

Abstract
Introduction
BRM, a key catalytic subunit of the SWI/SNF chromatin remodeling complex, is a putative tumor susceptibility gene that is silenced in 15% of non-small cell lung cancer (NSCLC). Two novel BRM promoter polymorphisms (BRM-741, BRM-1321) are associated with reversible epigenetic silencing of BRM protein expression.
Methods
Advanced NSCLC patients from the Princess Margaret (PM) cohort study and from the CCTG BR.24 clinical trial were genotyped for BRM promoter polymorphisms. Associations of BRM variants with survival were assessed using log-rank tests, the method of Kaplan and Meier, and Cox proportional hazards models. Promoter swap, luciferase assays, and chromatin immunoprecipitation (ChIP) experiments evaluated polymorphism function. In silico analysis of publicly available gene expression datasets with outcome were performed.
Results
Carrying the homozygous variants of both polymorphisms (“double homozygotes”, DH) when compared with those carrying the double wild-type, was associated with worse overall survival, with an adjusted hazard ratios (aHR) of 2.74 (95%CI: 1.9–4.0). This was confirmed in the BR.24 trial (aHR 8.97, 95%CI: 3.3–18.5). Lower BRM gene expression (by RNA-Seq or microarray) was associated with worse outcome (p<0.04). ChIP and promoter swap experiments confirmed binding of MEF2D and HDAC9 only to homozygotes of each polymorphism, associated with reduced promoter activity in the DH.
Conclusion
Epigenetic regulatory molecules bind to two BRM promoter sequence variants but not to their wild-type sequences. These variants are associated with adverse overall and progression free survival. Decreased BRM gene expression, seen with these variants, is also associated with worse overall survival.
Keywords: BRM, chromatin remodeling, genetic polymorphism, lung cancer, prognosis
INTRODUCTION
Lung cancer is the leading cause of cancer deaths in the industrialized world, even in this new era of screening1,2. The majority of non-small cell lung cancer (NSCLC) patients present at an advanced stage for which treatment is met with limited success. However, with considerable inter-individual variability in lung cancer development, outcomes and treatment response, heritable polymorphisms may play roles in lung cancer susceptibility/risk3,4 and outcome5.
The SWI/SNF (SWItch/sucrose non-fermentable) complex is a multimeric chromatin remodeling complex that plays a key role in regulating multiple cellular processes, including gene expression, differentiation, DNA repair and cell cycle control6–9. The complex, consisting of a catalytic subunit with helicase ATPase activity (either Brahma [BRM] or BRM-related gene 1 [BRG1]) is required for the function of a variety of signal transduction pathways and anticancer protein activities (retinoblastoma [Rb], p53 and BRCA17,9–11). BRM regulates the expression of 4–7% of mammalian genes, many of which have anticancer roles, among them, vimentin, E-cadherin, N-cadherin, estrogen receptor, progesterone receptor, and CD449, 12–16.
BRM protein expression is lost in 15–40% of many primary solid tumors, including in 17–30% of NSCLC17,18. Unlike most tumor suppressor genes, however, BRM is reversibly epigenetically silenced18–20. Initially, pan-histone deacetylase (HDAC) inhibitors were identified as compounds that could restore BRM, but these agents were found to also inactivate BRM by inducing its acetylation21. As these compounds showed that BRM is regulated by HDACs, further analysis showed that BRM silencing is regulated specifically by Class 1 HDAC (HDAC3) and Class 2 HDAC (HDAC9) enzymes. Further, HDAC9 is overexpressed in both BRM-deficient (>500 fold) cancer cell lines and (>20 fold) primary tumors22,23.
To determine how BRM was silenced, we previously sequenced the BRM promoter in cancer cell lines and identified the presence of two insertional/deletional polymorphisms (BRM-741:TTAAA) and (BRM-1321:TATTTTT) that correlated strongly with loss of BRM protein expression in cancer cell lines, and confirmed in primary lung cancers. The variant insertion alleles of both polymorphisms produce sequence variants that are highly homologous to myocyte enhancer factor-2 (MEF2) transcription factor binding sites, where MEF2 is known to recruit HDACs and silence genes24. shRNA knockdown of HDAC9, MEF2D in BRM-deficient cell lines results in the induction of BRM24,25. BRM demonstrates attributes of a tumor susceptibility gene, as the BRM-null mouse does not develop spontaneous tumors, but shows distinct abnormalities in cell cycle control; when combined with a carcinogen, tumor development is potentiated18. We reasoned, therefore, that the promoter insertion variants may be associated with cancer risk, and have confirmed that individuals carrying both BRM homozygous promoter insertion variants have a two-fold increase in the risk of lung cancer24, particularly early stage lung cancer25, and in other cancers26–28.
Prognostically, loss of BRM protein expression has been linked to adverse outcome across a varied mix of NSCLC stages17,29. However, the relationships between BRM promoter polymorphisms, BRM gene expression, and outcome have not been documented previously. Potential prognostic implications are important, as involvement of the BRM pathway in tumors may identify a subset of individuals with worse outcome that potentially could benefit from focused development of a class of drugs targeting BRM re-expression. That the BRM polymorphisms are associated with epigenetic factors is unique, but even more so when such epigenetic silencing has been reversed by a variety of compounds and drugs such as certain non-steroidal anti-inflammatory drugs (indoprofen), and flavonoids (genistein)30,31.
As cancer drug development focuses on advanced disease, we undertook this study to evaluate the role of these polymorphisms on survival outcomes in two independent patient cohorts. However, we first evaluated the functional significance of the polymorphisms through promoter swap and chromatin immunoprecipitation (ChIP) experiments; these analyses can provide important adjunctive evidence supporting a potential clinical role of this gene and specifically these two polymorphisms. Finally, we assessed the significance of BRM gene expression on clinical outcome, using publicly available databases, to provide the first evidence of a link between our putative biological mechanism and the associations found in this study.
METHODS
Study Cohorts
The study was approved by the institutional research ethics board and by the Lung Cancer Correlative Science and Tissue Banking Subcommittee of the Canadian Cancer Trials Group. Figure 1 shows the flow of patient specimens in the polymorphism-survival analyses.
Figure 1. CONSORT diagram of the Princess Margaret (PM) Cohort (left panel) and the Canadian Cancer Trials Group, BR.24 trial (right panel).
