

Abstract
Paragangliomas/pheochromocytomas comprise rare tumors that arise from the extra-adrenal paraganglia, with an incidence of about 2 to 8 per million people each year. Approximately 40% of cases are due to genetic mutations in at least one out of more than 30 causative genes. About 2530% of pheochromocytomas/paragangliomas develop under the conditions of a hereditary tumor syndrome a third of which are caused by mutations in the VHL gene. Together, the gene mutations in this disorder have implicated multiple processes including signaling pathways, translation initiation, hypoxia regulation, protein synthesis, differentiation, survival, proliferation, and cell growth. The present review contemplates the mutations associated with the development of pheochromocytomas/paragangliomas and their potential to serve as specific markers of these tumors and their progression. These data will improve our understanding of the pathogenesis of these tumors and likely reveal certain features that may be useful for early diagnostics, malignancy prognostics, and the determination of new targets for disease therapeutics.
Keywords: paragangliomas, pheochromocytomas, molecular markers, germline and somatic mutations, signaling pathways
INTRODUCTION
Paragangliomas comprise tumors of the paraganglia, which are organs derived from rudiments of the autonomic nervous system. Paraganglia are classified as chromaffin, which belong to the adrenal system and are capable of secretion, and nonchromaffin, most of which exhibit a chemoreceptor function. The largest chromaffin paraganglion is the adrenal medulla. The nonchromaffin paraganglia include the carotid, jugular, and other glomera. Paraganglion tumors are classified according to their origin. For example, adrenal tumors are referred to as pheochromocytomas, which are known to often secrete catecholamines (epinephrine and norepinephrine). Paragangliomas derived from cells of the sympathetic nervous system mostly occur in the paraxial/prevertebral regions including the pelvic organs and the organ of Zuckerkandl. Conversely, most extra-adrenal paragangliomas develop in the head and neck from cells of the parasympathetic nervous system. In the majority of cases these tumors have no secretory activity and manifest themselves as painless formations in the neck region. The head and neck paragangliomas are largely formed along the glossopharyngeal and vagus nerves (vagal paragangliomas) and are most often located at the carotid bifurcation, in the middle ear region, and at the jugular fossa. In particular, a tumor of the carotid body, comprising one of the head and neck paragangliomas, is referred to as a carotid paraganglioma.
Paragangliomas are presently among the most intensively studied tumors, despite their comparative rarity. The exact values are unknown but according to the data of the North American Neuroendocrine Tumor Society, the incidence of these tumors is estimated at 1:6500 to 1:2500 [1]. In comparison, the incidence of paragangliomas determined from autopsy data is 1:2000, indicating a high fraction of undetected tumors [2]. This phenomenon is likely due to the fact that most paragangliomas are benign [3], with only 10–15% undergoing malignant change as manifested by the abnormal presence of chromaffin tissue in the lymph nodes, liver, and lungs [4, 5]. Among paragangliomas, pheochromocytomas represent the most common identified subtype [6].
An important feature of these tumors is their high heritability rate as compared with other neoplasias. Almost 40% of paragangliomas exhibit germline mutations in at least one out of more than 30 potentially causative genes [7, 8]. Approximately 25–30% of these tumors develop under conditions of a hereditary tumor syndrome [9] a third of which are caused by mutations in the Von Hippel Lindau (VHL) gene. Additionally, 25–30% of the tumors carry somatic mutations in the RET, VHL, neurofibromin 1 (NF1), and MYC-associated factor X (MAX) genes among others [10–13]. Notably, the somatic and hereditary mutations are found in a mutually exclusive manner. A further characteristic feature of these tumors is that their originating mutations may occur in succinate dehydrogenase (SDH) genes [14] that encode metabolic enzymes. Conversely, other cancers generally involve disturbances of transcription factors and signaling pathways. Moreover, the presence of activating mutations in the hypoxia-inducible factor 2-α (HIF2A) gene was first shown in paragangliomas. Although such mutations had been previously detected in the course of tumor development, their role as oncogenes had not been demonstrated [13].
This review represents the most complete compilation of information available to date regarding the mechanisms of paraganglioma/pheochromocytoma development and the associated mutations. These data will improve the understanding of the pathogenesis of these tumors and likely reveal certain features that may be useful toward facilitating early tumor diagnosis, predicting their malignancy, and determining new targets for therapy.
GENETIC CLASSIFICATION OF PARAGANGLIOMAS/PHEOCHROMO-CYTOMAS
Paragangliomas/pheochromocytomas are traditionally classified according to their expression profiles and associated mutations based on transcriptomic and genomic data [15]. The first group (Group I) includes mutations in the VHL, SDH, and prolyl hydroxylase domain PHD genes as well as in markers of pseudohypoxia (EPAS1, NOX4, LOXL2), angiogenesis (vasoendothelial growth factor, VEGF), and reduced oxidative response [16]. The second group (Group II) includes mutations in the RET, NF1, transmembrane protein 127 (TMEM127), kinesin family member 1B-beta isoform (KIF1Bβ), and MAX genes. Furthermore, tumors of this group are characterized by the impaired regulation of several signaling pathways (PI3K/AKT, RAS/RAF/ERK, and mTORC1/p70S6 kinase (p70S6K)) as well as translation initiation, protein synthesis, and also neuronal (SHANK2 and RET) and neuroendocrine (PNMT, NCAM2, and CADPS) differentiation [16].
Tumors with mutations in the genes involved in the pseudohypoxic pathway of tumor development belong to Group I [15] (Figure 1). In the normal state these genes participate in the response to hypoxia; however, mutations impair the regulation of this response, leading to the activation of effector molecules in the absence of hypoxia. Group I tumors exhibit an increased rate of angiogenesis and elevated expression of VEGF and its receptors [17, 18]. Notably, these elevated expression levels have been observed in both benign and malignant paragangliomas [17]. In comparison, Group II tumors include those with mutations leading to the abnormal activation of various signaling pathways associated with kinase-like proteins; for example, PI3Kinase/AKT and mTOR [17] (Figure 2).
Figure 1. Impaired hypoxic status regulation owing to mutations in Group 1 genes.
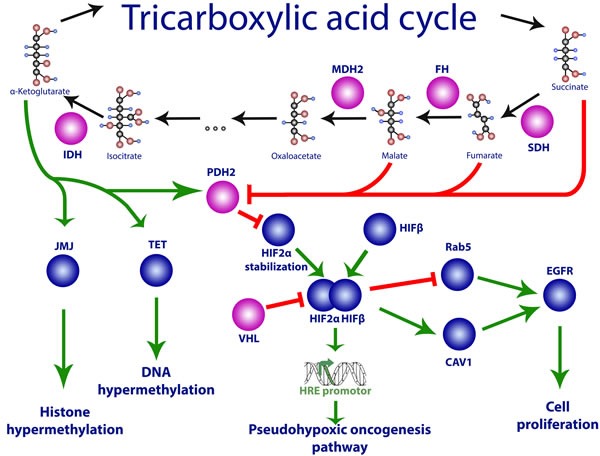
Mutations in VHL, SDH, HIF2A, PHD2, and FH genes may lead to activation of the transcription factor HIF-1 and its target genes that promote pseudohypoxic oncogenesis. See text for details.
Figure 2. Impaired growth factor signaling owing to mutations in Group 2 genes.
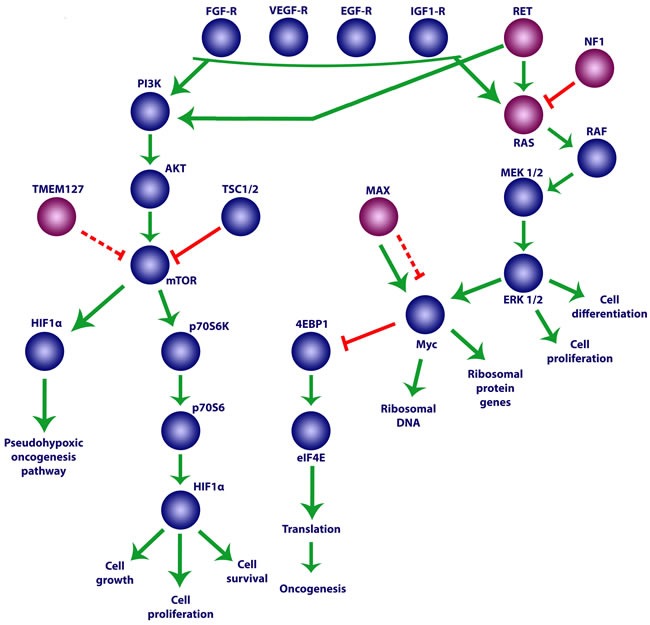
See text for details.
In turn, these groups are subdivided into subgroups according to differences in the corresponding transcription profiles. Group I comprises subgroup 1A, including paragangliomas with mutations in the SDH and fumarate hydratase (FH) genes, and subgroup 1B, representing those with mutations in the HIF2A and VHL genes [19]. Group II in turn comprises four subgroups: 2A-D [15]. Subgroup 2A includes tumors with mutations in the RET, MAX, NF1, and TMEM127 genes whereas subgroups 2B and 2C include sporadic tumors [15]. Subgroup 2D consists collectively of tumors that do not exhibit any known mutations associated with paragangliomas/pheochromocytomas.
Group I
The common trait of all tumors in Group I is the activation of HIFs. In the normal state, the induction of these transcription factors occurs in response to low oxygen content; i.e., hypoxia [20]. Constitutive activation of HIF regardless of the oxygen content results in pseudohypoxia [21] and inopportune activation of HIF target genes such as those encoding angiogenesis factors including VEGF and platelet-derived growth factor B chain [22, 23]. HIFs represent heterodimeric transcription factors, the inducible components of which are finely regulated by hydroxylation and proteasomal degradation [21, 24, 25]. They consist of an α subunit, which is sensitive to oxygen, and a β subunit. Furthermore, HIF-α has three isoforms: HIF1α is activated during short-term severe hypoxia, HIF2α functions during the longer period of moderate hypoxia, and HIF3α acts as an inhibitor of HIF1α [26].
At sufficient oxygen levels in the cell, the PHD protein hydroxylates HIF1α. Subsequently, the VHL protein recognizes hydroxylated HIF1α and ubiquitinates it, which leads to its degradation owing to 26S proteasome activity [27]. Hypoxia and pseudohypoxia are controlled in specific ways by the products of Group I tumor genes, thus explaining the mechanism underlying the involvement of mutations in these genes in paraganglioma development. Under normal conditions, HIF1α and HIF2α are inhibited through degradation controlled by the VHL protein. Furthermore, their degradation also requires hydroxylation of the proline in the HIF1α subunit, which is facilitated by proteins of the EGLN/PHD family (see below). Impairment of this process leads to the accumulation of HIF, which in turn activates genes involved in angiogenesis, hemopoiesis, cell growth, and migration [28] (Figure 1). Additionally, if the activity of FH is impaired, fumarate accumulation in the cell inactivates the EGLN/PHD proteins, which also leads to the accumulation of HIF [29]. Group I tumors most frequently exhibit mutations in the VHL, SDH, and HIF2A genes. However, as many tumors revealed no mutations in these genes, their apparent prevalence may result from insufficient data or undetected new mutations [30, 31].
Von hippel lindau (VHL)
VHL is a tumor suppressor gene located on chromosome 3p25.3 [32]. Its mutations result in von Hippel Lindau syndrome, an autosomal dominant genetic disorder that contributes to the development of clear cell renal cell carcinoma as well as cerebellar, spinal, and retinal hemangioblastomas in addition to paragangliomas/pheochromocytomas [24, 33]. Somatic mutations in this gene were first identified in cervical paragangliomas in 2013 [34].
VHL comprises the substrate-recognizing component of the E3 ubiquitin ligase complex that binds to HIF1α and HIF2α during their proteasomal degradation [35]. Thus, a mutation in the VHL gene reduces the efficiency of HIF1α and HIF2α degradation, which leads to the accumulation of these factors and constitutive activation of their targets. Although many targets are shared by HIF1α and HIF2α, others bind to only one of these factors. Notably, this selectivity might explain the higher oncogenicity reported for HIF2α [36, 37]. For example, HIF2α stimulates the activity of the protein encoded by the oncogene MYC, whereas HIF1α acts as an antagonist of MYC [38]. In particular, according to the results of in vitro experiments, most mutations in the VHL gene impair the degradation of the HIF2α subunit [39]. In addition, HIF2α has been shown to be constitutively activated in paragangliomas with VHL mutations [40, 41]; accordingly, such tumors were also shown to harbor mutations associated with unregulated activity of HIF2α [13, 42–44]. Similarly, elevated HIF1α activity has only been demonstrated in a study of paragangliomas carrying a VHL mutation [19]. However, the role of HIF1α in oncogenesis is poorly elucidated: this protein was found to suppress tumors under certain conditions, although deletions in the HIF1A gene were detected in renal carcinomas [45]. No mutations or deletions in the locus containing HIF1A have yet been identified in paragangliomas or pheochromocytomas [13].
In addition to the obvious relationships between mutations in the VHL gene, HIFs, and oncogenesis, an HIF-independent mechanism of pathogenesis involving VHL protein mutations has also been described [46]. Aside from HIF regulation, the VHL protein also takes part in controlling the cell cycle and in extracellular matrix formation [47, 48]. For example, VHL affects the assembly of the fibronectin matrix component through a direct interaction with fibronectin, thus influencing the secretion of metalloproteinases, the formation of integrin adhesive fibrils, and the regulation of other enzymes regulating extracellular matrix activity [46, 47, 49]. Consistent with this function, the morphology of cells with impaired VHL secretion differs greatly from that of normal cells. Specifically, in the complete absence of VHL the cells become spheroid and grow equally in all directions without any signs of differentiation [50] whereas in the presence of VHL, tumor cells form aggregates that exhibit some traits of epithelial differentiation and may even form monolayers [50, 51]. In order to bind to fibronectin, the VHL protein has to be modified by NEDD8. Notably, this modification does not change its E3 activity or its ability to inactivate HIF. Alternatively, mutations affecting the part of the VHL molecule that is involved in its modification instead impair the morphology of the differentiated cells and lead to oncogenesis [52].
Accordingly, two types of von Hippel Lindau syndrome are distinguished according to the kinds of tumors that develop and the underlying VHL gene mutations. In general, the pheochromocytomas that manifest in association with von Hippel Lindau syndrome are most often numerous and bilateral. Additionally, they produce norepinephrine owing to a deficiency of the enzyme phenylethanolamine N-methyltransferase, which converts norepinephrine to epinephrine in the adrenal glands [55]. Type 1 syndrome is associated with a low probability of pheochromocytomas (6–9% of all the patients with the syndrome) whereas type 2 carries a high probability (40–59%) of these particular tumors. Type 2 is further subdivided into type 2A and type 2B with a low and high probability of renal cancer, respectively. Moreover, in some cases, a syndrome of type 2C is also observed that is associated with pheochromocytomas but not with hemangioblastomas or renal cancer [53, 54]. Furthermore, mutations in the VHL gene have been found in both sporadic and hereditary head and neck paragangliomas/pheochromocytomas [56–62]. Type 1 corresponds to the complete absence of the VHL protein whereas tumors of type 2 exhibit small mutations leading to conformationally inactive protein forms [63–65]. In particular, these small mutations may impair the ability of the VHL protein to effect the formation of the fibronectin matrix while leaving the HIF interaction ability intact, which leads to the development of pheochromocytomas. However, further research is required to determine the reasons whereby pheochromocytomas do not occur in type 1 syndrome; i.e., in the complete absence of the VHL protein [46].
Succinate dehydrogenase (SDH)
The pseudohypoxic state that leads to the formation of paragangliomas/pheochromocytomas may be caused by mutations in SDH genes. SDH comprises a mitochondrial protein complex that participates both in the Krebs cycle and in the electron transport chain [66]. In the Krebs cycle, this complex oxidizes succinate to fumarate whereas in the transport chain it transfers electrons onto coenzyme Q [66]. The SDH enzyme complex consists of four subunits: SDHA, SDHB, SDHC, and SDHD [67]. Subunits A and B form the core of the complex whereas the two remaining subunits function as its structural fastening elements [68]. In addition, two factors are also involved in the assembly of the complex: SDHAF1 [69] and SDHAF2 [70]. Thus, a mutation in any of these genes, collectively termed SDHx genes, would impair the structure of the entire complex, leading to oncogenesis [71]. In accordance with this model, several studies have confirmed the role of germline mutations in the SDHAF1 [64, 72] and SDHAF2 genes [31, 59, 64, 65, 73–75] in the formation of head and neck paragangliomas/pheochromocytomas. Generally, germline mutations in the SDHx genes lead to heritable paragangliomas/pheochromocytomas, less frequently to renal cell carcinoma and gastrointestinal stromal tumors, and still less frequently to pituitary adenoma. This phenomenon is referred to as familial paraganglioma syndrome, an autosomal dominant heritable disease that is subdivided into 5 types according to the gene affected. The PGL1, PGL2, PGL3, PGL4, and PGL5 syndromes are associated with mutations in the SDHD, SDHAF2, SDHC, SDHB, and SDHA genes, respectively [76]. Analysis of data for 1045 patients in the Netherlands with this syndrome showed that mutations were most frequently found in SDHD (87.1%) but much less frequently in SDHAF2 (6.7%), SDHB (5.9%), and SDHC (0.3%) [271]. Mutation in the SDHD gene were also the most common in German patients [74], although among the germline mutations the most frequently recorded occurred in SDHB [61].
Among the SDH genes, homozygous germline mutations in the SDHA gene are associated with Leigh syndrome, which most commonly arises at an early age and is characterized by central nervous system impairment. In addition, a heterozygous mutation in this gene was first discovered in a patient with catecholamine-secreting abdominal paraganglioma [77]. Subsequently, germline mutations in SDHA were found in many paragangliomas [78, 79] including those of vagal origin [80]. Mutations in SDHB in paragangliomas/pheochromocytomas are associated with the highest mortality rate [81, 82] as well as with a high incidence of malignant tumors [81, 83, 84] and their metastasis [83, 85]. Additionally, germline mutations in this gene have been found in some samples of carotid [86, 87] and vagal [80] paragangliomas. In comparison, the PGL3 syndrome related to mutations in the SDHC gene consists of an autosomal dominant disease that is most often characterized by benign head and neck paragangliomas [88], although in rare cases paragangliomas/pheochromocytomas may occur in other body parts as well [76]. Mutations in SDHC were also found in samples of vagal paraganglioma [80]. PGL1 syndrome, which is associated with a mutation in the SDHD gene, also comprises an autosomal dominant disease that involves the development of multiple head and neck paragangliomas in patients aged from 28 to 31 years [81, 83, 89, 90], as well as the development of carotid body paraganglioma [91]. In contrast, although PGL2 syndrome is associated with paragangliomas, no cases of their metastasis or development of pheochromocytomas have been recorded [73, 75]. Furthermore, among the conditions related to mutations in the SDHx genes, the Carney diad (or Carney-Stratakis diad) syndrome is associated with mutations in the SDHB, SDHC, and SDHD genes [92] and manifests as paragangliomas and gastrointestinal stromal tumors in patients of either sex. Notably, this syndrome should be distinguished from the Carney triad, which mostly arises in young women and is characterized by sympathetic paragangliomas, gastrointestinal stromal tumors, and pulmonary chondromas. No SDHx mutations associated with the latter syndrome have yet been identified, although hypermethylation of the SDHC gene was observed in 3 out of 4 patients [93].
