 2291.4850 (05) | |
| H7 | 2670.7835 | ND | ND | |
| dH2 | 757.2156 | 757.1775 (34) | ND | |
| dH3 | 1136.3271 | 1136.2989 (75) | ND | |
| dH4 | 1515.4386 | 1515.4085 (66) | ND | |
| dH5 | 1894.5500 | 1894.5153 (60) | ND | |
| dH6 | 2273.6615 | 2273.6090 (42) | ND | |
| dH7 | 2652.7729 | 2652.7572 (07) | ND | |
Identification of candidate chitin-HA fragments
To facilitate description of the MS data and the large number of possible candidate chitin-HA oligos that might be present, we use a simplified GnHn nomenclature in which n is the independent number of GlcNAc residues (G) and of HA disaccharide units (H) in the m/z species. For example, G3H2 with three extra GlcNAc residues and two HA disaccharide units (a tetramer) has a predicted monoisotopic m/z of 1384.4643 for the [M−] species (Table II). Similarly, the m/z for G4H3 [M−] with four GlcNAc and three HA disaccharide units (hexamer) is 1966.6552. The G0H2–4 = H2–4 [M−] ions correspond to normal (not d-oligo) tetra-, hexa-, octa- oligos (Table I), which could only arise from a lyase cleavage product containing the NR-end.
Table II.
Monoisotopic m/z values of NR-end chitin-HA oligos in lyase digests of newly synthesized SeHAS HA. Observed values are the means of each GnHn species for the number of times (parentheses) each was observed in the Wash 1–3 and ACN fractions. ND, not detected
| Chitin-HA species GlcNAc—disaccharide units | [M]− Predicted | [M]− Observed in lyase digest | |
|---|---|---|---|
| (Gn) | (Hn) | ||
| G1 | H2 | 978.3056 | 978.2195 (38) |
| G1 | H3 | 1357.4170 | 1357.3708 (18) |
| G1 | H4 | 1736.5285 | 1736.5031 (06) |
| G1 | H5 | 2115.6400 | 2115.5738 (03) |
| G1 | H6 | 2494.7514 | ND |
| G2 | H2 | 1181.3850 | 1181.3118 (08) |
| G2 | H3 | 1560.4964 | 1560.4251 (07) |
| G2 | H4 | 1939.6079 | 1939.4068 (01) |
| G2 | H5 | 2318.7193 | ND |
| G2 | H6 | 2697.8308 | ND |
| G3 | H2 | 1384.4643 | 1384.4388 (84) |
| G3 | H3 | 1763.5758 | 1763.5414 (74) |
| G3 | H4 | 2142.6872 | 2142.6301 (55) |
| G3 | H5 | 2521.7987 | 2521.7333 (33) |
| G3 | H6 | 2900.9102 | 2900.7838 (19) |
| G4 | H2 | 1587.5437 | 1587.5135 (60) |
| G4 | H3 | 1966.6552 | 1966.6245 (57) |
| G4 | H4 | 2345.7666 | 2345.7011 (40) |
| G4 | H5 | 2724.8781 | 2724.8084 (22) |
| G4 | H6 | 3103.9895 | 3103.9354 (06) |
| G5 | H2 | 1790.6231 | 1790.5629 (23) |
| G5 | H3 | 2169.7345 | 2169.6508 (04) |
| G5 | H4 | 2548.8460 | ND |
| G5 | H5 | 2927.9575 | ND |
| G5 | H6 | 3307.0689 | ND |
We did not detect any normal (non-dHn) HA oligos in the starting digest or the eluted fractions (Table I), consistent with the absence or very low levels of normal HA oligos, but the presence of GnHn structures at the NR-end. Candidate species corresponding to G3H3, G3H2 and G1H2 were detected in the starting digest, although at lower signal intensity than dH5 (Figure 2A). However, in contrast to the fractionation of dHn oligos, candidate GnHn species, which would have a greater G:A ratio and lower charge:sugar ratio, might bind better to the solid phase and be eluted later in the Wash 2, Wash 3 or 10% ACN fractions (Figure 2C, D and E respectively). As expected, species with longer G stretches were bound more strongly to the solid phase and required organic solvent to elute. For example, G3H2–5 members were detected at different levels in all four fractions and G4H2–4 species were more prevalent in the ACN fraction compared to Wash 3 and not detected in Wash 1 or Wash 2. G5H2,3 species were detected only in the ACN fraction.
At least 19 candidate GnHn ions were observed, to varying extents, in the eluted fractions (Table II), corresponding to predicted m/z values for G1H2–5, G2H2–4, G3H2–6, G4H2–6 and G5H2,3. No m/z signals were observed for G1H6, G2H5,6 or G5H4-6, but these larger oligos are expected to be minor species, whose Hn size ranges likely vary among different lyase digests. Variations in the detection frequency of the various GnHn ions were reflected in the number of times a signal was observed in different fractions and digests (Table II; numbers in parentheses). For example, G3H2 (n = 84) and G3H3 (n = 74) were observed most frequently, whereas G5H3 (n = 04), G1H5 (n = 03) and G2H4 (n = 01) were found least frequently.
Hn NR-ends are detected if present in lyase digests
The lack of Hn oligos in lyase digests (Figure 3A) indicates that these structures are not present at the NR-end of new HA. Since this new information about the structure of HA and the mechanism of HAS action is important to verify, we performed a positive control to demonstrate that the protocol enabled detection of Hn oligos if present. Large HA (542 kDa; Lifecore) was partially digested with OTH to create 49 kDa HA with A–G–A–G- NR-ends; so 10 of the 11 products from each larger chain have Hn (G0Hn) at their NR-ends. After SEC purification, lyase digestion and fractionation, these oligos were readily detected (Figure 3B) in multiple fractions by MS (e.g. H2–4 in Wash 2). This result confirms that the undetectable levels of Hn NR-ends in SeHAS HA digests means that HAS does, in fact, start all new HA chains with G at the NR-end and not A.
Fig. 3.

NR-end Hn oligos created by OTH digestion are readily detected. Lifecore HA (542 kDa) was partially digested with OTH to a size of 49 kDa, to create ~11 times as many HA molecules with >90% having new A–G–A–G- NR-ends. The 49 kDa HA was purified by SEC, digested extensively with lyase and the final digest subjected to solid phase extraction as in Figure 2 and Methods. The indicated fractions were examined by MALDI-TOF MS in negative mode for m/z signals at 775 (H2), 1154 (H3), 1533 (H4) and 1912 (H5). (A) The four H2–5 species were not evident in digests of SeHAS HA. (B) In contrast, in the positive control-HA that had been predigested with OTH, all four H2–5 species were present in Wash 2 and H2–3 were found in Wash 3. Vertical dashed lines indicate positions where the four m/z species would be in samples with no signals for those species. Spuriously high G1Hn levels in lyase digests, first detected in this experiment, were confirmed in subsequent experiments (Table IV).
Digestion with JBH indicates that GlcNAc is at the NR-end of GnHn species
We next determined whether GlcNAc residues are at the NR-end of the candidate m/z species, which would not be the case for OTH- or lyase-generated oligo products that have GlcUA or dGlcUA, respectively, at their NR-ends. Wash 3 samples containing members of the GnH2–5 oligo series were treated with inactive (Figure 4A) or active (Figure 4B) JBH, which specifically cleaves HexNAc(β1-) linkages only at the NR-end (Li and Li 1970). To control for potential differences among samples, due to different amounts of associated JBH contaminants and buffer components, all samples in experiments using JBH had the same total amount of JBH as combinations of active and inactive enzyme. Control samples (Figure 4A, italic font), treated with heat-inactivated enzyme, showed strong M−1 signals for G3H2 (m/z 1384), G3H3 (m/z 1763), G3H4 (m/z 2142) and G4H3 (m/z 1966). Importantly, smaller less hydrophobic members of each series such as G1H2 (m/z 978), G1H3 (m/z 1357), G2H2 (m/z 1181), G2H3 (m/z 1560) and G2H4 (m/z 1939) were either very low abundance or not detected in the starting sample (Figure 4A, boldface font). However, after active JBH treatment, these five latter species were detected (Figure 4B), with strong signals for the two smallest at ~m/z 978 and 1181. In contrast, the five starting species with more G residues [G3H2,3 and G4H2–4] were either absent or greatly reduced, indicating that JBH treatment of these species, as predicted, removes at least one GlcNAc from the NR-end and might also remove additional Gs to create smaller members of each GnHn series.
Fig. 4.

GnHn oligos from SeHAS HA are sensitive to JBH. Wash 3 samples (1.0 µL), prepared as in Figure 2, were incubated overnight at 30°C with inactive (A) or active (B) JBH and examined by MALDI-TOF MS in negative mode as described in Methods. GnHn species are indicated by labels corresponding to the observed m/z values; predicted and mean observed m/z values are in Tables I and II. M−1 ions whose signals decrease or increase with active JBH treatment are indicated, respectively, in italic or boldface fonts.
JBH digestion demonstrates that multiple GlcNAc residues are at the NR-end of GnHn species
To determine whether multiple GlcNAc residues are at the NR-end of the candidate m/z species, pooled Wash 3 Fractions containing predominantly G3H2 (Figure 5A) or G3H3 (Figure 5B), and no detectable G0–2H2 or G0–2H3 respectively, were treated with varying amounts of JBH. All samples had the same amount of total JBH present as a combination of active and heat-treated inactive enzyme. Treatment with increasing amounts of active JBH caused increasingly greater losses of the G3H2 species, with a maximal 82% decrease (Figure 5A). As this species was lost, G2H2 increased dramatically and remained high across all JBH doses and G1H2 increased in a proportional manner with JBH level. The G0H2 (H2) signal increased, although to a lesser extent. Essentially identical results were obtained for JBH treatment of G3H3 (Figure 5B). Almost all samples treated with different amounts of active JBH were very significantly different than those treated with only inactive JBH. These results are consistent with the presence of multiple sequential GlcNAc(β1-4) linkages at the NR-end of the candidate GnHn species and confirm that these species are related members of the H2 or the H3 oligo series.
Fig. 5.

Dose-dependent JBH treatment converts larger GnHnoligos from SeHAS HA to smaller GnHn species indicating sequential GlcNAc linkages. Aliquots (0.75 µL) of Wash 3 were incubated at 30°C overnight with 4.0 µL of JBH as a combination of active and heat-inactivated enzyme. Samples had no active JBH (black bars; 0 µL active enzyme; 4.0 µL inactive enzyme) or increasing amounts of active JBH (µL: 0.5, white bars; 1.0, cross-lined bars; 2.0, light gray bars; 3.0, stippled bars; 4.0, dark gray bars), and decreasing amounts of inactive JBH, as described in Methods. Members of the G0–3H2 (A) and G0–3H3 (B) series were analyzed to monitor decreases or increases in each member as a function of increasing active JBH concentration. Significant differences for G3H2,3, assessed by paired Student t-tests, between samples with varying JBH activity vs. inactive JBH, are indicated for two independent experiments; values are the mean intensity ± SEM (n = 8): *P < 0.05; **P < 0.005; ***P < 0.0005; #P < 0.00005; ##P < 0.000005. Since starting values for G1,2H2,3 and H2 and H3 were zero (denoted by zero-line black rectangles), P values for the samples with 1.0–4.0 µL active JBH were compared to the sample with 0.5 µL, the lowest active JBH dose.
