

INTRODUCTION
Development of bispecific antibodies (BsAbs) as therapeutic agents has recently attracted significant attention, and investments in this modality have been steadily increasing. This review discusses challenges, and suggestions to overcome them, associated with the development of BsAbs, specifically those pertaining to clinical pharmacology, pharmacometrics, and bioanalysis. These challenges and possible solutions are discussed by presenting several case studies of BsAbs that have gained regulatory approval or that are currently in clinical development.
BsAbs, also termed “dual‐targeting” or “dual‐specificity” antibodies, have the ability to bind two different targets on the same or different cell(s); the targets may be cell‐surface receptors or soluble ligands, as shown in Figure 1. These dual‐nature antibodies have key advantages that can potentially enhance therapeutic efficacy compared with monotherapy or traditional combination therapies by: i) simultaneously blocking two different targets or mediators that have a primary role in the disease pathogenesis; ii) inducing cell signaling pathways (e.g., proliferation or inflammation); iii) retargeting to mediate antibody‐dependent cell‐mediated cytotoxicity (ADCC); iv) avoiding the development of resistance and increasing antiproliferative effects, specifically in oncology; and v) temporarily engaging a patient's own cytotoxic T cells to target cancer cells, thus activating cytotoxic T cells to cause tumor lysis (e.g., bispecific T‐cell engagers (BiTE)).
Figure 1.
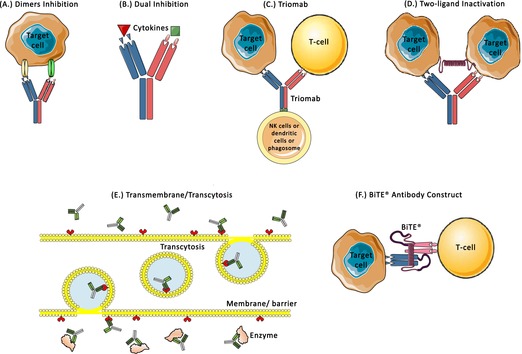
Various designs for BsAb molecules (a) Dimers inhibition: BsAbs can bind to two receptors/targets (HER2/HER3, HER2/HER4) on the same cell (e.g., MM‐111); (b) Dual inhibition: BsAbs can inhibit two different cytokines simultaneously, for example, COVA322 that inhibits TNF‐α and IL17A; (c) Triomabs: The antigen binding site binds to target cell receptors (EpCAM, HER2, or CD20) and the T‐cell receptors (CD3). The heavy chain site binds to NK cells or dendritic cells or macrophages/phagosome (e.g., catumaxomab, ertumaxomab, FBTA05); (d) Two‐ligand inactivation: two arms bind to different ligands on different cells belonging to the same population, such as DLL4 x VEGF, TNF‐α x IL17A, IL4 x IL13 (e.g., OMP‐305B83, COVA322, SAR156597); (e) Transmembrane/transcytosis: The BsAbs are designed specifically to cross the barriers/membrane via receptor transport (transferrin receptor) and bind to enzymes/receptors (BACE1) on the other side; (f) BiTE antibody construct: These are designed to bridge T cells and target cells by binding to CD3/CD28 or CD19/CD20/CD22/CEA/EpCAM, respectively (e.g., blinatumomab, MEDI‐565, MT110). The examples mentioned above can be found in Table 2 for further information. BACE1, β‐secretase 1; BiTE, bispecific T‐cell engagers; BsAbs, bispecific antibodies; DDL4, delta‐like ligand 4; EpCAM, epithelial cell adhesion molecule; HER, human epidermal growth factor receptor; IL, interleukin; NK, Natural Killer; TNF‐α, tumor necrosis factor‐alpha; VEGF, vascular endothelial growth factor.
Traditional combination therapies using monoclonal antibodies (mAbs) can also modulate multiple therapeutic targets. However, the development of mAbs presents challenges not encountered with BsAbs. For example, regulatory agencies have established stringent criteria for the codevelopment of new drugs that are intended for use as combination therapies. The sponsor has to demonstrate i) the rationale for use of the combination therapy rather than individual treatments; ii) a strong justification for why the individual drugs cannot be studied and developed independently; iii) that the nonclinical and clinical studies provide adequate evidence showing that the combination therapy provides significant therapeutic gain; and iv) a reasonable toxicity profile and more durable response than the monotherapy and existing standard of care.1 These guidelines can potentially make the drug development process for combination therapy lengthy and expensive. Conversely, BsAbs are able to address the biology associated with two different targets simultaneously via a similar regulatory pathway as that required for with a single‐target mAb. BsAbs may therefore offer the opportunity to benefit patients more quickly, and to access less costly development routes than can be afforded via classic combination therapies.
BsAbs also offer the opportunity to modulate unexplored biology in novel ways that may not be possible with single‐target mAbs. Avidity is defined as the measure of the overall strength of binding of an antigen with multiple antigenic determinants to multivalent antibodies. According to the “avidity hypothesis,” BsAbs may surpass combination therapy in terms of both biology and mechanistic behavior as a result of this theoretical concept. The theory states that avidity increases when two receptors are bound to a target cell, leading to efficacy greater than which could be expected from the additive combination each single mAb. A specific example is the development of JNJ‐61186372 (BsAb targeting epidermal growth factor receptor (EGFR) and c‐Met), which showed that the BsAb was more potent than the combination of single receptor‐binding antibodies.2 Furthermore, BsAbs are less likely than combination treatment to undergo off‐target binding in the presence of a surplus of decoy cells.3 BsAbs therefore have the theoretic potential to improve therapeutic window (safety and efficacy), selectivity, and regulatory efficiency as compared with a true combination therapy approach.
As a result of the aforementioned advantages of BsAbs (Table 1), BsAbs are one of the fastest growing classes of investigational drugs. In addition to the approved BsAbs, blinatumomab (BLINCYTO, Amgen, Thousand Oaks, CA) and catumaxomab (Removab, Fresenius Biotech, Homburg, Germany, initially marketed by Fresenius Biotech) for cancer immunotherapy, there are more than 50 additional BsAbs in clinical development4, 5, 6, 7, 8, 9, 10 (Table 2), with the potential for sales of up to $4.4 billion by 2023.11 Of note, almost all BsAbs that are currently in development target indications in oncology, with the following exceptions: COVA322 for plaque psoriasis, BsAbs targeting transferrin receptor (TfR) and β‐secretase 1 (BACE1) for central nervous system disorders, ABT981 for osteoarthritis, ALX‐0761 for psoriasis, AMG 570 for systemic lupus erythematosus, and JNJ‐61178104 and MDG010 for autoimmune diseases.12 In this review we aimed at increasing awareness of the multiple facets of translational and clinical development of BsAbs. We are presenting clinical pharmacology considerations (Table 3) and modeling simulation strategies with select examples as well as bioanalytical challenges and strategies, opportunities, and approaches supported by a variety of case studies. However, published information on some of the aspects discussed here is limited, thus restricting a broader selection of case studies and underscoring the need for a more extensive application of modeling simulation approaches to support efficient drug development.
Table 1.
Comparison of therapeutic modalities
| Properties | Small molecules | Peptides | mAbs | Antibody‐Drug Conjugate (ADC) | BsAb |
|---|---|---|---|---|---|
| Molecular weight | <1 kDa | <10 kDa (or <50 amino acids) | Few kDa to 150 kDa | Few kDa to 1,000 kDa | Few kDa to 1,000 kDa |
| Route of administration | PO, IV, SC, IM | PO (limited), IV, SC or IM | IV, SC or IM | IV, SC or IM | IV, SC or IM |
| PK | Linear at low doses; nonlinear at high doses | Linear at low doses; nonlinear at high doses | Mostly nonlinear at low doses. The linear PK is from FcR mediated clearance and nonlinear arises from TMDD. Linear at high doses | Nonlinear at low doses and linear at high doses. | May or may not be linear, dictated by the presence of Fc domain. Absence of Fc domain can lead to linear PK. |
| Distribution | Passive diffusion | Passive diffusion and convective transport | Convective transport | Convective transport | Convective transport |
| Metabolism | CYP's | Proteolytic degradation | Proteolytic degradation | Proteolytic degradation and CYPs | Proteolytic degradation |
| Serum T1/2 | Varies based on physicochemical properties | Short (<10 min) but can be increased with modifications | Usually long and depends on target mediated clearance and FcRn mediated antibody recycling | Usually long | Varies from h to days |
| Renal clearance | May or may not be a major route | Possible (if peptides are resistant to proteolysis) | Major route if Mol wt is <69 kDa | Possible route for both mAb and cytotoxic agent | Possible, very low to negligible |
| Hepatic clearance | May or may not be a major route | Not a major route with few exceptions | Target or site of action (liver or pancreas) dependent. | Route for cytotoxic agent | Unlikely |
| Target mediated clearance | No | Undergoes TMDD | May undergo TMDD depending on the target | Undergoes TMDD | Can possibly undergo TMDD for individual target |
| Intestinal clearance | Possible route driven by transporters and enzymes (transferases) | Not applicable | Not applicable | Not applicable | Not applicable |
| Typical dosing regimen | QD, BID or TID | Daily to weekly | Varying dosage and dosing regimens throughout the length of treatment. Regimen typically ranges from weekly to monthly to 6 months. | Weekly to monthly cycles | Weekly to monthly cycles |
| Toxicity | Mediated by Structure, physicochemical properties, metabolites, dose, and off‐target. | Limited | Immune‐mediated adverse events and immunotoxicity such as immunosuppression, immunostimulation, hypersensitivity and auto‐immunity | Immune‐mediated adverse events and immunotoxicity from antibody. Small molecules related toxicities form conjugated drug. | Immune‐mediated adverse events and immunotoxicity such as immunosuppression, immunostimulation, hypersensitivity and autoimmunity |
| Immunogenicity | Very rare | Low | May have non, low, or high ADA, neutralizing ADA can affect CL of mAbs | Very high, neutralizing ADA can affect CL of mAbs | Very likely, unless one of the targets is B‐cell. |
| DDI | High likelihood | Low | Implicit DDI resulting from the changes in immune system influencing the CYPs activities | Very low | Implicit short term DDI resulting from the changes in immune system influencing the CYPs activities |
Table 2.