Mutations in the SDHx genes increase the stability of HIFs and thus the expression of their targets [15, 19, 96]. This arises because functional inactivation of the SDH complex by mutations allows intracellular accumulation of its substrate, succinate, which is converted by PHDs from α-ketoglutarate. Accumulated succinate in turn inhibits PHDs owing to its structural similarity with α-ketoglutarate [95], one of several factors including oxygen, iron, and ascorbate that regulate the activity of the PHD enzymes PHD1 (EGLN2), PHD2 (EGLN1), and PHD3 (EGLN3) [25, 94]. As the stability of HIF α-subunits, an important component in determining HIF proteosomal degradation, depends on their hydroxylation by enzymes of the dioxygenase class (PHDs), SDHx mutation and the resultant PHD inhibition eventually hinders PHD-mediated degradation of HIF.
In addition, knockdown of SDHA or SDHB in cell lines also leads to the inhibition of other classes of α-ketoglutarate-dependent enzymes, such as Jumonji histone demethylases and TET hydroxylases [97]. This, in turn, results in histone and DNA hypermethylation in the setting of HIF accumulation and high expression of HIF targets (Figure 1). In particular, in vitro analyses demonstrated that SDH-mutant paragangliomas/pheochromocytomas exhibited extensive hypermethylation patterns and a lower level of 5-hydroxymethylcytosine expression, which indicated the impairment of DNA and histone demethylation in such tumors [98]. Similar epigenomic changes were also found in other SDH-mutant tumors, for example, in gastrointestinal stromal tumor cells [99]. In addition, some tumors characterized by hypermethylation (colon cancer, glioblastoma) were shown to carry mutations in various metabolic enzymes. It should be noted, however, that the imbalance between succinate and α-ketoglutarate resulting from SDH deficiency not only impairs the function of dioxygenases but may also cause other problems related to oncogenesis, as these metabolites have many different functions. To date, however, such pathways of tumor development have not been described.
Notably, VHL- and SDH-mutant tumors could be distinctly subdivided into two clusters according to their mutations following genomic analysis of 202 paraganglioma and pheochromocytoma samples. Furthermore, expression analysis also showed that the transcription profiles in these tumors were considerably different [96]. Mutations in the VHL gene lead to elevated expression of HIF1α target genes (ENO1, BNIP3, and CA9) and genes associated with glycolysis (ENO1 and SLC2A1), apoptosis (EGLn3), and metastasis (KISS1R). In comparison, the transcriptomes of SDH-mutant tumors are enriched in genes associated with transcription regulation (DDIT3, NR1H3, MEIS3, PAWR, SIX1, SIX4, and TRIB3), protein transport (GOSR2, HCN3, LAPTM4B, SLC16A10, and SLC35F2), proliferation (ESRRA), energy metabolism (NOXA1), and cell adhesion (DSP and CNTN4). These traits in association with mutations in SDHx genes were shown to be associated with metastasis, thereby indicating a high metastatic potential of SDH-mutant tumors. Additionally, analysis of the correlations between SDHB mutations, tumor malignancy, and poor prognosis has revealed some markers that could be used to predict the metastatic propensity of the tumor: MMP24 (coding for a metalloproteinase associated with metastatic transformation and invasiveness), DSP (coding for desmoplakin, a marker of poor prognosis in non-small cell lung carcinoma stage I), SIX1 (encoding a homeobox protein associated with proliferation and elevated invasiveness of hepatocellular carcinoma), LGR5 (encoding the target of β-catenin, which is highly expressed in several types of aggressive adrenocortical tumors), and LAPTM4B (encoding a lysosomal protein associated with recurrence and poor prognosis in a number of carcinomas) [96].
Fumarate hydratase (FH)
FH catalyzes the reversible conversion of fumarate to malate in the Krebs cycle (Figure 1). The deficiency of FH leads to an accumulation of fumarate, which is structurally similar to succinate and, correspondingly, affects the α-ketoglutarate-dependent enzymes in a similar manner [97]. However, under the conditions of FH deficiency, both the accumulation of HIF and inhibition of the Jumonji histone demethylase have been shown to depend on the level of reactive oxygen species (ROS) [100]. The ROS level is elevated in cells with mutant FH [101], whereas data on ROS concentration in SDH-mutant cells are contradictory [95, 102–104]. Mutations in FH are associated with hereditary leiomyomatosis and renal cancer [105]; additionally, mutations were also found in some pheochromocytomas that resembled SDH-mutant tumors in their transcription and methylation profiles [98]. An FH mutation associated with paraganglioma was first discovered in 2013 in one of 145 tumor samples exhibiting elevated methylation and no mutations in the SDHx genes [98, 106]. In another study that used 598 samples of paragangliomas/pheochromocytomas, mutations in FH were found in 0.83% of all the tumors, of which 60% of the FH-mutant tumors were malignant [107]. Notably, oncogenesis under the conditions of FH deficiency follows a genetic pathway similar to that in malignant SDHB-mutant paragangliomas/pheochromocytomas.
Prolyl hydroxylase domain (PHD) proteins
As PHD protein activity facilitates HIF degradation and, correspondingly, the development of pseudohypoxia and oncogenesis, cases of PHD gene mutations in paragangliomas/pheochromocytomas are worth noting. A germline mutation in the PHD2 gene was first recorded in a patient with erythrocytosis and paraganglioma in 2008 [108]. However, mutations of PHD genes in paragangliomas are rare. For example, no mutations in any of the three PHD genes were found among 82 patients with hereditary paragangliomas [109] and only a single sample with a mutation in the PHD2 gene was identified during the analysis of 72 pheochromocytomas and 14 paragangliomas [79].
Hypoxia-inducible factor 2-α (HIF2A)
HIF1α and HIF2α represent transcription factors of the bHLH-PAS protein family, which also includes the proteins participating in xenobiotic detoxification [110, 111], circadian rhythm regulation [112, 113], specification of tissue patterning during embryonic development [114, 115], and regulation of metabolic processes such as transport and handling of glucose and the Krebs cycle [116]. In particular, HIF2α is important during embryonic development of the sympathetic nervous system and the adrenal gland. It is expressed in the organ of Zuckerkandl (the main source of catecholamines in mammalian embryos), detects hypoxia during mid-gestational development, and regulates the expression of the genes responsible for the level of circulating catecholamines and normal performance of the heart [117]. Mutations in the HIF2A gene have recently been found in pheochromocytomas and paragangliomas [13, 42–44, 118, 119]. Notably, tumors with somatic mutations in HIF2A are characterized by elevated transcription of the genetic markers of immature chromaffin cells whereas the factors related to their differentiation are, by contrast, downregulated compared to their normal levels in adults [13]. Furthermore, genes encoding the MYC proteins and cyclin D1, which are associated with cell transformation in pseudohypoxic renal cancer, exhibit elevated expression in paragangliomas/pheochromocytomas with mutations in HIF2A [13]. Together, these data suggest that HIF2A-mutant paragangliomas may manifest a more aggressive phenotype.
A somatic HIF2A mutation in pheochromocytoma was first recorded in 2013 [44]; subsequently, both somatic and germline HIF2A mutations have been found in many tumors. Thus, HIF2A currently represent the second most frequent mutated gene (after the RET gene) associated with paragangliomas [79, 118, 120]. Specifically, these mutations affect one of the sites (Pro531) that facilitate the stability of the HIF2α molecule. Alteration of this site leads to conformational changes in HIF2α that hinder its binding with the PHD [121] and VHL proteins [122]. In vitro analyses confirmed that mutations in HIF2α were associated with the inability of PHD to recognize HIF2α, its lack of binding to VHL, and, correspondingly, with the prolonged activity of HIF2α and induction of its targets [13, 42–44, 119]. In addition, it was demonstrated in vivo that mutations at codon 531 caused oncogenesis [13]. Mutant alleles of HIF2A have been found in some other tumors as well [42, 123]. For example, a considerable fraction of patients exhibited somatostatinomas along with mutations in the HIF2A gene [44, 124] and approximately a half of those with HIF2α-mutant tumors demonstrated the early development of polycythemia [42, 124]. Furthermore, it was discovered in 2014 that HIF2A-mediated tumors were caused by postzygotic mutations in early development [125]. In particular, the resulting mosaicism led to the development of paragangliomas/pheochromocytoma, polycythemia, and somatostatinomas in the same patient.
Isocitrate dehydrogenase (IDH)
In the Krebs cycle, IDH comprises the oxidative decarboxylation enzyme that converts isocitrate to α-ketoglutarate [126]. In addition to its main function, under conditions of hypoxia or in tumor cells with defective mitochondria this enzyme participates in the reductive carboxylation of α-ketoglutarate to isocitrate during glutamine-dependent lipogenesis.
A mutation in the IDH1 gene was first detected in colorectal cancer cells [127], after which mutations in the IDH1 and IDH2 genes were found in various tumors of neural origin [128]. The mutant forms of the enzyme cannot perform oxidation in the normal way and produce 2-hydroxyglutarate instead of α-ketoglutarate [129]. Normal cells contain no 2-hydroxyglutarate; conversely, its accumulation in IDH1/IDH2-mutant cells activates the pseudohypoxic pathway of oncogenesis. Thus, 2-hydroxyglutarate may be regarded as an oncometabolite [130] although the exact mechanism of pseudohypoxia development owing to accumulation of 2-hydroxyglutarate remains unknown. It was initially assumed that mutations in IDH1 led to the stabilization of HIF1 and subsequent activation of its targets [131]. Subsequently, however, it was shown that 2-hydroxyglutarate was able to inhibit some 2-oxoglutarate-dependent dioxygenases although not PHD [132]. Furthermore, 2-hydroxyglutarate may facilitate the activity of PHD1 and PHD2, thus reducing the stability of HIF [132]. Alternatively, a more recent study demonstrated the possibility of nonenzymatic conversion of 2-hydroxyglutarate to 2-oxoglutarate, indicating that the earlier results may have been misinterpreted [133]. Thus, further research is needed to determine the exact mechanism of interaction between 2-hydroxyglutarate and the pseudohypoxic pathway of oncogenesis [134]. It has been suggested that the most important effect may be that resulting from the high level of 2-hydroxyglutarate on the epigenetic profile of the cell. Specifically, by interacting with dioxygenases instead of their usual substrate, 2-hydroxyglutarate promotes the inhibition of DNA demethylation and CpG hypermethylation. Notably, examples of hypermethylated phenotypes in leukemia and glioma accompanied by mutations in the IDH1 gene have been observed [135–138]. In paragangliomas, a mutation in IDH1 was first recorded during the analysis of 365 samples [30] and a somatic mutation in this gene was detected in carotid paraganglioma although no IDH mutations were found in pheochromocytomas. In another study, the analysis of 104 paragangliomas/pheochromocytomas did not reveal any mutation in the IDH genes [139]. It may therefore be concluded that such mutations are rare in pseudohypoxic paragangliomas.
Malate dehydrogenase 2 (MDH2)
MDH2 participates in the reversible oxidation of malate to oxaloacetate in the Krebs cycle (Figure 1) [140, 141]. In addition, this protein plays an important role in the malate-aspartate shuttle that is part of the metabolic interaction between the mitochondria and the cytoplasm [140]. Mutations in the MDH2 gene are usually associated with such conditions as sleeping sickness and 1-2-hydroxyglutaric aciduria [142]. A mutation in this gene in a patient with numerous malignant paragangliomas was first recorded in 2015; in particular, the transcription profile of the tumor cells resembled that of SDH-mutant tumors [143]. Among the relatives of this patient, two out of five carried the mutant MDH2 gene although they showed no symptoms of paragangliomas or other diseases associated with mutations in this gene. Notably, knockdown of this gene in HeLa cells results in the accumulation of malate and fumarate [143]. Similar to succinate and fumarate, malate also inhibits the hydroxylation of HIFs [144, 145], which may explain the involvement of MDH2 mutations in oncogenesis.
Group II
Genes associated with Group II tumors are associated with oncogenic signaling pathways. Primarily, these are associated with the PI3 kinase pathways PI3K/AKT/mTOR and RAS/RAF/ERK, whose activation or deregulation leads to the uncontrolled proliferation, growth, and survival of cells [16].
The somatic mutations involved in the PI3K/AKT/mTORC1 signaling pathway represent the most common disturbances in various types of cancer including breast, ovarian, prostate, endometrial, lung, brain, stomach, pancreatic, colon, and thyroid cancers, as well as hepatocellular carcinoma and malignant neuroendocrine tumors including paragangliomas [146–149]. The activation of tyrosine kinase receptors, such as RET, VEGF-R, epidermal growth factor receptor (EGF-R), FGF-R, and insulin-like growth factor 1 receptor (IGF1-R) by their respective growth factors leads to the activation of PI3K. In its turn, PI3K activates ATK, which initiates the activity of mTORC1 by reducing the level of its suppression by TSC1/2 (Figure 2). The mTORC1 complex consists of mTOR, the regulatory subunit Raptor, PRAS40, and mLST8. An important target of mTORC1 is the protein 4EBP1, which binds to the eukaryotic translation initiation factor 4E (eIF4E) [150]. Upon 4EBP1 phosphorylation by mTORC1, it loses its ability to inhibit eIF4E, allowing eIF4E to recruit the 40S ribosomal subunit to the 5′-end of the mRNA, facilitating the initiation of translation [151]. Additionally, the hyperactivation of eIF4E alone is sufficient for the onset of oncogenesis [152]. Furthermore, hyperactivation of eIF4E as the result of 4EBP1 inhibition is required for mTOR-mediated tumor development [153, 154].
Notably, the mTOR and MYC pathways intersect at the stage of eIF4E activity regulation [155]. The key function of MYC is the regulation of the protein synthesis apparatus by activating ribosomal DNA and the genes of e.g., ribosomal proteins and translation initiation factors. [156]. Among other factors, MYC transcriptionally activates eIF4E, the hyperexpression of which facilitates MYC-dependent oncogenesis [157]. It has recently been demonstrated that at the early stages of tumor development MYC not only enhances the overall protein synthesis but also specifically activates mTOR-mediated 4EBP1 phosphorylation, leading to eIF4E oncogene activation [155]. The mTORC1 complex also activates many additional proteins including p70S6K, which then phosphorylates p70S6. In turn, the activated p70S6 protein induces the growth, proliferation, and survival of mutant cells by activating HIF1α protein synthesis. Thus, inopportune activation of the PI3K/AKT/mTORC1 signaling pathway leads to tumor development both by the pseudohypoxic pathway and by activating cell growth and proliferation (Figure 2).
Activation of the RAS/RAF/ERK pathway is also often observed in oncogenesis [158]. The RAS protein is a protein kinase that phosphorylates and activates RAF kinase, which in turn activates MEK and then ERK. Stimulation of this pathway by the tyrosine kinase receptors RET, VEGF-R, EGF-R, FGF-R, and IGF1-R leads to the activation of cell cycle factors (cyclin D) and proto-oncogenes (c-MYC) [159]. For example, elevated expression of the main fibroblast growth factor and its receptor FGFR1 was detected in all of the 33 examined samples of head and neck paragangliomas [160]. Furthermore, uncontrolled activation of the RAS/RAF/ERK pathway is observed in paragangliomas/pheochromocytomas with mutations in the RET and NF1 genes [161–163] (Figure 2).
RET
RET constitutes a transmembrane tyrosine kinase receptor for extracellular signaling molecules of the GDNF family, which are largely expressed in cells of the urogenital system and in neural crest progenitor cells. Activation of this receptor is required for the development of the kidneys as well as the sympathetic, parasympathetic, and enteric nervous systems [164].
Germline mutations in the RET gene that enhance its activity are associated with Sipple syndrome (multiple endocrine neoplasia type 2, MEN2) comprising two subtypes, MEN2A and MEN2B. This syndrome consists of an autosomal dominant hereditary disease characterized by the development of pheochromocytomas and medullary thyroid cancer [165]. RET mutations of the MEN2A subtype affect the extracellular domain and result in ligand-independent homo-dimerization. This association is required for activation of the RET receptor and of the PI3-AKT, RAS, p38 MAPK, and JUN kinase pathway- stimulation of cell growth and proliferation [166, 167]. Furthermore, mutations associated with the MEN2A subtype affect the level of RET expression on the cell surface as an additional means of signal modification [166]. In contrast, the mutations associated with MEN2B affect the kinase catalytic site and result in the loss of substrate specificity [167]. Metastasis of pheochromocytomas in MEN2 syndrome is rare [168], although the MEN2B represents the more aggressive form. Approximately half of the patients with Sipple syndrome develop pheochromocytomas [169] whereas paragangliomas are extremely rare in this syndrome [9, 170].
Somatic mutations in RET have been detected in approximately 5% of sporadic pheochromocytomas and paragangliomas [96]. In particular, mutations that reduce RET activity, which lead to Hirschsprung's disease, disturb the endosomal processing of RET that in the normal state regulates the duration and specificity of its signal. However, no such impairments were detected resulting from the RET activating mutations associated with paragangliomas [171].
Neurofibromin 1 (NF1)
The NF1 gene encodes neurofibromin 1, which inhibits the GTPase HRAS and thus disrupts the RAS signaling pathway [172]. RAS constitutes the principal oncogene in malignant tumors and in the presence of mutant NF1 it becomes constitutively active [173–175]. Mice with complete absence of functional NF1 develop pheochromocytomas with high penetrance and exhibit higher expression levels of many genes including RET, which is responsible for early development of the central and peripheral nervous systems [176]. A similar transcription profile is observed in pheochromocytomas in human patients carrying a germline mutation in the NF1 gene [15, 96]. In particular, as the mTOR protein represents an important target of the RAS pathway; therefore its unregulated activation is typical of NF1-mutant paragangliomas/pheochromocytomas [177].
