

Summary
Advances in immune‐mediated targeted therapies have proved to be a double‐edged sword for patients by highlighting the risk of iatrogenic infective complications. This has been exemplified by progressive multi‐focal leucoencephalopathy (PML), a hitherto rare devastating viral infection of the brain caused by the neurotrophic JC polyoma virus. While PML achieved prominence during the first two decades of the HIV epidemic, effective anti‐retroviral treatment and restitution of T cell function has led to PML being less prominent in this population. HIV infection as a predisposing factor has now been supplanted by T cell immunodeficiency induced by a range of immune‐mediated therapies as a major cause of PML. This review focuses on PML in the context of therapeutic immunosuppression and encompasses therapeutic monoclonal antibodies, novel immunomodulatory agents such as Fingolimod and dimethyl fumarate, as well as emerging data on PML in primary immune deficiency.
Keywords: cell trafficking, EAE/MS, immunodeficiency diseases, neuroimmunology, viral
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Clinical challenges in the management of patients with B cell immunodeficiencies. Clinical and Experimental Immunology 2017, 188: 323–5.
The role of genomics in common variable immunodeficiency disorders. Clinical and Experimental Immunology 2017, 188: 326–32.
When to initiate immunoglobulin replacement therapy (IGRT) in antibody deficiency: a practical approach. Clinical and Experimental Immunology 2017, 188: 333–41.
Considerations for dosing immunoglobulin in obese patients. Clinical and Experimental Immunology 2017, 188: 353–62.
Chronic norovirus infection and common variable immunodeficiency. Clinical and Experimental Immunology 2017, 188: 363–70.
Introduction
The explosive growth in therapeutic monoclonal antibodies has propelled progressive multi‐focal leucoencephalopathy (PML) from the status of small print rarity in textbooks and journals to an iatrogenic complication that demands serious attention from clinicians and patients. Caused by the neurotrophic polyoma virus named JC virus (JCV) after its discoverer, John Cunningham, PML is a devastating infection of the central nervous system (CNS). By causing lytic infection of brain oligodendrocytes, and to a lesser extent astrocytes, JCV is responsible for causing widespread demyelination in the CNS. PML was first described in 1958 as a rare disorder complicating chemotherapy for patients with lymphoma and leukaemia 1, but it was only in 1971 that JCV was linked conclusively to PML following its isolation from the brain of a patient 2.
Its rarity either in the context of therapeutic immunosuppression or rare primary immunodeficiency disorders 3 merited frequent case reports during the ensuing decades, punctuated by the human immunodeficiency virus (HIV) epidemic in the 1980s, which firmly established impaired T cell immunity as an essential prerequisite for the development of PML. Although HIV‐1 infection remains an important predisposing condition, effective control of the virus and consequent improvement in T cell immunity by anti‐retroviral drugs has led to PML being less prominent in this population. It is noteworthy that JCV‐specific cellular immunity is a key determinant of clinical outcomes in HIV‐infected individuals with PML as evidenced by the correlation between JCV‐specific cytotoxic T cell responses and survival 4. In contrast, the advent of powerful biologicals interfering with leucocyte–endothelial interaction such as Natalizumab for multiple sclerosis and the short‐lived Efalizumab for psoriasis has led to a significant upsurge in cases of PML (Fig 1). While the incidence of PML associated with Efalizumab was deemed to be unacceptably high, and hence led to its withdrawal from the market, Natalizumab remains an important part of the therapeutic armamentarium for relapsing–remitting multiple sclerosis (RRMS). This review will focus on PML in the context of iatrogenic immunosuppression encompassing conventional immunosuppressive drugs, therapeutic monoclonals and novel immunomodulatory drugs such as Fingolimod and dimethyl fumarate, as well as emerging data on PML in primary immune deficiency.
Figure 1.
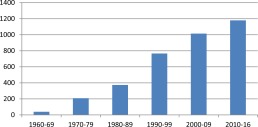
Time trends in reports of progressive multifocal leucoencephalopathy (PML) on PubMed 1960‐2016.
Epidemiology
Exposure to JCV is widespread, with recorded seroprevalence rates of 30–90% in the general population, depending on assay and geographical location (summarized in reference 5). While immunocompetent individuals are able to contain the virus successfully, impaired T cell immunity allows the virus to multiply and disseminate from its usual secluded niches to the brain. Following primary infection through the nose and mouth, JCV is believed to spread haematogenously via peripheral blood leucocytes 6 to the kidneys (and possibly bone marrow), where it is shed from tubular epithelial cells into the urine (Fig. 2). Cellular entry of JCV is mediated by initial attachment via the oligosaccharide, lactoseries tetrasaccharide (LSTc), followed by receptor‐mediated entry using the serotonin receptor, 5‐hydroxytryptamine (5‐HT2AR) 7. Evidence for the haematogenous route of spread is based on the demonstration of JCV DNA in all peripheral blood mononuclear populations and in bone marrow 8, 9, although the relevance of this observation for natalizumab‐induced PML has been questioned 10. Haematogenous spread of the virus alone is not sufficient to explain the development of clinical disease, given the demonstration of JCV DNA in the brains of normal individuals without PML 11. However, the true pathogenic significance of JCV DNA in the brain of asymptomatic individuals has been questioned in the absence of viable viral protein 12, thus emphasizing our limited understanding of the molecular and cellular events that lead to clinical disease. Differences in genomic structure between JCV found in the urine of immunocompetent individuals (highly conserved) and that isolated from the brain suggests that neurotrophism is associated with deletions, duplications and rearrangements in the non‐coding, control regions of the viral genome 13, 14.
Figure 2.
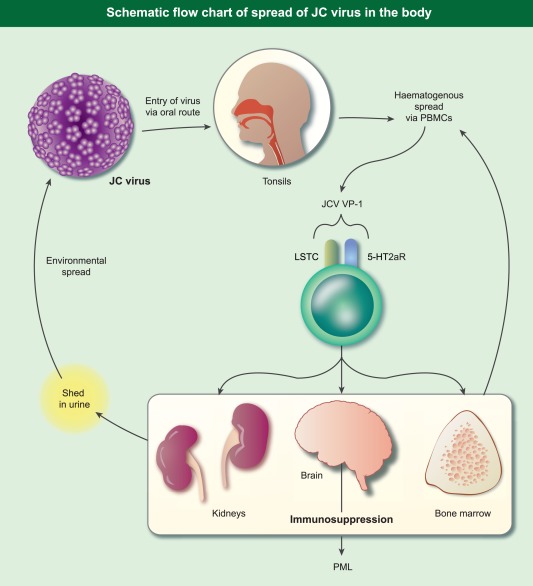
Schematic flowchart of spread of JC virus in the body.
Immune response to JC virus (Fig. 3)
Figure 3.
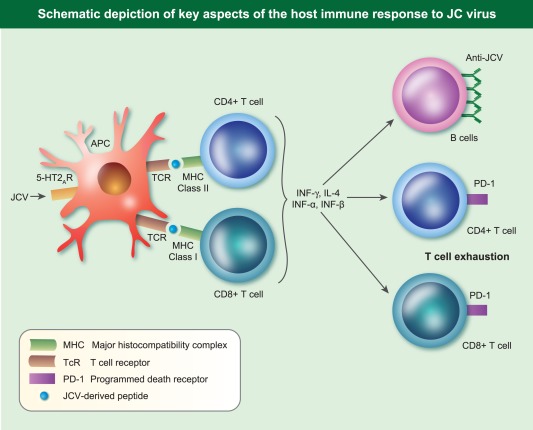
Schematic depiction of key aspects of the host immune response to JC virus.