JBH digestion kinetics show sequential product-substrate relationships among GnHn species, consistent with multiple GlcNAc residues at the NR-end
If multiple GlcNAc residues are at the NR-end of GnHnm/z species, then a series of sequential product-substrate relationships should be evident in the time-dependent changes of related species during JBH treatment. This was tested in JBH digestion-kinetics experiments with Wash 3 (Figure 6A and B) or ACN (Figure 6C) samples that showed large m/z signals for G3H3 (Figure 6A), G3H2 (Figure 6B) or G4H2 (Figure 6C), but little or no signal for the predicted smaller series members lacking GlcNAc. In each case, a time-dependent decrease in the larger starting species (e.g. G3H3 in Figure 6A) was rapidly followed by a large increase in the next smallest series member (lacking one GlcNAc; e.g. G2H3 in Figure 6A), which plateaued and then decreased substantially. The next species lacking a second GlcNAc appeared still more slowly (e.g. G1H3 in Figure 6A). The last species to arise during digestion (Figure 6A and B) were H3 and H2, respectively, which lack all extra GlcNAcs not associated with an expected HA oligo structure.
Fig. 6.

Kinetic changes during JBH treatment show that G3H2 and G3H3 species from SeHAS HA are sequentially converted to smaller members of the H2 and H3 series. Aliquots (5 µL) of Wash 3 were incubated with 22 µL inactive (time-zero) or active JBH at 30°C for up to 26 h (n = 4 except n = 3 at 26 h); values are the mean intensity ± SEM as indicated. Members of the G0–3H3 (A) and G0–3H2 (B) series were analyzed to monitor decreases or increases in each member as a function of increasing JBH digestion time, (G3H2,3, ●; G2H2,3, ▼; G1H2,3, ▲; G0H2,3, ■). Lines (r2 ≥ 0.97) for H2,3 (solid) and G3H2,3 (long dash) were calculated by linear regression or a 3-parameter hyperbolic decay function, respectively, using Sigma Plot v10. Lines for G2H2,3 (short dash) and G1H2,3 (dotted) are comprised of linear segments. (C) ACN fraction samples (10 μL) containing G4H2 and much less or no smaller members of this GnH2 series were incubated for up to 12 h, as indicated, with 22 µL of inactive (time-zero) or active JBH. Sequential and time-dependent changes in the levels of G3H2, G2H2, G1H2 and H2 are evident.
The changes in Figure 6C were more complex, with five curves due the presence of one more Gn series member. However, the same general pattern was observed, as in Figure 6A and B, with the starting species (G4H2) disappearing quickly, the three smaller species lacking 1, 2 or 3 additional GlcNAcs (G1–3H2) increasing after a sequential kinetic delay and then decreasing, and the H2 species, lacking all additional GlcNAcs, appearing last. The maximum level of H2 was almost twice that of the starting G4H2, which could be due to the presence of other G>4H2 species in the ACN sample or to a greater intrinsic signal intensity of H2 compared to other M−1 ions. In all three cases, the overall time-dependent pattern of GnH2,3 species changes indicates that the product of each digestion round is itself a substrate for further digestion. The sequential temporal relationship among species with successively fewer Gs was evident and significant (as assessed by the large number of time-paired samples whose error bars are nonoverlapping and well separated).
Extensive digestion of GnHn species with JBH generates normal HA oligos ranging from tetrasaccharides to dodecasaccharides
H2 and H3 were evident after JBH treatment in Figures 4–6. The presence of cryptic Hn oligos that are sensitive to JBH, since they are derived from the NR-end of chitin-capped HA chains, is integral to and predicted by the starting hypothesis that chitin primers remain at the NR-end of newly made HA (Figure 1B). To verify this key prediction, more extensive experiments were performed to release and identify these cryptic normal Hn oligos in Wash 3 (Figure 7A–F) and ACN (Figure 7G–J) fractions of lyase digests. Based on the greater frequency of occurrence observed for the GnH2, GnH3 and GnH4 series members compared to the GnH5,6 species (Table II), we focused on the M−1m/z signals corresponding to H2 (775; Figure 7A), H3 (1154; Figure 7C), and H4 (1533; Figure 7E), which were not detected in samples treated with inactive JBH. After active JBH treatment, however, m/z signals for H2 (Figure 7B), H3 (Figure 7D) and H4 (Figure 7F) were readily apparent. The use of increasing amounts of ACN Fraction sample confirmed dose-dependent increases, after JBH treatment, in the m/z signals for H2 (Figure 7G) and H3 (Figure 7H). As expected, parallel decreases occurred in the m/z signals for the larger G4H3 (1966; Figure 7I) and G4H2 (1587; Figure 7J) species; these M−1 signals were completely eliminated after treatment with active JBH.
Fig. 7.

JBH digestion of GnHn species from SeHAS HA releases nondehydro NR-end H2 and H3 oligos. A–F. Selected m/z ranges are shown for Wash 3 (0.5 μL) samples treated overnight at 30°C with inactive (A, C, E) or active (B, D, F) JBH and analyzed by MALDI-TOF MS in negative mode. GnHn species that decrease or increase, with active JBH treatment, are indicated in italic or boldface font, respectively. New m/z signals are evident corresponding to H2 (B compared to A), H3 (D compared to C) and H4 (F compared to E). (G–J) ACN samples (1, 2 or 3 µL), as indicated, were incubated overnight at 30°C with active or inactive JBH, and the presence of m/z signals, proportionally increasing with sample volume, was monitored for H2 (panel G) or H3 (panel H). The m/z signal decreases for the larger G4H3 (I) and G4H2 (J) ions were assessed in the 3 µL samples, treated with active (top) or inactive (bottom) JBH.
These important results were confirmed and quantified using replicate 1–3 µL ACN samples treated with active or inactive JBH (Figure 8A). The m/z signals for the two starting G4H2,3 species were proportionally greater with increasing sample volume, whereas H2 and H3 were absent in the presence of inactive JBH (Figure 8A, zero-line white boxes). In contrast, the starting species disappeared completely with active JBH (Figure 8A, zero-line black rectangles) and H2 and H3 increased greatly (black bars); in particular, H2 dramatically increased. In similar experiments (Figure 8B and C), samples of pooled ACN Fractions were treated overnight with active JBH and strong m/z signals were seen for normal HA oligos corresponding to H2 (tetra), H3 (hexa), H4 (octa), H5 (deca) and H6 (dodeca). No signals for these species were present in samples treated with inactive JBH (Figure 8, zero-line white rectangles). In separate MS Postsource Decay (PSD) experiments (not shown), the above Hn oligos released by JBH treatment gave the same fragmentation patterns as standard Hn oligos.
Fig. 8.

Lyase digests of SeHAS HA contain chitin-oligo NR-end fragments that conceal normal HA oligos ranging from tetra- to dodecyl-saccharides (H2-H6). (A) ACN samples (1–3 µL, as indicated) were treated in the same way as described in Figure 7 with inactive (white bars) or active (black bars) JBH and the m/z signal intensities of the four GnH2,3 species noted at the top were then determined. Rectangles on the zero-line indicate that there were no detected signals for H2 and H3 with inactive JBH (white rectangles) or for G4H3 or G4H2 after active JBH treatment (black rectangles). B and C. Several ACN fractions were pooled and samples (2 µL) were treated at 30°C for 26 h with 4 µL of active (black bars) or inactive (zero-line white rectangles, indicating no signals) JBH. The m/z signal intensities for H2 and H3 are shown at a different scale (B) than the signals for H4, H5 and H6 (C), which were less abundant; values are the mean intensity ± SEM (n = 6).
MS/MS PSD analysis of GnH2 and GnH3 species demonstrates a related series of fragments with either successively fewer sugars or corresponding to GlcNAc2–5 (chitin) and H1–3 oligos
PSD MS/MS analysis of three G3–5H2 and two G3,4H3 mono-sodiated M+1 ions in positive mode, which frequently shows many dehydro species, revealed strikingly similar fragmentation patterns that showed identities for many of the fragments released from each ion (Figure 9A–E). In control PSD analyses of commercial chitin oligo standards, we always observed greater signals corresponding to dGn compared to Gn species, indicating that dehydro fragments readily formed during PSD cleavage (not shown). Other groups have also observed dehydro cleavage of chitin oligos during various MS analyses, typically removing a reducing end GlcNAc first (Bahrke et al. 2002; Lattova and Perreault 2009; Vijayakrishnan et al. 2011; Lang et al. 2014). Consistent with this known fragmentation pattern, the largest fragment derived from each of the five GnHn parent ions resulted from loss of one GlcNAc (G) and one water. In each case, the successively smaller fragments showed identical patterns of losing an A residue (176 mu) and then a G residue (203 mu); three such apparent cycles were seen for the two ions predicted to contain three disaccharide units (G3,4H3; Figure 9D and E). Table III summarizes the range of fragments found in multiple PSD analyses of the five GnHn ions in Figure 9, as well as a sixth less abundant G5H3 ion (m/z 2193). In addition to the major species evident in Figure 9, many other m/z signals were observed, with varying frequencies, corresponding to other predicted fragments; overall, 33 fragments consistent with the proposed GnHn sequences of the six ions were identified after PSD (Table III).
Fig. 9.

MS/MS PSD fragmentation of related GnH2 and GnH3 series members from SeHAS HA. ACN fraction samples were subjected to MALDI-TOF PSD, in positive ion mode, for m/z species (Table II) corresponding to: G3H2 (A, m/z 1408), G4H2 (B, m/z 1611), G5H2 (C, m/z 1814), G3H3 (D, m/z 1787) and G4H3 (E, m/z 1990). The major fragment peaks are labeled with the species designation and m/z value (Tables I and III). Fragment m/z signal pairs corresponding to loss of one G (GlcNAc, 203 mu) or A (GlcUA, 176 mu) are indicated by double-headed arrows and fragments corresponding to chitin or HA oligos are indicated in boldface or italic fonts, respectively. All m/z species are mono-sodiated, the parent M+1 ion is on the far right and d denotes dehydro species (loss of water).
Table III.
