

Summary
Immune cells in the tumor microenvironment modulate cancer progression and are attractive therapeutic targets. Macrophages and T cells are key components of the microenvironment, yet their phenotypes and relationships in this ecosystem and to clinical outcomes are ill defined. We used mass cytometry with extensive antibody panels to perform in-depth immune profiling of samples from 73 clear cell renal cell carcinoma (ccRCC) patients and five healthy controls. In 3.5 million measured cells, we identified 17 tumor-associated macrophage phenotypes, 22 T cell phenotypes, and a distinct immune composition correlated with progression-free survival, thereby presenting an in-depth human atlas of the immune tumor microenvironment in this disease. This study revealed potential biomarkers and targets for immunotherapy development and validated tools that can be used for immune profiling of other tumor types.
Keywords: immune cell atlas, immunosuppression, clear cell renal cell carcinoma, T cell, macrophage, single-cell analysis, mass cytometry
Graphical Abstract
Highlights
-
•
Mass cytometry reveals the immune cell diversity of the ccRCC tumor ecosystem
-
•
PD-1+ cells display heterogeneous combinations of inhibitory receptors
-
•
CD38+CD204+CD206− tumor-associated macrophages correlate with immunosuppression
-
•
A specific immune signature is linked to shorter progression-free survival
Applying mass cytometry for high-dimensional single-cell analysis depicts an in-depth atlas of the immune microenvironment in clear cell renal cell carcinoma patients, thereby linking immune compositions with clinical features.
Introduction
Cells of the adaptive and innate immune systems infiltrate the tumor microenvironment (TME) and form an ecosystem that modulates all aspects of tumor development (Grivennikov et al., 2010). T cells are the most abundant and best-characterized population in the TME of solid tumors (Galon et al., 2006, Vesely et al., 2011). CD4+ helper T cells and cytotoxic CD8+ T cells can prevent tumor growth by targeting antigenic tumor cells, and high numbers of activated CD8+ T cells are associated with good prognosis in various cancers (Vesely et al., 2011). Tumors can lose their immunogenicity, however, and the tumor milieu can suppress T cell responses. Suppression may involve regulatory T cells, which secrete immunosuppressive cytokines and myeloid and stromal cells, which modulate immune checkpoints by activation of co-inhibitory receptors (e.g., PD-1, Tim-3, and CTLA-4) on T cells resulting in dysfunctional, exhausted T cell phenotypes (Speiser et al., 2016, Wherry and Kurachi, 2015).
Tumor-associated macrophages (TAMs) are another key immune population in the TME that can either block or facilitate tumor growth (Kitamura et al., 2015, Sica et al., 2008). Distinct TAM subsets can induce or repress anti-tumor immunity, angiogenesis, and cell migration (Murray et al., 2014, Qian and Pollard, 2010, Quatromoni and Eruslanov, 2012). TAM phenotypes are highly plastic, and recent reports show that the model distinguishing between classically polarized anti-tumor M1 and alternatively polarized pro-tumor M2 subtypes incompletely accounts for the phenotypic diversity in vivo (Ginhoux et al., 2016).
T cell and TAM phenotypes are attractive biomarkers (Daud et al., 2016, Galon et al., 2014, Ostuni et al., 2015), and both T cells and TAMs show promise as therapeutic targets (Maus and June, 2016, Ries et al., 2014, Shin and Ribas, 2015). Treatment with anti-PD-1 and anti-CTLA-4 antibodies can overcome T cell exhaustion in different cancer types (Shin and Ribas, 2015), and clinical trials are investigating the effect of depletion of TAMs and the repolarization of pro-tumor TAMs into anti-tumor TAMs (Pyonteck et al., 2013, Ries et al., 2014). Broader use of T cells and macrophages as biomarkers and drug targets has been hindered by the fact that human TAM and T cell phenotypes and relationships among them in the TME have not been comprehensively characterized. Characterization of TAMs in the TME has focused on a handful of markers (Komohara et al., 2013, Reinartz et al., 2014, Yang et al., 2015), and gene expression profiling has been done in bulk tissues or total macrophage populations (Dannenmann et al., 2013, Doedens et al., 2010). Single-cell RNA sequencing provides high-dimensional, single-cell data (Tirosh et al., 2016), yet the low numbers of cells analyzed mean that this technology currently cannot capture cellular complexity present in a tumor and is not yet suitable for studies of large patient cohorts.
In order to comprehensively analyze the immune landscape in the TME, millions of cells from human tumors must be characterized by simultaneous analysis of dozens of markers (Noy and Pollard, 2014, Quatromoni and Eruslanov, 2012). Mass cytometry enables the quantification of more than 50 readouts at the single-cell level by combining metal isotope-labeled antibodies with mass spectrometry detection (Bandura et al., 2009, Ornatsky et al., 2010). With its ability to analyze millions of cells in a short time at low cost, mass cytometry represents the method of choice to assess the phenotypic diversity of T cells and TAMs present in the TME in large patient cohorts.
In this study, we present a mass cytometry-based atlas of the immune landscape in tumor samples from 73 clear cell renal cell carcinoma (ccRCC) patients and five normal kidney controls. ccRCC is a common and lethal uro-genital cancer (Koul et al., 2011). Around 50% of ccRCC patients develop metastases despite treatment, and less than 10% of these patients survive 5 years. Targeted treatments such as tyrosine kinase inhibitors are only palliative (Koul et al., 2011), but novel checkpoint immunotherapies show considerable promise in some patients (Joseph et al., 2017). Thus new avenues to guide immunotherapy-based treatments are urgently needed (Shinohara and Abe, 2015). We used unsupervised computational approaches to reveal an unprecedented phenotypic complexity in TMEs in ccRCC patient samples. Our data expand the view of T cell immunosuppression phenotypes, suggest links between a TAM phenotype and populations of regulatory T cells and CD8+ immunosuppressed T cells, and identify an immune cell composition within the TME that is correlated with progression-free survival.
Results
In-Depth Immunophenotyping of ccRCC Tumor Samples Using Mass Cytometry
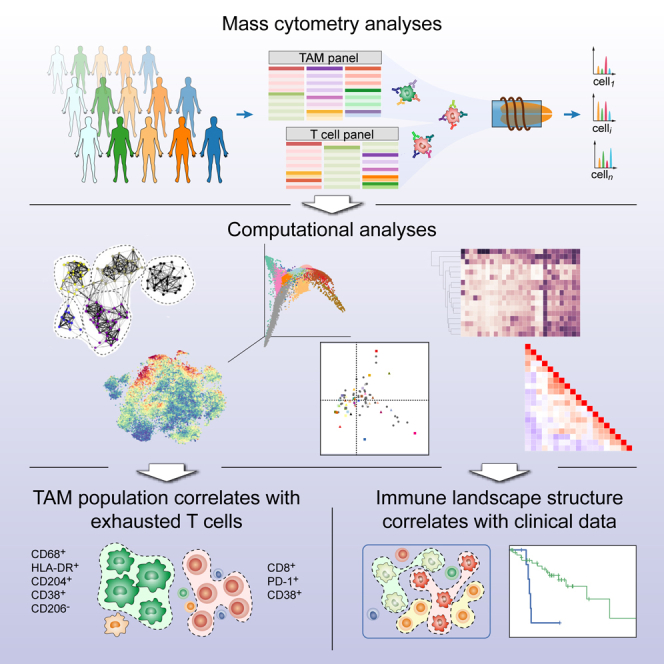
We performed a large-scale mass cytometry analysis of 73 tumor samples from patients with all grades of ccRCC and five healthy matched kidney samples (Figure 1A; Table S1). We stained cells with two antibody panels created for this study (Figures 1B and 1C; Table S2; STAR Methods). Since TAM phenotypes are little characterized in human, the TAM panel originated from an antibody screen (Figures 1B, S1A, and S1B), whereas the T cell panel (Figure 1C) was designed to identify different populations of naive, memory, effector, regulatory, and exhausted T cells. Comparison of mass cytometry to flow cytometry showed an average correlation of 0.88, confirming the reliability of the mass cytometry data (Figure S1C). Both panels included markers for the identification of B cells, natural killer cells, plasma cells, granulocytes, and myeloid cells. The samples were barcoded (Bodenmiller et al., 2012, Zunder et al., 2015), and standards analyzed on each plate (Figure S2A) demonstrated that the data were highly consistent across plates (Figures S2B and S2C).
Figure 1.

Characterization of T cells and TAMs in the ccRCC TME Using Mass Cytometry
(A) Experimental approach used in this study.
(B) Markers used to characterize TAM phenotypes.
(C) Markers used to characterize T cell phenotypes. Ag, antigen; DC, dendritic cells; pDC, plasmacytoid dendritic cells; NK, natural killer; R, receptor; SR, scavenger receptor; TCR, T cell receptor; TLR, toll-like receptor.
Figure S1.

Comparison of FACS and Mass Cytometry Analyses, Related to Figure 1
(A) Scheme illustrating the in vitro system used to generate the monocyte-derived macrophage (MDM) populations from monocytes isolated from the blood of healthy donors.
(B) Heatmap showing the mean expression of the indicated surface markers among the different MDMs and blood monocyte populations. Only markers showing a 2-fold change among the MDM populations were included.
(C) Correlations between FACS and mass cytometry measurements for the 33 antibodies present both in the mass cytometry antibody panel and in the cell surface flow cytometry antibody screen were assessed using a linear regression model. The coefficient of determination R2 and the linear model are shown for each antibody. MFI, mean fluorescent intensity; MC, metal count.
Figure S2.

Consistency of Mass Cytometry Data, Related to Figure 2
(A) Schematic representation of the experimental approach used to stain cells from all normal and patient samples with two antibody panels after barcoding on five plates.
(B) t-SNE maps derived from the standard cells measured on each of the five plates after staining with the T cell panel (upper panel) and the TAM panel (lower panel).
(C) Histogram overlays showing the expression of the markers included in the TAM and T cell panels on the standard cells measured on each of the five plates. Only markers with positive expression on the standard cells are shown.
(D) Gating strategy to identify cells (upper panel), live cells (middle panel), and gadolinium-negative cells (lower panel).
Immune Landscape in ccRCC
To generate a comprehensive view of the immune ecosystem of each tumor, we generated two-dimensional maps of the data using the dimensionality reduction algorithm t-SNE (for gating strategy, see Figure S2D) (Amir et al., 2013, van der Maaten and Hinton, 2008). This analysis showed a strong overlap between tumors of all grades (Figures 2A and 2B). To partition the cells into distinct phenotypes, we applied the PhenoGraph clustering algorithm (Levine et al., 2015), which takes into account phenotypic adjacency of cells in high-dimensional space and is suited for clustering of single-cell data (Levine et al., 2015). This analysis identified the main immune cell types in both T cell and TAM data (Figures 2C–2F, S3A, and S3B). Contour plots usually used to identify cell populations in manual gates confirmed identities (Figure 2G). PhenoGraph analyses revealed highly similar frequencies across plates and across panels, highlighting the robustness of the dataset and analysis (Figures S3C and S3D).
Figure 2.

