

Abstract
Histone deacetylases (HDACs) are key regulators of gene expression in cells and have been investigated as important therapeutic targets for cancer and other diseases. Different subtypes of HDACs appear to play disparate roles in the cells and are associated with specific diseases. Therefore, substantial effort has been made to develop subtype-selective HDAC inhibitors. In an effort to discover existing scaffolds with HDAC inhibitory activity, we screened a drug library approved by the US Food and Drug Administration and a National Institutes of Health Clinical Collection compound library in HDAC enzymatic assays. Ebselen, a clinical safe compound, was identified as a weak inhibitor of several HDACs, including HDAC1, HDAC3, HDAC4, HDAC5, HDAC6, HDAC7, HDAC8, and HDAC9 with half maximal inhibitory concentrations approximately single digit of µM. Two ebselen analogs, ebselen oxide and ebsulfur (a diselenide analog of ebselen), also inhibited these HDACs, however with improved potencies on HDAC8. Benzisothiazol, the core structure of ebsulfur, specifically inhibited HDAC6 at a single digit of µM but had no inhibition on other HDACs. Further efforts on structure–activity relationship based on the core structure of ebsulfur led to the discovery of a novel class of potent and selective HDAC6 inhibitors with RBC-2008 as the lead compound with single-digit nM potency. This class of histone deacetylase inhibitor features a novel pharmacophore with an ebsulfur scaffold selectively targeting HDAC6. Consistent with its inhibition on HDAC6, RBC-2008 significantly increased the acetylation levels of α-tubulin in PC-3 cells. Furthermore, treatment with these compounds led to cell death of multiple tumor cell lines in a dose-dependent manner. These results demonstrated that ebselen and ebsulfur analogs are inhibitors of HDACs, supporting further preclinical development of this class of compounds for potential therapeutic applications.
Keywords: HDAC, inhibitor, ebselen, RBC-2008, screening, compound library, drug discovery
Introduction
Histone deacetylases (HDACs) are a family of enzymes that mediate nucleosomal histone deacetylation and whose activities control the acetylation state of protein lysine residues, notably those contained in the N-terminal extensions of the core histones. The acetylation state of histones affects gene expression through its influence on chromatin conformation. In addition, the stability and biological function of several nonhistone proteins are regulated by the acetylation state of specific lysine residues.1 The overexpression of different isoforms of HDACs has been found in several types of cancer cells and also in neurological and inflammatory pathologies.2,3 HDACs have been demonstrated to intervene in a multitude of biological processes, such as cell cycle progression, cell survival, and differentiation. As these processes are affected by malignant transformation, HDAC inhibitors are developed as antineoplastic drugs and exhibit encouraging efficacy in cancer patients.4,5 Five pan HDAC inhibitors, that is, suberoylanilide hydroxamic acid (SAHA), romidepsin, chidamide, belinostat, and panobinostat, have already been approved for the treatment of cancers such as cutaneous T-cell lymphoma (CTCL), peripheral T-cell lymphoma (PTCL), and multiple myeloma. However, due to common side effects of these pan HDAC inhibitors, there are dramatic interests and efforts in identifying compounds that have selectivity within or between the human HDAC isoform classes for reducing the side effects and potential application in different therapeutic areas.6–8
In humans, HDAC proteins comprise a family of 18 members, which are separated into four classes based on the size, cellular localization, the number of catalytic active sites, and homology to yeast HDAC proteins.9 Class I includes HDAC1, HDAC2, HDAC3, and HDAC8. Class II consists of six HDAC proteins that are further divided into two subclasses: Class IIa includes HDAC4, HDAC5, HDAC7, and HDAC9, where each contains a single catalytic active site, and Class IIb includes HDAC6 and HDAC10, where each contains two active sites, although only HDAC6 has two catalytically competent active sites. HDAC11 is the sole member of Class IV, based on the phylogenetic analysis. Classes I, II, and IV HDAC proteins operate by a zinc (Zn) metal ion-dependent mechanism. In contrast, Class III HDAC proteins, referred to as sirtuins (ie, SIRT1 through SIRT7), operate by a nicotinamide adenine dinucleotide (NAD+)-dependent mechanism unrelated to the other HDAC proteins.10
Ebselen, 2-phenyl-1,2-benzisoselenazol-3(2H)-one, is a seleno-organic compound with antioxidant and anti-inflammatory properties.11 It is a substrate for human and mammalian thioredoxin reductase and a highly efficient oxidant of reduced thioredoxin. Ebselen has been reported to inhibit enzymes such as lipoxygenases,12 nicotinamide adenine dinucleotide phosphate (NADPH) oxidase,13 and H+/K+-adenosine triphosphatase (ATPase).14 It also inhibits nitric oxide synthesis and shows beneficial effects on the neurological outcome in animal models to elicit neuroprotective effects against ischemic brain injury.15 Recently, ebselen has been demonstrated to inhibit inositol monophosphatase (IMPase) and induce lithium-like effects on mouse behavior.16 Ebselen has been investigated as a therapeutic agent for the prevention of chemotherapy-induced hearing loss or ototoxicity and as a lithium alternative for the treatment of bipolar disease. Unlike many inorganic and aliphatic selenium (Se) compounds, ebselen has a low toxicity as the metabolism of the compound does not liberate the Se moiety, which remains within the ring structure. The results from placebo-controlled and double-blind clinical studies have revealed that ebselen significantly enhances outcome in patients who have experienced occlusive cerebral ischemia of limited duration.17 Safety and tolerability are good, and no major adverse effects have become apparent.
In the efforts to identify potential new HDAC inhibitors and inhibitory scaffolds, in the present study, we screened both Enzo Life Sciences US Food and Drug Administration (FDA)-approved drug library and National Institutes of Health (NIH) Clinical Collection library against all the HDACs in biochemical-based assays.18,19 The screenings identified ebselen as a weak inhibitor of several HDAC subtypes. Further efforts on structure–activity relationship (SAR) and chemical syntheses led to the discovery of a compound representing a novel class of potent and selective HDAC6 inhibitors. The activities of these compounds were further confirmed by cell-based assays.