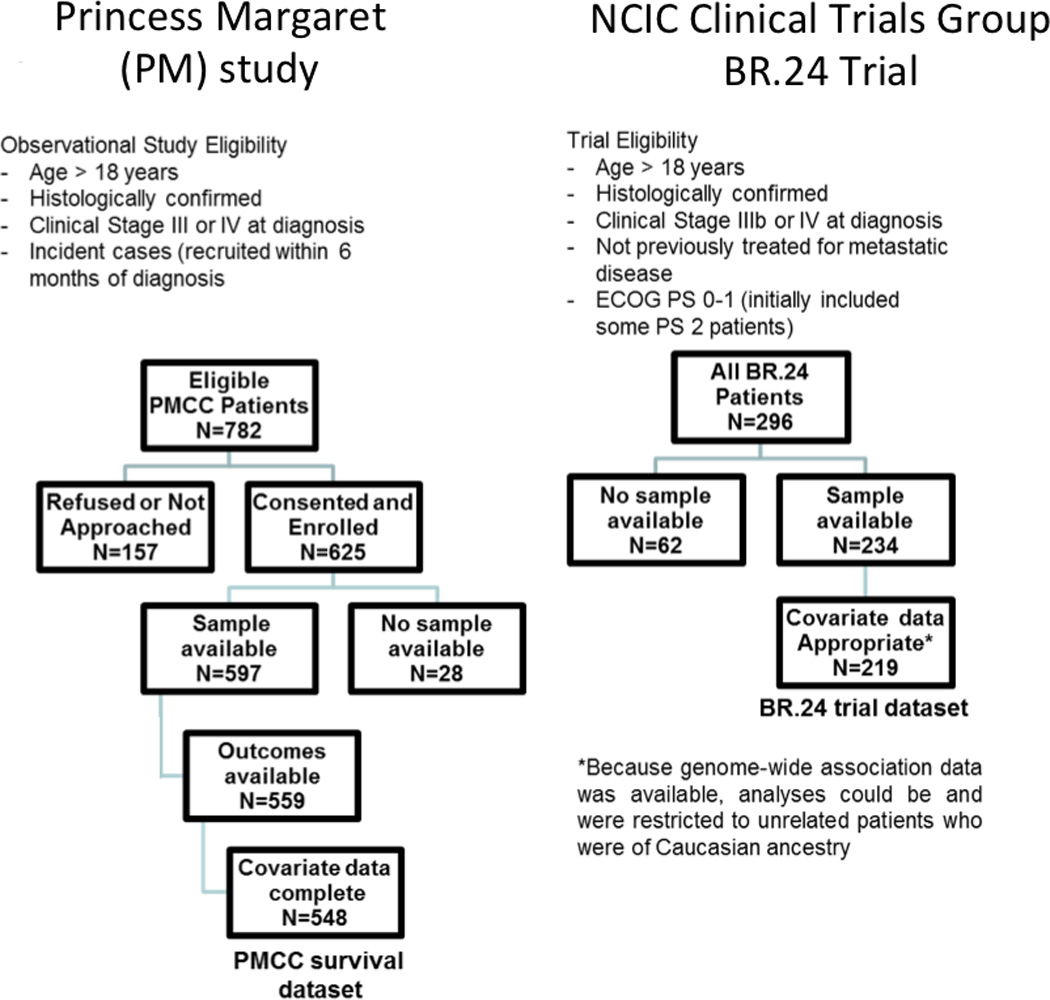
Princess Margaret (PM) Cohort Study
Cases were incident (diagnosis ≤ 6 months prior to enrollment) stage III-IV NSCLC patients who participated in a prospective study evaluating the molecular epidemiology of lung cancer at the PM Cancer Centre, Toronto, between 2006–2010. Eligibility included histological confirmation of NSCLC, and provision of written consent; exclusions were inability to communicate in English and cognitive deficits interfering with ability to understand consent. For cohort analysis of survival, cases were derived from the same underlying pool of advanced, incurable Stage III-IV NSCLCs as in the case-control analysis, with the following exceptions: smoking status was no longer part of the eligibility criteria, and all adult patients (≥ 18 year old) were eligible. Available treatment, covariate, and survival data for at least 6 months after diagnosis was an additional inclusion criterion.
BR.24 Clinical Trial
The Canadian Cancerl Trials Group (CTG) led the BR.24 clinical trial, a randomized double blinded, international Phase II trial that evaluated carboplatin (area under the curve, AUC 6) and paclitaxel (200 mg/m2) every three weeks for 6–8 cycles in combination with either daily oral cediranib or placebo in the first-line treatment of advanced stage IIIB-IV NSCLC patients, conducted between 2005 and 2008. Cediranib/placebo monotherapy afterwards in the absence of intolerable toxicity or disease progression was allowed. A pre-planned interim analysis determined an imbalance in the number of investigator-designated cause of deaths, and the trial was terminated without entering the Phase III portion.
DNA extraction and Genotyping
Germline DNA was extracted from the lymphocytes of whole blood of all patient cohorts using a commercially available DNA isolation kit (5Primer, Cat#2300740). Genotyping for BRM-741 and BRM-1321 was performed using two custom-designed Taqman assays24,25. For quality control, positive and negative controls and blinded duplicate samples were included.
Chromatin Immunoprecipitation (ChIP), Promoter Luciferase and Swap experiments
To demonstrate the proteins that bind the BRM polymorphic sequences, and altered promoter activity levels by these polymorphic variant, ChIP, promoter luciferase and swap experiments were performed. The insertion variants of the BRM polymorphisms are MEF2 binding sites (with >92% homology)24. Upon binding, MEF2D recruits class II HDACs, specifically HDAC9, which results in targeted gene silencing32,33. To demonstrate this, ChIP experiments were performed to determine specificity of MEF2D/HDAC9 binding to BRM insertion alleles, and not to wild-type deletion alleles. To further demonstrate that the specificity is polymorphism-dependent and not cell-line dependent, isogenic BRM promoter constructs (forming double-homozygous and double-wild-type genotypes) were placed stably in the SW13 cell line via homologous recombination. In these constructs (Figure 3), a luciferase reporter IRES-neomycin gene was inserted near exon 2 of the BRM gene, which effectively disrupted BRM expression, and allowed the inserted luciferase reporter gene to be under the control of the BRM promoter. These isogenic promoter swapped constructs were stably integrated into the SW13 cell line, and then assessed using ChIP assays. These same constructs were then used to determine the relative level of promoter activity through measurement of luciferase.
Figure 3. Functional assays.
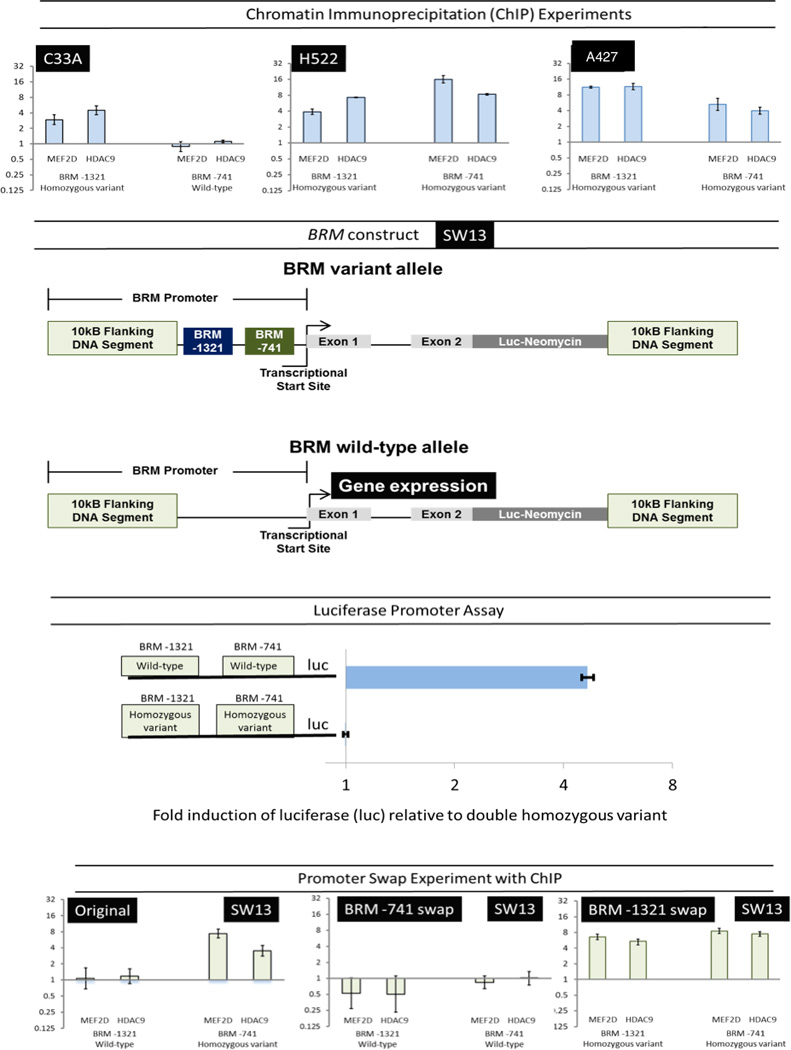
Top panel: Chromatin immunoprecipation (ChIP) experiments demonstrate that binding of MEF2D and HDAC9 occurs only in the presence of the homozygous variants (P<0.001; Student’s t-test) and not the wild-types (P>0.05). Vertical axes show the binding affinity relative to background binding by non-specific IgG in the C33A (cervical carcinoma), H552 (lung adenocarcinoma), and A547 (lung squamous carcinoma) cell lines (log-transformed ratio + standard error). Second panel: A BRM reporter construct was placed in the SW13 (adrenal cortical carcinoma) cell line via homologous recombination, where a luciferase (luc) gene linked to an IRES-(internal ribosime entry site)-neomycin gene was placed at the beginning of the transcriptional start site; the endogenous BRM gene is disrupted such that it is no longer expressed. Instead, the luciferase gene, now under the control of the BRM promoter is expressed as a measure of the BRM promoter activity. Third panel: We obtained a number of these SW13 daughter cell lines (with BRM reporter constructs) derived from single cells, by dilutional cloning both with and without these polymorphisms. By comparing luciferase activity from six clonal SW13 daughter cell lines (three each) which either did or did not harbor these BRM insertion variants, a five-fold higher luciferase activity was observed in the daughter cell lines harboring the BRM promoter wild-type construct when compared with the cell lines harboring the homozygous variants (P<0.001). Bottom panel: Using the same isogenic SW13 cell lines, ChIP evaluation of the swapped promoters demonstrated that MEF2D and HDAC9 bind only to the variant alleles (log-transformed data; P<0.001), and not to the wild-type alleles (P>0.05). Axes are the same as top panel.
