

Significance
Tubulin has emerged as a highly unexpected component of mitochondrial membranes involved in regulation of membrane permeability. This discovery has reawakened interest in the nature of the tubulin–membrane interaction to answer a new question: How does tubulin, a cytosolic protein famous for its role in microtubule structure and dynamics, come to target mitochondrial membranes? Here, using a combination of five biophysical methods, we study peripheral binding of tubulin to biomimetic membranes of different lipid compositions. We conclude that tubulin distinguishes between lamellar and nonlamellar lipids through a highly conserved amphipathic binding motif. Specifically, α-tubulin targets cell and organelle membranes by sensing lipid-packing defects via an amphipathic α-helix, with broad consequences for both normal cellular function and disease.
Keywords: dimeric tubulin, protein–lipid interactions, neutron reflectometry, molecular dynamics, peripheral membrane proteins
Abstract
Dimeric tubulin, an abundant water-soluble cytosolic protein known primarily for its role in the cytoskeleton, is routinely found to be associated with mitochondrial outer membranes, although the structure and physiological role of mitochondria-bound tubulin are still unknown. There is also no consensus on whether tubulin is a peripheral membrane protein or is integrated into the outer mitochondrial membrane. Here the results of five independent techniques—surface plasmon resonance, electrochemical impedance spectroscopy, bilayer overtone analysis, neutron reflectometry, and molecular dynamics simulations—suggest that α-tubulin’s amphipathic helix H10 is responsible for peripheral binding of dimeric tubulin to biomimetic “mitochondrial” membranes in a manner that differentiates between the two primary lipid headgroups found in mitochondrial membranes, phosphatidylethanolamine and phosphatidylcholine. The identification of the tubulin dimer orientation and membrane-binding domain represents an essential step toward our understanding of the complex mechanisms by which tubulin interacts with integral proteins of the mitochondrial outer membrane and is important for the structure-inspired design of tubulin-targeting agents.
Dimeric tubulin is an abundant water-soluble cytosolic protein comprising two subunits, α and β, with distinct primary structure but similar secondary structure. Tubulin is known primarily for its role in the cytoskeleton, where it is assembled into microtubules in which the exposed end is the β subunit. As a result, microtubule-targeting drugs (MTDs) used in chemotherapy bind exclusively to the β subunit (1). Until recently, any physiological role of unassembled, cytosol-solubilized dimeric tubulin remained unknown. Early clues involved the association of tubulin with various cellular membranes, including mitochondrial membranes (2), and led to the suggestion that tubulin is an intrinsic component of the mitochondrial outer membrane (MOM) (3). A variety of biochemical techniques gave conflicting views of the mechanism of membrane association (4): Tubulin-membrane binding to isolated cellular membranes was shown to be reversible under some conditions, suggesting that the tubulin heterodimer was peripherally bound (5); however, tryptophan fluorescence reported an increase in the hydrophobic environment of membrane-bound tubulin, indicating that it might be integrated into the membrane (6).
Emerging evidence, based on in situ experiments on respiration of isolated mitochondria (7, 8) and intact cancer cells (9, 10), now points to dimeric tubulin as a regulator of mitochondrial function via the voltage-dependent anion channel (VDAC) of the MOM. In vitro measurements suggest the insertion of tubulin’s polyanionic C terminus into the lumen of the VDAC β-barrel as the mechanism modulating the flow of ATP and ADP through VDAC and regulating metabolite fluxes across the MOM (11, 12). Both the VDAC–tubulin interaction rate and the amount of fluorescent tubulin bound to liposomes increased drastically if lamellar dioleoylphosphatidylcholine (DOPC) lipids were replaced with nonlamellar dioleoylphosphatidylethanolamine (DOPE) (13). These observations point to delivery of tubulin to VDAC via localization at the MOM as a crucial step in tubulin regulation of mitochondrial respiration. Despite decades of research into tubulin–membrane interactions, much remains unknown, including whether tubulin–membrane binding is peripheral or integral in nature, what the tubulin membrane-binding domain is, and why tubulin prefers PE to PC headgroups (14). These questions are of particular importance for the structure-inspired design of therapeutic agents targeting the VDAC–tubulin interaction and for understanding the interaction of MTDs with mitochondria (15).
In this paper we show that tubulin is a peripherally bound amphitropic protein (AP) and identify its membrane-binding domain. APs are the subfamily of peripheral membrane proteins that interact directly with the lipid membrane rather than with intrinsic membrane proteins and are therefore strongly influenced by lipid composition (16, 17). A number of diseases, such as atherosclerosis, type II diabetes, and lysosomal storage disorders, are associated with defects in maintaining the correct distribution of intracellular lipids (18). Although involvement of APs in various cell functions is soundly established (19–21), the mechanisms of their interaction with cellular membranes are only beginning to be understood because their characteristic reversible binding to the membranes creates obvious experimental difficulties in assessing binding conformations and kinetics (19, 20, 22).
Here, we surmount these experimental challenges using a combination of biophysical characterization methods that are optimized for peripheral membrane proteins, as summarized in Fig. 1. First, we use surface plasmon resonance (SPR), electrochemical impedance spectroscopy (EIS), and bilayer overtone analysis (BOA) to study the physicochemical properties of tubulin binding and its dependence on the lamellar (DOPC) vs. nonlamellar (DOPE) lipid composition of bilayer membranes. We show that in a binary mixture of these two zwitterionic lipids tubulin binding is strongly enhanced on membranes containing PE headgroups. Because all measurements were performed at room temperature, unsaturated lipids were used to preserve the membrane fluidity characteristic of the MOM (23, 24).
Fig. 1.

Schematic of techniques and bilayer architectures used in this work. (A) NR reports on the layered structure of the stBLM and any bound protein via the scattering length density SLD(z). (B) As protein binds to an stBLM on a gold substrate, SPR measures the shift in the angle of incident laser light required to excite plasmons in the gold. EIS is performed simultaneously and yields information about the capacitance and leakage resistance of the stBLM. (C) BOA uses the second harmonic of the current I(2f0) induced by an ac excitation potential V(f0) to monitor properties of the potential across a substrate-free planar lipid bilayer. When the transmembrane potential is offset by an applied dc potential , the amplitude of the second harmonic current is minimized (inset at the left), allowing to be tracked in time.
Second, we identify the insertion region of the tubulin protein that interacts with phospholipids by a combination of molecular dynamics (MD) simulations and neutron reflectometry (NR) data. Microsecond-long simulations of α- and β-tubulin monomers on the surfaces of DOPE and DOPC bilayers indicate unique monomer- and lipid-specific interaction modes, of which only α-tubulin on DOPE exhibits long-term stability. In neither NR experiments nor MD simulations is tubulin observed to penetrate deeply into the hydrophobic region of the bilayer. The combination of a highly conserved tryptophan residue and a nearby amphipathic helix of α-tubulin seems to be responsible for binding to both DOPC and DOPE surfaces, with apparent preference for DOPE bilayers. The mechanism of binding is likely to be adsorption of the binding helix into the headgroup region of PE-containing membranes; both hydrophobic and electrostatic interactions play a role in this process, explaining the preference of tubulin for membranes containing PE headgroups. These features identify tubulin as a member of a rare class of APs that sense lipid-packing defects without known involvement in lipid regulatory pathways; to our knowledge, the only other proteins in this category are the amyloid proteins containing class A amphipathic helical domains, such as α-synuclein (25), which also binds preferentially to PE headgroups (26). We conclude that the pronounced sensitivity of tubulin binding to the membrane PE content points to a regulatory interplay between the homeostatic lipid composition of the MOM and bound tubulin.
Results
Lipid Dependence of Tubulin Binding from SPR.
SPR (Fig. 1B) was chosen as the most direct method of obtaining the mass of protein that binds to a surface. The well-established sparsely tethered bilayer lipid membrane (stBLM) system (27) allows the formation of a membrane suitable for SPR measurement and is stable against buffer exchanges for days. Binding curves obtained from SPR experiments are shown in Fig. 2 for four different DOPC/DOPE lipid compositions corresponding to the PE mole fraction of 0, 0.25, 0.5, and 0.75. Three concentrations of tubulin were used: 100 nM, 300 nM, and 600 nM (M = moles per liter). Only membranes with show a significant amount of tubulin binding. The majority of the tubulin binding occurs slowly over several hours, with only a small amount [1 pixel (px) to 2 px] of fast binding, seen as small steps on the binding curves following the addition of tubulin. Due to the long time scales involved, the membrane was allowed to reach equilibrium only for 600 nM tubulin. The unbinding rate over 15 min was negligible.
Fig. 2.