Identification of the Main Immune Components of the ccRCC TME
(A and B) t-SNE maps displaying 100,000 cells from the ccRCC cohort analyzed with (A) T cell and (B) TAM panels and colored by grade.
(C and D) t-SNE maps displaying 100,000 cells from the ccRCC cohort analyzed with (C) T cell and (D) TAM panels and colored by the main cell populations based on manual annotation of PhenoGraph clustering.
(E and F) Heatmaps showing normalized expression of the markers from (E) T cell and (F) TAM panels for PhenoGraph clusters. Clusters are grouped by expression profiles and cell types are indicated by color. The cluster IDs and relative frequencies are displayed as a bar graph on the right.
(G) Contour plots for the indicated markers demonstrate the “purity” of the main cell types identified with the T cell panel and with the TAM panel.
(H) Dot plots showing the population frequency for each ccRCC sample among all immune cell types (left panel) and among T cell subsets (right panel). Dots are colored by grade. DC, dendritic cells; pDC, plasmacytoid dendritic cells; NK, natural killer; DP, double positive; DN, double negative; CD45-, CD45-CD3+ cells.
See also Figures S2 and S3.
Figure S3.

Consistency between Immune Populations Identified Using TAM and T Cell Panels, Related to Figure 2
(A) t-SNE map showing the expression of each marker included in the T cell panel after a 0 to 1 normalization based on the 99th percentile.
(B) t-SNE map showing the expression of each marker included in the TAM cell panel after a 0 to 1 normalization based on the 99th percentile.
(C) Scatterplot showing the frequencies of the indicated populations identified with the T cell panel (upper panel) and with the TAM panel (lower panel) for sample 26, which was loaded in duplicate on two different plates.
(D) Scatterplots showing the correlations for the indicated immune cell population frequencies established by automatic cell detection based on the T cell panel and the TAM panel. For each relationship, the R2 and the linear models are indicated. DC, dendritic cells, pDC, plasmacytoid dendritic cells, NK, natural killer.
T cells were the main immune cell population in the ccRCC TME, with a mean of 51% across samples (Figure 2H, left panel) (Geissler et al., 2015). The mean frequencies of myeloid cells, natural killer cells, and B cells were 31%, 9%, and 4%, respectively (Figure 2H, left panel). Granulocytes were present at very low levels in all but one sample. Plasma cells constituted a minor fraction in most samples. A double-positive CD8+/CD4+ population was observed in many samples, reaching up to 25% of the T cell compartment in some samples (Figure 2H, right panel).
T Cell Characterization Reveals Various Phenotypes Associated with Immunosuppression
When data have rich population structure, t-SNE and PhenoGraph find dominant phenotypes; finer granularity of structures is found by separate analyses of these dominant phenotypes. To exhaustively map cell phenotypes, we performed additional PhenoGraph analyses focused on each T cell and TAM subset defined in our initial analyses (Figures 2E and 2F). The expression profiles of the T cell PhenoGraph clusters were visualized in a heatmap (Figure 3A) and heterogeneity in marker level was assessed at the single-cell level using t-SNE (Figures 3B and 3C). This approach led to the identification of eight CD4+ phenotypes, 11 CD8+ phenotypes, one CD4+/CD8+ double-positive phenotype and one double-negative phenotype (Figure 3A). We observed highly similar phenotypes among CD4+ and CD8+ subsets including cells expressing CD127+, CD11b+, PD-1−/CD11b−, and CTLA-4+ (Figure 3D). Regulatory T cells (T-6) were mostly observed among the CD4+ subset and were defined by the co-expression of CD25, Foxp3, and CTLA-4 and the absence of CD127.
Figure 3.

In-Depth Characterization of the T Cell Compartment
(A) Heatmap showing normalized expression of the T cell panel markers for the 22 T cell clusters identified with PhenoGraph. Clusters are grouped by expression profile and were manually assigned to the main T cell subsets as indicated with the color code. The cluster IDs and relative frequencies are displayed as a bar graph on the right.
(B) t-SNE map displaying 2,000 cells from each PhenoGraph cluster identified in (A) colored by cluster.
(C) Cells colored by normalized expression of indicated markers on the t-SNE map.
(D) Contour plots showing expression of indicated markers for eight clusters with similar phenotypes in CD4+ and CD8+ T cell populations.
(E) Histograms showing expression of indicated costimulatory molecules, activation markers, and cell cycle markers of clusters of PD-1-expressing CD8+ cells (upper panel), CD4+ cells (middle panel), and CD8+/CD4+ double-positive cells (lower panel).
(F) Boxplots showing the frequencies of the indicated T cell clusters grouped into early stages (normal tissue and grade I) and late stages (grades II–IV and metastatic tissue). p values calculated with a Mann-Whitney-Wilcoxon test are shown for each cluster.
PD-1+ cells were observed in both CD8+ and CD4+ subsets (Figure 3E). The population T-0, characterized by the highest level of PD-1 expression among CD8+ cells, was also strongly positive for other co-inhibitory receptor (such as Tim-3), the activation markers CD38 and HLA-DR, and the co-stimulatory receptors ICOS and 4-1BB but had low levels of CD127 (Figures 3A and 3E). This phenotype was previously associated with exhausted T cells and anti-PD-1 treatment response (Ahmadzadeh et al., 2009, Daud et al., 2016).
Subsets with similar levels of PD-1 differed in expression of activation markers and co-stimulatory receptors (Figure 3E). The cluster T-1 had the same expression pattern as T-0 but markers were expressed at lower levels. The T-1 cells could correspond to precursors of a fully exhausted T-0 population, since during exhaustion inhibitory receptor expression progressively increases (Wherry, 2011). The other PD-1+ clusters (T-7, T-5, T-16, T-9, and T-19) were characterized by the absence of Tim-3 expression and by heterogeneity in expression of markers such as 4-1BB, CD38, CTLA-4, and OX-40. Except for T-16 and T-19, all clusters expressed CD38, a marker not previously associated with T cell exhaustion in cancer. Flow cytometry and transcriptome analysis of sorted populations confirmed co-expression of PD-1 with CD38 (Figures S4A–S4D; Table S3).
Figure S4.

In-Depth Characterization of T Cell Subsets, Related to Figure 3
(A) Histogram showing PD-1 expression on FACS-analyzed CD8 T cells identified as live cells, CD45+, CD3+, and CD8+ (top panel). The expression of CD38 among PD-1− and PD-1+ cells is shown as overlaid histograms (bottom panel).
(B) Dot plot showing the correlation between CD38 and PD-1 expression on 14 different PD-1+ CD8 T cell populations analyzed by FACS and gated as described in (A).
(C) Scatterplots showing the gating strategy used to sort CD8+/PD-1− and CD8+/PD-1+ cells.
(D) Boxplots showing the reads per kilobase of transcript per million mapped reads (rpkm) for PD-1 (PDCD1) and CD38 based on 6 samples of CD8+/PD-1+ and CD8+/PD-1- sorted cells analyzed by RNA-seq. The p values calculated with a Mann-Whitney-Wilcoxon test are shown.
(E) Boxplots showing the frequencies of the different T cell clusters by ccRCC grade and in normal and metastatic tissues.
Within the CD4+ T cell subset, one PD-1+ population (T-18) was observed (Figure 3E). Cells in the T-18 cluster co-expressed CD38 and Tim-3, but ICOS and 4-1BB were at lower levels than in the T-0 cluster. Populations of CD4+ cells that express CTLA-4 (T-13) and OX-40 (T-17) were also identified, but the level of PD-1 was lower on these cells than on their CD8+ counterparts (Figure 3E).
The marker expression pattern of the double-positive PD-1+ cluster T-8 was highly similar to that of the T-0 cluster (Figure 3E). Both cytotoxic and immunosuppressive roles have been suggested for this phenotype (Overgaard et al., 2015). In our ccRCC samples, this population exhibits an exhausted phenotype. Neither LAG-3 nor GITR were observed in any T cell population investigated, suggesting that the expression of these modulatory receptors is context specific (Figure 3A).
When cluster frequencies were analyzed based on tumor grade (Figures 3F and S4E), we observed that normal samples were positive for CD4+ central memory T cells (T-2), CD4+ and CD8+ effector memory cells (T-3, T-4), and CD8+/CD45RA+ T cells (T-14). Regulatory T cells (T-6) and PD-1+ clusters (T-0, T-1) were virtually absent from normal samples but were present in ccRCC samples at different levels depending on grade and in metastatic samples (Figures 3F and S4E). Collectively, these data reveal an unexpected phenotypic diversity among PD-1-expressing T cells present in samples from ccRCC tumors.
High-Resolution Analysis of TAMs Reveals Phenotypic Diversity of Tissue-Resident and Tumor-Specific Macrophages
TAM populations were characterized by more subtle differences in marker expression than T cell populations and were more challenging to categorize. To ensure a robust set of clusters, we developed a subsampling procedure to obtain a consensus PhenoGraph solution (STAR Methods) that led to the identification of 17 TAM phenotypes (Figures 4A and 4B). Visualization of single cells on the t-SNE map confirmed a continuum of expression for most markers; a few (CD123, CD169, CD163, CD204, and CD206) had bimodal expression (Figure 4C).
Figure 4.

In-Depth Characterization of TAM Phenotypes
(A) Heatmap showing normalized expression of markers from the TAM panel for the 17 cell clusters identified with PhenoGraph. Relative frequencies are displayed as a bar graph to the right.
(B) t-SNE map, colored by clusters, displaying 2,000 cells from each PhenoGraph cluster identified in (A).
(C) Normalized expression of indicated markers on the t-SNE map.
(D) Visualization of blood monocytes and TAM clusters using first, second, and third components of a diffusion map. Cells are colored by PhenoGraph clusters. The three main branches are indicated with solid arrows and two sets of clusters are circled with dashed lines.
(E) Boxplots showing the frequencies of the TAM clusters grouped in early stage (normal and grade 1) and advanced stage (grades 2–4 and metastatic) samples. p values calculated with a Mann-Whitney-Wilcoxon test are shown for each cluster.
(F) Conditional mean expression of the indicated markers along diffusion component one (top panel). Histograms displaying the TAM clusters used to calculate the conditional mean expression along diffusion component one (bottom panel).
(G) Histogram overlay showing expression of indicated markers of TAM clusters.
Others have suggested that macrophages form a spectrum of phenotypically related cell subsets (Ginhoux et al., 2016, Murray et al., 2014, Xue et al., 2014). To identify potential relationships among the TAM phenotypes, we used diffusion mapping, a method originally proposed to align cells along branching developmental paths (Haghverdi et al., 2015). Blood monocytes were included as a reference population. The resulting three-dimensional diffusion plot has three main branches (Figure 4D). The TAM groupings on these branches are consistent with the PhenoGraph clusters.
The first branch mostly consists of blood monocytes and the M-15 cluster, characterized by intermediate levels of HLA-DR, CD68, and CD64 and high levels of CD36 and CD11b (Figures 4A and S5A). This phenotype suggests that the M-15 cluster corresponds to circulating CD14+ monocytes from the tumor vasculature. Consistent with this hypothesis, this cluster was present in healthy samples and across all tumor grades (Figure 4E). This first branch expands along diffusion component one (DC1) to include clusters M-1 and M-14 (Figure 4D). Cells along this component were characterized by a progressive loss of CD36 and CD11b and increases in HLA-DR, CD4, CD68, and CD64 (Figure 4F), a trend culminating with cluster M-5, which showed one of the highest DC1 values. This marker regulation is consistent with changes observed during the monocyte to macrophage transition that occurs upon migration into tissue (Ostuni et al., 2015). Our data also suggest that CD54 and CD81 are associated with this transition.
Figure S5.