Materials and methods
Materials
Recombinant HDAC proteins and silent mating-type information regulation 2′ (SIRT) proteins were purified by Reaction Biology Corp. or purchased from BPS Bioscience (San Diego, CA, USA). Peptide RHKKAc-7-Amino-4-methylcoumarin (AMC), RHKAcKAc-AMC, and acetyl-Lys(trifluoroacetyl)-AMC were custom synthesized by CPC Scientific (Sunnyvale, CA, USA) and Biomer Technology LLC (Pleasanton, CA, USA). The NIH Clinical Collection library with 721 compounds was obtained from BioFocus DPI (San Francisco, CA, USA), under a contract from NIH. The FDA-approved drug library with 640 compounds was purchased from Enzo Life Sciences (Farmingdale, NY, USA). RBC-2004 (Ebsulfur), RBC-2005, RBC-2006, RBC-2007, RBC-2008, RBC-2009, RBC-2010, RBC-2011, and RBC-2012 were synthesized internally. Figures S1–S3 show the data of RBC-2008 obtained from nuclear magnetic resonance spectrometry and mass spectrometry. The HDAC inhibitors trichostatin A, TMP269, vorinostat (SAHA), and rocilinostat (ACY-1215) were obtained from Selleckchem.com. Ebselen and benzisothiazol were purchased from Sigma-Aldrich (Saint Louis, MI, USA), Ebselen oxide was obtained from Cayman Chemical (Ann Arbor, MI, USA), and Thr101 was purchased from EMD Millipore (Billerica, MI, USA). Bis-tris(hydroxymethyl)aminomethane (Tris) sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) ready gel, nitrocellulose membrane, Alexa Fluor 633 goat anti-rabbit IgG, and Alexa Fluor 532 anti-mouse IgG secondary antibodies were obtained from ThermoFisher Scientific (Waltham, MA, USA). Anti-acetylated tubulin antibody was obtained from Sigma-Aldrich. Anti-α-tubulin antibody was obtained from Santa Cruz Biotechnology (Dallas, TX, USA). All other chemicals were obtained from Sigma-Aldrich. All the cell culture media and reagents were purchased from ThermoFisher Scientific and American Type Culture Collection (ATCC; Manassas, VA, USA).
HDAC biochemical assay
The activities of all the 11 HDACs and 4 SIRTs were assessed by using acetylated AMC-labeled peptide substrates. The substrate RHKKAc-AMC was used for all Classes I and IIb HDACs (ie, HDAC1, HDAC2, HDAC3, HDAC6, HDAC10, and HDAC11) and SIRT1, SIRT2, and SIRT3. For HDAC8, the substrate used was RHKAcKAc-AMC. The activity against the Class IIa HDACs (ie, HDAC4, HDAC5, HDAC7, and HDAC9) was determined by using an acetyl-Lys(trifluoroacetyl)-AMC. For SIRT5 assay, an Ac-Lys(Succ.)-AMC substrate was used. All the assays were based on the fluorogenic substrates and developer combination as described previously.20 Briefly, HDAC enzyme was incubated in the assay buffer (50 mM Tris–HCl, pH 8.0, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, and 1 mg/mL bovine serum albumin). The substrate was then added to start the reaction. After the reaction was completed, a developer was added to digest the deacetylated substrate, and the fluorescence generated was detected with excitation (Ex) at 360 nM and emission (Em) at 460 nM by the EnVision Multilabel Plate Reader (PerkinElmer, Santa Clara, CA, USA).
Cell lines and cell culture
All the cell lines (A549, HTC116, MiaPaCa-2, SKOV-3, SW480, PC-3, SK-BR-3, K562, MDA-MB-231, A498, HT-29, Jurkat, KMS-11, RPMI-8226, MM.1S, REH, U266, and K562) were purchased from ATCC. As recommended by the manufacturer, the cells were cultured with Dulbecco’s Modified Eagle’s Medium, minimum essential media, or F-12K medium supplemented with 10% fetal bovine serum, penicillin to 100 units/mL, and streptomycin to 100 µg/mL. The cultures were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Cell viability assays
An assay kit from Promega (Madison, WI, USA) was used for CellTiter-Glo® cell viability assay. Briefly, 1,000 cells per well in 25 µL cell medium in the 384-well assay plates were treated with test compounds for 3 days at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Then, 25 µL of CellTiter-Glo® reagent was added to the wells of the assay plates and incubated at room temperature for 10 min with gentle shaking. The luminescent signal within plates was measured by using the EnVision Multilabel Plate Reader. The half maximal inhibitory concentration (IC50) curves were plotted, and IC50 values were calculated by using the GraphPad Prism 4 program based on a sigmoidal dose–response equation.
For [3-(4,5-dimethyl-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) cell viability assay, 25 µL of culture medium containing 2,000–5,000 of cells was added to the wells of the cell culture plates. The cells were incubated with the compounds at 37°C, 5% CO2 for 72 h; 5 µL of CellTiter 96® AQueous One Solution Reagent (MTS assay reagent) from Promega was added to each well and incubated at 37°C, 5% CO2 for 3 h. The absorbance at 492 nM was recorded by using Envision Multilabel Reader. The colored formazan product was used to determine the viable cells present in each well. The IC50 curves were plotted, and the IC50 values were calculated by using the GraphPad Prism 4 program based on a sigmoidal dose–response equation.
Western blot
PC-3 cells were seeded in 24-well plates with complete culture media overnight. The cells were treated with test compounds (10 doses with threefold dilution) for 18 h. After the cells had been washed once with phosphate-buffered saline, 1× SDS sample buffer (62.5 mM Tris–HCl, pH =6.8, 2% SDS, 10% glycerol, 0.01% bromophenol red, 50 mM dithiothreitol) was added to each well of the plates to lyse the cells. A 15 µL of cell lysate sample was separated using SDS-PAGE with 12% Bis-Tris SDS-PAGE and transferred onto nitrocellulose membranes by iBlot dry blotting system (Life Technologies). The membranes were blocked with 3% nonfat milk for 1 h and probed with primary anti-acetyl-tubulin (Sigma-Aldrich) overnight. Alexa Fluor 633 goat anti-rabbit IgG secondary antibody was used to detect the primary antibody. Then, the membranes were scanned by using Typhoon 9410 (GE Healthcare Life Sciences, Pittsburgh, PA, USA). In order to detect the total α-tubulin levels in each sample, the blots were then reprobed with anti-tubulin antibody and the Alexa Fluor 633 goat anti-rabbit IgG secondary antibody.
Results
Ebselen and analogs inhibit the functional activities of HDACs
In order to identify the inhibitors of HDACs, ~1,360 compounds were screened against 11 HDACs from Subtypes 1 to 11 in a functional enzymatic assay. These compounds were a combination of NIH Clinical Collection library with 721 compounds and the FDA-approved drug library with 640 compounds. Compounds in these collections have a history of use in human clinical trials with known safety profiles and may even be appropriate for potential use in new disease areas (www.nihclinicalcollection.com). A primary screening at 10 µM of compounds identified ebselen as one of the inhibitors of several HDAC subtypes with >50% inhibition. Further characterization of the compound in dose–response assays confirmed the inhibition of ebselen against HDACs. Ebselen inhibited HDAC5, HDAC6, HDAC8, and HDAC9 with IC50 values of 3.4 µM, 6.9 µM, 1.2 µM, and 3.7 µM respectively (Figure 1A). Ebselen had no significant inhibition on the other HDAC subtypes of up to 10 µM dose (Table 1). These results demonstrated that ebselen has inhibitory activities against several HDAC subtypes.