List of BsAbs currently undergoing clinical trials. (Source: clinicaltrials.gov)
| BsAb | Sponsor | Format | Target | Biological function | Clinical trial | Identifier | Conditions |
|---|---|---|---|---|---|---|---|
| Allogeneic cytomegalovirus‐specific cytotoxic T lymphocytes | Fred Hutchinson Cancer Research Center | Allogenic Hematopoietic stem cells (HSCT) | CD8 X CD19 | CAR‐T Cells | Completed phase I/II | NCT01475058 | Precursor Acute Lymphoblastic Leukemia, Lymphoma |
| AMG 330 | Amgen | BiTE antibody construct | CD3 X CD33 | T cell recruitment | Ongoing phase I (On hold) | NCT02520427 | Myeloid Leukemia, Acute non lymphoblastic leukemia |
| Anti CD3 X anti‐CD20 BsAb‐armed activated T cells | Barbara Ann Karmanos Cancer Institute | Activated T‐cells armed with BsAb | CD3 X CD20 | Activated T cells | Completed phase I | NCT00938626 | Multiple Myeloma and Plasma Cell Neoplasm |
| Anti‐CEA x anti‐DTPA and di‐DTPA‐131I peptide | Nantes University Hospital | scFv‐IgG | CEA X di‐DTPA‐131I | Radioimmunotherapy | Completed phase II | NCT00467506 | Thyroid Neoplasms |
| anti‐EpCAM x anti‐CD3 (removab) | AGO Study Group | Triomab | EpCAM X CD3 | T cell recruitment | Completed phase II | NCT00189345 | Ovarian Cancer |
| BAY2010112 | Bayer | BiTE antibody construct | CD3 X PSMA | T cell recruitment | Enrolling phase I | NCT01723475 | Prostatic Neoplasms |
| BI 836909 (AMG 420) | Boehringer Ingelheim | BiTE antibody construct | B‐cell maturation antigen (BCMA) | T cell recruitment | Ongoing phase I (recruiting participants) | NCT02514239 | Multiple myeloma |
| Blinatumomab | Amgen | BiTE antibody construct | CD3 X CD19 | T cell recruitment | Ongoing phase III | NCT02393859 | Leukemia, Acute Lymphoblastic |
| National Cancer Institute (NCI) | Not yet open (phase I) | NCT02568553 | B‐Cell Lymphoma (Unclassifiable with intermediate features) | ||||
| Ongoing (phase II) | NCT02143414 | B Acute Lymphoblastic Leukemia, Untreated Adult Acute Lymphoblastic Leukemia | |||||
| Ongoing (phase III) | NCT02101853 | B Acute Lymphoblastic Leukemia, Recurrent Adult/childhood Acute Lymphoblastic Leukemia | |||||
| Ongoing (phase III) | NCT02003222 | Adult B Acute Lymphoblastic Leukemia, Untreated Adult Acute Lymphoblastic Leukemia | |||||
| Amgen Research (Munich) | Ongoing phase I/II | NCT01471782 | Acute Lymphoblastic Leukemia | ||||
| Ongoing | NCT01741792 | Diffuse Large B‐cell Lymphoma | |||||
| Ongoing phase II | NCT01466179 | Acute Lymphoblastic Leukemia | |||||
| Ongoing phase II | NCT01209286 | B‐ALL | |||||
| Ongoing phase II | NCT01207388 | B‐cell Acute Lymphoblastic Leukemia | |||||
| Completed | NCT00560794 | Acute Lymphoblastic Leukemia | |||||
| Completed phase I | NCT00274742 | Non‐Hodgkin's Lymphoma, Relapsed | |||||
| Catumaxomab | Neovii Biotech | Triomab | EpCAM X CD3 | T cell recruitment | Completed phase II | NCT00464893 | Gastric Cancer, Gastric Adenocarcinoma |
| JSehouli | Completed phase II | NCT01815528 | Recurrent Epithelial Ovarian Cancer | ||||
| AIO‐Studien‐gGmbH | Ongoing phase II | NCT01504256 | Gastric Adenocarcinoma With Peritoneal Carcinomatosis, Siewert Type II/III Adenocarcinoma of Esophagogastric Junction With Peritoneal Carcinomatosis | ||||
| Grupo Español de Investigación en Cáncer de Ovario | phase II | NCT01246440 | Ovarian Cancer | ||||
| Neovii Biotech | Completed phase II | NCT01065246 | Malignant Ascites Due to Epithelial Carcinoma | ||||
| Completed phase III | NCT00822809 | Cancer, Neoplasms, Carcinoma, Malignant Ascites | |||||
| Completed phase II | NCT00377429 | Ovarian Cancer | |||||
| Completed phase II | NCT00326885 | Malignant Ascites | |||||
| Completed phase II/III | NCT00836654 | EpCam Positive Tumor (e.g.Ovarian, Gastric, Colon, Breast), Malignant Ascites | |||||
| Completed phase II | NCT00352833 | Gastric Cancer, Gastric Adenocarcinoma | |||||
| CD20Bi‐activated T cells (ATC) | Barbara Ann Karmanos Cancer Institute | Activated T‐cells armed with BsAb | CD3 X CD20 | Activated T cells | Completed phase I | NCT00244946 | Lymphoma |
| DT2219ARL | Masonic Cancer Center, University of Minnesota | 2 scFv linked to diphtheria toxin | CD19 X CD22 | Targeting of protein toxin to tumor | Ongoing phase I | NCT02370160 | Refractory and relapsed B‐Lineage Leukemia/Lymphoma |
| Ongoing phase I | NCT00889408 | Leukemia/Lymphoma | |||||
| EGFRBi‐Armed Autologous T Cells | Barbara Ann Karmanos Cancer Institute | T cells armed with BsAb | CD3 X EGFR | Autologous activated T cells to EGFR‐positive tumor | Withdrawn (phase I/II) | NCT02521090 | Adult Brain Glioblastoma, Adult Gliosarcoma, Recurrent Brain Neoplasm |
| GD2Bi‐aATC | Barbara Ann Karmanos Cancer Institute | Activated T‐cells armed with BsAb | CD3 X GD2 | Activated T cells | phase I/II | NCT02173093 | Desmoplastic Small Round Cell Tumor, Disseminated Neuroblastoma, Metastatic Childhood Soft Tissue Sarcoma, etc |
| HER2Bi‐aATC | Barbara Ann Karmanos Cancer Institute | Activated T‐cells armed with BsAb | CD3 X HER2 | Activated T cells | phase I | NCT02470559 | Malignant Ovarian Clear Cell Tumor, Malignant Ovarian Serous Tumor, Recurrent Fallopian Tube Carcinoma, Recurrent Ovarian Carcinoma, Recurrent Primary Peritoneal Carcinoma |
| Enrolling phase I | NCT02662348 | Esophageal, Gastric, Pancreatic, Liver, Gallbladder, Bowel Cancer | |||||
| IMCgp100 | Immunocore Ltd | ImmTAC | CD3 X gp100 | T cell recruitment | Ongoing phase I | NCT01211262 | Malignant Melanoma |
| phase I (Recruitment has not begun) | NCT02570308 | Uveal Melanoma | |||||
| Indium labeled IMP‐205xm734 | Radboud University | Radioimmunotherapy | Unknown (phase I) | NCT00185081 | Colonic Neoplasms | ||
| JNJ‐61186372 | Janssen Research & Development, LLC | Bispecific human IgG1 mAbs | EGFR X cMET | Inhibits receptor phosphorylation | Ongoing phase I (Recruitment has not started yet) | NCT02609776 | non‐small cell lung cancer (NSCLC) |
| LY3164530 | Eli Lilly and Company | OrthoFab‐IgG | MET X EGFR | Blockade of 2 receptors | Ongoing phase I | NCT02221882 | Neoplasms, Neoplasm Metastasis |
| MDX447 | Dartmouth‐Hitchcock Medical Center | 2 (Fab') crosslinked | CD64 x EGFR | Active monocytes to kill tumor | Completed phase I, | NCT00005813 | Brain and Central Nervous System Tumors |
| MEDI‐565 | MedImmune LLC | BiTE antibody construct | CEA X CD3 | T cell recruitment | Completed phase I | NCT01284231 | Gastrointestinal Adenocarcinomas |
| MGD006 | MacroGenics | Dual Affinity Re‐Targeting (DART) | CD123 x CD3 | Re‐targeting T cells to tumors | Ongoing phase I | NCT02152956 | AML |
| MGD007 | MacroGenics | DART | gpA33 X CD3 | Re‐targeting T cells to tumors | Ongoing phase I | NCT02248805 | Colorectal Carcinoma |
| MGD010 | MacroGenics | Dual Affinity Re‐Targeting (DART) | CD32B x CD79B | Safety assessment | Ongoing phase I | NCT02376036 | Healthy Subjects |
| Mitoxantrone packaged EDV (EnGeneIC Delivery Vehicle) | Dr David Ziegler | Delivery of nanoparticles | phase I (Recruitment has not begun) | NCT02687386 | Solid Tumor and CNS Tumor | ||
| MM‐111 | Merrimack Pharmaceuticals | HSA body | HER2 X HER4 | Blockade of 2 receptors | Completed phase I | NCT00911898 | Her2 Amplified Solid Tumors, Metastatic Breast Cancer |
| MM‐111 + Herceptin | Merrimack Pharmaceuticals | HSA body | HER2 X HER3 | Blockade of 2 receptors | Completed phase I | NCT01097460 | Breast Neoplasms |
| MOR209/ES414 | Emergent Product Development Seattle LLC | scFv‐IgG | PSMA X CD3 | T cell recruitment | Ongoing phase I | NCT02262910 | Prostate Cancer |
| MT 110 | Amgen Research (Munich) GmbH | BiTE antibody construct | CD3 X EpCAM | T‐cell recruitment | Completed phase I | NCT00635596 | Solid Tumors |
| OMP‐305B83 | OncoMed Pharmaceuticals, Inc. | DVD‐Ig | DLL4 X VEGF | 2‐ligand inactivation | Ongoing phase I | NCT02298387 | Advanced Solid Tumor Malignancies |
| REGN1979 | Regeneron Pharmaceuticals | CD20 X CD3 | T cell recruitment | Ongoing phase I | NCT02290951 | Non‐Hodgkin's Lymphoma, Chronic Lymphocytic Leukemia | |
| rM28 | University Hospital Tuebingen | Tandem scFv | CD28 X HMV‐MAA | Retargeting autologous lymphocytes to tumor | Completed phase I/II | NCT00204594 | Malignant Melanoma |
| RO6958688 | Hoffmann‐La Roche | Crossmab | CEA X CD3 | T cell recruitment | Ongoing phase I | NCT02324257 | Solid Cancers |
| TargomiRs | University of Sydney | EGFR X EDV | Delivery of nanoparticles | Ongoing phase I | NCT02369198 | Malignant Pleural Mesothelioma, Non‐Small Cell Lung Cancer | |
| TF2 | Radboud University | Dock and lock | CEA x HSG | Radioimmunotherapy | Completed phase I | NCT00860860 | Colorectal Neoplasms |
| Garden State Cancer Center at the Center for Molecular Medicine and Immunology | enzyme‐linked immunosorbent assay, pharmacological study | phase I | NCT00895323 | Colorectal Cancer | |||
| Nantes University Hospital | Immuno‐PET | Ongoing phase I/II | NCT01730638 | Medullary Thyroid Carcinoma | |||
| Centre René Gauducheau | Radioimmunotherapy | Ongoing (phase I/II) | NCT01221675 | Small Cell Lung Cancer, CEA‐expressing Non‐Small Cell Lung Carcinoma (NSCLC) | |||
| TF2 ‐ 68 Ga‐IMP‐288 | Nantes University Hospital | Dock and lock | CEA X HSG | Immuno‐PET | Ongoing phase I/II | NCT01730612 | HER2 Negative Breast Carcinoma Expressing CEA |
| TF2 antibody/68Ga‐IMP‐288 | Nantes University Hospital | Dock and lock | CEA X HSG | Radioimmunotherapy | Not yet open (phase II) | NCT02587247 | Metastatic Colorectal Cancer |
Table 3.