In-Depth Characterization of TAM Subsets, Related to Figure 4
(A) Contour plots showing the expression of the indicated markers for blood monocytes and the indicated TAM clusters.
(B) Histogram overlays showing expression levels of the indicated proteins on the surface of CD16- monocytes (blue) and CD16+ monocytes (red) isolated from the blood of a healthy donor.
(C and D) Diffusion maps showing the clusters M-1 and M-14 and the clusters constituting the groups I (C) and II (D) of branch three as defined in Figure 3D in three dimensional space defined by the first (DC1), second (DC2), and third (DC3) diffusion components.
(E) Boxplots showing the frequencies of the different T cell clusters by ccRCC grade and in normal and metastatic tissues.
The second branch was almost exclusively formed by cells of the M-16 cluster. The M-16 phenotype is consistent with that of inflammatory CD16+ circulating monocytes (Figure S5B). This cluster was present in healthy samples and across all tumor grades (Figure 4E).
The third branch has a complex structure (Figures 4D, S5C, and S5D). One grouping involves clusters M-5, M-11, M-12, and M-13 (Figures 4D and S5C). These clusters displayed the highest levels of HLA-DR, CD68, and CD64 of all clusters and no expression of CD11b or CD36, suggesting that the cells were mature (Ostuni et al., 2015). Consistent with this, these clusters were only present in tumors of grade II and higher (Figures 4E and S5E). These clusters were characterized by a previously unrecognized diversity of combinations of pro- (CD163, CD204, and CD206) and anti-tumor (CD169) TAM markers (Figure 4G; Table S4). M-5 and M-13 clusters expressed only CD204, the M-12 cluster was positive for CD204 and CD206, and the M-11 cluster was positive for all four markers. The M-5 cells also expressed high levels of CD38, a marker exclusively found upon M1 polarization in murine macrophages (Jablonski et al., 2015). Thus, macrophages in the TME can co-express anti-tumor and pro-tumor markers.
Another group in the third branch involves M-0, M-10, and M-7 clusters (Figures 4D and S5D). M-0 and M-10 clusters had high levels of CD206 expression. M-0 cells expressed low levels of CD163, CD169, and CD204, whereas M-10 cells were negative for these markers. M-0 and M-10 clusters were present in healthy tissues (Figure 4E), suggesting that these clusters correspond to tissue-resident macrophages.
Analysis of the Immune Landscape across Patients
To investigate heterogeneity of immune cell signatures across ccRCC patients, we determined frequencies of identified T cell and TAM phenotypes relative to parent populations for each individual patient. We performed a hierarchical clustering based on CD8+ T cells to order patients by compositional similarity (Figure 5, left). This analysis suggests compositional patterns in that in samples with the highest amount of T-0 and T-1 PD-1+ cells, the T-4, T-5, and T-11 populations, which express little or no PD-1, were absent, suggesting that these T cell phenotypes might be mutually exclusive. The analysis of the CD4+ T cells (Figure 5, middle) only revealed subtle patterns and no patterns were visible among the TAM clusters, which displayed the highest variability across and within samples (Figure 5, right). To confirm this visual observation, the Kullback-Leibler divergence of each patient composition relative to the mean composition was computed for TAM and CD4+ and CD8+ T cell compartments. Patients were most heterogeneous in their TAM composition, followed by CD8 composition, and were most uniform in their CD4 composition (Figure S6). Overall, these results suggest that relationships among the analyzed cell types can be identified within the TME.
Figure 5.

Frequencies of CD8+, CD4+, and TAM Clusters for Each ccRCC Sample
Data hierarchically clustered based on CD8+ subsets using Ward’s method. Sample types are indicated by color.
See also Figure S6.
Figure S6.

Variability across CD4+ and CD8+ T Cell and TAM Compartments, Related to Figure 5
Violin plot showing the Kullback-Leibler divergence computed for each patient for CD4+ and CD8+ T cell and TAM compartments. The Welsch t test was used to calculate differences between means, and the p value is shown for each relationship.
Systematic Correlation Analysis Reveals Relationships in the TME
To systematically quantify relationships between immune cell populations present in the TME, we calculated the frequencies for each immune cell phenotype of each individual tumor (Figure 6A) and used these frequencies in correlation analyses designed to exclude outlier effects (STAR Methods). Multiple robust relationships were identified (Figure 6B). All pairs of CD4+ and CD8+ correlations qualitatively identified (Figure 3D) were present among the top 20 strongest correlations (Figure S7A; Table S5), consistent with the idea that CD4+ and CD8+ T cells in the TME are exposed to a similar milieu and tend to follow similar polarization schemes.
Figure 6.

Relationships between TAM and T Cell Clusters in ccRCC Samples
(A) Schematic showing how clusters are related to parent populations.
(B) Heatmap showing Pearson coefficients of correlation for relationships between immune cell phenotypes.
(C) Scatterplots showing relationships between T-0 exhausted T cells and T-6 regulatory T cells with the pro-tumor TAM phenotypes M-5, M-11, and M-13. Pearson correlations and p values are indicated. For significant correlations, linear models are shown as blue lines.
(D) Pairwise relationships between the frequencies of pro-tumor TAM phenotypes M-5, M-11, and M-13.
(E) Boxplots showing RNA-sequencing data on sorted populations for indicated genes in control (n = 11) and M-5 (n = 6) macrophage populations. Mann-Whitney-Wilcoxon test p values are shown.
(F) Representative ccRCC tissue stained for CD68 (green), CD38 (red), CD8 (blue), and DNA (white). Arrows in selected area highlight cells positive for CD68 and CD38 (yellow) and for CD8 and CD38 (magenta). Scale bar, 100 μm.
Figure S7.

FACS Isolation of Macrophage Subsets and Deep-Sequencing Results, Related to Figure 6
(A) Scatterplots showing the relationships between pairs of phenotypically similar CD4+ and CD8+ subsets (see Figure 3D). For each relationship, the Pearson correlation score and the p value are indicated. The linear model describing the relationship is depicted as a blue line.
(B) Contour plots showing the gating strategy used to sort CD14+/HLA-DR+/CD204−/CD38−/CD206− (Control) and CD14+/HLA-DR+/CD204+/CD38+/CD206− (M-5) macrophage populations.
(C) Boxplots comparing the expression levels of the 30 macrophage-specific markers present in the TAM panel as assessed by mass cytometry (Protein) and by deep sequencing (mRNA). The surface phenotypes of the manually gated M-5 population (CD68+/HLA-DR+/CD204+/CD38+/CD206−) and the PhenoGraph identified M-5 cluster in the 6 populations used for deep sequencing were compared (M-5 gated versus M-5 PG cluster) highlighting the consistency of the different analysis modalities.
(D) Boxplots showing the expression level of the indicated genes in control (n = 11) and M-5 (n = 6) macrophage populations as assessed by deep sequencing. The p values calculated using a Mann-Whitney-Wilcoxon test are shown.
(E) Another antibody clone against CD274 provides a higher dynamic range and recapitulates the expression difference observed in (C). Top: Contour plot showing the gating strategy to identify CD68+/CD204+/CD38+ macrophage and CD68+/CD204−/CD38− control cells. Middle: Histogram overlay showing the expression of CD274 in control versus M-5 macrophage populations. Bottom: Boxplot showing the median intensity of CD274 on control cells (n = 6) and M-5 (n = 6) macrophage populations.
We were intrigued to find strong relationships among the exhausted T-0 cluster, the regulatory T-6 cluster, and the M-5 macrophage cluster. The frequency of T-0 cells was high in samples with high levels of M-5, and samples with high levels of T-0 and M-5 also contained high amounts of the T-6 CD4+ regulatory T cells (Figure 6C, left, and S7A). Not all samples containing high levels of regulatory T cells were enriched for M-5, however. Interestingly, the other two main pro-tumor macrophage phenotypes M-11 and M-13 displayed a mutually exclusive relationship with exhausted T cells (Figure 6C, top middle and top right) and did not show an association with regulatory T cells (Figure 6C, bottom middle and bottom right). We also observed that M-5, M-11, and M-13 pro-tumor macrophage populations tended not to co-occur (Figure 6D).
To explore whether the M-5 phenotype might directly trigger immunosuppression, we sorted these cells and analyzed their transcriptomes (Figures S7B and S7C; Table S3). The M-5 population expressed a mixture of pro-inflammatory markers, including TLR4, STAT1, STAT2, CIITA, and HLA-DP, and anti-inflammatory markers such as PPAR-γ, C/EBP-β, c-MAF, and CD200R compared to control (Figure 6E). However, some canonical markers of pro- and anti-inflammatory macrophages were not induced (Figures 6E and S7D). The M-5 population also exhibited direct immunosuppressive features including expression of PDCD1LG2 (CD273) and CD274, which encode ligands of PD-1 responsible for T cell exhaustion, and of CXCL10 and CCL8, which encode chemokines known to attract CD8+ T cells and regulatory T cells, respectively (Griffith et al., 2014) (Figure 6E). Imaging data confirmed that CD38 was co-expressed on both CD68+ and CD8+ cells and that these cells could co-localize (Figure 6F). Our imaging panel did not include enough markers to fully resolve the macrophage and the T cell phenotypes, but the samples used for imaging were highly enriched for the M-5 and T-0 populations (samples 37, 64, and 68; see Figure 5). Thus CD38 expressing CD68+ and CD8+ cells likely correspond to phenotypes M-5 and T-0, respectively. Taken together, these data emphasize that M-5 macrophages display pro-inflammatory and anti-tumor markers and suggest that this population might contribute to the immunosuppressed phenotypes of tumors containing high amounts of M-5 macrophages.
Structure of the Immune Landscape Correlates with Clinical Outcome
To obtain a global understanding of the relationships between all immune subsets and clinical parameters, we performed correspondence analysis (CA) (Härdle and Simar, 2003). CA is similar to principal component analysis but is applied to a frequency table (Table S6). CA reduces high-dimensional observations to a smaller set of “explanatory” components. In our case, visualizations based on CA display data on each patient and immune subsets in the same space. Patients are organized by their tendency to contain certain immune subsets, and immune subsets are organized by their tendency to co-occur in the same patients.
We found that the first component mainly captures the tendency of the exhausted T cells that express high levels of PD-1 (T-0) to co-occur with M-5 macrophages (Figures 7A and 7B). The second component provides a different ordering of immune subsets and patients, driven by co-occurrence patterns involving M-11 and M-13 clusters. Because the CA components represent the dominant co-occurrence patterns observed in these samples, we used these components to query statistical relationships between TME composition and clinical variables (Table S1). We found that only the second component was significantly associated with progression-free survival (hazard ratio = 7.5; p = 5.5 × 10−3; Table S6; STAR Methods). Examining the distribution of progression-free survival times across CA scores, we found that CA-2 assigned high scores to seven patients (Figure 7C; patient IDs 6, 28, 31, 38, 39, 40, and 53 [Table S1]). Among those patients one died from acute myeloid leukemia and was removed from subsequent statistical analyses. In this scenario, the CA-2 was again significantly associated with worse clinical outcome (Figure 7D; log-rank p = 0.013). Analysis of the clinical variables did not reveal any commonality that might explain the association with progression-free survival (Table S1).
Figure 7.