While containment of JCV in immunocompetent hosts testifies to the importance of the immune response, data on virus‐specific immune responses are limited. In addition to serum, JCV‐specific antibody responses can be demonstrated in CSF in the form of intrathecal anti‐VP1 (the major structural capsid protein of JCV) and JCV‐specific oligoclonal bands 15. Interestingly, low‐affinity intrathecal anti‐VP1 antibodies have also been reported, in the absence of PML, in a small proportion of patients with other immune‐mediated disorders such as MS, neuropsychiatric lupus, mumps meningitis and neuroborreliosis 16, 17, thus suggesting that background prevalence of anti‐JCV may extend to CSF.
Cellular immune responses during JCV infection are characterized by both CD4+ and CD8+ T cell responses. JCV‐specific CD8+ T cell responses in serum correlates with favourable clinical outcomes 18, 19. This observation has been strengthened by the demonstration of CD8+ T cells in close proximity to JCV‐infected glial cells in brain biopsies of PML patients 20. Similarly, CD4– T cell responses can be demonstrated in peripheral blood 21 and in the brain, as noted in patients developing PML‐associated immune reconstitution syndrome (IRIS) in the context of CD4+ T cell restitution with the use of plasmapheresis to remove Natalizumab in MS 21. The ability of brain‐infiltrating T cells to secrete both IFN‐γ and IL‐4 strengthens anti‐viral immune responses by the ability of the former to induce human leucocyte antigen (HLA) class II expression, thus enhancing antigen presentation of viral peptides, while IL‐4 drives B cell production of anti‐viral antibodies 22. These observations have been extended by the demonstration that CD4+ T cell recognition of VP1 occurs in the context of multiple HLA‐DR (HLA‐DR1*15) and HLA‐DQ molecules (HLA‐DQw6), a phenomenon referred to as HLA cross‐restriction 23.
In addition to IFN‐γ‐induced enhancement of anti‐viral responses, evidence from murine studies 24 and from in‐vivo studies using human glial cells 25 and renal epithelial cells 26 suggests a protective role for IFN‐β in containing JCV infection. These laboratory observations are mirrored by evidence of a reduction in JCV viraemia in MS patients treated with IFN‐β 27. Additional clinical evidence pointing to a protective role of an intact IFN pathway comes from the development of PML in patients with primary immunodeficiency disorders associated with gain of function (GOF) mutations in signal transducer and activator of transcription 1 (STAT‐1) 28.
From a clinical perspective, the demonstration of increased expression of the programmed cell death receptor (PD‐1), an inhibitory immune‐checkpoint molecule, on both CD4+ and CD8+ T cells from both HIV‐negative and ‐positive patients with PML suggests that immune exhaustion may play a role in the loss of T cell immunity to JCV 29, thus raising the possibility that therapeutic blockade of PD‐1 using a checkpoint inhibitor such as Pembrolizumab may be of benefit in the treatment of PML.
The combination of humoral and cellular immune responses as summarized above underlines the importance of the adaptive immune response to JCV. However, the occurrence of PML in the predominant setting of impaired cellular immunity clearly argues for a greater role for cell‐mediated immunity in viral containment.
Clinical features and diagnosis
The onset of PML is insidious, with subtle changes in cognition and loss of memory in association with a range of progressive neurological deficits. These are varied and include cognitive defects, sensory deficits, haemianopia, aphasia, difficulties in co‐ordination and gait, consistent with multi‐focal cerebral pathology. A high index of suspicion is required for early diagnosis, as these clinical features are not sufficiently distinctive to enable a definitive clinical diagnosis. Cranial imaging reveals characteristic multiple lesions in the subcortical hemispheric white matter or the cerebellar peduncles. Contrary to the implication in the title that only cerebral white matter is involved, PML lesions also occur in grey matter areas such as the basal ganglia or thalamus.
A typical patient would display the full complement of diagnostic features, comprising radiological changes, evidence of JCV infection [by detection of JCV DNA in situ and by polymerase chain reaction (PCR) in serum and CSF] and characteristic histology on brain biopsy. Recent guidance from the American Academy of Neurology clarifies this by stipulating that definitive diagnosis requires demonstration of the characteristic histological triad of demyelination, bizarre astrocytes and enlarged oligodendroglial nuclei coupled with demonstration of in‐situ JCV DNA or proteins. An alternative pathway to definitive diagnosis comprising the demonstration of JCV by PCR in CSF in combination with typical clinical and radiological features has also been recommended, thus precluding the need for brain biopsy 30. The development of a standardized case definition for the diagnosis of PML following treatment with monoclonal antibodies is based on similar principles but apportions different levels of diagnostic certainty, ranging from level 1 (highest level of certainty) to level 4 (lowest level), with an additional category where PML can be excluded (level 5) 31.
Role of therapeutic interventions in predisposing to PML
Selection of drugs and therapeutic monoclonal antibodies for discussion in this review was based on the results of a PubMed search. Drugs and therapeutic monoclonal antibodies were selected based on immunological interest and frequency of reports, although no minimum eligibility criteria were set.
Conventional immunosuppressive agents
Although sporadic case reports of PML in patients with systemic autoimmune disease treated with azathioprine, steroids, chlorambucil, methotrexate and cyclophosphamide have been documented 32, attempting to define the precise contribution of individual drugs has been confounded by the use of combination therapy, and in the case of systemic lupus erythematosus (SLE) by the possibility that disease‐related immunosuppression per se may predispose to PML 33. Equally, case reports of PML in the context of haematological malignancies were likely to be explained by dual immunosuppression induced by chemotherapy and underlying disease.
Therapeutic monoclonal antibodies
Natalizumab
The licensing and widespread use of Natalizumab has altered the epidemiology of PML dramatically. By binding to the α chains of both α4β1 and α4β7 integrins (Fig. 4), which are expressed on the cell surface of a range of haematopoietic cells (lymphocytes, monocytes, eosinophils), Natalizumab blocks lymphocyte interaction with its ligand, endothelial vascular cell adhesion protein 1 (VCAM‐1), thus interrupting lymphocyte trafficking into areas of neural inflammation. This was demonstrated elegantly in proof‐of‐principle studies in the murine model of experimental allergic encephalomyelitis (EAE), where in‐vivo injection of antibodies to α4β1 integrin 48 h after injection of pathogenic T cell clones ameliorated paralysis significantly 34. The prediction in this paper that ‘therapy based on inhibiting α4β1 integrin, or the ligand for this receptor on brain endothelium, may prove effective in treating inflammatory disorders of the CNS’ was shown to be prescient in phase III trials of Natalizumab in MS, leading to its accelerated approval by the US Food and Drugs Administration agency (FDA) in 2004 and by the European Medicines Agency (EMA) in 2006. Tellingly, however, within months of FDA approval PML was diagnosed in two patients from one of the pivotal phase III trials 35, 36 leading to voluntary marketing suspension. Following detailed analysis of all patients exposed to Natalizumab, a PML risk‐estimate of 1 : 1000 [95% confidence interval (CI) = 0·2–2·8 per 1000] 37 was felt to be acceptable for regulatory authorities to allow the reintroduction of Natalizumab for MS patients, albeit with enhanced pharmacovigilance measures.