Statistics
For patient cohorts, baseline demographic and clinico-pathological data were described and cross-tabulated. Additionally, for the BR.24 trial, baseline demographic, clinical information, and survival outcome were compared between individuals with data available for genetic analysis and those without. Departure from Hardy–Weinberg equilibrium (HWE) was tested using the Pearson chi-square test. All models assumed co-dominant genetic inheritance for both polymorphisms, and combined polymorphism analyses were performed using previously described categorizations24,25.
Survival was defined as the time from date of pathological diagnosis of stage III/IV (PM cohort) or randomization (BR.24 cohort) to either the date of death from any cause (overall survival, OS), or date of first disease progression/recurrence or death from any cause (progression free survival, PFS). Patients were censored when they were last known to be alive (for OS) and last assessed for progression/recurrence (for PFS).
Survival rates and median survival times were estimated using the Kaplan–Meier method. Cox proportional hazards models and the log rank test were used to test associations between the sequence variants and survival. Multivariate models were constructed with genetic markers after adjusting for individual covariates found to be associated with survival (at the p<0.10 level). P values, adjusted Hazard Ratios (aHR) and their 95% confidence intervals (95% CI) for survival were reported. All statistical tests were two-sided and were conducted using the SAS software (version 9.2, SAS Institute Inc.) and the R software (version 2.11.0, R Development Core Team). The Wald test was used for all the genetic models of inheritance. A two-sided p-value of less than <0.05 was considered significant.
For ChIP experiments, protein-DNA sequence binding affinity results were reported as specific anti-HDAC9 and anti-MEF2D antibody binding, normalized against background, non-specific IgG, which resulted in a measurement of fold-change (against the background). Log-transformed triplicate fold-change data were analysed using Student t-tests. Similar fold-change data were analysed for the promoter luciferase activity, comparing different promoter constructs.
Gene Expression Analysis of Publicly available databases, we used a publicly available tool (www.kmplot.com/lung) to evaluate the prognostic effect of low BRM (gene symbol SMARCA2) gene expression on OS in patients with NSCLC, using a lung cancer dataset encompassing 1715 samples (1405 NSCLC) with gene expression and survival data from 10 independent studies (January, 2015); analyses compared the bottom quartile of BRM expression with the top three quartiles. RNA-Seq lung cancer data was available from TCGA (n=431 adenocarcinomas; n=323 squamous cell carcinomas), and survival analysis by quartiles was performed. In all cases, the method of Kaplan-Meier was used, and log-rank tests performed.
RESULTS
Patient Cohort Demographics
Patient demographics for each study cohort are shown in Table 1. Patients from BR.24 included only those suitable for first line chemotherapy, rather than surgery or combined chemotherapy and radiation. Patients from the PM cohort included primarily unresectable patients. compared with BR.24 participants, PM cases were more likely to have adenocarcinoma and have a higher fraction of the Stage III patients. When comparing BR.24 patients analyzed with those not-analyzed due to lack of sample, excluded patients were more likely of later stage, have adenocarcinoma, and have unknown weight loss prior to randomisation, leading to decisions to adjust for these variables in the final BR.24 model. Despite these differences, the analysed population had similar survival outcomes and derived similar benefit from treatment compared to the total BR.24 cohort (test of homogeneity between treatment arm and availability of bio-specimens p=0.93). In comparison, the analysed and non-analyzed PM cohorts, however, were very similar (Supplemental Table 1).
Table 1.
Clinico-demographic and Genotyping information
| Survival Analysis | Comparison | ||||
|---|---|---|---|---|---|
| Characteristic | Category | PM Cohort | BR.24 Cohort Analyzed |
BR.24 Not Analyzed |
Analyzed vs Not Analyzed p-value |
| Total Number | 548 | 219 | 77 | ||
| Age (years) | Median (range) | 63 (31–90) | 60 (38–82) | 57 (36–72) | 0.10 |
| Sex | Male/Female | 52%/48% | 59%/41% | 56%/44% | 0.74 |
| Ethnicity | Caucasian/Asian/Other | 67%/15%/18% | Not reported3 | Not reported | |
| Smoking Status | Current/Former/Never-smoker | 30%/44%/26% | Not reported | Not reported | |
| Pack-Years1 | Median (Smokers) | 38 (1–216) | Not reported | Not reported | |
| ECOG Perfor- mance Status |
0/1–2/3+ | 15%/84%/1% | 23%/77%/0 | 27%/73%/0 | 0.59 |
| Weight Loss | ≥ 5%/< 5%/Unknown | Not reported | 28%/58%/13% | 18%/39%/43% | <0.001 |
| Histological | Adenocarcinoma | 67% | 46% | 64% | 0.01 |
| Subtype | Squamous Cell | 16% | 26% | 12% | |
| Other | 17% | 28% | 25% | ||
| Stage | III /IV | 46%/54% | 17%/83% | 6%/94% | 0.02 |
| Platinum Agent | Cisplatin/carboplatin based therapy | 82% | 100% | 100% | |
| No-platinum chemotherapy | 10% | 0% | 0% | ||
| Neither/Unknown | 8% | 0% | 0% | ||
| Follow-up Time | Median (months) | 42 | 18 | 17 | 0.91 |
| Overall Survival | Median (months) | 19 | 9 | 10 | 0.64 |
| Time to randomization2 |
< 6 months / ≥ 6 months | 89%/11% | 91%/9% | 0.89 | |
| Dose Cohort | 30 mg/ 45 mg of cediranib/placebo | 84%/16% | 86%/14% | 0.94 | |
| Treatment Arm | Conventional (placebo) Arm/ Experimental (cediranib) Arm |
50%/50% | 51%/49% | 0.99 | |
calculated in ever-smokers only;
from date of diagnosis;
majority are expected to be of Caucasian descent.