SPR response to tubulin binding to stBLMs, demonstrating that increase of DOPE content leads to elevated tubulin binding to the membrane surface. The baseline SPR response has been subtracted and the SPR signal at each tubulin concentration corrected for refractive index changes. stBLM lipid compositions are indicated. Dashed lines separate three time intervals during which the membrane was exposed to tubulin concentrations of 100 nM, 300 nM, and 600 nM tubulin, respectively.
These results can be understood in the context of a two-step binding model, in which a fast initial step is reversible and a slow second step has a very small unbinding rate:
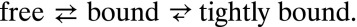 |
The solution to the kinetic equations is a sum of two exponentially decaying functions, a fast component from the reversible “bound” state and a slow component from transitions into the “tightly bound” state. The overall kinetics are dominated by the slow “off rate,” as is typical for APs (28).
An estimation shows that if we approximate the shape of the tubulin dimer by a rectangular prism of 4 nm × 4 nm × 8 nm, then a completely packed monolayer of tubulin bound by a 4-nm × 4-nm end would give a volume-filled layer 8 nm thick and a corresponding SPR response of 124 px. At the highest PE fraction () and protein concentration (600 nM) we observed about half this value.
Stability of stBLMs Confirmed by EIS.
EIS can be performed simultaneously with the SPR measurements (29) and, in addition to providing qualitative information about the mechanism of protein binding, reports on the degree of membrane disruption by protein binding. This allows the integrity of the stBLM to be monitored over the course of the experiment (30). EIS results are shown in Fig. 3 as a Cole–Cole plot, in which the imaginary part of the complex capacitance , where is the complex impedance, is plotted against the real part of the complex capacitance . The diameter of the semicircle is equal to the capacitance of a bilayer. For an ideal capacitor, the minimum shown in the inset would be zero; the nonzero imaginary component of this minimum is related to the density and structure of defects in the lipid bilayer (30).
Fig. 3.

Cole–Cole plots of EIS of tubulin-exposed stBLMs of (A) DOPC and (B) 1:1 DOPC:DOPE, showing little disruption of the bilayer electrical properties, even at the highest tubulin concentrations. The vertical and horizontal axes represent, respectively, the (negative) imaginary and real components of the complex capacitance , where is the complex impedance of the stBLM. The colored traces represent different tubulin concentrations, as indicated in the legend. The diameter of the half-circle corresponds to the capacitance of the membrane, which is similar for all lipid compositions.
For all lipid compositions, with only DOPC and 1:1 DOPC:DOPE shown in Fig. 3 as examples, the bilayers are mostly defect-free and essentially unaffected by the presence of tubulin, even after incubating at the highest concentrations for 12 h or more. This suggests that the tubulin is peripherally associated with the bilayers and does not create bilayer defects by embedding itself deep within the hydrophobic region. Note that tubulin at concentrations more than 300–500 nM (depending on lipid and the applied dc voltage) will often lead to pore formation and rupture of free planar lipid bilayers such as those used for BOA. By contrast, the unbiased stBLM system used here is stable at these tubulin concentrations.
Charge of the Tubulin Membrane Binding Domain from BOA.
In BOA (Fig. 1C), the second harmonic of an electrical signal reports on the bilayer surface potentials (31, 32), is a complementary technique to SPR and EIS because it reports on the electrical properties of both the bilayer and the membrane-bound species. BOA is performed on “free” planar lipid membranes, rather than the stBLM system, and is therefore free of the substrate-induced stresses that may influence membrane behavior. Forming planar lipid membranes from two opposed monolayers allows stable bilayers from pure DOPE to be obtained (33). The planar bilayer system directly corresponds to that used for single-channel recordings in studies of the VDAC–tubulin interaction.
The results of BOA experiments on pure DOPC and DOPE planar lipid bilayers are shown in Fig. 4A, which gives the time course of the potential across the bilayer. Note that the equilibration time required for tubulin binding varied with tubulin concentration. The significant amount of binding to DOPE membranes observed here at nanomolar concentrations (Fig. 4B) is consistent with previous observations using other techniques (2, 13), whereas no measurable binding to DOPC membranes was observed.
Fig. 4.

Bilayer overtone analysis of tubulin binding to DOPC (lower curve/points) and DOPE (upper curve/points). (A) Data from representative experiments. Tubulin additions are labeled by arrows; labels denote the total concentration of tubulin (in nanomolar) to which the bilayer was exposed until the next addition. (B) Binding curves for tubulin on DOPC and DOPE. Points and error bars represent the mean and SD from the mean for three independent experiments. The solid line, corresponding to a Hill equation with and a Hill coefficient of 6.8, is drawn to guide the eye.
The bilayer potential originates from a combination of at least two asymmetries between the two lipid leaflets: the difference in the net charge bound to the surface of each leaflet and the difference in lipid dipole potentials, which depends on the tilt angle of the lipid headgroups in the two leaflets. If the tilt angle is unaffected by protein binding, is expected to correspond only to the charges of the protein that fall within a Debye length of the surface; charges further away from the surface are screened by the electrolyte. If, however, the tilt angle of the lipid headgroups is affected by the binding of the protein, both the tilt angle effect (which can in principle be either positive or negative) and the bound protein charge effect contribute to the measured potential.
For the interpretation of these experiments, we assume that the signal we observe is due entirely to the charge of the membrane-binding region of tubulin. Notably, Fig. 4 shows that the change in is positive, despite the fact that tubulin carries a net negative charge at pH 7.4 (mostly in the acidic C-terminal tails). Because the electrostatic screening length is short in 1 M KCl (), we expect the observed positive charge to be due only to regions of the protein in close proximity to the membrane. Thus, any potential membrane-binding motif must be consistent with this constraint.
Orientation of Tubulin on the Bilayer Surface from NR.
NR (Fig. 1A) yields the density profile of proteins associated with stBLMs. Optimization of the contrast between bound protein, lipids, the substrate, and the bathing solvent is performed using mixtures of light and heavy water and is possible due to the large variability in the cross-section of neutron scattering from the various light elements composing water and biological materials. Previous NR studies with APs bound to tethered lipid bilayers have revealed the membrane-bound structures of amyloid proteins α-synuclein (34) and Aβ (35), determined the orientation of the relatively stiff HIV-1 matrix protein (36), and identified conformational changes in the disordered regions of the HIV-1 Gag extension (37). Availability of the tubulin αβ-heterodimer crystal structure (38) allows detailed reconstruction of tubulin dimer positioning on a membrane surface using NR spectra.
NR experiments were carried out on 1:1 DOPC:DOPE bilayers. The reflectivity curves (normalized by the Fresnel reflectivity between the silicon substrate and the buffer), along with the reflectivity curves expected for fits to the optimized model, are shown in Fig. 5A. Fig. 5B shows the scattering length density (SLD) profiles inferred from the reflectivity spectra and their correspondence to the real space model of the stBLM with bound tubulin. Model parameters are listed in Table S1.
Fig. 5.

