

Abstract
This phase I, randomized, 4‐period, 4‐sequence, double‐blind, active‐ and placebo‐controlled, crossover study assessed the effects of vonoprazan on the QT/QTc interval in healthy subjects. Subjects received single oral doses of vonoprazan 40 mg, vonoprazan 120 mg, moxifloxacin 400 mg (positive control/open label), and placebo. The primary end point was time‐matched, placebo‐corrected, baseline‐adjusted mean Fridericia‐corrected QT interval (ddQTcF). Of 64 subjects (mean age, 37.8 years; 50% male), 63 received all four regimens. One subject discontinued due to nondrug‐related adverse event of tonsillitis. Assay sensitivity was established; lower bound of the one‐sided 95% confidence interval (CI) for ddQTcF was >5 ms between 1.5 and 12 h following moxifloxacin administration. For both doses of vonoprazan, the one‐sided upper 95% CI ddQTcF did not exceed 10 ms. There was no correlation between plasma vonoprazan concentrations and increases in ddQTcF. Vonoprazan was well tolerated. No severe adverse events/deaths were reported. (European Clinical Trials Database Registry: 2011‐004003‐20.)
Study Highlights
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
✓ The effects of therapeutic and supratherapeutic doses of vonoprazan on QT/QTc interval in healthy male and female subjects are unknown.
WHAT QUESTION DID THIS STUDY ADDRESS?
✓ The effect of vonoprazan on the QT/QTc interval in healthy male and female subjects was investigated in accordance with ICH E14.
WHAT THIS STUDY ADDS TO OUR KNOWLEDGE
✓ Study results provided compelling evidence on the cardiovascular safety of vonoprazan at doses of 40 mg and 120 mg in healthy adult subjects.
HOW THIS MIGHT CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE
✓ The study supports the cardiovascular safety of vonoprazan, which belongs to a novel drug class for treatment of acid‐related diseases.
Potassium‐competitive acid blockers (P‐CABs) represent a novel class of acid‐suppressing agents that are a potential alternative to proton pump inhibitors (PPIs) for the treatment of gastroesophageal reflux disease.1 In contrast to PPIs, P‐CABs are acid‐stable H+, K+‐ATPase inhibitors that inhibit H+, K+‐ATPase noncovalently in a K+‐competitive manner.2
Vonoprazan (Takeda Pharmaceutical Company Ltd. Osaka, Japan), launched in Japan on 26 February 2015, is an orally available P‐CAB, approved for the treatment of adult patients with acid‐related diseases including gastric ulcer, duodenal ulcer, reflux esophagitis, and the prevention of recurrence of gastric or duodenal ulcer during low‐dose aspirin or nonsteroidal antiinflammatory drug (NSAID) administration. Vonoprazan is also approved as an adjunct to Helicobacter pylori eradication in patients with gastric ulcer, duodenal ulcer, gastric mucosa‐associated lymphatic tissue (MALT) lymphoma, idiopathic thrombocytopenic purpura, H. pylori gastritis, or after endoscopic resection of early‐stage gastric cancer.3
Clinical studies of single ascending doses4 (1–120 mg), as well as multiple repeated‐doses5 (10–40 mg) of vonoprazan, confirmed its tolerability in healthy subjects, with a rapid onset of action (<4 h) and prolonged duration (24 h) of action. Vonoprazan has a potent and long‐lasting antisecretory effect on H+, K+‐ATPase due to its high accumulation and slow clearance from gastric tissue.1
Cardiac safety studies have been an integral part of the development of investigational drugs, identifying drugs with potential cardiovascular liability as well as providing opportunities for safer drug administration regimens.6 QT prolongation is associated with cardiac arrhythmias, and may lead to torsades de pointes, which can be fatal.7 The International Conference on Harmonization (ICH) E14 guidelines recommend that all non‐antiarrhythmic drugs undergo clinical evaluation of absolute QT interval/corrected QT interval (QT/QTc) prolongation and proarrhythmic potential by means of a thorough QT/QTc study early in the course of their clinical development.8
Electrocardiogram (ECG) parameters for vonoprazan in clinical trials have not shown any clinically meaningful QT/QTc prolongation.4, 5, 9, 10, 11, 12, 13, 14 Despite the lack of cardiac arrhythmic events in the extensive clinical development program, a thorough QT study was conducted to provide a definitive assessment of the effects of vonoprazan on the QT interval. The aim of this study was to assess the effect of vonoprazan, at both therapeutic and supratherapeutic doses, compared with moxifloxacin as a positive control and placebo, on the QT interval in healthy subjects. The study design was based on the ICH E14 guidance.8
METHODS
Study design
This phase I, randomized, 4‐period, 4‐sequence, double‐blind, placebo‐ and active‐controlled (moxifloxacin/open label), crossover study examined the effect of vonoprazan 40 mg, vonoprazan 120 mg, and moxifloxacin 400 mg on the QT/QTc interval of healthy male and female subjects (Figure 1). The study was conducted between January and April 2012 at a single site in Rennes, France. The study design followed the general design principles outlined in the ICH‐E14 guidance for the clinical evaluation of the QT/QTc interval with non‐antiarrhythmic drugs (M. Hayashi, H. Jenkins, D. Crawford, and R. Jenkins, Takeda Global Investigator's Brochure TAK‐438, vonoprazan fumarate). This study was conducted in compliance with the Institutional Review Board regulations stated in Title 21 of the United States Code of Federal Regulations Part 56, Good Clinical Practice regulations and guidelines, the Declaration of Helsinki, and all applicable local regulations. All participants provided informed consent before initiation of any study‐related procedure. This trial was registered in the European Clinical Trials Database Registry (EudraCT) under registration number 2011‐004003‐20.