Germline mutations in the NF1 gene are associated with neurofibromatosis type 1, an autosomal dominant hereditary disease manifested by pigmented patches on the skin, neurofibromas, central nervous system tumors, and bone abnormalities [76]. The probability of pheochromocytomas in patients with mutant NF1 is approximately 0.1–5.7% whereas that of paragangliomas is very low [76, 83]. Notably, pheochromocytomas carrying this mutation are often malignant.
Somatic mutations in the NF1 gene have been detected in 20–25% of sporadic pheochromocytomas [10, 12]. Additionally, in several recorded cases, paragangliomas violated the “mutual exclusion of mutations” rule: these tumors simultaneously carried somatic mutations in the NF1 gene and in the RET or VHL genes [10]. This may have been the result of the tumor originating from two subclonal cell populations, each with a separate mutation. Another explanation may be that mutations in the genes of one group or interacting mutations in genes from different groups may provide certain advantages to the transforming cell [178]. In comparison, most other types of tumors show loss of heterozygosity in the NF1 locus [10, 12].
Transmembrane protein 127 (TMEM127)
TMEM127 is a transmembrane protein albeit with an as-yet unknown function. A study of its normal and mutant forms in pheochromocytomas showed this protein to function as a negative regulator of mTOR [177]. TMEM127 is expressed in various tissues and is located in the endoplasmic membranes and in the membranes of the numerous components of the endosomal system including endosomes at various stages of maturation, the Golgi apparatus, and lysosomes [177]. Mutant forms of TMEM127 occur diffusely in the cytoplasm regardless of the condition and are detected in very small quantities. In comparison, in the normal state, the relative content of wild-type TMEM127 in different cell components depends on pH and other factors [177]. An analysis of transcription profiles showed the expression of mutant TMEM127 forms to be reduced more that 4-fold in pheochromocytomas as compared to tumors with wild-type TMEM127 [177]. In addition, loss of heterozygosity for the mutant TMEM127 was also observed. These findings suggest that the mutant form of this protein is nonfunctional and that its transcript is less stable.
Germline missense mutations in the TMEM127 gene or mutations leading to truncated forms of this protein were detected among 103 patients with pheochromocytomas, wherein approximately 30% of hereditary tumors and 3% of sporadic tumors carried the mutant gene [177]. Subsequently, over 30 different mutations have been found, more than half of which result in a truncated TMEM127 gene product or affect one of its transmembrane domains [179–182]. Notably, pheochromocytomas were found in only 20% of patients or their relatives carrying mutations in this gene, which indicates low penetrance of the mutant alleles [139]. Furthermore, the examination of 990 patients with paragangliomas/pheochromocytomas identified mutations in the TMEM127 gene in 2% of these, although none exhibited paragangliomas [139].
The transcription profile of TMEM127-mutant pheochromocytomas was found to be similar to that of tumors with mutations in the RET and NF1 genes [177]. Additional support for the relationship between TMEM127 and mTOR may be surmised from the elevated phosphorylation of mTOR targets both in cell lines lacking TMEM127 and in human pheochromocytomas with mutations in this gene. A recent study has shown that mTOR is required to interact with late endosomes that are enriched in TMEM127 proteins, as binding to the mTOR activators Rheb and PA is necessary for its activation. [183]. Thus, impairing the interaction of mTOR with such endosomes or changes in their number may prevent mTOR activation. For example, data suggest that various growth factors affect the state of the TSC2 protein, a regulator of mTOR signaling, which leads to its translocation from the late endosomes and the activation of Rheb therein. In turn, Rheb activation results in the activation of mTOR [183]. Conversely, the activation of early endosomes through overexpression of the gene for Rab5, the product of which plays the key role in regulating membrane exchange as well as in the formation of early endosomes and their maturation to late endosomes, leads to the inhibition of mTOR signaling in response to different stimuli [184, 185]. In mice with inactivated TMEM127 it was shown that formation of hybrid “early-to-late” endosomes was impaired whereas the association of mTOR with late endosomes and lysosomes was enhanced in the absence of functional TMEM127 [186]. Thus, although the interaction between mTOR and TMEM127 has not been studied in detail, the available data suggest a possible mechanism of mTOR signaling inhibition by means of the TMEM127 protein. This supposition indicates the corresponding pathway of oncogenesis in cases with mutations in TMEM127.
It is worth noting that hyperactivation of HIF may facilitate cell proliferation through at least two endocytosis-dependent pathways. In renal cancer cells with a mutant VHL gene, activated HIF may inhibit Rab5 and thus disturb the membrane exchange, leading to the accumulation and activation of receptors of various growth factors such as EGF-R (Figure 1) [187]. The second mechanism is based on the elevation of HIF-dependent expression of the CAV1 gene that encodes caveolin, the main membrane component of caveolae, which participates in receptor-independent endocytosis. In particular, the elevated expression of caveolin results in an accumulation of EGFR in the caveolae and their activation by dimerization without a ligand [188].
MYC associated factor X (MAX)
The MAX gene encodes a transcription factor which, in association with the proto-oncogene MYC and the transcription factor MXD, participates in the regulation of cell proliferation, differentiation, and death [189, 190]. Impaired regulation of MYC/MAX interactions promotes the development of many neoplasmas including neuroblastomas [11, 191]. The MAX protein binds to the transcription factor MYC and this heterodimeric complex in turn binds to the E-box sequence of promoters of over a thousand genes with a wide range of functions including metabolism, angiogenesis, and cell growth [192]. MAX may also bind to other transcription factors of the families MXD1, MNT, and MGA that compete with MYC for the promoter E-box sequences and reduce the activity of the MYC target genes, thus potentially inhibiting cell growth and facilitating their terminal differentiation [193]. The balance of the MAX-MYC and MAX complexes with MYC inhibitors regulates the transcription of genes containing E-box sequences in their promoters [192]. Notably, although the interaction of MAX and MYC is considered to be activating, mutations in the MAX gene resulting in pheochromocytomas involve loss of function of this protein, whereas the activity of MYC in tumors is elevated [191]. Furthermore, however, it is also known that mutations that change the amino acid composition of the domains responsible for binding to other transcription factors and DNA may also affect the ability of MAX to bind to MYC and its repressors [11]. In addition, although the exact mechanism by which mutations in MAX lead to paraganglioma development is unknown, these tumors exhibit an elevated expression of the MYC target genes [191]. Certain characteristic traits of this mechanism have been suggested by a recent study, although this relationship has not been examined in paragangliomas/pheochromocytomas to date [155].
Both somatic and germline mutations in the MAX gene have been detected in pheochromocytomas [11, 191]. Mutations in this gene in paragangliomas were first discovered during the analysis of samples from 1694 patients [11]. This research also showed that somatic mutations in MAX occurred in 1.65% of paragangliomas/pheochromocytomas [11] but that only 7% of mutations in the MAX gene resulted in metastasis. Conversely, in a different study, 25% of MAX-mutant tumors were shown to have traits of metastasis [191]. Overall, the results of analysis of 2041 cases of paragangliomas/pheochromocytomas with mutations in the MAX gene are presently available in the literature [11, 65, 194, 195]. Based on these data, MAX mutations were found in 1.9% of the tumors and metastasis was observed in 8.5% of the tumors [11, 191]. Thus, mutations in the MAX gene appear to be rare and are unlikely to be associated with elevated tumor aggressiveness, making them unsuitable as targets of genetic screening for paragangliomas/pheochromocytomas.
Kinesin family member 1B (KIF1B)
The KIF1B gene consists of 50 exons and encodes for 2 protein isoforms, KIF1Bα and KIF1Bβ. These proteins play primary roles in the transport of mitochondria (KIF1Bα) and synaptic vesicle precursors (KIF1Bβ) [196, 197]. In addition, KIF1Bβ was shown to represent a target of the PHD3 protein and is involved in apoptosis. The absence of functional KIF1Bβ protein consequential to mutation protects neuroblasts from apoptosis and leads to oncogenesis [198].
Missense mutations in the KIF1B gene were first detected in two samples of pheochromocytoma in 2008 [198]. Transcription analysis showed these tumors to be similar to RET- and NF1-mutant pheochromocytomas and paragangliomas. In another study, a group of relatives was found who carried germline mutations in the KIF1B gene and showed an increased probability of developing not only pheochromocytomas but also neuroblastomas, ganglioneuromas, and lung tumors [199]. No record of paraganglioma with a mutation in the KIF1B gene has been published. Conversely, both germline and somatic mutations in this gene have been found in pheochromocytomas, occasionally occurring in combinations with mutations in other genes, such as NF1 or RET as well as VHL or SDHx [79, 199]. Furthermore, KIF1B-mutant tumors are considerably enriched in the genes associated with amino acid (glutamate, glutamine) metabolism and with oxidative stress response [199].
Menin (multiple endocrine neoplasia 1, MEN1)
The MEN1 gene codes for the protein menin [200], which is localized in the nucleus and interacts with a broad range of proteins involved in transcription regulation, genome stabilization, and cell division and proliferation [201]. For example, the interaction of menin with histone H3 methyltransferase affects the epigenetic profile of the cell [202].
Germline mutations in the MEN1 gene result in structural changes in menin, which lead to multiple endocrine neoplasia type 1 (the MEN1 syndrome), an autosomal dominant hereditary disease characterized by high penetrance (reaching 100% with age) and associated with the development of over 20 types of endocrine and non-endocrine tumors [203]. To date, seven cases of pheochromocytomas associated with MEN1 syndrome have been recorded [204, 205], with only one exhibiting malignancy. In addition, there is one known case of paraganglioma with MEN1 mutation [203]. Although mutations in the MEN1 gene are rare, they represent important objects of screening as they provide the means of early diagnostics of MEN1 syndrome [206].
Activation of the neuronal precursor cell pathway
In 2005, a process involving some genes from each of the different groups was shown to be related to the formation of paragangliomas/pheochromocytomas [207]. In particular, the genes VHL, SDHX (Group I), RET, and NF1 (Group II) are required for the regulation of apoptosis in neuronal precursor cells. The c-Jun protein is activated in the absence of signal from nerve growth factor (NGF) and causes neuronal cell apoptosis [208]. However, the NF1 gene product inhibits the NGF receptor TrkA and in the absence of neurofibromin the embryonic sympathetic neurons survive even without the NGF signal [209]. It was demonstrated in the pheochromocytoma-derived cell line PC12 that succinate accumulation induced cell growth not by its action on HIF1 but rather through inhibiting PHD3-dependent apoptosis, which led to the survival of embryonic neurons and the formation of paragangliomas/pheochromocytomas [207]. Inactivation of the VHL protein owing to mutation increases the level of Jun-B, which in turn is an antagonist of c-Jun. The PHD3 protein is necessary and sufficient for apoptosis induction after the cessation of the NGF signal; therefore, loss of function of this protein or impairment of its regulation, such as through succinate accumulation, prevents apoptosis and leads to oncogenesis [207]. Furthermore, the participation of menin in c-Jun activation and its suppression by the MYC protein suggest that mutations in MEN1 and MAX may play a certain role in paraganglioma development [210, 211] (Figure 3).
Figure 3. Activation of the neuronal precursor cell pathway in paragangliomas/pheochromocytomas by mutations in Group 1 and 2 genes.
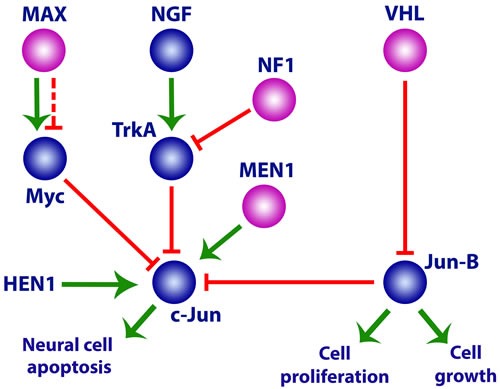
See text for details.
Genes that do not belong to any of the above groups but are associated with paraganglioma/pheochromocytoma formation
Glial cell line-derived neurotrophic factor (GDNF)
GDNF belongs to the transforming growth factor beta (TGF-β) superfamily [212]. GDNF is a ligand of the tyrosine kinase receptor RET [213] and their interaction leads to the activation of RET and RET-regulated pathways associated with cell survival, proliferation, and differentiation. GDNF was first described as a factor required for mesencephalic dopamine neuron survival.
Mutations in this gene are associated with Hirschsprung's disease, which is manifested by impaired innervation of the large intestine [214]. Considering the important role of RET in pheochromocytoma development, researchers further suggested that mutations in GDNF affecting its interaction with RET may be also associated with the disease [215]. However, whereas somatic mutations in the GDNF gene have been found in a few cases of pheochromocytomas, these are also present in healthy individuals. Thus, the role of GDNF mutations in tumor pathogenesis appears to be insignificant with low penetrance [215, 216].
RAS genes
As described previously, the impaired activation of the RAS/RAF/ERK pathway is often observed in oncogenesis. Ras is a membrane-bound guanosine triphosphate/diphosphate-binding protein that functions as a molecular switch conveying signals from the membrane to the nucleus. In addition, many so-called Ras proteins have been identified whose sequences are very similar at both the N- and C-ends. The Ras protein family includes H-Ras, K-Ras, M-Ras, N-Ras, R-Ras, Rap-1, Rap-2, and Ral. Mutations have been found in the genes for H-Ras, K-Ras, and N-Ras that result in constitutive activity of Ras, its resistance to inhibitors, and activation of the associated RAS/RAF/ERK and PI3K/AKT/mTOR pathways, which lead to uncontrolled cell proliferation and oncogenesis [217].
The earliest data regarding the state of Ras signaling pathway hyperactivation in pheochromocytoma pathogenesis were contradictory [218–220] However, an analysis of 169 endocrine tumor samples in 1992 revealed the first H-Ras mutation in pheochromocytoma [221]. Mutations associated with pheochromocytomas and paragangliomas were subsequently found only in the genes for H-Ras and K-Ras of the Ras family [61, 106, 222–224].
Germline mutations in the HRAS gene result in Costello syndrome, which is characterized by growth imbalance during prenatal and postnatal development, an increased probability of oncogenesis, mental retardation, and skin, musculoskeletal, and cardiovascular abnormalities [225]. A study carried out in 2013 revealed somatic H-Ras mutations in 6.9% of paragangliomas/pheochromocytomas among the 58 tumors analyzed [226]. Activation of the RAS/RAF/ERK signaling pathway was demonstrated in these mutant tumors. In a later study using more extensive material (271 samples), 5.2% of the tumors carried H-Ras mutations but no significant correlation of these mutations with pathological or clinical manifestations was observed [227]. Mutations in the HRAS gene that are associated with sporadic tumors are somatic; otherwise they result in the Costello syndrome phenotype [226, 227].
In comparison, K-Ras represents one of the most active oncogenes, as its activating mutations are found in 17–25% of all tumors [228, 229]. In the normal state K-Ras plays an important role in the signaling related to cell proliferation, differentiation, and aging. As for H-Ras, germline K-Ras mutations are associated with different syndromes, such as Noonan syndrome [230]. Pheochromocytomas were found to carry K-Ras mutations in 8 out of the 13 studied cases [231]; however, there is no data on such mutations in paragangliomas.
Guanine nucleotide binding protein (GNAS)
GNAS is a complex imprinted locus on chromosome 20q13.3 that produces many different transcripts owing to alternative promoter usage and alternative splicing [232]. In addition, some of the promoters are sensitive to parent-of-origin-specific methylation. For example, transcripts XLs, A/B, and AS are produced only from the unmethylated paternal allele of GNAS, whereas NESP55 is transcribed only from the unmethylated maternal allele [233]. In contrast, the promoter of one of the most functionally important proteins encoded in this locus, namely the α-subunit of the stimulating G protein (Gsα), is not imprinted, although only one of the parental alleles is expressed in some tissues.
As the GNAS locus encodes different transcripts with a wide range of functions, mutations in this locus lead to a variety of diseases and impairments including obesity, nervous system development disorders, and skeletal abnormalities, as reviewed in [234]. Mutations in the GNAS locus in paragangliomas were first detected in 1995 [235]. Subsequently, genomic and epigenomic analyses of malignant pheochromocytomas demonstrated the potential role of GNAS in the formation of these tumors [236].
Common oncogenes
Endocrine tumors, paragangliomas, and pheochromocytomas may also be caused by mutations in common oncogenes, in particular the genes encoding the cyclin-dependent kinase inhibitor (p16), transformation related protein 53 (p53), breast cancer associated protein 1 (BAP1), breast cancer 1 and breast cancer 2 (BRCA1 and BRCA2), α-thalassemia/mental retardation syndrome X-linked (ATRX), and lysine (K)-specific methyltransferase 2D (KMT2D).
The transformation related protein 53 gene (TP53) encodes the tumor suppressor protein p53, which includes domains of transcriptional activation, DNA interaction, and oligomerization [237]. Mutations in this gene are found in the majority of cancers [238–241]. Germline mutations in the TP53 gene are associated with hereditary cancer types; for example, Li-Fraumeni syndrome [242] and adrenocortical carcinoma in children [243]. Overexpression of the TP53 gene in paragangliomas/pheochromocytomas was detected in 2001 [244]. Additionally, somatic TP53 missense mutations were found in 2.35% of sporadic tumors [224], although no mutations were identified in a different study that analyzed tumors from 48 patients [245]. Thus, TP53 mutations appear to rarely occur in paragangliomas and do not represent a crucial factor of their pathogenesis.
The cyclin-dependent kinase inhibitor gene (p16) encodes a protein that regulates two pathways of particular importance for cell cycle regulation, p53 and retinoblastoma [246]. Mutations in this gene are associated with various nervous system tumors and with melanomas [247]. An analysis of 26 pheochromocytomas performed in 1996 revealed no deletions in the p16 gene [248]. Subsequently, however, hypermethylation of this gene was observed in 24% of the studied pheochromocytomas [249] and a decreased expression of p16 was demonstrated in 30 out of 31 tumors [249]. The available data therefore suggest the conclusion that the epigenetic state of the p16 gene and its inactivation are more significant for pheochromocytoma development than are mutations and deletions in this gene [250].
In comparison, BRCA1 and BRCA2 are regarded as oncosuppressors. They play an important role in DNA repair, cell cycle checkpoint regulation, and the maintenance of genome stability [251]. Germline mutations in these genes are associated with hereditary breast and ovarian cancer [252] and also with Fallopian tube, prostate, peritoneal, and pancreatic cancers [253]. The relationship between pheochromocytomas and BRCA1 and BRCA2 mutations was demonstrated by the detection of mutations in these genes in blood samples from two patients with pheochromocytoma [254]. Although no information is available from more detailed studies and a definite conclusion cannot be drawn from a set of only two cases, these findings suggest that an increased risk of pheochromocytomas may be associated with BRCA1 and BRCA2 mutations.