Figure 1.

Dose-dependent inhibition of HDACs by ebselen (A), ebselen oxide (B), Thr101 (C), and RBC-2008 (D) in a biochemical assay. The activity against all the 11 HDACs was assessed by using the acetylated AMC-labeled peptide substrates. HDAC enzyme was incubated with test compound for 10 min, and then, the substrate was added to start the reaction. After the reaction was completed, a developer was added to digest the deacetylated substrate, and the fluorescence generated was detected with excitation (Ex) at 360 nM and emission (Em) at 460 nM by using the EnVision Multilabel Plate Reader (PerkinElmer, Santa Clara, CA, USA). The IC50 curves were plotted, and IC50 values were calculated by using the GraphPad Prism 4 program based on a sigmoidal dose–response equation. Data shown here represent one of three independent experiments.
Abbreviations: AMC, 7-Amino-4-methylcoumarin; HDAC, histone deacetylase; IC50, half maximal inhibitory concentration.
Table 1.
Compound structures and IC50 values in the biochemical HDAC assays
| Compound ID | Compound structure | HDAC1
|
HDAC2
|
HDAC3
|
HDAC4
|
HDAC5
|
HDAC6
|
HDAC7
|
HDAC8
|
HDAC9
|
HDAC10
|
HDAC11
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 µM |
IC50 µM |
IC50 µM |
IC50 µM |
IC50 µM |
IC50 µM |
IC50 µM |
IC50 µM |
IC50 µM |
IC50 µM |
IC50 µM |
||
| Ebselen |
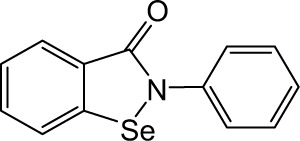
|
>10 | >10 | >10 | >10 | 3.49 | 6.99 | >10 | 1.21 | 3.75 | >10 | >10 |
| Ebselen oxide |
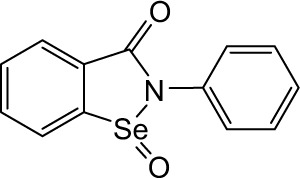
|
2.26 | >10 | 4.18 | 4.70 | 1.49 | 2.98 | 1.88 | 0.20 | 2.37 | >10 | >10 |
| Thr101 |
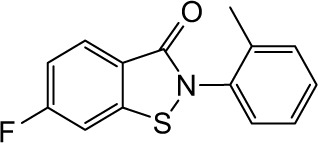
|
3.62 | >10 | 4.24 | 6.07 | 1.57 | 4.14 | 3.43 | 0.66 | 3.77 | >10 | >10 |
| RBC-2004 (Ebsulfur) |
|
1.65 | >10 | 2.72 | 4.12 | 1.49 | 2.89 | 1.51 | 0.19 | 1.70 | >10 | >10 |
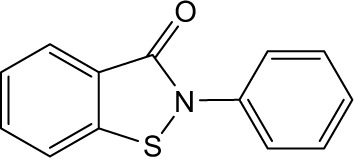
| Benzisothiazol |
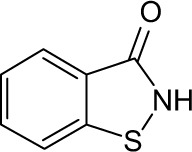
|
>10 | >10 | >10 | >10 | >10 | 3.20 | >10 | >10 | >10 | >10 | >10 |
| RBC-2005 |
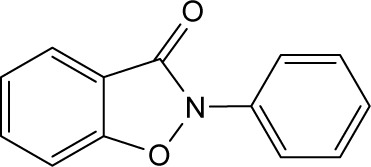
|
>10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| RBC-2006 |
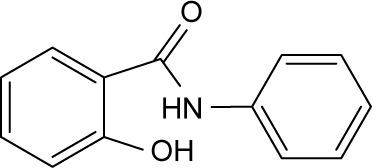
|
>10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| RBC-2008 |
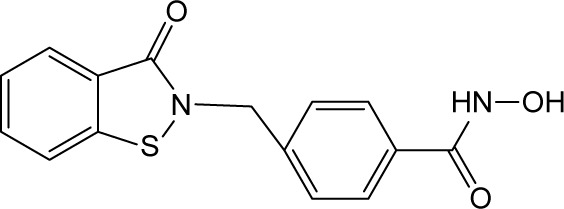
|
0.25 | >10 | 3.70 | 5.40 | 0.94 | 0.003 | 4.10 | 0.064 | 3.80 | >10 | >10 |
| RBC-2009 |
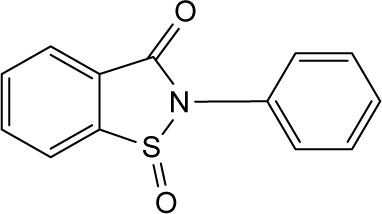
|
>10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 | >10 |
| RBC-2010 |
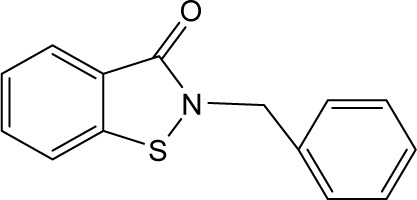
|
6.58 | >10 | 5.01 | >10 | 4.79 | 11.32 | 6.08 | 0.63 | 6.19 | >10 | >10 |
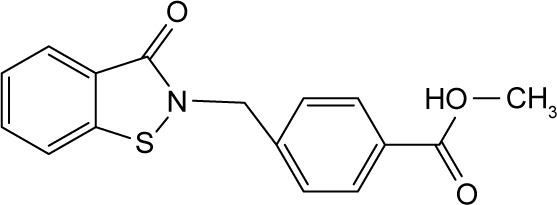
| RBC-2011 |
|
4.00 | >10 | 5.30 | 9.90 | 3.30 | >10 | 4.10 | 0.56 | 4.60 | >10 | >10 |
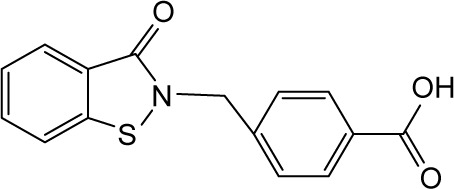
| RBC-2012 |
|
8.80 | >10 | 8.20 | >10 | 5.50 | >10 | 8.79 | 1.03 | >10 | >10 | >10 |
| SAHA |
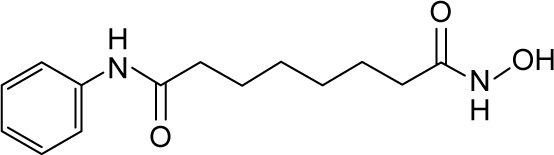
|
0.293 | 0.683 | 0.435 | 27.400 | 12.200 | 0.018 | >10 | 1.130 | >10 | 0.604 | 0.597 |
Abbreviations: HDAC, histone deacetylase; IC50, half maximal inhibitory concentration.