Results of NR experiment obtained on 1:1 DOPC:DOPE stBLM in the presence of 600 nM tubulin. (A) Neutron reflectivity spectra. R is the ratio between the reflected and incident neutron intensities, and Rfresnel is the Fresnel reflectivity expected from the buffer/silicon interface. The χ2 value for the simultaneous fit of all six spectra to the model described in the text was 1.197 ± 0.070. Lower panel shows the difference in the NR spectra after adding protein, normalized to the experimental error at each point. (B) SLD profiles determined from optimizing the tubulin orientation model to the reflectivity profiles, showing the correspondence of the profile to the layered structure of the stBLM; 95% CI on the SLD profiles are shown as dashed envelopes.
Table S1.
Parameters of the NR model
| Parameter* | Value | + 1 SD† | − 1 SD |
| SiO2 thickness, Å | 20.5 | 1.0 | 1.1 |
| Cr thickness, Å | 14.6 | 1.3 | 1.3 |
| Gold thickness, Å | 112.06 | 0.82 | 0.77 |
| SiO2 SLD, 10−6 Å−2 | 3.417 | 0.033 | 0.027 |
| Cr SLD, 10−6 Å−2 | 2.58 | 0.10 | 0.13 |
| Au SLD, 10−6 Å−2 | 4.612 | 0.005 | 0.006 |
| Tether length, Å | 4.53 | 0.54 | 1.77 |
| Inner leaflet lipid length, Å | 12.45 | 0.22 | 0.25 |
| Outer leaflet lipid length, Å | 13.42 | 0.25 | 0.29 |
| Change in inner leaflet lipid length with protein, Å | −0.399 | 0.059 | 0.061 |
| Change in outer leaflet lipid length with protein, Å | 0.123 | 0.057 | 0.063 |
| Fraction of exchangeable protons exchanged | 0.939 | 0.040 | 0.027 |
| No. fraction of tether molecules in inner leaflet | 0.964 | 0.026 | 0.044 |
| Ratio of β-mercaptoethanol to tether molecules | 5.0 | 3.4 | 3.4 |
| Fractional completeness of bare bilayer | 1.000 | 0.000 | 0.000 |
| Fractional completeness of bilayer with protein | 0.994 | 0.002 | 0.002 |
| Fractional completeness of overlayer | 0.019 | 0.002 | 0.002 |
| Overlayer leaflet thickness, Å | 13.20 | 0.25 | 0.26 |
| Distance of overlayer from bilayer, Å | 21.7 | 1.2 | 1.3 |
| No. of protein molecules per lipid | 0.0029 | 0.0003 | 0.0003 |
| Distance of protein from headgroups, Å | 0.5 | 1.7 | 2.2 |
| Euler angle α | See Fig. 6‡ | ||
| Euler angle β | |||
| Fraction of protein bound in H2O spectrum | 1.32 | 0.18 | 0.14 |
| Fraction of protein bound in CM4 spectrum§ | 0.99 | 0.15 | 0.13 |
| SLD of D2O solvent, bare bilayer, 10−6 Å−2 | 6.319 | 0.009 | 0.010 |
| SLD of H2O solvent, bare bilayer, 10−6 Å−2 | −0.481 | 0.023 | 0.027 |
| SLD of CM4 solvent, bare bilayer, 10−6 Å−2 | 4.020 | 0.006 | 0.007 |
| SLD of D2O solvent, bilayer with protein, 10−6 Å−2 | 6.397 | 0.002 | 0.007 |
| SLD of H2O solvent, bilayer with protein, 10−6 Å−2 | −0.224 | 0.034 | 0.026 |
| SLD of CM4 solvent, bilayer with protein, 10−6 Å−2 | 4.083 | 0.007 | 0.008 |
| Global roughness parameter, Å | 3.59 | 0.14 | 0.12 |
| Roughness of Cr/Au interface, Å | 10.51 | 0.55 | 0.56 |
| Roughness of bilayer, Å | 2.017 | 0.031 | 0.012 |
| Angle alignment correction, ° | 0.0092 | 0.0004 | 0.0004 |
| Fractional beam intensity | 1.005 | 0.003 | 0.004 |
| Log10 background, D2O solvent, bare bilayer | −8.3 | 1.0 | 1.2 |
| Log10 background, H2O solvent, bare bilayer | −8.63 | 0.95 | 0.94 |
| Log10 background, CM4 solvent, bare bilayer | −7.92 | 0.85 | 1.41 |
| Log10 background, D2O solvent, bilayer with protein | −8.85 | 0.83 | 0.79 |
| Log10 background, H2O solvent, bilayer with protein | −6.75 | 0.24 | 0.73 |
| Log10 background, CM4 solvent, bilayer with protein | −8.88 | 0.96 | 0.78 |
For clarity, parameters for the Catmull–Rom freeform spline are not shown.
SDs are calculated from the equilibrium solution to the MCMC optimization.
The two Euler angles α and β are highly correlated and are not reported as a value with SEs.
CM4 denotes 2:1 D2O:H2O.
The parameters of particular interest for this study are the rotation angles describing the orientation of tubulin dimer on the surface. Protein Data Bank (PDB) ID code 1TVK (39) was used to calculate the neutron reflectivity profile expected for the bound protein in various orientations on the surface. These orientations were parameterized using the Euler angles α and β (Fig. 6A). The angles are defined in an x–y–z extrinsic rotation scheme, where the z axis is codirectional with the surface normal. Because NR is sensitive to the SLD only in the z direction, it is not sensitive to the final rotation about the z axis, γ. Profiles were convolved with a width 6-Å Gaussian function to simulate a range of orientations of the surface-bound protein.
Fig. 6.

Determination of the orientation of bound tubulin by NR. In structures, α-tubulin is colored red, β-tubulin is yellow, the C termini of each chain are green, highlighted tryptophan residues are blue, and the putative binding regions are purple. (A) Euler angles used in orientation parameterizations according to an extrinsic x–y–z scheme, where z is the membrane normal. (B) Structure of the bilayer/protein system determined from fitting a composition model to the NR spectra. (Inset) Expanded view of protein region, showing comparison between free-form model, tubulin model using X-ray crystallographic structure (with a free-form control to reduce systematic error), and the coarse-grained MD simulation of α-tubulin. Dashed lines indicate 95% CIs on the protein density profiles. (C and D) Polar orientation plot of geometries consistent with NR data. Contours bound the 68% and 95% confidence regions. For β > 0°, α-tubulin is bound to the lipid surface (C); β-tubulin is bound for β < 0° (D). Selected orientations of bound tubulin are shown below the polar orientation plots. Orientations 1 and 2 are the orientations corresponding to the mean rotation angles, and orientation 1b most closely resembles the MARTINI orientation of α-tubulin (discussed in the text). Note that the identification of the binding surface does not change significantly with α rotations.
Fig. 6B shows the results of the Monte Carlo statistical analysis of the optimized model, showing the volume fraction occupied by each component of the model (lipid headgroups, acyl tails, tether molecules, protein, etc.) as a function of position. The dashed lines represent the 95% confidence intervals for the protein density profiles, demonstrating excellent correspondence between the Euler angle description and a freeform (Catmull–Rom spline) description of the protein density.
Fig. 6 C and D explore the highly correlated statistical properties of the Euler angle parameters. A heat map of the density of structures explored in the Markov Chain Monte Carlo (MCMC) algorithm at equilibrium is shown on polar plots, where the angular dimension is the azimuthal angle α, and the radial dimension is the polar angle β. Data for β < 0 (Fig. 6C) and β > 0 (Fig. 6D) are represented on two polar plots for clarity. There are two general orientations of tubulin bound to a 1:1 DOPC:DOPE membrane consistent with the NR data. These correspond to binding to the membrane either via the α-tubulin subunit (β > 0, orientations 1 and 1b in Fig. 6C) or the β-tubulin subunit (β < 0, orientation 2 in Fig. 6D). As shown by the shape of the confidence intervals (solid contours) in Fig. 6 C and D, NR is not very sensitive to the axis of rotation for the Euler angle α, which corresponds to the symmetry axis of the tubulin dimer; the tilt angle β is more tightly constrained.
Whereas the Euler angle α, Euler angle β, and the distance from the headgroup region are highly correlated, the other parameters are in general independent and can be interpreted as such. Importantly, the amount of tubulin on the bilayer does not significantly increase in H2O and 2:1 D2O:H2O solvent, despite the total incubation time of the tubulin on the surface exceeding 12 h while the spectra were collected, suggesting that the system is in equilibrium. The observed molar ratio of protein to lipid molecules on the surface, 0.0029 ± 0.0003, represents a protein–protein spacing of about 16 nm and a volume surface occupancy of about 10%.
Importantly, the NR results confirm that the tubulin is peripherally associated with the surface without large membrane-penetrating domains. This is consistent with the EIS measurements, which show little change in the bilayer electrical properties, even when significant amounts of tubulin are bound for long periods of time.
Identification of Membrane-Binding Tubulin Domain from Coarse-Grained MD Simulations.
To investigate the possible modes of binding for monomeric tubulin to different lipid bilayers, and to identify the membrane-binding domains, we performed MD simulations with the coarse-grained MARTINI force-field using both α-tubulin and β-tubulin on pure DOPE and DOPC bilayers. Using distance criteria as described in Materials and Methods, we observed a total of seven binding events from the combined 15 µs of trajectories for simulations performed with DOPC bilayers. For these bilayers, the bound α-tubulin resided on the surface for 55 ns on average and then rapidly dissociated into the solution. β-Tubulin behaved similarly on both DOPC and DOPE bilayers, with no binding events longer than 50 ns. By contrast, α-tubulin resided for the entire simulation time on the surface of DOPE bilayers in six independent runs with an average simulation time of 1.2 µs. These observations suggest that α-tubulin binds preferentially to DOPE bilayers without any significant penetration into their hydrophobic region, providing support to the SPR, BOA, EIS, and NR results discussed above.
The most stable (long-living) protein–membrane interface identified from MARTINI simulations and shown in Fig. 7A is formed predominantly by hydrophobic and basic residues from a short part of α-tubulin’s helix H10 (A330 to F343) and the loop between helix H10 and beta-sheet B9 as well as the S9 β-strand, particularly W346. A highly mobile structural element, H10 was proposed to play an important functional role in dimerization of tubulin (40) and is thought to be involved in conformational discrimination among nucleotide-bound states (41). Interestingly, the helix composition is reminiscent of a large group of cationic antimicrobial peptides (42) that bind to model bilayers. The helical wheel and net representations in Fig. 7B illustrate the amphipathic nature of this helical binding domain.
Fig. 7.