BAP1 also represents an oncosuppressor that participates in the regulation of such key processes as the cell cycle, cell differentiation and death, gluconeogenesis, and DNA damage response [255]. BRCA1 binds to BRCA1 associated RING domain 1 (BARD1) to form a complex that shows E3 ubiquitin ligase activity. In turn, this complex regulates the DNA damage response [256]. Activation of E3 ligase activity results from the deubiquitination of BARD1 via the BAP1 protein [257]. Conversely, the inhibition of BAP1 in vitro was shown to impair the DNA damage response and to determine radiation hypersensitivity [257]. Germline mutations in the BAP1 gene are associated with tumor predisposition syndrome as manifested by increased risks of malignant mesothelioma as well as of uveal and cutaneous melanoma [258], whereas somatic mutations in this gene have been detected in various types of tumors [259]. A germline BAP1 mutation in paraganglioma was first discovered in a family whose medical history included the presence of various tumors, in particular malignant uveal melanoma, mesothelioma, and breast cancer [260]. In addition, somatic loss of the wild-type allele of BAP1 has been detected in a patient with malignant uveal melanoma and paraganglioma [260].
The ATRX protein belongs to the SWitch/sucrose non fermentable (SWI/SNF) chromatin remodeler family, which plays an important role in supporting telomere and chromosome integrity [261]. Germline mutations in the ATRX gene are associated with X-linked α-thalassemia mental retardation syndrome and somatic mutations give rise to neuroblastomas and gliomas [262]. Recently, two samples of paraganglioma with mutant ATRX were identified [262]. The somatic ATRX mutation in these tumor samples was accompanied by an inherited mutation in the SDHB gene. The frequency of ATRX mutations was assessed by the analysis of two sets of samples, one with known mutant status and the other without any previous genetic analyses [262]. This study indicated that 12.6% of the studied samples carried somatic mutations in the ATRX gene.
The KMT2D (mixed-lineage leukemia 2, MLL2) protein participates in the regulation of DNA accessibility by histone H3K4 methylation and plays an important role in oogenesis and early development [263] as well as in spermatogenesis [264]. Combinatory analysis of proteins potentially interacting with KMT2D and the comparison of expression profiles of cells carrying the wild-type allele of KMT2D with isogenic cell lines lacking this gene revealed many KMT2D targets including proteins related to the p53 pathway, cAMP-mediated signaling, and cholestasis signaling [265]. Germline mutations in the KMT2D gene are associated with Kabuki syndrome, which is characterized by growth deficiency, peculiar facial features, and mental retardation [266]. In contrast, somatic mutations in this gene were found in medulloblastomas and lymphomas [267]; furthermore, somatic missense KMT2D mutations have also been detected in 11 out of 83 studied pheochromocytoma samples [267]. In the latter research, it was also shown that KMT2D-mutant tumors exceeded all others in size to a substantial degree.
Finally, the BRAF protein belongs to the family of RAF serine/threonine kinases, which also includes ARAF and RAF1 and comprises one of the targets of the RAS proteins. Thus, BRAF participates in activation of the RAS/RAF/ERK signal pathway [268]. Mutations in the BRAF gene were initially found in various tumors that are commonly associated with mutations in different isoforms of RAS, such as malignant melanoma or colon cancer. Furthermore, an activating mutation in the BRAF gene was recently found in one sample of pheochromocytoma [224]. The detected mutation often occurs in various neoplasms and has an increased kinase activity that, as shown in vitro, may induce cell transformation [269]. Although data from a single case are certainly not sufficient and more extensive transcriptome analysis is needed to classify the BRAF-mutant tumors into either of the two groups, the presently available findings indicate that pheochromocytomas with mutations in this gene should likely be placed in Group II [224].
CONCLUSIONS
Paragangliomas/pheochromocytomas result from genetic and/or epigenetic changes. This review considers all the genes that are known to be involved in the development of these tumors and provides detailed descriptions of the mechanisms by which mutations in these genes may lead to oncogenesis (Table 1). The genetic mutations associated with paragangliomas/pheochromocytomas may be classified into two main groups according to their expression profiles. In addition, classical oncogenes are also associated with these tumors as well as genes with specific mechanisms not resembling any of the mechanisms characteristic of the main groups.
Table 1. Summary of genes with mutations related to pheochromocytoma/paraganglioma formation.
| Gene | Gene name | Function | Classification | Malignancy risk | Relative mutation frequency | Predominant tumor site | Related hereditary disease | |
|---|---|---|---|---|---|---|---|---|
| somatic | germline | |||||||
| VHL | Von Hippel Lindau | regulates HIF1a and HIF2a proteasomal degradation | Group 1, neuronal precursor cell pathway | low | high | high | pheochromocytomas/paragangliomas | von Hippel Lindau syndrome, type1, type2 |
| SDHA | Succinate dehydrogenase subunit A | core subunit of the mitochondrial protein complex SDH | Group 1, neuronal precursor cell pathway | NA | NA | medium | paragangliomas | PGL5, Leigh syndrome (Homozygous germline mutations) |
| SDHB | Succinate dehydrogenase subunit B | core subunit of the mitochondrial protein complex SDH | Group 1, neuronal precursor cell pathway | high | NA | high | paragangliomas/pheochromocytomas | PGL4 syndrome |
| SDHC | Succinate dehydrogenase subunit C | structural subunit of the mitochondrial protein complex SDH | Group 1, neuronal precursor cell pathway | low | NA | medium | paragangliomas | PGL3 syndrome |
| SDHD | Succinate dehydrogenase subunit D | structural subunit of the mitochondrial protein complex SDH | Group 1, neuronal precursor cell pathway | low | NA | high | paragangliomas/pheochromocytomas | PGL1 syndrome |
| SDHAF2 | Succinate dehydrogenase complex assembly factor 1 | participates in SDH complex formation | Group 1 | NA | NA | low | paragangliomas | PGL2 syndrome |
| FH | Fumarate hydratase | catalyzes the conversion of fumarate to malate in the Krebs cycle | Group 1 | high | low | low | paragangliomas/pheochromocytomas | leiomyomatosis, renal cancer |
| PHD2 | Prolyl hydroxylase domain protein 2 | participates in the regulation of HIF activity | Group 1 | NA | NA | low | paragangliomas/pheochromocytomas | NA |
| HIF2α | Hypoxia-inducible factor 2-alpha | transcription factor of the bHLH–PAS protein family | Group 1 | NA | high | low | paragangliomas/pheochromocytomas | NA |
| IDH1 | Isocitrate dehydrogenase 1 | converts isocitrate to α-ketoglutarate | Group 1 | NA | low | NA | paragangliomas | NA |
| MDH2 | Malate dehydrogenase 2 | participates in oxidation of malate to oxaloacetate in the Krebs cycle | Group 1 | NA | NA | low | paragangliomas | NA |
| RET | Rearranged during transfection | transmembrane tyrosine kinase receptor for extracellular signal molecules of the GDNF family | Group 2, neuronal precursor cell pathway | low | high | high | pheochromocytomas/paragangliomas (rare) | Sipple syndrome |
| NF1 | Neurofibromin 1 | inhibits the GTPase HRAS and disrupts the RAS signaling pathway | Group 2, neuronal precursor cell pathway | high - of malignant tumors | very high | medium | pheochromocytomas/paragangliomas (rare) | neurofibromatosis type 1 |
| TMEM127 | Transmembrane protein 127 | unknown function | Group 2 | low | NA | low | pheochromocytomas | NA |
| MAX | MYC associated factor X | transcription factor participates in regulation of cell proliferation, differentiation, death | Group 2 | low | medium | low | pheochromocytomas/paragangliomas | NA |
| KIF1B | Kinesin family member 1B | transports mitochondria (KIF1Bα) and synaptic vesicle precursors (KIF1Bβ) | Group 2 | NA | low | low | pheochromocytomas | NA |
| MEN1 | Multiple endocrine neoplasia 1 | plays role in gene expression regulation | Group 2 | low | NA | low | pheochromocytomas/paragangliomas (rare) | multiple endocrine neoplasia type 1 |
| GDNF | Glial cell line derived neurotrophic factor | ligand of the tyrosine kinase receptor RET | no Group | NA | low | NA | pheochromocytomas | Hirschsprung's disease |
| HRAS | Harvey rat sarcoma viral oncogene homolog | molecular switch conveying signals from the membrane to the nucleus | no Group | NA | high | medium | pheochromocytomas/paragangliomas | Costello syndrome |
| KRAS | Kirsten rat sarcoma viral oncogene homolog | molecular switch conveying signals from the membrane to the nucleus | no Group | NA | NA | NA | pheochromocytomas | Noonan syndrome |
| GNAS | Guanine nucleotide binding Protein | complex imprinted locus on chromosome 20q13.3 | no Group | NA | low | NA | pheochromocytomas/paragangliomas | NA |
| TP53 | Tumor suppressor protein p53 | plays role in transcription activation, interaction with DNA, oligomerization | Common oncogene | NA | low | NA | paragangliomas | hereditary cancer types |
| p16 | Cyclin-dependent kinase inhibitor gene | regulates p53 and retinoblastoma pathways | Common oncogene | NA | NA | pheochromocytomas | ||
| BAP1 | Breast cancer associated protein 1 | participates in regulation of the cell cycle, cell differentiation, DNA damage response | Common oncogene | NA | very low | very low | paragangliomas | tumor predisposition syndrome |
| ATRX | Alpha thalassemia/mental retardation syndrome X-linked | plays an important role in supporting telomere and chromosome integrity | Common oncogene | NA | low | NA | pheochromocytomas/paragangliomas | X-linked alpha thalassemia mental retardation syndrome |
| KMT2D | Lysine (K)-specific methyltransferase 2D/mixed-lineage leukemia 2, MLL2 | participates in regulation of DNA accessibility by histone H3K4 methylation | Common oncogene | NA | low | NA | pheochromocytomas | Kabuki syndrome |
| BRAF | Murine sarcoma viral (v-raf) oncogene homolog B1 | participates in activation of the RAS/RAF/ERK signal pathway | Common oncogene | NA | NA | NA | pheochromocytomas | |
| BRCA1 | Breast cancer 1 gene | oncosupressor, plays role in cell cycle regulation, differentiation, DNA damage response | Common oncogene | NA | NA | NA | pheochromocytomas | breast, ovarian cancer |
| BRCA2 | Breast cancer 2 gene | oncosupressor, plays role in cell cycle regulation, differentiation, DNA damage response | Common oncogene | NA | NA | NA | pheochromocytomas | breast, ovarian cancer |
The identification of a germline mutation in a patient with paraganglioma/pheochromocytoma may help reveal other tumors typical of the syndrome associated with that particular mutation whereas the finding of a somatic mutation eliminates the necessity for examination of patient family members. Only somatic mutations associated with paragangliomas/pheochromocytomas have been detected in the HRAS, ATRX, TP53, and KMT2D genes whereas only germline mutations have been identified in the SDHA, SDHC, SDHAF2, FH, KIF1B, and TMEM127 genes. Mutations in some genes have been found only in single patients or families (e.g., MEN1, EGLN1, EGLN2, MDH2, IDH1, BAP1, BRAF); therefore, their role in the formation of paragangliomas/pheochromocytomas cannot be reliably confirmed.
At present, the main methods of treating these tumors constitute radiotherapy and surgery. Both methods have been described as highly efficient and safe; however, frequent cases of post-treatment complications have been reported. It is therefore clear that analyses of genetic and possibly epigenetic profiles should be carried out in order to estimate tumor risk, assess the possibility of malignant transformation, and to develop new, less invasive methods of paraganglioma/pheochromocytoma treatment. Overall, owing to the high degree of heritability of these tumors, their formation and behavior can be reliably predicted and their treatment can likely be optimized by using the newest techniques facilitated by the extensive, ongoing genetic research.
Acknowledgments
The authors thank the EIMB RAS “Genome” center (http://www.eimb.ru/rus/ckp/ccu_genome_c.php) for the use of computational resources.
Abbreviations
- ATRX
α-thalassemia/mental retardation syndrome X-linked
- BAP1
breast cancer associated protein 1
- BARD1
BRCA1 associated RING domain 1
- BRCA1,2
breast cancer 1,2
- DSP
desmoplakin
- EGF-R
epidermal growth factor receptor
- eIF4E
eukaryotic translation initiation factor 4E
- FH
fumarate hydratase
- GNAS
guanine nucleotide binding protein
- HIF2A
hypoxia-inducible factor 2-alpha
- IDH
isocitrate dehydrogenase
- IGF1-R
insulin-like growth factor 1 receptor
- KIF1Bβ
kinesin family member 1B-beta isoform
- KMT2D
lysine (K)-specific methyltransferase 2D
- MAX
MYC-associated factor X
- MDH2
malate dehydrogenase 2
- MEN1
menin, multiple endocrine neoplasia type 1
- MLL2
mixed-lineage leukemia 2
- NF1
neurofibromin 1
- NGF
nerve growth factor
- p70S6K
p70S6 kinase
- PHD
prolyl hydroxylase domain
- ROS
reactive oxygen species
- SDH
succinate dehydrogenase
- TMEM127
transmembrane protein 12
- TP53
transformation related protein 53
- VEGF
vasoendothelial growth factor
- VHL
Von Hippel Lindau
Footnotes
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
FUNDING
This work was financially supported by the Russian Foundation for Basic Research (grant 16-04-01521a) and ICGEB project CRP/RUS15-01.