Figure 1.
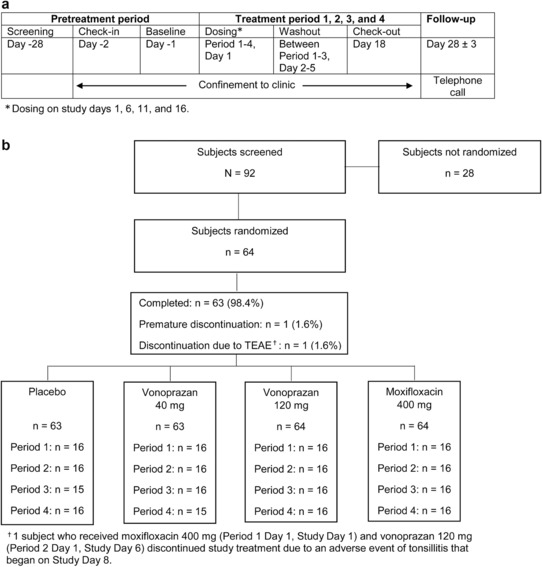
(a) Study design. (b) Patient disposition. TEAE, treatment‐emergent adverse event.
Study population
Eligible subjects for the phase I QT/QTc study included healthy males and females between 18 and 55 years of age with a body mass index (BMI) ≥18 and ≤30 kg/m2. Women were of nonchild‐bearing potential or agreed to protocol‐specified methods of contraception. All subjects had clinical laboratory evaluations within the reference range of the testing laboratory. Subjects were excluded if they had received any investigational compound within 90 days before the first dose of the study drug, or if they had previously received vonoprazan. Subjects were also excluded if there was evidence or a history of clinically significant cardiovascular, neurologic, hepatic, hematologic, respiratory, renal, allergic, metabolic, or endocrine disease; current or recent (within 6 months) gastrointestinal disease; any condition impairing optimal drug absorption; any additional risk factors for torsades de pointes; QT interval corrected by Fridericia formula (QTcF) >430 ms for males or >450 ms for females, PR interval >240 ms or QRS interval >110 ms, or bradycardia (<45 bpm) at the screening or baseline visit or before dosing; positive test for human immunodeficiency virus antibody, and for any other prespecified exclusion criterion.
Treatment protocol
Vonoprazan and placebo were provided by Takeda Pharmaceutical Company, and moxifloxacin was provided by Bayer HealthCare Pharmaceuticals (Whippany, NJ). Subjects were randomized to one of four sequences to receive each of the four treatments in a crossover fashion, using a Williams design. Subjects received single doses of blinded vonoprazan 40 mg (maximum therapeutic dose), vonoprazan 120 mg (supratherapeutic dose), placebo, or open‐label moxifloxacin 400 mg at the start of four treatment periods on study Days 1, 6, 11, and 16. All study drugs were administered orally. As the supratherapeutic dose of vonoprazan, a dose of 120 mg was selected and considered adequate, as patients with comorbidities may receive therapeutic dose adjustments in clinical settings. The supratherapeutic dose of vonoprazan provided at least ∼10 times higher exposure than the clinical dose (usually 20 mg or 10 mg, depending on indication) approved in Japan (Takecab Label). TAK‐438 is not expected to accumulate to any clinically relevant degree in the clinical setting.
Placebo‐control and positive‐control (moxifloxacin) groups were included to meet ICH E14 requirements and to demonstrate assay sensitivity.6 A dose of 40 mg was selected as the therapeutic dose for this study because in a previous phase II study,10 40 mg was the maximum dose used and doses ≥20 mg were expected to be used for different indications. The supratherapeutic dose in this study was 120 mg because it was the highest dose administered in healthy subjects in a phase I study4 and was well tolerated. Further, the exposures observed at this dose would be above the predicted “worst‐case scenario” exposures (the expected highest exposure) determined using physiologically based pharmacokinetic modeling. The 400 mg dose of moxifloxacin was selected because it is the recommended daily dose for adults in thorough QT studies.8, 16 All treatments were separated by a 4‐day washout period (Day 2 to Day 5 of each period). All study drugs were taken with 240 mL of water and subjects refrained from drinking water for at least 1 h postdose. Subjects fasted for 10 h before dosing and 4 h after dosing. The study duration, from screening to follow‐up, was a maximum of 59 days including the screening period through the follow‐up telephone call. Subjects stayed at the clinic from check‐in (Day –2) to check‐out (Day 18) of the study.
Randomization and blinding
The randomization schedule for this study was generated by the Takeda Global Research & Development Centre Europe (TGRD (EU)) Analytical Science Department. All randomization information was secured and housed in a locked storage area, accessible by the randomization officer at TGRD. The randomization schedule was also sent to the site so that the site pharmacist could access it. Subjects were assigned to a randomization sequence number by the order of enrollment. Equal numbers of male and female subjects were enrolled in each treatment sequence. Vonoprazan 40 mg, vonoprazan 120 mg, and placebo were administered in a double‐blind manner, and moxifloxacin 400 mg was administered open‐label.
Electrocardiographic methods
Study ECGs were acquired digitally from a 24‐h Holter recorder (H12+; Mortara Instrument, Milwaukee, WI) at baseline and on each dosing day. For baseline ECG parameters, there was 1 baseline day before Period 1, where measurements were made at the same timepoints as during a dosing day. Three 10‐s ECG recordings were extracted at predose (0 h) and at 0.5, 1, 1.5, 2, 3, 4, 5, 5.5, 6, 8, 12, 16, and 24 h after dosing. In addition to Holter monitoring, 12‐lead ECGs for standard assessment of safety were measured at screening, check‐in (Day –2), baseline (Day –1), and on dosing days (Days 1, 6, 11, and 16) at predose (0 h) and at 1, 2, 4, 8, 12, 24, and 48 h after dosing. Patients were supine and at rest during ECG recording. All ECG measurements were performed at a central laboratory (Biotrial Core Lab, France) using controlled standard procedures and interpreted by a trained cardiologist. All ECG readers and technicians were blinded to study treatment, timepoints, study day, and subject identifiers (including age and sex).
