

Abstract
Cerium oxide nanoparticles have widespread use in the materials industry, and have recently come into consideration for biomedical use due to their potent regenerative antioxidant properties. Given that the brain is one of the most highly oxidative organs in the body, it is subject to some of the greatest levels of oxidative stress, particularly in neurodegenerative disease. Therefore, cerium oxide nanoparticles are currently being investigated for efficacy in several neurodegenerative disorders and have shown promising levels of neuroprotection. This review discusses the basis for cerium oxide nanoparticle use in neurodegenerative disease and its hypothesized mechanism of action. The review focuses on an up-to-date summary of in vivo work with cerium oxide nanoparticles in animal models of neurodegenerative disease. Additionally, we examine the current state of information regarding biodistribution, toxicity, and safety for cerium oxide nanoparticles at the in vivo level. Finally, we discuss future directions that are necessary if this nanopharmaceutical is to move up from the bench to the bedside.
Keywords: cerium oxide nanoparticles, neurodegeneration, neuroprotection, toxicology, biodistribution, safety
Graphical Abstract
Introduction & Overview
Cerium oxide nanoparticles (CeONPs) have widespread use in the materials industry as glass polishing agents,1 fuel additives to improve combustion,2 electrolytes for solid oxide fuel cells,3 ultraviolet absorbents,4 and oxygen sensors.5 Their utility in these applications arises from their regenerative antioxidant functions and their ability to act as an “electron sponge”, shuffling ions through the nanoparticle matrix. However in addition to their materials applications, CeONPs show promising biomedical applications in treatment of diseases associated with oxidative stress, particularly those associated with neurodegeneration.6–16 This review will focus on the in vivo efficacy of CeONPs in treatment of diseases associated with the brain, including ischemia, traumatic brain injury, Alzheimer’s Disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), and other neurodegenerative disorders. Initially, reports on the biological utility of CeONPs in treating conditions associated with oxidative stress outpaced the literature on safety and potential toxicities, however this has slowly been changing. This review will also encompass reports of safety and toxicity of CeONPs, at the in vivo level, with an up-to-date discussion of what is known, and what remains to be elucidated.
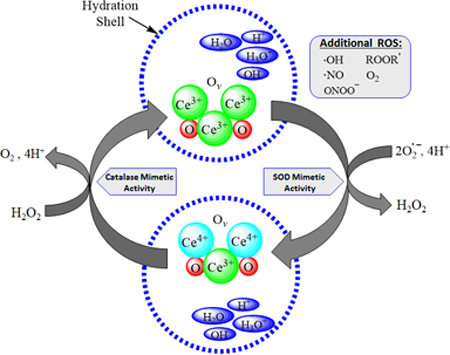
Cerium is a rare earth element of the lanthanide series, having 2 partially filled subshells of electrons, 4f and 5d, with several excited substates hypothesized.17 The cerium atom can exist in two valence states, either +4 (fully oxidized) or +3 (fully reduced), and alternates between the two in redox reactions.17–19 In the nanoparticle form, cerium is bound to oxygen in a crystalline fluorite lattice structure, which exhibits oxygen vacancies or defects (or simply put, electron holes) in the lattice structure, which arise through loss of oxygen and/or its electrons. Thus in addition to the cerium atom being capable of shifting between +3 and +4, the stoichiometry of the nanoparticle can change from CeO2 to CeO2-x.18 Based on changes in redox state and oxygen vacancies, CeONPs interact with numerous free radicals, detoxifying their deleterious activity. Further, because of the lattice structure and ease of electronic conversions with other ionic species at the quantum level, CeONPs are capable of regenerating their redox-active matrix, allowing repetitive free radical interactions, as shown in Figure 1. Couple these potent redox capabilities with the dramatically increased surface area and quantum effects of nano-scale particles, and the result is a highly efficient nano-scale free radical scavenging reactor.
Figure 1. Hypothesized Mechanism of Action of CeONPs.

In a given CeONP, the cerium atom exists in the 3+ and 4+ valence states, bound to oxygen and containing oxygen vacancies (Ov). When exposed to a superoxide radical, it exhibits SOD-mimetic activity, and Ce3+ is converted to Ce4+, with a corresponding change in oxygen vacancies. There is also likely a contribution to this reaction from the hydration shell around the CeONP. Superoxide is converted to H2O2. Via a catalase-mimetic activity involving Ce4+, H2O2 is converted to O2 + 4H+, and cerium valence to +3 (with corresponding changes in oxygen vacancies), regenerating the origin CeONP state. Again, there is a likely contribution from ions present in the water hydration shell. In the biological milieu, this action exists in a continuous cycle, depending on the ionic species exposed to the CeONPs, the hydration shell, oxygen partial pressure, and any surrounding ionic species. Although we utilized superoxide and H2O2 as examples, radicals scavenged could be any number of biologically relevant free radicals.
Although this review will focus primarily on in vivo activity of CeONPs, it is worth a brief recap of the in vitro findings that ultimately led to the biomedical applications of CeONPs in neurodegenerative diseases. Early studies by our group and others found that a single dose of CeONPs were readily taken up by brain cells.6 Through their potent and regenerative antioxidant effects, CeONPs extended the lifespan of cortical and spinal neurons in culture, decreased cell dysfunction associated with oxidative stressors, increased neurite outgrowth, increased transcription of genes associated with neuroprotection, and increased dopamine secretion.6–9, 15, 16, 20–24 In comparison to other free radical scavengers such as n-acetyl cysteine, vitamins E and C, and melatonin, CeONPs provided superior protection in terms of neuronal survival and maintenance of normal calcium signaling in response to oxidative stress.6, 7 These studies paved the ground for initiation of in vivo studies and movement of CeONPs along the bench to bedside pipeline.
Considerations for Use of CeONPs in Medicine
As movement of CeONPs into the realm of in vivo studies increases, we must remember that CeONPs are nanoparticles, and quite different from the standard pharmaceuticals. At the biochemical level, function is derived from the basic chemical structure. In contrast, the physical, electronic, and quantum effects take precedence with CeONPs and other nanoparticles. Additionally, we must realize we are not dealing with a simple chemical, per se, but rather are utilizing a physical entity with distinct structure, form, and properties – a mini-reactor.
Physico-Chemical Characterization is Critical
Size, surface area, valence states, bond lengths, and shape all convey different levels of activity to CeONPs.16, 19, 25 It cannot be stressed enough how critical physical and chemical particle characterization is to any experiment. In a 2011 study, we compared particle size, valence state, oxygen vacancies, and chemical reactivity (rate constant for dissociation of H2O2) to cellular activity in terms of protection from challenge with superoxide or H2O2, using mixed organotypic neuronal cultures.9 Interestingly, the beneficial biological effects were not solely related to particle size. Several samples of 5 and 7 nm particles, prepared by university engineering labs, had a small toxic effect as measured by increased propidium iodide uptake in cells exposed to these nanoparticles. We speculate that these small but significant deleterious effects may be due to alterations in lattice parameters that promote radical generation, rather than radical scavenging. Alternatively, the presence of endotoxin or other protein contaminants, which are readily adsorbed by CeONPs, may have caused these effects. For the particles tested, the beneficial biological effects on hydroxyl or superoxide radical scavenging were also not clearly related to Ce3+ content, but there appeared to be a range of Ce3+ content, from 31–33%, which produced the best general antioxidant activity. Likewise, 7–10 nm particle sizes also appeared to provide the most in vitro neuroprotection.
Mechanism of Action and Cerium Valence State
A basic mechanism of action for CeONPs is shown in Figure 1. However in vivo few conditions of oxidative stress are likely to produce only superoxide and H2O2. CeONPs have also demonstrated the ability to scavenge hydroxyl, nitric oxide radicals, and peroxynitrite, which would also fuel the cycle shown in Figure 1.6, 9, 12, 26, 27 In vitro, CeONPs display multiple redox enzyme mimetic properties including superoxide dismutase,28 catalase,29 peroxidase,30 oxidase,31 and phosphatase.32 However there has been some controversy regarding the contribution of the 3+ vs. 4+ valence states of cerium and its biological activity and toxicity. In vitro studies reported that smaller sized particles (≥5 nm), having a higher content of Ce3+, scavenge primarily superoxide,28 while those containing a higher Ce4+ content scavenge primarily H2O2.29 Several reports suggested that the cerium in the 3+ state imparts enhanced toxicity 33, 34. So a somewhat closer look at the in vitro physical chemistry involved is warranted.
In the case of purely stoichiometric CeO2 nanoparticles, cerium is octahedrally coordinated to the nearest eight oxygen atoms with each oxygen tetrahedrally coordinated to the nearest four cerium atoms in a fluorite crystal structure.19 Defects in CeONPs exist in the form of oxygen vacancies present at the surface to compensate for deficiencies in positive charge from the presence of Ce3+.35 Particle size plays a role in the amount of Ce3+ and Ce4+ present in CeONPs in that as particle size decreases, Ce3+ content increases while Ce4+ declines.19, 25
Celardo and coworkers proposed that superoxide interacts with available oxygen vacancies at the surface36, 37, in the presence of available protons possibly from the hydration shell, 18, 38 to produce hydrogen peroxide. Pirmohamed and coworkers discovered that in vitro, CeONPs exhibit catalase mimetic activity dependent upon the +4 valence state.29 An improved ability for hydrogen peroxide degradation occurred with CeONPs having a higher +4 concentration, a result opposite of findings presented when CeONPs act as SOD mimics (a higher ratio of +3/+4 cerium atoms acted as a more efficient SOD mimic). H2O2 produced by CeONPs acting as SOD mimics could then enter into the catalase mimetic cycle to produce less harmful molecular oxygen (O2) and H2O,37 with the caveat that both enzyme mimetic cycles are working in tandem and the degradation rate of H2O2 is equal to or greater than its formation (Figure 1).