REFERENCES
- 1.Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV, Pacak K. North American Neuroendocrine Tumor Society (NANETS). The North American Neuroendocrine Tumor Society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–783. doi: 10.1097/MPA.0b013e3181ebb4f0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.McNeil AR, Blok BH, Koelmeyer TD, Burke MP, Hilton JM. Phaeochromocytomas discovered during coronial autopsies in Sydney, Melbourne and Auckland. Aust N Z J Med. 2000;30:648–652. doi: 10.1111/j.1445-5994.2000.tb04358.x. [DOI] [PubMed] [Google Scholar]
- 3.Jansen JC, van den Berg R, Kuiper A, van der Mey AG, Zwinderman AH, Cornelisse CJ. Estimation of growth rate in patients with head and neck paragangliomas influences the treatment proposal. Cancer. 2000;88:2811–2816. [PubMed] [Google Scholar]
- 4.Granger JK, Houn HY. Head and neck paragangliomas: a clinicopathologic study with DNA flow cytometric analysis. South Med J. 1990;83:1407–1412. [PubMed] [Google Scholar]
- 5.Scholz T, Schulz C, Klose S, Lehnert H. Diagnostic management of benign and malignant pheochromocytoma. Exp Clin Endocrinol Diabetes. 2007;115:155–159. doi: 10.1055/s-2007-970410. [DOI] [PubMed] [Google Scholar]
- 6.Mannelli M, Castellano M, Schiavi F, Filetti S, Giacchè M, Mori L, Pignataro V, Bernini G, Giachè V, Bacca A, Biondi B, Corona G, Di Trapani G, et al. Clinically guided genetic screening in a large cohort of italian patients with pheochromocytomas and/or functional or nonfunctional paragangliomas. J Clin Endocrinol Metab. 2009;94:1541–1547. doi: 10.1210/jc.2008-2419. [DOI] [PubMed] [Google Scholar]
- 7.Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44:328–333. doi: 10.1055/s-0031-1301302. [DOI] [PubMed] [Google Scholar]
- 8.Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. [DOI] [PubMed] [Google Scholar]
- 9.Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, Niccoli-Sire P, Richard S, Rohmer V, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–8818. doi: 10.1200/JCO.2005.03.1484. [DOI] [PubMed] [Google Scholar]
- 10.Burnichon N, Buffet A, Parfait B, Letouze E, Laurendeau I, Loriot C, Pasmant E, Abermil N, Valeyrie-Allanore L, Bertherat J, Amar L, Vidaud D, Favier J, et al. Somatic NF1 inactivation is a frequent event in sporadic pheochromocytoma. Hum Mol Genet. 2012;21:5397–5405. doi: 10.1093/hmg/dds374. [DOI] [PubMed] [Google Scholar]
- 11.Burnichon N, Cascón A, Schiavi F, Morales NP, Comino-Méndez I, Abermil N, Inglada-Pérez L, de Cubas AA, Amar L, Barontini M, de Quirós SB, Bertherat J, Bignon YJ, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. 2012;18:2828–2837. doi: 10.1158/1078-0432.CCR-12-0160. [DOI] [PubMed] [Google Scholar]
- 12.Welander J, Larsson C, Bäckdahl M, Hareni N, Sivlér T, Brauckhoff M, Söderkvist P, Gimm O. Integrative genomics reveals frequent somatic NF1 mutations in sporadic pheochromocytomas. Hum Mol Genet. 2012;21:5406–5416. doi: 10.1093/hmg/dds402. [DOI] [PubMed] [Google Scholar]
- 13.Toledo RA, Qin Y, Srikantan S, Morales NP, Li Q, Deng Y, Kim SW, Pereira MA, Toledo SP, Su X, Aguiar RC, Dahia PL. In vivo and in vitro oncogenic effects of HIF2A mutations in pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2013;20:349–359. doi: 10.1530/ERC-13-0101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der Mey A, Taschner PE, Rubinstein WS, Myers EN, Richard CW, 3rd, Cornelisse CJ, Devilee P, et al. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 2000;287:848–851. doi: 10.1126/science.287.5454.848. [DOI] [PubMed] [Google Scholar]
- 15.Dahia PL, Ross KN, Wright ME, Hayashida CY, Santagata S, Barontini M, Kung AL, Sanso G, Powers JF, Tischler AS, Hodin R, Heitritter S, Moore F, et al. A HIF1alpha regulatory loop links hypoxia and mitochondrial signals in pheochromocytomas. PLoS Genet. 2005;1:72–80. doi: 10.1371/journal.pgen.0010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nölting S, Grossman AB. Signaling pathways in pheochromocytomas and paragangliomas: prospects for future therapies. Endocr Pathol. 2012;23:21–33. doi: 10.1007/s12022-012-9199-6. [DOI] [PubMed] [Google Scholar]
- 17.Favier J, Gimenez-Roqueplo AP. [Genetics of paragangliomas and pheochromocytomas] Med Sci (Paris) 2012;28:625–632. doi: 10.1051/medsci/2012286016. [DOI] [PubMed] [Google Scholar]
- 18.Favier J, Gimenez-Roqueplo AP. Pheochromocytomas: the (pseudo)-hypoxia hypothesis. Best Pract Res Clin Endocrinol Metab. 2010;24:957–968. doi: 10.1016/j.beem.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 19.López-Jiménez E, Gómez-López G, Leandro-García LJ, Muñoz I, Schiavi F, Montero-Conde C, de Cubas AA, Ramires R, Landa I, Leskelä S, Maliszewska A, Inglada-Pérez L, de la Vega L, et al. Research resource: Transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol. 2010;24:2382–2391. doi: 10.1210/me.2010-0256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Semenza GL. HIF-1, O(2), and the 3 PHDs: how animal cells signal hypoxia to the nucleus. Cell. 2001;107:1–3. doi: 10.1016/s0092-8674(01)00518-9. [DOI] [PubMed] [Google Scholar]
- 21.Gruber M, Simon MC. Hypoxia-inducible factors, hypoxia, and tumor angiogenesis. Curr Opin Hematol. 2006;13:169–174. doi: 10.1097/01.moh.0000219663.88409.35. [DOI] [PubMed] [Google Scholar]
- 22.Gnarra JR, Zhou S, Merrill MJ, Wagner JR, Krumm A, Papavassiliou E, Oldfield EH, Klausner RD, Linehan WM. Post-transcriptional regulation of vascular endothelial growth factor mRNA by the product of the VHL tumor suppressor gene. Proc Natl Acad Sci U S A. 1996;93:10589–10594. doi: 10.1073/pnas.93.20.10589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iliopoulos O, Levy AP, Jiang C, Kaelin WG, Jr, Goldberg MA. Negative regulation of hypoxia-inducible genes by the von Hippel-Lindau protein. Proc Natl Acad Sci U S A. 1996;93:10595–10599. doi: 10.1073/pnas.93.20.10595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaelin WG., Jr The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8:865–873. doi: 10.1038/nrc2502. [DOI] [PubMed] [Google Scholar]
- 25.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 26.Semenza GL. Defining the role of hypoxia-inducible factor 1 in cancer biology and therapeutics. Oncogene. 2010;29:625–634. doi: 10.1038/onc.2009.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- 28.Jochmanová I, Zelinka T, Widimský J, Jr, Pacak K. HIF signaling pathway in pheochromocytoma and other neuroendocrine tumors. Physiol Res. 2014;63:S251–262. doi: 10.33549/physiolres.932789. [DOI] [PubMed] [Google Scholar]
- 29.Yang C, Zhuang Z, Fliedner SM, Shankavaram U, Sun MG, Bullova P, Zhu R, Elkahloun AG, Kourlas PJ, Merino M, Kebebew E, Pacak K. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J Mol Med (Berl) 2015;93:93–104. doi: 10.1007/s00109-014-1205-7. [DOI] [PubMed] [Google Scholar]
- 30.Gaal J, Burnichon N, Korpershoek E, Roncelin I, Bertherat J, Plouin PF, de Krijger RR, Gimenez-Roqueplo AP, Dinjens WN. Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2010;95:1274–1278. doi: 10.1210/jc.2009-2170. [DOI] [PubMed] [Google Scholar]
- 31.Yao L, Barontini M, Niederle B, Jech M, Pfragner R, Dahia PL. Mutations of the metabolic genes IDH1, IDH2, and SDHAF2 are not major determinants of the pseudohypoxic phenotype of sporadic pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2010;95:1469–1472. doi: 10.1210/jc.2009-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, Stackhouse T, Kuzmin I, Modi W, Geil L, Schmidt L, Zhou F, Li H, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science. 1993;260:1317–1320. doi: 10.1126/science.8493574. [DOI] [PubMed] [Google Scholar]
- 33.Cherkasova E, Malinzak E, Rao S, Takahashi Y, Senchenko VN, Kudryavtseva AV, Nickerson ML, Merino M, Hong JA, Schrump DS, Srinivasan R, Linehan WM, Tian X, et al. Inactivation of the von Hippel-Lindau tumor suppressor leads to selective expression of a human endogenous retrovirus in kidney cancer. Oncogene. 2011;30:4697–4706. doi: 10.1038/onc.2011.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Menara M, Oudijk L, Badoual C, Bertherat J, Lepoutre-Lussey C, Amar L, Iturrioz X, Sibony M, Zinzindohoué F, de Krijger R, Gimenez-Roqueplo AP, Favier J. SDHD immunohistochemistry: a new tool to validate SDHx mutations in pheochromocytoma/paraganglioma. J Clin Endocrinol Metab. 2015;100:E287–291. doi: 10.1210/jc.2014-1870. [DOI] [PubMed] [Google Scholar]
- 35.Shen C, Kaelin WG., Jr The VHL/HIF axis in clear cell renal carcinoma. Semin Cancer Biol. 2013;23:18–25. doi: 10.1016/j.semcancer.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Contrasting properties of hypoxia-inducible factor 1 (HIF-1) and HIF-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol. 2005;25:5675–5686. doi: 10.1128/MCB.25.13.5675-5686.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Keith B, Johnson RS, Simon MC. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat Rev Cancer. 2012;12:9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dang CV, Kim JW, Gao P, Yustein J. The interplay between MYC and HIF in cancer. Nat Rev Cancer. 2008;8:51–56. doi: 10.1038/nrc2274. [DOI] [PubMed] [Google Scholar]
- 39.Rechsteiner MP, von Teichman A, Nowicka A, Sulser T, Schraml P, Moch H. VHL gene mutations and their effects on hypoxia inducible factor HIFalpha: identification of potential driver and passenger mutations. Cancer Res. 2011;71:5500–5511. doi: 10.1158/0008-5472.CAN-11-0757. [DOI] [PubMed] [Google Scholar]
- 40.Pollard PJ, El-Bahrawy M, Poulsom R, Elia G, Killick P, Kelly G, Hunt T, Jeffery R, Seedhar P, Barwell J, Latif F, Gleeson MJ, Hodgson SV, et al. Expression of HIF-1alpha, HIF-2alpha (EPAS1), and their target genes in paraganglioma and pheochromocytoma with VHL and SDH mutations. J Clin Endocrinol Metab. 2006;91:4593–4598. doi: 10.1210/jc.2006-0920. [DOI] [PubMed] [Google Scholar]
- 41.Favier J, Briere JJ, Burnichon N, Rivière J, Vescovo L, Benit P, Giscos-Douriez I, De Reyniès A, Bertherat J, Badoual C, Tissier F, Amar L, Libé R, et al. The Warburg effect is genetically determined in inherited pheochromocytomas. PloS One. 2009;4:e7094. doi: 10.1371/journal.pone.0007094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Comino-Méndez I, de Cubas AA, Bernal C, Álvarez-Escolá C, Sánchez-Malo C, Ramirez-Tortosa CL, Pedrinaci S, Rapizzi E, Ercolino T, Bernini G, Bacca A, Letón R, Pita G, et al. Tumoral EPAS1 (HIF2A) mutations explain sporadic pheochromocytoma and paraganglioma in the absence of erythrocytosis. Hum Mol Genet. 2013;22:2169–2176. doi: 10.1093/hmg/ddt069. [DOI] [PubMed] [Google Scholar]
- 43.Favier J, Buffet A, Gimenez-Roqueplo AP. HIF2A mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367:2161. doi: 10.1056/NEJMc1211953. author reply 2161-2162. [DOI] [PubMed] [Google Scholar]
- 44.Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, Popovic V, Stratakis CA, Prchal JT, Pacak K. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367:922–930. doi: 10.1056/NEJMoa1205119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shen C, Beroukhim R, Schumacher SE, Zhou J, Chang M, Signoretti S, Kaelin WG., Jr Genetic and functional studies implicate HIF1alpha as a 14q kidney cancer suppressor gene. Cancer Discov. 2011;1:222–235. doi: 10.1158/2159-8290.CD-11-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WG., Jr von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet. 2001;10:1019–1027. doi: 10.1093/hmg/10.10.1019. [DOI] [PubMed] [Google Scholar]
- 47.Ohh M, Yauch RL, Lonergan KM, Whaley JM, Stemmer-Rachamimov AO, Louis DN, Gavin BJ, Kley N, Kaelin WG, Jr, Iliopoulos O. The von Hippel-Lindau tumor suppressor protein is required for proper assembly of an extracellular fibronectin matrix. Mol Cell. 1998;1:959–968. doi: 10.1016/s1097-2765(00)80096-9. [DOI] [PubMed] [Google Scholar]
- 48.Pause A, Lee S, Lonergan KM, Klausner RD. The von Hippel-Lindau tumor suppressor gene is required for cell cycle exit upon serum withdrawal. Proc Natl Acad Sci U S A. 1998;95:993–998. doi: 10.1073/pnas.95.3.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Elder EE, Elder G, Larsson C. Pheochromocytoma and functional paraganglioma syndrome: no longer the 10% tumor. J Surg Oncol. 2005;89:193–201. doi: 10.1002/jso.20177. [DOI] [PubMed] [Google Scholar]
- 50.Lieubeau-Teillet B, Rak J, Jothy S, Iliopoulos O, Kaelin W, Kerbel RS. von Hippel-Lindau gene-mediated growth suppression and induction of differentiation in renal cell carcinoma cells grown as multicellular tumor spheroids. Cancer Res. 1998;58:4957–4962. [PubMed] [Google Scholar]
- 51.Davidowitz EJ, Schoenfeld AR, Burk RD. VHL induces renal cell differentiation and growth arrest through integration of cell-cell and cell-extracellular matrix signaling. Mol Cell Biol. 2001;21:865–874. doi: 10.1128/MCB.21.3.865-874.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stickle NH, Chung J, Klco JM, Hill RP, Kaelin WG, Jr, Ohh M. pVHL modification by NEDD8 is required for fibronectin matrix assembly and suppression of tumor development. Mol Cell Biol. 2004;24:3251–3261. doi: 10.1128/MCB.24.8.3251-3261.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Neumann HP, Eng C, Mulligan LM, Glavac D, Zauner I, Ponder BA, Crossey PA, Maher ER, Brauch H. Consequences of direct genetic testing for germline mutations in the clinical management of families with multiple endocrine neoplasia, type II. JAMA. 1995;274:1149–1151. [PubMed] [Google Scholar]
- 54.Crossey PA, Eng C, Ginalska-Malinowska M, Lennard TW, Wheeler DC, Ponder BA, Maher ER. Molecular genetic diagnosis of von Hippel-Lindau disease in familial phaeochromocytoma. J Med Genet. 1995;32:885–886. doi: 10.1136/jmg.32.11.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiménez C, Cote G, Arnold A, Gagel RF. Review: Should patients with apparently sporadic pheochromocytomas or paragangliomas be screened for hereditary syndromes? J Clin Endocrinol Metab. 2006;91:2851–2858. doi: 10.1210/jc.2005-2178. [DOI] [PubMed] [Google Scholar]
- 56.Rattenberry E, Vialard L, Yeung A, Bair H, McKay K, Jafri M, Canham N, Cole TR, Denes J, Hodgson SV, Irving R, Izatt L, Korbonits M, et al. A comprehensive next generation sequencing-based genetic testing strategy to improve diagnosis of inherited pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2013;98:E1248–1256. doi: 10.1210/jc.2013-1319. [DOI] [PubMed] [Google Scholar]
- 57.Kim JH, Seong MW, Lee KE, Choi HJ, Ku EJ, Bae JH, Park SS, Choi SH, Kim SW, Shin C, Kim SY. Germline mutations and genotype-phenotype correlations in patients with apparently sporadic pheochromocytoma/paraganglioma in Korea. Clin Genet. 2014;86:482–486. doi: 10.1111/cge.12304. [DOI] [PubMed] [Google Scholar]
- 58.Currás-Freixes M, Inglada-Pérez L, Mancikova V, Montero-Conde C, Letón R, Comino-Méndez I, Apellániz-Ruiz M, Sánchez-Barroso L, Aguirre Sánchez-Covisa M, Alcázar V, Aller J, Álvarez-Escolá C, Andia-Melero VM, et al. Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J Med Genet. 2015;52:647–656. doi: 10.1136/jmedgenet-2015-103218. [DOI] [PubMed] [Google Scholar]
- 59.Merlo A, de Quirós SB, de Santa-María IS, Pitiot AS, Balbin M, Astudillo A, Scola B, Aristegui M, Quer M, Suarez C, Chiara MD. Identification of somatic VHL gene mutations in sporadic head and neck paragangliomas in association with activation of the HIF-1alpha/miR-210 signaling pathway. J Clin Endocrinol Metab. 2013;98:E1661–1666. doi: 10.1210/jc.2013-1636. [DOI] [PubMed] [Google Scholar]
- 60.Jafri M, Whitworth J, Rattenberry E, Vialard L, Kilby G, Kumar AV, Izatt L, Lalloo F, Brennan P, Cook J, Morrison PJ, Canham N, Armstrong R, et al. Evaluation of SDHB, SDHD and VHL gene susceptibility testing in the assessment of individuals with non-syndromic phaeochromocytoma, paraganglioma and head and neck paraganglioma. Clin Endocrinol (Oxf) 2013;78:898–906. doi: 10.1111/cen.12074. [DOI] [PubMed] [Google Scholar]
- 61.Zhu WD, Wang ZY, Chai YC, Wang XW, Chen DY, Wu H. Germline mutations and genotype-phenotype associations in head and neck paraganglioma patients with negative family history in China. Eur J Med Genet. 2015;58:433–438. doi: 10.1016/j.ejmg.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 62.Piccini V, Rapizzi E, Bacca A, Di Trapani G, Pulli R, Giachè V, Zampetti B, Lucci-Cordisco E, Canu L, Corsini E, Faggiano A, Deiana L, Carrara D, et al. Head and neck paragangliomas: genetic spectrum and clinical variability in 79 consecutive patients. Endocr Relat Cancer. 2012;19:149–155. doi: 10.1530/ERC-11-0369. [DOI] [PubMed] [Google Scholar]
- 63.Crossey PA, Richards FM, Foster K, Green JS, Prowse A, Latif F, Lerman MI, Zbar B, Affara NA, Ferguson-Smith MA, Maher ER. Identification of intragenic mutations in the von Hippel-Lindau disease tumour suppressor gene and correlation with disease phenotype. Hum Mol Genet. 1994;3:1303–1308. doi: 10.1093/hmg/3.8.1303. [DOI] [PubMed] [Google Scholar]
- 64.Maher ER, Webster AR, Richards FM, Green JS, Crossey PA, Payne SJ, Moore AT. Phenotypic expression in von Hippel-Lindau disease: correlations with germline VHL gene mutations. J Med Genet. 1996;33:328–332. doi: 10.1136/jmg.33.4.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stebbins CE, Kaelin WG, Jr, Pavletich NP. Structure of the VHL-ElonginC-ElonginB complex: implications for VHL tumor suppressor function. Science. 1999;284:455–461. doi: 10.1126/science.284.5413.455. [DOI] [PubMed] [Google Scholar]
- 66.Rustin P, Rötig A. Inborn errors of complex II—unusual human mitochondrial diseases. Biochim Biophys Acta. 2002;1553:117–122. doi: 10.1016/s0005-2728(01)00228-6. [DOI] [PubMed] [Google Scholar]