Pharmacokinetic methods
To determine drug concentrations in plasma, serial venous blood samples were collected into chilled vacutainers at predose (0 h) on the day of dosing and at 0.5, 1, 1.5, 2, 3, 4, 5, 5.5, 6, 8, 12, 16, 24, and 48 h after dosing. Concentrations of vonoprazan and moxifloxacin in plasma were measured by a fully validated liquid chromatographic method with tandem mass spectrometry detection (LC‐MS/MS). The assays were validated over the vonoprazan concentration range of 0.100–100 ng/mL, and moxifloxacin concentration range of 25–5,000 ng/ml in human plasma. Pharmacokinetic parameters were derived using noncompartmental methods with WinNonlin Enterprise v. 5.2 (Pharsight, Mountain View, CA). Pharmacokinetic parameters of interest included the maximum observed concentration (Cmax), time to reach Cmax (Tmax), area under the plasma concentration–time curve (AUC) from time 0 to time of the last quantifiable concentration (AUC0‐tlqc), AUC from time 0 to infinity (AUC0‐inf), terminal elimination half‐life (T1/2), terminal elimination rate constant (λz), apparent volume of distribution (Vz/F), and apparent clearance after oral administration (CL/F). Pharmacokinetic calculations were based on actual collection times recorded during the study.
Outcome measures
The primary end point was the mean difference in the postdose, time‐matched, baseline‐adjusted QTcF between vonoprazan and placebo and between moxifloxacin and placebo (ddQTcF).
Secondary end points included the concentration of vonoprazan in plasma, primary pharmacokinetic variables derived from the concentrations of moxifloxacin and vonoprazan, the mean difference in the postdose, time‐matched, QT interval corrected by Bazett's formula (QTcB), QT interval corrected by individual (QTcI), and QT interval study‐specific correction (QTcss), and QT between vonoprazan and placebo and between moxifloxacin and placebo (ddQTcB, ddQTcI, ddQTcss). For each treatment, QT intervals (QTcF, QTcB, QTcI, QTcss, and QT) categorized with respect to the number and percentage of subjects having time‐matched increases >30 ms and >60 ms from baseline were recorded, as were the number and percentage of subjects having QT intervals >450 ms, >480 ms, and >500 ms, and the number and percentage of subjects with >30 ms change from baseline and >450 ms; and for each treatment heart rates, PR intervals, QRS intervals, RR intervals, and T and U wave complex morphology were also recorded.
Safety end points included treatment‐emergent adverse events (TEAEs), clinical laboratory parameters, vital signs (blood pressure, heart rate, tympanic temperature, and body weight), and 12‐lead ECG.
Statistical analysis
Sample size
Assuming the true difference in the largest time‐matched mean change of QTcF from baseline between vonoprazan and placebo was no more than 5 ms, 60 evaluable subjects were needed to provide 93% power to establish no QT prolongation effect (defined as the upper limit of the two‐sided 90% confidence interval (CI) for the comparison of vonoprazan vs. placebo was <10 ms) of vonoprazan. This sample size also provided at least 93% power to demonstrate assay sensitivity (defined as the lower limit of the two‐sided 90% CI for the comparison of moxifloxacin vs. placebo being greater than 5 ms for at least 1 timepoint between 1 h and 4 h postdose). This assumed that the true difference in the largest time‐matched mean change from baseline between moxifloxacin and placebo was ≥10 ms. A total of 64 subjects were planned and randomized in this study to account for potential dropouts.
Electrocardiography
Baseline ECG measurements were used for time‐matched baselines. QTc values were derived using four different correction methods (QTcF, QTcB, QTcI, and QTcss). QTcF was selected as the primary correction method. For QT and each QT correction method, the difference between the study drug and placebo time‐matched baseline‐adjusted QT (ddQTc) was calculated. The change from baseline in QTc interval at each timepoint (dQTc) was used as the dependent variable. This analysis was conducted using a repeated‐measures, mixed‐effects model with time, sequence, period, treatment, and time‐by‐treatment interaction as fixed effects and subjects nested within sequence as a random effect, with average baseline QTc as covariate. The analysis included the following timepoints: 0.5, 1, 1.5, 2, 3, 4, 5, 5.5, 6, 8, 12, 16, and 24 h postdose. For the primary end point, ddQTcF, no QT prolongation effect of vonoprazan was declared if the upper limit of one‐sided 95% CI at each timepoint was below 10 ms. The study assay sensitivity analysis required moxifloxacin's lower limit of one‐sided 95% CI to exceed 5 ms at 1 or more of the timepoints from 1 to 4 h postdose. There was no adjustment for multiplicity. Uncorrected and corrected QT intervals and heart rates, PR intervals, QRS intervals, and RR intervals were summarized at each timepoint for both vonoprazan doses, for moxifloxacin, and for placebo.
Categorical analysis of QTcF was performed to determine the number and percentage of subjects per group who had an increase from baseline at any postdose timepoint of >30 ms and >60 ms. In addition, the number and percentage of subjects per group who had a postdose QTcF of >450 ms, >480 ms, and >500 ms at any postdose timepoint were summarized.