Postsource decay of G3–5H2 and G3–5H3 from SeHAS HA show similar fragment patterns and release chitin and HA oligos. Wash 3 or ACN samples were spotted with matrix on an MS plate and 7–11 PSD analyses were performed for the first five M+1 parent ions indicated, as noted in Figure 9 and Methods. Three PSD analyses were performed on the weaker m/z 2193 signal. Numbers in parentheses indicate the number of times the particular fragment was observed. All m/z values are for monoisotopic mono-sodiated ions; d, dehydro. The noted m/z values only have the digits following the decimal; the preceding numbers are identical to the predicted values. Several minor peaks that could be fragments that occur with standard HA oligos or involving unusual breaks across a sugar ring are indicated by asterisks (*). Although some m/z ions could represent two different sequences, the noted GnHn species are consistent with the predicted parent ion sequence
| GnHn species | Predicted value | Parent [M•Na]+1 Ions | |||||
|---|---|---|---|---|---|---|---|
| G3H2 1408 | G4H2 1611 | G3H3 1787 | G5H2 1814 | G4H3 1990 | G5H3 2193 | ||
| dH1 | 402.10 | 0.30 (7) | 0.31 (10) | 0.30 (8) | 0.27 (7) | 0.24 (9) | ND |
| H1 | 420.11 | 0.45 (2) | 0.28 (5) | 0.36 (3) | ND | 0.28 (5) | ND |
| dH1A | 578.13 | 0.43 (7) | 0.44 (3) | 0.52 (7) | ND | 0.40 (5) | ND |
| dH2 | 781.21 | 0.46 (7) | 0.53 (7) | 0.63 (8) | ND | 0.54 (10) | ND |
| H2 | 799.22 | 0.50 (7) | 0.51 (7) | 0.60 (5) | ND | 0.65 (7) | ND |
| dH2A | 957.24 | ND | ND | 0.59 (7) | ND | ND | ND |
| dH3 | 1160.32 | ND | ND | ND | ND | 0.56 (4) | ND |
| H3 | 1178.33 | ND | ND | 0.64 (8) | ND | 0.63 (10) | ND |
| dG1 | 226.07 | 0.26 (4) | 0.32 (5) | 0.30 (3) | ND | 0.16 (1) | 0.19 (2) |
| G1 | 244.08 | 0.18 (6) | 0.22 (8) | 0.24 (4) | 0.21 (1) | 0.23 (2) | ND |
| dG1H1 | 605.18 | 0.42 (7) | 0.51 (9) | 0.48 (7) | 0.50 (3) | 0.45 (9) | 0.31 (1) |
| G1H1 | 623.19 | 0.45 (6) | 0.50 (6) | 0.50 (4) | ND | 0.53 (4) | ND |
| dG1H2 | 984.29 | ND | ND | 0.57 (6) | ND | 0.65 (8) | ND |
| G1H2 | 1002.30 | 0.58 (7) | 0.59 (9) | 0.72 (8) | ND | ND | ND |
| G1H3 | 1381.41 | ND | ND | ND | ND | 0.67 (10) | ND |
| dG2 | 429.15 | 0.34 (7) | 0.47 (3) | ND | ND | ND | ND |
| dG2H1 | 808.26 | 0.56 (7) | 0.56 (10) | 0.68 (8) | 0.67 (4) | 0.61 (9) | ND |
| dG2H2 | 1187.37 | 0.67 (7) | ND | 0.74 (8) | ND | ND | ND |
| G2H2 | 1205.38 | ND | 0.95 (1) | ND | ND | ND | ND |
| dG2H3 | 1566.48 | ND | ND | 0.71 (8) | ND | ND | ND |
| dG3 | 632.23 | 0.51 (7) | 0.54 (10) | 0.55 (8) | 0.51 (7) | 0.54 (9) | ND |
| G3 | 650.24 | 0.47 (2) | 0.44 (1) | 0.35 (1) | 0.30 (1) | ND | |
| dG3H1 | 1011.34 | 0.63 (7) | 0.69 (10) | 0.76 (8) | 0.57 (10) | 0.71 (10) | ND |
| dG3H2 | 1390.45 | ND | 0.82 (10) | 0.72 (8) | ND | 0.74 (10) | ND |
| dG3H3 | 1769.56 | ND | ND | ND | 0.52 (3) | 0.80 (10) | ND |
| dG4 | 835.31 | 0.61 (7)* | 0.65 (10) | 0.75 (8)* | 0.65 (11) | 0.65 (10) | 0.77 (1) |
| G4 | 853.32 | 0.67 (3)* | ND | ND | ND | ND | ND |
| dG4H1 | 1214.42 | ND | 0.76 (10) | 0.80 (8) | 0.73 (11) | 0.74 (10) | 0.63 (2) |
| dG4H2 | 1593.53 | ND | ND | ND | ND | ND | 0.65 (3) |
| dG4H3 | 1972.64 | ND | ND | ND | ND | ND | 0.89 (1) |
| dG5 | 1038.39 | ND | 0.72 (2)* | ND | 0.71 (11) | ND | 0.63 (2) |
| dG5H1 | 1417.50 | ND | ND | ND | 0.71 (11) | ND | 0.77 (2) |
| dG5H2 | 1796.61 | ND | ND | ND | ND | ND | 0.86 (1) |
Although several m/z ions could represent two different sequences (e.g. dG4A and dG3H1 both have m/z values of 1011; Figure 9A, B, D and E), based on the following findings and conclusions, these ambiguous ions are labeled according to the predicted parent ion sequence (Table III). Among the smallest fragments identified in PSD of all five ions in Figure 9, were an HA disaccharide dH1 (m/z 402) and a chitin trisaccharide dG3 (m/z 632). Further supporting the important result of identifying chitin oligo fragments derived from these M+1 ions, dG4 (m/z 835) was a major peak in the PSD analyses of the three species predicted to contain ≥G4; G4H2 (Figure 9B), G5H2 (Figure 9C) and G4H3 (Figure 9E). The largest chitin peak, the penta-saccharide dG5 (m/z 1038) was a major peak only in the G5H2 parent ion (Figure 9C). In addition, and consistent with the predicted GnHn mono-sodiated ion structures in positive mode (Table III), HA tetrasaccharide fragments (dH2, m/z 781 and H2, m/z 799) were found with four of the six parent ions and the hexa-saccharide H3 (m/z 1178) was seen for two of three ions predicted to contain H3 (Table III). Negative mode PSD analyses (not shown) also showed strong signals for H2–5 oligos from the appropriate parent ions.
More than 90% of G1Hn in lyase digests is spurious
Unexpectedly, the Hn positive control (Figure 3B) revealed high levels of G1Hn species that should not have been present, based on the lyase cleavage mechanism. New Hn NR-ends in the 49 kDa HA (made by OTH treatment of 542 kDa HA) should be at least 91% of the total NR-ends. Lifecore made this 542 kDa HA by heat treatment of larger HA, so the native NR-ends were already diluted by new ends made from breaks of larger chains. Since the ratio of Hn-to-other NR-ends in the OTH-treated HA is 10:1, all other NR-ends can only be 9% of the total.
To quantify signal intensities in these lyase digests, we determined the linear response ranges (LRRs) and cumulative signal intensities in all fractions for each of the dHn species and the three NR-end types: e.g. G3H3 (Figure 10A), G1H2 (Figure 10B) and G0H2 (Figure 10C). We found 95.8% of total signal intensity in dHn oligos, close to the predicted 97% for an average fragment size of seven sugars. In contrast, the intensities for Hn and G1Hn species were 2.8% and 1.5%, respectively, a nearly 2:1 ratio rather than the predicted 10:1. Recovery of Hn as 2.8% of total fragments is essentially the predicted value for a 49 kDa HA digested to 1 NR-end and 35 internal oligos (2.9%). However, production of G1Hn species was ≥5-times the expected level, indicating there is some other reason or mechanism for how these fragments are generated.
Fig. 10.

Determination of MS linear response ranges for individual GnH2–5 species from SeHAS HA. Lyase digests were serially diluted (1:1) and all dilutions were spotted and analyzed by MS in negative mode as described in Methods. Signal intensity vs. relative concentration (with 1.0 being maximal; undiluted) are shown for the GnHn species G3H3 (A), G1H2, (B) and G0H2 (C) from a typical lyase digest. Data were curve-fit by the same hyperbolic function, using Sigma Plot v10. The insert graphs show expanded linear response ranges at lower concentrations with linear regression lines (r2 ≥ 0.97), whose intercepts at an undiluted concentration of 1.0 give the calculated theoretical intensities in the undiluted starting samples. For all such analyses, 95% of the r2 values were ≥ 0.90.
To test if abnormal G1Hn generation is due to lyase activity itself, we digested larger HA (730 kDa; Lifecore, heat-treated) with either lyase or OTH and normalized the LRR signal intensities of G1Hn to total signal intensity of dHn or Hn oligos in the respective digests (Table IV). If G1Hn species are bona fide in both OTH and lyase digests, then the ratio of each G1Hn signal intensity compared to the signal intensity of internal oligos should be essentially identical. This was not the case, however, since lyase digests showed 14.4 ± 4.3 times higher G1Hn levels than OTH digests. Thus, the G1Hn signal intensities in lyase digests overestimate by 14-fold the presence of these sequences as original NR-ends present prior to lyase digestion. In subsequent lyase experiments, therefore, actual G1Hn content was estimated using a 10-fold correction (90% reduction), to account for its over-production by lyase.
Table IV.
G1Hn HA oligos are artifactually created by lyase digestion. Three paired digestions of 730 kDa HA (Lifecore) with lyase or OTH, experiment (exp) 1–3, were fractionated and analyzed to determine LRRs and calculated total signal intensities for all G1Hn species and the appropriate Hn or dHn oligos as described in Methods and Figure 10. Total G1Hn signal intensity is expressed as a percent of the total Hn or dHn oligo signal intensity in each digest type. The mean fold-increase of G1Hn species in lyase digests compared to OTH digests was 14.4 ± 4.3 (SEM); au, arbitrary units
| Exp | HAase | HA oligo total intensity (×10−6 au) | Total G1Hn (% total HA oligos) |
G1Hn ratio (Lyase:OTH) |
|---|---|---|---|---|
| 1 | Lyase — dHn | 917 | 0.348 | 17.4 |
| OTH — Hn | 650 | 0.020 | ||
| 2 | Lyase — dHn | 1470 | 0.338 | 6.0 |
| OTH — Hn | 3802 | 0.056 | ||
| 3 | Lyase — dHn | 3691 | 2.58 | 19.8 |
| OTH — Hn | 9345 | 0.13 |
The stoichiometry of chitin-HA NR-ends
Before assessing stoichiometry, we identified all possible NR-ends by MS analysis of lyase digests of SeHAS HA processed as in Figure 1B and then assessed their detection frequency in undiluted Fractions. Among the possible NR-end types (i.e. AGAG-, GAGAG- and GnGAGAG- oligos), the NR-ends detected most often (81%) began with a chitin sequence, G2–5GAGAG- and a smaller fraction (19%) began with GAGAG- (Figure 11A). This abundance frequency of chitin NR-ends isolated from a cohort of HA chains indicates that at least 80% of new HA-UDP chains are initiated on a chitin-UDP primer.
Fig. 11.

The apparent stoichiometry of chitin-oligo NR-ends in SeHAS HA is 0.97. (A) GnHn oligos show different distributions and abundance patterns during fractionation of lyase digests of SeHAS HA (Table II). The frequencies of finding a GnH2–6 series member containing 1 (white bars) or 2–5 (black bars) GlcNAc residues (G), as indicated, is shown as a percent of all the candidate m/z signals observed in the Wash 1 (W1), Wash 2 (W2), Wash 3 (W3) and ACN fractions (n = 5–8). The summary numbers (totals to right of vertical dashed line) were then calculated from these values as an average of the observed number- and weight-average signal frequencies. The most readily detected species were G3H2–5 (50%), G4H2–5 (25%), and G1H2–5 (19%; uncorrected for lyase over-production) with G2H2–5 and G5H2–5 each at 3%. (B) LRR values and calculated theoretical m/z signal intensities in the original digests, determined as in Figure 10, are shown for G0–5 NR-end types with 2–5 HA disaccharide units (H); G1 (white bar) and G0,2–5 (black bars). Values are mean signal intensity ± SEM (n = 4) in arbitrary units (au). (C) Chitin-HA oligos (i.e. G2–5H2–5) account for 96.5% of the total signal intensity of all NR-end fragments identified and quantified (as in B); G0H2–5 and G1H2–5 were 1.6 and 1.9% of total signal intensity, respectively. Values are mean percent ± SEM (n = 4) of total LRR signal intensity. Based on the 14-fold over-estimation of G1H2–5 species in lyase digests (Table IV), these initial values were reduced 10-fold (90%) in (B) and (C).