Binding residues identified by MARTINI MD simulation. (A) The proposed binding residues in α-tubulin (shown as vdW spheres) on the DOPE membrane. The binding residues were in direct contact with lipids (highlighted in yellow) for the entire duration of the simulation. (B) Helical wheel (Left) and helical net (Right) diagrams for the putative amphipathic binding region at the α end of the tubulin heterodimer. Hydrophobic residues are denoted by open squares, polar uncharged residues by orange circles, and positively and negatively charged residues by blue and red circles, respectively. Dashed loops indicate membrane-binding surfaces.
Exploration of Orientation Fluctuations and Lipid Specificity with All-Atom MD Simulations.
The coarseness of the MARTINI model sets some limits to understanding how tubulin interacts with bilayers at the atomic scale. To explore the mechanism of binding in detail we used the structures from our MARTINI simulations to construct all-atom systems and to assess the stability of the two tubulin subunits on the surface of model membranes. As with the NR analysis, the orientations were parameterized with the Euler angles in an x–y–z extrinsic rotation scheme and aligned by minimizing rms distances (RMSD) between each α carbon in the polypeptide backbones of the simulated and reference structures (43–45). After a total of ≊1,000 ns of all-atom MD simulations, of which only the last 750–850 ns were used for analysis, peripherally inserted α-tubulin and β-tubulin monomers exhibited behavior qualitatively similar to that observed for simulations with the MARTINI force field. Fig. S1 shows that all four simulations reached a stable plateau in rms values for heavy atoms. The overall flexibility in MD simulations is illustrated with per-residue rms fluctuation (RMSF) values collected in Fig. S2. The most flexible regions are loops and unstructured elements (for example, the C-terminal tails) present in the monomers; interestingly, the proposed insertion region for α-tubulin (A330 to W346) displays very low RMSFs, between 1.0 and 2.1 Å, indicating overall structural stability. In all MD simulations, β-tubulin unbinds and tumbles away from the membrane surface (Figs. S3 and S4), and the α-tubulin/DOPE system is the only one that displays persistent peripheral insertion. As shown in Fig. S5, the membrane-bound helix H10 exhibits significant dynamics relative to the bilayer normal; the angle between the vector defining the helical axis and the bilayer normal adopts a range of values from –30° to 60°. The size of the system and length of the simulation accessed in this study make it difficult to achieve complete sampling of tumbling dynamics for the bound tubulin; the distributions of α-tubulin orientations observed are shown in Fig. 8A. Despite the range of α-tubulin orientations on the surface, the binding residues located in the helix itself and an unstructured loop region were found to form long-living contacts with lipids (Fig. 8B). Several of the hydrophobic (underlined) amino acids in the binding region A330AIATIKTKRSIQFVDW346 are interacting with lipids, whereas the KTK338R339 fragment is stably coordinated by the phosphate moiety of the polar headgroups.
Fig. S1.

RMSD for backbone atoms (relative to 1TVK coordinates) from ANTON production of all-atom MD simulations for α-tubulin and β-tubulin monomers starting from membrane-attached states. RMSD values for α-tubulin with DOPE and DOPC bilayers are shown in black and red solid lines, respectively. RMSD values for β-tubulin with DOPE and DOPC bilayers are shown in green and blue solid lines, respectively.
Fig. S2.

(Top) Per-residue RMSF for the α-tubulin monomer in DOPE and DOPC systems. (Bottom) Per-residue RMSF for the β-tubulin monomer in DOPE and DOPC systems.
Fig. S3.

(Top) The distance (Z-component only) from the α-tubulin center of mass (solid lines) and that of W346 (dotted lines) to the center of DOPE (black) and DOPC bilayers (red). (Bottom) The distance (Z-component only) from the β-tubulin center of mass (solid lines) and that of W407 (dotted lines) to the center of DOPE (green) and DOPC bilayers (blue). The idealized position of the hydrophobic interface (17.5 Å from center of the bilayer) is shown as a violet band on both graphs.
Fig. S4.

Example of tumbling dynamics of the β-tubulin monomer on DOPE surface from simulations with ANTON. The tentative insertion region from MARTINI simulations (330–345) is labeled in red, and W407 is shown in sphere mode to illustrate unbinding movements. No stable insertion (lasting more than 50 ns) was recorded in MARTINI runs or 790 ns of production ANTON MD simulations.
Fig. S5.

Distribution of two helical tilt angles for proposed insertion region 1 (solid line) A330–W346 in DOPE (black) and DOPC (red) bilayers. We used the definitions of Chothia et al. (77) to define the principal axis of the helical element in the insertion region and then determined tilt angles in respect to the bilayer normal.
Fig. 8.

Rotational dynamics for stably bound α-tubulin on DOPE membranes from ANTON MD simulations. (A) Probability distribution of Euler rotation angles for α-tubulin. (B) Time evolution of the stable α-tubulin tethering on the DOPE membrane surfaces from all-atom MD simulations starting from the most abundant cluster in MARTINI simulations and its subsequent evolution over 880 ns of all-atom MD simulations. The insertion helix identified in MARTINI simulations is color-coded for chemical moieties as follows: blue, positively charged residues; red, negatively charged residues; orange, hydrophobic residues; and yellow, asparagine or glutamine residues. W346 is shown in molecular spheres mode. The bilayer normal and axis vector for insertion helix are shown. The unstructured C-terminal tail is shown in a ribbon mode.
The reorientation of the insertion region observed in microsecond MD simulations for this system underscores the importance of the two building blocks of the insertion machinery—hydrophobic residues and positive charges—to maximize interactions with the bilayer surface. To form such an interaction, tubulin does not have to penetrate deeply, in accordance with our MARTINI and MD simulations, but should be able to access the hydrophobic interface. This finding may explain preferential targeting to DOPE-containing membranes, because the bulkier and more ordered headgroups of DOPC lead to an additional penalty for helix penetration.
Discussion
Orientation of Membrane-Bound Tubulin.
NR measures the density profile arising from the time- and space-averaged ensemble of membrane-bound tubulin structures. The freeform description of the protein density profile (Fig. 6B) identifies a significant amount of protein density at a distance from the surface that cannot be described by a simple model in which both monomers are simultaneously bound to the surface. As a result, the orientation model reports a dimer tilt angle of β ≈ 60°. Both of these results are also consistent with a highly dynamic membrane-bound structure in which tubulin is bound by one or more flexible domains, which allows the dimer to explore a large range of tilt angles and rotations relative to the membrane surface.
This picture of α-tubulin binding to DOPC and DOPE lipids is supported by the MD simulations, in which the binding region, helix H10, is connected by flexible loops to the more rigid tubulin body. Because H10 is on one end of the dimer, a large range of tilt angles relative to the surface can in principle be explored, consistent with the NR observations. It is important to stress that the MD simulations were performed with a monomeric tubulin and hence the degree of protein dynamics on bilayer surfaces may be overestimated. Fig. S6 illustrates the idea that some angles explored by the α-tubulin monomer alone are in fact sterically “forbidden” for tubulin dimers due to overlap between the β-tubulin monomer and the membrane.
Fig. S6.