Pharmacokinetic analyses
For pharmacokinetic parameters of interest, descriptive statistics (number of subjects (N), arithmetic mean, standard deviation (SD), percent coefficient of variation (%CV), median, minimum, and maximum) were used to summarize plasma vonoprazan concentrations for both doses and moxifloxacin concentrations at each scheduled timepoint.
Pharmacokinetic and pharmacodynamics analyses
A linear mixed effects model of the plasma vonoprazan concentration–QT response relationship was built, assuming random slope and intercept terms. The dependent variable was ddQTc calculated for each subject.
Safety analyses
All safety assessments were summarized by treatment group; TEAEs were coded by system organ class (SOC) and preferred term (PT) using the Medical Dictionary for Regulatory Activities (MedDRA) version (M. Hayashi, H. Jenkins, D. Crawford, and R. Jenkins, Takeda Global Investigator's Brochure TAK‐438, vonoprazan fumarate). Subjects were assessed for events throughout the study, and they could report events at any time during the study. All data analyses, tables, graphs, and listings were generated using SAS v. 9.2 (SAS Institute, Cary, NC). All statistical tests were two‐tailed with significance at α = 0.05 unless otherwise stated.
RESULTS
Demographics and baseline clinical characteristics
Between January 2012 and April 2012, 92 subjects were screened. Of these 92 subjects, 64 were randomly assigned to one of the four treatment sequences and received at least one dose of the study drug (Figure 1 a). Sixty‐three subjects received all four regimens (Figure 1 b); one subject discontinued the study due to a nondrug‐related adverse event of tonsillitis following administration of moxifloxacin 400 mg in Period 1 and vonoprazan 120 mg in Period 2.
Demographics and other baseline characteristics were similar among the four treatment sequence groups (Table 1). The mean age of all subjects was 37.8 (SD ± 11.2 years), and mean BMI was 24.0 (SD ± 2.89) kg/m2. Male and female subjects were evenly divided (32 males and 32 females). A majority (93.8%) of the subjects were White, 4.69% were African American, and 1.56% were Asian.
Table 1.
Demographic and baseline characteristics
| Treatment sequencea | |||||
|---|---|---|---|---|---|
| Characteristic | Sequence ABDC n = 16 | Sequence BCAD n = 16 | Sequence CDBA n = 16 | Sequence DACB n = 16 | Total N = 64 |
| Age, mean (SD), years | 33.2 (11.9) | 42.9 (8.52) | 38.1 (11.1) | 36.9 (11.6) | 37.8 (11.2) |
| Sex, n (%) | |||||
| Male | 8 (50.0) | 8 (50.0) | 8 (50.0) | 8 (50.0) | 32 (50.0) |
| Female | 8 (50.0) | 8 (50.0) | 8 (50.0) | 8 (50.0) | 32 (50.0) |
| Race, n (%) | |||||
| White | 15 (93.8) | 15 (93.8) | 15 (93.8) | 15 (93.8) | 60 (93.8) |
| Asian | 0 | 0 | 1 (6.25) | 0 | 1 (1.56) |
| Black or African American | 1 (6.25) | 1 (6.25) | 0 | 1 (6.25) | 3 (4.69) |
| Height,b mean (SD), cm | 171 (8.87) | 170 (9.10) | 168 (10.4) | 170 (8.68) | 170 (9.13) |
| Weight,c mean (SD), kg | 66.3 (8.82) | 70.1 (11.8) | 67.1 (8.48) | 72.4 (12.7) | 69.0 (10.7) |
| BMI,d mean (SD), kg/m2 | 22.6 (1.82) | 24.3 (3.21) | 23.9 (2.874) | 25.0 (3.15) | 24.0 (2.89) |
| Smoking status, n (%) | |||||
| Never smoked | 8 (50.0) | 11 (68.8) | 10 (62.5) | 12 (75.0) | 41 (64.1) |
| Current smoker | 0 | 0 | 0 | 0 | 0 |
| Exsmoker | 8 (50.0) | 5 (31.3) | 6 (37.5) | 4 (25.0) | 23 (35.9) |
| Alcohol consumption, n (%) | |||||
| Never consumed | 15 (93.8) | 14 (87.5) | 15 (93.8) | 16 (100) | 60 (93.8) |
| Current consumer | 1 (6.3) | 2 (12.5) | 1 (6.3) | 0 | 4 (6.3) |
| Ex consumer | 0 | 0 | 0 | 0 | 0 |
BMI, body mass index; SD, standard deviation.
aFor all treatment sequences, A = vonoprazan 120 mg, B = vonoprazan 40 mg, C = placebo,
and D = moxifloxacin 400 mg.
bCollected/measured at screening.
cMeasured prior to the first dose of study drug.
dBMI is calculated from the weight taken prior to the first dose of study drug and height taken at screening.
Electrocardiographic results
The time‐matched least squares (LS) mean difference in dQTcF between moxifloxacin and placebo (ddQTcF) increased at 1 h after dosing of moxifloxacin, and the lower bound of the one‐sided 95% CI of the dQTcF difference from 1.5 h to 12 h after dosing of moxifloxacin was >5 ms (Figure 2). The peak effect occurred at 4 h after dosing with moxifloxacin (12.0 ms, with a lower bound of 9.90 ms for the one‐sided 95% CI).
Figure 2.