Celardo et al. synthesized CeONPs with altered Ce3+/Ce4+ ratios, without changing the existing oxygen vacancies on the surface, by doping with samarium (Sm).36, 37 Ce3+ concentration and antioxidant efficacy decreased as a function of Sm content. The authors concluded this was a function of decrease in Ce3+ content since oxygen vacancies were unchanged at the surface by X-Ray Diffraction analysis (XRD).36 In a related study Dunnick and coworkers synthesized CeONPs doped with gadolinium (Gd2O3) resulting in an increase in Ce3+/Ce4+ concentration by XRD (contrary to Sm doped CeONPs but explained by the ability of Gd2O3 to incorporate more Ce3+ content). Despite the increased Ce3+ content, these particles demonstrated a decrease in antioxidant behavior.39 The decreased efficacy as an antioxidant was attributed to the inability of Gd-doped CeONPs to oscillate between valence states, and not the ratio of Ce3+/Ce4+ in that anti-oxidant behavior decreased with increasing Ce3+ content and increasing dopant.39 To complicate matters further, a study by Cafun et al. 40 indicated that Ce3+ may not truly form with any stability at all, due to rapid redox cycling. This underscores the cycling between cerium valence states and oxygen vacancies in the effective function of CeONPs as a biological antioxidant, rather than the absolute concentrations of 3+ vs. 4+.
A most important concept that we see emerging is that redox cycling and thus the antioxidant efficacy or toxicity of CeONPs is directly related to its environment. For example, the formation of oxygen vacancies at the surface of CeONPs is both dynamic and variable, resulting from changes in oxygen partial pressure,19 electrical field variations,41 temperature,17 surface stresses,19 and ionic species present. This has demonstrated by Kuchibhatla and coworkers who showed that by tuning environmental conditions, the structure of nanoparticles could be reversibly altered.42
Furthermore, and perhaps somewhat more important, the biological environment in terms of ionic species plays a critical role in CeONP activity. Even though one may administer a CeONP with a high +3/+4 ratio, the cycling shown in Figure 1 will still occur. It is unlikely to remain as stable 3+ in the biological environment, but rather will cycle depending on the local ionic environment shown in Figure 1. Biologically, what may cause a preponderance of +3 vs. +4 would be the types of radicals present. For example, looking at Figure 1, if a CeONP were in an area with high superoxide concentrations and little H2O2, the redox cycle could not be completed and more cerium would remain in the +4 state and H2O2 may accumulate, affecting toxicity. Likewise, if more H2O2 were present, the reactions on the left side of the cycle shown would predominate, causing more of the +3 state. These considerations should be taken into account, rather than the absolute 3+/4+ ratio of the starting material, as they are likely to regulate biological effect in vivo. Importantly, the free radicals available to fuel the CeONP cycle may differ in disease states vs. normal conditions, in that disease states may have abundant radicals to fuel the redox cycle of the nanoparticle.
This structural dependence of CeONPs on environmental conditions makes it difficult to pinpoint one particular feature affecting antioxidant ability, and invariably leads to inconsistencies in the literature. These considerations may be responsible for the seemingly differential in vivo activities of CeONPs. For example, the Minarchick group found differences in CeONP activity in different vascular beds, particularly when given to animals with high oxidative stress, as compared to normal levels of oxidative stress.43, 44 It may be that the beneficial biological activity of CeONPs are most evident in cases of high oxidative stress, as occurs in disease.
In summary, we must move our thinking outside the box and realize that we are dealing with a mini-reactor that requires input from all elements of the cycle shown in Figure 1 to function efficiently. If one element of the cycle is perturbed, then aberrant activity and toxicity may occur. Further, as discussed by Fronzi et al. 38 and Aneggi et al., 18 the hydration shell which forms around CeONPs is also critical to its radical scavenging and regenerative properties, as ionic species in water may serve as final electron or proton acceptors. Once again, this underscores the need for consideration of the complex biological environment encountered in vivo, when considering mechanism of action of CeONPs and potential beneficial vs. toxic effects.
Oxygen Storage Capacity
In the materials industry, CeONPs are known for their oxygen storage capacity – their ability to take up and release oxygen in a redox reaction, through the creation and annihilation of oxygen vacancies.45, 46 In the biomedical realm, we appear to have neglected to think about this aspect of CeONPs when considering their action in vivo. When a valence change from +4 to +3 occurs, oxygen is usually released to the environment, depending on the oxygen partial pressure.47 This capacity of CeONPs needs to be considered when examining in vivo activity, as release of oxygen can be important in conditions such as ischemia, where tissue oxygen is lacking. Alternatively, production of excess oxygen can be associated with an increase in oxidative stress. Oxygen partial pressure will also dictate the reformation of Ce4+ from Ce3+, which may be different in the cellular compartment vs the arterial or venous circulation.
Agglomeration
Depending on the solution CeONPs are in, it is well known that nanoparticles may agglomerate into larger clumps. Agglomeration produces larger sized particles of CeONPs that may not have the same in vivo properties as their smaller counterparts. Agglomerated particles may block vasculature or display altered delivery to tissues, and may interfere with cellular uptake. In any in vivo experiment, aggregation in the delivery vehicle must be avoided. By examining transmission electron micrographs (TEM) of 7 nm CeONP in water mixed by vortexing vs. sonication6 it was shown that the non-sonicated suspension formed large clumps of unevenly dispersed CeONPs, while the sonicated suspension had a more even distribution of particles. For this reason, adequate sonication just prior to delivery is utilized by many to assure a uniform delivery suspension of non-agglomerated particles.
However in addition to sonication, uniform size dispersions for in vivo applications can be improved by attention to delivery vehicle. Xue et al. demonstrated that CeONPs bind phosphate when suspended in phosphate buffers, forming cerium phosphate on the surface of the nanoparticle.48 This interferes with cerium cycling between the +3 and +4 states, dampening redox activity. We have found that suspension of stock CeONP in saline-citrate buffer produces a uniform, non-agglomerated delivery solution that effectively distributes in vivo.9 Other groups have utilized citrate capping and citrate-EDTA coating to eliminate agglomeration during injection49, 50. Regardless, the “as delivered” CeONP solution should always be assessed for agglomeration prior to in vivo delivery, and of course fully characterized as to physico-chemical properties.
CeONPs as Neuroprotectants In Vivo
The brain in the most highly oxidative organ in the body and is subjected to some of the highest levels of oxidative stress.51 Multiple endogenous pathways detoxify oxidative stress, including superoxide dismutase, catalase, glutathione/glutathione peroxidase, and single molecule antioxidants such as vitamins C and E. However in many disease states, the production of free radicals exceeds the endogenous defense mechanisms, resulting in states of high oxidative stress. Most neurodegenerative diseases including Alzheimer’s, Parkinson’s, Huntington’s, Multiple Sclerosis, ischemia, traumatic brain injury and aging itself are associated with excessive oxidative stress in the brain. However to date, the use of antioxidants to improve outcome in neurodegenerative disease has met with only limited success. Our current pharmacological armament of antioxidants utilizes compounds which scavenge a single free radical and are destroyed in the process. Thus repetitive dosing is required, which is still not sufficient to handle the level of radicals generated. Based on CeONPs superior role as a regenerative antioxidant, it was logical to make the progression to utilization of CeONPs in neurodegenerative disease.
Experimental Autoimmune Encephalomyelitis (EAE)
Eitan et al. tested CeONPs (3–5 nm) in a mouse model of EAE, which is representative of the disease Multiple Sclerosis (MS) in humans.11 CeONPs were delivered after induction of EAE, in multiple IV doses of 1 mg/kg in phosphate buffered saline, and were compared to lenalidomide, a drug used to decrease EAE severity. In some animals, a combined treatment of CeONPs + lenalidomide was used. When given alone, lenalidomide delayed symptom onset, but did not prevent the eventual development of disease. Administration of CeONPs alone had no effect on symptom onset, but significantly improved recovery late in the disease. However combination of CeONPs + lenalidomide eliminated development of clinical symptoms, reduced white matter damage, and decreased CNS inflammation, making CeONPs a good potential choice for combination therapy in MS. However we would note in this study, CeONPs were delivered in phosphate buffer, which is known to interfere with redox activity of CeONPs as discussed above. In this case, it did not appear to interfere with disease-modifying effects of CeONPs, but further improvement in efficacy may be found with delivery in a vehicle that promotes non-aggregation, such as saline-citrate.
Heckman et al. also examined the utility of CeONPs in the mouse EAE model.49 For these experiments, a citrate/EDTA “stabilized” CeONP was used, which presumably would result in greater delivery to the brain. However the precise mechanism regarding the “stabilization” was not described. CeONPs utilized were uniform 2.9 nm particles and were delivered as either a preventive or therapeutic dose. The preventative dose consisted of one dose delivered IV before disease induction, followed by maintenance doses every 7 days thereafter. The therapeutic dose was initiated 3 days after disease induction and followed by maintenance doses thereafter. Doses utilized were 10, 20, and 30 mg/kg, higher than in the Eitan study.11 CeONPs were compared to a standard MS treatment drug, fingolimod, and to vehicle-treated animals. Both fingolimod and the preventative 30 mg/kg treatment regimen delayed disease onset and decreased disease severity, with CeONPs being equivalent to fingolimod, although the two work by different mechanisms. The 30 mg/kg dose of CeONPs also improved motor function using rotarod, hanging wire, and balance beam tasks for both preventive and therapeutic treatment, and again, were similar to fingolimod. In addition, CeONPs reduced the levels of reactive oxygen species in the brain late in the disease (day 42) whereas fingolimod had no effect on levels of reactive oxygen species, but decreased inflammatory cell populations in the brain. So in this case, the CeONPs were equivalent to a standard drug, fingolimod. However it would be interesting to see the results of studies combining the two drug treatments, to determine if effects were additive, as in the Eiten study.11
Although the citrate/EDTA coating did increase levels of cerium in the brain as compared to a prior study by the authors, the primary sites of CeONP distribution were in the liver, spleen and kidney. Deposition in those tissues remained some 10–100 fold higher than in brain. It is interesting to note that in the Eitan study 11, CeONPs were given at a 1 mg/kg dose in phosphate buffer, which should have theoretically inactivated some of the CeONP redox activity. The particles used in the Heckman study49 were citrate/EDTA stabilized to hopefully improve brain distribution, yet produced seemingly equivalent results. This underscores importance of another variable, protein coating during biodistribution, discussed below. Despite our efforts to maintain stabilized, well dispersed CeONPs, protein coating in the body may redirect our best efforts. Taken together, these results present a clear potential for the use of CeONPs in treatment of MS.