BRM Polymorphisms and Survival Analyses
Both polymorphisms were in Hardy Weinberg Equilibrium (p>0.05), and in mild linkage disequilibrium (D’= 0.48 for PM cohort, and 0.39 for BR.24 trial). Genotyping was successful in >99.8% of cases. Although ~20% carried at least one homozygous variant of these two polymorphisms, 11–15% of patients carried homozygous variants of both polymorphisms (termed the “double homozygote” or DH). Because of a higher proportion of Stage III patients and the inclusion of unresectable Stage IIIA patients, the median OS for the PM cohort is substantially higher than that of BR.24 (Table 1).
Table 2 (OS), Table 3 (PFS), and Figure 2 (Kaplan-Meier curves) report strongly significant survival relationships with the individual polymorphisms, and further specifically with the DH variants of these two BRM polymorphisms (when compared with the double-wildtype polymorphisms). The strongest survival relationship was found in the placebo (conventional therapy) arm of BR.24, where there was uniform treatment of all patients with carboplatin-paclitaxel, and adjusted hazard ratios exceeded 8 (PFS) and 16 (OS). Subgroup prognostic analyses (Supplementary Table 2) showed similar results in never-smokers and smokers, in patients with adenocarcinomas vs. squamous cell carcinomas, by age and gender, for both the PM cohort and the BR.24 trial participants. For the PM cohort, subset analysis of the 82% of patients treated with platinum-based chemotherapy, found virtually identical survival associations with these two BRM polymorphisms as the entire PM cohort; further subset analyses found consistent associations regardless of whether the therapy was cisplatin or carboplatin-based doublet therapy.
Table 2.
BRM polymorphisms and overall survival in advanced stage non-small cell lung cancer
| Poly- morphism |
Category | PM Cohort (N=548) |
BR.24 Conventional Arm (N=109) |
BR.24 Experimental Arm (N=110) |
BR.24 All Patients (N=219) |
||||
|---|---|---|---|---|---|---|---|---|---|
| % | aHR (95%CI)1 | % | aHR (95%CI)2 | % | aHR (95%CI)2 | % | aHR (95%CI)2 | ||
| Wild-type | 29% | Reference | 30% | Reference | 27% | Reference | 28% | Reference | |
| Heterozygote | 48% | 1.60 (1.2–2.2) | 43% | 2.81 (1.5–5.4) | 49% | 2.21 (1.1–4.5) | 45% | 2.75 (1.7–4.4) | |
| BRM -741 | Homozygote | 23% | 2.49 (1.9–3.3) | 29% | 5.52 (2.7–11.2) | 25% | 3.91 (1.8–8.4) | 27% | 4.98 (3.0–8.3) |
| P value3 | <0.001 | <0.001 | 0.002 | <0.001 | |||||
| Minor allele frequency |
46% | 50% | 49% | 50% | |||||
| Wild-type | 34% | Reference | 28% | Reference | 30% | Reference | 29% | Reference | |
| Heterozygote | 44% | 1.18 (0.9–1.5) | 55% | 3.14 (1.6–6.0) | 46% | 2.01 (1.1–3.8) | 50% | 2.53 (1.6–3.9) | |
| BRM -1321 | Homozygote | 22% | 1.99 (1.5–2.7) | 18% | 7.64 (3.5–16.7) | 24% | 3.54 (1.8–7.1) | 21% | 4.98 (3.0–8.3) |
| P value3 | <0.001 | <0.001 | 0.002 | <0.001 | |||||
| Minor allele Frequency |
44% | 44% | 46% | 45% | |||||
| Combined Poly- morphisms |
Double Wild-type | 21% | Reference | 19% | Reference | 18% | Reference | 19% | Reference |
| No homozygote | 47% | 1.40 (1.0–1.8) | 46% | 2.90 (1.3–6.7) | 48% | 3.46 (1.3–9.3) | 47% | 3.40 (1.8–6.3) | |
| One Homozygote | 21% | 2.17 (1.6–3.1) | 24% | 5.05 (2.09–12.3) | 18% | 9.26 (3.2–27.0) | 21% | 6.62 (3.4–13.1) | |
| Two homozygotes | 12% | 2.74 (1.9–4.0) | 11% | 16.8 (6.0–46.7) | 15% | 6.36 (2.2–19.1) | 13% | 8.97 (3.3–18.5) | |
| P value3 | <0.001 | <0.001 | 0.001 | <0.001 | |||||
adjusted for sex, stage, ECOG performance status at diagnosis, number. of lines of systemic therapy received, and prior surgical resection of lung primary or site of oligometastases;
adjusted for sex, stage, ECOG performance status, weight loss, and time from diagnosis to randomization; in the “all patients” analysis, there was also stratification by treatment arm.
p-values are from Cox proportional hazard models comparing BRM polymorphisms and survival.
PM – Princess Margaret
Table 3.
BRM polymorphisms and progression-free survival in advanced stage non-small cell lung cancer
| Poly- morphism |
Category | PM Cohort (N=548) |
BR.24 Conventional Arm (N=109) |
BR.24 Experimental Arm (N=110) |
BR.24 All Patients (N=219) |
||||
|---|---|---|---|---|---|---|---|---|---|
| % | aHR (95%CI)1 | % | aHR (95%CI)2 | % | aHR (95%CI)2 | % | aHR (95%CI)2 | ||
| Wild-type | 29% | Reference | 30% | Reference | 27% | Reference | 28% | Reference | |
| Heterozygote | 48% | 1.57 (1.2–1.9) | 43% | 1.63 (0.9–3.0) | 49% | 1.45 (0.8–2.7) | 45% | 1.69 (1.1–2.6) | |
| BRM -741 | Homozygote | 23% | 2.05 (1.6–2.7) | 29% | 3.75 (1.9–7.4) | 25% | 2.25 (1.1–4.5) | 27% | 3.02 (1.9–4.8) |
| P value3 | <0.001 | <0.001 | 0.07 | <0.001 | |||||
| Minor allele frequency |
46% | 50% | 49% | 50% | |||||
| Wild-type | 34% | Reference | 28% | Reference | 30% | Reference | 29% | Reference | |
| Heterozygote | 44% | 1.22 (1.0–1.5) | 55% | 2.04 (1.1–3.7) | 46% | 1.10 (0.6–2.0) | 50% | 1.48 (1.0–2.2) | |
| BRM -1321 | Homozygote | 22% | 1.86 (1.5–2.4) | 18% | 3.37 (1.6–7.2) | 24% | 1.96 (1.0–3.9) | 21% | 2.57 (1.6–4.2) |
| P value3 | <0.001 | 0.006 | 0.07 | <0.001 | |||||
| Minor allele Frequency |
44% | 44% | 46% | 45% | |||||
| Combined Poly- morphisms |
Double Wild-type | 21% | Reference | 19% | Reference | 18% | Reference | 19% | Reference |
| No homozygote | 47% | 1.52 (1.2–1.9) | 46% | 1.71 (0.8–3.7) | 48% | 1.27 (0.6–2.7) | 47% | 1.62 (1.0–2.7) | |
| One Homozygote | 21% | 1.86 (1.4–2.5) | 24% | 2.87 (1.3–6.6) | 18% | 2.96 (1.2–7.0) | 21% | 3.11 (1.7–5.6) | |
| Two homozygotes | 12% | 2.71 (1.9–3.8) | 11% | 8.30 (3.2–21.9) | 15% | 2.08 (0.8–5.3) | 13% | 3.75 (1.9–7.3) | |
| P value3 | <0.001 | 0.001 | 0.02 | <0.001 | |||||
adjusted for sex, stage, ECOG performance status at diagnosis, number. of lines of systemic therapy received, and prior surgical resection of lung primary or site of oligometastases;
adjusted for sex, stage, ECOG performance status, weight loss, and time from diagnosis to randomization; in the “all patients” analysis, there was also stratification by treatment arm.
p-values are from Cox proportional hazard models comparing BRM polymorphisms and survival.