In order to identify the core structure of ebselen as an HDAC inhibitor, several ebselen analogs, including the ebselen oxide (1-oxide-2-phenyl-1,2-benzisoselenazol-3 (2H)-one) and Thr101 (6-Fluoro-2-o-tolylbenzo[d] isothiazol-3(2H)-one, 6-Fluoro-2-(2-methylphenyl)-1, 2-benzothiazol-3(2H)-one), which contains a sulfur group substitution of Se (Table 1), were tested by using HDAC assays. As shown in Figure 1, both ebselen oxide and Thr101 dose dependently inhibited HDAC1, HDAC3, HDAC4, HDAC5, HDAC6, HDAC7, HDAC8, and HDAC9 (Figure 1B and C). It was notably observed that these two ebselen analogs exhibited increased potency on HDAC8 in comparison with ebselen. Ebselen oxide and Thr101 inhibited HDAC8 with IC50 values of 0.2 and 0.6 µM, respectively, compared to the IC50 value of 1.2 µM for ebselen (Table 1). Furthermore, RBC-2004 (ebsulfur) (structure shown in Table 1) exhibited very similar inhibitory profiles against all the HDACs as that of Thr101 (Table 1). Benzisothiazol, another ebsulfur analog without the phenyl moiety on the right of the molecule, was further tested by the HDAC assays. It was interestingly found that benzisothiazol specifically inhibited HDAC6 with IC50 of 3.2 µM and had no significant effect on all the other 10 HDACs tested (Table 1). These results were consistent with the hypothesis that replacement of Se with S and modification of other moieties may significantly change the inhibitory profiles and selectivity of the compounds on HDACs. These results also suggested that benzisothiazol may represent a core structure of new HDAC6 selective inhibitors. In order to further elucidate the relationship between the structure and activity, several analogs of ebsulfur were synthesized, and their activities on HDACs were assessed. As shown in Table 1, RBC-2010 (an analog of RBC-2004 with a longer aliphatic linker) showed similar inhibitory potency as RBC-2004 on HDACs, suggesting that the length of linker does not significantly affect the compound activities on HDACs. RBC-2011 (an ester analog of RBC-2010) and RBC-2012 (an acid analog of RBC-2010) inhibited HDACs in similar potencies as RBC-2010. However, RBC-2005 (an oxygen substitution of sulfur on RBC-2004) and RBC-2009 (a sulfonyl group substitution of sulfur on the RBC-2004) did not significantly inhibit all the 11 HDACs of up to 10 µM concentrations.
RBC-2008 selectively inhibits HDAC6
All the HDACs in the Classes I, II, and IV are Zn-binding proteins, and their deacetylase function is Zn2+ dependent.9 Most HDAC inhibitors discovered so far contain a Zn-binding motif such as hydroxamic acid or benzamide moiety, which significantly enhances compound inhibitory potency. By following this strategy to increase the compound potency on HDACs, a new compound RBC-2008 was designed and synthesized based on the structure of RBC-2004 by adding a hydroxamic acid to the benzoyl moiety (structure shown in Table 1). As expected, RBC-2008 significantly increased its inhibition on all the 11 HDACs (Figure 1). Importantly, it selectively inhibited HDAC6 with IC50 of 3.2 nM, which was ~20-fold more selective than HDAC8 (IC50 of 64 nM), >70-fold more selective than HDAC1 (with IC50 of 245 nM), and >1,000-fold more selective than the rest of the HDAC isoforms (Table 1).
Ebselen and analogs inhibit the activities of Class III HDACs
Sirtuins belong to the third class of deacetylase enzymes, which are dependent on NAD(+) for activity. Sirtuins activity is linked to gene repression, metabolic control, apoptosis and cell survival, DNA repair, development, inflammation, neuroprotection, and healthy aging.10 The activities of ebselen and analogs including RBC-2008 on SIRT1, SIRT2, SIRT3, and SIRT5 activities were further tested. The results showed that ebselen and analogs dose dependently inhibited SIRT1, SIRT2, SIRT3, and SIRT5 activities also in the biochemical assays. The IC50 values of these three compounds on SIRTs were in the range of 0.3 to 10 µM (Table 2). The IC50 values of RBC-2008 on SIRT1, SIRT2, SIRT3, and SIRT5 were 1.2 µM, 7.3 µM, >10 µM, and 3.2 µM, respectively, which was more than 400-fold separation compared with its IC50 on HDAC6.
Table 2.
Effects of ebselen and ebsulfur analogs on SIRT functional activities
| Compound ID | SIRT1
|
SIRT2
|
SIRT3
|
SIRT5
|
|---|---|---|---|---|
| IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | |
| Ebselen | 1.12E–06 | 4.96E–06 | >1.00E–05 | 6.06E–06 |
| Ebselen oxide | 3.78E–07 | 3.74E–06 | >1.00E–05 | 4.39E–06 |
| Thr101 | 1.23E–07 | 2.75E–06 | >1.00E–05 | 4.07E–06 |
| RBC-2004 | 3.43E–07 | 2.99E–06 | >1.00E–05 | 4.88E–06 |
| RBC-2008 | 1.19E–06 | 7.35E–06 | >1.00E–05 | 7.55E–06 |
| RBC-2010 | 2.83E–07 | 3.70E–06 | >1.00E–05 | 3.22E–06 |
Note: Data shown here represent one of three independent experiments.
Abbreviations: IC50, half maximal inhibitory concentration; SIRT, silent mating-type information regulation 2′.
RBC-2008 reversibly inhibits HDAC6
Ebselen contains electrophilic Se moiety, which could form a selenylsulfide (–Se–S–) bond with a cysteine residue on the target protein. It has been reported that this covalent linkage leads to irreversible binding of ebselen on proteins.20 By comparison, the RBC-2008 that contains a sulfur group substitution of Se could be reversible on HDAC. In order to examine the mechanism of action of RBC-2008 on HDACs, an enzyme kinetic analysis of RBC-2008 on HDAC6 was performed as an example. The results showed that RBC-2008 did not significantly affect Vmax but dose dependently decreased the Km value, suggesting that it is a competitive inhibitor of HDAC6 (Figure 2A). Furthermore, a compound dilution assay analysis of RBC-2008 on HDAC6 was performed. If an inhibitor binds to the enzyme reversibly, the complex of the inhibitor and enzyme would be unstable to dilution. The results showed that the inhibition of HDAC6 by RBC-2008 was nearly fully relieved by 100-fold dilution (100 nM to 1 nM) in a time course for recovery extending to 3 h (Figure 2B), thus indicating that the inhibition of RBC-2008 on HDAC6 is reversible.