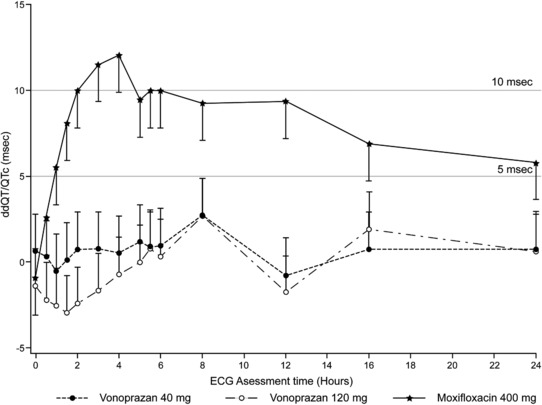
Least‐square means (95% CI) of ddQT/QTcF vs. electrogardiographic assessment time by treatment. CI, confidence interval; ddQT/QTcF, mean change from baseline QT interval corrected by Fridericia method.
For both vonoprazan 40 mg and 120 mg, the maximum upper bound of the one‐sided 95% CI of the difference in the means of dQTcF estimated using the linear mixed effect model from placebo (ddQTcF) was 4.9 ms, which occurred at 8 h after dosing of vonoprazan.
For vonoprazan or placebo, no subject experienced an absolute QTcF value >450 ms at any postdose timepoint or had changes from baseline in QTcF >30 ms at any postdose timepoint. For moxifloxacin 400 mg, one subject had maximum QTcF >450 ms postdose, and four subjects had changes from baseline in QTcF >30 ms postdose (Table 2). Results of QTcB, QTcI, and QTcss were consistent with that of QTcF (Supplementary Table S1).
Table 2.
QT/QTc interval categorical analysisa
| Number of subjects (%) | |||||
|---|---|---|---|---|---|
| QTcF (ms) | Baseline (Day –1) n = 64 | Placebo n = 63 | Vonoprazan 40 mg n = 63 | Vonoprazan 120 mg n = 64 | Moxifloxacin 400 mg n = 64 |
| >450 ms | 0 | 0 | 0 | 0 | 1 (1.56) |
| >480 ms | 0 | 0 | 0 | 0 | 0 |
| >500 ms | 0 | 0 | 0 | 0 | 0 |
| >30 ms increase from Baseline | – | 0 | 0 | 0 | 4 (6.25) |
| >60 ms increase from Baseline | – | 0 | 0 | 0 | 0 |
| >30 ms increase from Baseline and | – | 0 | 0 | 0 | 0 |
| >450 ms | |||||
QTcF, QT interval corrected by Fridericia formula.
aResults of QTcB, QTcI, and QTcss (Supplementary Material) were consistent with that of QTcF.
For vonoprazan 40 mg and 120 mg, there was a decrease in baseline heart rate with a mean range from 2 to 6 bpm starting from 4 h postdose to 6 h postdose. No U‐waves were observed in any subject. Similar percentages of T‐wave abnormalities were observed for all treatments and were similar to baseline measurements.
Pharmacokinetic results
Following single doses of vonoprazan 40 mg and 120 mg, the median Tmax values were reached at 2 h postdose. The mean plasma Cmax and exposure increased in more than a dose‐proportional manner (Table 3).
Table 3.
Pharmacokinetic properties
| Pharmacokinetic parameter | Vonoprazan 40 mg (n = 63) | Vonoprazan 120 mg (n = 64) | Moxifloxacin 400 mg (n = 64) |
|---|---|---|---|
| AUC0‐tlqc a (hr·ng/mL) | 490 (181) | 2,420 (720) | 33,000 (6,670) |
| AUC0‐inf a (hr·ng/mL) | 498 (187) | 2,470 (745) | 35,400 (7,350) |
| Cmax a (ng/mL) | 47.1 (15.8) | 226 (76.5) | 2,350 (560) |
| Tmax b (hr) | 2.00 (1.00, 5.00) | 2.00 (1.00, 6.00) | 2.00 (0.500, 5.00) |
| λza (1/hr) | 0.0910 (0.0130) | 0.0910 (0.0150) | 0.0560 (0.00700) |
| T1/2 a (hr) | 7.80 (1.17) | 7.90 (1.47) | 12.6 (1.69) |
| CL/Fa (L/hr) | 91.9 (34.3) | 53.2 (16.1) | 11.7 (2.19) |
| Vz/Fa (L) | 1,000 (327) | 592 (160) | 213 (48.6) |
λz, terminal elimination rate constant; AUC0‐tlqc, area under the plasma concentration–time curve from time 0 to time of the last quantifiable concentration; AUC0‐inf, area under the plasma concentration–time curve from time 0 to infinity; CL/F, apparent clearance after oral administration; Cmax, maximum observed concentration; T1/2, terminal elimination half‐life, Tmax, time to reach maximum observed concentration; Vz/F, apparent volume of distribution.
aMean and standard deviation values are presented.
bMedian and range (min, max) are presented.
The median plasma moxifloxacin Tmax (2.0 h) was within the 24‐h pharmacodynamic assessment period, indicating that the pharmacodynamic measurements occurred during peak plasma moxifloxacin levels.
After a 5‐day wash out, carryover was detected in 3 of the 64 subjects for TAK‐438. On each occasion, vonoprazan 120 mg had been administered in the prior period. Two of the subjects were receiving placebo and had verifiable concentrations above the limit of quantitation (LoQ). The other subject had TAK‐438 predose concentrations above the LoQ, but which were <0.3% of the Cmax of the subsequent profile (40 mg) for the subject.