Relationships between Immune Landscape and Clinical Outcomes
(A) The first two components of correspondence analysis, accounting for 30% of the co-association structure between immune subsets and patients in the cohort, are shown. Immune phenotypes are displayed as squares (TAMs) and triangles (T cells), and patient samples as circles.
(B) Contributions of the immune subsets to CA-1 and CA-2.
(C) Projection of each patient onto the first and second component of the correspondence analysis. Circle size represents progression-free survival time. CA-2 assigns high scores (right of dotted line and labeled in blue) to six patients with low progression-free survival times.
(D) Kaplan-Meier analysis of the six patients with low progression-free survival times (blue curve) versus the remaining patients (green curve). Shaded regions represent 95% confidence intervals.
The most important immune subpopulations for CA-2 are the pro-tumor macrophage subpopulations M-11, M-13, and M-5 (Figure 7B), in that patients with high CA-2 scores and poor progression-free survival times had high frequencies of either M-11 or M-13 and low frequencies of M-5 macrophages. In fact, we found that a score computed as %M-11 + %M-13 – %M-5 produced a close approximation of the CA-2 score and generated similar progression-free survival statistics. Thus, the association between CA-2 and progression-free survival is obtained by capturing higher order relationships between immune subsets. These results show that the comprehensive mapping of the immune landscape across 73 patients by mass cytometry not only identified relationships among immune cell phenotypes in the TME but also stratified patients in a meaningful way as reflected in the concordance with clinical outcomes. The latter association would not have been possible if only individual markers or cell types had been analyzed.
Discussion
Immune cells of the TME are part of a complex and intricate ecosystem. Even after tumors evade immune elimination (Schreiber et al., 2011), immune cells are able to modulate tumor progression. With the success of immune checkpoint therapies, immune cells have become a focus of cancer research and pharmaceutical development. To improve understanding of the TME, we performed a mass cytometry analysis of the immune cells present in normal renal tissue and ccRCC samples. This high-quality dataset validates prior observations and affords many insights into the immune landscape of the TME.
T cell profiling expanded the view on CD8+/PD-1+ T cell phenotypes and found that these cells are characterized by combinatorial expressions of inhibitory receptors. Whereas PD-1 had broad expression, TIM-3, CTLA-4 and 4-1BB were expressed only by few PD-1+ clusters; thus, targeting those molecules might be less effective than targeting PD-1 in ccRCC. Clinical trials should assess whether targeting specific combinations of inhibitory receptors will enable personalization of therapy and improve patient outcome. We also identified CD38 as a potential T cell exhaustion marker in ccRCC. CD38 activates nitric oxide synthetase, leading to the release of nitric oxide, a mediator of T cell immunosuppression (Mayo et al., 2008). In HIV patients, CD38 is co-expressed with PD-1 on exhausted T cells (Hoffmann et al., 2016). There are differences in exhausted T cells in patients with chronic infection versus those with cancer (Speiser et al., 2016), but we propose here that expression of CD38, which shows the highest overlap with PD-1, is a hallmark of exhausted T cells in ccRCC.
Based on clustering of data generated with our antibody panel, we identified 17 major TAM phenotypes. Using a diffusion map to characterize how TAM clusters are related, we identified a developmental trajectory that recapitulates the transition between circulating monocytes and early immigrant and mature macrophages in the tissue. Although CD163, CD204, and CD206 have been interchangeably used to define pro-tumorigenic TAM populations (Komohara et al., 2013, Shigeoka et al., 2013, Xu et al., 2014), our results suggest that these markers distinguish subsets. Previous reports suggested that CD169+ macrophages are linked to anti-tumorigenic function due to a cross-presentation of tumor antigens to CD8+ T cells (Asano et al., 2011, Martinez-Pomares and Gordon, 2012, Ohnishi et al., 2013). We observed that in most subsets CD169 was co-expressed with CD163, CD204, or CD206. This raises the question of whether CD169 has anti-tumorigenic functions in ccRCC.
As a first step toward understanding the functional diversity of these TAM phenotypes, we studied their relationships with other immune cell populations and to clinical data. We observed a relationship between the immunosuppressed T cell compartment, characterized by high levels of PD-1-expressing CD8+ T cells and a high frequency of regulatory CD4+ T cells, and the M-5 TAM subpopulation. The specificity of this TAM cluster is mostly due to the expression of CD38. Depletion or inhibition of CD38 slows progression of glioblastoma in animal models (Blacher et al., 2015, Levy et al., 2012), and CD38-positive myeloid-derived suppressor cells suppress T cell activity and promote tumor growth in esophageal cancer models (Karakasheva et al., 2015). Our results demonstrate that CD38 expression is not restricted to myeloid-derived suppressor cells but is also present on TAM subsets. The cluster M-13, which shares most features with M-5 but does not express CD38, does not show any association with immunosuppression. This suggests that CD38 has an important role in modulating T cell activity.
The correspondence analysis performed on all immune cell subsets demonstrated that taking into consideration the frequencies of all immune infiltrates provides a more powerful way to predict patient outcome than does looking at subsets individually. We observed that the frequencies of the M-11, M-13, and M-5 TAM subsets showed a relationship with progression-free survival. Specifically, patients whose TMEs had higher frequencies of M-11 and/or M-13 and lower frequencies of M-5 had shorter progression-free survival.
Our study has limitations. First, mass cytometry relies on high-quality, informative antibodies. The TAM panel is based on an antibody screen against human monocyte-derived macrophages and might be biased toward these in vitro polarized cells. Second, we analyzed dissociated ccRCC biopsy samples, and thus we cannot distinguish immune cell phenotypes specific to the tumor from those specific to healthy adjacent tissue and vessels. Third, the presented immune cell phenotypes are based on single-cell measurements coupled to computational analysis. Our approach recapitulated the known immune landscape, but follow-up studies to define roles of the phenotypes identified here are needed. The question of what should ultimately be defined as a cell type must be resolved. Fourth, cell phenotype correlations and associations with clinical outcome are based on a cohort of 73 patients and five matched healthy samples. Larger and independent cohorts need to be analyzed to yield statistical power sufficient to identify relationships between clinical outcome and additional immune phenotypes (e.g., previous work proposed that exhausted T cells correlate with outcome (Kang et al., 2013)) and to confirm the relationships proposed in our study. Clinical trials are needed to assess the value of targeting phenotypes described in this study. Fifth, highly multiplexed tissue imaging is necessary to determine whether cell type frequency correlations are reflections of direct cell-cell interactions (Giesen et al., 2014). However, given the influence of the tissue milieu over space and time on tissue-resident cells, the influence of one cell type on another cell type does not necessarily depend on direct cell-cell interactions.
Our fine-grained analysis of the immune cells in the ccRCC TME revealed well-defined TAM and T cell phenotypes and showed that distinct subsets are associated with clinical outcome. Immunotherapy targeting immune checkpoints is revolutionizing cancer treatment but only for subsets of patients. The mechanisms of treatment failures are poorly understood, and monophasic biomarkers such as PD-L1 (CD274) expression do not identify all responders. The first step in improving immunotherapies and supporting the development of novel treatments is to map immune cell infiltrate of tumors and to study the relationships among these cells. The immune cell atlas reported here will provide a valuable resource to accelerate research in this direction.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD8a (RPA-T8) – purified | Biolegend | Cat# 301002 |
| CD64 (10.1) – purified | Biolegend | Cat# 305002 |
| CD38 (HIT2) – purified | Biolegend | Cat# 303502 |
| CD68 (Y1/82A) – purified | Biolegend | Cat# 333802 |
| CD36 (5-271) – purified | Biolegend | Cat# 336215 |
| CD71 (CY1G4) – purified | Biolegend | Cat# 334102 |
| CD20 (H1(FB1)) – purified | Becton Dickinson | Cat# 555677 |
| CD206 (15-2) – purified | Biolegend | Cat# 321112 |
| CD13 (WM15) – purified | Biolegend | Cat# 301702 |
| CD204 (351615) – purified | R&D Systems | Cat# MAB2708 |
| CD123 (6H6) – purified | Biolegend | Cat# 306002 |
| CD11b (M1/70) – purified | Biolegend | Cat# 101202 |
| CD40 (5c3) – purified | eBioscience | Cat# 14-0409 |
| Slamf7 (162.1) – purified | Biolegend | Cat# 331802 |
| CD273/PD-L2 (MIH18) – purified | Biolegend | Cat# 345502 |
| Neuropilin/CD304 (446921) – purified | R&D Systems | Cat# MAB38701 |
| CD82 (ASL-24) – purified | Biolegend | Cat# 342102 |
| CD274/PD-L1 (130021) – purified | R&D Systems | Cat# MAB1561 |
| CD274/PD-L1 (E1L3N) – purified | Cell Signaling Technologies | Cat# 13684S |
| CD119 (GIR-208) – purified | Biolegend | Cat# 308604 |
| CXCR4 (12G5) – purified | Biolegend | Cat# 306502 |
| CD279/PD-1 (EH12.2H7) – purified | Biolegend | Cat# 329902 |
| CD7 (M-T701) – purified | Becton Dickinson | Cat# 555359 |
| CD4 (RPA-T4) – purified | Biolegend | Cat# 300516 |
| CD32 (FUN-2) – purified | Biolegend | Cat# 303202 |
| CD16 (3G8) – purified | Biolegend | Cat# 302002 |
| CD14 (RMO52) – purified | Beckman Coulter | Cat# IM2580U |
| CD163 (GHI/61) – purified | Biolegend | Cat# 333602 |
| CD169 (7-239) – purified | Biolegend | Cat# 346002 |
| CD86 (233(FUN-1)) – purified | Becton Dickinson | Cat# 555655 |
| HLA-ABC (W6/32) – purified | Biolegend | Cat# 311402 |
| CD81 (5A6) – purified | Biolegend | Cat# 349502 |
| CD88 (S5/1) – purified | Biolegend | Cat# 344302 |
| HLA-DR (L243) – purified | Biolegend | Cat# 307602 |