Figure 2.

Mechanisms of HDAC inhibition by RBC-2008.
Notes: (A) Kinetic analysis. 2.6 ng of purified HDAC6 was incubated with a wide range of concentrations of substrate from 0 to 100 µM in the presence of the vehicle DMSO or RBC-2008 at 10 nM and 50 nM for 1 h of reaction. The data were then fitted based on the kinetics model of Michaelis–Menten plot. The Km of HDAC6 was determined to be 29 µM with DMSO, 65 µM with 10 nM RBC-2008, and 266 µM in the presence of 50 nM of the compound. The Vmax was not dramatically changed by the RBC-2008. The Vmax was ~12 (nM/min/mg) with DMSO, in comparison with 6.8 and 5.9 nM/min/mg in the presence of 10 nM or 50 nM of compound, respectively. (B) Effect of dilution on the inhibition of HDAC6 by RBC-2008. 2.6 ng of HDAC6 was incubated with DMSO vehicle or 300 nM RBC-2008 for 1 h. Then, a portion of the mixtures was diluted 100-fold, and a portion of the mixtures was not diluted to serve as a positive inhibition control for RBC-2008. Then, 50 µM substrate was added to the mixtures to start the reaction. The reactions were stopped at indicated time points up to 3 h by adding 10 µM trichostatin A, and a developer was added to digest the deacetylated substrate, and the fluorescence generated was detected with excitation (Ex) at 360 nM and emission (Em) at 460 nM by using the EnVision Multilabel Plate Reader (PerkinElmer). Data shown here represent one of two independent experiments.
Abbreviations: DMSO, dimethyl sulfoxide; HDAC, histone deacetylase.
Modulation of α-tubulin acetylation in cells by RBC-2008
α-Tubulin has been demonstrated to be one of major substrates of HDAC6 in cells.21 In order to detect the activity of RBC-2008 on the acetylation level of α-tubulin in cells, human prostate cancer PC-3 cells were treated with RBC-2008 or known HDAC6 inhibitors (tubastatin and ACY-1215) for 18 h. The whole cell lysates were subject to Western blot analyses with anti-acetylated-α-tubulin and normalized by the total α-tubulin. The results showed that RBC-2008, as well as known HDAC6-specific inhibitor tubastatin and ACY-1215, significantly increased the acetylated α-tubulin level in PC-3 cells, whereas there were no significant changes in total α-tubulin (Figure 3). These results suggested that the activities of RBC-2008 on α-tubulin acetylation seem to be correlated with their inhibition on the HDAC6 in enzymatic assays. Consistent with its selective inhibition on HDAC6, RBC-2008 had no visible effect on the deacetylation of histone H3 in PC-3 cells (data not shown). Any visible activities of ebselen on the acetylation level on both α-tubulin and histone H3 in PC-3 cells were not observed by Western blot (data not shown), which may partly result from its relatively weak in vitro potency on HDACs and/or poor cellular stability.
Figure 3.

Modulation of α-tubulin acetylation by RBC-2008 in PC-3 cells.
Notes: PC-3 cells were treated with the indicated concentrations of RBC-2008, HDAC inhibitor tubastatin A, or ACY-1215 for 18 h. The whole cell lysates were subject to Western blot analyses with anti-acetylated-tubulin antibody and anti-α-tubulin antibody. Data shown here represent one of three independent experiments.
Abbreviations: DMSO, dimethyl sulfoxide; HDAC, histone deacetylase.
Effects of ebselen and RBC-2008 on human cancer cell viability
In order to assess the effects of ebselen and ebsulfur analogs, including RBC-2008, on cancer cell viability, these compounds were tested in 12 human tumor cell lines from different tumor types, including lung carcinoma A549 cells, colon carcinoma HTC116 cells, pancreas carcinoma MiaPaCa-2 cells, ovary carcinoma SKOV-3 cells, colon adenocarcinoma SW480 cells, prostate carcinoma PC-3 cells, breast adenocarcinoma SK-BR-3 cell, leukemia K562 cells, breast carcinoma MDA-MB-231 cells, kidney carcinoma A498 cells, colon adenocarcinoma HT-29 cells, and leukemia Jurkat cells. The tumor cells were treated with the compounds for 3 days, and then, cell viability was measured by MTS cell viability assay. Several reference compounds such as SAHA (a pan HDAC inhibitor) and ACY-1215 (a HDAC6 selective inhibitor) were also included in the present study. As shown in Table 3, ebselen decreased cell viability on several cancer cell lines with EC50 values from single to double digits of µM, which in general was less potent than SAHA (Table 3). Ebsulfur and analogs including RBC-2008 showed similar potency to ACY1215 in impairing the cell viability on these cancer cells.
Table 3.
Activities of compounds on human tumor cell viability
| Compound ID | A549
|
HTC116
|
MiaPaCa-2
|
SKOV-3
|
SW480
|
PC-3
|
SK-BR-3
|
K562
|
MDA-MB-231
|
A498
|
HT-29
|
Jurkat
|
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | IC50 (M) | |
| Ebselen | >1.00E–04 | 2.59E–05 | 4.03E–05 | 7.81E–05 | 9.24E–05 | >1.00E–04 | 9.27E–05 | 1.30E–05 | >1.00E–04 | >1.00E–04 | 6.35E–05 | 1.38E–05 |
| Ebselen oxide | >1.00E–04 | >1.00E–04 | 3.41E–05 | >1.00E–04 | 1.10E–04 | >1.00E–04 | >1.00E–04 | 1.51E–05 | 8.36E–05 | 4.71E–05 | 9.35E–05 | 7.95E–05 |
| Thr101 | 1.82E–05 | 9.03E–06 | 1.07E–05 | 1.98E–05 | 6.29E–06 | 2.52E–05 | 1.58E–05 | 4.50E–06 | 2.17E–05 | 3.00E–05 | 2.06E–05 | 1.24E–05 |
| RBC-2004 | 3.89E–05 | 9.64E–06 | 3.00E–05 | 2.44E–05 | 7.53E–06 | >1.00E–04 | 3.37E–05 | 5.94E–06 | 2.67E–05 | 4.01E–05 | 2.61E–05 | 1.36E–05 |
| RBC-2008 | >1.00E–04 | 2.96E–05 | 1.81E–05 | >1.00E–04 | >1.00E–04 | >1.00E–04 | 2.81E–05 | 1.71E–05 | 3.16E–05 | >1.00E–04 | 7.93E–05 | 6.40E–06 |
| RBC-2010 | >1.00E–04 | 2.04E–05 | 1.82E–05 | 2.20E–05 | 8.35E–06 | 2.08E–05 | 4.10E–05 | 4.90E–06 | 1.86E–05 | 4.51E–05 | 3.12E–05 | 7.57E–06 |
| ACY-1215 | >1.00E–04 | 6.56E–06 | 7.38E–06 | 1.42E–05 | 9.55E–06 | >1.00E–04 | 1.09E–05 | 1.12E–05 | 1.58E–05 | 6.21E–05 | 1.86E–05 | 4.40E–06 |
| Tubastatin A | >1.00E–04 | 2.31E–05 | 1.96E–05 | 3.86E–05 | 3.31E–04 | >1.00E–04 | 6.28E–06 | 3.42E–05 | 1.68E–05 | >1.00E–04 | >1.00E–04 | .1.00E–04 |
| SAHA | 2.00E–05 | 6.55E–07 | 3.23E–06 | 1.28E–05 | 3.54E–06 | 2.15E–05 | 1.23E–06 | 2.36E–06 | 3.85E–06 | 1.38E–05 | 3.74E–06 | 7.92E–07 |
Abbreviations: IC50, half maximal inhibitory concentration; SAHA, suberoylanilide hydroxamic acid.