PM - Princess Margaret.
Figure 2. Kaplan Meier survival curves by BRM polymorphisms and BRM gene expression.
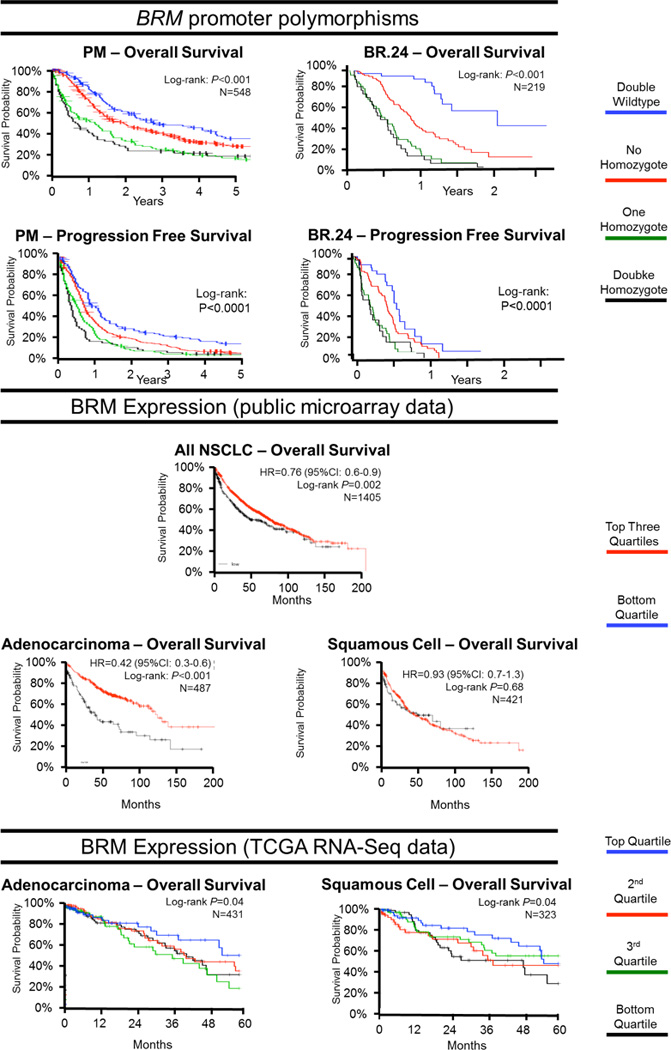
P values are derived from log-rank tests. Where reported, hazard ratios (HRs) with 95% confidence intervals are from univariable Cox proportional hazard models. N = number of patients assessed in each analysis. Top panel: Overall and progression-free survival of Princess Margaret (PM) Cohort (463/548 (84%) had died at a median follow-up of 42 months) and BR.24 trial patients (153/219 (70%) had died at a median follow-up of 18 months) by BRM polymorphism combinations. Middle Panel: Overall survival curves of non-small cell lung cancer (NSCLC) patients by BRM gene expression levels in tumor tissue from ten publicly available microarray gene expression datasets (www.kmplot.com/lung). Bottom panel: The Cancer Genome Atlas (TCGA) lung adenocarcinoma and squamous cell carcinoma datasets using RNA-Seq data (February, 2014). To avoid duplication of results, 67 squamous cell carcinoma cases were removed from the TCGA analysis, as these samples were already included in the microarray gene expression analysis.
Chromatin Immunoprecipitation (ChIP) and Promoter Swap Experiments
Figure 3 shows ChIP experiments of representative cell lines (including several lung cancer cell lines) carrying different BRM polymorphism combinations. In brief, of the cell lines evaluated, all BRM wild-type genotypes resulted in little to no binding of MEF2/HDAC9, while there was significant binding observed in all homozygous variants (p<0.0001, all comparisons), including homozygous variants created through the promoter construct swap experiment of the SW13 cell line. In addition, the same promoter constructs demonstrated a 7.8-fold increased luciferase expression when these insertion alleles are absent as compared to when they are present (Figure 3), thereby demonstrating that these BRM polymorphic variants bind specific proteins, and are functionally involved in regulating BRM expression.
BRM Gene Expression and Survival
Gene expression data from publicly available sources of expression microarrays (Figure 2, middle panels) and RNA-Seq (Figure 2, right panels) of mainly early and locally-advanced stage NSCLC are presented. Significant association was observed between lower BRM expression levels and poorer survival in expression microarray data, with a HR of 0.42 (95%CI: 0.32–0.84); p=0.02). lower BRM expression was associated inversely with prognosis consistently in adenocarcinomas regardless of the method of assessing BRM gene expression: from RNA-Seq data, p=0.04 and expression microarray data, p=0.01. In contrast, two different sets of data obtained through two different assessments of BRM gene expression differed on the relationships with survival in squamous cell carcinoma, where there were significant associations in the RNA-Seq data (p=0.04) but not in expression microarray data (p=0.68). Separate analyses of expression microarray data by smoking status suggested that BRM expression was strongly associated with NSCLC in never-smokers but not in smokers; however, further multivariable analyses determined that this difference was largely driven by histology (p<0.02) rather than smoking status (p=0.62).
DISCUSSION
Two novel BRM promoter insertion deletion polymorphisms, BRM-741 and BRM-1321, are linked to epigenetic silencing of BRM expression and have been linked to lung cancer risk24,25. The presence of both homozygous variants strongly correlates with loss of BRM expression in lung cancer, a loss that, in turn, has been linked to adverse clinical outcomes17,29,34. Whilst previously we had documented these polymorphisms as being associated with risk of early stage NSCLC27, in the present analysis, we further describe that individuals carrying the homozygous variants of one or both polymorphisms had substantially worse survival outcomes compared to their wild-type counterparts in two independent datasets, one an observational cohort, and the other, a clinical trial. We performed molecular experiments to explain the functional significance of these polymorphisms, demonstrating that the presence of these polymorphic variants is physically connected to binding of regulatory proteins important in BRM expression. Finally, we reported that BRM gene expression was also prognostic for NSCLC survival, through analysis of publicly available data. Each of these findings incrementally builds on our understanding of these BRM polymorphisms and its clinical relevance. These findings include the mechanism of polymorphism function, and consistent findings across DNA and RNA levels of BRM with prognosis, compatible with proposed mechanisms of BRM function. Such cumulative evidence supports the importance of BRM in NSCLC outcome.
BRM germline polymorphisms may be useful not only for risk stratification, but also for prognostication. This is of importance since our prior research suggests that BRM is a potentially druggable target21. In one clinical setting, CT screening of lung cancer may benefit from further refinement by incorporation of molecular risk markers35. Yet in a contrasting set of circumstances, other markers such as somatic ALK translocations already help identify patients with aggressive (poor prognosis) tumors that respond well to drugs that target the rearranged ALK protein36. If a somatic genomic alteration such as ALK can lead to drug targets, perhaps the concept of a germline epigenetic target may not be so far-fetched. In the past, a barrier to adoption of polymorphic variants in risk/prognostic models has been a lack of clear biological or functional significance of such polymorphisms, a flaw that is addressed in this study. In fact, putting all the available evidence from the literature and the results of this study together, truly functional consequences of these polymorphisms may affect tumor biology and drug sensitivity, whilst our prior research suggested BRM is a druggable target21,24.