| CD54 (HA58) – purified | Biolegend | Cat# 353102 |
| CD15 (HI98) – purified | Biolegend | Cat# 301902 |
| CD80 (2D10) – purified | Biolegend | Cat# 305202 |
| CD28 (CD28.2) – purified | eBioscience | Cat# 16-0289 |
| Tim-3 (F38-2E2) – purified | Biolegend | Cat# 345035 |
| LAG-3 (333210) – purified | R&D Systems | Cat# MAB23193 |
| CTLA-4 (L3D10) – purified | Biolegend | Cat# 349902 |
| CD278/ICOS (C398.4A) – purified | Biolegend | Cat# 313502 |
| CD134/OX40 (Ber-ACT35) – purified | Biolegend | Cat# 350002 |
| 4-1BB ((4B4-1) – purified | Biolegend | Cat# 309802 |
| GITR (621) – purified | Biolegend | Cat# 311602 |
| CD3 (UCHT1) – purified | Biolegend | Cat# 300402 |
| CD25 (M-A251) – purified | Biolegend | Cat# 356102 |
| Ki-67 (8D5) – purified | Cell Signaling Technologies | Cat# 9449 |
| CD45 (HI30) – purified | Biolegend | Cat# 304002 |
| CD45RA (HI100) – purified | Biolegend | Cat# 304102 |
| CD11c (Bu15) – purified | Biolegend | Cat# 337221 |
| CD127 (A019D5) – purified | Biolegend | Cat# 351302 |
| Foxp3 (PCH101) - Dy162 | Fluidigm | Cat# 3162011A |
| CD197/CCR7 (G043H7) - Sm159 | Fluidigm | Cat# 3159003A |
| CD38 (EPR4106) – purified | Abcam | Cat# ab176886 |
| Foxp3 (236A/E7) – purified | eBioscience | Cat# 14-4777-82 |
| PD-1 (D4W2J) – purified | Cell Signaling | Cat# 86163S |
| CD8 (4B11) – purified | Bio-Rad | Cat# MCA1817T |
| Anti-mouse IgG1, 2a, 3 (Fc fragment) - HRP | Jackson Immuno | Cat# 115-035-164 |
| anti-goat IgG (H+L) - HRP | SantaCruz | Cat# sc-2020 |
| anti-rabbit IgG (minimal x-reactivity) - HRP | Biolegend | Cat# 406401 |
| Biological Samples | ||
| Buffy Coat | Zurich Blood Transfusion Service | N/A |
| Clear cell renal cell carcinoma samples | University Health Network (UHN) Biospecimen Sciences Program and the Cooperative Health Tissue Network (CHTN) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| M-CSF | PeproTech | Cat# 300-25 |
| LPS | Sigmal Aldrich | Cat# L2654 |
| IL10 | Peprotech | Cat# 200-10 |
| Ovalbumin | Sigma Aldrich | Cat# C6534 |
| α-chicken | Sigma Aldrich | Cat# A5503 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15710 |
| Alexa Fluor 700 NHS Ester | Molecular Probes | Cat# 20010 |
| Alexa Fluor 750 NHS Ester | Molecular Probes | Cat# 20011 |
| Monocyte isolation Kit | Miltenyi Biotech | Cat# 130-096-537 |
| Bromoacetamidobenzyl-EDTA (BABE) | Dojindo Laboratories | Cat# B437-10 |
| Maleimido mono amide DOTA (mDOTA) | Macrocyclics | Cat# B-272 |
| Cisplatin | Fluidigm | Cat# 201064 |
| Iridium | Fluidigm | Cat# 201192A |
| Maxpar X8 Multimetal labeling kit | Fluidigm | Cat# 201300 |
| Lanthanide (III) metal isotopes as chloride salts | Fluidigm | N/A |
| FcR Blocking Reagent, human | Miltenyi Biotech | Cat# 130-059-901 |
| Zombie NIR dye | Biolegend | Cat# 423105 |
| Trilogy buffer | CellMarque | Cat# 920P-04 |
| Critical Commercial Assays | ||
| LEGENDScreen Human Cell Screening (PE) Kit | Biolegend | cat # 700001 |
| SMARTer Stranded Total RNA-Seq Kit - Pico Input Mammalian | ThermoFischer | Cat# KIT0204 |
| High Sensitivity NGS Fragment Analysis Kit | Advanced Analytical | Cat# DNF-474 |
| Opal 7-Color Fluorescent IHC Kit | PerkinElmer | Cat# OP7DS1001KT |
| Deposited Data | ||
| RNA-sequencing data | This paper | ArrayExpress: E-MTAB-5640 |
| Mass cytometry data | This paper | https://premium.cytobank.org/cytobank/projects/875 |
| Software and Algorithms | ||
| FlowJo v10.0.7 | N/A | https://www.flowjo.com/ |
| Cytobank | Kotecha et al., 2010 | https://www.cytobank.org/ |
| Concatenation tool | Cytobank | https://support.cytobank.org/hc/en-us/articles/206336147-FCS-file-concatenation-tool |
| Normalizer | Finck et al., 2013 | https://github.com/nolanlab/bead-normalization/releases |
| Single cell debarcoder | Zunder et al., 2015 | https://github.com/nolanlab/single-cell-debarcoder |
| t-SNE | van der Maaten and Hinton, 2008 | https://github.com/jkrijthe/Rtsne |
| PhenoGraph | Levine et al., 2015 | https://github.com/jacoblevine/PhenoGraph |
| Diffusion map | Angerer et al., 2016 | https://cran.r-project.org/web/packages/diffusionMap/index.html |
| Correspondence Analysis | Härdle and Simar, 2003 | https://gist.github.com/jacoblevine/2e91ff5ccad671d4ba716c3f941ec296 |
| Lifelines | Davidson-Pilon, 2016 | https://github.com/CamDavidsonPilon/lifelines/ |
| Survival (R package) | N/A | https://cran.r-project.org/web/packages/survival/index.html |
| STAR 2.5.2b | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| fastq-screen 0.9.5 | N/A | http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/ |
| QuasR 1.14.0 | Gaidatzis et al., 2015 | http://bioconductor.org/packages/release/bioc/html/QuasR.html |
| edgeR 3.16.5 | Robinson et al., 2010 | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| R 3.3.2 | R Development Core Team, 2015 | https://www.R-project.org |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by Lead Contact Bernd Bodenmiller (bernd.bodenmiller@imls.uzh.ch).
Experimental Model and Subject Details
Buffy coats from healthy donors were obtained from the Zurich Blood Transfusion Service. Primary ccRCC tissue samples were obtained from the University Health Network (UHN) Biospecimen Sciences Program and from the Cooperative Health Tissue Network (CHTN) from patients providing written consent under UHN Research Ethics Board approval, protocol #09-0828-T. Tumor grades were histologically determined by a pathologist and are reported in Table S1. 78 samples grouped as normal kidney (n = 5), grade I (n = 5), grade II (n = 34), grade III (n = 22), grade IV (n = 8) and metastasis (n = 4) were used in this study. Further clinical data were available only for the 47 tumor samples collected via the UHN, since additional clinical information associated to the CHTN samples could not be retrieved. 15 tumors were collected from women and 32 from men. 14 samples were from patients under 60, 16 from patients between 60 and 70 and 17 from patients above 70.
Method Details
MDM Culture
Monocytes were isolated by histopaque (Sigma Aldrich) density gradient centrifugation followed by a MACS purification using the pan monocyte isolation kit (Miltenyi Biotech) according to manufacturer’s instructions. Monocytes were differentiated into immature macrophages by culture for 5 days in 75-cm2 tissue culture dishes in presence of 30 ng/ml M-CSF (PeproTech). Cells were subsequently polarized for 24 hr as indicated in Table S2. For mass cytometry analysis, cells were stained for viability with 10 μM cisplatin (Enzo Life Sciences) in a 1 min pulse before quenching with 10% FBS as previously described (Fienberg et al., 2012). Cells were then fixed with 1.6% paraformaldehyde (Electron Microscopy Sciences) for 10 min at room temperature and stored at −80°C.
Fluorescence Cell Barcoding and Antibody Screening
Fixed MDM populations were barcoded with 0, 0.01, 0.1, or 1 μg/ml Alexa Fluor 700 carboxylic acid, succinimidyl ester (Molecular Probes) and 0, 0.1, or 10 μg/ml of Alexa Fluor 750 carboxylic acid, succinimidyl ester (Molecular Probes) as previously described (Krutzik and Nolan, 2006). The combined samples were screened with the BioLegend human cell screening kit containing 342 pre-titrated PE-conjugated antibodies arrayed on four 96-well plates according to manufacturer’s instructions. Immediately after staining, data were acquired on a Canto II (BD Biosciences) using the auto sampler. We identified 87 differentially expressed markers (Figure S1B), and a subset of these was selected for the TAM panel (Figure 1C).
Tumor and Standard Sample Preparation
Tumor samples were digested to generate single-cell suspensions as previously described (Gedye and Ailles, 2013). Cells were then viably frozen in 10% DMSO in D-SDCM complemented with 10% FBS. Cryopreserved cells were resuscitated for mass cytometry analyses by rapid thawing and slow dilution. Cells were stained for viability, fixed, and stored as indicated for MDMs. A standard was generated by mixing PBMCs with three populations of monocytes obtained after activation with LPS, with LPS and immune complex, and with IL-10 in a 3:1:1:1 ratio.
Gadolinium Contamination Test
Some patients were scanned during diagnosis with magnetic resonance imaging using a gadolinium-containing contrast agent. To identify samples positive for gadolinium, a small aliquot of each sample was collected after fixation and analyzed individually in the mass cytometer.
Mass Cytometry Barcoding
To ensure homogeneous staining, 0.3 × 106 to 0.8 × 106 cells from each tumor sample were barcoded using a 20-well barcoding scheme consisting of unique combinations of three out of six barcoding reagents as previously described (Zunder et al., 2015). Four palladium isotopes (104Pd, 106Pd, 108Pd, and 110Pd, Fluidigm) were conjugated to bromoacetamidobenzyl-EDTA (BABE) and two indium isotopes (113In and 115In, Fluidigm) were conjugated to 1,4,7,10-tetraazacy-clododecane-1,4,7-tris-acetic acid 10-maleimide ethylacetamide (mDOTA) following standard procedures (Zivanovic et al., 2013). Mass tag barcoding reagents were titrated to ensure an equivalent staining for each reagent, and the final concentrations were between 50 nM and 200 nM. Cells were barcoded using the transient partial permeabilization protocol (Behbehani et al., 2014). The samples were grouped as gadolinium-positive and gadolinium-negative, and samples were randomly loaded in five 96-well plates. One standard sample was loaded in one well of each plate. Cells were washed with 0.03% saponin in PBS (Sigma Aldrich) and incubated for 30 min with 200 μL of mass tag barcoding reagents. Cells were washed twice with PBS-S and twice with cell staining medium (CSM, PBS with 0.5% bovine serum albumin and 0.02% sodium azide). At this point, samples from each plate were pooled and split into two tubes for subsequent cell staining with the two antibody panels.
Antibodies and Antibody Labeling
Provider, clone, and metal tag of each antibody used in this study are listed in Table S2. Antibody labeling with the indicated metal tag was performed using the MaxPAR antibody conjugation kit (Fluidigm). The concentration of each antibody was assessed after metal conjugation using a Nanodrop (Thermo Scientific). The concentration was adjusted to 200 μg/ml in Candor Antibody Stabilizer. Conjugated antibodies were titrated for optimal concentration for use. All antibodies used in this study were managed using the cloud-based platform AirLab (Catena et al., 2016).
Antibody Staining