Opportunities for translational and clinical pharmacology in drug development for BsAbs
| Functional areas | Question | Possible approaches |
|---|---|---|
| Bioanalytical | What are the key fundamental points in selecting a bioanalytical strategy and optimizing the assay for BsAb? | The key rationale is around what to measure for interrogating the exposure response relationship (or safety) of the BsAb. If bifunctional form assay is not available, subsequent assay risk assessment should be considered. There is a timing aspect as well. The approaches are then dictated often by the nature of the BsAb which then can steer towards an LBA as appropriate or LC‐MS for example. The method optimization in terms of what are measured (free, partially bound or total) may be required along with program progression during development. |
| Preclinical and translational | What are the considerations for the receptor occupancy calculation applied in dose determination for BsAbs? | It is dictated by the affinity and avidity of the BsAbs with the target and any prior information from the mAbs agent. |
| What is the basis of selecting the dose and dosing regimen for the FIH study? | The doses are selected from the dose range finding studies in cynomolgus monkeys using MABEL approach. PKPD modeling is performed with preclinical studies and in vitro cytotoxicity data to project the FIH doses. | |
| What are the considerations for the PD end points that can influence the optimal dose and dosing regimen decision? |
|
|
| How does modeling and simulation inform the selection and design of BsAbs? | M&S approaches can elucidate the conditions under which the BsAb modality is superior to a traditional combination therapy and inform the design of a BsAb molecule with optimal characteristics for efficacy. | |
| Clinical | What is the rationale for determining doses in combination treatment involving BsAbs as one of the therapeutics? | The rationale is based on the prior knowledge of clinical data with individual targets, toxicity and efficacy studies, and may be different than the equivalent combination of individual molecules due to factors such as avidity. |
| How are DDI studies mitigated? | Based on the known existing potential and interactions with the individual targets, in vitro data and PBPK modeling. | |
| How are safety and efficacy end points selected? | The efficacy and safety end points may be specific for a BsAb and may not be applied horizontally across all the existing BsAbs. This can often vary from molecule to molecule. |
STRUCTURAL FORMATS OF BsAbs
The functional domain architecture of mAbs have been extensively exploited to create a number of different BsAbs formats. Spiess et al. originally classified them into five distinct structural groups:7 i) bispecific IgG,13, 14 ii) IgG appended with an additional antigen‐binding moiety,15, 16 iii) BsAbs fragments,17, 18 iv) bispecific fusion proteins,19 and v) BsAb conjugates.20 The new formats are categorized based on the Fc‐mediated effector functions and are classified as immunoglobulin G (IgG)–like molecules and non‐IgG–like molecules, as shown in Table 4. The IgG formats are larger and undergo FcRn recycling, which results in a longer serum half‐life, whereas non‐IgG formats have a smaller size, which enables increased tissue penetration.
Table 4.
Structural format categories for BsAbs
| IgG‐like formats | Non‐IgG‐like formats |
|---|---|
| Quadroma | scFv based BsAbs |
| Knob‐into‐holes | Nanobodies |
| Dual variable domains Ig | Dock and lock methods |
| IgG‐single‐chain Fv (scFv) | Dual affinity retargeting molecules (DARTs) |
| Two‐in‐one Fab (or Dual action Fab) | |
| Half molecule exchange | |
| Κλ‐bodies |
These unique formats vary in antigen‐binding valency properties, thereby offering a potential opportunity to optimize valency of each component antibody based on the biology of the mechanism perturbed by the therapeutic.7 Because these formats are comprised of individual functional domains, the activities of the domains can be monitored via quantification of the single domain. Elucidating the exposure of the active therapeutic for informing model‐based drug development in the context of a BsAb is complex. The molecular variant chosen to illicit target interrogation can affect the likelihood of biotransformation of the molecule, a critical factor in translating active exposure to a pharmacological response. The physiochemical BsAb features must be further coupled with structural variants that may exist in free, partially bound, and bound forms21, 22 resulting from binding to a soluble target, for example. BsAbs are furthermore confounded by valency properties of each antibody component binding to its respective antigen. Partially bound forms of the molecule may alter the stoichiometry associated with the dual‐target binding of the biotherapeutic. Innovative bioanalytical approaches are required to fully understand the active exposure of a BsAb, which is dependent on both its unique physical/chemical properties and the dual‐targeting strategy represented by the molecule. These distinct structural groups serve to illustrate the structural complexity and diversity of BsAbs, which raise unique challenges related to the bioanalytical strategy.
CLINICAL PHARMACOLOGY CONSIDERATIONS
Several global regulatory agencies have published guidelines for the development of both small molecules and biologics. However, development strategies may differ between BsAbs, mAbs, and traditional combination therapies.
Immunogenicity, biomarkers, and imaging
As with any biologic molecule, BsAbs have the potential to elicit an immune reaction. Immunogenicity is typically assessed by detection of antidrug antibodies (ADA). Formation of ADA is not always associated with mAbs. There are cases of mAbs without ADA formation (e.g., rilotumumab, which has an ADA incidence of 0%). Therefore, the incidence of ADA associated with BsAbs cannot be lower than zero. Among BsAbs, catumaxomab has an ADA incidence of 0%, while blinatumomab has a low ADA incidence of ∼1%. Our experience in the field suggests that key factors of ADA formation related to BsAbs are similar to those associated with other mAbs. This may include, but is not limited to, the structure of BsAbs (i.e., whether it can be recognized by the immune system as “foreign”), the presence of foreign sequences (e.g., asymmetric rat‐mouse hybrid BsAb), route of administration (higher incidence of ADA with subcutaneous than with intravenous (IV) administration), dose, and characteristics of the patient's immune system. It is well known that the formation of ADA may alter pharmacokinetics (PK), leading to subsequent changes in the pharmacodynamic (PD) properties of a BsAb. However, the drug‐binding characteristics of an ADA may lead to differentiating impact on the individual target binding.23
Biomarker development programs for BsAbs often face several unique challenges. Standard approaches to evaluate for biological/biochemical impact via target engagement and PD assays are routinely focused on specific analytes to ensure that an appropriate dose and schedule are selected to advance farther along into clinical testing. BsAbs often present the challenge of dissecting the biologic complexity and PD coverage associated with two targets simultaneously. A BsAb targeting two antigens or ligands may create the need to define two target coverage thresholds (unique to each marker), all the while dealing with the reality that the BsAb provides a fixed ratio of exposure to either target at a given exposure. In the case of a BiTE antibody construct, it is critical in early clinical trials to demonstrate engagement of T cells (via the CD3 binder) as well as the tumor‐specific target (CD19 in the case of blinatumomab). Such T‐cell activation has been explored with blinatumomab by evaluating CD69 and CD25 upregulation posttreatment in acute lymphocytic leukemia patients.24
Similarly, patient stratification hypotheses for sensitivity or resistance are generally formulated in the preclinical development and then tested throughout development with the ultimate goal of identifying a predictive marker for response to a single biologic process. Patient stratification marker(s) for BsAbs may need to be tailored to each binding arm of the BsAb, or the proposed combinatorial biology elicited via simultaneous dual target PD.
Molecular imaging can play an important role in identifying and improving the success rate of promising new drug candidates, including BsAbs. Goldenberg et al. described various examples of the utility of BsAbs as tools for imaging in preclinical and clinical settings. Imaging studies have also been demonstrated to be of value in identifying the immunogenicity effect of BsAbs.25 These tools can be utilized in oncology for diagnosis and detection of small tumors or lesions using differential approaches and rationales, such as Dock and Lock, click chemistry,26 or heterodimer conjugated nanomaterials.27
Drug–drug interaction of BsAbs
Drug–drug interactions (DDIs) for BsAbs are less well understood than for monotherapies or traditional combination of mAbs. If the investigational therapeutic protein (TP) is a cytokine or modulates cytokine biology, studies should be conducted to determine its effects on CYP enzymes or transporters. Lee et al. conducted a survey aimed at systematically reviewing US Food and Drug Administration (FDA)–approved therapeutic proteins and the implications of therapeutic protein–drug interactions. The survey encompassed 68 new therapeutic proteins that had been approved by the FDA by the end of 2008.28 The results showed that cytokine release followed by administration of an mAb with cytokine‐modulating properties was a major reason for DDI after mAb administration. Cytokines are involved in the pathophysiology of multiple human diseases, and their levels are increased during infection and inflammation. Therefore, biologics that modulate cytokine activities can indirectly influence the expression of specific CYP enzymes and drug transporters by affecting cytokine concentrations. This concept is described extensively elsewhere.29, 30 The magnitude of cytokine‐induced effects on CYP450 depends on the level of cytokine elevation, the type of cytokine (e.g., IL‐6 being important), and the duration of cytokine elevation. The potential for DDI with transient cytokine elevation may be very different from that with chronic cytokine elevation.31
Kenny et al. published an article to facilitate better understanding of the current science, investigative approaches, and knowledge gaps in this field.32 Key issues discussed included translating in vitro to in vivo knowledge in DDI along with questions of whether in vitro data could add value in defining the need for a clinical DDI study, whether the acute phase response protein C‐reactive protein (CRP) could be used as a potential biomarker for CYP modulation in inflammatory disease, whether TP‐DDI could be quantitatively predicted from preclinical data, and how a clinical DDI study can be designed appropriately.