To better understand why these polymorphisms might underlie clinically relevant associations, we examined potential roles of BRM in cancer. Loss of BRM expression has been implicated in the progression of lung adenocarcinoma into solid predominant tumors with features of epithelial mesenchymal transition (EMT), which is consistent the fact the SWI/SNF regulates key genes involved in EMT changes such as E-cadherin, N-cadherin and vimentin, and loss of bronchial epithelial phenotype34. Similarly, a link between BRM loss and cancer progression has been suggested among patients with gastric37 and skin cancer38. Loss of BRM protein expression has been linked to poorer outcome among patients with NSCLC17,29, and other cancers39.
BRM can impact cancer evolution in a number of ways. Its functional linkage to such proteins as Rb and p53 may in part explain how BRM silencing can result in the loss of cellular growth control, a central mechanism by which BRM is thought to potentiate the risk of cancer12, 40,41. However, BRM and SWI/SNF are also involved in DNA repair, and the aberration of DNA repair proteins such as GADD45, BRCA1 and p53, functionally linked to BRM and SWI/SNF, are known to potentiate cancer development10,42. Loss of BRG1 and BRM has been linked to the epithelium-mesenchymal transition (EMT), thought to be a major step towards more aggressive disease; BRM and SWI/SNF have been linked to E-cadherin, N-cadherin, vimentin, CD44, CEACAM1, and integrin expression, which are also linked to EMT12,34,43,44. In addition, SWI/SNF in general has been shown to play roles in cellular differentiation and organ development45–49. Interestingly, the SWI/SNF complex may also increase sensitivity to cisplatin50 but increase resistance to EGFR inhibitors. Indeed, there is no shortage of feasible mechanisms by which BRM loss could impact cancer development and clinical outcomes.
Studies to evaluate lung cancer risk or prognosis as a function of a given germline polymorphism have been pursued vigorously. Prototypical polymorphisms that lie within the coding region of a protein are surmised to alter the protein's function by changing its primary sequence. This change in protein sequence alters the function of the protein, the cell, and the organism, thus yielding a significant clinical association. When these polymorphisms are not specifically related to a protein sequence, the value of a given polymorphism is strengthened by evidence showing that it impacts on gene function. To this end, the ChIP and luciferase data presented herein show that two key BRM regulating proteins, MEF2D and HDAC9, bind in or near the polymorphisms, in the presence of the variant insertion allele. Luciferase assays suggest that the BRM variants are also associated with lower BRM expression.
In our study design, the consistency of our BRM findings across different study populations of risk24,25 and prognosis (herein) is a strength of our findings. Limitations are present, though. Firstly, because we lacked sufficient quantities of research tumor specimens in our advanced stage cohorts, we were unable to perform matched polymorphism-gene expression-protein expression assessments of BRM. Previously published protein expression-outcome analyses17,29 and the current gene expression-outcome analyses confirm a consistent association with clinical outcome, but utilized samples from earlier stage NSCLC patients. Gene expression analysis of public databases may be confounded by differences in stage and other prognostic variables, possibly explaining the significant relationship in squamous cell carcinoma by RNA-Seq but not by expression microarray. Secondly, we are pursuing a parallel analysis of these polymorphisms in a randomized clinical trial of adjuvant cisplatin-chemotherapy in early stage NSCLC, where we will be able to answer questions of potential BRM-platinum drug interactions50 (all BR.24 patients were treated with carboplatin), and to evaluate a uniformly cisplatin-treated patient cohort. Nonetheless, the PM cohort was treated with a platinum doublet in 82% of patients, of which 61% were cisplatin-based doublets (primarily cisplatin-etoposide for Stage III and cisplatin-vinorelbine for Stage IV), where subset analyses found similar results regardless of use of platinum agents or the specific platinum agent itself. Thirdly, the interplay between EGFR, ALK, and other somatic changes with BRM on prognosis is unknown; we had too few patients with these molecular alterations to perform molecular-specific analyses of BRM relationships. We also acknowledge that the dual roles of BRM polymorphisms on risk and prognosis may not be consistent: in other tumours and settings, a risk association may not translate into a survival association, and vice versa; evaluation in each specific cancer disease site is necessary51. We did not have the power or tumor samples to explore the effect of these polymorphisms in difference milieu (e.g. different BRG expression patterns, modifying effect of other chromatin remodeling members). In the TCGA analysis, survival data could have been confounded by other known prognostic variables (e.g. stage); however, TCGA clinical data quality is heterogeneous, and thus these data are offered only as weak supporting evidence. Nonetheless, the totality of our findings have opened additional routes of exploration.
In summary, the same two BRM promoter polymorphisms that were previously associated with increased risk of developing NSCLC are also strongly associated with adverse survival in two different advanced NSCLC patient samples. This is a clinically important discovery given the high magnitude of prognostic association found, with values that could conceivably lead to clinically meaningful germline molecular prognostication. Further, pharmacological reversal of the epigenetic silencing of BRM has been shown to be a potentially viable therapeutic strategy18,21. With one in five patients homozygous for either BRM-741 or BRM-1321, and one in ten homozygous for both, a sizeable population of NSCLC may benefit either from future targeted therapy or prognostic stratification to allow decision-making on utilizing more aggressive or different therapeutic strategies. Additional studies are needed to further validate our study findings in other lung cancer populations, specifically the relationships between these polymorphisms and somatic lung cancer molecular alterations, and between these polymorphisms and other members of these chromatin remodelling complexes.
Supplementary Material
Statement of Translational Relevance.
The role of germline biomarkers of chromatic remodeling has not been well defined. The identification of two promoter polymorphisms in the BRM gene has focused previously on lung cancer risk associations. In this study, we first demonstrate using expression data and functional assays, the potential importance of this pathway and these specific polymorphisms to the clinical outcomes of lung cancer patients. We demonstrate a novel and consistent prognostic association of these polymorphisms with overall and progression-free survival across clinical trial and observational datasets. These results help identify high-risk poor prognosis patients that potentially could benefit from future targeting of this pathway by reversing the epigenetic phenomenon, a process we have demonstrated previously using in vitro models.
Acknowledgments
FINANCIAL SUPPORT
Canadian Cancer Trials Group conduct of this trial was supported by funding received from the Canadian Cancer Society Research Institute (Grant #021039 and #015469) and through a contract with AstraZeneca. Pharmacogenetic analyses were also supported by the Ontario Institute for Cancer Research High Impact Clinical Trials Program, and the Cancer Care Ontario Chair in Experimental Therapeutics and Population Studies (to G.L.). Support was also provided by the Princess Margaret Cancer Centre Lusi Wong Early Detection of Lung Cancer Program, Alan Brown Chair in Molecular Genomics, and the Posluns Family Fund. The functional evaluations were supported by 1R01CA136683-01A1 (PI Reisman) and 1R01CA127636-01 (PI Reisman).
We acknowledge the support of Alan B. Brown Chair in Molecular Genomics (to G.L.), the Scott Taylor Chair in Lung Cancer Research (to F.A.S.), the Ontario Institute for Cancer Research High Impact Clinical Trials Network (to G.L.), the OSI Pharmaceuticals Foundation Chair in Cancer New Drug Development (to N.L.), the Qasim Choksi Chair of Lung Cancer Translational Research (to M.S.T.), and Rachel Dresbeck for her contribution in editing. This work was supported by the Canadian Cancer Trials Group Tumour Tissue Data Repository (TTDR), and in particular, Shakeel Virk and Lois E. Shepherd. The CCTG TTDR is a member of the Canadian Tumour Repository Network. We also acknowledge use of data from the The Cancer Genome Atlas (TCGA) lung adenocarcinoma and lung squamous cell carcinoma projects.