Figure 4.
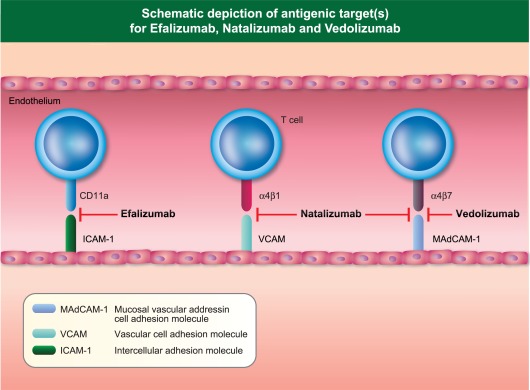
Schematic depiction of antigenic target(s) of Efalizumab, Natalizumab and Vedolizumab.
Sadly, these risk estimates have proved to be substantially inaccurate. Based on systematic collection of data, three key risk factors for Natalizumab‐induced PML have been identified: (1) duration of treatment of at least or greater than 24 months, (2) JC virus seropositivity and (3) prior immunosuppressive therapy preceding commencement of natalizumab. For patients having all three risk factors, the risk of PML was revised to 1 : 90, followed more recently by an estimated risk of 1 : 44 38. In contrast, the risk of PML in JCV‐seronegative patients is estimated at 0·1 in 1000 39. In the context of MS, impaired T cell trafficking by Natalizumab appears to be a double‐edged sword responsible for disease amelioration as well as increasing the risk of PML.
Based on changes in integrin and selectin expression, a number of immunological biomarkers have been proposed to monitor treatment efficacy and improve current risk stratification in patients receiving Natalizumab. Sustained down‐regulation of α4 (CD49d) and, to a lesser extent, β1 (CD29) integrin expression is a feature of Natalizumab therapy 40. A reduction in the proportion of T cells expressing CD62L (L‐selectin) in long‐term recipients of Natalizumab has been proposed as an additional marker of PML risk 41, 42, but this test requires the use of frozen mononuclear cells, and hence raises the possibility that reduction in CD62L represents assay artefact. For this reason, the validity of this assay has been questioned 43.
The striking risk of PML associated with Natalizumab is in contrast to that with Vedolizumab, a gut‐selective α4 β7 integrin inhibitor, which has a licence in Crohn's disease and specifically interrupts lymphocyte trafficking into sites of bowel inflammation 44. In more than 2800 patients treated to date, PML has not been reported to date with Vedolizumab 44, thus emphasizing the importance of α4 β1 inhibition as a mechanistic risk factor.
Efalizumab
Efalizumab is a humanized therapeutic monoclonal antibody directed against CD11a (expressed on all leucocytes), the α‐subunit of the β2‐integrin leucocyte functional antigen‐1 (LFA‐1). By binding to CD11a, it inhibits T cell interaction with its cognate endothelial ligand, intercellular adhesion molecule‐1 (ICAM‐1), thus inhibiting leucocyte trafficking through the vasculature to areas of tissue inflammation (Fig. 4). Alongside its inhibitory effects on T cell activation and proliferation, Efalizumab's efficacy in psoriasis was underpinned by its ability to inhibit T cell trafficking, leading to its approval by the FDA in 2003, and by the EMEA in 2004 for moderate to severe plaque psoriasis. However, the occurrence of PML in four psoriasis patients in 2008 45, 46, 47 receiving Efalizumab monotherapy led to its voluntary withdrawal by the manufacturer in 2009.
It is noteworthy that at the time of market withdrawal in 2009, no other cases of PML had been reported in psoriasis patients. This was to change with subsequent reports of PML in 2015 in association with dimethyl fumarate treatment in patients with psoriasis 48, 49 (dimethyl fumarate is discussed later in this review). The estimated risk of PML with Efalizumab in 2009 was 1 : 400, in contrast to the 1 : 1000 risk of PML associated with Natalizumab at this time 45. Given that both Efalizumab and Natalizumab's propensity to trigger PML is likely to be based on inhibition of T cell trafficking, coupled with the documented increasing risk of Natalizumab with continued use in MS, it is likely that market withdrawal of Efalizumab in 2009 has prevented many more cases of PML.
Rituximab
Concerns about rituximab‐associated PML (RAP) first emerged in 2006 within a decade of its licensing for B cell lymphoma (1997), when the US FDA issued a safety alert to highlight the development of PML in two patients with SLE who had received rituximab alongside other immunosuppressive therapy 50. This prompted the inclusion of PML as a risk in the summary of product characteristics (SPC) for rituximab. A further safety alert from the FDA in October 2009, accompanied by a letter to health‐care professionals, highlighted the development of PML in a patient with rheumatoid arthritis (RA), who had not received prior treatment with a tumour necrosis factor (TNF) antagonist 51.
Current estimates of the risk of RAP vary with the underlying disease indication. In patients with B cell lymphoproliferative disease, the incidence of RAP is substantially higher that that associated with the use of rituximab in RA and SLE 52. This difference is plausible in the context of disease‐related immunodeficiency associated with B cell lymphoproliferative disease compounded by the use of rituximab‐based chemotherapy regimens. Analysis of a large US health insurer database for the period January 2000–June 2008 involving 138 469 patients with autoimmune diseases and 25 706 patients with non‐Hodgkin lymphoma (NHL) or chronic lymphocytic leukaemia (CLL) revealed an incidence rate of PML of 8·3 (95% CI = 1·7–24·2) and 11·1 (95% CI = 0·28–61·7) per 100 000 person‐years for NHL and CLL, respectively. In contrast, there were no cases of PML in a cohort of 72 494 patients with RA (95% CI = 0.0–2.2) 52. More definitive data on the risk of RAP in RA have emerged from an international study based on analysis of 129 000 patients treated with rituximab, four of whom developed PML. Based on this, and the inclusion of a fifth patient, the risk of RAP in RA is estimated at 1 : 25 000 53. For patients with SLE, the risk of RAP has been estimated at 1 : 4000 which, in part, is likely to reflect the higher risk of PML associated with lupus per se 45.
While the mechanism of rituximab‐associated PML is unclear, it is unlikely to be attributable to B cell depletion alone given the ability of rituximab to deplete circulating T cells 54, including T helper type 17 (Th17) cells producing IL‐17 55. Whether the risk of PML associated with rituximab extends to other anti‐CD20 therapeutic antibodies is unclear, but it is noteworthy that PML is an emerging signal for ofatumumab (fully human anti‐CD20 with more pronounced complement‐mediated cytotoxicity) and obinutuzumab (humanized, glycoengineered anti‐CD20) in the FDA's adverse event reporting database 56.
Brentuximab–vedotin
Brentuximab–vedotin (BV) is an antibody drug conjugate composed of a chimeric immunoglobulin (Ig)G monoclonal antibody directed against CD30 conjugated to a cytotoxic agent, monomethyl auristan E. CD30 is a member of the TNF receptor (TNFR) superfamily and is expressed on activated T and B lymphocytes. It was granted accelerated approval in 2011 by the FDA and EMEA for the treatment of recurrent or refractory anaplastic large cell lymphoma and Hodgkin lymphoma.