Illustration of allowed and forbidden rotations for dimeric tubulin. The insertion region (A330–W346) is colored according to chemical functionality (negative residues in red, positive residues in blue, and hydrophobic/aromatic in orange). The location of W346 is shown in a sphere mode. β-Tubulin monomer was placed using the 1TVK dimer and RMS orientation operation relative to the position of α-tubulin heavy atoms. The β-tubulin is shown as a magenta-colored ribbon. The following ranges of the two major Euler angles would result in a collision between β-tubulin and membrane bilayer: α from −35 to −45° and β from −10 to −20°.
The amphipathic structure of helix H10 (α-tubulin residues 325–337), including hydrophobic residues F343 and W346, is clear from Fig. 7B. Note that we assume the helix to extend to residue W346, which is not observed in the crystal structure but may occur upon association with the membrane. Tellingly, W346 is extremely well-conserved across eukaryotic species (Table S2). Helix H10 is identified as a low-complexity region, which is typically associated with less specificity of binding (46). The significantly tighter binding of this motif to DOPE membranes than DOPC membranes is also consistent with the BOA and SPR results. The net positive charge of helix H10 satisfies the charge constraint from the BOA experiments (Fig. 4). As shown in Fig. S7 and Table S3, β-tubulin does not contain such a clear lipid-binding motif on the binding surface compatible with the NR data, consistent with the results of the MD simulations.
Table S2.
Putative α-tubulin binding region surrounding H10 (A330 to W346) shows broad conservation across species, particularly W346
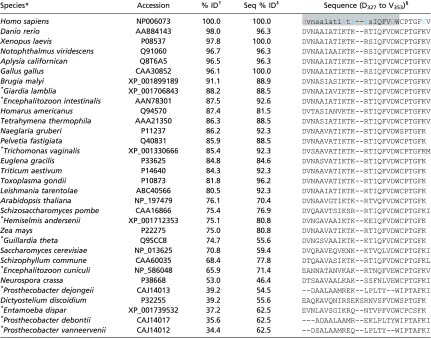 |
Species marked with an asterisk do not contain mitochondria.
Species are sorted by α-tubulin similarity to H. sapiens, expressed as percent identity (% ID).
The percent identity of the binding region is shown as Seq % ID. The correlation coefficient between % ID and Seq % ID is 0.84.
In the H. sapiens structure, lowercase residues denote a sequence considered a low-complexity region by the BLASTP alignment algorithm (79). Red represents anionic and blue cationic residues, and the gray region is helix H10.
Fig. S7.

Helical wheel (Left) and helical net (Right) diagrams for a possible β-tubulin binding surface identified by NR (Fig. 6D). This surface comprises several distinct regions, including helix H2, which is quite polar; a polar loop from 94 to 101; a group of four hydrophobic valine residues between 171–182; helix H6, which has quite a lot of hydrophobic character, but in the 1TVK structure is not clearly facing the membrane; and helix H11, which like α-tubulin has a hydrophobic patch with conserved tryptophan W407 (see Table S3 and helical wheel and net diagrams here). Of these, only helix H11 is net cationic. Fig. S3 shows that in all-atom MD simulations all contacts between W407 and lipids are short-lived and β-tubulin drifts away from the bilayer surface. Helix H11 forms only transient interactions with lipids as well (Fig. S4).
Table S3.
Possible β-tubulin binding region surrounding H11 (F399 to T409) shows broad conservation across species, particularly W407
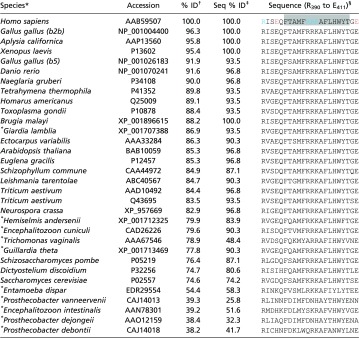 |
Species marked with an asterisk do not contain mitochondria.
Species are sorted by similarity of entire β-tubulin sequence to H. sapiens, as ranked by the BLASTP algorithm (79), and tabulated as percent identity (% ID).
The percent identity of the binding region is shown as Seq %ID. The correlation coefficient between % ID and Seq % ID is 0.92.
In the H. sapiens sequence, the gray region marks the putative membrane-binding domain. Red represents anionic and blue cationic residues.
Equilibrium Properties of Membrane-Bound Tubulin.
APs bind to membranes in a variety of ways. Anionic lipids typically enhance the strength of the electrostatic interaction and promote AP binding (28, 47). Tubulin, remarkably, distinguishes between two zwitterionic lipid species, DOPC and DOPE. The acidic character of the tubulin C-terminal tails may in fact be responsible for a reduction in tubulin’s interaction with anionic membranes (4, 13). In our present experiments, therefore, we suppressed the electrostatic attraction by using a high salt concentration (1 M KCl) and neutral lipids, thus highlighting hydrophobic effects, although the MD simulations clearly show electrostatic attraction between the charged lysine and arginine residues of the α-tubulin binding domain and the zwitterionic headgroups at 150 mM KCl. Under the experimental conditions, tubulin binds strongly to membranes with high PE content. This observation is consistent with a hydrophobicity-dominated amphipathic helix binding mechanism, with the consequence that tubulin differentiates between different zwitterionic lipids rather than between zwitterionic and anionic lipids. This unusual feature suggests that the amphipathic binding helix of tubulin senses lipid-packing defects arising from the presence of nonlamellar lipids such as DOPE (48). A handful of other eukaryotic proteins, all of which are involved in regulation of lipid composition, curvature, or metabolism, share this binding mechanism (25). It remains to be seen whether tubulin also regulates membrane properties.
Tubulin, like many APs (47), binds to membranes with apparent cooperativity. The SPR results show a sharp increase in tubulin binding above , immediately suggesting that each tubulin molecule binds multiple DOPE lipids. Previously published data on the rate of interaction of tubulin with single VDAC channels reconstituted in DOPC/DOPE mixtures (13) also show cooperative binding to PE lipids. This kind of behavior has been extensively treated before; here, we follow the approach of Mosior and McLaughlin (47) to estimate the number of PE lipids bound to each tubulin molecule and the binding affinity for a single lipid. This formulation assumes equal binding energies for all associated lipids. As shown in Fig. S8 A–C, the SPR response and VDAC–tubulin binding kinetics response as a function of , as well as the BOA data in which the protein concentration is varied, can be jointly fit to the Mosior–McLaughlin sequential lipid binding model with two common parameters: the single-lipid dissociation constant and the number of PE lipid binding sites . The fit converges with . The 68% confidence interval (CI) constrains these parameters as and . The median values are and . Because the binding per lipid is weak, not all of the binding sites are occupied on average (Fig. S8D); the average occupancy calculated from the Mosior–McLaughlin model is nine lipids (68% CI: ± 1). At the maximum interaction area per lipid of ≊70 Å2, this number of bound lipids is commensurate with the hemicylindrical area of the putative α-tubulin binding helix (≊480 Å2, assuming a 12-Å helical outer diameter and that each of the 17 aa in the helix contribute 1.5 Å translation per residue).
Fig. S8.