Ischemia
The production of free radicals after stroke is substantial, and has been associated with a cascade of free radical events, as recently reviewed, 14 making CeONPs of potential utility in treatment.
Although not purely an in vivo study per se, Esteves et al.12 tested CeONPs in a rat stroke model, using brain slices. Here, commercially prepared 10 nm CeONPs were utilized, suspended in distilled water with sonication. Using concentrations of 0.2–1µg/ml, CeONPs dose-dependently reduced ischemic cell death in brain slices by over 50%, and reduced the concentrations of NO and superoxide by 15%. TEM showed CeONPs localized to lipid membranes, mitochondria, and neurofilaments, all sites of potential free radical damage. One of the most significant findings in this study was the reduction of 3-nitrosotyrosine, a protein adduct formed by interaction of peroxynitrite (or its precursors NO and superoxide) with tyrosine residues on proteins. These results imply that CeONPs may reduce free radical damage to cellular molecules in ischemia. Importantly, administration of CeONPs up to 4 hrs post-stroke still provided significant neuroprotection. Interestingly, CeONPs also improved cell survival in control brain slices not exposed to stroke, likely by decreasing oxidative stress incurred by slice preparation.
In a study by Kim et al.13 3.3 nm CeONPs prepared in their laboratory were PEGylated (polyethylene glycol conjugated) to improve circulation time in the blood stream, and tested in a rat stroke model (although the article does not describe the precise model used).13 CeONPs were given at IV doses of 0.1 – 0.5 mg/kg (vehicle was not described) prior to stroke. CeONP pretreatment significantly reduced infarct volumes at the 0.5 mg/kg dose, and decreased the number of TUNEL positive cells in brain sections. Higher doses of 1 and 1.5 mg/kg showed no protection. Possibly, the higher doses altered biodistribution, as discussed below. However one may question why a 0.5 mg/kg dose worked, and a 1 mg/kg dose did not, since this is only a 2- fold change. When thinking of dose, we must remember that the reactive sites of a nanoparticle come from its surface area, which is large and highly irregular. So although going from 0.5 to 1.0 mg/kg is a doubling in weight, the surface area change is likely over 100–1000 fold more. In fact, Sayes and Warheit52 have proposed a change in dose-metrics for nanoparticles, based on surface area, which at this point appears warranted.
One interesting point of the Kim study13 is that the authors found that CeONPs did not penetrate the brain of normal animals. Rather, CeONPs entered the brain after stroke, suggesting that a possible breakdown of the blood-brain barrier enabled the nanoparticles to enter the brain tissue. However PEGylation may not be the best method to promote brain entry of nanoparticles, as studies with PEG-conjugated superoxide dismutase also exhibited difficulties crossing the blood brain barrier, and were elevated only after the blood-brain barrier had been damaged.53
Retina
Athough the retina is not considered the brain, per se, retinal neurons project directly to the brain, and the two are in close contact. Numerous studies have been done examining the effects of CeONPs on retinal degeneration in several models, which have been reviewed in depth15 and will be summarized briefly here. A single intravitreous injection of 2 nMols CeONP (15 nm size) delivered 3 weeks prior to light damage in the rat, reduced photoreceptor cell death and decreased production of TNFα and microglial activation in the retina.54 The mutant tubby mouse undergoes extensive photoreceptor loss during its lifetime, with 2/3 cell loss by postnatal day 49.55 In this model, CeONP (3–5 nm) delivered as a single intravitreal injection of 172 ng in saline, preserved photoreceptor cells and increased expression of several genes associated with oxidative stress and antioxidant defense. The Vldr−/− mouse (low density lipoprotein receptor knockout) is a model for human retinal angiomatous proliferation, a form of “wet” macular degeneration.56 Again, using a single 172 ng intravitreal injection in saline, CeONPs prevented retinal photoreceptor cell death and reduced abnormal levels of VEGF observed in this knockout mouse, decreasing it to levels seen in wild type controls.56, 57 The P23H-1 rat, another photoreceptor degeneration model, is autosomal dominant for retinitis pigmentosa.58 Intravitreal injection of CeONP as described above increased rod and cone cell functions for up to 3 weeks after injection, and reduced lipid peroxidation in the retinas of CeONP-treated animals.58 Wong et al. examined the residence time of CeONPs in the retina, finding 90% retained for at least 120 days, decreasing somewhat after that.58 Given the low doses of CeONPs used, the intravitreal injection delivery, and relative retention in the retina, CeONPs show excellent potential as a future treatment of diseases of retinal degeneration.
Alzheimer’s Disease (AD) and Parkinson’s Disease (PD)
Both AD and PD are associated with high levels of oxidative stress, making them a target for treatment with CeONPs. The hallmark of AD is initial death of cortical cholinergic neurons, while in PD, the hallmark is death of dopaminergic nigrostriatal neurons. To date, no in vivo studies with CeONPs have been performed in AD animal models, but several in vitro studies show excellent potential and will be mentioned here. We first described the potential for benefit of CeONPs in AD, using electron paramagnetic resonance to demonstrate that it scavenged free radicals produced in vitro during Aβ1–42 aggregation.7 In these studies, aggregated Aβ1–42 induced rapid death of pure rat cortical neuronal cultures, which was blocked by 10 nm CeONPs (10 nM), strongly suggesting the utility of CeONPs in AD animal models. Subsequently, Dowding et al.27 showed similar results for 3–5 nm CeONPs, and demonstrated that they also blocked mitochondrial fragmentation produced by Aβ1–42. Further experimentation will be necessary to determine any beneficial effects in animal models of AD.
PD is also associated with high levels of oxidative stress, causing death in neurons of the substantia nigra and striatum. Our group has examined the utility of CeONPs in treating PD, with in vivo models. Using a Drosophila model of PD which involves exposure to the redox cycling agent paraquat, our group demonstrated that pretreatment of flies with 10 nM and 1 µM CeONPs improved survival and preserved motor function.6, 9 For these studies we used 10 nm CeONPs, commercially prepared, pharmaceutical grade, from Nanophase, Inc. (Romeoville, Illionois). The particles were received as a non-agglomerated 1.2% suspension in water, with a pH of 3.5. Particles were tested to be endotoxin-free, an important criteria for our studies as described below. For Drosophila studies, nanoparticles were diluted in water containing 0.01% docusate sodium to assure adequate dispersal in Drosphila feeding medium.
We also conducted studies using the MPTP mouse model of PD. For these studies, we utilized the same CeONPs as described above, but the particles were dissolved in saline-citrate buffer for delivery (without docusate sodium), which we have shown to eliminate agglomeration.6 In our initial studies, mice were pretreated with 0.05 – 50 mg/kg CeONP in saline citrate via IV injection. Five days after the last dose, we induced PD with MPTP, followed by examination of brain dopamine and histochemistry at 7 days after disease induction. We found that CeONPs from 0.05–5 mg/kg dose dependently blocked the MPTP-induced decline in striatal dopamine, and preserved dopaminergic neurons in the substantia nigra. Levels of striatal dopamine and dopaminergic neuronal number in the substantia nigra were equivalent to controls with the 0.5 and 5.0 mg/kg dose. However the 50 mg/kg dose was without effect, possibly due to particle agglomeration in the blood and lack of delivery to the brain. A representative result of preservation of dopaminergic neurons in the substantia nigra is shown in Figure 2.
Figure 2. CeONP Preserves TH+ Neurons in the Substantia Nigra of MPTP-Challenged Mice.

Mice (C57Bl/6) were pretreated with the indicated dose of 10 nm CeONP in saline-citrate, followed by MPTP challenge 5 days later (6 animals per group). Animals were euthanized 7 days after MPTP administration, brains were perfusion fixed, and stained for tyrosine hydroxylase (TH+, brown), a marker of dopaminergic neurons. Nuclei are counterstained with Nissl (blue). Note the almost complete destruction of dopaminergic neurons by MPTP (upper rt panel), which was abrogated in CeONP-treated mice (lower 2 panels).
We went on to test the efficacy of CeONPs when delivered 24 hrs after disease induction with MPTP. We found that CeONPs preserved striatal dopamine by 52%, when delivered 24 hrs after MPTP, and preserved dopaminergic neurons in the substantia nigra after MPTP challenge, to 84–87% of the levels seen in controls.60 These results suggest that CeONPs may be a disease-modifying future therapy for PD.
Traumatic Brain Injury
Our in vivo studies on CeONPs in traumatic brain injury (TBI) were prompted by our in vitro studies using a tissue culture model of TBI. In the tissue culture model, pretreatment, or post-injury treatment up to 6 hrs after injury, preserved neuronal number and improved dysfunctional calcium signaling normally observed in injured cultures.7, 9 Therefore, we tested the efficacy of CeONPs in improving the outcome of lateral fluid percussion brain injury in the rat, a model for human TBI. In these studies, we pretreated rats with 0.14 mg/kg CeONPs (10 nm) in saline-citrate, and delivered a moderate head injury 48 hrs later.61 Cognitive function was assessed with the Morris water maze, examining the rate at which rats learn the location of a hidden platform, shown in Figure 3A. Vestibulomotor function was tested after injury by a simple beam balance task, shown in Figure 3B. CeONP pretreatment enhanced performance in both tasks, compared to injured vehicle-treated animals, and the rate of recovery improved substantially. By 4 days post-injury, injured animals treated with CeONPs demonstrated motor performance on the beam balance similar to sham animals. In the water maze, animals treated with CeONPs displayed enhanced cognitive recovery as indicated by reduced goal proximity across 5 days of water maze data acquisition.