While the SPC for BV included a warning of PML, its actual development in five patients with lymphoid malignancies (three with Hodgkin lymphoma, one patient with cutaneous T cell lymphoma, one patient with mycosis fungoides) reinforced this risk 57. The absence of data on BV prescriptions does not enable precise risk estimates to be calculated, although the risk of PML has also been flagged up recently in the FDA's adverse event reporting database 56. A particularly noteworthy feature of BV‐induced PML was the rapidity of onset of symptoms within weeks of treatment in contrast to a median of 63 weeks after rituximab, a median of 26 months after treatment with Natalizumab 58 and more than 3 years for Efalizumab 59.
While preceding chemotherapy in four of these patients are potential confounding factors in assessing the precise contributory role of BV, the rapidity of onset of disease, coupled with the development of an immune reconstitution inflammatory syndrome (IRIS) on drug discontinuation, point to a causal relationship 57.
Alemtuzumab
As a potent humanized monoclonal antibody targeted at the CD52 antigen (expressed on lymphocytes and monocytes), Alemtuzumab induces pronounced sustained immunosuppression characterized by global lymphopenia. It is licensed for use in resistant CLL and for RRMS. Given its pronounced immunosuppressive effects on T cells, it is surprising that PML has not been reported more frequently. Currently, reports of PML have been confined to case reports in patients with CLL and a lung transplant recipient 60, 61, 62, but not in patients with MS 63. One possible explanation for the lack of PML to date in MS patients may be the use of Alemtuzumab in a less immunosuppressed patient population in contrast to CLL patients, who are likely to have been heavily pretreated with other chemo‐immunotherapeutic regimens. More prominent than opportunistic infections has been the occurrence of systemic autoimmune diseases such as thyrotoxicosis, immune thrombocytopenic purpura and Goodpasture's syndrome in approximately 47% of patients with MS treated with Alemtuzumab 64. The propensity of Alemtuzumab to trigger autoimmunity has been attributed to the generation of oligoclonal, chronically activated (CD28–, CD57+) T cells with a proliferative phenotype (Ki67+) 65.
Eculizumab
PML in association with Eculizumab (humanized monoclonal anti‐C5 antibody) has been reported in a patient who had undergone a double intestinal‐kidney transplant for atypical haemolytic–uraemic syndrome 66. While there are difficulties in attributing causality because of the confounding effects of concomitant immunosuppressive therapy and the lack of evidence for a role for complement in host defence against the JC virus, it would be important not to rule out this possibility.
Anti‐tumour necrosis factor biologicals
Cases of PML have been reported in three patients with RA treated with infliximab 67, 68 and adalimumab 69, but the strength of evidence regarding causality is confounded by the use of concomitant treatment with methotrexate in at least two of these cases. A single case of PML associated with the use of etanercept in a patient with lupus and erosive polyarthritis has been reported 70. These reports have not prompted the inclusion of PML as a possible risk in the summary of product characteristics for any anti‐TNF agent. Coupled with this, the rarity of PML in millions of patients treated with anti‐TNF biologicals to date casts further doubt on the strength of this association.
Dimethylfumarate and fumaric acid esters
While fumaric acid esters (FAE) have been long established as a treatment for psoriasis, dimethyl fumarate (DMF) was licensed for RRMS in 2013. DMF has wide‐ranging immunological effects, including induction of apoptosis 71, accentuation of Th2 immune responses 72, inhibition of nuclear factor κ‐B signalling 73 and inhibition of lymphocyte trafficking 74. Both these drugs have now been associated with PML in patients in both disease groups, prompting updated safety advice from the UK Medicines and Healthcare Products Regulatory Agency in 2016. To date, there are at least six published cases of PML in recipients of FAE and DMF 48, 49, 75, 76, 77, 78, which does not enable precise risk‐estimate calculations in the absence of a reliable denominator.
Although the precise mechanism(s) by which these drugs predispose patients to PML are unclear, both FAE and DMF cause significant lymphopenia. In an integrated analysis of 2513 MS patients enrolled in longitudinal studies of DMF, mean absolute lymphocyte counts (ALC) fell by 30% during the first year, with 2·2% of patients developing lymphocyte counts below 0·5 × 109/l 79. Lymphocyte subset analysis reveals a disproportionate fall in CD8+ T cells 80, a finding of particular interest in the context of PML, where JCV‐specific CD8+ T cell responses influence clinical outcomes 18. This observation has been supported by reports of PML associated with CD4+ and CD8+ lymphopenia, despite absolute lymphocyte counts between 0·5 and 1·0 × 109/l 49. Consequently, monitoring of lymphocyte counts is crucial as a risk‐mitigation strategy in patients receiving FAE and DMF.
Fingolimod
Fingolimod was the first oral immunomodulatory agent to be licensed for RRMS in 2010 by the FDA. Its novel mechanism of action in preventing lymphocyte egress from lymph nodes by binding and blocking sphingosine 1‐phosphate receptors (S1P) underlies its immunosuppressive effects 81.
By December 2015, three confirmed cases of PML associated with Fingolimod treatment in patients who had not been exposed to Natalizumab had been reported to regulatory authorities 82. In contrast, 17 cases of PML have been reported in approximately 20 000 patients who switched to Fingolimod following previous treatment with Natalizumab 82. Notwithstanding the washout period of 2–3 months between cessation of Natalizumab and commencement of Fingolimod, the totality of available evidence suggests that Fingolimod per se increases the risk of PML. In determining the duration of pharmacodynamic effects of Natalizumab, it is pertinent to note that changes in circulating T and B lymphocyte numbers had normalized at 14 months following cessation of therapy in a group of MS patients 83.
Ibrutinib
Reports of PML in patients with CLL treated with Ibrutinib, a Bruton tyrosine kinase inhibitor, have emerged recently 84, 85. Because many of these patients had been treated with Ibrutinib as part of a broader chemo‐immunotherapeutic regimen (including rituximab), there is insufficient evidence to implicate Ibrutinib as the definitive causal trigger. However, from a mechanistic point of view, impaired T cell function through inhibition of IL‐2 kinase (ITK) 86, coupled with recent reports of Pneumocystis jirovecii pneumonia in treatment‐naive CLL patients treated with Ibrutinib 87, lends credence to the possibility that Ibrutinib predisposes to PML.
Other drugs
There is a single report of PML in a patient with myelofibrosis receiving Ruxolitinib, an inhibitor of Janus kinase (JAK)‐1 and JAK‐2 kinases 88, while recent analysis of the World Health Organization's adverse drug reaction database reveals a newly emergent signal for PML in association with muromonab (anti‐CD3) and basiliximab (anti‐CD25) 89. Because inhibition of the therapeutic targets of these agents suggests some biological plausibility for the development of opportunistic infection, the results of enhanced pharmacovigilance initiatives will be required in order to assess the risk of PML with these drugs.