After barcoding, pooled cells were incubated with FcR blocking reagent (Miltenyi Biotech) for 10 min at 4°C. Samples were stained with 300 μl of the antibody panel per 107 cells for 20 min at 4°C. Cells were washed twice in CSM and resuspended in 1 mL of nucleic acid Ir-Intercalator (Fluidigm) for 1 hr at room temperature. Cells were then washed once in CSM, once in PBS, and twice in water. Cells were then diluted to 0.5 × 106 cells/ml in H2O containing 10% of EQ Four Element Calibration Beads (Fluidigm). Samples were acquired on a CyTOF 2 mass cytometer (TAM panel) and a Helios upgraded CyTOF 2 (T cell panel). Individual .fcs files collected from each set of samples were concatenated using the .fcs concatenation tool from Cytobank, and data were normalized using the executable MATLAB version of the Normalizer tool (Finck et al., 2013). Individual samples were debarcoded using the executable MATLAB version of the single-cell debarcoder tool (Zunder et al., 2015).
Cell Sorting, RNA Extraction and Deep Sequencing
M-5, control macrophages, PD-1- and CD8+/PD-1+ cells were isolated from tumor samples enriched for those subsets (Table S3). Viably frozen samples were resuscitated as previously described, incubated with the Zombie NIR dye (Biolegend) for dead cell exclusion for 15 min at room temperature, blocked with FcR blocking reagent (Miltenyi Biotech) for 10 min at 4°C, and stained for 30 min at 4°C with the antibodies indicated in Table S3. As shown in Figures S4C and S7B, cell subsets were sorted as CD14+/HLA-DR+/CD204-/CD38-/CD206- (Control) and CD14+/HLA-DR+/CD204+/CD38+/CD206- (M-5), CD3+/CD8+/PD-1- (CD8+/PD-1- T cells) and CD3+/CD8+/PD-1+ (CD8+/ PD-1+ T cells) using a 100-μm nozzle on a FACS Aria III 5L Cell sorter. Cells were harvested in RPMI-FBS (10%) medium and immediately used for RNA purification using the PicoPure RNA Isolation Kit (ThermoFischer) according to manufacturer’s instructions. RNA was resuspended in 15 μL of elution buffer, quality-checked on the Bioanalyzer instrument (Agilent Technologies) using the RNA 6000 Pico Chip (Agilent Technologies), and quantified by fluorometry using Quant-iT RiboGreen RNA Assay Kit (Life Technologies). Of each sample, 4 ng RNA was used to generate the sequencing libraries using the SMARTer Stranded Total RNA-Seq Kit - Pico Input Mammalian (Takara Bio) and Nextera XT (Illumina) according to manufacturer’s instructions. Libraries were quality-checked on the Fragment Analyzer (Advanced Analytical) using the High Sensitivity NGS Fragment Analysis Kit (Advanced Analytical). Libraries were pooled and sequenced SR75 on a NextSeq 500 system (Illumina) using the High Output Kit v2 (75 cycles) following the manufacturer’s protocols.
Multiplexed Imaging
Formalin-fixed paraffin embedded sections from M-5 positive and control ccRCC samples were stained using the Opal 7-Color Fluorescent IHC Kit (PerkinElmer) according to manufacturer’s protocol. Slides were deparaffinized and rehydrated and antigen retrieved using Trilogy buffer (CellMarque) by autoclaving for 15 min. Slides were treated with 3% H2O2 for 15 min, washed, and blocked using 4% BSA/PBS/0.1% Triton X-100 (all from Sigma). Primary antibodies and consecutive HRP-conjugated secondary antibodies (Table S7) were diluted in 1% BSA/PBS/0.1% Triton X-100 and incubated for 1 hr at room temperature. Slides were then incubated in Amplification diluent containing a tyramide-conjugated fluorophore for 10 min. Prior to the second primary antibody incubation, the slides were heated for 10 min in 10 mM citric acid, pH 6.0 at 95°C to strip the antibodies of the first staining round. The protocol was repeated from the blocking step 3 to 4 times to co-stain several markers.
Quantification and Statistical Analysis
Deep sequencing Data Analysis
Sequences were aligned against the human GRCh37/hg19 genome assembly using STAR (Dobin et al., 2013) version 2.5.2b using parameters:–outFilterType BySJout,–outFilterMultimapNmax 20,–outMultimapperOrder Random,–alignSJoverhangMin 8,–alignSJDBoverhangMin 1,–outFilterMismatchNmax 999,–alignIntronMin 20,–alignIntronMax 1000000,–alignMatesGapMax 1000000,–outSAMmultNmax 1, and–clip5pNbases 3. These parameters will clip the first three bases from each read and align reads with up to 20 hits in the genome, reporting a single randomly selected hit per read, resulting in average mapping rates of 70% and 95% for uniquely and total mapped reads, respectively, corresponding to an average of 30.5 Mio. aligned reads per sample (ranging from 22.7 Mio. to 40.9 Mio.). Ribosomal RNA contamination was estimated using fastq-screen version 0.9.5 http://www.bioinformatics.babraham.ac.uk/projects/fastq_screen/ using a collection of mammalian ribosomal RNA sequences downloaded from GenBank (https://www.ncbi.nlm.nih.gov/genbank/) and was found to be on average about 4% (ranging from 0.5% to 8.1%). Counts representing gene expression levels were obtained using the qCount function from the QuasR Bioconductor package (Gaidatzis et al., 2015) with parameters reportLevel = ”gene,” orientation = ”same” and gene annotation from the TxDb.Hsapiens.UCSC.hg19.knownGene package, which counts all reads that overlap with any exon from a gene on the sense strand. Differentially expressed genes were identified using the edgeR bioconductor package (Robinson et al., 2010), specifically the quasi-likelihood generalized linear model framework (Lun et al., 2016)
To control for possible patient effects that would similarly affect all cell populations isolated from the same patient donor and confound the comparison of cell populations across patients, we fit the data using glmQLFit and a model of the form ∼0 + cell.population + patient. In this model, the resulting coefficient cell.population represents average gene expression of each cell population, whereas patient-specific effects are absorbed by the patient coefficient. For visualization, normalized gene expression levels were calculated as read per kilobase and million as: rpkmg,i = ng,i /Ni ∗106 /lg ∗103, where ng,i is the read count for gene g in sample i, Ni the total number of read counts for all genes in sample i, and lg is the number of exonic bases in gene g.
Mass Cytometry Data Analysis
Files (.fcs) were uploaded into Cytobank, populations of interest were manually gated, and events of interest were exported as .fcs files. For visualization of biaxial marker expression we used FlowJo. For downstream analysis, .fcs files were loaded into R (R Development Core Team, 2015). Signal intensities for each channel were arcsinh transformed with a cofactor of 5 (x_transf = asinh(x/5)). To visualize the high-dimensional data in two dimensions, the t-SNE algorithm was applied on data from 2,000 randomly selected cells from each sample. The total cell population was used when less than 2,000 cells were available. The R t-SNE package for Barnes-Hut implementation of t-SNE was used. Data were displayed using the ggplot2 R package (Wickham, 2009).
To visualize marker expression analyses on t-SNE maps, the top percentile was excluded, and the maximum intensity was defined as the 99th percentile. The data from all samples were divided by this value leading to signal intensities ranging between 0 and 1 for each channel. For hierarchical clustering, pairwise distances between samples were calculated using the Spearman correlation. Dendrograms were generated using Ward’s method. Heatmaps were displayed in R using the heatmap.2 function from the ggplot package. Clustering analysis was performed using the Python implementation of PhenoGraph run on all samples simultaneously. To identify the main cell subsets in the datasets generated with TAM and T cell panels, PhenoGraph was run with the parameter k, defining the number of nearest neighbors, set to 100 (Levine et al., 2015). Sample #61 was excluded as an outlier for the myeloid compartment and samples #25 and #17 were excluded due to insufficient immune cell number.
To specifically define the T cell clusters, PhenoGraph was run on the T cell subsets defined in the first PhenoGraph analysis after principal component analysis preprocessing on the components accounting for 99% of the variance. CD86, CD20, CD206, CD68, and CD15 were excluded from the analysis to remove markers not expressed on T cells and likely to add noise in the cluster generation process. CD7 was also excluded from the PhenoGraph clustering since this marker split most clusters into CD7+ and CD7- fractions without a clear biological meaning and simultaneously reduced the impact of more biologically relevant markers on the clustering. The parameter k to define the nearest neighbors was set to 100. Because TAM subsets were less well-characterized than T cell subsets and because they tend to form less discrete structures in phenotypic space, we deployed a subsampling procedure in order to identify robust TAM clusters. PhenoGraph was run on 500 random subsamples of the myeloid cells identified by the initial application of the algorithm (Figure 2C). Each subsample retained 90% of the original cells and the value of the parameter k was also set to a random value between 40 and 80 for each subsample. The purpose of this procedure was to identify substructures that occur consistently despite perturbations to the underlying data and parameter k. In order to obtain a consensus solution based on these subsamples, each intermediate result was compared using an overlap score, which quantifies how well each result represents the others. For two partitions and , the score is defined as:
where and index clusters in and and the following quantities are defined:
and is the total number of cells. Note that for random and and when . The intermediate solution with the highest mean score was taken as the best consensus solution. Because this solution contained cluster assignments for 90% of cells, the remaining 10% of cells were assigned to clusters using the PhenoGraph classification procedure as previously described (Levine et al., 2015).
To assess single-cell trajectories among TAMs, we used the R implementation of the diffusion map algorithm destiny (Angerer et al., 2016). A maximum of 2,000 cells randomly selected from each cluster were included in the analysis. Diffusion distances were calculated based on arcsinh transformed data (cofactor of 5). The width sigma of the Gaussian kernel was defined using the heuristic estimation provided in the destiny implementation, the k nearest neighbor was set to 100, and Euclidean distance was used as a metric.