Assessment of DDI generated with a BsAbs should be approached the same way as with any large molecule. If clinical studies are restricted to patients instead of healthy volunteers, population PK modeling provides a feasible approach for TP‐DDI assessment. Population PK modeling allows less intensive sampling, incorporation of TP‐DDI assessment in larger phase II and III trials involving relevant patient populations, and integration of data generated from multiple studies during different development phases. Population PK modeling also supports evaluation of the effects of combined “perpetrators” on a TP and, potentially, the effect of a TP on comedications when the analysis is prespecified and concentrations of the comedications are evaluated. Trends identified in an exploratory population PK analysis can be used to guide decisions for the need of additional DDI studies.
Regulatory agencies have included recommendations in their guidance on TP‐DDI assessment during drug development. The European Medicines Agency (EMA) guideline,33 published in July 2007, entitled “Guideline on the Clinical Investigation of the Pharmacokinetics of Therapeutic Proteins,” describes concerns about immunomodulators such as cytokines, which have shown a potential for inhibition or induction of CYP enzymes, thereby altering the metabolism of coadministered small‐molecules that are substrates of these enzymes. The guideline suggests that the in vitro and/or in vivo studies should be considered on a case‐by‐case basis. The 2012 FDA draft guidance on DDI similarly expands the US agency's current recommendation on TP‐DDI assessment.34
MODELING AND SIMULATION APPROACHES
Model‐based approaches increasingly support decisions spanning the entire drug development process, from preclinical development through postmarketing, as shown in Figure 2. The application of such approaches to the development of BsAbs follows this standard paradigm and is applied at various stages during the development of biologic therapeutics.
Figure 2.
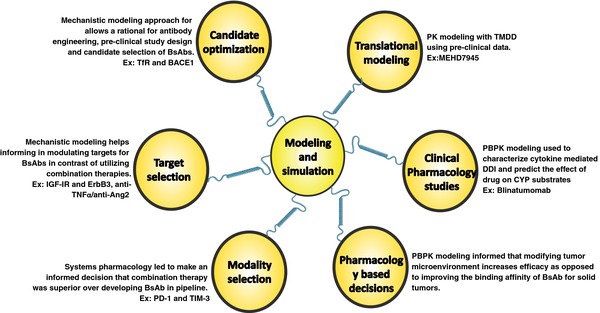
Various applications of modeling and simulation approaches used for BsAbs drug development. Ang2, angiopoietin‐2; BACE1, β‐secretase 1; BsAb, bispecific antibodies; IGF‐IR, insulin‐like growth factor‐I receptor; PBPK, physiologically based pharmacokinetic; PD‐1, programmed death‐1; Tfr, transferrin receptor; TIM‐3, T cell immunoglobulin and mucin domain; TMDD, target‐mediated drug disposition; TNF‐α, tumor necrosis factor‐alpha.
Translational modeling
Predicting the PK of BsAbs generally follows the same paradigm as mAbs; i.e., allometrically scaling preclinical PK parameters to predict human PK. Although this tends to work best for antibodies with linear PK, models incorporating nonlinear clearance mechanisms have been developed. For example, preclinical PK of the BsAb MEHD7945 in cynomolgus monkeys were fitted to a standard two‐compartment PK model with nonlinear and linear clearance components, and the resulting PK parameters were translated using a common method for scaling.35 More complicated target‐mediated drug disposition (TMDD) models that mechanistically describe simultaneous binding to two targets have also been proposed, although it is not straightforward to translate such models from preclinical species to humans because of a lack of critical information, such as the relative density of the target in preclinical species compared with humans.36 While this is true of TMDD models in general, these challenges are exacerbated for BsAb because they bind two targets simultaneously. Still, successful development of a TMDD model that describes the PK of a BsAb along with an understanding of species differences impacting the model may help guide first‐in‐human (FIH) dose selection, because such mechanistic models predict the degree of target engagement for each BsAb arm. Projection of target engagement is a key application of modeling and simulation approaches, particularly for development programs that lack biomarker data. Clinical decisions regarding the FIH starting dose are based on results from toxicology studies and on expected pharmacology, a comprehensive approach recommended by the FDA.37 However, such approaches have had mixed success for traditional mAb and their application has been limited. Advances in this area represent a significant opportunity for modeling and simulation to contribute to the transition from preclinical space to the clinic.38
Pharmacology‐based decisions
Mechanistic modeling and simulation approaches tailored to the interrogation of BsAb pharmacology have yielded critical insights into the mechanism of action of BsAbs and the conditions under which they offer advantages. A physiologically based pharmacokinetic (PBPK) model developed by Friedrich et al. provided a rationale for the lack of clinical success of molecules that retarget effector cells in solid tumor indications, and a platform for designing molecules with a greater potential for success. Their PBPK model suggested that effector cells dictate the distribution of the BsAb and that modifications to the tumor microenvironment were more promising approaches to boosting efficacy in solid tumor indications than optimizing the binding parameters of the BsAb.39
Modality selection
For a BsAb targeting both programmed death‐1 (PD‐1) and T‐cell immunoglobulin and mucin domain (TIM)‐3 to manipulate a patient's immune system into attacking a foreign entity (such as a cancer), a systems pharmacology approach was used to assess the combinations potential to provide enhanced efficacy over monotherapy or traditional combinations. The investigators used a mechanistic model and, based on its predictions suggesting that a BsAb offered no advantages over a fixed‐dose combination, ultimately decided not to develop the BsAb. The model predictions also allowed for accelerated project timelines by informing the selection of individual target candidates with optimal pharmacological properties.40
Target selection
The development of MM‐141, a tetravalent BsAb targeting the IGF‐IR and ErbB3 pathways (for the treatment of cancer), was also informed by mechanistic modeling.41 A mechanistic model that integrates information across pathways can provide insight into both the selection of the best targets for overcoming resistance mechanisms and the means by which those targets should be modulated. The simulations for MM‐141 showed that a BsAb should be used to modulate the targets, predicting a superior outcome with simultaneous target binding compared with a combination of individual antibodies, for any ratio of IGF‐IR and ErbB3 receptors.41 Indeed, this prediction was validated by preclinical studies comparing the antitumor potency of MEHD7945A, a BsAb also targeting IGF‐IR and ErbB3, with a combination of cetuximab and an anti‐ErbB3 antibody.42 Models of a similar nature were also developed for rheumatoid arthritis to elucidate the effects of an anti‐TNFα/anti‐Ang2 BsAb43 and for relapsed/refractory acute lymphoblastic leukemia (ALL) to provide insight into the mechanisms governing response and nonresponse after administration of blinatumomab.44
Candidate optimization
Mechanistic modeling approaches have also been used to determine the optimal characteristics of a BsAb. For example, a mathematical model addressing PK, PD, and safety outcomes was developed for a BsAb that targets the TfR and BACE1. This model was used to support preclinical candidate selection. While it may seem intuitive that increasing affinity of the TfR arm would promote delivery of the molecule to the target site (maximizing the biologic effect), the modeling showed that there is an optimal TfR affinity range for maximum reduction of Aβ peptides. Degradation of the molecule during transcytosis increased with increasing affinity, resulting in lower average brain exposures over time and reduced pharmacologic effect. Reducing affinity therefore undermined TfR‐mediated transport through the blood–brain barrier. Similarly, the model suggested the relationship between TfR affinity and reticulocyte depletion was biphasic in nature. This model allowed for a rational approach to antibody engineering, preclinical study design, and candidate selection, resulting in BsAbs with optimal safety and PK/PD profiles.45
These examples show how critical model‐based approaches allow for an improved understanding of the mechanism of action of BsAbs (compared with corresponding combination therapies), the conditions under which they offer advantages, and the optimal pharmacologic properties required of such agents to maximize the therapeutic effectiveness. A combination of traditional PK/PD modeling approaches and newly emerging quantitative systems pharmacology provide the best path forward for maximizing the probability of success and for accelerating the development of these molecules.
BIOANALYTICAL STRATEGY FOR BISPECIFIC ANTIBODIES
Informing meaningful PK and PD assessments of both efficacy and safety in the development of these molecules demands an appropriate bioanalytical strategy with careful consideration applied to method suitability for measuring functionally relevant forms of the BsAbs. The majority of the bioanalytical methods for BsAbs are based on the principles of ligand binding assays (LBA). The advantages and principles of LBA for measuring biotherapeutics are well described.46 The choice of an appropriate LBA includes the selection of assay platform, format, and critical reagents. It also requires appropriate assessment of other contributing factors that may cause bioanalytical error and mislead the evaluation (risk assessment). Although LBA constitutes the most commonly used approach, there are other assay platforms, such as flow cytometry and mass spectrometry, which are also well suited for addressing bioanalysis questions for BsAbs.
BsAbs can bind to various circulating partners, such as free target proteins, ADAs, and other endogenous serum components. In addition, BsAbs may lose their binding ability as a result of biotransformation. Consequently, BsAbs can exist in a pharmacologically active form, characterized by dual‐target antigen binding sites, and in an inactive form, which has partial or no target binding sites. Whether to measure the active concentration or the total concentration (active plus inactive/partially active forms) for PK/PD assessment continues to be debated as part of the bioanalytical strategy.14, 47 The recent white paper by the AAPS Ligand‐Binding Assay Bioanalytical Focus Group discussed the challenges and issues of measuring free (active), total drugs and target proteins and how these data should be used to support drug discovery and development.48 Typically, when considering which form of the therapeutic protein should be measured to achieve the intended purpose of the study, a “fit‐for‐purpose” approach is adopted. A working bioanalytical strategy and the selection of appropriate assay technologies to measure the intended forms is presented in Figure 3 as well as in two examples discussed later.
Figure 3.
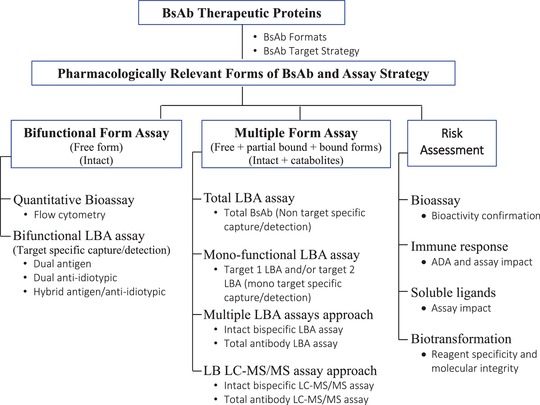
Bioanalytical strategy and the selection of appropriate assay technologies to measure the intended forms for BsAb. BsAb, bispecific antibodies; LBA, ligand binding assay; LC‐MS/MS, liquid chromatography‐tandem mass spectrometry.