PML and primary immunodeficiency
To date, 26 cases of PML have been described in patients with primary immunodeficiency disorders (PID), of whom only 54% were characterized molecularly 28. The identification of GOF mutations in STAT‐1 in four of these patients raises the possibility that STAT‐1‐mediated IFN (types I, II and III) responses are a key part of the genetic pathway controlling JCV in immunocompetent patients. In support of this hypothesis, three of the four patients with STAT‐1 GOF developed PML unassociated with immunosuppressive therapeutic intervention on a background of recurrent infections with candida and other pathogens. While IFN‐γ is an important regulator of JCV replication in vitro 90, the clinical features of patients with STAT‐1 GOF mutations appear to be more in keeping with a blunted IFN‐γ response. Among other PIDs, the occurrence of PML in patients with dedicator of cytokinesis 8 (DOCK‐8) deficiency 91 and hyper‐IgE syndrome 92 raises the possibility that control of JCV may also require an intact IL‐17 pathway.
Lessons learnt from drug‐induced PML
It is noteworthy that PML had not been described in MS until the advent of Natalizumab, thus highlighting the modest immune‐modulatory effects of previous therapies such as IFN‐β and the trade‐off between clinical improvement associated with potent immune‐mediated treatment and the risk of PML. Given the different mechanisms of action of the various biologicals and drugs discussed in this review, it is not possible to develop a single unifying explanation for drug‐induced PML, apart from impaired T cell immunity. Interestingly, if the strength of T cell inhibition was the major contributory factor it would have been reasonable to expect an increased risk of PML in recipients of abatacept, a cytotoxic T lymphocyte‐associated protein 4 (CTLA‐4)–Ig fusion receptor licensed for the use of rheumatoid arthritis. However, there are no reports to date of abatacept‐associated PML, and this accords with the absence of a signal in the FDA's adverse drug reporting system for PML 56.
The differential risk between integrin inhibitors depending on specificity, as exemplified by the absence of PML to date in patients treated with Vedolizumab, a gut‐specific α4β7 integrin inhibitor 44, contrasts with the high risk associated with Natalizumab as a pan‐α4 integrin inhibitor (Fig. 4), suggesting that interruption of leucocyte endothelial trafficking within the central nervous system is a key predisposing factor.
Management
Because of the lack of effective treatments and associated dismal prognosis, risk mitigation and early recognition of disease is vital to reduce mortality and minimize neurological disability. The principles of treatment of PML are based on attempts to restore the host immune response, blocking viral entry into cells, treatment of underlying disease (e.g. HIV infection) and/or removal of the iatrogenic trigger. The latter is exemplified by the use of plasma exchange in patients with Natalizumab‐associated PML 93, which suggests that disease amelioration is possible, but this is associated frequently with an immune reconstitution inflammatory syndrome (IRIS) 93. The use of IL‐7 94 and Maraviroc, a CCR5 blocker, are examples of attempts to restore T cell immunity 95 while Mirtazapine has been used to block cellular entry of JCV via the 5‐HT2 receptor (Fig. 2) 96. Another approach to treatment has been based on drugs with anti‐viral activity against JCV. These have included Cytarabine (chemotherapeutic agent), Cidofovir (anti‐viral agent used to treat cytomegalovirus infection), Mefloquine (anti‐malarial) and Topotecan, a topoisomerase inhibitor used in oncology 97, 98, 99, 100.
Despite these different therapeutic approaches, the fatality rate in PML remains worryingly high, with only 52–58% surviving at 1 year 101. For patients with underlying HIV‐negative lymphoproliferative disease, median survival has been reported to be 3 months, with only five of 42 patients surviving beyond 10 months 102.
Concluding remarks
The challenge of PML as an iatrogenic complication begs the question: is it possible to develop effective, targeted immune‐mediated treatments without incurring a significant risk of serious infection? Given that the raison d’être of the immune system is prevention of infection, designing effective immunological interventions without increasing the risk of opportunistic infections may well be an insurmountable challenge. If this is the case, better understanding of the immunopathogenesis, patient selection based on careful risk–benefit analysis and the development of effective treatments for infective complications will help to alleviate the iatrogenic burden of targeted immunotherapy for patients.
Disclosure
None.
Acknowledgements
The author thanks Alison Schroeer for transforming his hand‐drawn diagrams into clear and comprehensible figures suitable for publication.
References
- 1. Astrom KE, Mansell EL, Richardson EP Jr. Progressive multifocal leukoencephalopathy: a hitherto unrecognised complication of chronic lymphocytic leukaemia and Hogkin's disease. Brain 1958; 81:93–111. [DOI] [PubMed] [Google Scholar]
- 2. Padgett BL, Walker DL, ZuRhein GM, Eckroade RJ, Dessel BH. Cultivation of papova‐like virus from human brain with progressive multifocal leukoencephalopathy. Lancet 1971; 1:1257–60. [DOI] [PubMed] [Google Scholar]
- 3. Misbah SA, Spickett GP, Zeman A et al Progressive multifocal leukoencephalopathy, sclerosing cholangitis, bronchiectasis and disseminated warts in a patient with primary combined immune deficiency. J Clin Path 1992; 45:624–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Koralnik IJ, Du Pasquier RA, Letvin NL. JC virus‐specific cytotoxic T lymphocytes in individuals with progressive multifocal leukoencephalopathy. J Virol 2001; 75:3483–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wollebo HS, White MK, Gordon J, Berger JR, Khalili K. Persistence and pathogenesis of the neurotropic polyoma virus JC. Ann Neurol 2015; 77:560–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dorries K, Vogel E, Gunther S, Czub S. Infection of human polyoma viruses JC and BK in peripheral blood leucocytes from immunocompetent individuals. Virology 1994; 198:59–70. [DOI] [PubMed] [Google Scholar]
- 7. Assetta B, Maginnis MS, Ahufinger IG et al 5‐HT2 receptors facilitate JC polyoma virus entry. J Virol 2013; 87:13490–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frohman EM, Monaco MC, Remington G et al JC virus in CD34+ and CD19+ cells in patients with multiple sclerosis treated with natalizumab. JAMA Neurol 2014; 71:596–602. [DOI] [PubMed] [Google Scholar]
- 9. Chalkias S, Dang X, Bord E et al JC virus reactivation during prolonged natalizumab monotherapy for multiple sclerosis. Ann Neurol 2014; 75:925–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Warnke C, Adams O, Kieseier B. Relevance of CD34+ cells as a reservoir for JC virus in patients with multiple sclerosis. JAMA Neurol 2014; 71:1192. [DOI] [PubMed] [Google Scholar]
- 11. Perez‐Liz G, Del Valle L, Gentilella A, Croul S, Khalili K. Detection of JC virus DNA fragments but not proteins in normal brain tissue. Ann Neurol 2008; 64:379–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Major EO. Progressive multifocal leukoencephalopathy in patients on immunomodulatory therapies. Ann Rev Med 2010; 61:35–47. [DOI] [PubMed] [Google Scholar]
- 13. Ciappi S, Azzi A, De Santis R et al Archetypal and rearranged sequences of human polyoma virus JC transcription control region in peripheral blood leucocytes and in cerebrospinal fluid. J Gen Virol 1999; 80:1017–23. [DOI] [PubMed] [Google Scholar]