Pharmacokinetic and pharmacodynamic relationship analysis
Results from the linear model to evaluate the relationship between ddQTc and plasma vonoprazan concentration showed that there was no increase in the QTc interval with increasing concentration of vonoprazan (Table 4 , Figure 3). After administration of a single 40‐mg dose of vonoprazan, the model‐based estimates of ddQTcF, ddQTcB, ddQTcI, and ddQTcss at the mean plasma vonoprazan 40 mg Cmax (47.1 ng/mL) ranged from –1.10 (90% CI –2.16, –0.0454) to 0.274 (90% CI –0.781, 1.33). After administration of a single 120‐mg dose of vonoprazan, the model‐based estimates of ddQTcF, ddQTcB, ddQTcI, and ddQTcss at the mean plasma vonoprazan 120 mg Cmax (226 ng/mL) ranged from –4.49 (90% CI –6.26, –2.73) to –1.82 (90% CI –3.39, –0.246).
Table 4.
Linear relationship between ddQTc and plasma vonoprazan concentration
| Solution for fixed effects | Estimate of ddQTc at Cmax of vonoprazan | |||||||
|---|---|---|---|---|---|---|---|---|
| Intercept | Slope | Vonoprazan 40 mg | Vonoprazan 120 mg | |||||
| ECG parameter (ms) | Estimate | P‐Value | Estimate | P‐Value | Estimate | 90% CIa | Estimate | 90% CI |
| QTcFb | 0.814 | 0.204 | –0.0117 | 0.00160d | 0.263 | –0.787, 1.31 | –1.83 | –3.39, –0.263 |
| QTcB | –0.209 | 0.747 | –0.0190 | ≤.0001e | –1.10 | –2.16, –0.0454 | –4.49 | –6.26, –2.73 |
| QTcI | 0.812 | 0.206 | –0.0129 | 0.00140d | 0.204 | –0.853, 1.26 | –2.11 | –3.77, –0.445 |
| QTcss | 0.825 | 0.200 | –0.0117 | 0.00160d | 0.274 | –0.781, 1.33 | –1.82 | –3.39, –0.246 |
| QT | 2.71 | 0.0217c | 0.00230 | 0.697 | 2.82 | 0.920, 4.72 | 3.24 | 0.541, 5.93 |
CI, confidence interval; Cmax, maximum observed concentration; ddQTc, change from baseline, placebo‐corrected QT; ECG, electrocardiogram; QTcB, QT interval corrected by Bazett's formula; QTcF, QT interval corrected by Fridericia formula; QTcI, QT interval corrected by individual; QTcss, QT interval study specific corrected.
aTwo‐sided 90% CI.
bddQTcF at a particular concentration = 0.8139 – 0.0117 × concentration.
cIndicates statistical significance at the 0.05 level.
dIndicates statistical significance at the 0.01 level.
eIndicates statistical significance at the 0.001 level.
Figure 3.
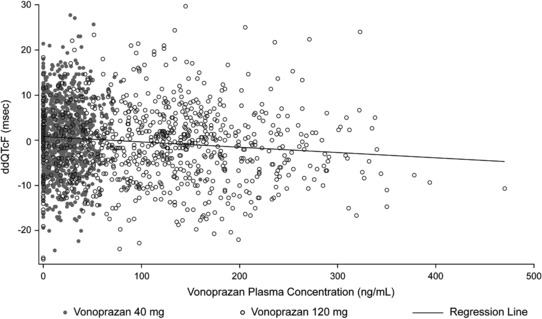
Linear relationship between ddQTcF and plasma vonoprazan concentration. ddQTcF, mean change from baseline QT interval corrected by Fridericia method.
Safety and tolerability measures
Overall, 31 of 64 subjects (48.4%) experienced ≥1 TEAE during the study (Supplementary Table S2). Similar incidences of adverse events were reported during vonoprazan 40 mg, vonoprazan 120 mg, and moxifloxacin 400 mg treatments (17.5%, 18.8%, and 18.8%, respectively). The majority of the TEAEs experienced during the study were mild (44 of 52 events); 42 of 52 events were study drug‐related. No events were considered severe, and no deaths were reported.
The most common TEAEs in subjects who were administered vonoprazan 40 mg and 120 mg were headache (9.52% and 7.81%, respectively) and nausea (1.6% and 7.8%, respectively). A greater percentage of subjects had markedly abnormal QT values following moxifloxacin administration (6.25%) than with placebo (1.59%), vonoprazan 40 mg (3.17%), or vonoprazan 120 mg (3.13%). No markedly abnormal values were considered clinically significant.
No findings from safety laboratory tests (serum chemistry, hematology, and liver function tests), vital signs, or urinalysis values were reported as adverse events, and no clinically significant changes from baseline were observed.
DISCUSSION
Vonoprazan is shown to have effects on hERG channel current at the doses studied with an IC50 value of 4.8 μg/mL. Although TAK‐438 showed a statistically significant inhibitory effect on the hERG channel current at all concentrations examined (0.5, 5, and 50 μg/mL), based on a Cmax value of 59.9 ng/mL seen at the highest expected human dose (40 mg) of TAK‐438 and the percentage human plasma protein binding of TAK‐438 ranging from 85.2–88.0%, the safety margin based on free drug is calculated to be >540‐fold. The metabolites of vonoprazan are inactive and have not been tested in vitro for their inhibitory effect on hERG. A single administration of TAK‐438 at doses up to 20 mg/kg had no acute effects on the dog cardiovascular system (M. Hayashi, H. Jenkins, D. Crawford, and R. Jenkins, Takeda Global Investigator's Brochure TAK‐438, vonoprazan fumarate).