Figure 3. Pretreatment with CeONPs improve latency to goal in the Morris Water Maze, and Beam Balance performance after moderate lateral fluid percussion brain injury.

Male Long-Evans rats were pretreated with 0.14mg/kg CeONPs followed by a moderate lateral fluid percussion brain injury 3 days later (6 animals per group). Sham animals received CeONPs and the surgery for lateral fluid percussion brain injury, without delivery of the brain injury. On the indicated day post-injury, rats were tested for latency to platform in the Morris Water Maze task (A) and for Beam Balance Latency To Fall (B). Note the significant improvement, to near-sham levels, with CeONP pretreatment. Results represent mean ± SE for 6 animals per group. *Significant from sham, p<0.01; **Significant from Sham and Injury+CeONP; #Significant from Injury+Vehicle and Sham.
Experiments completed to date demonstrated that post-injury administration of CeONPs reduced oxidative stress, protein nitrosylation, and lipid peroxidation, as well as led to cognitive improvement (Rzigalinski & VandeVord, in submission). These studies suggest that CeONPs may be a promising treatment for human head injury, resulting in decreased brain damage and improved functional recovery.
Considering the in vivo evidence thus far, CeONPs are a promising target for neurodegenerative diseases in which oxidative stress plays a major role. However forward progress requires the appropriate supporting studies. Given an IV dose of CeONPs, distribution is likely to the entire organism, so biodistribution, safety, and toxicity merit equal consideration.
Biodistribution and Pharmacokinetics
Although the target for treatment of neurodegenerative disorders is the brain, biodistribution to other organs must be examined, since concentrations in these organs were higher than concentrations in brain. For CeONPs, biodistribution is generally measured by inductively coupled plasma mass spectrometry (ICP-MS) for cerium. Of course, this does not directly measure the nanoparticle itself, but measures its presence by virtue of the element cerium. It should be noted that all tissues usually have some background level of cerium. Although many studies convert ICP-MS cerium levels to weight levels of CeONPs, this derivation is not necessarily appropriate and should be used with caution. In addition, the detection limits of the particular instrument should be established using CeONP spiked samples, prior to experimentation. Failure to do so may result in underestimation of tissue cerium.
The Yokel group compared biodistribution of a range of CeONP sizes (5, 15, 30 and 55) infused into rats at doses of 85–100 mg/kg, and analyzed 1, 20 or 720 hrs later.62 Liver and spleen were the primary organs of deposition, with little clearance at the 720 hr time point. The larger particles (15, 30, and 55 nm) were rapidly cleared from the blood, while the 5 nm particles remained in the circulation longer. In this study, although 5 nm particles were found in the brain in higher concentrations than larger sized particles, the authors state that they were not in the brain per se, but were in the vasculature, and had not crossed the blood brain barrier, as evidenced from electron microscopy (EM). Similar results were reported in a subsequent paper in which citrate capped 5 nm CeONPs were given at the 85 mg/kg dose and rats were examined 30 days after exposure.50
A study by Hirst et al. examined CeONPs biodistibution in mice using oral, intraperitoneal (IP), and IV routes, with once weekly dosing for 2 or 5 weeks.63 A dose of 0.5 mg/kg was used, with PBS as vehicle. PBS is known to interfere with CeONP valence cycling in vitro, as phosphate groups bind the surface cerium ions of the nanoparticle causing agglomeration. Their findings indicated that the most tissue distribution arose from the IP and IV routes, with little absorption orally. Tissue accumulations persisted through the 5 weeks of this study.
An examination of distribution, retention, and effectiveness of CeONPs after IV administration was reported by Heckman et al., 49 using 2.9 nm CeONP and ICP-MS. Major organs of biodistribution were again liver, kidney and spleen. CeONPs (as cerium) could still be detected 5 months post-administration, including levels of < 0.1 µg/g in brain, consistent with our low dose studies discussed below.
We examined biodistribution of CeONPs specifically at the doses used in our head injury and PD studies, to ascertain the biodistribution of biomedically relevant doses. Rats were utilized, as mouse tissues did not provide enough material for ICP-MS analysis at these low dose ranges. ICP-MS analyses were performed by Cerium Labs (Austin, Tx). Prior to analysis, the “as injected” volumes were exhaustively analyzed, as well as spiked tissue samples to assure detection limits were adequate and CeONPs were appropriately detected. Rats were injected IV with 0.05–5 mg/kg CeONP (10 nm, in saline-citrate). Tissues were collected for biodistribution studies 2 days or 6 months after injection into the rat. As shown in Figure 4, the biodistribution of CeONP differs depending on the dose, but remains in the low ng/g range. We would note that tissue cerium was detected even in control rats, animals not subjected to CeONPs and sequestered from CeONP treated rats. Two days after delivery in the rat, the 0.5 µg/g dose (black bars on Figure 4a) produced a 10 fold elevation in brain cerium, a 3 fold elevation in heart, and a 1.6 fold elevation in the lung. The most substantial elevations in tissue cerium were the liver (28 fold), kidney (10 fold) and spleen (53 fold), consistent with other studies that found extensive accumulation in these organs. A ten-fold higher dose, 5 µg/g, did not produce significantly higher levels of cerium in the brain or heart (light grey bars). In contrast to the 0.5 µg/g dose, the 5.0 µg/g dose appeared to distribute primarily to lung, liver, kidney, and spleen, where cerium accumulation was highest. This is consistent with several published reports in which injection of milligram quantities of CeONP accumulate primarily in liver, lung, and spleen, with little brain accumulation detectable.50, 63–65
Figure 4. Biodistribution of CeONPs to selected organs 2 days (A) and 6 months (B) after IV injection.

Rats were treated with the indicated doses of CeONPs as described in the text. Two days or 6 months after treatment, animals were euthanized and tissues collected for ICP-MS analysis of cerium. Results represent mean ± SE for 6 animals.
Thus, low therapeutic single doses may bioaccumulate somewhat differently. The hypothesis for the differential accumulation with dose may relate to removal by the reticuloendothelial system. It is known that upon entering the blood, nanoparticles are rapidly coated with plasma proteins.66 We hypothesize that low doses (5 mg/kg and below) are coated with such proteins allowing their ready distribution to tissues. However once the quantity of particles delivered at one time exceeds the capacity of plasma protein coating, or they are coated with different proteins, the nanoparticles become subject to interaction with the reticuloendothelial and immune systems, and move to different organs such as liver and spleen. Alternatively, the protein coating that results in brain delivery may be limited, and higher doses exceed the coating capacity. This altered biodistribution at high doses may explain why higher doses do not work as effectively in neuroprotection paradigms, as we have observed in our previous studies. It would be interesting to examine the biodistribution with repetitive sub-micromolar doses, to determine if a low repetitive dosing parameter would promote bioaccumulation in the brain, or possibly cause a more even biodistribution, without the majority of material moving to liver and spleen. It is interesting to note that even in cases where nanoparticles were coated, the low distribution to the brain, as opposed to spleen, liver, and kidney, was still evident.49
Our early reports on biodistribution show persistence of CeONPs for at least 3 months.8 Biodistribution was also assessed 6 months after CeONP injection, as shown in Figure 4B, using 0.5 and 5.0 mg/kg doses. In 6 month old controls (which received saline-citrate vehicle), tissue cerium content, except for the lung, was below detection limit. An obvious question that arises is why cerium could be detected in control animals from the 2-day study above, and not in 6 month controls. However one must remember that we are measuring cerium, not the nanoparticles directly. Tissue cerium can arise from many sources including glass leachings from water bottles, water sources, air (particularly if exhaust fumes are present), catalytic converter residues, combustion of fossil fuels, food, bedding, and numerous other sources. Once present in our animal housing facility, we try to maintain a low cerium environment. So during the 6 month housing in our facility, background cerium declined in these animals. It remains unknown as to whether this represents other cerium compounds, or CeONPs from outside sources.
In animals treated with CeONPs at both low (<5 mg/kg) and high (>50 mg/kg) doses, we see persistence of cerium in the tissues at 6 months. In fact, the cerium content of all tissues analyzed for animals treated with CeONP is increased at 6 months, compared to the 2 day study. This is likely due to redistribution from interstitial fluids or other areas of the body not initially analyzed. As can be seen from Figure 4, cerium content of all organs analyzed increased during the 6 month post-administration period. For the low dose, 0.5 mg/kg, the largest increases are in brain, heart, and lung.
At the 5.0 mg/kg dose, we see little change in brain and liver cerium content, but large increases in kidney and spleen concentrations (although there was high variability between animals as evidenced by the large SE). Again, this may be due to redistribution or removal by the reticuloendothelial system. Nonetheless, the biodistribution appears to be dose-dependent.
In summary, although biomedical efficacy for treatment of neurodegenerative disorders is high for CeONPs, the major sites of bioaccumulation of an administered IV dose are liver, kidney, and/or spleen. Again, this may be related to dose, delivery vehicle, functionalizatiom, or nanoparticle size. Nonetheless it appears prudent that further toxicological investigation into CeONPs effects on these organs is necessary.