Several studies reported that HDAC6 selective inhibitors such as ACY-1215 exhibited better potency on the killing of myeloma and leukemia tumor cell lines and showed promise in potential treatment in multiple myeloma and certain leukemia.22 The effects of RBC-2008 in a few more myeloma and leukemia cell lines such as KMS-11, RPMI-8226, MM.1S, REH cells, U266 cells, and K562 cells was further tested by using a second cell viability assay of Cell Titer-Glo® that measures intracellular ATP levels and has different readout from the MTS assay in a previous test (Table 3). As shown in Figure 4, RBC-2008 dose dependently reduced cell viability on the six myeloma and leukemia cell lines, and IC50 values varied from 1.8 µM to 15 µM. The results confirmed that RBC-2008 significantly reduced cell viability on human tumor cell lines including leukemia and myeloma cells.
Figure 4.
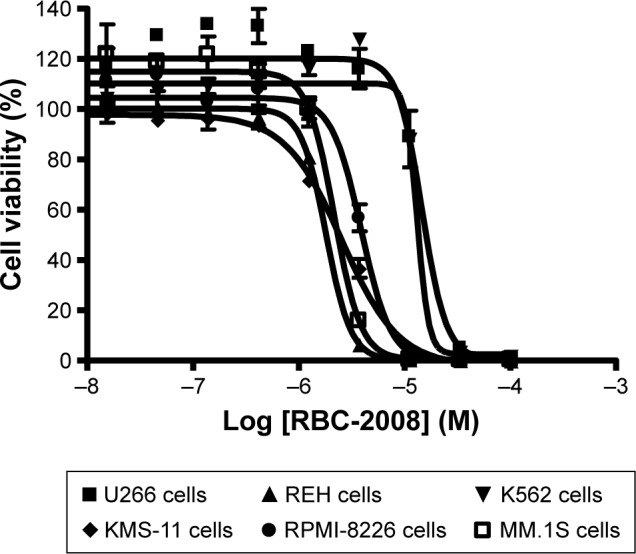
Effects of RBC-2008 on the human tumor cell viability in CellTiter-Glo® assay. Approximately 1,000 of U266 cells, REH cells, K562 cells, KMS-11 cells, RPMI-8226 cells, and MM.1S cells were incubated with the indicated concentrations of RBC-2008 in 384-well assay plates at 37°C, 5% CO2 for 72 h. Then, 25 µL of CellTiter-Glo® reagent was added to the wells of the assay plates and incubated at a room temperature for 10 min with gentle shaking. The luminescent signal within plates were measured by using EnVision Multilabel Reader. The IC50 curves were plotted, and the IC50 values were calculated by using the GraphPad Prism 4 program based on sigmoidal dose–response equation. The IC50 values of RBC-2008 were calculated to be 14.5 µM, 1.8 µM, 13.1 µM, 2.5 µM, 3.8 µM, and 2.1 µM on U266 cells, REH cells, K562 cells, KMS-11 cells, RPMI-8226 cells, and MM.1S cells, respectively. Data shown here represent one of three independent experiments.
Abbreviation: IC50, half maximal inhibitory concentration.
Discussion
HDAC inhibitors have been aggressively pursued as antineoplastic drugs. To date, four pan HDAC inhibitors, including SAHA, romidepsin, belinostat, and panobinostat, have been approved by FDA for the treatment of cancers such as CTCL, PTCL, and multiple myeloma.7 However, it has been observed that these broad HDAC inhibitors have very common side effects and toxicities such as thrombocytopenia, diarrhea, and fatigue, which significantly limit their therapeutic applications in patients. At present, there are dramatic interests and efforts in identifying compounds that have selectivity within or between the human HDAC isoform classes with a hope for reducing side effects and for potential application in different therapeutic areas. By screening the FDA-approved drugs and clinically relevant compounds from commercial libraries, a pharmacophore structure from ebselen and ebsulfur, which exhibited selective inhibition on HDAC subtypes, was identified. This structure represents a novel class of HDAC inhibitors with RBC-2008 as the first in-class lead compound. This class of HDAC inhibitors features a novel pharmacophore specifically targeting HDAC6 with an ebsulfur-like scaffold.
Current pharmacophore model of well-known HDAC inhibitors includes a cap group able to interact with the rim of the catalytic tunnel of the enzyme, a polar connection unit linked to a hydrophobic spacer, which allows the molecule to lie into the tunnel, and a Zn-binding group able to complex the Zn2+ at the bottom of the cavity.4,6,8,23 The preliminary SAR studies on RBC-2008 confirmed such a tripartite structure of this scaffold consisting of a benzisothiazol scaffold as “cap” pharmacophore, a short aliphatic phenyl hydrophobic group as “linker,” and a hydroxamic amide as “Zn chelator”. The data demonstrated that RBC-2008 displays a unique HDAC inhibition pharmacology. Consistent with its selective inhibition on HDAC6, RBC-2008 was very potent in suppressing the deacetylation of α-tubulin but had no visible inhibition of the deacetylation of histone H3 in cell-based assays.