Prolongation of cardiac repolarization, as measured by the QT interval, can potentially increase the probability of fatal cardiac arrhythmias.15 The ICH E14 guidance recommends conducting a thorough QT/QTc study for all new non‐antiarrhythmic drugs to characterize their effects on QT intervals.8, 16 As part of the clinical development program, this phase I, randomized, single‐center, 4‐period, 4‐sequence, crossover study was designed to examine the effect of therapeutic and supratherapeutic doses of vonoprazan, a novel orally active P‐CAB, on the QT/QTc interval in healthy male and female subjects by analyzing the mean difference in the postdose, time‐matched, baseline‐adjusted QTcF between vonoprazan and placebo and between moxifloxacin and placebo. To prevent reader bias and to maintain consistency in QT measurement, the ICH E14 guidance states that ECGs should be read in a central laboratory by a small group of trained readers blinded to both treatment and patient identity.8 In the present study, continuous Holter ECG recordings were performed for 24 h at baseline and following each treatment regimen. ECGs were extracted at scheduled pharmacokinetic timepoints for blinded review by a trained cardiologist at the central laboratory. The maximum upper bound of the one‐sided 95% CI for ddQTcF for TAK‐438 40 and 120 mg were below the threshold of regulatory concern of 10 ms.8 The findings from this study demonstrate that, in healthy subjects, vonoprazan did not cause concern about QTc prolongation according to the ICH E 14 criteria, and was well tolerated.
In accordance with the ICH E14 guidelines, a positive control was used in this study to verify the assay sensitivity. This study included moxifloxacin 400 mg administered as a single dose at one of the four periods to each subject open‐label. Assay sensitivity was established in this study because the lower bound of the one‐sided 95% CI for ddQTcF was >5 ms between 1.5 and 12 h postdose. Therefore, QT data for vonoprazan could be interpreted, and following 40 mg and 120 mg doses of vonoprazan, the maximum upper bound of the one‐sided 95% CI for ddQTcF was 4.9 ms. This is less than the threshold of concern when the maximum upper bound of the one‐sided 95% CI for ddQTcF is >10 ms.
Since the QT interval shortens with increasing heart rate, QT correction methods are used.17 In this study, QT interval correction methods that were used included QTcF, QTcB, QTcss, and QTcI. An assessment of the QT correction methods using Spearman correlation coefficients indicated that the most appropriate intervals for assessment were QTcF, QTcI, and QTcss. Assessment and correction of QT/QTc by multiple correction methods confirmed that vonoprazan did not significantly prolong the QT/QTc in healthy subjects. There were no subjects with a QTcF interval >450 ms or an increase from baseline >30 ms after administration of vonoprazan.
For vonoprazan 40 mg and 120 mg, there was a small decrease from baseline in heart rate with a mean range of 2 to 6 bpm between 4 h and 6 h postdose. This decrease was considered not clinically significant and was not expected to confound heart‐rate correction of QT values. No clinically significant U‐waves or T‐waves were observed in any subject. In addition, no cardiac adverse events were reported. The results obtained from ECG recordings in this study are consistent with a lack of clinically meaningful changes for any ECG parameters observed during previous clinical studies of vonoprazan to date.4, 5, 9 In addition, an analysis of dQTcF data from earlier phase I single rising dose PK studies (data on file) showed no statistical evidence of QT prolongation, with the rate of change of dQTcF per unit of concentration of TAK‐438 of –0.023 (ms/ng/mL) (95% CI –0.65, 0.19).
Similar to data from previous studies,4, 5 plasma concentrations of vonoprazan increased in a slightly more than dose‐proportional manner. Thorough QT studies should ensure that dose–response and concentration–response relationships for QTc prolongation are fully characterized, including exploration of concentrations higher than those anticipated in therapeutic use. In this study, there was no evidence of QT/QTc prolongation or arrhythmogenic effects with maximum clinical and supratherapeutic doses of vonoprazan, and results from the linear model to evaluate the relationship between ddQTc and plasma vonoprazan concentration showed that there was no increase in the QTc interval with increasing concentration of vonoprazan.
The safety profile of vonoprazan in this study was consistent with previous studies of vonoprazan in healthy subjects.4, 5, 9 Single doses of vonoprazan at 40 mg and 120 mg were well tolerated and most adverse events were considered mild or moderate in intensity. No deaths or serious adverse events were reported during the study. No abnormal laboratory values were reported. Hepatic laboratory values were within normal limits.
A potential limitation of this study is that the study was conducted in healthy volunteers, rather than the target population. Another limitation is that the current study evaluated the effects on the QT interval after only a single dose of vonoprazan, while the recommended oral dose of vonoprazan is 10 mg or 20 mg once daily for 4 weeks to 8 weeks, depending on the indication.3 However, in a phase I study of vonoprazan 10 mg to 40 mg once daily for 7 consecutive days in healthy Japanese males (N = 60), there was no clinically relevant accumulation of vonoprazan or its metabolites with similar pharmacokinetic parameters on Days 1 and 7.5 Although the long‐term effects of vonoprazan on QT/QTc in patients cannot be determined from the current study, long‐term clinical studies with vonoprazan lasting up to 2 years have not identified clinically meaningful changes in serum electrolyte levels or ECG parameters (unpublished data; OCT‐301, Mizokami Y, Oda K, Funao N, Nishimura A, Soen S, Kawai T, Ashida K, et al.; and OCT‐302, Kawai T, Oda K, Funao N, Nishimura A, Matsumoto Y, Mizokami Y, Ashida K, et al.)
The results of this study are strengthened by its randomized, 4‐period, 4‐sequence, placebo‐ and active‐controlled crossover study design outlined in the ICH E14 guidelines for the evaluation of the QT/QTc interval with non‐antiarrhythmic drugs. Assay sensitivity was established in the study by the use of moxifloxacin as a positive control to detect clinically significant changes from baseline in QT/QTc. The present study utilized a supratherapeutic dose of vonoprazan in addition to a potential maximum therapeutic dose. Given that there was no increase in the QTc interval at the supratherapeutic dose, it can be expected that there will not be an effect of vonoprazan on the QTc interval at the dose targeted for clinical use.