Biodistribution and the Protein Corona
A critical component of biodistribution, for which there is little information regarding CeONP, is the protein corona. When a nanoparticle enters a biological system, it is well-accepted that it is immediately surrounded by a protein coating.67 However “protein corona” may not be quite descriptive, as the coating likely also contains lipids and sugars. Although a comprehensive discussion of protein coating of nanoparticles is beyond the subject of this review, detailed information is presented in several excellent review publications. 66–69
The lack of translation from in vitro effects of CeONPs to in vivo actions may be due, in part, to the biological coatings that a nanoparticle encounters as it enters the circulation. Such coatings may drive the accumulation of the nanoparticle into different tissues, including crossing the blood-brain barrier.66 Although we know that nanoparticles are coated by plasma proteins and other biomolecules, we have no idea what these proteins, lipids and sugars are in vivo, and it is likely they differ with different nanoparticles, physicochemical characteristics, and coatings. Thus, regardless of the coatings we place on nanoparticles of various sizes dispensed in various vehicles, once they enter body systems, the corona is likely to determine its fate. Some speculate that complement activation, opsonization, and acute phase proteins may coat nanoparticles and mediate inflammation.15 Yet the biological and toxicological studies do not demonstrate inflammation at IV biomedical doses (see below), and no in vivo evidence suggests these coatings will predominate. Adsorption of large, bulky, hydrophilic proteins such as serum albumin have been shown to stabilize some nanoparticles against aggregation, and may be responsible for delivery to the brain.66, 68 Future work will undoubtedly shed light on this issue, but at present, we must be mindful of this phenomenon when comparing different studies with different CeONP constructs.
Toxicity / Safety
Due to their widespread use, need for exposure and toxicity studies with CeONP has been noted. The review by Cassee et al.2 placed emphasis on both CeO microparticles and CeONPs, noting that differences in effects could be expected due to differences in physicochemical properties.
For the present review, in vitro safety studies, even when using neural cells, are not discussed because they are not relevant when whole organism effects are endpoints of value or concern. When applied to cultured cells, consideration of absorption, distribution and elimination are not part of the evaluation, yet it is the contribution of these pharmacokinetic factors that are of importance to the patients that could be given CeONPs as neuroprotectants. In vitro experiments have value, however, for identifying and describing molecular and cellular mechanisms associated with the protective and detrimental effects of CeONP, even if they only provide a small window about what may be occurring in the body as a whole. For example, cytotoxicity, oxidative stress and changes in intracellular signaling have been noted and reported to be different in tissues extracted from exposed animals and then compared to tissues from control rodents.44
Mechanism of toxicity in vivo have been suggested in several reports. This includes measureable endpoints of oxidative stress 30 days after IV administration of 70–85 mg/kg CeONP (ceria) to rats.70,71 Multiple endpoints have been used to identify oxidative stress as the mechanism of toxicity in high-dose studies. For example, increased oxidative stress in the brain was demonstrated as CeONP administration elevated Hsp70 levels in the hippocampus and cerebellum, and 3-nitrostyrosine and iNOS levels in the cortex. Further, the GSH:GSSG ratios were decreased in the hippocampus and cerebellum.72,73 Hardas et al.70 reported that endpoints of oxidative stress were no longer present 90 days after exposure, and recognized that CeONPs have both oxidative and antioxidative properties. In contrast, only antioxidant properties were reported in mice given CeONP by the IV or the intraperitoneal routes at much lower dosages (0.5 mg/kg weekly for 2 to 5 weeks).63
The Yokel group has performed some extensive studies using well-characterized CeONPs at concentrations much higher than those used for neurodegenerative disease, but necessary to establish toxicological limits.50,62,64,65,71–73 These investigators thoroughly characterized the particles they administered by IV infusion, noting difference in effects based on size and time after administration. For their studies, doses of 100 mg/kg were often used, with recognition that this high dose allowed detection by ICP-MS of the administered compound up to 90 days after administration, as that group has interest in CeONP persistence. The product infused was laboratory synthesized, citrate-stabilized to prevent agglomeration, and characterized by transmission electron microscopy (TEM). This dosing paradigm was not detrimental to the rats, as the investigators noted only lower body weight shortly after administration and some white spots seen on the spleen at the time of necropsy.
Yokel et al.74 provide a comprehensive review summarizing systemic effects following CeONP exposure by IV, intraperitoneal, pulmonary, oral, dermal and ocular routes, including identification of gaps in available data collected following single and multiple exposures. This review and other reports70,75–77 noted that CeONP could be detected in brain after inhalation exposure, a most likely route for environmental exposures, but quantities were much less than those measured in lung.
Many toxicity studies with nanomaterials in general have examined effects after the environmentally relative respiratory route of exposure, often by use of bronchoalveolar lavage.78–80 Inflammation and pulmonary fibrosis have been reported. Much of this information is included in the extensive review by Yokel et al.74
Most published studies on the toxicity and/or distribution of CeONPs after administration by extra-respiratory routes have used doses considerably higher than those that are neuroprotective (compare studies described in this section with those described above in neuroprotective section of this review). Oral exposure studies, for example, looked at comparative toxicity of cerium oxide micro and nanoparticles (~190 nm) in rats following high doses (30, 300 and 600 mg/kg/day) for 28 days.81 This would provide cumulative exposures of 0.84, 8.4 or 16.8 grams, which is far above what has been demonstrated to be neuroprotective (see above). The two highest doses of the nanoparticulates resulted in some adverse effects on liver and spleen but nothing was noted after administration of microparticles. However, rats did lose weight as feed intake was reduced. To have such a large proportion of rat food intake consist of CeONPs could very likely diminish their intake of feed with nutrients. Distribution of CeONP to brain occurred after administration of these high doses, genotoxicity of leukocytes and liver cells was reported, and considerable excretion in the urine early after administration was noted. These authors also reported genotoxicity to leuokocytes after a single oral dose of 1 gm/kg, and again noted detrimental effects on liver after CeONP administration.82
The need for high oral doses to demonstrate any deleterious effects suggests that CeONP are not well absorbed from the gastrointestinal tract. This is not unexpected, and has been reported before,74,77 as suspensions in aqueous vehicles used in experiments described in the previous paragraph, are generally less likely to be systemically absorbed than test substance in solution.83 Crossing cell membranes is required for absorption into the systemic circulation, and this usually requires at least limited solubility in the aqueous-based secretions of the gastrointestinal tract or in the vehicle used to administer a drug in solid form. For example, in our Drosophila studies with CeONPs, the CeONPs are mixed with the food. To assure adequate dosing to the flies, we incorporate 0.1% docusate sodium into the CeONP suspension, to ensure adequate dispersal and oral absorption by Drosphila, as discussed above.
Additional studies in which CeONPs have been administered by extrapulmonary routes have been done in mice as well as rats. For example, Poma et al.84 noted increases in weight of Peyer’s patches and numbers of lymphocytes and used these as indicators of inflammation after single oral doses that they stated were 2000 – 5000 mg/kg CeONP. However, review of information presented in tabular form in this manuscript, which provided data on mouse weights, concentration of CeONPs in stock solution and µL injected, suggests that doses administered for acute toxicity testing were actually between 2 and 5 mg/kg. No lethalities occurred during the 7 day post-administration observation period following the single doses used to provide acute toxicity data. Repeated-day experiments used mice given doses the authors noted to be between 0.05 – 5 mg/kg, prepared as a suspension in phosphate buffered saline and given by the intraperitoneal route for 14 days. Volume of injection and concentration of the dosing solution were not provided for these experiments. Following repeated days of dosing, some indices of possible effects on kidneys were reported. Dose response relationships were not linear. Source and size of the ceria dosed were not noted in this manuscript, and CeONP concentrations in tissues were not quantitated. Clinical pathology endpoints were expressed as percentage of variation compared to negative control, making it difficult to interpret results.
The doses used by Hirst et al.63 in their IV, intraperitoneal and oral biodistribution studies presented above are lower than those used in many studies, and are within the range considered neuroprotective as discussed in the previous section of this review. We recently examined safety of CeONP in C557BL/6 mice, which, when dosed with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), provide the animal model for Parkinson’s Disease.85 Mice were given, by IV administration, a single efficacious (neuroprotective) dose of 0.5 µg/g (0.5 mg/kg) and a dose 10-fold higher (5 mg/kg). They were assessed for CeONP-induced neurotoxicity by evaluation of behavioral changes 2, 4, 7, 9, 11 and 14 days after dosing, with tissues collected for histopathological examination 2 and 14 days after CeONP administration. Behavioral assessment was based on a Functional Observational Battery designed for rats, but modified for use in mice.86 The evaluation included comparison of untreated and CeONP-mice for activity, coat and tail condition, presence or absence of ataxia, presence or absence of tremors/convulsions, vocalizations, stool consistency, presence or absence of oral and nasal discharges, piloerection, cyanosis, ability to stay on a rotating wooden rod, body temperature and weight. No differences between control and treated mice were observed; results did not differ whether mice were given 0.5 or 5 mg/kg. Tissues collected for histopathological examination at 2 and 14 days included heart, lung, ribs and mediastinum (containing thymus and/or mediastinal lymph nodes), kidney, adrenal, spleen, liver, large intestine and brain. No CeONP-related lesions were noted at the light microscopic level.
Rats given 0.5 and 5 mg/kg IV CeONP in our laboratory were also examined histopathologically two days and 6 months after IV dosing with the same techniques used for mice given these doses. Other than common background changes seen in adult/older rats, no evidence of CeONP-induced injury was detected.
In summary, studies to date suggest that CeONP have a low profile of toxicity, as single IV doses as high as 100 mg/kg or oral doses even higher did not appear to cause serious detrimental effects in rodents. CeONP appear to have potential, however, to be retained in body organs such as liver and spleen, especially after multiple and/or high doses, but even in such cases notable toxicity to exposed rodents was not clinically evident. Toxicity of nanomaterials such as CeONPs that have potential to be medically useful as neuroprotectants, however, still need evaluation for distribution and interactions inside the body at efficacious doses.87 With neuroprotective doses so much lower (0.5 mg/kg) than those used in toxicity studies, potential to evaluate retention, especially over long term, becomes far more difficult as concentrations approach limits of detection of current analytical instrumentation.