Ebselen inhibits many enzymes such as lipoxygenases,12 NADPH oxidase,13 protein kinase C, H+/K+-ATPase,14 and IMPase.16 Interestingly, ebselen has been reported to also inhibit multiple subtypes of HDAC in both biochemical and cellular assays.24 Ebselen contains Se, which can form a selenylsulfide (–Se–S–) bond with the target proteins. Ebselen has been demonstrated to covalently modify the cysteine residue on IMPase and lead to an irreversible inhibition of the enzyme.16 Although covalent drugs have proved to be successful therapies for various indications in some cases, they are rarely considered when initiating a target-directed drug discovery project largely owing to potential safety concerns. Therefore, the irreversible inhibition of ebselen on many targets increases concerns of potential risks and challenges in drug development. In order to overcome these issues, in this study, RBC-2008 that contains a sulfur group substitution of Se was designed, and it was demonstrated that the compound is reversible HDAC6 inhibitor in compound dilution assay analysis. Further enzyme kinetic analysis suggested that RBC-2008 is a competitive inhibitor of HDAC6. These results support RBC-2008 series to be a good candidate in the early stage of drug discovery for various indications.
The activities of HDACs repress gene transcription by modulating chromatin structure in a global manner to affect thousands of genes in the genome.1–3 However, the broad effects of HDAC inhibition on chromatin can be viewed as a double-edged sword. The good part is that HDAC inhibition could drive the reexpression of silenced genes, including those that encode tumor-suppressing proteins. However, at the same time, some genes may not be as beneficial as one would like and are likely to underpin the clinical toxicities.5 HDAC family contains >18 members divided into four classes, and each HDAC subtype has its preference and specificity on different substrates. Therefore, there are hopes that more selective HDAC inhibitors would have better safety profiles and better therapeutic indexes than the pan HDAC inhibitors that have been approved by FDA so far. An example is the selective HDAC6 inhibitor ricolinostat developed by Acetylon Pharmaceuticals in Phase II trials in multiple myeloma and lymphoma. Ricolinostat can be dosed more frequently with better tolerability than that of panobinostat or vorinostat and can be combined more easily with other drugs such as immunomodulatory agents.4
HDAC6 is one of the best characterized Class IIb deacetylases that regulate many important biological processes in cells.21 HDAC6 possesses two catalytic domains and a C-terminal Zn finger domain that binds free ubiquitin as well as monoubiquitinated and polyubiquitinated proteins with a high affinity.9 HDAC6 localizes predominantly in the cytoplasm and less in the nucleus. It has unique substrate specificity for nonhistone proteins such as α-tubulin, Heat Shock Protein 90 (HSP90), cortactin, peroxiredoxins, chaperone proteins, β-catenin, and hypoxia inducible factor-lα.4,25 HDAC6 has been reported as a tubulin deacetylase that has effects on microtubule-mediated processes through both deacetylase-dependent and deacetylase-independent mechanisms.21 Consequently, it regulates cell migration, cell spread, viral infection, degradation of misfolded proteins, and stress granule formation through complexes with partner proteins. It has been reported that HDAC6 does not catalyze histone deacetylation in vivo.26 Therefore, it may represent a safer drug target for HDAC selective inhibition as it does not directly impact DNA biology. Mice lacking HDAC6 are viable and have greatly elevated tubulin acetylation in multiple organs.27 In addition, they exhibit a moderately impaired immune response and bone homeostasis. Such diverse functions of HDAC6 suggested that it serves as a potential therapeutic target for the treatment of a wide range of diseases such as cancers, inflammatory disorders, and neurodegenerative diseases. To date, HDAC6 selective inhibitors have been tested in preclinical indications for cancers, neurology, inflammation, Gaucher’s disease, Parkinson’s disease, Huntington’s disease, Alzheimer’s diseases, depression and anxiety, pain, and so on.4,5,22,26 Despite extensive efforts, however, very few clinical results for HDAC6-selective inhibitors have been reported, and the safety profiling remains undefined. Acetylon Pharmaceuticals, Inc. has developed a pipeline of optimized, orally administered, HDAC6 selective compounds (http://www.acetylon.com). The selective HDAC6 inhibitor ricolinostat (ACY-1215) is targeted for the treatment of hematologic and solid tumor cancers and is currently being evaluated in several clinical trials for the treatment of relapsed or refractory multiple myeloma and a Phase 1b trial for the treatment of relapsed or refractory lymphoma.23 Experimental results also suggested that selective HDAC6 inhibitors including ricolinostat could have potential in combination with proven immunotherapeutic drugs such as immune checkpoint inhibitor monoclonal antibodies and in combination with proteasome inhibitors.28,29 For nononcology indications, several selective and potent inhibitors of HDAC6 are in the late-stage preclinical development. Such compounds have demonstrated biological activity in the areas of autoimmunity and neurodegeneration as well as in neurological and other diseases in laboratory studies.30–32 HDAC6 selective inhibitors have also demonstrated enhancement of the oncolytic viral activities in different types of cancers.33,34
Conclusion
In summary, we have identified a new HDAC inhibitor chemotype that selectively inhibits HDAC6 protein that could potentially overcome some of the noted limitations of currently known pan HDAC inhibitor. The lead compound RBC-2008’s selectivity coupled with interesting biological activities in suppressing tumor cell proliferation can support further preclinical development of the series of compounds for potential therapeutic applications.
Supplementary materials
1H NMR spectrum for RBC-2008.
Abbreviation: NMR, nuclear magnetic resonance.
13C PENDANT NMR spectrum for RBC-2008.
Abbreviation: NMR, nuclear magnetic resonance.
Matrix-assisted ionization-high-resolution mass spectrum for RBC-2008.
Note: Predicted spectrum for C15H12N2O3S+H is shown at the bottom.
Acknowledgments
The authors thank Charles N McEwen and Khoa Hoang for their help with high-resolution mass spectrometry of compounds. The current affiliation of Jeffrey Wang is Ernest Mario School of Pharmacy, Rutgers University, Piscataway, NJ, USA.