Determination of tubulin–DOPE dissociation constant and the maximum number of DOPE molecules that can be bound to each tubulin dimer , using the methods in Mosior and McLaughlin (47). Briefly, the mass balance equations for tubulin and lipid were numerically solved for the concentrations of unbound lipid, unbound protein, and all protein–lipid complexes in which lipids are bound. Solid lines represent best-fit curves to experimental data, using and . Other free parameters include the maximum SPR response, maximum BOA response, the maximum and minimum VDAC interaction rates, and a fixed ratio between the interaction rates at the two voltages, and the volume/area ratio for the BOA and single-channel measurements. The volume/area ratio for the SPR measurements is fixed by the experimental geometry. (A) Difference in tubulin binding measured by SPR to binary DOPC/DOPE membranes with PE fraction relative to , showing clear positive cooperativity. Error bars are mean ± one SD from the mean for two experiments per lipid composition. (B) Tubulin binding measured by BOA (reproduced from Fig. 4). (C) On rate of the tubulin/VDAC interaction (data from ref. 13) in binary DOPC/DOPE membranes at 20 mV and 25 mV transmembrane potential, showing similar positive cooperativity. (D) Distribution of bound species in the dilute limit at equilibrium, using best fit parameters. Average occupancy is nine bound PE lipids.
Implications for Mitochondrial Regulation via VDAC–Tubulin Interaction.
The foregoing discussion and the analysis in Fig. S8 suggest that the highest sensitivity to membrane PE content occurs around . Interestingly, the outer mitochondrial membrane contains lipids in a 58:42 PC:PE ratio (49). The high sensitivity of tubulin binding to the PE content of the membrane at this ratio supports the notion that the outer membrane composition may be involved in regulating the VDAC–tubulin interaction (13) and, consequently, mitochondrial respiration by controlling VDAC permeability to ATP and ADP.
In addition, the orientation of the tubulin dimer on the membrane surface has direct implications for the availability of each tubulin CTT to block VDAC and thus participate in mitochondrial regulation. For binding occurring via either the α- or β-subunit, there is a clear difference in the accessibility of the CTTs to the VDAC pore embedded into the lipid membrane. Whereas for both α- and β-tubulin-bound dimers the 17-aa (≊6.8 nm long) β-tubulin CTT is close enough to the surface to be captured by the VDAC channel, the shorter α-tubulin CTT (11 amino acids, ≊4.4 nm long) is unlikely to be accessible to the VDAC pore for β-tubulin–bound dimers. In the latter case, if β-tubulin could be prevented from binding to the membrane, less interaction with the VDAC channel should be observed. To test this, we measured tubulin interaction with the VDAC pore when the β-tubulin end was blocked by a DARPin that is known to bind to the β-tubulin putative binding surface (50). These single-channel measurements showed no decrease in VDAC–tubulin interaction for VDAC reconstituted in planar lipid bilayers (Fig. S9), further confirming that β-tubulin site is unlikely to be responsible for membrane binding. Further exploration of these effects will require the use of recently developed expression systems for recombinant tubulin (51–53) and chimeric systems for presenting tubulin CTTs to the VDAC channel (54).
Fig. S9.