Summary and Future Directions
The research thus far demonstrates a clear utility for CeONPs in neurodegenerative disease, at therapeutic doses. These are seriously debilitating diseases, for which we do not have highly effective treatment. But the question of safety also arises. To date, there is little evidence of toxicity at doses used to treat neurodegenerative diseases. However biodistribution studies clearly demonstrate that the major organs of accumulation are liver, kidney, and spleen. Nonetheless, there has been little to no toxicity demonstrated in these organs at therapeutic doses and accumulated doses in these organs remain low, in the ng/g range. Future studies should encompass investigation of biochemical changes in these organs, such as alterations of signaling pathways, particularly since CeONPs have been shown to persist in these organs, possibly for years. In contrast such persistence is a facet of what enables CeONPs to be highly protective agents in neurodegenerative diseases.
Another issue that needs to be addressed regards the question of how much free radical scavenging is too much? Free radicals are known to participate in many normal signaling events, NO contribution to blood pressure control being one of them.88–90 To date, aside from a series of studies done by Minarchik et al.,43,44 there are few studies examining the effects of CeONPs in the vasculature. Activation of protein phosphatases and function of skeletal muscle also employ free radicals for normal signaling.90 The effects of therapeutic doses on these systems is in need of investigation, particularly in long term studies.
As discussed in a previous section, the actions of CeONPs depend on completion of a reactive cycle, and disruption or stalling of that cycle could theoretically result in accumulation of radical species. Therefore, the biochemical environment of the tissue in which CeONPs reside must also be considered. For example, CeONPs may have different effects on the redox environment in normal brain, where oxidative stress is low, compared to a brain with neurodegenerative disease, where oxidative stress is high. Additional examples comes from studies by Minarchick et al.43 who found differing actions of CeONPs in different vascular beds, some resulting in vasodilation while other displayed vasconstriction and distinct vascular dysfunction. In one study, CeONPs improved vascular function in rats that already had high levels of oxidative stress (the spontaneously hypertensive rat), while having more detrimental actions on vascular dysfunction in controls.43,44 The beneficial effects of CeONPs may be limited to those conditions in which high oxidative stress predominates, such as neurodegenerative disease or hypertension. In this regard, they do not differ from most pharmaceutical agents used today – they are used to treat a disease state. In the case of CeONPs, this state would be high oxidative stress, and benefits of having a superior antioxidant nanopharmaceutical may outweigh the risks of disease. In summary, although addition work needs to be done, CeONPs show promise as a potential nanopharmaceutical in the treatment of neurodegenerative diseases in the brain.
Acknowledgments
Research reported in this publication was supported by the National Institute of Neurological Disorders and Stroke of the National Institutes of Health under award number 7R15NS072873 and by a grant from the Michael J. Fox Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Contributor Information
Beverly A. Rzigalinski, Via College of Osteopathic Medicine, Blacksburg, VA 24060
Charles S. Carfagna, Molecular Materials Discovery Center, Macromolecular Innovations Institute, Virginia Tech, Blacksburg, VA 24061
Marion Ehrich, Virginia-Maryland College of Veterinary Medicine, Blacksburg, Va 24061.
References
- 1.Reed K, Cormack A, Kulkarni A, Mayton M, Savie D, Klaessiq F, Stadler B. Exploring the properties and applications of nanoceria: is there still plenty of room at the bottom? Env Sci Nano. 2014;1:390–405. [Google Scholar]
- 2.Cassee FR, van Balen EC, Singh C, Green D, Muijser H, Weinstein J, Dreher K. Exposure, health and ecological effects review of engineered nanoscale cerium and cerium oxide associated with its use as a fuel additive. Crit Rev Toxicol. 2011;41:213–229. doi: 10.3109/10408444.2010.529105. [DOI] [PubMed] [Google Scholar]
- 3.Eguchi K, Setoguchi T, Inoue T, Arai H. Electrical properties of ceria-based oxides and their applications to solid oxide fuel-cells. Solid-State Ion. 1992;52:165–172. [Google Scholar]
- 4.Tsunekawa S, Sivamohan R, Ohsuna T, Kasuya A, Takahashi H, Tohji K. Ultraviolet absorption spectra of CeO2 nano-particles. Mater Sci Forum. 1999;315–317:439–445. [Google Scholar]
- 5.Izu N, Shin W, Matsubara I, Murayama N. Development of resistive oxygen sensors based on cerium oxide thick film. J Electroceramics. 2004;13:703–706. [Google Scholar]
- 6.Rzigalinski BA, Meehan C, Davis RM, Miles WC, Cohen CA. Radical nanomedicine. Nanomedicine. 2006;1:399–412. doi: 10.2217/17435889.1.4.399. [DOI] [PubMed] [Google Scholar]
- 7.Singh N, Cohen CA, Rzigalinski BA. Cerium oxide nanoparticles are neuroprotective for free radical injury and enhance neuronal longevity. Proc NY Acad Sci. 2007;1122:219–230. [Google Scholar]
- 8.Rzigalinski BA, Danelisen I, Strawn E, Cohen C, Liang C. Biological nanoparticles for cell engineering – a radical concept. In: Kumar C, editor. Nanotechnologies for Life Sciences – Tissue Cell and Organ Engineering. Vol. 9. NY: Wiley & Sons; 2006. pp. 361–380. [Google Scholar]
- 9.Rzigalinski BA, Meehan K, Whiting MD, Dillon CE, Hockey K, Brewer M. Antioxidant nanoparticles. In: Hunter RJ, Preedy VR, editors. Antioxidant Nanoparticles in Nanomedicine in Health and Disease. NY: CRC Press; 2011. [Google Scholar]
- 10.Rzigalinski BA. Nanoparticles & cell longevity. Tech Cancer Res Treatment. 2005;4:651–660. doi: 10.1177/153303460500400609. [DOI] [PubMed] [Google Scholar]
- 11.Eitan E, Hutchinson ER, Nigel HG, Tweedie D, Celik H, Ghosh S, Fishbein KW, Spencer RG, Sasaki CY, Ghosh P, et al. Combination therapy with lenalidomide and nanoceria ameliorates CNS autoimmunity. Exptl Neurol. 2015;273:151–160. doi: 10.1016/j.expneurol.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Estevez AY, Pritchard S, Harper K, Aston JY, Lynch A, Lucky JJ, Ludington JS, Chatani P, Mosental WP, Leiter JC, et al. Neuroprotective mechanisms of cerium oxide nanoparticles in a mouse hippocampal brain slice model of ischemia. Free Rad Biol Med. 2011;51:1155–1163. doi: 10.1016/j.freeradbiomed.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Kim CK, Kim T, Choi I-Y, Soh M, Kim D, Kim Y-J, Jang H, Yang H-S, Kim JY, Park H-K, et al. Ceria nanoparticles that can protect against ischemic stroke. Angewandte Chem. 2012;51:11039–11043. doi: 10.1002/anie.201203780. [DOI] [PubMed] [Google Scholar]
- 14.Estevez AY, Erlichman JS. Cerium oxide nanoparticles for the treatment of neurological oxidative stress diseases. In: Adreescu S, et al., editors. Oxidative Stress: Diagnostics, Prevention, & Therapy. Washington DC: ACS Symposium Series, Amer Chem Soc; 2011. [Google Scholar]
- 15.Walkey C, Das S, Seal S, Erlichman J, Heckman K, Ghibelli L, Taversa E, McGinnis JF, Self WT. Catalytic properties and biomedical applications of cerium oxide nanoparticles. Environ Sci Nano. 2015;2:33–53. doi: 10.1039/C4EN00138A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He L, Su Y, Lanhong J, Shi S. Recent advances of cerium oxide nanoparticles in synthesis, luminescence, and biomedical studies: a review. J Rare Earths. 2015;33:791–799. [Google Scholar]
- 17.Suzuki KT, Kosacki I, Anderson HU. Electrical conductivity and lattice defects in nanocrystalline cerium oxide thin films. J Am Cer Soc. 2001;84:2001–2014. [Google Scholar]
- 18.Aneggi E, Boaro M, de Leitenberg C, Dolcetti G, Trovarellis A. Insights into the redox properties of ceria-based oxides and their implications in catalysis. J Alloys Compounds. 205:406–412. 1096–1102. [Google Scholar]
- 19.Trovarelli A. Catalysis by Ceria and Related materials. London, UK: Imperial College Press; 2002. [Google Scholar]
- 20.Schubert D, Dargusch R, Raitano J, Chan S-W. Cerium and yttrium oxide nanoparticles are neuroprotective. Biochem Biophys Res Comm. 2006;342:86–96. doi: 10.1016/j.bbrc.2006.01.129. [DOI] [PubMed] [Google Scholar]
- 21.Das M, Patil S, Bhargava N, Kang J-F, Riedel LM, Seal S, Hickman JJ. Auto-catalytic ceria nanoparticles offer neuroprotection to adult rat spinal cord neurons. Biomaterials. 2007;28:1918–1925. doi: 10.1016/j.biomaterials.2006.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailey D, Chow L, Merchant S, Kuiry SC, Patil S, Seal S, Rzigalinski BA. Cerium Oxide nanoparticles extend cell longevity and act as free radical scavengers. Nature Biotechnology. 2003;14:112. [Google Scholar]