There are no biochemical methods to determine the stoichiometry of an HA NR-end type. Radiolabel methods can identify reducing- but not NR-sugars. The short chitin sequences (3–4 sugars) likely preclude binding or capture assays by a chitin-binding protein or lectin or the release of chitin fragments by a chitinase able to recognize chitin-HA, even if such reagents were available. In addition, the very low percentage of total sugar in an NR-end fragment means their quantification is not attainable biochemically. Therefore, we used two strategies to estimate stoichiometry by MS analysis that were necessarily indirect.
First, we determined the LRRs and calculated cumulative signal intensities of the NR-end types in lyase digests of SeHAS HA (Figure 11B and C), with a correction (i.e. a 10-fold reduction) for the over-estimation of G1Hn signal intensity noted above (Table IV). Not surprisingly for MS analyses, the quantitative assessment of signal intensity, based on LRR values, showed a different trend than the signal frequency in undiluted fractions. G3Hn oligo signals were >37-fold more intense than the G0Hn, G1Hn, G2Hn, G4Hn or G5Hn species (Figure 11B). The predominant NR-end structure was G3Hn representing 96.5% of all sequence intensities (Figure 11C). The corrected amount of G1Hn oligos was 1.9% and the amount of G0Hn oligos was 1.6% of total signal intensity. Thus, the most abundant chitin-HA NR-ends were G3Hn structures, representing ~97% of total LRR intensity and indicating that SeHAS initiates all new HA chains on a (GlcNAc)3-UDP chitin primer.
Second, we determined the signal intensity ratio of chitin-HA NR-ends to total dHn species, which reflects the relative amount of NR-ends compared to internal oligos in lyase digests of SeHAS HA. The LRR intensity of all G2–5Hn species in lyase digest fractions was 5.99% ± 1.24% (n = 3, SEM) of total dHn species, indicating that these chitin-HA sequences are, in fact, the majority of NR-ends in new HA chains. In typical lyase digests dH3 was the major product, with dH2 and dH4 LRR intensities at 60% and 50% of the dH3 value, respectively; dH5 and dH6 intensities were 7% and 1% of the dH3 value, respectively. It is problematic to account for species larger than dH4, since they do not excite and vaporize as well in MS and their m/z signals are spread over many sodiated species. If an HA chain 264 sugars long (50 kDa) yields an average of 35 7.5-sugar fragments, then the intensity ratio of NR-ends to total dHn (1 ÷ 35) is 0.029; the NR-end is ~3% of total fragments released from an HA chain. The LRR-based estimate of NR-end frequency (6%) is within a factor of two, which may be reasonable agreement, especially considering the caveat that the intrinsic behavior for dHn vs. G2–5Hn during MS analysis may or may not be the same.
G4Hn chitin NR-ends are present in HA made by live cells
Lifecore produces HA made by a fermentation process using live Streptococcus pyogenes cells. Some of their commercial batches are treated to create smaller HA size ranges, while other batches are untreated and represent native HA that is minimally broken. We digested large native untreated 730 kDa Lifecore HA with lyase and fractionated the digest using our standard protocol. Fractions were analyzed by negative mode MS and, since this HA was ~15-times larger than our in-house 50 kDa SeHAS HA, the frequency and signal intensity of NR-ends was much lower than in Figures 2, 3, 7 or 9. Nonetheless, we detected m/z signals in ACN fractions (Figure 12A) for G4H2 (1587) G4H3 (1966), G4H4 (2345) and G4H5 (2724). Signal intensities did not enable PSD MS/MS in positive mode to identify chitin fragments (not shown). However, negative mode PSD MS/MS analysis of the m/z 1966 signal (Figure 12B) showed the presence of five chitin-related fragments expected for the G4H3 structure. Additional PSD analyses revealed 14 predicted chitin-HA fragments derived from the m/z 1966 M−1 ion (Table V), confirming its identity as a chitin tetrasaccharide-HA trisaccharide hybrid species (G4H3). PSD analyses of the m/z 1587 (G4H2) and 2345 (G4H4) M−1 ions (Table V) also generated 10 and 8 predicted chitin-HA fragments, respectively. Overall, 18 different chitin-HA fragments with ≥2 successive Gs were found in multiple scans (n = 25) of the three M−1 ions, confirming that these species are the expected chitin tetramer-HA oligos. Thus, native HA made by live S. pyogenes cells contains NR-ends that are chitin tetramers: G–G–G–G–A–G–A–G-.
Fig. 12.

Live S. pyogenes cells make HA with a chitin oligo at the NR-end. Native HA (Lifecore, 730 kDa), of the original size made by fermentation, was digested extensively with lyase and fractionated as in Figure 2 and Methods; ACN fractions are shown. (A) MALDI-TOF MS in negative mode, shows m/z signals for four G4Hn species at 1587.4577 (G4H2), 1966.5301 (G4H3), 2345.6168 (G4H4) and 2724.7179 (G4H5). (B) PSD analysis of the m/z 1966 parent M−1 ion shows five breakdown fragments (indicated by labels) expected for this chitin-HA oligo structure. Table V summarizes results from multiple PSD analyses of the m/z 1587, 1966 and 2345 M−1 species.
Table V.
PSD MS/MS analysis of G4Hn NR-ends from HA made by live cells. Large native HA (730 kDa; Lifecore) made by live S. pyogenes cells was lyase-digested, purified and analyzed by MS in negative mode as described in Figure 12 and Methods. The m/z 1587 (G4H2), 1966 (G4H3) and 2345 (G4H4) parent ions (M−1) in ACN fractions were subjected to PSD fragmentations in negative mode. Numbers in brackets indicate the number of independent PSD scans performed on each parent ion and numbers in parentheses indicate the number of scans in which the chitin-cap fragment was observed. All m/z values are for monoisotopic unsodiated ions; d, dehydro. The fragment m/z values only have digits following the decimal; the preceding numbers are identical to the predicted values. ND, not detected
| GnHn species | Predicted value | Parent M−1 ions | ||
|---|---|---|---|---|
| G4H2 1587 [25] | G4H3 1966 [21] | G4H4 2345 [10] | ||
| dG2H1 | 784.26 | 0.25 (5) | 0.34 (2) | ND |
| G2H1 | 802.27 | 0.46 (6) | 0.17 (5) | ND |
| dG2H2 | 1163.37 | 0.32 (3) | 0.27 (2) | ND |
| G2H2 | 1181.39 | 0.21 (12) | 0.14 (4) | 0.17 (4) |
| dG3H1 | 987.34 | 0.23 (12) | 0.02 (1) | ND |
| G3H1 | 1005.35 | 0.26 (8) | 0.06 (2) | ND |
| dG3H2 | 1366.45 | 0.17 (7) | 0.41 (7) | ND |
| G3H2 | 1384.46 | 0.36 (25) | 0.32 (15) | ND |
| dG3H3 | 1745.57 | ND | 0.50 (11) | ND |
| G3H3 | 1763.58 | ND | 0.55 (21) | 0.54 (1) |
| dG3H4 | 2124.68 | ND | ND | 0.18 (3) |
| G3H4 | 2142.69 | ND | ND | 0.54 (10) |
| dG4H1 | 1190.42 | 0.34 (5) | 0.15 (2) | ND |
| G4H1 | 1208.43 | 0.31 (12) | 0.27 (3) | ND |
| dG4H2 | 1569.53 | ND | 0.46 (21) | 0.28 (4) |
| G4H2 | 1587.54 | Parent ion | 0.37 (10) | 0.50 (3) |
| dG4H3 | 1948.64 | – | ND | 0.38 (5) |
| G4H3 | 1966.66 | – | Parent ion | 0.23 (3) |
Discussion
Essentially all newly synthesized HA chains have a NR-end chitin cap
The present study confirms the prediction that the NR-end of newly made HA has a chitin oligo present as a NR-end cap, which must be derived from use of a chitin-UDP oligo as a primer to start HA synthesis. Stoichiometry was estimated in two ways. First, based on relative signal intensities of all NR-end types, virtually all (97%) HA chains had a chitin trimer as the first three NR-end sugars. Thus, GlcNAc but not GlcUA is at the NR-end of all native HA. The ~3% of G0Hn and G1Hn nonchitin sequences likely represent new NR-ends created by breakage of native HA chains during the clean-up and purification protocol. A second estimate of NR-end stoichiometry was to determine the ratio of chitin NR-ends (G2–5Hn species) to all internal dHn digestion oligos. The expected percent of NR-ends (for 50 kDa HA) is ~3% of total fragments, assuming an average size of 7–8 sugars. The calculated value based on quantified signal intensities was ~6%, which may be good agreement for an MS analysis. The two values might differ for several reasons: (i) the behavior and comparative signal intensities of the two structurally different species during MS may be intrinsically different and (ii) the average fragment size is likely greater than the estimated 7–8 sugars, since many larger dHn species may be present but not revealed by MS (e.g. due to increasing sodiated forms); larger species would not have to be as abundant as smaller species to increase average fragment size substantially and to increase the ratio of NR-ends-to-internal oligos.
A key finding for assessing NR-end prevalence was that lyase preferentially creates G1Hn species, products that are not made by OTH; G1Hn oligos were 14-fold more abundant in lyase compared to OTH digests (Table IV). One possibility for how additional G1Hn species, might be created (as an artifact) is as a lyase-mediated side reaction during HA cleavage. The lyase mechanism involves loss of the GlcUA C4 O-glycosyl (HA-GlcNAc-) leaving group and –H abstraction at C5 to create a –C4 = C5– double bond that twists the pyranose ring (Shimada and Matsumura 1980). This distortion could make enzyme-bound intermediates of the reaction unstable enough that a few percent of HA fragments hydrolyze at C1 of the dA residue before product release from the enzyme. Loss of a dA sugar from a dHn fragment (dAGAG-) would result in creation of a G1Hn oligo and thus overestimate by ~14-fold the presence of these sequences as bona fide NR-ends present prior to digestion. The very low levels of spurious G1Hn oligos would not be easy to detect biochemically, but are readily found by MS.