In conclusion, this study met the requirements of a negative QT/QTc study and further supports the assertion that there is a limited likelihood that a clinically meaningful QT prolongation would occur in a clinical setting during treatment with vonoprazan.
Supporting information
Supplementary Table S1. QT/QTc interval categorical analysis.
Supplementary Table S2. Summary of TEAEs reported in ≥2 subjects during the study.
Acknowledgments
This study was sponsored by Takeda Global Research & Development Centre (Europe) Ltd. Medical writing assistance was provided by Sneha D'Silva, MD, and Tania Dickson, PhD, of ProScribe – Envision Pharma Group, and was funded by Takeda Pharmaceutical Company.
Author Contributions
H.J., B.A., and R.J., designed the research; H.J., B.A., and R.J. performed the research; H.J., B.A., and R.J. analyzed the data.
Conflict of Interest
R.J. and H.J. are employees of Takeda Pharmaceutical Company and have no other relevant financial disclosures. BA is an employee of Biotrial which received research funding from Takeda Pharmaceutical Company to perform the study.
References
- 1. Hori, Y. et al 1‐[5‐(2‐Fluorophenyl)‐1‐(pyridin‐3‐ylsulfonyl)‐1H‐pyrrol‐3‐yl]‐N‐methylmethanamin e monofumarate (TAK‐438), a novel and potent potassium‐competitive acid blocker for the treatment of acid‐related diseases. J. Pharmacol. Exp. Ther. 335, 231–238 (2010). [DOI] [PubMed] [Google Scholar]
- 2. Andersson, K. & Carlsson, E. Potassium‐competitive acid blockade: a new therapeutic strategy in acid‐related diseases. Pharmacol, Ther, 108, 294–307 (2005). [DOI] [PubMed] [Google Scholar]
- 3. Takeda . New drug application approval of TAKECAB® for the treatment of acid‐related diseases in Japan (press release). http://www.takeda.com. (26 Dec 2014).
- 4. Sakurai, Y. et al Safety, tolerability, pharmacokinetics, and pharmacodynamics of single rising TAK‐438 (vonoprazan) doses in healthy male Japanese/non‐Japanese subjects. Clin. Transl. Gastroenterol. 6, e94 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jenkins, H. et al Randomised clinical trial: safety, tolerability, pharmacokinetics and pharmacodynamics of repeated doses of TAK‐438 (vonoprazan), a novel potassium‐competitive acid blocker, in healthy male subjects. Aliment. Pharmacol. Ther. 41, 636–648 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Salvi, V. , Karnad, D.R. , Panicker, G.K. & Kothari, S. Update on the evaluation of a new drug for effects on cardiac repolarization in humans: issues in early drug development. Br. J. Pharmacol. 159, 34–48 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cubeddu, L.X. QT prolongation and fatal arrhythmias: a review of clinical implications and effects of drugs. Am. J. Ther. 10, 452–457 (2003). [DOI] [PubMed] [Google Scholar]
- 8. U.S. Food and Drug Administration . Guidance for Industry: E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐Antiarrhythmic Drugs. Rockville, MD: Center for Drug Evaluation and Research; (2005). [Google Scholar]
- 9. Sakurai, Y. et al Acid‐inhibitory effects of vonoprazan 20 mg compared with esomeprazole 20 mg or rabeprazole 10 mg in healthy adult male subjects ‐ a randomised open‐label cross‐over study. Aliment. Pharmacol. Ther. 42, 719–730 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Ashida, K. et al Randomised clinical trial: a dose‐ranging study of vonoprazan, a novel potassium‐competitive acid blocker, vs. lansoprazole for the treatment of erosive oesophagitis. Aliment. Pharmacol. Ther. 42, 685–695 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iwakiri, K. et al Tu1059 A phase 3, randomized, double‐blind, multicenter study to evaluate the efficacy and safety of TAK‐438 (20 mg once‐daily) compared to lansoprazole (30 mg once‐daily) in patients with erosive esophagitis. Gastroenterology 146, S‐741. [Google Scholar]
- 12. Mizokami, Y. et al Tu1054 TAK‐438 versus lansoprazole 15 mg for secondary prevention of peptic ulcers associated with non‐steroidal anti‐inflammatory drug (NSAID) therapy: results of a phase 3 trial. Gastroenterology 146, S‐739. [Google Scholar]
- 13. Kawai, T. et al Tu1055 TAK‐438 versus lansoprazole 15 mg for secondary prevention of peptic ulcers associated with low‐dose aspirin therapy: results of a phase 3 trial. Gastroenterology 146, S‐739. [Google Scholar]
- 14. Umegaki, E. et al Tu1052 A phase 3, randomized, double‐blind, multicenter study to evaluate the efficacy and safety of TAK‐438 (10 mg or 20 mg once‐daily) compared to lansoprazole (15 mg once‐daily) in a 24‐week maintenance treatment for healed erosive esophagitis. Gastroenterology 146, S‐738. [Google Scholar]
- 15. Shah, R.R. The significance of QT interval in drug development. Br J Clin Pharmacol 54, 188–202 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Darpo, B. The thorough QT/QTc study 4 years after the implementation of the ICH E14 guidance. Br. J. Pharmacol. 159, 49–57 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Viitasalo, M. & Karjalainen, J. QT intervals at heart rates from 50 to 120 beats per minute during 24‐h electrocardiographic recordings in 100 healthy men. Effects of atenolol. Circulation 86, 1439–1442 (1992). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. QT/QTc interval categorical analysis.
Supplementary Table S2. Summary of TEAEs reported in ≥2 subjects during the study.