Tubulin–DARPin complex (50) does not induce less blockage of VDAC than free tubulin. (A) Representative current traces of the same single VDAC reconstituted into a planar membrane in the presence of 15 nM tubulin/DARPin (2:1) complex in the cis side of the membrane and 15 nM of free tubulin in the opposite, trans, side at −5 and + 25 mV of applied voltage, respectively. Tubulin induces characteristic blockages of VDAC conductance only when negative potential is applied at the side of tubulin addition. In our experimental setup negative potential is referred to as the cis side, or side of VDAC addition. Dashed lines indicate zero-current level and dotted lines indicate VDAC open and tubulin-blocked states. Current records were additionally filtered using a 1-kHz eight-pole digital Bessel filter. Medium consisted of 1 M KCl buffered with 5 mM Tris at pH 7.4. The planar membrane was formed from diphytanoylphosphatidylcholine (13). Recombinant mouse VDAC1 was isolated following the published protocol (78). Fifteen nanomolar tubulin was preincubated with 30 nM DARPin [purified using the published protocol (50)] on ice for 30 min before addition to the membrane-bathing solution. The current records were taken 75 min after addition of tubulin/DARPin or tubulin to the experimental chamber. Current measurements were performed as described previously (13). (B) The on-rate constants of tubulin–VDAC binding in the presence of tubulin/DARPin complex or free tubulin at 25 mV applied voltage. All experimental conditions are as in A. The data are mean of two experiments ± SE (error bars). On rates were obtained from kinetic analysis of channel blockage events as shown in A using ClampFit 10.5 software (Molecular Devices, LLC) and described previously (13).
Conclusions
In addition to its structural role in microtubules, tubulin also seems to function in the capacity of a transiently bound peripheral membrane protein. This study aims to elucidate how tubulin becomes a functional component of cellular membranes and, in particular, mitochondrial membranes. The mechanism of tubulin–membrane association, which involves highly conserved hydrophobic residues and nearby amphipathic domains, allows tubulin to differentiate between zwitterionic lipid species. Together, these ideas suggest that the regulation of the integral membrane protein VDAC by the AP tubulin is controlled by the lipid composition of the MOM. Other scenarios where tubulin associates with different cellular membranes and plays so far unknown functional roles, in addition to VDAC regulation in mitochondria, are quite plausible.
Finally, the likelihood that the α-subunit binds preferentially to membrane surfaces has implications for identifying tubulin-targeting agents. Although we cannot completely rule out more complex binding schemes, our long-time-scale MD simulations showing very rapid dissociation for β-tubulin, in stark contrast to a system containing α-tubulin, channel experiments in the presence of DARPin; and helical wheel analysis strongly favor α-tubulin membrane binding. Although microtubule-targeting compounds associate exclusively with sites on β-tubulin, the data presented here suggest that α-tubulin, particularly the binding helix H10, is also a promising target for therapeutics. Additionally, any strategy for controlling the regulation of VDAC by tubulin via the CTTs must account for the fact that both the α- and β-tubulin CTTs are likely involved, because they are both accessible to the VDAC pore for α-tubulin–bound dimers.
Materials and Methods
stBLM Preparation for SPR and NR.
stBLMs were prepared as previously described (27). A schematic of the prepared bilayer is shown in Fig. 1A. Briefly, a substrate (sapphire for SPR and silicon for NR) was coated with thin films of Cr ≊5 Å and Au ≊400 Å for SPR, Cr ≊25 Å and Au ≊130 Å for NR) using magnetron sputtering at the National Institute of Standards and Technology (NIST) Center for Nanoscale Science and Technology. The substrate was then immersed for 8 h in an ethanolic solution of the thiol-lipid linking molecule HC18 [Z20-(Z-octadec-9-enyloxy)-3,6,9,12,15,18,22-heptaoxatetracont-31-ene-1-thiol)] (55) and β-mercaptoethanol in a 3:7 molar ratio and a total concentration of 0.2 mM. HC18 was a kind gift of David Vanderah, Institute for Bioscience and Biotechnology Research, University of Maryland, Rockville, MD. The resulting self-assembled monolayer (SAM) was rinsed in ethanol and dried in a nitrogen stream.
For NR, the dry SAM-coated substrate was exposed to a 10 mg/mL ethanolic solution of DOPC and DOPE in the desired molar ratio. After 15 min the stBLM was formed by rapid solvent exchange (27) with pure H2O. To ensure a single bilayer on the surface, the stBLM was rinsed with gradually increasing concentrations of EtOH in H2O from 15 to 30% vol/vol until no change was observed in the NR spectrum. This method reliably yields stable stBLMs up to 50% PE content by mass.
For SPR, the dry SAM-coated substrate was then mounted in the instrument. A 12.5 mg/mL solution of the desired molar ratio of DOPC (1,2-dioleoyl-sn-glycero-3-phospho-choline) and DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine) dissolved in 1:1 ethanol:chloroform was added to the substrate. All lipids were purchased from Avanti Polar Lipids, Inc. After 3 min the stBLM was formed by rapid solvent exchange with 100 mM KCl buffered by 5 mM Hepes at pH 7.4. The membrane was allowed to mature for 45 min then rinsed with 25% EtOH in H2O to ensure a single bilayer on the surface. This method allowed formation of stBLMs containing up to 75% PE.
Tubulin Preparation.
Lyophilized bovine brain tubulin (Cytoskeleton) was reconstituted in an 80 mM Pipes, 1 mM MgCl2, and 1 mM EGTA aqueous buffer at pH 6.9, aliquoted at 10 μM, and immediately flash-frozen. The reconstitution buffer for tubulin in NR experiments contained the D2O:H2O ratio used at each contrast condition. Tubulin aliquots were thawed immediately before use and kept on ice over the course of the experiment. All experiments were performed at room temperature.
SPR.
SPR was performed as previously described with a custom-built instrument (SPR Biosystems) in the Kretschmann configuration (29). The exposed area of the substrate is ≊0.3 cm2 and the total cell volume about 1.0 mL. The instrument response was calibrated to be 1 px = 0.003972° = 0.647 Å of a complete organic layer with refractive index 1.45. After formation of the stBLM, the solution in the sample cell was replaced with a 1 M KCl and 5 mM Hepes buffer at pH 7.4. The instrument response was allowed to equilibrate for up to 2 h. Tubulin additions were performed sequentially at concentrations from 100 to 600 nM, with about 30 min between additions.
EIS.
EIS measurements (30) were performed in the SPR instrument using a three-electrode configuration in which an Ag/AgCl reference electrode and platinum counter electrode were suspended in 1 M KCl buffered at pH 7.4 by 5 mM Hepes. The gold substrate served as a working electrode. The impedance of the stBLM with tubulin was measured from 10 kHz to 10 Hz using a Solartron Analytical Modulab 2100A potentiostat equipped with a 1-MHz FRA frequency analyzer. The impedance spectrum was analyzed for each tubulin concentration measured by SPR.
BOA.
BOA measurements were performed using free planar lipid bilayers formed from two opposing lipid monolayers across ≊70 μm aperture in a 15-μm-thick Teflon partition separating two ≊1.2-mL compartments, as previously described (56). The two compartments were filled with 1 M KCl buffered at pH 7.4 by 5 mM Hepes and connected to a Stanford Research Systems 830 lock-in amplifier via Ag/AgCl electrodes in 2 M KCl-bathed agarose bridges. An excitation potential with frequency , amplitude , and variable dc potential was applied across the membrane. The ac current was measured at , that is, the frequency of the second harmonic of the excitation potential. For a lipid bilayer membrane with a voltage-dependent capacitance given by , where is the capacitance in the absence of an applied potential and is related to the compressibility of the membrane, the second harmonic current is (31)
Here , the intrinsic membrane potential, depends on the asymmetry between two monolayer leaflets of the bilayer membrane and changes in response to binding an adsorbent. Experimentally, was determined from measuring as a function of , which was swept from –50 to 50 mV in steps of 10 mV; is minimized at (Fig. 1C). Data are represented as , with the convention that positive corresponds to the addition of positive charge to the cis side of the membrane. Tubulin was added stepwise to the cis compartment under constant stirring. and membrane capacitance C were measured approximately once per minute. Each data point is an average of 10 subsequent measurements taken when the signal reached a steady-state level, as defined by variations within ±0.5 mV.
NR.
NR measurements were carried out at the NG7 horizontal reflectometer at the NIST Center for Neutron Research. The sample cell was mounted in a collimated 35-mm neutron beam in such a way that the neutrons traveled through the silicon wafer to reflect from the silicon/solvent interface (Fig. 1A). Specular reflectivity spectra were collected using cold neutrons (wavelength λ = 4.768 Å) over incident angles from θ = 0.174° to 5.46° (Q = 0.008 Å−1 to Q = 0.251 Å−1). The momentum transfer is used in all data representations. The beam collimation was varied to maintain a constant illuminated area on the interface.
Contrast variation is achieved in NR by changing the environment of the lipid-protein system from H2O to D2O, taking advantage of the large difference in scattering of cold neutrons from hydrogen and deuterium (57). For these measurements, three contrasts were measured at each experimental condition: D2O, H2O, and a 2:1 D2O:H2O mixture. For each mixture, background spectra collected at reflection angles above and below the incident angle were subtracted, and direct beam measurements were used to achieve proper normalization of the reflectivity spectrum. All solutions contained 1 M KCl buffered at pH 7.4 by 5 mM Hepes.
Reflectivity spectra were obtained at two experimental conditions: before and after addition of 600 nM tubulin. To change contrasts, the cell was flushed with the buffer solution in the new D2O:H2O mixture; then 600 nM tubulin in the new solvent was reintroduced. The six normalized reflectivity spectra (two conditions, each with three contrasts) were then simultaneously fit to a bilayer/tubulin α/β-heterodimer composition space model (58) using an MCMC algorithm implemented in the software package Refl1D (59). The MCMC optimization was performed on 10 parallel populations and run to at least 50,000 steps, 10 times the estimated relaxation time for convergence of about 5,000 steps. The final 500 steps were used to perform a statistical analysis of the equilibrium solution, yielding the CIs on the parameters and density profiles. Calculations were run on the Darter Cray XC30 supercomputer (60). Protein visualizations were generated using the software package VMD (61).
Coarse-Grained MD Simulations.