COVA322 ((tumor necrosis factor) TNF x (interleukin) IL‐17A), also known as a FynomAb, is a bispecific fusion protein consisting of an antibody and a Fynomer.19 Fynomers are small binding proteins derived from the human Fyn SH3 domain. Fynomers can be engineered to bind to target molecules with the same affinity and specificity as antibodies. It is critical to maintain the activity of COVA322 and monitor that the Fynomer is not cleaved in vivo by undergoing potential biotransformation. An enzyme‐linked immunosorbent assay (ELISA) format utilizing the antigen specificity of the BsAbs is normally used to directly measure the concentration of the bioactive BsAbs. Two ELISA assays were developed: bifunctional ELISA, detecting intact and bioactive COVA322 using dual antigen, and monofunctional ELISA, detecting the anti‐TNF binding antibody portion of COVA322. The assays demonstrated that the plasma concentrations obtained from PK samples by both assays were comparable, indicating that COVA322 stayed fully functional (bioactive) and that there were no indications of in vivo biotransformation. However, this may not be representative of the results observed for target binding in the bispecific formats. For instance, glucagon‐like peptide‐1 (GLP‐1) is a 37‐amino acid peptide for the treatment of type II diabetes via GLP‐1 receptor binding. A CovX‐Body was generated by conjugating the GLP‐1 peptide to the N‐terminus of a carrier mAb. After in vivo administration of the GLP‐1 CovX‐Body to mice,49 quantification of the therapeutic was achieved by two different ELISA assays that each used an anti‐idiotypic antibody capture of the mAb portion of the construct. Two different detection antibodies were used: antihuman IgG, to measure the mAb only (total assay), and anti‐GLP‐1 antibody (N‐terminus specific), to measure the intact GLP‐1 CovX‐Body. The intact assay produced drug concentrations that were significantly lower than the total assay throughout the PK time course. Unlike the FynomAb, these results indicate that, although the concentration of the mAb portion of the construct was sustained, the N‐terminal region of the GLP‐1 moiety was quickly degraded, demonstrating a high degree of in vivo biotransformation (e.g., cleavage of GLP‐1 moiety from the antibody).
Because BsAbs may present as a mixture of biologically active and inactive forms, it is important to identify the BsAb form that is most pharmacologically relevant to PK/PD assessment and to develop a validated assay that measures the appropriate form accordingly.
CASE STUDIES
The following case studies illustrate specific examples for each of the issues that are pertinent to the development of BsAbs.
Case study 1: Catumaxomab
Mechanism of action
The epithelial cell adhesion molecule (EpCAM) represents a potentially attractive antigen for antitumor therapies, because the transmembrane glycoprotein is highly expressed in a broad range of solid tumor indications and correlates with a poor patient prognosis in most of them.50 Catumaxomab is a BsAb targeting EpCAM and CD3 that was approved in the EU in 2009 for the treatment of malignant ascites. Exudative ascites is fluid accumulation in the peritoneal cavity (abdominal space) that can be caused by the presence of solid tumors. Treatment with catumaxomab along with paracentesis (manual drainage) can significantly delay the need for repeated manual drainage thereafter in patients with malignant ascites caused by ovarian or nonovarian cancer.51
Catumaxomab has been generated by fusing two different hybridoma cells, a technique called “quadroma technology.” The hybrid Fc region exerts binding capacity to human FcγR I, FcγR IIa, and FcγR III enabling activation of NK cells, dendritic cells, monocytes, and macrophages.52 In addition, the targeting of EpCAM on tumor cells and CD3 on T cells allows T‐cell–mediated redirected lysis of EpCAM‐positive target cells, which results in a more than 1,000‐fold higher potency compared with monospecific anti‐EpCAM antibodies.
Challenges in FIH studies
Catumaxomab is hypothesized to exert its effect via a combination of T‐cell–mediated tumor cell killing, ADCC, and phagocytosis via FcγR‐activated accessory immune cells. Delivery of catumaxomab in the clinical setting is limited to localized intraperitoneal (IP) administration. In an FIH study where catumaxomab was administered IV, fatal acute liver failure and cytokine release‐associated systemic toxicity were observed even at low doses.53 In a pilot study, the feasibility of IP administration at increasing doses was explored; however, no information on starting dose selection was provided.54 IP administration of catumaxomab delivered sufficient compartmental concentrations observable antitumor activity with mitigation of the systemic side effects from significant unspecific T‐cell activation caused by intravenous administration.
Bioanalytical approaches
Plasma concentrations of catumaxomab were measured by a validated two‐site ELISA. Catumaxomab was captured by an antirat IgG λ light chain‐specific antibody. Bound catumaxomab was then detected via an antimouse IgG2a‐specific biotin‐labeled detection antibody followed by colorimetric measurement. The assay format suggests measurement of the total drug concentration by utilizing the chimeric composition of catumaxomab rather than antigen specificity; therefore, the assay results do not allow for conclusions about the BsAbs maintenance of in vivo binding activity. A functional bioassay was performed using PK samples to characterize the functional in vivo binding activity by testing for the killing activity against EpCAM‐positive tumor cells.55
PK/PD
In a PK study, patients with symptomatic malignant ascites received four IP infusions of increasing doses (10, 20, 50, and 150 μg) over 6 h each on days 0, 3, 7, and 10. High concentrations of catumaxomab were observed in ascites, approaching effective concentrations. The concentration levels increased with the number of infusion doses, and peak concentrations were detected ∼19 h after completion of the last administration. Interestingly, these concentrations were sufficient to induce tumor cell killing in spiking experiments, and this antitumor activity outside the peritoneum was confirmed by the observation of systemic responses to catumaxomab therapy described in various case reports.56, 57 The mean terminal elimination half‐life was 2.13 days. PD indication of antitumor activity was observed by cytokine release (TNF‐α, (interferon) IFN‐γ, IL‐2, IL‐6, and IL‐10). In the phase II/III study, a relative lymphocyte count above 13% at baseline appeared to be a potential biomarker with prognostic significance.58
In a phase II study, immuno‐monitoring revealed i) redistribution of effector T cells from blood into the peripheral tissue, ii) expansion and shaping of a preexisting EpCAM‐specific T‐cell repertoire, and iii) spreading of antitumor immunity to different tumor antigens. EpCAM‐specific T cells disappeared completely from the peripheral blood immediately after completion of catumaxomab administration and reappeared with even higher numerical amounts 4 weeks later. Shaping of immune responses of EpCAM‐specific T cells was characterized by the occurrence of a T‐cell repertoire more restricted to certain epitopes with the EpCAM sequences, and expression of Th1‐type effector T cells.59
Immunogenicity and impact on PK/PD
As expected from the murine/rat nature of the antibody, ADA responses were observed after last administration of catumaxomab in the PK study.14 However, none of the patients developed significant ADA responses before the last infusion was administered. Interestingly, the phase II/III study revealed longer paracentesis‐free survival for human antimouse antibody (HAMA)‐positive patients vs. HAMA‐negative patients.60 The authors discussed various possible reasons for this observation. A more intact immune response may lead to a better responsiveness to catumaxomab, given that ADA responses require a functional immune system. Moreover, the anti‐idiotypic network hypothesis postulates that i) anti‐idiotypic antibodies itself can induce humoral responses; and further potentiation of the immune response can be achieved by ii) internalization of complexes comprised of murine therapeutic antibodies and human anti‐idiotype antibodies, with its presentation by antigen‐presenting cells possibly triggering further T‐cell activation, which can result in antitumor response. Besides the initial humoral response, a direct cellular immune response could also be induced by cytotoxic T cells recognizing a neoantigen in the presentation of the murine antibody via the major histocompatibility complex (MHC) of the antigen‐presenting cells.
CASE STUDIES: BiTE ANTIBODY CONSTRUCTS
BiTE‐mediated killing of cancer cells is independent of common immune escape mechanisms, such as expression of MHC class 1 molecules, antigen presentation, and activation of costimulatory molecules. Clinical studies have been conducted or are ongoing for several BiTE antibody constructs. Some of those studies will be presented in more detail in the following section.
Case study 2: Blinatumomab
Mechanism of action
Blinatumomab is a BiTE antibody construct that redirects CD‐3‐positive T cells to CD19‐expressing target cells.61, 62 The targeted CD19 antigen is constitutively expressed on normal B cells throughout a person's lifetime63 and is highly conserved in B‐cell malignancies.64, 65 Blinatumomab has an innovative mechanism of action that utilizes a patient's own T cells to attack CD19‐positive B cells, including malignant cells such as those in ALL. It transiently connects T cells and B cells, inducing T‐cell‐mediated killing of the bound B cell. A single activated T cell can trigger a serial lysis of multiple malignant or normal cells, a process that resembles a natural cytotoxic T‐cell reaction.
Blinatumomab activates T cells at picomolar concentrations, mimicking a natural MHC class I/peptide interaction with the T‐cell receptor. Expression of MHC class I and the presence of T‐cell receptor are not required for the redirected lysis, indicating that the effect of blinatumomab is independent of peptide antigen presentation and the presence of a T‐cell receptor.66 An advantage of this approach is that engaged T cells will be less susceptible to major immune escape mechanisms of tumor cells, thereby improving the effectiveness of T‐cell‐mediated killing.
Challenges in FIH studies
Blinatumomab does not cross‐react with mice, rats, or dogs. Therefore, a surrogate molecule (muS103new, binding to equivalent murine target antigens, CD3 and CD19) was constructed to conduct formal nonclinical toxicology and safety pharmacology investigations. Although the surrogate mediated redirected lysis of murine B cells and induced activation of murine T cells along with cytokine release analogous to the mechanism of blinatumomab in human cells, the blinatumomab FIH dose calculation did not follow conventional methods. Instead, a “trial and error” approach was applied.
Since blinatumomab was a new class of immunotherapy agent, no true “best schedule” information could be generated in preclinical studies prior to clinical introduction, owing to the lack of an appropriate animal model. Hence, three pilot phase I studies were conducted before a more extensive clinical study was initiated. Blinatumomab was first tested under 2‐ or 4‐h IV infusion 1, 2, or 3 times weekly. Of note, blinatumomab has a short half‐life of 1 to 2 h. All three short‐term infusion studies were terminated because of lack of clinical benefit in the face of central nervous system and cytokine release‐related adverse events (AEs). Some mechanism‐based biologic activities, such as cytokine release, minimal T‐cell activation, and selective decreases in peripheral B‐cell counts, were observed. The AEs appeared to be dose‐dependent and occurred mainly at the beginning of treatment.67
Due to the fast elimination of blinatumomab and the requirement for higher exposures to achieve efficacy, it was hypothesized that maintaining a prolonged steady‐state concentration with continuous IV infusion might be advantageous; a 4‐ or 8‐week continuous IV infusion for sustained T‐cell activation and B‐cell depletion was initiated. To mitigate the early AEs related to immune activation, step dosing regimens were introduced along with pretreatment cytoreduction regimens (mainly via steroids) to better manage AEs.