- 67.Li M, Kim WY. Two sides to every story: the HIF-dependent and HIF-independent functions of pVHL. J Cell Mol Med. 2011;15:187–195. doi: 10.1111/j.1582-4934.2010.01238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yankovskaya V, Horsefield R, Törnroth S, Luna-Chavez C, Miyoshi H, Léger C, Byrne B, Cecchini G, Iwata S. Architecture of succinate dehydrogenase and reactive oxygen species generation. Science. 2003;299:700–704. doi: 10.1126/science.1079605. [DOI] [PubMed] [Google Scholar]
- 69.Ghezzi D, Goffrini P, Uziel G, Horvath R, Klopstock T, Lochmüller H, D’Adamo P, Gasparini P, Strom TM, Prokisch H, Invernizzi F, Ferrero I, Zeviani M. SDHAF1, encoding a LYR complex-II specific assembly factor, is mutated in SDH-defective infantile leukoencephalopathy. Nat Genet. 2009;41:654–656. doi: 10.1038/ng.378. [DOI] [PubMed] [Google Scholar]
- 70.Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, et al. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 2009;325:1139–1142. doi: 10.1126/science.1175689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Castelblanco E, Santacana M, Valls J, de Cubas A, Cascón A, Robledo M, Matias-Guiu X. Usefulness of negative and weak-diffuse pattern of SDHB immunostaining in assessment of SDH mutations in paragangliomas and pheochromocytomas. Endocr Pathol. 2013;24:199–205. doi: 10.1007/s12022-013-9269-4. [DOI] [PubMed] [Google Scholar]
- 72.Hensen EF, Bayley JP. Recent advances in the genetics of SDH-related paraganglioma and pheochromocytoma. Fam Cancer. 2011;10:355–363. doi: 10.1007/s10689-010-9402-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bayley JP, Kunst HP, Cascon A, Sampietro ML, Gaal J, Korpershoek E, Hinojar-Gutierrez A, Timmers HJ, Hoefsloot LH, Hermsen MA, Suarez C, Hussain AK, Vriends AH, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11:366–372. doi: 10.1016/S1470-2045(10)70007-3. [DOI] [PubMed] [Google Scholar]
- 74.Hensen EF, Siemers MD, Jansen JC, Corssmit EP, Romijn JA, Tops CM, van der Mey AG, Devilee P, Cornelisse CJ, Bayley JP, Vriends AH. Mutations in SDHD are the major determinants of the clinical characteristics of Dutch head and neck paraganglioma patients. Clin Endocrinol (Oxf) 2011;75:650–655. doi: 10.1111/j.1365-2265.2011.04097.x. [DOI] [PubMed] [Google Scholar]
- 75.Kunst HP, Rutten MH, de Mönnink JP, Hoefsloot LH, Timmers HJ, Marres HA, Jansen JC, Kremer H, Bayley JP, Cremers CW. SDHAF2 (PGL2-SDH5) and hereditary head and neck paraganglioma. Clin Cancer Res. 2011;17:247–254. doi: 10.1158/1078-0432.CCR-10-0420. [DOI] [PubMed] [Google Scholar]
- 76.Santos P, Pimenta T, Taveira-Gomes A. Hereditary Pheochromocytoma. Int J Surg Pathol. 2014;22:393–400. doi: 10.1177/1066896914537683. [DOI] [PubMed] [Google Scholar]
- 77.Burnichon N, Brière JJ, Libé R, Vescovo L, Rivière J, Tissier F, Jouanno E, Jeunemaitre X, Bénit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–3020. doi: 10.1093/hmg/ddq206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Korpershoek E, Favier J, Gaal J, Burnichon N, van Gessel B, Oudijk L, Badoual C, Gadessaud N, Venisse A, Bayley JP, van Dooren MF, de Herder WW, Tissier F, et al. SDHA immunohistochemistry detects germline SDHA gene mutations in apparently sporadic paragangliomas and pheochromocytomas. J Clin Endocrinol Metab. 2011;96:E1472–1476. doi: 10.1210/jc.2011-1043. [DOI] [PubMed] [Google Scholar]
- 79.Welander J, Andreasson A, Juhlin CC, Wiseman RW, Bäckdahl M, Höög A, Larsson C, Gimm O, Söderkvist P. Rare germline mutations identified by targeted next-generation sequencing of susceptibility genes in pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2014;99:E1352–1360. doi: 10.1210/jc.2013-4375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.González-Orús Álvarez-Morujo R, Aristegui Ruiz M, Martin Oviedo C, Álvarez Palacios I, Scola Yurrita B. Management of vagal paragangliomas: review of 17 patients. Eur Arch Otorhinolaryngol. 2015;272:2403–2414. doi: 10.1007/s00405-014-3141-0. [DOI] [PubMed] [Google Scholar]
- 81.Gimenez-Roqueplo AP, Favier J, Rustin P, Rieubland C, Crespin M, Nau V, Khau Van Kien P, Corvol P, Plouin PF, Jeunemaitre X. COMETE Network. Mutations in the SDHB gene are associated with extra-adrenal and/or malignant phaeochromocytomas. Cancer Res. 2003;63:5615–5621. [PubMed] [Google Scholar]
- 82.Benn DE, Gimenez-Roqueplo AP, Reilly JR, Bertherat J, Burgess J, Byth K, Croxson M, Dahia PL, Elston M, Gimm O, Henley D, Herman P, Murday V, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab. 2006;91:827–836. doi: 10.1210/jc.2005-1862. [DOI] [PubMed] [Google Scholar]
- 83.Fishbein L, Nathanson KL. Pheochromocytoma and paraganglioma: understanding the complexities of the genetic background. Cancer Genet. 2012;205:1–11. doi: 10.1016/j.cancergen.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mediouni A, Ammari S, Wassef M, Gimenez-Roqueplo AP, Laredo JD, Duet M, Tran Ba, Huy P, Oker N. Malignant head/neck paragangliomas. Comparative study. Eur Ann Otorhinolaryngol Head Neck Dis. 2014;131:159–166. doi: 10.1016/j.anorl.2013.05.003. [DOI] [PubMed] [Google Scholar]
- 85.Jafri M, Maher ER. The genetics of phaeochromocytoma: using clinical features to guide genetic testing. Eur J Endocrinol. 2012;166:151–158. doi: 10.1530/EJE-11-0497. [DOI] [PubMed] [Google Scholar]
- 86.Peterson LA, Litzendorf M, Ringel MD, Vaccaro PS. SDHB gene mutation in a carotid body paraganglioma: case report and review of the paraganglioma syndromes. Ann Vasc Surg. 2014;28:1321–e9-12. doi: 10.1016/j.avsg.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 87.Ellis RJ, Patel D, Prodanov T, Nilubol N, Pacak K, Kebebew E. The presence of SDHB mutations should modify surgical indications for carotid body paragangliomas. Ann Surg. 2014;260:158–162. doi: 10.1097/SLA.0000000000000283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schiavi F, Boedeker CC, Bausch B, Peczkowska M, Gomez CF, Strassburg T, Pawlu C, Buchta M, Salzmann M, Hoffmann MM, Berlis A, Brink I, Cybulla M, et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA. 2005;294:2057–2063. doi: 10.1001/jama.294.16.2057. [DOI] [PubMed] [Google Scholar]
- 89.Boedeker CC, Neumann HP, Offergeld C, Maier W, Falcioni M, Berlis A, Schipper J. Clinical features of paraganglioma syndromes. Skull Base. 2009;19:17–25. doi: 10.1055/s-0028-1103123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Douwes Dekker PB, Hogendoorn PC, Kuipers-Dijkshoorn N, Prins FA, van Duinen SG, Taschner PE, van der Mey AG, Cornelisse CJ. SDHD mutations in head and neck paragangliomas result in destabilization of complex II in the mitochondrial respiratory chain with loss of enzymatic activity and abnormal mitochondrial morphology. J Pathol. 2003;201:480–486. doi: 10.1002/path.1461. [DOI] [PubMed] [Google Scholar]
- 91.Kim ES, Kim SY, Mo EY, Jang DK, Moon SD, Han JH. Novel germline SDHD mutation in a patient with recurrent familial carotid body tumor and concomitant pheochromocytoma. Head Neck. 2014;36:E131–135. doi: 10.1002/hed.23670. [DOI] [PubMed] [Google Scholar]
- 92.McWhinney SR, Pasini B, Stratakis CA. International Carney Triad and Carney-Stratakis Syndrome Consortium. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med. 2007;357:1054–1056. doi: 10.1056/NEJMc071191. [DOI] [PubMed] [Google Scholar]
- 93.Haller F, Moskalev EA, Faucz FR, Barthelmeß S, Wiemann S, Bieg M, Assie G, Bertherat J, Schaefer IM, Otto C, Rattenberry E, Maher ER, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer. 2014;21:567–577. doi: 10.1530/ERC-14-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Burnichon N, Vescovo L, Amar L, Libé R, de Reynies A, Venisse A, Jouanno E, Laurendeau I, Parfait B, Bertherat J, Plouin PF, Jeunemaitre X, Favier J, et al. Integrative genomic analysis reveals somatic mutations in pheochromocytoma and paraganglioma. Hum Mol Genet. 2011;20:3974–3985. doi: 10.1093/hmg/ddr324. [DOI] [PubMed] [Google Scholar]
- 95.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7:77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 96.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 97.Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, et al. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 2012;26:1326–1338. doi: 10.1101/gad.191056.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Letouzé E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, et al. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 2013;23:739–752. doi: 10.1016/j.ccr.2013.04.018. [DOI] [PubMed] [Google Scholar]
- 99.Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jr, Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, et al. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013;3:648–657. doi: 10.1158/2159-8290.CD-13-0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, Licht JD, Deberardinis RJ, Chandel NS. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51:236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sudarshan S, Sourbier C, Kong HS, Block K, Valera Romero VA, Yang Y, Galindo C, Mollapour M, Scroggins B, Goode N, Lee MJ, Gourlay CW, Trepel J, et al. Fumarate hydratase deficiency in renal cancer induces glycolytic addiction and hypoxia-inducible transcription factor 1alpha stabilization by glucose-dependent generation of reactive oxygen species. Mol Cell Biol. 2009;29:4080–4090. doi: 10.1128/MCB.00483-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Smith EH, Janknecht R, Maher LJ., 3rd Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet. 2007;16:3136–3148. doi: 10.1093/hmg/ddm275. [DOI] [PubMed] [Google Scholar]
- 103.Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res. 2005;65:203–209. [PubMed] [Google Scholar]
- 104.Guzy RD, Sharma B, Bell E, Chandel NS, Schumacker PT. Loss of the SdhB, but Not the SdhA, subunit of complex II triggers reactive oxygen species-dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet. 2002;30:406–410. doi: 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 106.Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, Ras C, Seifar RM, van Minderhout I, Cornelisse CJ, Hogendoorn PC, Breuning MH, Suijker J, Korpershoek E, Kunst HP, Frizzell N, et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget. 2015;6:38777–38788. doi: 10.18632/oncotarget.6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Castro-Vega LJ, Buffet A, De Cubas AA, Cascón A, Menara M, Khalifa E, Amar L, Azriel S, Bourdeau I, Chabre O, Currás-Freixes M, Franco-Vidal V, Guillaud-Bataille M, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23:2440–2446. doi: 10.1093/hmg/ddt639. [DOI] [PubMed] [Google Scholar]
- 108.Ladroue C, Carcenac R, Leporrier M, Gad S, Le Hello C, Galateau-Salle F, Feunteun J, Pouysségur J, Richard S, Gardie B. PHD2 mutation and congenital erythrocytosis with paraganglioma. N Engl J Med. 2008;359:2685–2692. doi: 10.1056/NEJMoa0806277. [DOI] [PubMed] [Google Scholar]
- 109.Astuti D, Ricketts CJ, Chowdhury R, McDonough MA, Gentle D, Kirby G, Schlisio S, Kenchappa RS, Carter BD, Kaelin WG, Jr, Ratcliffe PJ, Schofield CJ, Latif F, et al. Mutation analysis of HIF prolyl hydroxylases (PHD/EGLN) in individuals with features of phaeochromocytoma and renal cell carcinoma susceptibility. Endocr Relat Cancer. 2011;18:73–83. doi: 10.1677/ERC-10-0113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Schmidt JV, Bradfield CA. Ah receptor signaling pathways. Annu Rev Cell Dev Biol. 1996;12:55–89. doi: 10.1146/annurev.cellbio.12.1.55. [DOI] [PubMed] [Google Scholar]
- 111.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 112.Kay SA, Millar AJ. New models in vogue for circadian clocks. Cell. 1995;83:361–364. doi: 10.1016/0092-8674(95)90113-2. [DOI] [PubMed] [Google Scholar]
- 113.King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, Steeves TD, Vitaterna MH, Kornhauser JM, Lowrey PL, Turek FW, Takahashi JS. Positional cloning of the mouse circadian clock gene. Cell. 1997;89:641–653. doi: 10.1016/s0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wilk R, Weizman I, Shilo BZ. trachealess encodes a bHLH-PAS protein that is an inducer of tracheal cell fates in Drosophila. Genes Dev. 1996;10:93–102. doi: 10.1101/gad.10.1.93. [DOI] [PubMed] [Google Scholar]
- 115.Isaac DD, Andrew DJ. Tubulogenesis in Drosophila: a requirement for the trachealess gene product. Genes Dev. 1996;10:103–117. doi: 10.1101/gad.10.1.103. [DOI] [PubMed] [Google Scholar]
- 116.Krasnov GS, Dmitriev AA, Snezhkina AV, Kudryavtseva AV. Deregulation of glycolysis in cancer: glyceraldehyde-3-phosphate dehydrogenase as a therapeutic target. Expert Opin Ther Targets. 2013;17:681–693. doi: 10.1517/14728222.2013.775253. [DOI] [PubMed] [Google Scholar]
- 117.Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL. The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev. 1998;12:3320–3324. doi: 10.1101/gad.12.21.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Taieb D, Yang C, Delenne B, Zhuang Z, Barlier A, Sebag F, Pacak K. First report of bilateral pheochromocytoma in the clinical spectrum of HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab. 2013;98:E908–913. doi: 10.1210/jc.2013-1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lorenzo FR, Yang C, Tang Ng, Fui M, Vankayalapati H, Zhuang Z, Huynh T, Grossmann M, Pacak K, Prchal JT. A novel EPAS1/HIF2A germline mutation in a congenital polycythemia with paraganglioma. J Mol Med (Berl) 2013;91:507–512. doi: 10.1007/s00109-012-0967-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Favier J, Amar L, Gimenez-Roqueplo AP. Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol. 2015;11:101–111. doi: 10.1038/nrendo.2014.188. [DOI] [PubMed] [Google Scholar]
- 121.McDonough MA, Li V, Flashman E, Chowdhury R, Mohr C, Lienard BM, Zondlo J, Oldham NJ, Clifton IJ, Lewis J, McNeill LA, Kurzeja RJ, Hewitson KS, et al. Cellular oxygen sensing: Crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2) Proc Natl Acad Sci U S A. 2006;103:9814–9819. doi: 10.1073/pnas.0601283103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Min JH, Yang H, Ivan M, Gertler F, Kaelin WG, Jr, Pavletich NP. Structure of an HIF-1alpha -pVHL complex: hydroxyproline recognition in signaling. Science. 2002;296:1886–1889. doi: 10.1126/science.1073440. [DOI] [PubMed] [Google Scholar]
- 123.Dahia PL. The genetic landscape of pheochromocytomas and paragangliomas: somatic mutations take center stage. J Clin Endocrinol Metab. 2013;98:2679–2681. doi: 10.1210/jc.2013-2191. [DOI] [PubMed] [Google Scholar]
- 124.Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T, Prchal JT, Tischler AS, Lechan RM, Zhuang Z. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol. 2013;31:1690–1698. doi: 10.1200/JCO.2012.47.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Buffet A, Smati S, Mansuy L, Ménara M, Lebras M, Heymann MF, Simian C, Favier J, Murat A, Cariou B, Gimenez-Roqueplo AP. Mosaicism in HIF2A-related polycythemia-paraganglioma syndrome. J Clin Endocrinol Metab. 2014;99:E369–373. doi: 10.1210/jc.2013-2600. [DOI] [PubMed] [Google Scholar]
- 126.Winkler BS, DeSantis N, Solomon F. Multiple NADPH-producing pathways control glutathione (GSH) content in retina. Exp Eye Res. 1986;43:829–847. doi: 10.1016/s0014-4835(86)80013-6. [DOI] [PubMed] [Google Scholar]
- 127.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, Silliman N, Szabo S, Buckhaults P, Farrell C, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 128.Parsons DW, Jones S, Zhang X, Lin JC, Leary RJ, Angenendt P, Mankoo P, Carter H, Siu IM, Gallia GL, Olivi A, McLendon R, Rasheed BA, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–1812. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ward PS, Patel J, Wise DR, Abdel-Wahab O, Bennett BD, Coller HA, Cross JR, Fantin VR, Hedvat CV, Perl AE, Rabinowitz JD, Carroll M, Su SM, et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell. 2010;17:225–234. doi: 10.1016/j.ccr.2010.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rodriguez-Cuevas S, López-Garza J, Labastida-Almendaro S. Carotid body tumors in inhabitants of altitudes higher than 2000 meters above sea level. Head Neck. 1998;20:374–378. doi: 10.1002/(sici)1097-0347(199808)20:5<374::aid-hed3>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 131.Zhao S, Lin Y, Xu W, Jiang W, Zha Z, Wang P, Yu W, Li Z, Gong L, Peng Y, Ding J, Lei Q, Guan KL, et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1alpha. Science. 2009;324:261–265. doi: 10.1126/science.1170944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Koivunen P, Lee S, Duncan CG, Lopez G, Lu G, Ramkissoon S, Losman JA, Joensuu P, Bergmann U, Gross S, Travins J, Weiss S, Looper R, et al. Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature. 2012;483:484–488. doi: 10.1038/nature10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tarhonskaya H, Rydzik AM, Leung IK, Loik ND, Chan MC, Kawamura A, McCullagh JS, Claridge TD, Flashman E, Schofield CJ. Non-enzymatic chemistry enables 2-hydroxyglutarate-mediated activation of 2-oxoglutarate oxygenases. Nat Commun. 2014;5:3423. doi: 10.1038/ncomms4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Morin A, Letouzé E, Gimenez-Roqueplo AP, Favier J. Oncometabolites-driven tumorigenesis: From genetics to targeted therapy. Int J Cancer. 2014;135:2237–2248. doi: 10.1002/ijc.29080. [DOI] [PubMed] [Google Scholar]