The following key findings support the conclusion that chitin is present as a NR-end cap on HA. (i) As predicted, a small fraction of lyase digestion m/z products of SeHAS HA was observed corresponding to 19 candidate GnHn products, each with no dA, 1–6 extra GlcNAc residues and normal HA oligos with 1–5 disaccharides. The observed m/z values for these M−1 ions were nearly identical to the predicted values (Tables I and II). It is important to note that we looked for but found no m/z species that might correspond to oligos with internal chitin sequences (e.g. AGAGnAG). (ii) All of the G2–5H2–6 lyase-generated products had multiple GlcNAc(β1) residues that were removed by JBH, which is specific for HexNAcβ1 linkages at the NR-end. (iii) JBH digestion created a series of smaller products with a time-dependent appearance and then disappearance, as expected for the sequential generation of products that then become new substrates. As predicted, the GnHn products treated with JBH behaved as a series of related Hn oligo family members with varying numbers of GlcNAc(β1) residues at the NR-end. (iv) Evidence for a (GlcNAcβ1,4)n structure includes: (a) the normal GlcNAc linkage HAS creates in HA is (β1,4); (b) chitin oligos containing 2–5 GlcNAc residues were generated for all the GnHn products analyzed by PSD MS/MS (Figure 9); (c) larger (GlcNAcβ1,4)n-UDP oligos, made in the absence of GlcUA-UDP were sensitive to Streptomyces griseus chitinase (Weigel et al. 2015), which first releases and then hydrolyzes disaccharide units (Eijsink et al. 2010), confirming these are chitin oligos. In contrast, the GnHn species were not cleaved effectively; only sporadic and modest signals were seen for cleavage products; too low to be useful. Since chitin-UDP species made by HAS in the absence of UDP-GlcUA are larger (up to 15 residues) than the chitin cap on HA (3–4 sugars), they may be more readily recognized by chitinase as substrates. The chitinase substrate binding region likely interacts with 4–6 sugars, so binding to short (GlcNAcβ1,4)3–4-HA oligos may be poor or be hindered by the proximity of GlcUA. (v) Extensive JBH digestion of GnHn oligos that removes all NR-end GlcNAc(β1) residues releases normal (non-dHn) HA oligos ranging from tetra- to dodeca-saccharides. The only part of an HA chain that does not give rise to a dHn product in a lyase digest is the single unique fragment containing the NR-end of the polymer (Figure 1B).
The Class I HAS family is thought to be most related to, and likely derived from, the chitin synthases (DeAngelis 1999; Itano and Kimata 2002; McDonald and Hascall 2002; Weigel and DeAngelis 2007), an idea supported by the present results. HAS appears to be a chitin-like synthase limited to making only UDP-linked oligomers, rather than longer chitin, and with an evolved ability to make the HA repeating disaccharide by addition to short chitin-UDP oligos. The HAS synthetic mechanism is processive and the enzyme does not release the growing HA-UDP chain during sugar elongation (DeAngelis and Weigel 1994; Baggenstoss and Weigel 2006). This processive mechanism is achieved by a topological “lock” in which the growing chain is within an intraprotein pore (Hubbard et al. 2012; Medina et al. 2012) that enables it to be translocated across the membrane without contacting the lipid bilayer. Not surprisingly, we found that purified exogenous HA chains made by SeHAS or obtained commercially could not be bound and extended by fresh enzyme (data not shown). It remains to be determined if HAS can rebind free chitin-UDP and initiate HA synthesis or if the enzyme only initiates HA-UDP synthesis in a continuous and concerted fashion on an already synthesized and bound chitin-UDP.
Live cells make HA with a chitin tetramer at the NR-end
SpHAS initiates HA synthesis on chitin-UDP primers in S. pyogenes cells, supporting the in vitro findings with HA made in vitro by SeHAS membranes. Interestingly, only G4H2–5 species were detected in lyase digests of native HA, whereas the predominant NR-end made by SeHAS membranes was G3H2–5. The inability to detect G2–3H2–5 NR-ends in native HA could be due to technical issues but, if present, G3H2–5 species should have co-purified in the ACN fraction with G4H2–5. Not finding G3H2–5 species likely means that the HA NR-ends made by SpHAS in cells are chitin tetramers. The in vitro results show that SeHAS makes both G3Hn and G4Hn species but that 97% of all HA NR-ends are a chitin trimer. This difference in chitin NR-end length could be due to intrinsic differences between SeHAS and SpHAS or differences for either HAS between HA synthesis in cells vs. membranes. We suspect the latter is more likely than the former, based on the high degree of conserved functional characteristics between the two streptococcal HASs (Weigel 2002, 2015; Weigel and DeAngelis 2007) and on considerations of how the chitin-UDP primer might interact with the intra-HAS pore in membranes to enable HA assemble to begin (discussed below). Finally, since the native SpHAS HA was almost 15-times larger than the SeHAS HA, we did not expect to detect NR-ends readily. Thus, the ability to detect and characterize the G4H2–5 NR-ends in digests of native 730 kDa HA indicates that the frequency of these structures may be relatively high; that all new HA chains made in vivo by SpHAS likely have a chitin tetramer at their NR-end.
The structure of HA
The discovery of HA in bovine vitreous humor was reported in 1934 by Meyer and Palmer (Meyer and Palmer 1934), who named it and identified its two component sugars, a uronic acid and an amino sugar and later elucidated the alternating β1,4 and β1,3 linkages (Weissmann and Meyer, 1952). Since then, the only other change to our understanding of the alternating GlcNAc(β1,4)GlcUAβ1,3 (G–A) disaccharide structure of HA was the discovery that the heavy chain of inter-α1-trypsin inhibitor can be covalently transferred by TSG6 from chondroitin sulfate to HA carboxyl groups (Jessen et al. 1994; Zhao et al. 1995; Mukhopadhyay et al. 2004; Rugg et al. 2005; Evanko et al. 2007). The present study demonstrates that the structure of growing or newly released HA chains is not entirely comprised of only repeating GlcNAc(β1,4)GlcUAβ1,3 disaccharides. The NR-end of each chain is not HA but rather a chitin trimer or tetramer, 3–4 GlcNAc residues, that HAS assembles before it makes the first HA disaccharide. Therefore, (GlcNAcβ1,4)2,3[GlcNAc(β1,4)GlcUA(β1,3)]n-UDP is the overall structure of a growing HA chain. Since it is unknown if HAS adds each sugar unit separately or makes disaccharides in a concerted reaction, structures [1] and [2] more accurately represent a growing (as shown) or completed (lacking UDP) HA chain:
[1] (GlcNAcβ1,4)2,3[GlcNAc(β1,4)GlcUA(β1,3)]n-GlcNAc(α1→)UDP
[2] (GlcNAcβ1,4)2,3[GlcNAc(β1,4)GlcUA(β1,3)]nGlcNAc(β1,4)-GlcUA(α1→)UDP
Like DNA, large HA is very shear-labile and readily broken so that preparations of commercially processed HA or HA purified from tissues will likely have many broken chains, with fragments whose NR-ends lack the chitin-cap present on the original intact HA molecule released from HAS.
HAS uses 12 substrate binding and GTase activities to make a chitin-UDP primer, add one GlcUA and then synthesize the HA copolymer
HAS possesses four recently recognized functions (Weigel et al. 2015) that enable the enzyme to synthesize chitin-UDP oligos (Table VIA). We found these free hybrid oligos only when UDP-GlcUA was absent, but the present results show that chitin-UDP oligos must be made in the presence of both UDP sugars. Most likely, these primers are not detected during active HA synthesis because they are effectively used rather than being released. Mouse HAS1 (Yoshida et al. 2000), frog HAS1 (Semino et al. 1996) and SeHAS (Weigel et al. 2015) make [GlcNAc(β1,4)]n linkages during synthesis of chitin-UDP oligos, indicating that all Class I HAS family members make chitin-UDP. Free chitin oligos arise without HAS being able to assemble them directly, since they are produced by the loss of UDP from the initial GlcNAc(β1,4)nGlcNAc(α1→)UDP product. HAS adds β(1,4)-linked GlcNAc residues, from GlcNAc(α1→)UDP to a GlcNAc(β1,4)GlcNAc(α1→)UDP acceptor creating (GlcNAcβ1,4)nGlcNAc(α1→)UDP. The most prevalent chitin-UDP species made by SeHAS, in the absence of GlcUA-UDP, contain 2–3 β-linked and one α-linked GlcNAc residues, corresponding to 3–4 total GlcNAc residues (Weigel et al. 2015).
Table VI.
Three stages of synthesis catalyzed by HAS creates additional polarity to the HA polymer. (A) The overall reaction for the synthesis of a chitin-UDP oligo utilizes two donor binding site functions and two GTase activities; functions number 8–11, respectively, in (Weigel et al. 2015). (B) Due to the presence of an oligo-GlcNAc cap at the NR-end, the first HA disaccharide unit synthesized is necessarily GlcNAc(β1,4)GlcUA(α1→3), rather than GlcUA(α1→3)GlcNAc(β1,4), and is made by GlcNAc(β1,4)nGlcNAc(α1→)UDP: GlcUA(α1→)UDP, [GlcNAc(β1,4)]n + 1 transferase; this novel function is the 12th discrete binding or catalytic activity identified for HAS. Based on the present study, n in this reaction is 2 or 3. (C) Synthesis of the repeating GlcNAc(β1,4)GlcUA(β1,3) disaccharide units of HA then proceeds using previously recognized substrate binding and GTase functions, numbers 1–7 (Weigel, 2002). (D) The NR-end of new HA is not an HA sequence (Figure 1B). The mechanism of HA initiation creates a unique NR-end hybrid sequence that is part chitin and part HA
| A. HA synthase: overall reaction for synthesis of a GlcNAcn-UDP primer. |
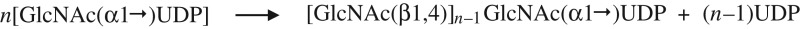 |
| B. HA synthase: assembly of the first hyaluronan G–A disaccharide at the NR-end. |
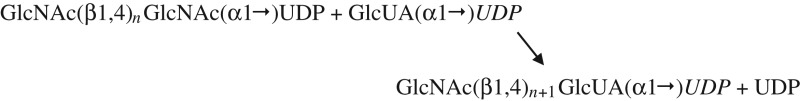 |
| C. HA synthase: overall reaction for ongoing HA disaccharide synthesis |
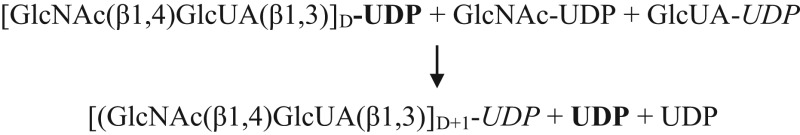 |
| D. Non-HA chitin sequences are present at the nonreducing ends of new HA chains. |
| GlcNAc(β1,4)GlcNAc(β1,4)GlcNAc(β1,4)GlcNAc(β1,4)GlcUA(α1,3)GlcNAc(β1,4)GlcUA(α1,3)-- |
| GlcNAc(β1,4)GlcNAc(β1,4)GlcNAc(β1,4)GlcUA(α1,3)GlcNAc(β1,4)GlcUA(α1,3)-- |
The present findings are highly consistent with this latter result: the chitin cap length at the NR-end of new SeHAS HA chains was three sugars (Figure 11B and C) and the chitin cap length at the NR-end of SpHAS HA made by live cells was 4 sugars (Figure 12). Like threading a needle with thread, inserting a growing chitin-UDP chain into the HAS pore would likely occur only at a particular length (i.e. number of sugars); too long and the chain can no longer access the pore, too short and it might be aligned correctly but not yet engaged with the pore. Pore engagement is likely a key step in initiation of HA synthesis because if chain engagement with the pore cannot occur, then either HA would be made intracellularly or (more likely) cells would degrade HAS in order to avoid this situation. We propose that a mechanistic requirement for HAS enzymes to initiate alternating HA disaccharide assembly is that the chitin-UDP primer must first become engaged by and be within the HAS pore.