Geoffrey Liu has received honoraria for advisory boards for AstraZeneca, Roche, Novartis, Pfizer, EMD-Serono, Millennium and Takeda. Ming-Sound Tsao has received honoraria for advisory boards for Pfizer, AstraZeneca, Hoffman LaRoche, Bristol-Meiers Squibb and Merck Canada, and has received research grant funding from Pfizer and AstraZeneca. Natasha B. Leighl has institutional funding from Novartis, and has received honoraria from AstraZeneca. Frances A. Shepherd has received honoraria for advisory boards for AstraZeneca, Boehringer Ingelheim, Bristol-Meiers Squibb, Eli Lilly, Merck Canada, Pfizer and Roche-Genentech, has received payment for consulting for Eli Lilly, and holds investments in Eli Lilly and AstraZeneca. Penelope Bradbury has received honoraria for advisory boards for Pfizer.
Footnotes
Conflicts of Interest:
For the remaining authors none was declared.
REFERENCES
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016;66:7–30. doi: 10.3322/caac.21332. [DOI] [PubMed] [Google Scholar]
- 2.National Lung Screening Trial Research Team. Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. 2011;365:395–409. doi: 10.1056/NEJMoa1102873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rafnar T, Sulem P, Stacey SN, Geller F, Gudmundsson J, Sigurdsson A, et al. Sequence variants at the TERT-CLPTM1L locus associate with many cancer types. Nat Genet. 2009;41:221–227. doi: 10.1038/ng.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Broderick P, Wang Y, Vijayakrishnan J, Matakidou A, Spitz MR, Elsen T, et al. Deciphering the impact of common genetic variation on lung cancer risk: a genome-wide association study. Cancer Res. 2009;69:6633–6641. doi: 10.1158/0008-5472.CAN-09-0680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Horgan AM, Yang B, Azad AK, Amir E, John T, Cescon DW, et al. Pharmacogenetic and germline prognostic markers of lung cancer. J Thorac Oncol. 2011;6:296–304. doi: 10.1097/JTO.0b013e3181ffe909. [DOI] [PubMed] [Google Scholar]
- 6.Peterson CL, Workman JL. Promoter targeting and chromatin remodeling by the SWI/SNF complex. Curr Opin Genet Dev. 2000;10:187–192. doi: 10.1016/s0959-437x(00)00068-x. [DOI] [PubMed] [Google Scholar]
- 7.Klochendler-Yeivin A, Muchardt C, Yaniv M. SWI/SNF chromatin remodeling and cancer. Curr Opin Genet Dev. 2002;12:73–79. doi: 10.1016/s0959-437x(01)00267-2. [DOI] [PubMed] [Google Scholar]
- 8.Simone C. SWI/SNF: the crossroads where extracellular signaling pathways meet chromatin. J Cell Physiol. 2006;207:309–314. doi: 10.1002/jcp.20514. [DOI] [PubMed] [Google Scholar]
- 9.Reisman D, Glaros S, Thompson EA. The SWI/SNF complex and cancer. Oncogene. 2009;28:1653–1668. doi: 10.1038/onc.2009.4. [DOI] [PubMed] [Google Scholar]
- 10.Bochar DA, Wang L, Beniya H, Kinev A, Xue Y, Lane WS, et al. BRCA1 is associated with a human SWI/SNF-related complex: linking chromatin remodeling to breast cancer. Cell. 2000;102:257–265. doi: 10.1016/s0092-8674(00)00030-1. [DOI] [PubMed] [Google Scholar]
- 11.Muchardt C, Yaniv M. When the SWI/SNF complex remodels...the cell cycle. Oncogene. 2001;20:3067–3075. doi: 10.1038/sj.onc.1204331. [DOI] [PubMed] [Google Scholar]
- 12.Reisman DN, Strobeck MW, Betz BL, Sciariotta J, Funkhouser W, Jr, Muchardt C, et al. Concomitant down-regulation of BRM and BRG1 in human tumor cell lines: differential effects on RB-mediated growth arrest vs CD44 expression. Oncogene. 2002;21:1196–1207. doi: 10.1038/sj.onc.1205188. [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Pedrero JM, Kiskinis E, Parker MG, Belandia B. The SWI/SNF chromatin remodeling subunit BAF57 is a critical regulator of estrogen receptor function in breast cancer cells. J Biol Chem. 2006;281:22656–22664. doi: 10.1074/jbc.M602561200. [DOI] [PubMed] [Google Scholar]
- 14.Marquez SB, Thompson KW, Lu L, Reisman D. Beyond Mutations: Additional Mechanisms and Implications of SWI/SNF Complex Inactivation. Front Oncol. 2015;4:372. doi: 10.3389/fonc.2014.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sánchez-Tilló E, Lázaro A, Torrent R, Cuatrecasas M, Vaquero EC, Castells A, et al. ZEB1 represses E-cadherin and induces an EMT by recruiting the SWI/SNF chromatin-remodeling protein BRG1. Oncogene. 2010;29:3490–3500. doi: 10.1038/onc.2010.102. [DOI] [PubMed] [Google Scholar]
- 16.Matsubara D, Kishaba Y, Ishikawa S, Sakatani T, Oguni S, Tamura T, et al. Lung cancer with loss of BRG1/BRM, shows epithelial mesenchymal transition phenotype and distinct histologic and genetic features. Cancer Sci. 2013;104:266–273. doi: 10.1111/cas.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reisman DN, Sciarrotta J, Wang W, Funkhouser WK, Weissman BE. Loss of BRG1/BRM in human lung cancer cell lines and primary lung cancers: correlation with poor prognosis. Cancer Res. 2003;63:560–566. [PubMed] [Google Scholar]
- 18.Glaros S, Cirrincione GM, Muchardt C, Kleer CG, Michael CW, Reisman D. The reversible epigenetic silencing of BRM: implications for clinical targeted therapy. Oncogene. 2007;26:7058–7066. doi: 10.1038/sj.onc.1210514. [DOI] [PubMed] [Google Scholar]
- 19.Yamamichi N, Yamamichi-Nishina M, Mizutani T, Watanabe H, Minoguchi S, Kobayashi N, et al. The Brm gene suppressed at the post-transcriptional level in various human cell lines is inducible by transient HDAC inhibitor treatment, which exhibits antioncogenic potential. Oncogene. 2005;24:5471–5481. doi: 10.1038/sj.onc.1208716. [DOI] [PubMed] [Google Scholar]
- 20.Bourachot B, Yaniv M, Muchardt C. Growth inhibition by the mammalian SWI-SNF subunit Brm is regulated by acetylation. EMBO J. 2003;22:6505–6515. doi: 10.1093/emboj/cdg621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gramling S, Rogers C, Liu G, Reisman D. Pharmacologic reversal of epigenetic silencing of the anticancer protein BRM: a novel targeted treatment strategy. Oncogene. 2011;30:3289–3294. doi: 10.1038/onc.2011.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kahali B, Yu J, Marquez SB, Thompson KW, Liang SY, Lu L, et al. The silencing of the SWI/SNF subunit and anticancer gene BRM in Rhabdoid tumors. Oncotarget. 2014;5:3316–3332. doi: 10.18632/oncotarget.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kahali B, Gramling SJ, Marquez SB, Thompson K, Lu L, Reisman D. Identifying targets for the restoration and reactivation of BRM. Oncogene. 2014;33:653–664. doi: 10.1038/onc.2012.613. [DOI] [PubMed] [Google Scholar]