In this expanded phase I study, adult patients with relapsed non‐Hodgkin's lymphoma (NHL) received blinatumomab at doses ranging from 0.5–90 μg/m2/day by continuous IV infusion for 4 or 8 weeks. In this trial, multiple regimens were tested, including 0.5, 1.5, 5, 15, 30, 60, and 90 μg/m2/day flat dosing; 5–15, 5–30, 5–60, 15–60 μg/m2/day 1‐step dosing; and 5–15–60 μg/m2/day 2‐step dosing. The minimal effective dose and maximal tolerable dose were established at 5 and 60 μg/m2/day, respectively, and step dosing regimens were more effective and tolerable than the flat dosing in most cases.68
Bioanalytical approaches
The concentration of blinatumomab in human serum was determined by fluorescence activated cell sorting (FACS) analysis. The assay is based on the upregulation of CD69 on the activated T‐cell surface upon dual binding of blinatumomab to CD3 and CD19. The activation of CD69 was concentration dependent, which can be monitored by FACS after labeling with a fluorescent anti‐CD69 antibody. This activity‐based assay likely provides advantages over ELISA in terms of detecting the presence of the drug in its active form and at low concentrations.67
PK/PD
Blinatumomab is a recombinant non‐glycosylated protein that does not have an Fc domain, thus it does not undergo FcRn‐mediated recycling. Blinatumomab is rapidly catabolized into simple amino acids and cleared from the circulation, much like small proteins. The drug exhibits a linear and time‐independent PK and has not shown signs of target‐mediated clearance. Blinatumomab mainly remains in the blood circulation with a volume of distribution around 3–5 L, similar to normal blood volume. It has a short elimination half‐life of roughly 2 h and negligible renal clearance.69
Blinatumomab acts as a short adaptor, forcing T cells and tumor cells into close proximity, which results in a transient formation of a cytolytic synapse between a cytotoxic T cell and the cancer target cell. T cells are only activated by blinatumomab when a target cell is present.70 Following blinatumomab‐induced T‐cell proliferation, granzyme‐containing granules and the pore‐forming protein perforin fuse with the T‐cell membrane to discharge their toxic content into the target cell. Blinatumomab‐induced T‐cell activation and proliferation locally increases T‐cell numbers in the target tissue.71 The PD effects of blinatumomab include T‐cell redistribution, activation, and expansion; dose‐dependent B‐cell depletion; and transient cytokine elevation.69, 72
Considerations for dose selection
Blinatumomab dose selection was primarily based on its PK, PD, safety, and efficacy profiles. Drug administration with continuous IV infusion was supported by PK and efficacy profiles. Efficacy appeared to be dose‐dependent, requiring steady blinatumomab concentrations in the serum.
The requirement for step dosing depended on safety profiles, baseline B‐cell levels, and target effective dose levels for selected indications. If baseline B‐cell levels and the target dose level were low, AEs related to immune reaction appeared less prevalent, potentially negating the need for step dosing, as shown for the treatment of minimal residual disease (MRD)‐positive ALL.73 If baseline B‐cell levels were high but the target dose level was low, one‐step dosing was needed for better management of AEs, as indicated for the treatment for relapsed/refractory ALL.74 Lastly, if both baseline B‐cell levels and target dose level were high, two‐step dosing was needed for AE management, as reported for the treatment of relapsed/refractory diffuse large B‐cell lymphoma.75
Immunogenicity and impact on PK
Blinatumomab showed a low incidence of immunogenicity (∼1%) across studies. As the formation of ADA requires B cells, and depletion of B cells is the primary outcome of blinatumomab treatment, blinatumomab would prevent B cells from differentiating into plasma cells and producing ADA. The impact of formation of ADA on PK (i.e., reduction of drug concentrations) was observed in some cases, whereas the impact on efficacy and safety cannot be concluded owing to the low number of cases in which ADA developed.69
Drug–drug interaction
Blinatumomab did not affect CYP450 enzyme activities based on in vitro assays with human hepatocytes. As part of immune reactions, blinatumomab mediates transient cytokine elevations in patients during the first 1 to 2 days of treatment. The effects of a cytokine cocktail on P450 isozymes were examined via in vitro experiments, and the results showed that cytokines suppressed CYP3A4, CYP1A2, and CYP2C9 enzyme activities in a time‐ and concentration‐dependent manner; the effect was maximized at clinically observed cytokine peak concentrations.31
It is known that cytokines, especially IL‐6, can suppress CYP450 activities.76 A PBPK model for IL‐6 was established to evaluate magnitude and duration of IL‐6 suppression on hepatic CYP450 activities. The results suggested that transient IL‐6 elevation (1 to 2 days) up to clinically observed peak concentrations could suppress CYP3A4, CYP1A2, and CYP2C9 activities by as much as 30% for a week. This may in turn cause a < twofold increase of drug exposure to sensitive substrates of CYP3A4, CYP1A2, and CYP2C9. This evaluation indicated that the blinatumomab‐induced transient cytokine elevation may have a low risk for clinical drug interaction.31 Safety monitoring, especially for CYP3A4 substrates that have narrow therapeutic windows, is suggested in the early part of treatment.77
Case study 3: Solitomab (AMG 110, MT110)
Mechanism of action
The EpCAM‐targeting BiTE antibody construct consists of two single‐chain Fv domains derived from two different antibodies, one targeting EpCAM on epithelial‐derived cancer cells and the other targeting CD3ε on T cells, to form a single polypeptide chain. The mechanism of action is similar to other BiTE antibody constructs, as described previously. Solitomab showed potent in vitro antitumor activity and prevented tumor outgrowth completely, or resulted in durable eradication of established tumors in NOD/SCID mouse models.78
Challenges in FIH studies
An FIH study at doses between 1 and 96 μg/day was conducted in patients with refractory solid tumors known to frequently express EpCAM.79 The starting dose of 1 μg/day during weeks 1–4 was selected based on the minimal anticipated biological effect level (MABEL) established with a murine surrogate in mice. Dose‐limiting toxicities were mainly changes in gastrointestinal system (liver function abnormalities and severe diarrhea). Transient liver enzyme elevations during the first days of treatment demanded a low starting dose of 3 μg/day and slow, stepwise, intrapatient escalation. A prophylactic corticosteroid treatment during the first 3 days of solitomab therapy was instituted to prevent the initial changes in liver parameters. Severe diarrhea limited longer infusions for more than 3 or 4 weeks at higher doses. The nature and severity of dose‐limiting toxicities required multiple adjustments to the dosing regimen and made it difficult to identify a therapeutic window. Preclinical studies in cynomolgus monkey using a crossreactive molecule instead of rodent studies with a mouse surrogate may help identify possible severe adverse reactions more clearly.
PK/PD
PK analysis suggested linear PK over the tested dose range, with the steady‐state plasma level reached within 24 h.80 The half‐life of solitomab was 4.5 h. The mean maximum serum concentration (Cmax) was reported as ∼6 ng/mL at a dose of 96 μg/day. Preclinical studies with a murine version of solitomab showed that a 1‐week adaptation at a lower dose permitted prolonged treatment at a high dose thereafter by blunting the initial cytokine release. Furthermore, repeated long‐term dosing did not cause T‐cell anergy or compromise the effector function of T cells.80, 81
Immunogenicity and impact on PK/PD
ADA were detected in 7 of 63 (11%) tested patients; two patients had altered PK. None of the ADA‐positive patients showed signs or symptoms that could be attributed to anaphylactic or other hypersensitivity‐type reactions in the phase I study (Amgen data on file).
Case study 4: Other BiTE antibody constructs
Challenges in FIH and PK Considerations
Ryan et al. reported development of a CEA/CD3ε‐specific BiTE antibody construct that targets carcinoembryonic antigen (CEA), which is known to be found on epithelial cell membranes and in the cytoplasm of gastrointestinal adenocarcinomas, breast, and lung cancers.82 The molecule exerts the same BiTE antibody construct‐associated mechanism of action. Low BiTE antibody construct concentrations were sufficient to induce killing of CEA and tumor cells after administration of T cells from patients or healthy donors.83 An FIH study was conducted in adult patients with advanced gastrointestinal adenocarcinomas. Since no relevant animal model could be identified to perform in vivo toxicology studies, the selection of the FIH start dose was solely based on assessment of dose–response correlations in vitro. The MABEL that demonstrated 20% maximal effect (EC20) of BiTE antibody construct‐induced tumor cell lysis as the most sensitive measure of biologic activity was used to select the starting dose for the FIH study. In addition, a nonterminal PK study conducted in cynomolgus monkeys was used to predict human PK parameters based on allometric scaling. The exposure ranges around the identified MABEL concentration guided selection of the start dose and administration schedule for the phase I study.84 PK considerations may also inform phase I studies. As seen with blinatumomab and solitomab, antitumor activity may require a long‐term, steady‐state exposure above a threshold with trough levels maintained above the range of EC50 or EC90. In addition, by avoiding fluctuations in Cmax exposures, a sustained target coverage with less Cmax‐driven toxicity can be expected. The start dose selection for additional phase Ia or phase Ib studies can also be based on a simulation of average total area‐under‐the‐curve with cIV administration utilizing PK information from intermittent dosing of the FIH study.
Friedrich et al. reported another example of a BiTE antibody construct targeting prostate‐specific membrane antigen (PSMA) via binding to PSMA and CD3ε of human and macaque origin. It is the first BiTE antibody construct that contains an amino acid sequence very close to human germline Ig‐V segments, allowing crossreactivity to human and nonhuman primate PSMA and CD3 antigens. In preclinical experiments, the PMSA/CD3ε BiTE antibody construct was considerably more potent and able to mediate rapid tumor shrinkage and complete remissions of established human 22Rv1 prostate cancer xenografts after subcutaneous (s.c.) administration of the PMSA/CD3ε BiTE antibody construct. A serum half‐life of approximately 8 h after single bolus or s.c. administration and 18% bioavailability after s.c. administration in mice was found.85 These results formed the rationale for the selected route of administration in the FIH study (ClinicalTrials.gov NCT01723475).