Since one other distinct GTase activity is needed in order to create the first novel GlcNAc(β1,4)GlcUA(β1,3) disaccharide at the NR-end, the present results confirm that HAS possesses a proposed 12th function (Table VIB; Weigel et al. 2015) needed to start disaccharide synthesis. This novel HA-initiating activity is a GlcNAc(β1,4)2,3GlcNAc(α1→)UDP: GlcUA(α1→)UDP, [GlcNAc(β1,4)]3,4 GTase. Synthesis of the repeating disaccharide units can then proceed using the previously recognized seven binding and catalytic functions (Table VIC; Weigel 2002; Weigel et al. 2015). Thus, overall HAS uses 12 discrete substrate binding and catalytic functions, first to make (GlcNAc-β1,4)2,3GlcNAc(α1→)UDP oligos, then to add the first GlcUA and then to assemble the repeating linear [GlcNAc(β1,4)GlcUA(β1,3)]n copolymer, HA (Weigel 2002, 2015).
Based on the present results, therefore, it is appropriate to consider the best representation of the repeating disaccharide structure of HA as [GlcNAc(β1,4)GlcUA(β1,3)]n rather than [GlcUA(β1,3)GlcNAc(β1,4)]n, since this first disaccharide that defines a unique (not random) register of the alternating sugars in all HA chains, starting from the NR-end, is G–A.
The results also provide new insight into the mechanism of HA synthesis and how HAS begins the molecular process of HA assembly (Figure 13). HAS cannot directly begin alternate sugar addition to make the repeating HA disaccharide GlcNAc(β1,4)GlcUA(β1,3). The enzyme requires [GlcNAc(β1,4)]3,4-UDP, a chitin oligo, upon which it can then polymerize HA. Native (i.e. newly made) HA chains are thus comprised of two types of GlcNAc(β1,4) linkages; one in the chitin cap with the glycoside bond to another GlcNAc and a second throughout the HA portion of the molecule with the glycoside bond to GlcUA.
Fig. 13.

The HAS mechanism for the sugar assembly order and linkage during HA chain initiation. HAS always starts the NR-end of a new HA molecule with GlcNAc-UDP (blue squares) and then adds a second GlcNAc-UDP. The enzyme bound to this G–G-UDP product then only accepts another GlcNAc-UDP as the third sugar to make G–G–G-UDP (middle line). After this critical length of three GlcNAc sugars is attained (e.g. to ensure that the nascent chain inserts into the intraprotein pore for successful chain translocation: Tlapak-Simmons et al. 1999; Hubbard et al. 2012), HAS adds either GlcUA (A; blue/white diamonds) to initiate alternating HA disaccharide assembly on a chitin trisaccharide (bottom line, black solid arrows) or HAS adds a fourth GlcNAc to make G–G–G–G-UDP (top line, gray dashed arrows). HAS then adds only GlcUA to initiate alternating HA disaccharide assembly on this chitin tetrasaccharide. Both branched pathways for assembly of G3-UDP or G4-UDP primers allow HAS to start alternating sugar assembly and produce HA chains, the vast majority of which contain a G3 or G4 chitin oligo at their NR-end. In the present studies using SeHAS containing membranes, the chitin trimer was the predominant (97%) NR-end. Initial studies, using HA made by S. pyogenes HAS in live cells, show that a chitin tetramer may be the predominant NR-end. These two results indicate that HAS may be able to create an appropriate length tri- or tetra- GlcNAc primer depending on local membrane conditions (e.g. fluidity changes due to altered temperature or lipid composition) that might influence HAS conformation, pore proximity or the optimal length for entry of a nascent chitin-UDP chain into the intra-HAS pore and effective priming to initiate subsequent HA synthesis.
Cellular and physiologic implications for the presence of a chitin cap at the NR-ends of HA
The presence of the novel biosynthetic pathway for chitin-UDP oligo synthesis and the NR-end chitin cap on HA has multiple possible molecular and physiological implications. Their main function is to serve as primers for initiating HA synthesis. In addition, other possible functions for chitin-UDP oligos made by any HAS could include the following:
If GlcUA-UDP levels are low or the primer and HA synthetic activities are differentially regulated by modulators, post-translational modifications or different lipid requirements (Evanko et al. 2007; Sakr et al. 2008; Viola et al. 2008; Karousou et al. 2010; Tammi et al. 2011; Hascall et al. 2014; Ontong et al. 2014; Vigetti et al. 2014; Moretto et al. 2015), then HAS could produce excess chitin-UDP to generate free chitin for as yet unknown processes, such as development (Semino and Allende 2000; Van der Holst et al. 2001).
A novel unknown GTase could transfer activated chitin oligos from chitin-UDP, released by HAS, to specific cellular acceptors (e.g. protein, glycans).
HAS synthesis of a chitin-UDP primer could be a key regulatory step, a cell-protection and gate-keeper mechanism, before commitment to HA synthesis. Class I HAS Km values for GlcNAc-UDP are generally two to three times higher than for GlcUA-UDP, consistent with the relative abundance of the two sugars in cellular glycoconjugates and the likely evolutionary pressure to not allow HAS to over-consume GlcNAc-UDP at the expense of cellular energy, metabolite loss, synthesis of other glycans (e.g. N-glycosylation) or regulatory modifications (e.g. O-GlcNAcylation). For example, putting HAS into bacterial cells that do not normally make HA prevents them from growing because GlcNAc-dependent cell wall biosynthesis is impaired as HA synthesis captures GlcNAc-UDP (DeAngelis et al. 1993; Weigel 2004). Our prediction in this scenario is that the HAS Km for GlcNAc-UDP will be higher for chitin-UDP primer synthesis than for HA synthesis (i.e. primer synthesis is the rate-limiting step in HA synthesis). A higher Km for primer synthesis ensures that ample GlcNAc-UDP is available for initiating subsequent HA-UDP synthesis.
A chitin-UDP oligo would plug the intraprotein HAS pore and thus minimize ion leakage and dissipation of electrochemical gradients needed for important cellular processes. Protein biosynthesis in the ER inserts HAS with its active sites in the cytoplasm, where UDP-GlcNAc is available, so correctly folded HAS could immediately make a chitin-UDP primer that plugs the HAS pore. Although much is known about sugar nucleotide synthesis in the cytoplasm and transport into ER and Golgi within cells, the distribution of these precursors in various compartments is unknown (Coates et al. 1980; Hirschberg et al. 1998). HA synthesis is activated at the plasma membrane (Torronen et al. 2014; Koistinen et al. 2015; Viola et al. 2015), where high-flux synthesis of UDP-sugars is needed to support HA production. Considering this and the above possible function (iii), a corollary prediction is that if a chitin-UDP primer plug cannot be made (indicating low GlcNAc-UDP levels), then HAS will be degraded so that HA cannot be made in a metabolically vulnerable cell and HAS cannot uncouple ion gradients.
The residual chitin oligo primer at the NR-ends of HA chains presents a unique hybrid sequence (Figure 1B and Table VID) that might be recognized by novel extracellular chitin-HA binding proteins. If such proteins exist and are also oligomeric, they could bind to this unique site in an HA chain and organize multiple chains in register to form larger complexes of HA chains or cables (Jokela et al. 2008) anchored at their NR-ends. Though short, the NR-end chitin oligos might also weakly self-associate, and be stabilized by extracellular binding partners that recognize such sequences. Complexes of organized HA chains in register could influence, or have unexpected effects on, cell behavior or extracellular matrix organization and function.
The frequency of the unique NR-end oligos in lyase digests of MDa HA will be much lower than observed in the present study and, therefore, more difficult to detect. It will thus be difficult to confirm the presence of chitin at the NR-end of large mammalian HA. However, based on their conserved topology, mechanism of synthesis, lipid-dependence, the production of chitin and other common molecular and mechanistic features, all Class I mammalian HASs are expected to make and use chitin-UDP primers to initiate HA synthesis, although the optimal chitin-UDP oligomer length may vary among HAS1, HAS2 and HAS3 or among different species.
Materials and methods
Materials, reagents and buffers
All water used was purified by reverse osmosis or distillation followed by deionization. Jack bean β-N-acetylhexosaminidase (JBH, #A2264), Streptomyces hyalurolyticus hyaluronidase (HA lyase, #H1136), OTH (#H-6254), and ACN (HPLC grade) were from Sigma-Aldrich (St. Louis, MO). HA preparations of 542 kDa (heat-treated) and 730 kDa (native, not heat-treated), based on SEC-multi angle light scattering analysis in our lab, were from Lifecore Biomedical (Chaska, MN); #HA700k-1, lots 016066 and 025317, respectively. Bradford reagent (#23239) was from Pierce (Rockford, IL). Sep-Pak C18-cartridges (360 mg) used for solid phase extraction were from Waters (Milford, MA). Chitin oligosaccharides were from Vector Labs (Burlingame, CA). MgCl2 and methanol were from EMD (Billerica, MA). Ethanol was from Pharmco Aaper (Brookfield, CT). All other reagents were from Sigma-Aldrich. Prep Buffer contained 5 mM sodium phosphate, pH 7.4, 50 mM NaCl, 2 mM dithiothreitol, and 0.02% sodium azide. JBH Buffer, for digestion reactions, contained 20 mM ammonium acetate, pH 5.2 and 10 mM dithiothreitol.
Membrane preparation
Escherichia coli SURE2 cells expressing recombinant SeHAS were grown, induced, and membranes were isolated as described previously (Tlapak-Simmons et al. 1999; Baggenstoss and Weigel 2006). Overall, 40 centrifuged membrane pellets were obtained from 18 L of cell culture and two pellets were then scraped into each of 20 tubes, overlaid with 50 mM Tris, pH 7.3, 500 mM NaCl, 10% glycerol with 1 mM PMSF and stored frozen at −80°C until use. All steps were at 4°C unless noted otherwise. Ten tubes of membrane pellets were each dispersed in 1 mL Prep Buffer by sonication using a 1/8 inch micro-tip at a setting of 20% with alternating 20 s pulses and 1 min intervals until suspensions appeared uniform. The pool was divided into two ultracentrifuge tubes and centrifuged at 100,000 × g for 30 min. Supernatants were removed and each membrane pellet was resuspended by sonication at 20% intensity for 20 sec using three successive 2 mL portions of Prep Buffer. The suspended membrane fractions (6 mL from each pellet) were combined, protein was determined by the Bradford method and the protein concentration was adjusted to 10 mg/mL with Prep Buffer. Aliquots (6 mL) were frozen at −80°C until the time of use.