- 14. Van Loy T, Thys K, Ryschkewitsch C et al JC virus quasispecies analysis reveals a complex viral population underlying progressive multifocal leukoencephalopathy and supports viral dissemination via the haematogenous route. J Virol 2015; 89:1340–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sindic CJ, Trebst C, Van Antwerpen MP et al Detection of CSF‐specific oligoclonal antibodies to recombinant JC virus VP1 in patients with progressive multifocal leukoencephalopathy. J Neuroimmunol 1997; 76:100–4. [DOI] [PubMed] [Google Scholar]
- 16. Sindic CJ, Monteyne P, Laterre EC. The intrathecal synthesis of virus‐specific oligoclonal IgG in multiple sclerosis. J Neuroimmunol 1994; 54:75–80. [DOI] [PubMed] [Google Scholar]
- 17. Luxton RW, Zeman A, Holzel H et al Affinity of antigen‐specific IgG distinguishes multiple sclerosis from encephalitis. J Neurol Sci 1995; 132:11–9. [DOI] [PubMed] [Google Scholar]
- 18. Koralnik IJ, Du Pasquier RA, Kuroda MJ et al Association of prolonged survival in HLA‐A2+ progressive multifocal leucoencephalopathy patients with a cytotoxic T lymphocyte response specific for a commonly recognised JC virus epitope. J Immunol 2002; 168:499–504. [DOI] [PubMed] [Google Scholar]
- 19. Du Pasquier RA, Kuroda MJ, Zheng Y, Jean‐Jacques J, Letvin NL, Koralnik IJ. A prospective study demonstrates an association between JC virus‐specific cytotoxic T lymphocytes and the early control of progressive multifocal leucoencephalopathy. Brain 2004; 127:1970–8. [DOI] [PubMed] [Google Scholar]
- 20. Wuthrich C, Kesari S, Kim WK et al Characterization of lymphocytic infiltrates in progressive multifocal leucoencephalopathy: co‐localization of CD8 T cells with JCV‐infected glial cells. J Neurovirol 2006; 12:116–28. [DOI] [PubMed] [Google Scholar]
- 21. Gheuens S, Bard E, Kesari S et al Role of CD4+ and CD8+ T cell responses against JC virus in the outcome of patients with progressive multifocal leucoencephalopathy (PML) and PML with immune reconstitution syndrome. J Virol 2011; 85:7256–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Aly L, Yousef S, Schippling S et al Central role of JC virus‐specific CD4+ lymphocytes in PML‐immune reconstitution syndrome. Brain 2011; 134:2687–702. [DOI] [PubMed] [Google Scholar]
- 23. Yousef S, Planas R, Chakroun K et al TCR bias and HLA cross‐restriction are strategies of human brain‐infiltrating JC virus specific CD4+ T cells during viral infection. J Immunol 2012; 189:3618–30. [DOI] [PubMed] [Google Scholar]
- 24. Qin Q, Shwetank, Frost EL, Maru S, Lukacher AE. Type I interferons regulate the magnitude and functionality of mouse polyomavirus‐specific CD8 T cells in a virus strain‐dependent manner. J Virol 2016; 90:5187–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. O'Hara BA, Atwood WJ. Interferon β1‐a and selective anti‐5HT2a receptor antagonists inhibit infection of human glial cells by JC virus. Virus Research 2008; 132:97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Assetta B, De Cecco M, O'Hara B, Atwood WJ. JC polyomavirus infection of primary human renal epithelial cells is controlled by a type I IFN‐induced response. mBio 2016; 7:e00903‐16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delbue S, Guerini FR, Mancuso R et al JC virus viraemia in interferon‐β‐treated and untreated Italian multiple sclerosis patients and healthy controls. J Neurovirol 2007; 13:73–7. [DOI] [PubMed] [Google Scholar]
- 28. Zerbe CS, Marciano BE, Katial RK et al Progressive multifocal leukoencephalopathy in primary immune deficiencies: STAT‐1 gain of function and review of the literature. Clin Infect Dis 2016; 62:986–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tan CS, Bord E, Broge TA Jr et al Increased programmed cell death‐1 expression on T lymphocytes of patients with progressive multifocal leucoencephalopathy. J Acquir Immune Def Syn 2012; 60:244–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Berger JR, Aksamit AJ, Clifford DB et al PML diagnostic criteria. Consensus statement from the American Academy for Neurology Neuroinfectious Disease Section. Neurology 2013; 80:1430–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mentzer D, Prestel J, Adams O et al Case definition for progressive multifocal leucoencephalopathy following treatment with monoclonal antibodies. J Neurol Neurosurg Psychiatry 2012; 83:927–33. [DOI] [PubMed] [Google Scholar]
- 32. Calabrese LH, Molloy ES, Huang D, Ransohoff RM. PML in rheumatic diseases: evolving clinical and pathologic patterns of disease. Arthritis Rheum 2007; 56:2116–28. [DOI] [PubMed] [Google Scholar]
- 33. Molloy ES, Calabrese LH. PML in patients with rheumatic diseases: are patients with SLE at particular risk? Autoimmunity Rev 2008; 8:144–6. [DOI] [PubMed] [Google Scholar]
- 34. Yednock TA, Cannon C, Fritz LC, Sanchez‐Madrid F, Steinman L, Karin N. Prevention of experimental autoimmune encephalomyelitis by antibodies against α4β1 integrin. Nature 1992; 356:63–6. [DOI] [PubMed] [Google Scholar]
- 35. Langer‐ Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. PML in a patient treated with Natalizumab. N Engl J Med 2005; 353:375–81. [DOI] [PubMed] [Google Scholar]
- 36. Kleinschmidt‐De Masters BK, Tyler KL. PML complicating treatment with Natalizumab and interferon β‐1a for multiple sclerosis. N Engl J Med 2005; 353:369–74. [DOI] [PubMed] [Google Scholar]
- 37. Yousry TA, Major EO, Ryschkewitsch C et al Evaluation of patients treated with Natalizumab for PML. N Engl J Med 2006; 354:924–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berger JR, Fox RJ. Reassessing the risk of natalizumab‐associated PML. J Neurovirol 2016; 22:533–5. [DOI] [PubMed] [Google Scholar]
- 39. Chalkley JJ, Berger JR. Progressive multifocal leukoencephalopathy in multiple sclerosis. Curr Neurol Neurosci Rep 2013; 13:408. [DOI] [PubMed] [Google Scholar]
- 40. Defer G, Mariotte D, Derache N et al CD49d expression as a promising biomarker to monitor natalizumab efficacy. J Neurol Sci 2012; 314:138–42. [DOI] [PubMed] [Google Scholar]
- 41. Schwab N, Schneider‐Hohendorf T, Posevitz V et al L‐selectin is a possible biomarker for individual PML risk in natalizumab‐treated MS patients. Neurology 2013; 81:865–71. [DOI] [PubMed] [Google Scholar]
- 42. Pignolet B, Schwab N, Schneider‐Hohendorf T et al CD62L test at 2 years of Natalizumab predicts progressive multifocal leukoencephalopathy. Neurology 2016; 87:1–3. [DOI] [PubMed] [Google Scholar]
- 43. Lieberman LA, Zeng W, Singh C et al CD62L is not a reliable marker for predicting PML risk in natalizumab‐treated relapsing–remitting multiple sclerosis patients. Neurology 2016; 86:375–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Colombel JF, Sands BE, Rutgeerts P et al The safety of vedolizumab for ulcerative colitis and Crohn's disease. Gut 2016; pii: gutjnl-2015-311079. doi: 10.1136/gutjnl-2015-311079. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Crason KR, Focosi D, Major EO et al Monoclonal antibody‐associated progressive multifocal leukoencephalopathy in patients treated with rituximab, natalizumab and efalizumab: a review from the research on adverse drug events and reports (RADAR) project. Lancet Oncol 2009; 10:816–24. [DOI] [PubMed] [Google Scholar]
- 46. Schwab N, Ulzheimer JC, Fox RJ et al Fatal PML associated with efalizumab therapy – insights into integrin αLβ2 in JC virus control. Neurology 2012; 78:458–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Korman BD, Tyler KL, Korman NJ. Progressive multifocal leukoencephalopathy, efalizumab and immunosuppression – a cautionary tale for dermatologists. Arch Dermatol 2009; 145:937–42. [DOI] [PubMed] [Google Scholar]
- 48. Bartsch T, Rempe T, Wrede A et al Progressive neurologic dysfunction in a psoriasis patient treated with dimethyl fumarate. Ann Neurol 2015; 78:501–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nieuwkamp DJ, Murk JL, van Oosten BW et al PML in a patient without severe lymphocytopenia receiving dimethyl fumarate. N Engl J Med 2015; 372:1474–6. [DOI] [PubMed] [Google Scholar]
- 50. US Food and Drug Administration . FDA Alert Rituximab‐PML [12/2006]. Available at: www.fda.gov/Drugs/DrugSafety/…/ucm126519.htm (accessed 1 November 2016).