- 24.Liu G, Gramling S, Munoz D, Cheng D, Azad AK, Mirshams M, et al. Two novel BRM insertion promoter sequence variants are associated with loss of BRM expression and lung cancer risk. Oncogene. 2011;30:3295–3304. doi: 10.1038/onc.2011.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wong KM, Qiu X, Cheng D, Azad AK, Habbous S, Palepu P, et al. Two BRM promoter insertion polymorphisms increase the risk of early-stage upper aerodigestive tract cancers. Cancer Med. 2014;3:426–433. doi: 10.1002/cam4.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang JR, Gramling SJ, Goldstein DP, Cheng D, Chen D, Azad AK, et al. Association of Two BRM Promoter Polymorphisms with Head and Neck Squamous Cell Carcinoma Risk. Carcinogenesis. 2013;34:1012–1017. doi: 10.1093/carcin/bgt008. [DOI] [PubMed] [Google Scholar]
- 27.Wong KM, Qiu X, Cheng D, Azad AK, Palepu P, Mirshams M, et al. The effect of two BRM promoter variants on the risk of stage I/II upper aerodigestive tract cancers. Journal of Clinical Oncology. 2012;30 [Google Scholar]
- 28.Gao X, Huang M, Liu L, He Y, Yu Q, Zhao H, et al. Insertion/deletion polymorphisms in the promoter region of BRM contribute to risk of hepatocellular carcinoma in Chinese populations. PLoS One. 2013;8:e55169. doi: 10.1371/journal.pone.0055169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukuoka J, Fujii T, Shih JH, Dracheva T, Meerzaman D, Player A, et al. Chromatin remodeling factors and BRM/BRG1 expression as prognostic indicators in non-small cell lung cancer. Clin Cancer Res. 2004;10:4314–4324. doi: 10.1158/1078-0432.CCR-03-0489. [DOI] [PubMed] [Google Scholar]
- 30.Gramling S, Reisman D. Discovery of BRM Targeted Therapies, Novel Reactivation of an Anti-cancer Gene. Lett Drug Des Discov. 2011;8:93–99. doi: 10.2174/157018011793663840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kahali B, Marquez SB, Thompson KW, Yu J, Gramling SJ, Lu L, et al. Flavonoids from each of the six structural groups reactivate BRM, a possible cofactor for the anticancer effects of flavonoids. Carcinogenesis. 2014;35:2183–2193. doi: 10.1093/carcin/bgu117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertos NR, Wang AH, Yang XJ. Class II histone deacetylases: structure, function, and regulation. Biochem Cell Biol. 2001;79:243–252. [PubMed] [Google Scholar]
- 33.Haberland M, Arnold MA, McAnally J, Phan D, Kim Y, Olson EN. Regulation of HDAC9 gene expression by MEF2 establishes a negative-feedback loop in the transcriptional circuitry of muscle differentiation. Mol Cell Biol. 2007;27:518–525. doi: 10.1128/MCB.01415-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsubara D, Kishaba Y, Ishikawa S, Sakatani T, Oguni S, Tamura T, et al. Lung cancer with loss of BRG1/BRM, shows epithelial mesenchymal transition phenotype and distinct histologic and genetic features. Cancer Sci. 2013;104:266–273. doi: 10.1111/cas.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hassanein M, Callison JC, Callaway-Lane C, Aldrich MC, Grogan EL, Massion PP. The state of molecular biomarkers for the early detection of lung cancer. Cancer Prev Res (Phila) 2012;5:992–1006. doi: 10.1158/1940-6207.CAPR-11-0441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yamamichi N, Inada K, Ichinose M, Yamamichi-Nishina M, Mizutane T, Watanabe H, et al. Frequent loss of Brm expression in gastric cancer correlates with histologic features and differentiation state. Cancer Res. 2007;67:10727–10735. doi: 10.1158/0008-5472.CAN-07-2601. [DOI] [PubMed] [Google Scholar]
- 38.Bock VL, Lyons JG, Huang XX, Jones AM, McDonald LA, Scoyler RA, et al. BRM and BRG1 subunits of the SWI/SNF chromatin remodelling complex are downregulated upon progression of benign skin lesions into invasive tumours. Br J Dermatol. 2011;164:1221–1227. doi: 10.1111/j.1365-2133.2011.10267.x. [DOI] [PubMed] [Google Scholar]
- 39.Endo M, Yasui K, Zen Y, Gen Y, Zen K, Tsuji K, et al. Alterations of the SWI/SNF chromatin remodelling subunit-BRG1 and BRM in hepatocellular carcinoma. Liver Int. 2013;33:105–117. doi: 10.1111/liv.12005. [DOI] [PubMed] [Google Scholar]
- 40.Strober BE, Dunaief JL, Guha, Goff SP. Functional interactions between the hBRM/hBRG1 transcriptional activators and the pRB family of proteins. Mol Cell Biol. 1996;16:1576–1583. doi: 10.1128/mcb.16.4.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu Y, Zhang J, Chen X. The activity of p53 is differentially regulated by Brm- and Brg1-containing SWI/SNF chromatin remodeling complexes. J Biol Chem. 2007;282:37429–37435. doi: 10.1074/jbc.M706039200. [DOI] [PubMed] [Google Scholar]
- 42.Wang M, Gu C, Qi T, Tang W, Wang L, Wang S, et al. BAF53 interacts with p53 and functions in p53-mediated p21-gene transcription. J Biochem. 2007;142:613–620. doi: 10.1093/jb/mvm176. [DOI] [PubMed] [Google Scholar]
- 43.Damiano L, Stewart KM, Cohet N, Mouw JK, Lakins JN, Debnath J, et al. Oncogenic targeting of BRM drives malignancy through C/EBPbeta-dependent induction of alpha5 integrin. Oncogene. 2014;33:2441–2453. doi: 10.1038/onc.2013.220. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M, Knudsen ES, et al. SWI/SNF chromatin-remodeling factors induce changes in DNA methylation to promote transcriptional activation. Cancer Res. 2005;65:3542–3547. doi: 10.1158/0008-5472.CAN-04-3554. [DOI] [PubMed] [Google Scholar]
- 45.de la Serna IL, Carlson KA, Imbalzano AN. Mammalian SWI/SNF complexes promote MyoD-mediated muscle differentiation. Nat Genet. 2001;27:187–190. doi: 10.1038/84826. [DOI] [PubMed] [Google Scholar]
- 46.Seo S, Herr A, Lim JW, Richardson GA, Richardson H, Kroll KL. Geminin regulates neuronal differentiation by antagonizing Brg1 activity. Genes Dev. 2005;19:1723–1734. doi: 10.1101/gad.1319105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lickert H, Takeuchi JK, Von Both I, Walls JR, McAuliffe F, Adamson SL, et al. Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 2004;432:107–112. doi: 10.1038/nature03071. [DOI] [PubMed] [Google Scholar]
- 48.Vradii D, Wagner S, Doan DN, Nickerson JA, Montecino M, Lian JB, et al. Brg1, the ATPase subunit of the SWI/SNF chromatin remodeling complex, is required for myeloid differentiation to granulocytes. J Cell Physiol. 2006;206:112–118. doi: 10.1002/jcp.20432. [DOI] [PubMed] [Google Scholar]
- 49.Gao X, Tate P, Hu P, Tijan R, Skarnes WC, Wang Z. ES cell pluripotency and germ-layer formation require the SWI/SNF chromatin remodeling component BAF250a. Proc Natl Acad Sci U S A. 2008;105:6656–6661. doi: 10.1073/pnas.0801802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kothandapani A, Gopalakrishnan K, Kahali B, Reisman D, Patrick SM. Downregulation of SWI/SNF chromatin remodeling factor subunits modulates cisplatin cytotoxicity. Exp Cell Res. 2012;318:1973–1986. doi: 10.1016/j.yexcr.2012.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Segedi M, Anderson LN, Espin-Garcia O, Borgida O, Bianco T, Cheng D, Chen Z, Patel D, Brown MC, Xu W, Reisman DR, Gallinger S, Cotterchio M, Hung R, Liu G, Cleary SP. BRM polymorphisms, pancreatic cancer risk and survival. Int J Cancer. doi: 10.1002/ijc.30369. (in press) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.