- 23.Ciofani G, Genchi GG, Mazzolai B, Mattoli V. Transcriptional profile of genes involved in oxidative stress and antioxidant defense in PC12 cells following treatment with cerium oxide nanoparticles. Biochim Biophys Acta. 2014;1840:495–506. doi: 10.1016/j.bbagen.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 24.Ciofani G, Genchi GG, Liakos I, Cappello V, Gemmi M, Athanassiou A, Mazzolai B, Mattoli V. Effects of cerium oxide nanoparticles on PC12 neuronal-like cells: proliferation, differentiation, and dopamine secretion. Pharm Res. 2013;30:2133–2145. doi: 10.1007/s11095-013-1071-y. [DOI] [PubMed] [Google Scholar]
- 25.Deshpande S, Patil S, Kuchibhatla S, Seal S. Size dependency variation in lattice parameter and valency states in nanocrystalline cerium oxide. Applied Phys Lett. 2005;87:133113. [Google Scholar]
- 26.Sardesai SP, Andreescu D, Andreescu S. Electroanalytical evaluation of antioxidant activity of cerium oxide nanoparticles by nanoparticle collisions at microelectrodes. J Am Chem Soc. 2013;135:16770–16773. doi: 10.1021/ja408087s. [DOI] [PubMed] [Google Scholar]
- 27.Dowding JM, Dosai T, Kumar A, Seal S, Self WT. Cerium oxide nanoparticles scavenge nitric oxide radical (NO) Chem Commun. 2012;48:4896–4898. doi: 10.1039/c2cc30485f. [DOI] [PubMed] [Google Scholar]
- 28.Korsvik C, Patil S, Seal S, Self WT. Superoxide dismutase mimetic properties exhibited by vacancy engineered ceria nanoparticles. Chem Commun. 2007;14:1056–1058. doi: 10.1039/b615134e. [DOI] [PubMed] [Google Scholar]
- 29.Pirmohamed T, Dowding JM, Singh S, Wasserman B, Heckert E, Karakoti AS, King JES, Seal S, Self WT. Nanoceria exhibit redox state-dependent catalase mimetic activity. Chem Commun. 2010;46:2736–2738. doi: 10.1039/b922024k. 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heckert EG, Seal S, Self WT. Fenton-like reaction catalyzed by the rare earth inner transition metal cerium. Env Sci & Tech. 2008;42:5014–5019. doi: 10.1021/es8001508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiao X, Song H, Zhao H, Bai W, Zhang L, Li Y. Well-redispersed ceria nanoparticles: promising peroxidase mimetics for H2O2 and glucose detection. Anal Meth. 2012;4:3261. [Google Scholar]
- 32.Tan F, Zhang Y, Wang J, Wei J, Cai Y, Qian X. An efficient method for dephosphorylation of phosphopeptides by cerium oxide. J Mass Spec. 2008;43:2308–2312. doi: 10.1002/jms.1362. [DOI] [PubMed] [Google Scholar]
- 33.Pulido-Reyes G, Rodea-Palomares I, Das S, Sakthivel TS, Leganes F, Rosal R, Seal S, Fernandez-Pinas F. Untangling the biological effects of cerium oxide nanoparticles: the role of surface valence states. Sci Rep. 2015;5:15613–15620. doi: 10.1038/srep15613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng L, He X, Zhang P, Zhang J, Ma Y, Ding Y, Wu Z, Chai Z, Zhang Z. Comparative pulmonary toxicity of two ceria nanoparticles with the same primary size. Int J Mol Sci. 2014;15:6072–6085. doi: 10.3390/ijms15046072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skorodumova NV, Simak SI, Lundqvist BI, Abrikosov IA, Johansson B. Quantum origin of the oxygen storage capability of ceria. Phys Rev Lett. 2002;89:166601. doi: 10.1103/PhysRevLett.89.166601. [DOI] [PubMed] [Google Scholar]
- 36.Celardo I, De Nicola M, Mandoli C, Pedersen JZ, Traversa E, Ghibelli L. Ce3+ ions determine redox-dependent anti-apoptotic effect of cerium oxide nanoparticles. ACS Nano. 2011;5:4537–4549. doi: 10.1021/nn200126a. [DOI] [PubMed] [Google Scholar]
- 37.Celardo I, Pedersen JZ, Traversa E, Ghibelli L. Pharmacological potential of cerium oxide nanoparticles. Nanoscale. 2011;3:1411. doi: 10.1039/c0nr00875c. [DOI] [PubMed] [Google Scholar]
- 38.Fronzi M, Piccinin S, Delley B, Traversa E, Stampfl C. Water adsorption on the stoichiometric and reduced CeO2(111) surface: a first principles investigation. Phys Chem Chem Phys. 2009;11:9188–9199. doi: 10.1039/b901831j. [DOI] [PubMed] [Google Scholar]
- 39.Dunnick KM, Pillai R, Pisane KL, Stefaniak AB, Sabolsky EM, Leonard SS. The effect of cerium oxide nanoparticle valence state on reactive oxygen species and toxicity. Biol Trace Elem Res. 2014;166:96–107. doi: 10.1007/s12011-015-0297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cafun J-D, Kvashnina KO, Casals E, Puntes VT, Glatzel P. Absence of Ce3+ sites in chemically active colloidal ceria nanoparticles. ACS Nano. 2013;7:10726–10732. doi: 10.1021/nn403542p. [DOI] [PubMed] [Google Scholar]
- 41.Gao P, Kang Z, Fu W, Wng W, Bai X, Wang E. Electrically driven redox process in cerium oxides. J Am Chem Soc. 2010;132:4197–4201. doi: 10.1021/ja9086616. [DOI] [PubMed] [Google Scholar]
- 42.Kuchibhatia SVNT, Karakoti AS, Baer DR, Samudrala S, Engelhard MH, Amonette JE, Thevuthasan S, Seal S. Influence of aging and environment on nanoparticle chemistry - Implication to confinement effects in nanoceria. J Phys Chem C Nanomater Interfaces. 2015;116:14108–14114. doi: 10.1021/jp300725s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minarchick VC, Stapleton PA, Fix NR, Leonard SS, Sabolsky EM, Nurkiewicz TR. Intravenous and gastric cerium dioxide nanoparticle exposure disrupts microvascular smooth muscle signaling. Toxicol Sci. 2015;144:77–89. doi: 10.1093/toxsci/kfu256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Minarchick VC, Stapleton PA, Sabolsky EM, Nurkiewicz TR. Cerium dioxide nanoparticle exposure improves microvascular dysfunction and reduces oxidative stress in spontaneously hypertensive rats. Front Physiol. 2015;6:1–12. doi: 10.3389/fphys.2015.00339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang S, Li L, Van der Biest G, Vleugels J. Influence of oxygen partial pressure on the reduction of CeO2 and CeO2-ZrO2 ceramics. Solid State Sci. 2005 [Google Scholar]
- 46.Spulber M, Baumann P, Liu J, Palivan G. Ceria loaded nanoreactors: a nontoxic superantioxidant system with high stability and efficacy. Nanoscale. 2015;7:1411–1420. doi: 10.1039/c4nr02748e. [DOI] [PubMed] [Google Scholar]
- 47.Esch F, Fabris S, Zhou L, Montini T, Africh C, Fornasiero P, Comelli G, Rosei R. Electron localization determines defect formation on ceria substrates. Science. 2005;309:752–755. doi: 10.1126/science.1111568. [DOI] [PubMed] [Google Scholar]
- 48.Xue Y, Zhai Y, Zhou K, Wang L, Tan H, Luan Q, Yao X. The vital role of buffer anions in the antioxidant activity of CeO2 nanoparticles. Chem Eur J. 2012;18:11115–11122. doi: 10.1002/chem.201200983. [DOI] [PubMed] [Google Scholar]
- 49.Heckman KL, DeCoteau W, Estevez A, Reed KJ, Costanzo W, Sanford D, Letter JC, Clauss J, Knapp K, Gomez C, Mullen P, et al. Custom cerium oxide nanoparticles protect against a free radical mediated autoimmune degenerative disease in the brain. ACS Nano. 2013;7:10582–10596. doi: 10.1021/nn403743b. [DOI] [PubMed] [Google Scholar]
- 50.Hardas SS, Sultana R, Warrier G, Dan M, Florence RL, Wu P, Grulke EA, Tseng MT, Unrine JM, Graham UM, Yokel RA, Butterfield DA. Rat brain pro-oxidant effects of peripheraaly administered 5 nm ceria 30 days after exposure. Nanotoxicology. 2012;33:1147–1155. doi: 10.1016/j.neuro.2012.06.007. [DOI] [PubMed] [Google Scholar]
- 51.Halliwell B. Oxidative stress in neurodegeneration: where are we now? J Neurochem. 2006;97:1634–1658. doi: 10.1111/j.1471-4159.2006.03907.x. [DOI] [PubMed] [Google Scholar]
- 52.Sayes CM, Warheit DB. Characterization of nanomaterials for toxicity assessment. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2009 Nov-Dec;(6):660–670. doi: 10.1002/wnan.58. [DOI] [PubMed] [Google Scholar]
- 53.Yoshida K, Burton GF, McKinney JS, Young H, Ellis EF. Brain and tissue distribution of polyethylene glycol-conjugated superoxde dismutase in rats. Stroke. 1992;6:865–869. doi: 10.1161/01.str.23.6.865. [DOI] [PubMed] [Google Scholar]