Footnotes
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Mohammad HP, Baylin SB. Linking cell signaling and the epigenetic machinery. Nat Biotechnol. 2010;28:1033–1038. doi: 10.1038/nbt1010-1033. [DOI] [PubMed] [Google Scholar]
- 2.Portela1 A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28:1057–1068. doi: 10.1038/nbt.1685. [DOI] [PubMed] [Google Scholar]
- 3.Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Falkenberg KJ, Johnstone RW. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat Rev Drug Discov. 2014;13:673–691. doi: 10.1038/nrd4360. [DOI] [PubMed] [Google Scholar]
- 5.West AC, Johnstone RW. New and emerging HDAC inhibitors for cancer treatment. J Clin Invest. 2014;12(1):30–39. doi: 10.1172/JCI69738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mottamal M, Zheng S, Huang TL, Wang G. Histone deacetylase inhibitors in clinical studies as templates for new anticancer agents. Molecules. 2015;20:3898–3941. doi: 10.3390/molecules20033898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guha M. HDAC inhibitors still need a home run, despite recent approval. Nat Rev Drug Discov. 2015;14:225–226. doi: 10.1038/nrd4583. [DOI] [PubMed] [Google Scholar]
- 8.Micelli C, Rastelli G. Histone deacetylases: structural determinants of inhibitor selectivity. Drug Discov Today. 2015;20(6):718–735. doi: 10.1016/j.drudis.2015.01.007. [DOI] [PubMed] [Google Scholar]
- 9.De Ruijter AJM, Van Gennip AH, Caron HN, Kemp S, Van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochem J. 2003;370:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol. 2012;13:225–238. doi: 10.1038/nrm3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao R, Masayasu H, Holmgren A. Ebselen: a substrate for human thioredoxin reductase. Proc Natl Acad Sci U S A. 2002;99(13):8579–8584. doi: 10.1073/pnas.122061399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schewe C, Schewe T, Wendel A. Strong inhibition of mammalian lipoxygenases by the antiinflammatory seleno-organic compound ebselen in the absence of glutathione. Biochem Pharmacol. 1994;48(1):65–74. doi: 10.1016/0006-2952(94)90224-0. [DOI] [PubMed] [Google Scholar]
- 13.Smith SM, Min J, Ganesh T, Diebold B, et al. Ebselen and congeners inhibit NADPH oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem Biol. 2012;19(6):752–763. doi: 10.1016/j.chembiol.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beil W, Staar U, Sewing KF. Interaction of the anti-inflammatory seleno-organic compound ebselen with acid secretion in isolated parietal cells and gastric H+/K+-ATPase. Biochem Pharmacol. 1990;40(9):1997–2003. doi: 10.1016/0006-2952(90)90229-e. [DOI] [PubMed] [Google Scholar]
- 15.Zembowicz A, Hatchett RJ, Radziszewski W, Gryglewski RJ. Inhibition of endothelial nitric oxide synthase by ebselen. J Pharmacol Exp Ther. 1993;267(3):1112–1118. [PubMed] [Google Scholar]
- 16.Singh N, Halliday AC, Thomas JM, et al. A safe lithium mimetic for bipolar disorder. Nat Commun. 2013;4:1332. doi: 10.1038/ncomms2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Parnham M, Sies H. Ebselen: prospective therapy for cerebral ischaemia. Expert Opin Investig Drugs. 2000;9(3):607–619. doi: 10.1517/13543784.9.3.607. [DOI] [PubMed] [Google Scholar]
- 18.Enzo. SCREEN-WELL FDA approved drug library V2 Accelerates drug optimization with a unique collection of compounds of known safety and bioavailability for diverse targets. [Accessed March 27, 2017]. Available from: http://www.enzolifesciences.com/BML-2843/screen-well-fda-approved-drug-library-v2/
- 19.National Institutes of Health (NIH) Clinical Collection library [Accessed March 27, 2017]. Available from: www.nihclinicalcollection.com.
- 20.Wegener D, Wirsching F, Riester D, Schwienhorst A. A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem Biol. 2003;10:61–68. doi: 10.1016/s1074-5521(02)00305-8. [DOI] [PubMed] [Google Scholar]
- 21.Li Y, Shin H, Kwon SH. Histone deacetylase 6 plays a role as a distinct regulator of diverse cellular processes. FEBS J. 2013;280:775–793. doi: 10.1111/febs.12079. [DOI] [PubMed] [Google Scholar]
- 22.Santo L, Hideshima T, Kung AL, et al. Preclinical activity, pharmacodynamic and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood. 2012;119(11):2579–2589. doi: 10.1182/blood-2011-10-387365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Raje N, Vogl DT, Hari PN, et al. ACY-1215, a selective histone deacetylase (HDAC) 6 inhibitor: interim results of combination therapy with bortezomib in patients with multiple myeloma (MM) Blood. 2013;122(21):759. [Google Scholar]
- 24.Inks ES, Josey BJ, Jesinkey SR, Chou CJ. A novel class of small molecule inhibitors of HDAC6. ACS Chem Biol. 2012;7:331–339. doi: 10.1021/cb200134p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shankar S, Srivastava RK. Histone deacetylase inhibitors: mechanisms and clinical significance in cancer: HDAC inhibitor-induced apoptosis. Adv Exp Med Biol. 2008;615:261–298. doi: 10.1007/978-1-4020-6554-5_13. [DOI] [PubMed] [Google Scholar]
- 26.Lee JH, Mahendranb A, Yaob Y, et al. Development of a histone deacetylase 6 inhibitor and its biological effects. Proc Natl Acad Sci U S A. 2013;110(39):15704–15709. doi: 10.1073/pnas.1313893110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Kwon SH, Yamaguchi T, et al. Mice lacking histone deacetylase 6 have hyperacetylated tubulin but are viable and develop normally. Mol Cell Biol. 2008;28(5):1688–1701. doi: 10.1128/MCB.01154-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hideshima T, Cottini F, Ohguchi H, et al. Rational combination treatment with histone deacetylase inhibitors and immunomodulatory drugs in multiple myeloma. Blood Cancer J. 2015;5:e312. doi: 10.1038/bcj.2015.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vogl DT, Raje N, Hari P, et al. Phase 1B results of ricolinostat (ACY-1215) combination therapy with bortezomib and dexamethasone in patients with relapsed or relapsed and refractory multiple myeloma (MM) Blood. 2014;124(21):4764. [Google Scholar]
- 30.Butler KV, Kalin J, Brochier C, Vistoli G, Langley B, Kozikowski AP. Rational design and simple chemistry yield a superior, neuroprotective hdac6 inhibitor, Tubastatin A. J Am Chem Soc. 2010;132(31):10842–10846. doi: 10.1021/ja102758v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dietz KC, Casaccia P. HDAC inhibitors and neurodegeneration: at the edge between protection and damage. Pharmacol Res. 2010;62(1):11–17. doi: 10.1016/j.phrs.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Regna NL, Reillya CM. Isoform-selective HDAC inhibition in autoimmune disease. J Clin Cell Immunol. 2014;5:207. [Google Scholar]
- 33.Marchini A, Scott EM, Rommelaere J. Overcoming barriers in oncolytic virotherapy with HDAC inhibitors and immune checkpoint blockade. Viruses. 2016;8(1):9. doi: 10.3390/v8010009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakashima H, Kaufmann JK, Wang PY, et al. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J Clin Invest. 2015;125(11):4269–4280. doi: 10.1172/JCI80713. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1H NMR spectrum for RBC-2008.
Abbreviation: NMR, nuclear magnetic resonance.
13C PENDANT NMR spectrum for RBC-2008.
Abbreviation: NMR, nuclear magnetic resonance.
Matrix-assisted ionization-high-resolution mass spectrum for RBC-2008.
Note: Predicted spectrum for C15H12N2O3S+H is shown at the bottom.