In order to rank the correlations between immune cell type frequencies across patients while ignoring inflated correlations driven by individual patients, we report the Pearson correlation coefficient and associated p value corresponding to the worst performing subset of all leave-one-out patient subsets. The correlation was computed for each pair of immune cell types on 73 subsets of samples, each subset lacking one of each of the 73 samples included in the analysis. The correlation for each cell type pair was then defined as the result among subsets for which the p value was largest.
Correspondence analysis was performed on a frequency table in which each row represents a patient and each column represents the frequency of an immune cell type in that patient (Table S6). Calculation of the projections, variance explained, and absolute contributions was performed exactly as described (Härdle and Simar, 2003).
Progression-free survival was defined as the number of days from diagnosis until the first of locoregional recurrence, distant recurrence, or death if any of these occurred. Survival analysis was conducted in R and in Python using the ‘survival’ and ‘lifelines’ libraries, for Cox proportional hazards regression and Kaplan-Meier analysis, respectively (Davidson-Pilon, 2016). The association between CA score and progression-free survival was assessed in several contexts. First, each of the first 25 component scores—accounting collectively for 98% of the variance in the co-association structure of the data—was tested in a univariate context (with age and gender included as possible confounding variables); only CA-2 was significant in this setting (p = 0.006). Next, we considered multivariate models including up to six CA components; CA-2 was the only component associated with progression-free survival in any context (Table S6). The same statistical analyses were repeated after exclusion of patient ID #53 who died from acute myeloid leukemia. The association between CA-2 and progression-free survival was the only significant relationship in any univariate context (with age and gender included as possible confounding variables) and in a multivariate context until and including CA-3 (Table S6). The dataset was under-powered to include stage and grade in the regression models, but an in-depth assessment of the seven patients highlighted for Kaplan-Meier analysis did not reveal a common covariate—such as stage or grade—that might explain the association with progression-free survival.
Data and Software Availability
The accession number for the sequencing data reported in this paper is ArrayExpress: E-MTAB-5640, and the mass cytometry data are accessible from https://premium.cytobank.org/cytobank/projects/875. All data can be downloaded from http://www.bodenmillerlab.org.
Author Contributions
S.C., B.R., and B.B. conceived the study. S.C. performed all experiments with help from D.S., L.A., M.A.S.J., and C.G. K.S. performed the multiplexed IHC experiments, and C.B. performed the sequencing. S.C., J.H.L., V.R.T.Z., M.B.S., and D.P. performed the data analysis. J.H.L. and D.P. conceived the correspondence analysis and analysis of clinical data. S.C., K.S., M.v.d.B., M.B., C.R., L.A., M.A.S.J., H.M., C.G., B.R., D.P., and B.B. performed the biological analysis and interpretation. S.C. and B.B. wrote the manuscript with input from all authors.
Acknowledgments
We are very grateful for the generous donation of tumor samples by patients undergoing surgery that was collected through the University Health Network Biobank and Cooperative Health Tissue Network. We thank Manfred Kopf, Luigi Tortola, Sabine Hoves, Sylvia Herter, and the Bodenmiller lab for fruitful discussions; Andrew Evans for pathological analysis of tissue samples; Andrea Jacobs and Stefanie Engler for technical support; Christina Ewald for cell sorting; the CyTOF facility and the Center for Microscopy and Image Analysis (University of Zürich); and the Genomics Facility Basel. B.B.’s research is funded by a SNSF R’Equip grant, a SNSF Assistant Professorship grant, the SystemsX Transfer Project “Friends and Foes,” the SystemsX MetastasiX grant, and the European Research Council (ERC) under the European Union’s Seventh Framework Program (FP/2007-2013/ERC grant agreement no. 336921). This work was funded by a Roche Postdoctoral Fellowship (to S.C.); an EMBO fellowship (ALTF 970-2014) co-funded by the European Commission (LTFCOFUND2013, GA-2013-609409) (to D.S.); an RACP CSL Fellowship (to C.G.); a CIHR/KCC SHOPP Fellowship (to C.G.), an NHMRC Early Career Fellowship (628939 to C.G.), NIH grants (DP1-HD084071 and R01CA164729), a SNSF grant (31003A_152851 to M.v.d.B.), a Zurich Cancer League grant (to M.v.d.B. and K.S.), a Stiftung für Forschung in der Onkologie grant (to M.v.d.B.), a University Research Priority Project “Translational Cancer Research” grant (to M.v.d.B.), and a MSK Cancer Center Support Grant/Core Grant (P30 CA008748) (to D.P.). This work was in part supported by a fellowship from F. Hoffmann-La Roche AG. M.B., C.R., and B.R. are employees for F. Hoffmann-La Roche AG.
Published: May 4, 2017
Footnotes
Supplemental Information includes seven figures and seven tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2017.04.016.
Supplemental Information
References
- Ahmadzadeh M., Johnson L.A., Heemskerk B., Wunderlich J.R., Dudley M.E., White D.E., Rosenberg S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–1544. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amir A.D., Davis K.L., Tadmor M.D., Simonds E.F., Levine J.H., Bendall S.C., Shenfeld D.K., Krishnaswamy S., Nolan G.P., Pe’er D. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat. Biotechnol. 2013;31:545–552. doi: 10.1038/nbt.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angerer P., Haghverdi L., Büttner M., Theis F.J., Marr C., Buettner F. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics. 2016;32:1241–1243. doi: 10.1093/bioinformatics/btv715. [DOI] [PubMed] [Google Scholar]
- Asano K., Nabeyama A., Miyake Y., Qiu C.H., Kurita A., Tomura M., Kanagawa O., Fujii S., Tanaka M. CD169-positive macrophages dominate antitumor immunity by crosspresenting dead cell-associated antigens. Immunity. 2011;34:85–95. doi: 10.1016/j.immuni.2010.12.011. [DOI] [PubMed] [Google Scholar]
- Bandura D.R., Baranov V.I., Ornatsky O.I., Antonov A., Kinach R., Lou X., Pavlov S., Vorobiev S., Dick J.E., Tanner S.D. Mass cytometry: technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009;81:6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- Behbehani G.K., Thom C., Zunder E.R., Finck R., Gaudilliere B., Fragiadakis G.K., Fantl W.J., Nolan G.P. Transient partial permeabilization with saponin enables cellular barcoding prior to surface marker staining. Cytometry A. 2014;85:1011–1019. doi: 10.1002/cyto.a.22573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacher E., Ben Baruch B., Levy A., Geva N., Green K.D., Garneau-Tsodikova S., Fridman M., Stein R. Inhibition of glioma progression by a newly discovered CD38 inhibitor. Int. J. Cancer. 2015;136:1422–1433. doi: 10.1002/ijc.29095. [DOI] [PubMed] [Google Scholar]
- Bodenmiller B., Zunder E.R., Finck R., Chen T.J., Savig E.S., Bruggner R.V., Simonds E.F., Bendall S.C., Sachs K., Krutzik P.O., Nolan G.P. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol. 2012;30:858–867. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catena R., Özcan A., Jacobs A., Chevrier S., Bodenmiller B. AirLab: a cloud-based platform to manage and share antibody-based single-cell research. Genome Biol. 2016;17:142. doi: 10.1186/s13059-016-1006-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dannenmann S.R., Thielicke J., Stöckli M., Matter C., von Boehmer L., Cecconi V., Hermanns T., Hefermehl L., Schraml P., Moch H. Tumor-associated macrophages subvert T-cell function and correlate with reduced survival in clear cell renal cell carcinoma. OncoImmunology. 2013;2:e23562. doi: 10.4161/onci.23562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daud A.I., Loo K., Pauli M.L., Sanchez-Rodriguez R., Sandoval P.M., Taravati K., Tsai K., Nosrati A., Nardo L., Alvarado M.D. Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J. Clin. Invest. 2016;126:3447–3452. doi: 10.1172/JCI87324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson-Pilon C. GitHub Repos; 2016. Lifelines. [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doedens A.L., Stockmann C., Rubinstein M.P., Liao D., Zhang N., DeNardo D.G., Coussens L.M., Karin M., Goldrath A.W., Johnson R.S. Macrophage expression of hypoxia-inducible factor-1 alpha suppresses T-cell function and promotes tumor progression. Cancer Res. 2010;70:7465–7475. doi: 10.1158/0008-5472.CAN-10-1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fienberg H.G., Simonds E.F., Fantl W.J., Nolan G.P., Bodenmiller B. A platinum-based covalent viability reagent for single-cell mass cytometry. Cytometry A. 2012;81:467–475. doi: 10.1002/cyto.a.22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finck R., Simonds E.F., Jager A., Krishnaswamy S., Sachs K., Fantl W., Pe’er D., Nolan G.P., Bendall S.C. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83:483–494. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaidatzis D., Lerch A., Hahne F., Stadler M.B. QuasR: quantification and annotation of short reads in R. Bioinformatics. 2015;31:1130–1132. doi: 10.1093/bioinformatics/btu781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J., Costes A., Sanchez-Cabo F., Kirilovsky A., Mlecnik B., Lagorce-Pagès C., Tosolini M., Camus M., Berger A., Wind P. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- Galon J., Mlecnik B., Bindea G., Angell H.K., Berger A., Lagorce C., Lugli A., Zlobec I., Hartmann A., Bifulco C. Towards the introduction of the ‘Immunoscore’ in the classification of malignant tumours. J. Pathol. 2014;232:199–209. doi: 10.1002/path.4287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gedye C., Ailles L. Humana Press; 2013. Basic Cell Culture Protocols. [Google Scholar]
- Geissler K., Fornara P., Lautenschläger C., Holzhausen H.-J., Seliger B., Riemann D. Immune signature of tumor infiltrating immune cells in renal cancer. OncoImmunology. 2015;4:e985082. doi: 10.4161/2162402X.2014.985082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giesen C., Wang H.A.O., Schapiro D., Zivanovic N., Jacobs A., Hattendorf B., Schüffler P.J., Grolimund D., Buhmann J.M., Brandt S. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods. 2014;11:417–422. doi: 10.1038/nmeth.2869. [DOI] [PubMed] [Google Scholar]
- Ginhoux F., Schultze J.L., Murray P.J., Ochando J., Biswas S.K. New insights into the multidimensional concept of macrophage ontogeny, activation and function. Nat. Immunol. 2016;17:34–40. doi: 10.1038/ni.3324. [DOI] [PubMed] [Google Scholar]