CONCLUSION
BsAbs are an emerging drug modality with significant therapeutic value. While the development of BsAbs is an evolving field, the growing experience and application of BsAbs will become increasingly important for informing development steps. We have attempted to expand the body of existing knowledge in the space of BsAb.
Acknowledgments
Editorial support was provided by Jim Slade and Tonya Goodman of Fishawack Communications, which was funded by Amgen. Gregory Friberg, Global product general manager at Amgen Inc. provided scientific suggestions and comments in preparing this article.
Conflict of Interest
A.T., M.Z., H.L., T.Y., T.H., and R.L. are full‐time employees of and shareholders in Amgen. S.S. is a full‐time employee of Amgen Research (Munich) and a shareholder in Amgen. J.G. and S.K. were a full‐time employee of Amgen at the time of article preparation and is a current shareholder in Amgen.
References
- 1. U.S. Department of Health and Human Services FaDA , Center for Drug Evaluation and Research (CDER) Codevelopment of Two or More New Investigational Drugs for Use in Combination http://wwwfdagov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/defaulthtm. June 2013.
- 2. Moores, S.L. et al A novel bispecific antibody targeting EGFR and cMet is effective against EGFR inhibitor‐resistant lung tumors. Cancer Res. 76, 3942–3953 (2016). [DOI] [PubMed] [Google Scholar]
- 3. van Steeg T.J., Bergmann, K.R. , Dimasi, N. , Sachsenmeier, K.F. & Agoram, B. The application of mathematical modelling to the design of bispecific monoclonal antibodies. mAbs. 8, 585–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Byrne, H. , Conroy, P.J. , Whisstock, J.C. & O'Kennedy, R.J. A tale of two specificities: bispecific antibodies for therapeutic and diagnostic applications. Trends Biotechnol. 31, 621–632 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jost, C. & Pluckthun, A. Engineered proteins with desired specificity: DARPins, other alternative scaffolds and bispecific IgGs. Current Opin. Struct. Biol. 27, 102–112 (2014). [DOI] [PubMed] [Google Scholar]
- 6. Huehls, A.M. , Coupet, T.A. & Sentman, C.L. Bispecific T‐cell engagers for cancer immunotherapy. Immunol. Cell Biol. 93, 290–296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Spiess, C. , Zhai, Q. & Carter, P.J. Alternative molecular formats and therapeutic applications for bispecific antibodies. Mol. Immunol. 67(2 Pt A), 95–106 (2015). [DOI] [PubMed] [Google Scholar]
- 8. Kontermann, R.E. & Brinkmann, U. Bispecific antibodies. Drug Discov. Today. 20, 838–847 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Fan, G. , Wang, Z. , Hao, M. & Li, J. Bispecific antibodies and their applications. J. Hematol. Oncol. 8, 130 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lameris, R. et al Bispecific antibody platforms for cancer immunotherapy. Crit. Rev. Oncol. Hematol. 92, 153–165 (2014). [DOI] [PubMed] [Google Scholar]
- 11. Release, P. Bispecific antibodies market to be worth USD 4.4 billion by 2023, predicts roots analysis. Reuters. (2013). [Google Scholar]
- 12. Sheridan, C. Despite slow progress, bispecifics generate buzz. Nat. Biotechnol. 34, 1215–1217 (2016). [DOI] [PubMed] [Google Scholar]
- 13. Gunasekaran, K. et al Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG. J. Biol. Chem. 285, 19637–19646 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ruf, P. et al Pharmacokinetics, immunogenicity and bioactivity of the therapeutic antibody catumaxomab intraperitoneally administered to cancer patients. Br. J. Clin. Pharmacol. 69, 617–625 (2010) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kienast, Y. et al Ang‐2‐VEGF‐A CrossMab, a novel bispecific human IgG1 antibody blocking VEGF‐A and Ang‐2 functions simultaneously, mediates potent antitumor, antiangiogenic, and antimetastatic efficacy. Clin. Cancer Res. 19, 6730–6740 (2013). [DOI] [PubMed] [Google Scholar]
- 16. Wu, C. et al Simultaneous targeting of multiple disease mediators by a dual‐variable‐domain immunoglobulin. Nat. Biotechnol. 25, 1290–1297 (2007). [DOI] [PubMed] [Google Scholar]
- 17. Bargou, R. et al Tumor regression in cancer patients by very low doses of a T cell‐engaging antibody. Science 321, 974–977 (2008). [DOI] [PubMed] [Google Scholar]
- 18. Friedrich, M. et al Preclinical characterization of AMG 330, a CD3/CD33‐bispecific T‐cell‐engaging antibody with potential for treatment of acute myelogenous leukemia. Mol. Cancer Ther. 13, 1549–1557 (2014). [DOI] [PubMed] [Google Scholar]
- 19. Silacci, M. et al Discovery and characterization of COVA322, a clinical‐stage bispecific TNF/IL‐17A inhibitor for the treatment of inflammatory diseases. mAbs. 8, 141–149 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Doppalapudi, V.R. et al Chemical generation of bispecific antibodies. Proc. Natl. Acad. Sci. U.S.A. 107, 22611–22616 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ahene, A.B. Application and interpretation of free and total drug measurements in the development of biologics. Bioanalysis. 3, 1287–1295 (2011). [DOI] [PubMed] [Google Scholar]
- 22. Zhang, Y.J. , Luo, L. & Desai, D.D. Overview on biotherapeutic proteins: impact on bioanalysis. Bioanalysis. 8, 1–9 (2016). [DOI] [PubMed] [Google Scholar]
- 23. Wang, Y.M. , Jawa, V. & Ma, M. Immunogenicity and PK/PD evaluation in biotherapeutic drug development: scientific considerations for bioanalytical methods and data analysis. Bioanalysis. 6, 79–87 (2014) [DOI] [PubMed] [Google Scholar]
- 24. Han, Y. , Guo, Q. , Zhang, M. , Chen, Z. & Cao, X. CD69+ CD4+ CD25‐ T cells, a new subset of regulatory T cells, suppress T cell proliferation through membrane‐bound TGF‐beta 1. J Immunol. 182, 111–120 (2009). [DOI] [PubMed] [Google Scholar]
- 25. Goldenberg, D.M. , Chatal, J.F. , Barbet, J. , Boerman, O. & Sharkey, R.M. Cancer imaging and therapy with bispecific antibody pretargeting. Update Cancer Ther. 2, 19–31 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luo, H. et al Noninvasive brain cancer imaging with a bispecific antibody fragment, generated via click chemistry. Proc. Natl. Acad. Sci. U.S.A. 112, 12806–12811 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo, H. , Hong, H. , Yang, S.P. & Cai, W. Design and applications of bispecific heterodimers: molecular imaging and beyond. Mol. Pharm. 11, 1750–1761 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee, J.I. , Zhang, L. , Men, A.Y. , Kenna, L.A. & Huang, S.M. CYP‐mediated therapeutic protein‐drug interactions: clinical findings, proposed mechanisms and regulatory implications. Clinic Pharmaco. 49, 295–310 (2010). [DOI] [PubMed] [Google Scholar]
- 29. Zhang X, S.C. & Grange, S. Disease‐drug interaction studies of tocilizumab with cytochrome p450 substrates in vitro and in vivo . Clin. Pharmacol. Ther. 85, S59 (2009) [Google Scholar]
- 30. Le Vee, M. , Lecureur, V. , Stieger, B. & Fardel, O. Regulation of drug transporter expression in human hepatocytes exposed to the proinflammatory cytokines tumor necrosis factor‐alpha or interleukin‐6. Drug Metab. Dispos. 37, 685–693 (2009). [DOI] [PubMed] [Google Scholar]
- 31. Xu, Y. et al Physiologically based pharmacokinetic model to assess the influence of blinatumomab‐mediated cytokine elevations on cytochrome P450 enzyme activity. CPT Pharmacometrics Syst. Pharmacol. 4, 507–515 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kenny, J.R. , et al Therapeutic protein drug‐drug interactions: navigating the knowledge gaps‐highlights from the 2012 AAPS NBC Roundtable and IQ Consortium/FDA workshop. AAPS J. 15, 933–940 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Agency, E.M. Guideline on the Clinical Investigation of the Pharmacokinetics of Therapeutic Proteins. http://wwwemaeuropaeu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003029pdf. 2007.
- 34. (CDER) USDoHaHSFaDACfDEaR . Drug Interaction Studies — Study Design, Data Analysis, Implications for Dosing, and Labeling Recommendations. http://wwwfdagov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/defaulthtm 2012.
- 35. Kamath, A.V. et al Preclinical pharmacokinetics of MEHD7945A, a novel EGFR/HER3 dual‐action antibody, and prediction of its human pharmacokinetics and efficacious clinical dose. Cancer Chemother. Pharmacol. 69, 1063–1069 (2012). [DOI] [PubMed] [Google Scholar]
- 36. Gibiansky, L. & Gibiansky, E. Target‐mediated drug disposition model for drugs that bind to more than one target. J. Pharmacokinet. Pharmacodyn. 37, 323–346 (2010). [DOI] [PubMed] [Google Scholar]
- 37. U.S. Department of Health and Human Services FaDA , Center for Drug evaluation and research (CDER) guidance for industry: Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers. July 2005.
- 38. Hu, L. & Hansen, R.J. Issues, challenges, and opportunities in model‐based drug development for monoclonal antibodies. J. Pharm. Sci. 102, 2898–2908 (2013). [DOI] [PubMed] [Google Scholar]
- 39. Friedrich, S.W. et al Antibody‐directed effector cell therapy of tumors: Analysis and optimization using a physiologically based pharmacokinetic model. Neoplasia. 4, 449–463 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Apgar, J.F. , Wong, J. , Ryan Phennicie, R. , Briskin, M. & Burke, J.M. Abstract A151: Quantitative systems pharmacology approaches accelerate lead generation and optimization of a PD‐1 x TIM‐3 therapeutic in immuno‐oncology. Mol. Cancer Ther. 14(12 Supplement 2), A151 (2015). [Google Scholar]
- 41. Fitzgerald, J.B. et al MM‐141, an IGF‐IR– and ErbB3‐directed bispecific antibody, overcomes network adaptations that limit activity of IGF‐IR Inhibitors. Mol. Cancer Ther. 13, 410–425 (2014). [DOI] [PubMed] [Google Scholar]
- 42. Huang, S. et al Dual targeting of EGFR and HER3 with MEHD7945A overcomes acquired resistance to EGFR inhibitors and radiation. Cancer Res. 73, 824–833 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yan, L. et al Quantitative systems pharmacology modeling to evaluate clinical response of an anti‐TNFα‐anti‐Ang2 bispecific antibody in rheumatoid arthritis ASCPT 2014 Annual Meeting. 2014;March 18‐22, 2014(Atlanta, GA).