- 51. US Food and Drug Administration . FDA Alert Rituximab‐PML posted 23.10.2009. https://www.fda.gov/downloads/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/UCM187792.pdf (accessed 1 November 2016).
- 52. Amend KL, Turnbull B, Foskett N, Napalkov P, Kurth T, Seeger J. Incidence of progressive multifocal leukoencephalopathy in patients without HIV. Neurology 2010; 75:1326–32. [DOI] [PubMed] [Google Scholar]
- 53. Clifford DB, Ances B, Costello C et al Rituximab‐associated progressive multifocal leukoencephalopathy in rheumatoid arthritis. Arch Neurol 2011; 68:1156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Melet J, Mulleman D, Goupille P, Ribourtout B, Watier H, Thibault G. Rituximab‐induced T cell depletion in patients with rheumatoid arthritis: association with clinical response. Arthritis Rheum 2013; 65:2783–90. [DOI] [PubMed] [Google Scholar]
- 55. Wilk E, Witte T, Marquardt N et al Depletion of functionally active CD20+ T cells by rituximab treatment. Arthritis Rheum 2009; 60:3563–71. [DOI] [PubMed] [Google Scholar]
- 56. Raisch DW, Rafi JA, Cheng C, Bennett CL. Detection of cases of PML associated with new biologicals and targeted cancer therapies from the FDA's adverse drug reporting system. Expert Opin Drug Saf 2016; 15:1003–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Carson KR, Newsome SD, Kim EJ et al Progressive multifocal leukoencephalopathy associated with brentuximab vedotin therapy – a report of 5 cases from the Southern Network on Adverse Reactions (SONAR) project. Cancer 2014; 120:2464–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bloomgren G, Richman S, Hotermans C et al Risk of natalizumab‐associated PML. N Engl J Med 2012; 366:1870–80. [DOI] [PubMed] [Google Scholar]
- 59. Molloy ES, Calabrese LH. Therapy: targeted but not trouble‐free. Nat Rev Rheumatol 2009; 5:418–9. [DOI] [PubMed] [Google Scholar]
- 60. Isidoro L, Pires P, Rito L, Cordeiro G. Progressive multifocal leukoencephalopathy in a patient with CLL treated with alemtuzumab. BMJ Case Rep 2014; 2014: bcr2013201781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. D'Souza A, Wilson J, Mukherjee S, Jaiyesimi I. Progressive multifocal leukoencephalopathy in CLL: a report of three cases and review of the literature. Clin Lymphoma Myeloma Leuk 2010; 4:e2012043. [DOI] [PubMed] [Google Scholar]
- 62. Waggoner J, Martinu T, Palmer SM. Progressive multifocal leukoencephalopathy following heightened immunosuppression after lung transplant. J Heart Lung Transplant 2009; 28:395–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wingerchuk DM, Weinshenker BG. Disease modifying therapies for relapsing multiple sclerosis. Br Med J 2016; 354:i3518. [DOI] [PubMed] [Google Scholar]
- 64. Tuohy O, Costelloe L, Hill‐Cawthorne G et al Alemtuzumab treatment of multiple sclerosis: long term safety and efficacy. J Neurol Neurosurg Psychiatry 2015; 86:208–15. [DOI] [PubMed] [Google Scholar]
- 65. Jones JL, Thompson SA, Loh P et al Human autoimmunity after lymphocyte depletion is caused by homeostatic T cell proliferation. Proc Natl Acad Sci USA 2013; 110:20200–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gomez‐Cibeira E, Ivanovic‐Barbeito Y, Gutierrez‐ Martinez E et al Eculizumab‐related progressive multifocal leukoencephalopathy. Neurology 2016; 86:399–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kumar D, Bouldin TW, Berger RG. A case of progressive multifocal leucoencephalopathy in a patient treated with infliximab. Arthritis Rheum 2010; 62:3191–5. [DOI] [PubMed] [Google Scholar]
- 68. Sammut L, Wallis D, Holroyd C. Progressive multifocal leucoencephalopathy associated with infliximab. J R Coll Physicians Edinb 2016; 46:163–5. [DOI] [PubMed] [Google Scholar]
- 69. Ray M, Curtis JR, Baddley JW. A case report of progressive multifocal leucoencephalopathy (PML) associated with adalimumab. Ann Rheum Dis 2014; 73:1429–30. [DOI] [PubMed] [Google Scholar]
- 70. Graff‐Radford J, Robinson MJ, Warsame RM, Matteson EL, Eggers SD, Keegan BM. Progressive multifocal leukoencephalopathy in a patient treated with etanercept. Neurologist 2012; 18:85–7. [DOI] [PubMed] [Google Scholar]
- 71. Treumer F, Zhu K, Glaser R, Mrowietz U. Dimethyl fumarate is a potent inducer of apoptosis in human T cells. J Invest Dermatol 2003; 121:1383–8. [DOI] [PubMed] [Google Scholar]
- 72. de Jong R, Bezemer AC, Zomerdijk TP, van de Pouw‐Kraan T, Ottenhoff TH, Nibbering PH. Selective stimulation of T helper 2 cytokine responses by the anti‐psoriasis agent monomethylfumarate. Eur J Immunol 1996; 26:2067–74. [DOI] [PubMed] [Google Scholar]
- 73. Vandermeeren M, Janssens S, Wouters H et al Dimethylfumarate is an inhibitor of cytokine‐induced nuclear translocation of NF‐kappaB1, but not ReIA in normal human dermal fibroblast cells. J Invest Dermatol 2001; 116:124–30. [DOI] [PubMed] [Google Scholar]
- 74. Rubant SA, Ludwig RJ, Diehl S et al Dimethy fumarate reduces leukocyte rolling in vivo through modulation of adhesion molecule expression. J Invest Dermatol 2008; 128:326–31. [DOI] [PubMed] [Google Scholar]
- 75. Ermis U, Weis J, Schulz JB. PML in a patient treated with fumaric acid. N Engl J Med 2013; 368:1657–8. [DOI] [PubMed] [Google Scholar]
- 76. van Oosten BW, Killestein J, Barkhof F, Polman CH, Wattjes MP. PML in a patient treated with dimethylfumarate from a compounding pharmacy. N Engl J Med 2013; 368:1658–9. [DOI] [PubMed] [Google Scholar]
- 77. Rosenkranz T, Novas M, Terborg C. PML in a patient with lymphocytopenia treated with dimethyl fumarate. N Engl J Med 2015; 372:1476–8. [DOI] [PubMed] [Google Scholar]