- 54.Fiorani L, Passacantando M, Santucci S, Di Marco S, Bisti S, Maccarone R. Cerium oxide nanoparticles reduced microglial activatgion and neurodegeneration events in light damaged retina. PLOS One. 2010;10:e0140387. doi: 10.1371/journal.pone.0140387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cai X, Sezate SA, Seal S, McGinnins JF. Sustained protection against photoreceptor degeneration in tubby mice by intravitreal injection of nanoceria. Biomaterials. 2012;33:8771–8781. doi: 10.1016/j.biomaterials.2012.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cai X, Seal S, McGinnis JF. Sustained inhibition of neovascularization in vldlr−/− mice following intravitreal injection of cerium oxide nanoparticles and the role of the ASK1-P38/JNK-NF-kB pathway. Biomaterials. 2014;35:249–258. doi: 10.1016/j.biomaterials.2013.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou X, Wong LL, Karakoti AS, Seal S, McGinnis JF. Nanoceria inhibit the development and promote the regression of pathologic retinal neovascularization in the Vldlr knockout mouse. PLOS One. 2011;6:e16733. doi: 10.1371/journal.pone.0016733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wong LL, Pye QN, Chen L, Seal S, McGinnis JF. Defining the catalytic activity of nanoceria in the P23H-1 rat, a photoreceptor degeneration model. PLOS One. 2015;10:e0121977. doi: 10.1371/journal.pone.0121977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dowding JM, Song W, Bossy K, Karakoti A, Kumar A, Kim A, Bossy B, Seal S, Ellisman MH, Self WT, Bossy-Wetzel E. Cerium oxide nanoparticles protect against Ab-induced mitochondrial fragmentation and neuronal cell death. Cell Death Diff. 2105;21:1622–1632. doi: 10.1038/cdd.2014.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Frey A, Bates JA, Sholar CA, Hockey KS, Rzigalinski BA. Cerium oxide nanoparticles as a disease-modifying therapy for Parkinson’s disease (#199.01) Neuroscience. 2014 [Google Scholar]
- 61.Whiting MD, Rzigalinski BA, Ross JS. Cerium oxide nanoparticles improve neuropathological and functional outcome following traumatic brain injury. J Neurotrauma. 2009;26:101. [Google Scholar]
- 62.Yokel RA, Tseng MT, Dan M, Unrine JM, Graham UM, Peng Wu, Grulke EA. Biodistribution and biopersistence of ceria engineered nanomaterials: size dependence. Nanomedicine: Nanotech Biol Med. 2013;9:398–407. doi: 10.1016/j.nano.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 63.Hirst SM, Karakota A, Sing S, Self W, Tyler R, Seal S, Reilly CM. Bio-distribution and in vivo antioxidant effects of cerium oxide nanoparticles in mice. Environ Toxicol. 2013;28:107–118. doi: 10.1002/tox.20704. [DOI] [PubMed] [Google Scholar]
- 64.Tseng MT, Fu Q, Lor K, Fernandez-Botran R, Deng Z-H, Graham U, Butterfield DA, Grulke EA, Yokel RA. Resistant hepatic structural alterations following nanoceria vascular infusion in the rat. Toxicologic Pathol. 2014;42:984–996. doi: 10.1177/0192623313505780. [DOI] [PubMed] [Google Scholar]
- 65.Graham UM, Tseng MT, Jasinski JB, Yokel RA, Unrine JM, Davis BH, Dozier AK, Hardas SS, Sultana R, Grulke EA, Butterfield DA. In vivo processing of ceria nanoparticles inside liver: Impact on free-radical scavenging activity and oxidative stress. Chempluschem. 2014;79:1083–1088. doi: 10.1002/cplu.201402080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Monopoli MP, Aberg C, Salvati A, Dawson KA. Biomolecular coronas provide the biological identity of nanosized materials. Nat Nano. 2012;7:779–786. doi: 10.1038/nnano.2012.207. [DOI] [PubMed] [Google Scholar]
- 67.Lynch I, Cedervall T, Lundqvist M, Cabaleiro-Lago C, Linse S, Dawson KA. The nanoparticle-protein complex as a biological entity: a complex fluids and surface science challenge for the 21st century. Adv Colloid Interface Sci. 2007:167–174. doi: 10.1016/j.cis.2007.04.021. [DOI] [PubMed] [Google Scholar]
- 68.Cedervall T, Lynch I, Lindman S, Berggard, Thulin E, Nilsson H, Dawson KA, Linse S. Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparricles. Proc Natl Acad Sci. 2007;104:2050–2055. doi: 10.1073/pnas.0608582104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, Dawson KA. Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc Natl Acad Sci. 2008;105:14265–14270. doi: 10.1073/pnas.0805135105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hardas SS, Sultana S, Warrier G, Dan M, Wu P, Grulke EA, Tseng MT, Unrine JM, Graham UM, Yokel RA, Butterfield DA. Rat hippocampal responses up to 90 days after a single nanoceria dose extends a hierarchical oxidative stress model for nanoparticle toxicity. Nanotoxicology. 2014;8:153–166. doi: 10.3109/17435390.2013.868059. [DOI] [PubMed] [Google Scholar]
- 71.Tseng MT, Lu X, Duan X, Hardas SS, Sultana R, Wu P, Unrine JM, Graham U, Butterfield DA, Grulke EA, Yokel RA. Alteration of hepatic structure and oxidative stress induced by intravenous nanoceria. Toxicol Appl Pharmacol. 2012;260:173–182. doi: 10.1016/j.taap.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 72.Hardas SS, Butterfield DA, Sultana R, Tseng MT, Dan M, Florence RL, Unrine JM, Graham UM, Pen W, Grulke EA, Yokel RA. Brain distribution and toxicological evaluation of a systemically delivered engineered nanosacale ceria. Toxicol Sci. 2010;116:562–576. doi: 10.1093/toxsci/kfq137. [DOI] [PubMed] [Google Scholar]
- 73.Yokel RA, Au TC, MacPhail R, Hardas SS, Butterfield DA, Sultana R, Goodman M, Tseng MT, Dan M, Haghnazu H, Unrine JM, Graham UM, Wu P, Grulke EA. Distribution, elimination and biopersistence to 90 of a systemically introduced 30 nm ceria-engineered nanomaterial in rats. Toxicol Sci. 2012;127:256–268. doi: 10.1093/toxsci/kfs067. [DOI] [PubMed] [Google Scholar]
- 74.Yokel RA, Hussain S, Garantziotis S, Demokritou P, Castranova V, Cassee FR. The yin: an adverse health perspective of nanoceria: uptake, distribution, accumulation, and mechanisms of its toxicity. Environ Sci Nano. 2014;1:406–428. doi: 10.1039/C4EN00039K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aalapati S, Ganapathy S, Manapuram S, Anumolu G, Prakya BM. Toxicity and bio-accumulation of inhaled cerium oxide nanoparticles in CD1 mice. Nanotoxicology. 2014;48:786–798. doi: 10.3109/17435390.2013.829877. [DOI] [PubMed] [Google Scholar]
- 76.Geraets L, Oomen AG, Schroeter JD, Coleman VA, Cassee FR. Tissue distribution of inhaled micro- and nano-sized cerium oxide particles in rats: results from a 28-day exposure study. Toxicol. Sci. 2012;127:463–473. doi: 10.1093/toxsci/kfs113. [DOI] [PubMed] [Google Scholar]
- 77.He X, Zhang H, Ma Y, Bai W, Zhang Z, Lu K, Ding Y, Zhao Y, Chai Z. Lung deposition and extrapulmonary translocation of nano-ceria after intratracheal instillation. Nanotechnology. 2010;21:285103. doi: 10.1088/0957-4484/21/28/285103. 8 pp (on line) [DOI] [PubMed] [Google Scholar]
- 78.Ma JY, Mercer RR, Barger M, Schwegler-Berry D, Scabilloni J, Ma JK, Castranova V. Induction of pulmonary fibrosis by cerium oxide nanoparticles. Toxicol Appl Pharmacol. 2012;262:255–264. doi: 10.1016/j.taap.2012.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ma JY, Young SH, Mercer RR, Barger M, Schwegler-Berry D, Ma JK, Castranova V. Interactive effects of cerium oxide and diesel exhaust nanoparticles on inducing pulmonary fibrosis. Toxicol Appl Pharmacol. 2014;278:135–147. doi: 10.1016/j.taap.2014.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ma J, Mercer RR, Barger M, Schwegler-Berry D, Cohen JM, Demokritou P, Castranova V. Effects of amorphous silica coating on cerium oxide nanoparticles induced pulmonary resonses. Toxicol Appl Pharmacol. 2015;88:63–73. doi: 10.1016/j.taap.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kumari M, Kumari I, Grover P. Genotoxicity analysis of cerium oxide micro and nanoparticles in Wistar rats after 28 days of repeated oral administration. Mutagenesis. 2014;29:467–479. doi: 10.1093/mutage/geu038. [DOI] [PubMed] [Google Scholar]
- 82.Kumari M, Kumari SI, Kamal SSK, Grover P. Genotoxicity assessment of cerium oxide nanoparticles in female Wistar rats after acute oral exposure. Mutation Res. 2014;775–775:7–19. doi: 10.1016/j.mrgentox.2014.09.009. [DOI] [PubMed] [Google Scholar]
- 83.Buxton ILO, Benet LZ. Pharmacokinetics: the dynamics of drug absorption, distribution, metabolism, and elimination. In: Brunton LL, Chabner BS, Knollman BC, editors. Goodman & Gilman’s The Pharmacological Basis of Therapeutics. 2011. pp. 17–39. [Google Scholar]
- 84.Poma A, Ragnelli AM, de Lapuente J, Ramos D, Borras M, Aimola P, Di Gioacchino M, Santucci S, De Marzi L. J Immunol Res Article ID. 2014:361419. doi: 10.1155/2014/361419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Jortner BS, Ehrich M, Hinckley J, Rzigalinski BA. Safety studies on a cerium containing nanomaterial. The Toxicologist. 144:507. (abstract 2357) [Google Scholar]
- 86.Moser VC. Neurobehavioral screen in rodents. In: Maines MD, editor. Current Protocols in Toxicology. NY: John Wiley & Sons; 1999. pp. 11.2.1–11.2.16. [Google Scholar]
- 87.Howes RM. The free radical fantasy: a panoply of paradoxes. Ann NY Acad Sci. 2006;1067:22–26. doi: 10.1196/annals.1354.004. [DOI] [PubMed] [Google Scholar]
- 88.Zhao J, Castranova V. Toxicology of nanomaterials used in nanomedicine. J. Toxicol Environ Health B Crit Rev. 2011;14:593–632. doi: 10.1080/10937404.2011.615113. [DOI] [PubMed] [Google Scholar]
- 89.Tonks NK. Redox redux: revisiting PTP’s and the control of cell signaling. Cell. 2005;121:667–670. doi: 10.1016/j.cell.2005.05.016. [DOI] [PubMed] [Google Scholar]
- 90.Barbieri E, Sestili P. Reactive oxygen species in skeletal muscle signaling. J Signal Transduc. 2012:982794–928299. doi: 10.1155/2012/982794. [DOI] [PMC free article] [PubMed] [Google Scholar]