- 44. Singh, I. et al A systems pharmacology model to characterize the effect of blinatumomab in patients with adult B precursor acute lymphoblastic leukemia. March 2014;Poster presented at the ASCPT annual meeting, Atlanta, GA.
- 45. Gadkar, K. et al Mathematical PKPD and safety model of bispecific TfR/BACE1 antibodies for the optimization of antibody uptake in brain. Eur. J. Pharm. Biopharm. 101, 53–61 (2016). [DOI] [PubMed] [Google Scholar]
- 46. Findlay, J.W. et al Validation of immunoassays for bioanalysis: a pharmaceutical industry perspective. J. Pharm. Biomed. Anal. 21, 1249–1273 (2000). [DOI] [PubMed] [Google Scholar]
- 47. Sampei, Z. et al Identification and multidimensional optimization of an asymmetric bispecific IgG antibody mimicking the function of factor VIII cofactor activity. PLoS One. 8, e57479 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stevenson, L. et al 2014 White Paper on recent issues in bioanalysis: a full immersion in bioanalysis (Part 3 ‐ LBA and immunogenicity). Bioanalysis. 6, 3355–3368 (2014). [DOI] [PubMed] [Google Scholar]
- 49. Murphy, R.E. et al Combined use of immunoassay and two‐dimensional liquid chromatography mass spectrometry for the detection and identification of metabolites from biotherapeutic pharmacokinetic samples. J. Pharm. Biomed. Anal. 53, 221–227 (2010). [DOI] [PubMed] [Google Scholar]
- 50. Baeuerle, P.A. & Gires, O. EpCAM (CD326) finding its role in cancer. Br. J. Cancer. 96, 417–423 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Heiss, M.M. et al The trifunctional antibody catumaxomab for the treatment of malignant ascites due to epithelial cancer: Results of a prospective randomized phase II/III trial. Int. J. Cancer. 127, 2209–2221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Seimetz, D. , Lindhofer, H. & Bokemeyer, C. Development and approval of the trifunctional antibody catumaxomab (anti‐EpCAM x anti‐CD3) as a targeted cancer immunotherapy. Cancer Treat. Rev. 36, 458–467 (2010). [DOI] [PubMed] [Google Scholar]
- 53. Mau‐Sorensen, M. et al A phase I trial of intravenous catumaxomab: a bispecific monoclonal antibody targeting EpCAM and the T cell coreceptor CD3. Cancer Chemother. Pharmacol. 75, 1065–1073 (2015). [DOI] [PubMed] [Google Scholar]
- 54. Heiss, M.M. et al Immunotherapy of malignant ascites with trifunctional antibodies. Int J Cancer. 117, 435–443 (2005). [DOI] [PubMed] [Google Scholar]
- 55. Burges, A. et al Effective relief of malignant ascites in patients with advanced ovarian cancer by a trifunctional anti‐EpCAM x anti‐CD3 antibody: a phase I/II study. Clin. Cancer Res. 13, 3899–3905 (2007). [DOI] [PubMed] [Google Scholar]
- 56. Bezan, A. , Hohla, F. , Meissnitzer, T. & Greil, R. Systemic effect of catumaxomab in a patient with metastasized colorectal cancer: a case report. BMC Cancer. 13, 618 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Woopen, H. , Pietzner, K. , Darb‐Esfahani, S. , Oskay‐Oezcelik, G. & Sehouli, J. Extraperitoneal response to intraperitoneal immunotherapy with catumaxomab in a patient with cutaneous lymphangiosis carcinomatosa from ovarian cancer: a case report and review of the literature. Med. Oncol. 29, 3416–3420 (2012). [DOI] [PubMed] [Google Scholar]
- 58. Heiss, M.M. et al The role of relative lymphocyte count as a biomarker for the effect of catumaxomab on survival in malignant ascites patients: results from a phase II/III study. Clin. Cancer Res. 20, 3348–3357 (2014). [DOI] [PubMed] [Google Scholar]
- 59. Atanackovic, D. et al The trifunctional antibody catumaxomab amplifies and shapes tumor‐specific immunity when applied to gastric cancer patients in the adjuvant setting. Hum. Vaccin. Immunother. 9, 2533–2542 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ott, M.G. et al Humoral response to catumaxomab correlates with clinical outcome: results of the pivotal phase II/III study in patients with malignant ascites. Int. J. Cancer. 130, 2195–2203 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Hoffmann, P. et al Serial killing of tumor cells by cytotoxic T cells redirected with a CD19‐/CD3‐bispecific single‐chain antibody construct. Int. J. Cancer. 115, 98–104 (2005). [DOI] [PubMed] [Google Scholar]
- 62. Dreier, T. et al Extremely potent, rapid and costimulation‐independent cytotoxic T‐cell response against lymphoma cells catalyzed by a single‐chain bispecific antibody. Int. J. Cancer. 100, 690–697 (2002). [DOI] [PubMed] [Google Scholar]
- 63. Smet, J. , Mascart, F. & Schandene, L. Are the reference values of B cell subpopulations used in adults for classification of common variable immunodeficiencies appropriate for children? Clin. Immunol. 138, 266–273 (2011). [DOI] [PubMed] [Google Scholar]
- 64. Tedder, T.F. CD19: a promising B cell target for rheumatoid arthritis. Nat. Rev. Rheumatol. 5, 572–577 (2009). [DOI] [PubMed] [Google Scholar]
- 65. Wang, K. , Wei, G. & Liu, D. CD19: a biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 1, 36 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Offner, S. , Hofmeister, R. , Romaniuk, A. , Kufer, P. & Baeuerle, P.A. Induction of regular cytolytic T cell synapses by bispecific single‐chain antibody constructs on MHC class I‐negative tumor cells. Mol. Immunol. 43, 763–771 (2006). [DOI] [PubMed] [Google Scholar]
- 67. Nagorsen, D. , Kufer, P. , Baeuerle, P.A. & Bargou, R. Blinatumomab: a historical perspective. Pharmacol. Ther. 136, 334–342 (2012). [DOI] [PubMed] [Google Scholar]
- 68. Goebeler, M.E. et al Bispecific T‐Cell Engager (BiTE) antibody construct blinatumomab for the treatment of patients with relapsed/refractory non‐Hodgkin lymphoma: final results from a phase I study. J Clin Oncol. 34, 1104–1111 (2016). [DOI] [PubMed] [Google Scholar]
- 69. Zhu, M. et al Blinatumomab, a Bispecific T‐cell Engager (BiTE) for CD‐19 targeted cancer immunotherapy: clinical pharmacology and its implications. Clin. Pharmacokinet. 2016. [DOI] [PubMed] [Google Scholar]
- 70. Baeuerle, P.A. , Reinhardt, C. & Kufer, P. BiTE: A new class of antibodies that recruit T‐cells. Drugs Fut. 33, 137 (2008). [Google Scholar]
- 71. Nagorsen, D. & Baeuerle, P.A. Immunomodulatory therapy of cancer with T cell‐engaging BiTE antibody blinatumomab. Exp. Cell Res. 317, 1255–1260 (2011). [DOI] [PubMed] [Google Scholar]
- 72. Klinger, M. et al Immunopharmacologic response of patients with B‐lineage acute lymphoblastic leukemia to continuous infusion of T cell‐engaging CD19/CD3‐bispecific BiTE antibody blinatumomab. Blood. 119, 6226–6233 (2012). [DOI] [PubMed] [Google Scholar]
- 73. Gökbuget, N.D.H. et al Long‐term outcomes after blinatumomab treatment: follow‐up of a phase 2 study in patients (Pts) with minimal residual disease (MRD) positive B‐cell precursor acute lymphoblastic leukemia (ALL). Oral presentation at 58th American Society of Hematology Anuual Meeting, Orlando, FL, Dec 2015. 2015.
- 74. Topp, M.S. et al Safety and activity of blinatumomab for adult patients with relapsed or refractory B‐precursor acute lymphoblastic leukaemia: a multicentre, single‐arm, phase 2 study. Lancet Oncol. 16, 57–66 (2015). [DOI] [PubMed] [Google Scholar]
- 75. Viardot, A. et al Phase 2 study of the bispecific T‐cell engager (BiTE) antibody blinatumomab in relapsed/refractory diffuse large B‐cell lymphoma. Blood. 127, 1410–1416 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Huang, S.M. et al Therapeutic protein‐drug interactions and implications for drug development. Clin. Pharmacol. Ther. 87, 497–503 (2010). [DOI] [PubMed] [Google Scholar]
- 77. Amgen Inc TO, CA, USA 2014 . BlincytoTM (blinatumomab) prescribing information. 2014.
- 78. Brischwein, K. et al MT110: a novel bispecific single‐chain antibody construct with high efficacy in eradicating established tumors. Mol. Immunol. 43, 1129–1143 (2006). [DOI] [PubMed] [Google Scholar]
- 79. Fiedler, W.M. et al, editors. A phase I study of EpCAM/CD3‐bispecific antibody (MT110) in patients with advanced solid tumors. ASCO Annual Meeting Proceedings; 2012.
- 80. Amann, M. et al Therapeutic window of an EpCAM/CD3‐specific BiTE antibody in mice is determined by a subpopulation of EpCAM‐expressing lymphocytes that is absent in humans. Cancer Immunol. Immunother. 58, 95–109 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Amann, M. et al Antitumor activity of an EpCAM/CD3‐bispecific BiTE antibody during long‐term treatment of mice in the absence of T‐cell anergy and sustained cytokine release. J Immunother. 32, 452–464 (2009). [DOI] [PubMed] [Google Scholar]
- 82. Hammarstrom, S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin. Cancer Biol. 9, 67–81 (1999). [DOI] [PubMed] [Google Scholar]
- 83. Osada, T. et al Metastatic colorectal cancer cells from patients previously treated with chemotherapy are sensitive to T‐cell killing mediated by CEA/CD3‐bispecific T‐cell‐engaging BiTE antibody. Br. J. Cancer. 102, 124–133 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ryan, P.C. et al In vitro MABEL approach for nonclinical safety assessment of MEDI‐565 (MT111). ALTEX Proc. 1, 85–87 (2012). [Google Scholar]
- 85. Friedrich, M. et al Regression of human prostate cancer xenografts in mice by AMG 212/BAY2010112, a novel PSMA/CD3‐Bispecific BiTE antibody cross‐reactive with non‐human primate antigens. Mol. Cancer Ther. 11, 2664–2673 (2012). [DOI] [PubMed] [Google Scholar]