Synthesis and purification of SeHAS HA
One membrane suspension aliquot was thawed, fresh dithiothreitol was added to 2 mM and MgCl2 was added to 30 mM. The suspension was mixed well and 380 µL aliquots were put into multiple 2 mL flat bottom tubes (Phenix, MAX 820), which were preincubated at 30°C for 10 min. A mixture of UDP-GlcNAc and UDP-GlcUA was added to final concentrations of 5 and 1 mM, respectively; one tube was set up at a time and each tube was immediately mixed by vortexing and placed in a heating block at 100°C for 20 min. The samples were placed on ice for at least 5 min before pooling all samples; the tubes were then rinsed serially with 1 mL of 50 mM ammonium acetate. The pooled samples and rinse(s) were centrifuged at 100,000 × g for 30 min at 4°C to remove membrane debris. The supernatant was removed and subjected to two successive Folch extractions (Folch et al. 1957), each using three volumes of chloroform-methanol (2:1). The pooled aqueous phases were centrifuged in a speed-vacuum to remove organic solvents and concentrated to half the starting volume, and then mixed with three volumes of 95% ethanol. After 2 h at −20°C, the precipitated HA was collected by centrifugation at 20,000 × g for 30 min at 1°C. The pellet was dissolved in 400 μL 50 mM ammonium acetate and 0.005% sodium azide and subjected to a third Folch extraction to remove residual lipid. The aqueous phase was treated as above to remove organics and then loaded onto a 90 mL Sephacryl 300HR SEC column (1.5 cm × 45 cm) at room temperature and eluted at a flow rate of 0.5 mL/min for 3 h in 50 mM ammonium acetate with 20% ethanol. Fractions (2.4 mL) were collected, monitored for absorbance at 210 nm and 280 nm and analyzed by agarose gel electrophoresis and Stains-All staining to identify HA (Cowman et al. 2011; Weigel et al. 2013). Fractions containing HA (typically #14–22; Figure 1A) were pooled and lyophilized to remove ammonium acetate.
HA digestion with lyase or OTH and enrichment of candidate chitin-HA oligos
HA (2 mg in 2 mL) in 50 mM ammonium acetate, pH 5.6, was digested overnight at 30°C with 100U of streptomyces lyase or 1000U of OTH. The pH was then adjusted to 6.5 with NaOH and the digest was applied to a Sep-Pak C18 cartridge pre-equilibrated using four sequential 4 mL washes of methanol, 100% ACN, 50% ACN and then 50 mM ammonium acetate, pH 6.5. The cartridge was washed twice with 1 mL (Wash 1 and Wash 2) and once with 1.5 mL (Wash 3) of 50 mM ammonium acetate, pH 6.5 followed by elution with 1 mL of 2% or 10% ACN and finally with 1 mL 50% ACN. The fractions containing ACN were centrifuged in a speed-vacuum to remove organics, all fractions were frozen at −80°C then lyophilized overnight. The lyophilized fractions were taken up in 10% ACN to ensure solubilization from microfuge or glass tube walls prior to MS or enzymatic analyses.
Digestion of chitin-HA oligos from SeHAS HA with JBH
For overnight JBH digestions, 1 µL of JBH Buffer was added to 2 µL of the indicated Wash or ACN Fraction sample and 4 µL JBH was mixed with 1.0 µL of JBH Buffer and added to the sample, which was incubated overnight at 30°C. For kinetic digestion assays 22 µL JBH, diluted 20% as above with JBH Buffer, was added to 5 or 10 µL of the indicated Wash or ACN Fraction samples and incubated at 30°C with shaking as above. After sample addition, final concentrations were 4 mM ammonium acetate, 2.5 mM dithiothreitiol and 1.25 M ammonium sulfate. Aliquots (2–3 µL) were removed at the indicated times, diluted 10-fold with water, heated at 100°C for 10 min (to inactivate JBH), and then stored on ice or at 4°C until MS analysis. Inactive JBH controls for each type of assay were performed in parallel samples by first heating the JBH solutions at 100°C for 10 min prior to addition to samples. This control allows for identification of unrelated JBH-associated background peaks that are within the m/z range of interest and also ensures that each sample is exposed to the same components of ammonium sulfate buffer and enzyme throughout an experiment (since commercial JBH suspensions are 2.5 M ammonium sulfate). For experiments with different amounts of active JBH, all samples had the same amount of total JBH present as a combination of active and heat-treated inactive enzyme (e.g. samples with 0 µL active JBH had 4 µL inactive JBH and samples with 1.0 µL active JBH also had 3.0 µL inactive JBH). Values presented in graphs are the mean intensity ± SEM.
LRR analysis of m/z species
For LRR analysis, each Sep-Pak fraction was serially diluted (1:1) 11–18 times, as needed, with distilled, deionized water and each dilution set was analyzed by MS during the same instrument session. For each dilution scan, the m/z peak intensities were noted for all species of interest. These intensity data were then plotted (using Sigma Plot v10) and analyzed to determine the dilution region with a linear slope (r2 values for 95% of all regression lines were >0.90). The slope intercepts at 1.0, which are the theoretical undiluted starting concentrations, were then compared among species to calculate relative abundance.
Analysis of chitin NR-ends from HA made by live cells
A total of ~55 mg of native HA (730 kDa, Lifecore), made by live S. pyogenes cells and not degraded to smaller size, was digested extensively with lyase, as noted above, as 10 independent batches and processed over 10C-18 SepPaks. Since most GnHn signals, observed by MS in negative mode, were in the ACN fractions, these were further analyzed by PSD MS/MS in negative mode separately and as different combinations of pooled and concentrated ACN fractions (to increase the concentration of low abundance ions). PSD analyses were performed in negative mode.
MALDI-TOF MS analysis
Samples (0.4–0.8 µL) were spotted on a polished steel target plate, 0.8 µL of 6-aza-2-thiothymine as matrix (6 mg/mL in 50% ACN with 0.1% trifluoracetic acid) was immediately added and mixed by pipetting up and down twice. The plate was put under vacuum to induce rapid crystallization and analyzed in Reflectron mode in a Bruker Ultraflex II MALDI-TOF-TOF (Billerica, MA). For each sample, ≥1000 individual shots, collected in 200 shot sections usually from the center areas of sample spots, were accumulated and summed. Peaks were chosen using centroid peak identification with baseline subtraction by the Bruker Daltonics program. For LRR calculations of signal intensity, the intensities of the parent and all sodiated peaks were added together to determine LRR and total intensity for the parent m/z species. MS/MS analyses using PSD were performed in negative or positive mode with 6-aza-2-thiothymine as matrix, as above, and with up to 100 mM ammonium sulfate (final concentration) added to samples to facilitate ionization and enhance intensities. For each positive mode PSD spectrum 2000–4000 individual laser shots, collected in 200 shot increments (usually from the center areas of sample spots), were accumulated and summed. For negative mode PSD samples, 4000–6000 individual laser shots were collected in 400 shot groups (usually from center areas of sample spots). M/Z scans were processed using Bruker Flex Analysis v3.4 software for peak identifications and intensity data. GlycoWorkbench v2.1 was used to calculate m/z values.
Acknowledgments
We thank Dr. Virginie Sjoelund, Huaiwen Wang and The Laboratory for Molecular Biology and Cytometry Research at OUHSC for the use of the Core Facility and assistance with, and access to, the mass spectrometry equipment.
Author contributions
P.H.W. conceived and planned the project, secured funding and designed experiments. B.A.B. and J.L.W. designed and performed experiments, helped interpret data and generate figures and tables. All authors participated in writing and editing the article.
Funding sources
National Institute of General Medical Sciences grant R01 GM035978 from the National Institutes of Health (to P.H.W.) and Ed Miller Chair in Molecular Biology (to P.H.W.).
Conflicts of interest
The authors declare no competing financial interests.
Abbreviations
A, GlcUA or d-glucuronic acid; ACN, acetonitrile; d, dehydro (unsaturated); G, GlcNAc or N-acetyl-d-glucosamine; d-oligo, dehydro-oligosaccharide; GTase, glycosyltransferase; H, a hyaluronan disaccharide unit; HA, hyaluronic acid, hyaluronate, hyaluronan; HAS, HA synthase; JBH, jack bean N-acetylhexosaminidase; LRR, linear response range; m/z mass:charge ratio; oligos, oligosaccharides; OTH, ovine testicular hyaluronidase; PSD, postsource decay; SEC, size exclusion chromatography; SeHAS, Streptococcus equisimilis HAS; SpHAS, Streptococcus pyogenes HAS
References
- Asplund T, Brinck J, Suzuki M, Briskin MJ, Heldin P. 1998. Characterization of hyalur-onan synthase from a human glioma cell line. Biochim Biophys Acta. 1380:377–388. [DOI] [PubMed] [Google Scholar]
- Baggenstoss BA, Weigel PH. 2006. Size exclusion chromatography-multiangle laser light scattering analysis of hyaluronan size distributions made by membrane-bound hyaluronan synthase. Anal Biochem. 352:243–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrke S, Einarsson JM, Gislason J, Haebel S, Letzel MC, Peter-Katalinic J, Peter MG. 2002. Sequence analysis of chitooligosaccharides by matrix-assisted laser desorption ionization postsource decay mass spectrometry. Biomacromolecules. 3:696–704. [DOI] [PubMed] [Google Scholar]
- Bodevin-Authelet S, Kusche-Gullberg M, Pummill PE, DeAngelis PL, Lindahl U. 2005. Biosynthesis of hyaluronan: Direction of chain elongation. J Biol Chem. 280:8813–8818. [DOI] [PubMed] [Google Scholar]
- Campbell JA, Davies GJ, Bulone V, Henrissat B. 1997. A classification of nucleotide-diphospho-sugar glycosyltransferases based on amino acid sequence similarities. Biochem J. 326:929–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coates SW, Gurney T Jr., Sommers LW, Yeh M, Hirschberg CB. 1980. Subcellular localization of sugar nucleotide synthetases. J Biol Chem. 255:9225–9229. [PubMed] [Google Scholar]
- Cowman MK, Chen CC, Pandya M, Yuan H, Ramkishun D, LoBello J, Bhilocha S, Russell-Puleri S, Skendaj E, Mijovic J et al.. 2011. Improved agarose gel electrophoresis method and molecular mass calculation for high molecular mass hyaluronan. Anal Biochem. 417:50–56. [DOI] [PubMed] [Google Scholar]
- DeAngelis PL. 1999. Hyaluronan synthases: Fascinating glycosyltransferases from vertebrates, bacterial pathogens, and algal viruses. Cell Mol Life Sci. 56:670–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeAngelis PL, Weigel PH. 1994. Immunochemical confirmation of the primary structure of streptococcal hyaluronan synthase and synthesis of high molecular weight product by recombinant enzyme. Biochemistry. 33:9033–9039. [DOI] [PubMed] [Google Scholar]