- Griffith J.W., Sokol C.L., Luster A.D. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu. Rev. Immunol. 2014;32:659–702. doi: 10.1146/annurev-immunol-032713-120145. [DOI] [PubMed] [Google Scholar]
- Grivennikov S.I., Greten F.R., Karin M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. doi: 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghverdi L., Buettner F., Theis F.J. Diffusion maps for high-dimensional single-cell analysis of differentiation data. Bioinformatics. 2015;31:2989–2998. doi: 10.1093/bioinformatics/btv325. [DOI] [PubMed] [Google Scholar]
- Härdle W., Simar L. Applied multivariate statistical analysis applied multivariate statistical analysis. Technometrics. 2003;47:486. [Google Scholar]
- Hoffmann M., Pantazis N., Martin G.E., Hickling S., Hurst J., Meyerowitz J., Willberg C.B., Robinson N., Brown H., Fisher M., SPARTAC and CHERUB Investigators Exhaustion of activated CD8 T cells predicts disease progression in primary HIV-1 infection. PLoS Pathog. 2016;12:e1005661. doi: 10.1371/journal.ppat.1005661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonski K.A., Amici S.A., Webb L.M., Ruiz-Rosado Jde.D., Popovich P.G., Partida-Sanchez S., Guerau-de-Arellano M. Novel markers to delineate murine M1 and M2 macrophages. PLoS ONE. 2015;10:e0145342. doi: 10.1371/journal.pone.0145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph R.W., Chatta G., Vaishampayan U. Nivolumab treatment for advanced renal cell carcinoma: Considerations for clinical practice. Urol. Oncol. 2017;35:142–148. doi: 10.1016/j.urolonc.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M.J., Kim K.M., Bae J.S., Park H.S., Lee H., Chung M.J., Moon W.S., Lee D.G., Jang K.Y. Tumor-infiltrating PD1-positive lymphocytes and FoxP3-positive regulatory T cells predict distant metastatic relapse and survival of clear cell renal cell carcinoma. Transl. Oncol. 2013;6:282–289. doi: 10.1593/tlo.13256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karakasheva T.A., Waldron T.J., Eruslanov E., Kim S.-B., Lee J.-S., O’Brien S., Hicks P.D., Basu D., Singhal S., Malavasi F., Rustgi A.K. CD38-expressing myeloid-derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer Res. 2015;75:4074–4085. doi: 10.1158/0008-5472.CAN-14-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T., Qian B.-Z., Pollard J.W. Immune cell promotion of metastasis. Nat. Rev. Immunol. 2015;15:73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komohara Y., Niino D., Saito Y., Ohnishi K., Horlad H., Ohshima K., Takeya M. Clinical significance of CD163+ tumor-associated macrophages in patients with adult T-cell leukemia/lymphoma. Cancer Sci. 2013;104:945–951. doi: 10.1111/cas.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N., Krutzik P.O., Irish J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010;10 doi: 10.1002/0471142956.cy1017s53. Unit10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul H., Huh J.S., Rove K.O., Crompton L., Koul S., Meacham R.B., Kim F.J. Molecular aspects of renal cell carcinoma: a review. Am. J. Cancer Res. 2011;1:240–254. [PMC free article] [PubMed] [Google Scholar]
- Krutzik P.O., Nolan G.P. Fluorescent cell barcoding in flow cytometry allows high-throughput drug screening and signaling profiling. Nat. Methods. 2006;3:361–368. doi: 10.1038/nmeth872. [DOI] [PubMed] [Google Scholar]
- Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir A.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy A., Blacher E., Vaknine H., Lund F.E., Stein R., Mayo L. CD38 deficiency in the tumor microenvironment attenuates glioma progression and modulates features of tumor-associated microglia/macrophages. Neuro Oncol. 2012;14:1037–1049. doi: 10.1093/neuonc/nos121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lun A.T., Chen Y., Smyth G.K. It’s DE-licious: A recipe for differential expression analyses of RNA-seq experiments using quasi-likelihood methods in edgeR. Methods Mol Biol. 2016;1418:391–416. doi: 10.1007/978-1-4939-3578-9_19. [DOI] [PubMed] [Google Scholar]
- Martinez-Pomares L., Gordon S. CD169+ macrophages at the crossroads of antigen presentation. Trends Immunol. 2012;33:66–70. doi: 10.1016/j.it.2011.11.001. [DOI] [PubMed] [Google Scholar]
- Maus M.V., June C.H. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin. Cancer Res. 2016;22:1875–1884. doi: 10.1158/1078-0432.CCR-15-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo L., Jacob-Hirsch J., Amariglio N., Rechavi G., Moutin M.-J., Lund F.E., Stein R. Dual role of CD38 in microglial activation and activation-induced cell death. J. Immunol. 2008;181:92–103. doi: 10.4049/jimmunol.181.1.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray P.J., Allen J.E., Biswas S.K., Fisher E.A., Gilroy D.W., Goerdt S., Gordon S., Hamilton J.A., Ivashkiv L.B., Lawrence T. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41:14–20. doi: 10.1016/j.immuni.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noy R., Pollard J.W. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohnishi K., Komohara Y., Saito Y., Miyamoto Y., Watanabe M., Baba H., Takeya M. CD169-positive macrophages in regional lymph nodes are associated with a favorable prognosis in patients with colorectal carcinoma. Cancer Sci. 2013;104:1237–1244. doi: 10.1111/cas.12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornatsky O., Bandura D., Baranov V., Nitz M., Winnik M.A., Tanner S. Highly multiparametric analysis by mass cytometry. J. Immunol. Methods. 2010;361:1–20. doi: 10.1016/j.jim.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Ostuni R., Kratochvill F., Murray P.J., Natoli G. Macrophages and cancer: from mechanisms to therapeutic implications. Trends Immunol. 2015;36:229–239. doi: 10.1016/j.it.2015.02.004. [DOI] [PubMed] [Google Scholar]
- Overgaard N.H., Jung J.-W., Steptoe R.J., Wells J.W. CD4+/CD8+ double-positive T cells: more than just a developmental stage? J. Leukoc. Biol. 2015;97:31–38. doi: 10.1189/jlb.1RU0814-382. [DOI] [PubMed] [Google Scholar]
- Pyonteck S.M., Akkari L., Schuhmacher A.J., Bowman R.L., Sevenich L., Quail D.F., Olson O.C., Quick M.L., Huse J.T., Teijeiro V. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat. Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian B.-Z., Pollard J.W. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quatromoni J.G., Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am. J. Transl. Res. 2012;4:376–389. [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team R: a language and environment for statistical computing. R Found. Stat. Comput. 2015;1:409. [Google Scholar]
- Reinartz S., Schumann T., Finkernagel F., Wortmann A., Jansen J.M., Meissner W., Krause M., Schwörer A.-M., Wagner U., Müller-Brüsselbach S., Müller R. Mixed-polarization phenotype of ascites-associated macrophages in human ovarian carcinoma: correlation of CD163 expression, cytokine levels and early relapse. Int. J. Cancer. 2014;134:32–42. doi: 10.1002/ijc.28335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ries C.H., Cannarile M.A., Hoves S., Benz J., Wartha K., Runza V., Rey-Giraud F., Pradel L.P., Feuerhake F., Klaman I. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell. 2014;25:846–859. doi: 10.1016/j.ccr.2014.05.016. [DOI] [PubMed] [Google Scholar]
- Robinson M.D., McCarthy D.J., Smyth G.K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber R.D., Old L.J., Smyth M.J. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–1570. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Shigeoka M., Urakawa N., Nakamura T., Nishio M., Watajima T., Kuroda D., Komori T., Kakeji Y., Semba S., Yokozaki H. Tumor associated macrophage expressing CD204 is associated with tumor aggressiveness of esophageal squamous cell carcinoma. Cancer Sci. 2013;104:1112–1119. doi: 10.1111/cas.12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D.S., Ribas A. The evolution of checkpoint blockade as a cancer therapy: what’s here, what’s next? Curr. Opin. Immunol. 2015;33:23–35. doi: 10.1016/j.coi.2015.01.006. [DOI] [PubMed] [Google Scholar]
- Shinohara N., Abe T. Prognostic factors and risk classifications for patients with metastatic renal cell carcinoma. Int. J. Urol. 2015;22:888–897. doi: 10.1111/iju.12858. [DOI] [PubMed] [Google Scholar]
- Sica A., Larghi P., Mancino A., Rubino L., Porta C., Totaro M.G., Rimoldi M., Biswas S.K., Allavena P., Mantovani A. Macrophage polarization in tumour progression. Semin. Cancer Biol. 2008;18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Speiser D.E., Ho P.-C., Verdeil G. Regulatory circuits of T cell function in cancer. Nat. Rev. Immunol. 2016;16:599–611. doi: 10.1038/nri.2016.80. [DOI] [PubMed] [Google Scholar]
- Tirosh I., Izar B., Prakadan S.M., Wadsworth M.H., 2nd, Treacy D., Trombetta J.J., Rotem A., Rodman C., Lian C., Murphy G. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten L., Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- Vesely M.D., Kershaw M.H., Schreiber R.D., Smyth M.J. Natural innate and adaptive immunity to cancer. Annu. Rev. Immunol. 2011;29:235–271. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- Wherry E.J. T cell exhaustion. Nat. Immunol. 2011;12:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. Springer; 2009. ggplot2: Elegant Graphics for Data Analysis. [Google Scholar]
- Xu L., Zhu Y., Chen L., An H., Zhang W., Wang G., Lin Z., Xu J. Prognostic value of diametrically polarized tumor-associated macrophages in renal cell carcinoma. Ann. Surg. Oncol. 2014;21:3142–3150. doi: 10.1245/s10434-014-3601-1. [DOI] [PubMed] [Google Scholar]
- Xue J., Schmidt S.V., Sander J., Draffehn A., Krebs W., Quester I., De Nardo D., Gohel T.D., Emde M., Schmidleithner L. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014;40:274–288. doi: 10.1016/j.immuni.2014.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L., Wang F., Wang L., Huang L., Wang J., Zhang B., Zhang Y. CD163+ tumor-associated macrophage is a prognostic biomarker and is associated with therapeutic effect on malignant pleural effusion of lung cancer patients. Oncotarget. 2015;6:10592–10603. doi: 10.18632/oncotarget.3547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivanovic N., Jacobs A., Bodenmiller B. A practical guide to multiplexed mass cytometry. Curr. Top. Microbiol. Immunol. 2013;377:95–109. doi: 10.1007/82_2013_335. [DOI] [PubMed] [Google Scholar]
- Zunder E.R., Finck R., Behbehani G.K., Amir A.D., Krishnaswamy S., Gonzalez V.D., Lorang C.G., Bjornson Z., Spitzer M.H., Bodenmiller B. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 2015;10:316–333. doi: 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.