- 135.Pansuriya TC, van Eijk R, d’Adamo P, van Ruler MA, Kuijjer ML, Oosting J, Cleton-Jansen AM, van Oosterwijk JG, Verbeke SL, Meijer D, van Wezel T, Nord KH, Sangiorgi L, et al. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat Genet. 2011;43:1256–1261. doi: 10.1038/ng.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, Campos C, Fabius AW, Lu C, Ward PS, Thompson CB, Kaufman A, Guryanova O, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature. 2012;483:479–483. doi: 10.1038/nature10866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, Li Y, Bhagwat N, Vasanthakumar A, Fernandez HF, Tallman MS, Sun Z, Wolniak K, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell. 2010;18:553–567. doi: 10.1016/j.ccr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 2011;19:17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yao L, Schiavi F, Cascon A, Qin Y, Inglada-Perez L, King EE, Toledo RA, Ercolino T, Rapizzi E, Ricketts CJ, Mori L, Giacche M, Mendola A, et al. Spectrum and prevalence of FP/TMEM127 gene mutations in pheochromocytomas and paragangliomas. JAMA. 2010;304:2611–2619. doi: 10.1001/jama.2010.1830. [DOI] [PubMed] [Google Scholar]
- 140.Minárik P, Tomásková N, Kollárová M, Antalik M. Malate dehydrogenases—structure and function. Gen Physiol Biophys. 2002;21:257–265. [PubMed] [Google Scholar]
- 141.Kudryavtseva AV, Krasnov GS, Dmitriev AA, Alekseev BY, Kardymon OL, Sadritdinova AF, Fedorova MS, Pokrovsky AV, Melnikova NV, Kaprin AD, Moskalev AA, Snezhkina AV. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget. 2016;7:44879–44905. doi: 10.18632/oncotarget.9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Van Schaftingen E, Rzem R, Veiga-da-Cunha M. L:-2-Hydroxyglutaric aciduria, a disorder of metabolite repair. J Inherit Metab Dis. 2009;32:135–142. doi: 10.1007/s10545-008-1042-3. [DOI] [PubMed] [Google Scholar]
- 143.Cascón A, Comino-Méndez I, Currás-Freixes M, de Cubas AA, Contreras L, Richter S, Peitzsch M, Mancikova V, Inglada-Pérez L, Pérez-Barrios A, Calatayud M, Azriel S, Villar-Vicente R, et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst. 2015:107. doi: 10.1093/jnci/djv053. [DOI] [PubMed] [Google Scholar]
- 144.Philip B, Ito K, Moreno-Sánchez R, Ralph SJ. HIF expression and the role of hypoxic microenvironments within primary tumours as protective sites driving cancer stem cell renewal and metastatic progression. Carcinogenesis. 2013;34:1699–1707. doi: 10.1093/carcin/bgt209. [DOI] [PubMed] [Google Scholar]
- 145.Pan Y, Mansfield KD, Bertozzi CC, Rudenko V, Chan DA, Giaccia AJ, Simon MC. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27:912–925. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221–1224. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 147.Grozinsky-Glasberg S, Franchi G, Teng M, Leontiou CA, Ribeiro de Oliveira A, Jr, Dalino P, Salahuddin N, Korbonits M, Grossman AB. Octreotide and the mTOR inhibitor RAD001 (everolimus) block proliferation and interact with the Akt-mTOR-p70S6K pathway in a neuro-endocrine tumour cell line. Neuroendocrinology. 2008;87:168–181. doi: 10.1159/000111501. [DOI] [PubMed] [Google Scholar]
- 148.Grozinsky-Glasberg S, Rubinfeld H, Nordenberg Y, Gorshtein A, Praiss M, Kendler E, Feinmesser R, Grossman AB, Shimon I. The rapamycin-derivative RAD001 (everolimus) inhibits cell viability and interacts with the Akt-mTOR-p70S6K pathway in human medullary thyroid carcinoma cells. Mol Cell Endocrinol. 2010;315:87–94. doi: 10.1016/j.mce.2009.09.027. [DOI] [PubMed] [Google Scholar]
- 149.Harthill JE, Pozuelo Rubio M, Milne FC, MacKintosh C. Regulation of the 14-3-3-binding protein p39 by growth factors and nutrients in rat PC12 pheochromocytoma cells. Biochem J. 2002;368:565–572. doi: 10.1042/BJ20020838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gingras AC, Raught B, Sonenberg N. Regulation of translation initiation by FRAP/mTOR. Genes Dev. 2001;15:807–826. doi: 10.1101/gad.887201. [DOI] [PubMed] [Google Scholar]
- 151.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–745. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Ruggero D, Montanaro L, Ma L, Xu W, Londei P, Cordon-Cardo C, Pandolfi PP. The translation factor eIF-4E promotes tumor formation and cooperates with c-Myc in lymphomagenesis. Nat Med. 2004;10:484–486. doi: 10.1038/nm1042. [DOI] [PubMed] [Google Scholar]
- 153.Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR, Meyuhas O, Shokat KM, Ruggero D. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer Cell. 2010;17:249–261. doi: 10.1016/j.ccr.2010.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Dumstorf CA, Konicek BW, McNulty AM, Parsons SH, Furic L, Sonenberg N, Graff JR. Modulation of 4E-BP1 function as a critical determinant of enzastaurin-induced apoptosis. Mol Cancer Ther. 2010;9:3158–3163. doi: 10.1158/1535-7163.MCT-10-0413. [DOI] [PubMed] [Google Scholar]
- 155.Pourdehnad M, Truitt ML, Siddiqi IN, Ducker GS, Shokat KM, Ruggero D. Myc and mTOR converge on a common node in protein synthesis control that confers synthetic lethality in Myc-driven cancers. Proc Natl Acad Sci U S A. 2013;110:11988–11993. doi: 10.1073/pnas.1310230110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.van Riggelen J, Yetil A, Felsher DW. MYC as a regulator of ribosome biogenesis and protein synthesis. Nat Rev Cancer. 2010;10:301–309. doi: 10.1038/nrc2819. [DOI] [PubMed] [Google Scholar]
- 157.Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, Cordon-Cardo C, Pelletier J, Lowe SW. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–337. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 158.Blank A, Schmitt AM, Korpershoek E, van Nederveen F, Rudolph T, Weber N, Strebel RT, de Krijger R, Komminoth P, Perren A. SDHB loss predicts malignancy in pheochromocytomas/sympathethic paragangliomas, but not through hypoxia signalling. Endocr Relat Cancer. 2010;17:919–928. doi: 10.1677/ERC-09-0316. [DOI] [PubMed] [Google Scholar]
- 159.Zhu J, Blenis J, Yuan J. Activation of PI3K/Akt and MAPK pathways regulates Myc-mediated transcription by phosphorylating and promoting the degradation of Mad1. Proc Natl Acad Sci U S A. 2008;105:6584–6589. doi: 10.1073/pnas.0802785105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Douwes Dekker PB, Kuipers-Dijkshoorn NJ, Baelde HJ, van der Mey AG, Hogendoorn PC, Cornelisse CJ. Basic fibroblast growth factor and fibroblastic growth factor receptor-1 may contribute to head and neck paraganglioma development by an autocrine or paracrine mechanism. Hum Pathol. 2007;38:79–85. doi: 10.1016/j.humpath.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 161.Califano D, Rizzo C, D’Alessio A, Colucci-D’Amato GL, Cali G, Bartoli PC, Santelli G, Vecchio G, de Franciscis V. Signaling through Ras is essential for ret oncogene-induced cell differentiation in PC12 cells. J Biol Chem. 2000;275:19297–19305. doi: 10.1074/jbc.M905866199. [DOI] [PubMed] [Google Scholar]
- 162.Rossel M, Pasini A, Chappuis S, Geneste O, Fournier L, Schuffenecker I, Takahashi M, van Grunsven LA, Urdiales JL, Rudkin BB, Lenoir GM, Billaud M. Distinct biological properties of two RET isoforms activated by MEN 2A and MEN 2B mutations. Oncogene. 1997;14:265–275. doi: 10.1038/sj.onc.1200831. [DOI] [PubMed] [Google Scholar]
- 163.Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O’Connell P, Cawthon RM, Innis MA, McCormick F. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell. 1990;63:843–849. doi: 10.1016/0092-8674(90)90150-d. [DOI] [PubMed] [Google Scholar]
- 164.Ichihara M, Murakumo Y, Takahashi M. RET and neuroendocrine tumors. Cancer Lett. 2004;204:197–211. doi: 10.1016/S0304-3835(03)00456-7. [DOI] [PubMed] [Google Scholar]
- 165.Mulligan LM, Kwok JB, Healey CS, Elsdon MJ, Eng C, Gardner E, Love DR, Mole SE, Moore JK, Papi L, Ponder MA, Telenius H, Tunnacliffe A, et al. Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature. 1993;363:458–460. doi: 10.1038/363458a0. [DOI] [PubMed] [Google Scholar]
- 166.Asai N, Iwashita T, Matsuyama M, Takahashi M. Mechanism of activation of the ret proto-oncogene by multiple endocrine neoplasia 2A mutations. Mol Cell Biol. 1995;15:1613–1619. doi: 10.1128/mcb.15.3.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Santoro M, Carlomagno F, Romano A, Bottaro DP, Dathan NA, Grieco M, Fusco A, Vecchio G, Matoskova B, Kraus MH, Di Fiore PP. Activation of RET as a dominant transforming gene by germline mutations of MEN2A and MEN2B. Science. 1995;267:381–383. doi: 10.1126/science.7824936. [DOI] [PubMed] [Google Scholar]
- 168.Thosani S, Ayala-Ramirez M, Palmer L, Hu MI, Rich T, Gagel RF, Cote G, Waguespack SG, Habra MA, Jimenez C. The characterization of pheochromocytoma and its impact on overall survival in multiple endocrine neoplasia type 2. J Clin Endocrinol Metab. 2013;98:E1813–1819. doi: 10.1210/jc.2013-1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Leboulleux S, Travagli JP, Caillou B, Laplanche A, Bidart JM, Schlumberger M, Baudin E. Medullary thyroid carcinoma as part of a multiple endocrine neoplasia type 2B syndrome: influence of the stage on the clinical course. Cancer. 2002;94:44–50. doi: 10.1002/cncr.10205. [DOI] [PubMed] [Google Scholar]
- 170.Pasquali D, Di Matteo FM, Renzullo A, Accardo G, Esposito D, Barbato F, Colantuoni V, Circelli L, Conzo G. Multiple endocrine neoplasia, the old and the new: a mini review. Il Giornale di chirurgia. 2012;33:370–373. [PubMed] [Google Scholar]
- 171.Hyndman BD, Gujral TS, Krieger JR, Cockburn JG, Mulligan LM. Multiple functional effects of RET kinase domain sequence variants in Hirschsprung disease. Hum Mutat. 2013;34:132–142. doi: 10.1002/humu.22170. [DOI] [PubMed] [Google Scholar]
- 172.Viskochil D, Buchberg AM, Xu G, Cawthon RM, Stevens J, Wolff RK, Culver M, Carey JC, Copeland NG, Jenkins NA, White R, O’Connell P. Deletions and a translocation interrupt a cloned gene at the neurofibromatosis type 1 locus. Cell. 1990;62:187–192. doi: 10.1016/0092-8674(90)90252-a. [DOI] [PubMed] [Google Scholar]
- 173.Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell. 1990;63:851–859. doi: 10.1016/0092-8674(90)90151-4. [DOI] [PubMed] [Google Scholar]
- 174.Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–715. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- 175.Cichowski K, Santiago S, Jardim M, Johnson BW, Jacks T. Dynamic regulation of the Ras pathway via proteolysis of the NF1 tumor suppressor. Genes Dev. 2003;17:449–454. doi: 10.1101/gad.1054703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Powers JF, Evinger MJ, Zhi J, Picard KL, Tischler AS. Pheochromocytomas in Nf1 knockout mice express a neural progenitor gene expression profile. Neuroscience. 2007;147:928–937. doi: 10.1016/j.neuroscience.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 177.Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, Lechleiter JD, Sass M, Aronin N, Schiavi F, Boaretto F, Opocher G, Toledo RA, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–233. doi: 10.1038/ng.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Dahia PL. Pheochromocytoma and paraganglioma pathogenesis: learning from genetic heterogeneity. Nat Rev Cancer. 2014;14:108–119. doi: 10.1038/nrc3648. [DOI] [PubMed] [Google Scholar]
- 179.Burnichon N, Lepoutre-Lussey C, Laffaire J, Gadessaud N, Molinie V, Hernigou A, Plouin PF, Jeunemaitre X, Favier J, Gimenez-Roqueplo AP. A novel TMEM127 mutation in a patient with familial bilateral pheochromocytoma. Eur J Endocrinol. 2011;164:141–145. doi: 10.1530/EJE-10-0758. [DOI] [PubMed] [Google Scholar]
- 180.Takeichi N, Midorikawa S, Watanabe A, Naing BT, Tamura H, Wakakuri-Kano T, Ishizaki A, Sugihara H, Nissato S, Saito Y, Aita Y, Ishii KA, Igarashi T, et al. Identical germline mutations in the TMEM127 gene in two unrelated Japanese patients with bilateral pheochromocytoma. Clin Endocrinol (Oxf) 2012;77:707–714. doi: 10.1111/j.1365-2265.2012.04421.x. [DOI] [PubMed] [Google Scholar]
- 181.Elston MS, Meyer-Rochow GY, Prosser D, Love DR, Conaglen JV. Novel mutation in the TMEM127 gene associated with phaeochromocytoma. Intern Med J. 2013;43:449–451. doi: 10.1111/imj.12088. [DOI] [PubMed] [Google Scholar]
- 182.Neumann HP, Sullivan M, Winter A, Malinoc A, Hoffmann MM, Boedeker CC, Bertz H, Walz MK, Moeller LC, Schmid KW, Eng C. Germline mutations of the TMEM127 gene in patients with paraganglioma of head and neck and extraadrenal abdominal sites. J Clin Endocrinol Metab. 2011;96:E1279–1282. doi: 10.1210/jc.2011-0114. [DOI] [PubMed] [Google Scholar]
- 183.Jacobs BL, Goodman CA, Hornberger TA. The mechanical activation of mTOR signaling: an emerging role for late endosome/lysosomal targeting. J Muscle Res Cell Motil. 2014;35:11–21. doi: 10.1007/s10974-013-9367-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Flinn RJ, Yan Y, Goswami S, Parker PJ, Backer JM. The late endosome is essential for mTORC1 signaling. Mol Biol Cell. 2010;21:833–841. doi: 10.1091/mbc.E09-09-0756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bridges D, Fisher K, Zolov SN, Xiong T, Inoki K, Weisman LS, Saltiel AR. Rab5 proteins regulate activation and localization of target of rapamycin complex 1. J Biol Chem. 2012;287:20913–20921. doi: 10.1074/jbc.M111.334060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Qin Y, Deng Y, Ricketts CJ, Srikantan S, Wang E, Maher ER, Dahia PL. The tumor susceptibility gene TMEM127 is mutated in renal cell carcinomas and modulates endolysosomal function. Hum Mol Genet. 2014;23:2428–2439. doi: 10.1093/hmg/ddt638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wang Y, Roche O, Yan MS, Finak G, Evans AJ, Metcalf JL, Hast BE, Hanna SC, Wondergem B, Furge KA, Irwin MS, Kim WY, Teh BT, et al. Regulation of endocytosis via the oxygen-sensing pathway. Nat Med. 2009;15:319–324. doi: 10.1038/nm.1922. [DOI] [PubMed] [Google Scholar]
- 188.Wang Y, Roche O, Xu C, Moriyama EH, Heir P, Chung J, Roos FC, Chen Y, Finak G, Milosevic M, Wilson BC, Teh BT, Park M, et al. Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc Natl Acad Sci U S A. 2012;109:4892–4897. doi: 10.1073/pnas.1112129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Grandori C, Cowley SM, James LP, Eisenman RN. The Myc/Max/Mad network and the transcriptional control of cell behavior. Annu Rev Cell Dev Biol. 2000;16:653–699. doi: 10.1146/annurev.cellbio.16.1.653. [DOI] [PubMed] [Google Scholar]
- 190.Snezhkina AV, Krasnov GS, Lipatova AV, Sadritdinova AF, Kardymon OL, Fedorova MS, Melnikova NV, Stepanov OA, Zaretsky AR, Kaprin AD, Alekseev BY, Dmitriev AA, Kudryavtseva AV. The dysregulation of polyamine metabolism in colorectal cancer is associated with overexpression of c-Myc and C/EBPbeta rather than enterotoxigenic Bacteroides fragilis infection. Oxid Med Cell Longev. 2016;2016:2353560. doi: 10.1155/2016/2353560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, Landa I, Leandro-Garcia LJ, Letón R, Honrado E, Ramos-Medina R, Caronia D, Pita G, Gómez-Graña A, de Cubas AA, Inglada-Pérez L, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–667. doi: 10.1038/ng.861. [DOI] [PubMed] [Google Scholar]
- 192.Blackwood EM, Lüscher B, Eisenman RN. Myc and Max associate in vivo. Genes Dev. 1992;6:71–80. doi: 10.1101/gad.6.1.71. [DOI] [PubMed] [Google Scholar]
- 193.Cascón A, Robledo M. MAX. MYC: a heritable breakup. Cancer Res. 2012;72:3119–3124. doi: 10.1158/0008-5472.CAN-11-3891. [DOI] [PubMed] [Google Scholar]
- 194.Peczkowska M, Kowalska A, Sygut J, Waligórski D, Malinoc A, Janaszek-Sitkowska H, Prejbisz A, Januszewicz A, Neumann HP. Testing new susceptibility genes in the cohort of apparently sporadic phaeochromocytoma/paraganglioma patients with clinical characteristics of hereditary syndromes. Clin Endocrinol (Oxf) 2013;79:817–823. doi: 10.1111/cen.12218. [DOI] [PubMed] [Google Scholar]
- 195.Crona J, Maharjan R, Delgado Verdugo A, Stálberg P, Granberg D, Hellman P, Björklund P. MAX mutations status in Swedish patients with pheochromocytoma and paraganglioma tumours. Fam Cancer. 2014;13:121–125. doi: 10.1007/s10689-013-9666-3. [DOI] [PubMed] [Google Scholar]
- 196.Nangaku M, Sato-Yoshitake R, Okada Y, Noda Y, Takemura R, Yamazaki H, Hirokawa N. KIF1B, a novel microtubule plus end-directed monomeric motor protein for transport of mitochondria. Cell. 1994;79:1209–1220. doi: 10.1016/0092-8674(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 197.Zhao C, Takita J, Tanaka Y, Setou M, Nakagawa T, Takeda S, Yang HW, Terada S, Nakata T, Takei Y, Saito M, Tsuji S, Hayashi Y, et al. Charcot-Marie-Tooth disease type 2A caused by mutation in a microtubule motor KIF1Bbeta. Cell. 2001;105:587–597. doi: 10.1016/s0092-8674(01)00363-4. [DOI] [PubMed] [Google Scholar]
- 198.Schlisio S, Kenchappa RS, Vredeveld LC, George RE, Stewart R, Greulich H, Shahriari K, Nguyen NV, Pigny P, Dahia PL, Pomeroy SL, Maris JM, Look AT, et al. The kinesin KIF1Bbeta acts downstream from EglN3 to induce apoptosis and is a potential 1p36 tumor suppressor. Genes Dev. 2008;22:884–893. doi: 10.1101/gad.1648608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Yeh IT, Lenci RE, Qin Y, Buddavarapu K, Ligon AH, Leteurtre E, Do Cao C, Cardot-Bauters C, Pigny P, Dahia PL. A germline mutation of the KIF1B beta gene on 1p36 in a family with neural and nonneural tumors. Hum Genet. 2008;124:279–285. doi: 10.1007/s00439-008-0553-1. [DOI] [PubMed] [Google Scholar]
- 200.Larsson C, Skogseid B, Oberg K, Nakamura Y, Nordenskjold M. Multiple endocrine neoplasia type 1 gene maps to chromosome 11 and is lost in insulinoma. Nature. 1988;332:85–87. doi: 10.1038/332085a0. [DOI] [PubMed] [Google Scholar]
- 201.Agarwal SK, Kester MB, Debelenko LV, Heppner C, Emmert-Buck MR, Skarulis MC, Doppman JL, Kim YS, Lubensky IA, Zhuang Z, Green JS, Guru SC, Manickam P, et al. Germline mutations of the MEN1 gene in familial multiple endocrine neoplasia type 1 and related states. Hum Mol Genet. 1997;6:1169–1175. doi: 10.1093/hmg/6.7.1169. [DOI] [PubMed] [Google Scholar]