- DeAngelis PL, Papaconstantinou J, Weigel PH. 1993. Molecular cloning, identification and sequence of the hyaluronan synthase gene from group A streptococcus pyogenes. J Biol Chem. 268:19181–19184. [PubMed] [Google Scholar]
- Eijsink V, Hoell I, Vaaje-Kolstada G. 2010. Structure and function of enzymes acting on chitin and chitosan. Biotechnol Genet Eng Rev. 27:331–366. [DOI] [PubMed] [Google Scholar]
- Evanko SP, Tammi MI, Tammi RH, Wight TN. 2007. Hyaluronan-dependent pericellular matrix. Adv Drug Deliv Rev. 59:1351–1365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH. 1957. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 226:497–509. [PubMed] [Google Scholar]
- Hascall VC, Wang A, Tammi M, Oikari S, Tammi R, Passi A, Vigetti D, Hanson RW, Hart GW. 2014. The dynamic metabolism of hyaluronan regulates the cytosolic concentration of UDP-GlcNAc. Matrix Biol. 35:14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg CB, Robbins PW, Abeijon C. 1998. Transporters of nucleotide sugars, ATP, and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annu Rev Biochem. 67:49–69. [DOI] [PubMed] [Google Scholar]
- Hubbard C, McNamara JT, Azumaya C, Patel MS, Zimmer J. 2012. The hyaluronan synthase catalyzes the synthesis and membrane translocation of hyaluronan. J Mol Biol. 418:21–31. [DOI] [PubMed] [Google Scholar]
- Itano N, Kimata K. 2002. Mammalian hyaluronan synthases. IUBMB Life. 54:195–199. [DOI] [PubMed] [Google Scholar]
- Jessen TE, Odum L, Johnsen AH. 1994. In vivo binding of human inter-alpha-trypsin inhibitor free heavy chains to hyaluronic acid. Biol Chem Hoppe Seyler. 375:521–526. [DOI] [PubMed] [Google Scholar]
- Jokela TA, Lindgren A, Rilla K, Maytin E, Hascall VC, Tammi RH, Tammi MI. 2008. Induction of hyaluronan cables and monocyte adherence in epidermal keratinocytes. Connect Tissue Res. 49:115–119. doi:10.1080/03008200802148439. [DOI] [PubMed] [Google Scholar]
- Karousou E, Kamiryo M, Skandalis SS, Ruusala A, Asteriou T, Passi A, Yamashita H, Hellman U, Heldin CH, Heldin P. 2010. The activity of hyaluronan synthase 2 is regulated by dimerization and ubiquitination. J Biol Chem. 285:23647–23654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinen V, Karna R, Koistinen A, Arjonen A, Tammi M, Rilla K. 2015. Cell protrusions induced by hyaluronan synthase 3 (HAS3) resemble mesothelial microvilli and share cytoskeletal features of filopodia. Exp Cell Res. 337:179–191. [DOI] [PubMed] [Google Scholar]
- Kultti A, Karna R, Rilla K, Nurminen P, Koli E, Makkonen KM, Si J, Tammi MI, Tammi RH. 2010. Methyl-beta-cyclodextrin suppresses hyaluronan synthesis by down-regulation of hyaluronan synthase 2 through inhibition of Akt. J Biol Chem. 285:22901–22910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang YZ, Zhao X, Liu LL, Yu GL. 2014. Applications of mass spectrometry to structural analysis of marine oligosaccharides. Mar Drugs. 12:4005–4030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lattova E, Perreault H. 2009. Method for investigation of oligosaccharides using phenylhydrazine derivatization. Methods Mol Biol. 534:65–77. [DOI] [PubMed] [Google Scholar]
- Li S-C, Li Y-T. 1970. Studies on the glycosidases of Jack bean meal. III. Crystallization and properties of B-N-acetylhexosaminidase. J. Biol. Chem. 245:5153–5160. [PubMed] [Google Scholar]
- McDonald J, Hascall VC. 2002. Hyaluronan minireview series. J Biol Chem. 277:4575–4579. [DOI] [PubMed] [Google Scholar]
- Medina AP, Lin J, Weigel PH. 2012. Hyaluronan synthase mediates dye translocation across liposomal membranes. BMC Biochem. 13:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K, Palmer JW. 1934. The polysaccharide of the vitreous humor. J Biol Chem. 107:629–634. [Google Scholar]
- Moretto P, Karousou E, Viola M, Caon I, D'Angelo ML, De Luca G, Passi A, Vigetti D. 2015. Regulation of hyaluronan synthesis in vascular diseases and diabetes. J Diabetes Res. 2015:167283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukhopadhyay D, Asari A, Rugg MS, Day AJ, Fulop C. 2004. Specificity of the tumor necrosis factor-induced protein 6-mediated heavy chain transfer from inter-{alpha}-trypsin inhibitor to hyaluronan: Implications fo rthe assembly of the cumulus extracellular matrix. J Biol Chem. 279:11119–11128. [DOI] [PubMed] [Google Scholar]
- Ontong P, Hatada Y, Taniguchi S, Kakizaki I, Itano N. 2014. Effect of a cholesterol-rich lipid environment on the enzymatic activity of reconstituted hyaluronan synthase. Biochem Biophys Res Commun. 443:666–671. [DOI] [PubMed] [Google Scholar]
- Prehm P. 1983. Synthesis of hyaluronate in differentiated teratocarcinoma cells. Mechanism of chain growth. Biochem J. 211:191–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prehm P. 2006. Biosynthesis of hyaluronan: Direction of chain elongation. Biochem J. 398:469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rugg MS, Willis AC, Mukhopadhyay D, Hascall VC, Fries E, Fulop C, Milner CM, Day AJ. 2005. Characterization of complexes formed between TSG-6 and inter-alpha -inhibitor that act as intermediates in the covalent transfer of heavy chains on to hyaluronan. J Biol Chem. 280:25674–25686. [DOI] [PubMed] [Google Scholar]
- Sakr SW, Potter-Perigo S, Kinsella MG, Johnson PY, Braun KR, Goueffic Y, Rosenfeld ME, Wight TN. 2008. Hyaluronan accumulation is elevated in cultures of low density lipoprotein receptor-deficient cells and is altered by manipulation of cell cholesterol content. J Biol Chem. 283:36195–36204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semino CE, Allende ML. 2000. Chitin oligosaccharides as candidate patterning agents in zebrafish embryogenesis. Int J Dev Biol. 44:183–193. [PubMed] [Google Scholar]
- Semino CE, Specht CA, Raimondi A, Robbins PW. 1996. Homologs of the Xenopus developmental gene DG42 are present in zebrafish and mouse and are involved in the synthesis of Nod-like chitin oligosaccharides during early embryogenesis. Proc Natl Acad Sci USA. 93:4548–4553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada E, Matsumura G. 1980. Degradation process of hyaluronic acid by streptomyces hyaluronidase. J Biochem. 88:1015–1023. [DOI] [PubMed] [Google Scholar]
- Spicer AP, McDonald JA. 1998. Characterization and molecular evolution of a vertebrate hyaluronan synthase gene family. J Biol Chem. 273:1923–1932. [DOI] [PubMed] [Google Scholar]
- Tammi RH, Passi AG, Rilla K, Karousou E, Vigetti D, Makkonen K, Tammi MI. 2011. Transcriptional and post-translational regulation of hyaluronan synthesis. FEBS J. 278:1419–1428. [DOI] [PubMed] [Google Scholar]
- Tlapak-Simmons VL, Baggenstoss BA, Clyne T, Weigel PH. 1999. Purification and lipid dependence of the recombinant hyaluronan synthases from Streptococcus pyogenes and Streptococcus equisimilis. J Biol Chem. 274:4239–4245. [DOI] [PubMed] [Google Scholar]
- Tlapak-Simmons VL, Baron CA, Gotschall R, Haque D, Canfield WM, Weigel PH. 2005. Hyaluronan biosynthesis by Class I streptococcal hyaluronan synthases occurs at the reducing end. J Biol Chem. 280:13012–13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlapak-Simmons VL, Baron CA, Weigel PH. 2004. Characterization of the purified hyaluronan synthase from Streptococcus equisimilis. Biochem. 43:9234–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torronen K, Nikunen K, Karna R, Tammi M, Tammi R, Rilla K. 2014. Tissue distribution and subcellular localization of hyaluronan synthase isoenzymes. Histochem Cell Biol. 141:17–31. [DOI] [PubMed] [Google Scholar]
- Van der Holst PPG, Schlaman HRM, Spaink HP. 2001. Proteins involved in the production and peception of oligosaccharides in relation to plant and animal development. Curr Opin Struct Biol. 11:608–616. [DOI] [PubMed] [Google Scholar]
- Vigetti D, Deleonibus S, Moretto P, Bowen T, Fischer JW, Grandoch M, Oberhuber A, Love DC, Hanover JA, Cinquetti R et al. . 2014. Natural antisense transcript for hyaluronan synthase 2 (HAS2-AS1) induces transcription of HAS2 via protein O-GlcNAcylation. J Biol Chem. 289:28816–28826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakrishnan B, Issaree A, Corilo YE, Ferreira CR, Eberlin MN, Peter MG. 2011. MSn of the six isomers of (GlcN)(2)(GlcNAc)(2) aminoglucan tetrasaccharides (diacetylchitotetraoses): Rules of fragmentation for the sodiated molecules and application to sequence analysis of hetero-chitooligosaccharides. Carbohydr Polym. 84:713–726. [Google Scholar]
- Viola M, Karousou E, D'Angelo ML, Caon I, De Luca G, Passi A, Vigetti D. 2015. Regulated hyaluronan synthesis by vascular cells. Int J Cell Biol. 2015:208303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viola M, Vigetti D, Genasetti A, Rizzi M, Karousou E, Moretto P, Clerici M, Bartolini B, Pallotti F, De Luca G et al. . 2008. Molecular control of the hyaluronan biosynthesis. Connect Tissue Res. 49:111–114. [DOI] [PubMed] [Google Scholar]
- Weigel PH. 2002. Functional characteristics and catalytic mechanisms of the bacterial hyaluronan synthases. Int Union Biochem Mol Biol. 54:201–210. [DOI] [PubMed] [Google Scholar]
- Weigel PH. 2004. The bacterial hyaluronan synthases—An update. In: Science of Hyaluronan Today (Hascall, V.C. and Yanagishita, M. editors), chapter 6, www.GlycoForum.gr.jp.
- Weigel PH. 2015. Hyaluronan synthase: The mechanism of initiation at the reducing end and a pendulum model for polysaccharide translocation to the cell exterior. Int J Cell Biol. 2015:367579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weigel PH, DeAngelis PL. 2007. Hyaluronan synthases: A decade-plus of novel glycosyltransferases. J Biol Chem. 282:36777–36781. [DOI] [PubMed] [Google Scholar]
- Weigel PH, Kyossev Z, Torres LC. 2006. Phospholipid dependence and liposome reconstitution of purified hyaluronan synthase. J Biol Chem. 281:36542–36551. [DOI] [PubMed] [Google Scholar]
- Weigel PH, Padgett-McCue AJ, Baggenstoss BA. 2013. Methods for measuring class I membrane-bound hyaluronan synthase activity. Methods Mol. Biol. 1022:229–247. [DOI] [PubMed] [Google Scholar]
- Weigel PH, West CM, Zhao P, Wells L, Baggenstoss BA, Washburn JL. 2015. Hyaluronan synthase assembles chitin oligomers with -GlcNAc(alpha1-->)UDP at the reducing end. Glycobiology. 25:632–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissmann B, Meyer K. 1952. Structure of hyaluronic acid—The glucuronidic linkage. J Am Chem Soc. 74:4729. [Google Scholar]
- Yoshida M, Itano N, Yamada Y, Kimata K. 2000. In vitro synthesis of hyaluronan by a single protein derived from mouse HAS1 gene and characterization of amino acid residues essential for the activity. J Biol Chem. 275:497–506. [DOI] [PubMed] [Google Scholar]
- Zhao M, Yoneda M, Ohashi Y, Kurono S, Iwata H, Ohnuki Y, Kimata K. 1995. Evidence for the covalent binding of SHAP, heavy chains of inter- alpha-trypsin inhibitor, to hyaluronan. J Biol Chem. 270:26657–26663. [DOI] [PubMed] [Google Scholar]