- 78. Baharnoori M, Lyons J, Dastagir A, Koralnik I, Stankiewicz JM. Non fatal PML in a patient with multiple sclerosis treated with dimethyl fumarate. Nuerol Neuroimmunol Neuroinflamm 2016; 3:e274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Fox RJ, Chan A, Gold R et al Characterizing absolute lymphocyte count profiles in dimethyl fumarate‐treated patients with MS. Neurology Clin Pract 2016; 6:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Spencer CM, Crabtree‐Hartman EC, Lehmann‐Horn K, Cree BA, Zamvil SS. Reduction of CD8+ T lymphocytes in multiple sclerosis patients treated with dimethyl fumarate. Neurol Neuroimmunol Neurinflamm 2015; 2:e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Draayer YM, Sarazin J, Fox D, Schiopu E. The sphingosine‐1‐ phosphate receptor: a novel therapeutic target for multiple sclerosis and other autoimmune diseases. Clin Immunol 2017; 175:10–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. European Medicines Agency . New recommendations to minimise risk of the rare brain infection PML and a type of skin cancer with Gilenya. Available at: http://www.ema.europa.eu/ema/index.jsp?curl=pages/news_and_events/news/2015/12/news_detail_002447.jsp&mid=WC0b01ac058004d5c1 (accessed 13 December 2016).
- 83. Stuve O, Cravens PD, Frohman EM et al Immunologic, clinical and radiologic status 14 months after cessation of Natalizumab therapy. Neurology 2009; 72:396–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Lutz M, Schulze AB, Rebber E et al Progressive multifocal leukoencephalopathy after Ibrutinib treatment for chronic lymphocytic leukaemia. Cancer Res Treat 2016; doi 10.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bennett CL, Berger JR, Sartor O et al Progressive multifocal leukoencephalopathy among ibrutinib‐treated patients with chronic lymphocytic leukaemia. Br J Haem 2016; doi 10.1111/bjh 14322 [Google Scholar]
- 86. Dobovsky JA, Beckwith KA, Natarajan G et al Ibrutinib is an irreversible inhibitor of ITK driving a TH1‐selective pressure in T lymphocytes. Blood 2013; 122:2539–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Ahn IE, Jerussi T, Farooqui M, Wiestner A, Gea‐Banacloche J. Atypical pneumocystis jirovecii pneumonia in previously untreated patients with CLL on single agent ibrutinib. Blood 2016; 128:1940–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Wathes R, Moule S, Milojkovic D. Progressive multifocal leukoencephalopathy associated with ruxolitinib. N Engl J Med 2013; 369:197–8. [DOI] [PubMed] [Google Scholar]
- 89. Melis M, Biagi C, Smabrekke L et al Drug‐induced progressive multifocal leukoencephalopathy: a comprehensive analysis of the WHO adverse drug database. CNS Drugs 2015; 29:879–91. [DOI] [PubMed] [Google Scholar]
- 90. De‐Simone FI, Sariyar R, Otalora YL et al IFN‐gamma inhibits JC virus replication in glial cells by suppressing T‐antigen expression. PLOS ONE 2015; 10:e0129694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Day‐Williams AG, Sun C, Jelcic I et al Whole genome sequencing reveals a chromosome 9p deletion causing DOCK8 deficiency in an adult with diagnosed with hyper‐IgE syndrome who developed progressive multifocal leukoencephalopathy. J Clin Immunol 2015; 35:92–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Angelini L, Pietrogrande MC, Delle Paine MR et al Progressive multifocal leukoencephalopathy in a child with hyperimmunoglobulin E recurrent infection syndrome and review of the literature. Neuropediatrics 2001; 32:250–5. [DOI] [PubMed] [Google Scholar]
- 93. Schroder A, Lee DH, Hellwig K, Lukas C, Linker RA, Gold R. Successful management of natalizumab‐associated progressive multifocal leukoencephalopathy and immune reconstitution syndrome in a patient with multiple sclerosis. Arch Neurol 2010; 67:1391–4. [DOI] [PubMed] [Google Scholar]
- 94. Miskin DP, Chalkias SG, Dang X, Bord E, Batson S, Koralnik IJ. Interleukin‐7 treatment of PML in a patient with idiopathic lymphocytopenia. Neurol Neuroimmunol Neuroinflamm 2016; 3:e213.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Middel A, Arends JE, van Lelyveld SFL et al Clinical and immunologic effects of maraviroc in progressive multifocal leukoencephalopathy. Neurology 2015; 85:104–6. [DOI] [PubMed] [Google Scholar]
- 96. Cettomai D, McArthur JC. Mirtazapine use in human immunodeficiency virus‐infected patients with progressive multifocal leukoencephalopathy. Arch Neurol 2009; 66:255–8. [DOI] [PubMed] [Google Scholar]
- 97. Hall CD, Dafni U, Simpson D et al Failure of cytarabine in progressive multifocal leukoencephalopathy associated with human immunodeficiency virus infection. N Engl J Med 1998; 338:1345–51. [DOI] [PubMed] [Google Scholar]
- 98. De Luca A, Ammassari A, Pezzotti P et al Cidofovir in addition to antiretroviral treatment is not effective for AIDS‐associated progressive multifocal leukoencephalopathy: a multi‐cohort analysis. AIDS 2008; 22:1759–67. [DOI] [PubMed] [Google Scholar]
- 99. Brickelmaier M, Lugovsky A, Kartikeyan R et al Identification and characterisation of mefloquine efficacy against JC virus in‐vitro . Antimicrobial Agents Chemother 2009; 53:1840–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nukuzuma S, Nakamichi K, Kameoka M et al Suppressive effects of topoisomerase inhibitors on JC polyomavirus propagation in human neuroblastoma cells. Microbiol Immunol 2016; 60:253–60. [DOI] [PubMed] [Google Scholar]
- 101. Marzocchetti A, Tompkins T, Clifford DB et al Determinants of survival in progressive multifocal leukoencephalopathy. Neurology 2009; 73:1551–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Garcia‐Suarez J, de Miguel D, Krsnik I, Bañas H, Arribas I, Burgaleta C. Changes in the natural history of progressive multifocal leukoencephalopathy in HIV‐negative lymphoproliferative disorders: impact of novel therapies. Am J Hematol 2005; 80:271–81. [DOI] [PubMed] [Google Scholar]