- 202.Wu X, Hua X. Menin, histone h3 methyltransferases, and regulation of cell proliferation: current knowledge and perspective. Curr Mol Med. 2008;8:805–815. doi: 10.2174/156652408786733702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Jamilloux Y, Favier J, Pertuit M, Delage-Corre M, Lopez S, Teissier MP, Mathonnet M, Galinat S, Barlier A, Archambeaud F. A MEN1 syndrome with a paraganglioma. Eur J Hum Genet. 2014;22:283–285. doi: 10.1038/ejhg.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Schussheim DH, Skarulis MC, Agarwal SK, Simonds WF, Burns AL, Spiegel AM, Marx SJ. Multiple endocrine neoplasia type 1: new clinical and basic findings. Trends Endocrinol Metab. 2001;12:173–178. [Google Scholar]
- 205.Welander J, Söderkvist P, Gimm O. Genetics and clinical characteristics of hereditary pheochromocytomas and paragangliomas. Endocr Relat Cancer. 2011;18:R253–276. doi: 10.1530/ERC-11-0170. [DOI] [PubMed] [Google Scholar]
- 206.Dackiw AP, Cote GJ, Fleming JB, Schultz PN, Stanford P, Vassilopoulou-Sellin R, Evans DB, Gagel RF, Lee JE. Screening for MEN1 mutations in patients with atypical endocrine neoplasia. Surgery. 1999;126:1097–1103. doi: 10.1067/msy.2099.101376. discussion 1103-1094. [DOI] [PubMed] [Google Scholar]
- 207.Lee S, Nakamura E, Yang H, Wei W, Linggi MS, Sajan MP, Farese RV, Freeman RS, Carter BD, Kaelin WG, Jr, Schlisio S. Neuronal apoptosis linked to EglN3 prolyl hydroxylase and familial pheochromocytoma genes: developmental culling and cancer. Cancer Cell. 2005;8:155–167. doi: 10.1016/j.ccr.2005.06.015. [DOI] [PubMed] [Google Scholar]
- 208.Palmada M, Kanwal S, Rutkoski NJ, Gustafson-Brown C, Johnson RS, Wisdom R, Carter BD. c-jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J Cell Biol. 2002;158:453–461. doi: 10.1083/jcb.200112129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Vogel KS, Brannan CI, Jenkins NA, Copeland NG, Parada LF. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell. 1995;82:733–742. doi: 10.1016/0092-8674(95)90470-0. [DOI] [PubMed] [Google Scholar]
- 210.Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- 211.Vaque JP, Fernández-Garcia B, García-Sanz P, Ferrandiz N, Bretones G, Calvo F, Crespo P, Marin MC, León J. c-Myc inhibits Ras-mediated differentiation of pheochromocytoma cells by blocking c-Jun up-regulation. Mol Cancer Res. 2008;6:325–339. doi: 10.1158/1541-7786.MCR-07-0180. [DOI] [PubMed] [Google Scholar]
- 212.Trupp M, Arenas E, Fainzilber M, Nilsson AS, Sieber BA, Grigoriou M, Kilkenny C, Salazar-Grueso E, Pachnis V, Arumäe U. Functional receptor for GDNF encoded by the c-ret proto-oncogene. Nature. 1996;381:785–789. doi: 10.1038/381785a0. [DOI] [PubMed] [Google Scholar]
- 213.Baloh RH, Enomoto H, Johnson EM, Jr, Milbrandt J. The GDNF family ligands and receptors - implications for neural development. Curr Opin Neurobiol. 2000;10:103–110. doi: 10.1016/s0959-4388(99)00048-3. [DOI] [PubMed] [Google Scholar]
- 214.Ivanchuk SM, Myers SM, Eng C, Mulligan LM. De novo mutation of GDNF, ligand for the RET/GDNFR-alpha receptor complex, in Hirschsprung disease. Hum Mol Genet. 1996;5:2023–2026. doi: 10.1093/hmg/5.12.2023. [DOI] [PubMed] [Google Scholar]
- 215.Woodward ER, Eng C, McMahon R, Voutilainen R, Affara NA, Ponder BA, Maher ER. Genetic predisposition to phaeochromocytoma: analysis of candidate genes GDNF, RET and VHL. Hum Mol Genet. 1997;6:1051–1056. doi: 10.1093/hmg/6.7.1051. [DOI] [PubMed] [Google Scholar]
- 216.Dahia PL, Toledo SP, Mulligan LM, Maher ER, Grossman AB, Eng C. Mutation analysis of glial cell line-derived neurotrophic factor (GDNF), a ligand for the RET/GDNF receptor alpha complex, in sporadic phaeochromocytomas. Cancer Res. 1997;57:310–313. [PubMed] [Google Scholar]
- 217.Adjei AA. Blocking oncogenic Ras signaling for cancer therapy. J Natl Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- 218.Lin SR, Tsai JH, Yang YC, Lee SC. Mutations of K-ras oncogene in human adrenal tumours in Taiwan. Br J Cancer. 1998;77:1060–1065. doi: 10.1038/bjc.1998.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Moley JF, Brother MB, Wells SA, Spengler BA, Biedler JL, Brodeur GM. Low frequency of ras gene mutations in neuroblastomas, pheochromocytomas, and medullary thyroid cancers. Cancer Res. 1991;51:1596–1599. [PubMed] [Google Scholar]
- 220.Moul JW, Bishoff JT, Theune SM, Chang EH. Absent ras gene mutations in human adrenal cortical neoplasms and pheochromocytomas. J Urol. 1993;149:1389–1394. doi: 10.1016/s0022-5347(17)36397-8. [DOI] [PubMed] [Google Scholar]
- 221.Yoshimoto K, Iwahana H, Fukuda A, Sano T, Katsuragi K, Kinoshita M, Saito S, Itakura M. ras mutations in endocrine tumors: mutation detection by polymerase chain reaction-single strand conformation polymorphism. Jpn J Cancer Res. 1992;83:1057–1062. doi: 10.1111/j.1349-7006.1992.tb02722.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Clark GR, Sciacovelli M, Gaude E, Walsh DM, Kirby G, Simpson MA, Trembath RC, Berg JN, Woodward ER, Kinning E, Morrison PJ, Frezza C, Maher ER. Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab. 2014;99:E2046–2050. doi: 10.1210/jc.2014-1659. [DOI] [PubMed] [Google Scholar]
- 223.Martins R, Bugalho MJ. Paragangliomas/Pheochromocytomas: clinically oriented genetic testing. Int J Endocrinol. 2014;2014:794187. doi: 10.1155/2014/794187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Luchetti A, Walsh D, Rodger F, Clark G, Martin T, Irving R, Sanna M, Yao M, Robledo M, Neumann HP, Woodward ER, Latif F, Abbs S, et al. Profiling of somatic mutations in phaeochromocytoma and paraganglioma by targeted next generation sequencing analysis. Int J Endocrinol. 2015;2015:138573. doi: 10.1155/2015/138573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Costello JM. A new syndrome: mental subnormality and nasal papillomata. Aust Paediatr J. 1977;13:114–118. doi: 10.1111/j.1440-1754.1977.tb01135.x. [DOI] [PubMed] [Google Scholar]
- 226.Crona J, Delgado Verdugo A, Maharjan R, Stalberg P, Granberg D, Hellman P, Björklund P. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab. 2013;98:E1266–1271. doi: 10.1210/jc.2012-4257. [DOI] [PubMed] [Google Scholar]
- 227.Oudijk L, de Krijger RR, Rapa I, Beuschlein F, de Cubas AA, Dei Tos AP, Dinjens WN, Korpershoek E, Mancikova V, Mannelli M, Papotti M, Vatrano S, Robledo M, et al. H-RAS mutations are restricted to sporadic pheochromocytomas lacking specific clinical or pathological features: data from a multi-institutional series. J Clin Endocrinol Metab. 2014;99:E1376–1380. doi: 10.1210/jc.2013-3879. [DOI] [PubMed] [Google Scholar]
- 228.Kranenburg O. The KRAS oncogene: past, present, and future. Biochim Biophys Acta. 2005;1756:81–82. doi: 10.1016/j.bbcan.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 229.Kudryavtseva AV, Lipatova AV, Zaretsky AR, Moskalev AA, Fedorova MS, Rasskazova AS, Shibukhova GA, Snezhkina AV, Kaprin AD, Alekseev BY, Dmitriev AA, Krasnov GS. Important molecular genetic markers of colorectal cancer. Oncotarget. 2016;7:53959–53983. doi: 10.18632/oncotarget.979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Schubbert S, Zenker M, Rowe SL, Böll S, Klein C, Bollag G, van der Burgt I, Musante L, Kalscheuer V, Wehner LE, Nguyen H, West B, Zhang KY, et al. Germline KRAS mutations cause Noonan syndrome. Nat Genet. 2006;38:331–336. doi: 10.1038/ng1748. [DOI] [PubMed] [Google Scholar]
- 231.Hrasćan R, Pećina-Slaus N, Martić TN, Colić JF, Gall-Troselj K, Pavelić K, Karapandza N. Analysis of selected genes in neuroendocrine tumours: insulinomas and phaeochromocytomas. J Neuroendocrinol. 2008;20:1015–1022. doi: 10.1111/j.1365-2826.2008.01755.x. [DOI] [PubMed] [Google Scholar]
- 232.Bastepe M, Jüppner H. GNAS locus and pseudohypoparathyroidism. Horm Res. 2005;63:65–74. doi: 10.1159/000083895. [DOI] [PubMed] [Google Scholar]
- 233.Jüppner H. Genetic and epigenetic defects at the GNAS locus cause different forms of pseudohypoparathyroidism. Ann Endocrinol (Paris) 2015;76:92–97. doi: 10.1016/j.ando.2015.03.011. [DOI] [PubMed] [Google Scholar]
- 234.Turan S, Bastepe M. GNAS spectrum of disorders. Curr Osteoporos Rep. 2015;13:146–158. doi: 10.1007/s11914-015-0268-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Williamson EA, Johnson SJ, Foster S, Kendall-Taylor P, Harris PE. G protein gene mutations in patients with multiple endocrinopathies. J Clin Endocrinol Metab. 1995;80:1702–1705. doi: 10.1210/jcem.80.5.7745022. [DOI] [PubMed] [Google Scholar]
- 236.Sandgren J, Andersson R, Rada-Iglesias A, Enroth S, Akerstrom G, Dumanski JP, Komorowski J, Westin G, Wadelius C. Integrative epigenomic and genomic analysis of malignant pheochromocytoma. Exp Mol Med. 2010;42:484–502. doi: 10.3858/emm.2010.42.7.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Chen PL, Chen YM, Bookstein R, Lee WH. Genetic mechanisms of tumor suppression by the human p53 gene. Science. 1990;250:1576–1580. doi: 10.1126/science.2274789. [DOI] [PubMed] [Google Scholar]
- 238.Boslooper K, King-Yin Lam A, Gao J, Weinstein S, Johnson N. The clinicopathological roles of alpha-B-crystallin and p53 expression in patients with head and neck squamous cell carcinoma. Pathology. 2008;40:500–504. doi: 10.1080/00313020802198010. [DOI] [PubMed] [Google Scholar]
- 239.Lam AK, Ong K, Ho YH. hTERT expression in colorectal adenocarcinoma: correlations with p21, p53 expressions and clinicopathological features. Int J Colorectal Dis. 2008;23:587–594. doi: 10.1007/s00384-008-0455-7. [DOI] [PubMed] [Google Scholar]
- 240.Lam KY, Chan AC, Chan KW, Leung ML, Srivastava G. Expression of p53 and its relationship with human papillomavirus in penile carcinomas. Eur J Surg Oncol. 1995;21:613–616. doi: 10.1016/s0748-7983(95)95262-4. [DOI] [PubMed] [Google Scholar]
- 241.Lam KY, Tsao SW, Zhang D, Law S, He D, Ma L, Wong J. Prevalence and predictive value of p53 mutation in patients with oesophageal squamous cell carcinomas: a prospective clinico-pathological study and survival analysis of 70 patients. Int J Cancer. 1997;74:212–219. doi: 10.1002/(sici)1097-0215(19970422)74:2<212::aid-ijc13>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 242.Li FP, Fraumeni JF, Jr, Mulvihill JJ, Blattner WA, Dreyfus MG, Tucker MA, Miller RW. A cancer family syndrome in twenty-four kindreds. Cancer Res. 1988;48:5358–5362. [PubMed] [Google Scholar]
- 243.Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty AR, DeLacerda L, Rabin M, Cadwell C, Sampaio G, Cat I, Stratakis CA, Sandrini R. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A. 2001;98:9330–9335. doi: 10.1073/pnas.161479898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Lam KY, Lo CY, Wat NM, Luk JM, Lam KS. The clinicopathological features and importance of p53, Rb, and mdm2 expression in phaeochromocytomas and paragangliomas. J Clin Pathol. 2001;54:443–448. doi: 10.1136/jcp.54.6.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Petri BJ, Speel EJ, Korpershoek E, Claessen SM, van Nederveen FH, Giesen V, Dannenberg H, van der Harst E, Dinjens WN, de Krijger RR. Frequent loss of 17p, but no p53 mutations or protein overexpression in benign and malignant pheochromocytomas. Mod Pathol. 2008;21:407–413. doi: 10.1038/modpathol.3801013. [DOI] [PubMed] [Google Scholar]
- 246.Robertson KD, Jones PA. Tissue-specific alternative splicing in the human INK4a/ARF cell cycle regulatory locus. Oncogene. 1999;18:3810–3820. doi: 10.1038/sj.onc.1202737. [DOI] [PubMed] [Google Scholar]
- 247.Bahuau M, Vidaud D, Jenkins RB, Bièche I, Kimmel DW, Assouline B, Smith JS, Alderete B, Cayuela JM, Harpey JP, Caille B, Vidaud M. Germ-line deletion involving the INK4 locus in familial proneness to melanoma and nervous system tumors. Cancer Res. 1998;58:2298–2303. [PubMed] [Google Scholar]
- 248.Aguiar RC, Dahia PL, Sill H, Toledo SP, Goldman JM, Cross NC. Deletion analysis of the p16 tumour suppressor gene in phaeochromocytomas. Clin Endocrinol (Oxf) 1996;45:93–96. [PubMed] [Google Scholar]
- 249.Dammann R, Schagdarsurengin U, Seidel C, Trumpler C, Hoang-Vu C, Gimm O, Dralle H, Pfeifer GP, Brauckhoff M. Frequent promoter methylation of tumor-related genes in sporadic and men2-associated pheochromocytomas. Exp Clin Endocrinol Diabetes. 2005;113:1–7. doi: 10.1055/s-2004-830522. [DOI] [PubMed] [Google Scholar]
- 250.Pillai S, Gopalan V, Smith RA, Lam AK. Updates on the genetics and the clinical impacts on phaeochromocytoma and paraganglioma in the new era. Crit Rev Oncol Hematol. 2016;100:190–208. doi: 10.1016/j.critrevonc.2016.01.022. [DOI] [PubMed] [Google Scholar]
- 251.Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature. 1997;386(761):763. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 252.Larsen MJ, Thomassen M, Gerdes AM, Kruse TA. Hereditary breast cancer: clinical, pathological and molecular characteristics. Breast Cancer (Auckl) 2014;8:145–155. doi: 10.4137/BCBCR.S18715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Mersch J, Jackson MA, Park M, Nebgen D, Peterson SK, Singletary C, Arun BK, Litton JK. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer. Cancer. 2015;2015121(121):269–275. 2474–2475. doi: 10.1002/cncr.29041. Erratum. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Barak F, Shiri-Svredlov R, Bruchim-Bar Sade R, Kruglikova A, Friedman E, Ben-Dor D, Goldberg I. Adrenal tumors in BRCA1/BRCA2 mutation carriers. Am J Med Genet. 2001;98:277–279. doi: 10.1002/1096-8628(20010122)98:3<277::aid-ajmg1082>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 255.Carbone M, Yang H, Pass HI, Krausz T, Testa JR, Gaudino G. BAP1 and cancer. Nat Rev Cancer. 2013;13:153–159. doi: 10.1038/nrc3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Greenberg RA, Sobhian B, Pathania S, Cantor SB, Nakatani Y, Livingston DM. Multifactorial contributions to an acute DNA damage response by BRCA1/BARD1-containing complexes. Genes Dev. 2006;20:34–46. doi: 10.1101/gad.1381306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Nishikawa H, Wu W, Koike A, Kojima R, Gomi H, Fukuda M, Ohta T. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res. 2009;69:111–119. doi: 10.1158/0008-5472.CAN-08-3355. [DOI] [PubMed] [Google Scholar]
- 258.Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, Windpassinger C, Wackernagel W, Loy S, Wolf I, Viale A, Lash AE, Pirun M, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Murali R, Wiesner T, Scolyer RA. Tumours associated with BAP1 mutations. Pathology. 2013;45:116–126. doi: 10.1097/PAT.0b013e32835d0efb. [DOI] [PubMed] [Google Scholar]
- 260.Wadt K, Choi J, Chung JY, Kiilgaard J, Heegaard S, Drzewiecki KT, Trent JM, Hewitt SM, Hayward NK, Gerdes AM, Brown KM. A cryptic BAP1 splice mutation in a family with uveal and cutaneous melanoma, and paraganglioma. Pigment Cell Melanoma Res. 2012;25:815–818. doi: 10.1111/pcmr.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Ratnakumar K, Bernstein E. ATRX: the case of a peculiar chromatin remodeler. Epigenetics. 2013;8:3–9. doi: 10.4161/epi.23271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D’Andrea K, Merrill S, Cho NW, Greenberg RA, Else T, Montone K, LiVolsi V, Fraker D, Daber R, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6140. doi: 10.1038/ncomms7140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Andreu-Vieyra CV, Chen R, Agno JE, Glaser S, Anastassiadis K, Stewart AF, Matzuk MM. MLL2 is required in oocytes for bulk histone 3 lysine 4 trimethylation and transcriptional silencing. PLoS Biol. 2010:8. doi: 10.1371/journal.pbio.1000453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Glaser S, Lubitz S, Loveland KL, Ohbo K, Robb L, Schwenk F, Seibler J, Roellig D, Kranz A, Anastassiadis K, Stewart AF. The histone 3 lysine 4 methyltransferase, Mll2, is only required briefly in development and spermatogenesis. Epigenetics Chromatin. 2009;2:5. doi: 10.1186/1756-8935-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Guo C, Chang CC, Wortham M, Chen LH, Kernagis DN, Qin X, Cho YW, Chi JT, Grant GA, McLendon RE, Yan H, Ge K, Papadopoulos N, et al. Global identification of MLL2-targeted loci reveals MLL2’s role in diverse signaling pathways. Proc Natl Acad Sci U S A. 2012;109:17603–17608. doi: 10.1073/pnas.1208807109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, et al. Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet. 2010;42:790–793. doi: 10.1038/ng.646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Juhlin CC, Stenman A, Haglund F, Clark VE, Brown TC, Baranoski J, Bilguvar K, Goh G, Welander J, Svahn F, Rubinstein JC, Caramuta S, Yasuno K, et al. Whole-exome sequencing defines the mutational landscape of pheochromocytoma and identifies KMT2D as a recurrently mutated gene. Genes Chromosomes Cancer. 2015;54:542–554. doi: 10.1002/gcc.22267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Hoeflich KP, Gray DC, Eby MT, Tien JY, Wong L, Bower J, Gogineni A, Zha J, Cole MJ, Stern HM, Murray LJ, Davis DP, Seshagiri S. Oncogenic BRAF is required for tumor growth and maintenance in melanoma models. Cancer Res. 2006;66:999–1006. doi: 10.1158/0008-5472.CAN-05-2720. [DOI] [PubMed] [Google Scholar]
- 269.Kumar SM, Dai J, Li S, Yang R, Yu H, Nathanson KL, Liu S, Zhou H, Guo J, Xu X. Human skin neural crest progenitor cells are susceptible to BRAF(V600E)-induced transformation. Oncogene. 2014;33:832–841. doi: 10.1038/onc.2012.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC. HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell. 2007;11:335–47. doi: 10.1016/j.ccr.2007.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Hensen EF, van Duinen N, Jansen JC, Corssmit EP, Tops CM, Romijn JA, Vriends AH, van der Mey AG, Cornelisse CJ, Devilee P, Bayley JP. High prevalence of founder mutations of the succinate dehydrogenase genes in the Netherlands. Clin Genet. 2012;81:284–8. doi: 10.1111/j.1399-0004.2011.01653.x. [DOI] [PubMed] [Google Scholar]