The α-tubulin and β-tubulin coordinates were taken from PDB ID code 1TVK (39). To test whether tubulin monomers could be sequestered by the membrane surface we used an established coarse-grained MARTINI model for lipids and amino acids (62, 63) and the recently developed polarizable MARTINI water model (64). We chose to work with the 1TVK structure to take advantage of substantial prior efforts in rebuilding starting structures containing the C-terminal tails (40). All MD simulations were run with a 20-fs time step and standard cutoff scheme. Four bilayer patches, one with 512 DOPC and the other with 512 DOPE lipids, were used to test the dependence of the adsorption process on the lipid type for each of the monomers. The protein structures were constrained in all coarse-grained simulations using protocols recommended by the developers. The particle mesh Ewald method was used to account for long-range electrostatic interactions. The tubulin monomers were positioned 15 Å away from the center of the bilayer. Fifteen starting structures for each of the system of interest were created by random rotations of the protein with respect to the bilayer. An equilibration run of 100 ns was followed by the production run of 2,500 ns for each starting structure. We observed stable surface localization of α-tubulin in two different positions and then used these trajectories for the analysis and subsequent construction of an all-atom system (DOPC/DOPE bilayers) using a previously developed back-mapping protocol (discussed below). Because the β-tubulin monomer exhibited only transient insertions, we used the k-means clustering algorithm to define a starting membrane-tethered structure for β-tubulin, similar to a previously used protocol (65). All simulations with the coarse-grained force field were performed with the GROMACS program package (66).
All-Atom MD Simulations.
The stable binding pose extracted from the MARTINI simulation was used to rebuild the all-atom system. The protein coordinates from coarse-grained simulations were obtained using the protocol by Rzepiela et al. (67). Positions of the lipid chains from MARTINI simulations were used to rebuild the lipid bilayer around the protein. The protein–membrane system was solvated using the stepwise protocol from CHARMM-GUI server (68). The resulting simulation systems contained α-tubulin and β-tubulin peripherally bound to the 512-lipid DOPC and DOPE bilayers (69) in the orientation extracted from coarse-grained MD simulations. The protein–membrane system was solvated by a 150 mM KCl aqueous salt solution. All of the equilibrating MD simulations were performed using the biomolecular simulation program NAMD 2.9 (70). The all-atom energy function CHARMM-36 for proteins and phospholipids was used (71). The resulting systems contained over 230,000 atoms. All-atom MD simulations were performed with periodic boundary conditions in an NPaT (constant membrane area) ensemble. The temperature was set to 315 K with Lowe–Anderson thermostat, and the SHAKE algorithm was used to maintain the bond lengths that involve hydrogen atoms (72). Electrostatic interactions were calculated using particle-mesh Ewald summation. The system was equilibrated for 10 ns in the presence of harmonic constraints acting on the heavy atoms of the tubulin monomer, which was followed by 180–200 ns of equilibration MD simulations. We gradually released the harmonic constraints acting on the protein backbone atoms during an equilibration phase, allowing for membrane relaxation. Four systems (α-tubulin and β-tubulin monomers with DOPE and DOPC bilayers) were further simulated for 780–880 ns on Anton (73) using the CHARMM36 force field. The NPT ensemble was used with the temperature maintained at 315 K using the Nosé–Hoover scheme. The time step was 2 fs and trajectories were saved every 240 ps. The short-range nonbonded and electrostatic interactions were calculated with a cutoff of 9.5 Å. The long-range electrostatic interactions were calculated with the split Ewald method (74) with a 72 × 72 × 72 grid and the SHAKE algorithm was used for all simulations. The stability of all simulations for membrane–protein–solvent systems was monitored with global RMSD values for heavy atoms and an RMSF per-residue decomposition. The results of these analyses and pertinent discussions are summarized in Figs. S1 and S2. Briefly, the atoms in the flexible elements (unstructured C-terminal tail and flexible loops) are the most dynamic ones, but global RMSD values from 0.8 μs to 0.9 μs plateau between 3.2 Å to 3.7 Å. Contact analysis between the tubulin and lipids was carried out as described previously (75, 76) with a heavy–heavy atom cutoff distance of 3.7 Å. For Euler angles calculations proteins were reoriented (without rotation) relative to the structures from crystallographic studies (reference frame orientation is shown in Fig. 6A) and all simulations were recentered.
Acknowledgments
We thank Frank Heinrich, Ken Rubinson, and Felipe Montecinos-Franjola for helpful discussions; Guangcui Yuan, Sushil Sajita, Brian Maranville, Paul Kienzle, and Oscar Teijido-Hermida for technical assistance; Mathias Lösche for the generous use of Anton time and helpful discussions; D. Peter Tieleman, Drew Bennett, Seonghoon Kim, and Wonpil Im for generously sharing scripts and numerical receipts for MARTINI and ANTON simulations, respectively; Matthew Robinson for his role in creating samples for SPR and NR at the NIST Center for Nanoscale Science and Technology; Minhajuddin Sirajuddin for providing DARPin protein; and NIST for providing the neutron research facilities used in this work. This work used the Extreme Science and Engineering Discovery Environment, which is supported by NSF Grant ACI-1053575. Specifically, it used the Darter Cray XC30 system and the Bridges system, which is supported by NSF Award ACI-1445606, at the Pittsburgh Supercomputing Center (PSC). Molecular dynamics simulations were performed on the West-Grid/Compute Canada facilities. Anton computer time was provided by the National Center for Multiscale Modeling of Biological Systems through Grant R01GM116961 from the NIH and PSC. The Anton machine at PSC was generously made available by D. E. Shaw Research. This work was supported by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH (T.K.R., D.J., and S.M.B.) and by the National Sciences and Engineering Research Council Discovery Grant RGPIN-315019 (to S.Y.N.) and the Alberta Innovates Technology Futures Strategic Chair in (Bio)Molecular Simulation. Certain commercial materials, equipment, and instruments are identified in this work to describe the experimental procedure as completely as possible. In no case does such an identification imply a recommendation or endorsement by NIST, nor does it imply that the materials, equipment, or instrument identified are necessarily the best available for the purpose.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1619806114/-/DCSupplemental.
References
- 1.Field JJ, Waight AB, Senter PD. A previously undescribed tubulin binder. Proc Natl Acad Sci USA. 2014;111:13684–13685. doi: 10.1073/pnas.1414572111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bernier-Valentin F, Aunis D, Rousset B. Evidence for tubulin-binding sites on cellular membranes: Plasma membranes, mitochondrial membranes, and secretory granule membranes. J Cell Biol. 1983;97:209–216. doi: 10.1083/jcb.97.1.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carré M, et al. Tubulin is an inherent component of mitochondrial membranes that interacts with the voltage-dependent anion channel. J Biol Chem. 2002;277:33664–33669. doi: 10.1074/jbc.M203834200. [DOI] [PubMed] [Google Scholar]
- 4.Wolff J. Plasma membrane tubulin. Biochim Biophys Acta. 2009;1788:1415–1433. doi: 10.1016/j.bbamem.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 5.Caron JM, Berlin RD. Reversible adsorption of microtubule protein to phospholipid-vesicles. J Cell Biol. 1980;87:A255. doi: 10.1083/jcb.81.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar N, Klausner RD, Weinstein JN, Blumenthal R, Flavin M. Interaction of tubulin with phospholipid vesicles. II. Physical changes of the protein. J Biol Chem. 1981;256:5886–5889. [PubMed] [Google Scholar]
- 7.Rostovtseva TK, Bezrukov SM. VDAC regulation: Role of cytosolic proteins and mitochondrial lipids. J Bioenerg Biomembr. 2008;40:163–170. doi: 10.1007/s10863-008-9145-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Monge C, et al. Regulation of respiration in brain mitochondria and synaptosomes: Restrictions of ADP diffusion in situ, roles of tubulin, and mitochondrial creatine kinase. Mol Cell Biochem. 2008;318:147–165. doi: 10.1007/s11010-008-9865-7. [DOI] [PubMed] [Google Scholar]
- 9.Maldonado EN, et al. Voltage-dependent anion channels modulate mitochondrial metabolism in cancer cells: regulation by free tubulin and erastin. J Biol Chem. 2013;288:11920–11929. doi: 10.1074/jbc.M112.433847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maldonado EN, Patnaik J, Mullins MR, Lemasters JJ. Free tubulin modulates mitochondrial membrane potential in cancer cells. Cancer Res. 2010;70:10192–10201. doi: 10.1158/0008-5472.CAN-10-2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rostovtseva TK, Bezrukov SM. VDAC inhibition by tubulin and its physiological implications. Biochim Biophys Acta. 2012;1818:1526–1535. doi: 10.1016/j.bbamem.2011.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Noskov SY, Rostovtseva TK, Bezrukov SM. ATP transport through VDAC and the VDAC-tubulin complex probed by equilibrium and nonequilibrium MD simulations. Biochemistry. 2013;52:9246–9256. doi: 10.1021/bi4011495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rostovtseva TK, Gurnev PA, Chen MY, Bezrukov SM. Membrane lipid composition regulates tubulin interaction with mitochondrial voltage-dependent anion channel. J Biol Chem. 2012;287:29589–29598. doi: 10.1074/jbc.M112.378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Noskov SY, et al. Current state of theoretical and experimental studies of the voltage-dependent anion channel (VDAC) Biochim Biophys Acta. 2016;1858:1778–1790. doi: 10.1016/j.bbamem.2016.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rovini A, Savry A, Braguer D, Carré M. Microtubule-targeted agents: When mitochondria become essential to chemotherapy. Biochim Biophys Acta. 2011;1807:679–688. doi: 10.1016/j.bbabio.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Prinz WA. Lipid trafficking sans vesicles: Where, why, how? Cell. 2010;143:870–874. doi: 10.1016/j.cell.2010.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holthuis JC, Menon AK. Lipid landscapes and pipelines in membrane homeostasis. Nature. 2014;510:48–57. doi: 10.1038/nature13474. [DOI] [PubMed] [Google Scholar]
- 18.Hanada K, Voelker D. Interorganelle trafficking of lipids: Preface for the thematic review series. Traffic. 2014;15:889–894. doi: 10.1111/tra.12193. [DOI] [PubMed] [Google Scholar]
- 19.Cho W, Stahelin RV. Membrane-protein interactions in cell signaling and membrane trafficking. Annu Rev Biophys Biomol Struct. 2005;34:119–151. doi: 10.1146/annurev.biophys.33.110502.133337. [DOI] [PubMed] [Google Scholar]
- 20.Pu M, Orr A, Redfield AG, Roberts MF. Defining specific lipid binding sites for a peripheral membrane protein in situ using subtesla field-cycling NMR. J Biol Chem. 2010;285:26916–26922. doi: 10.1074/jbc.M110.123083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stahelin RV. Lipid binding domains: More than simple lipid effectors. J Lipid Res. 2009;50:S299–S304. doi: 10.1194/jlr.R800078-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Monje-Galvan V, Klauda JB. Peripheral membrane proteins: Tying the knot between experiment and computation. Biochim Biophys Acta. 2016;1858:1584–1593. doi: 10.1016/j.bbamem.2016.02.018. [DOI] [PubMed] [Google Scholar]
- 23.Schenkel LC, Bakovic M. Formation and regulation of mitochondrial membranes. Int J Cell Biol. 2014;2014:709828. doi: 10.1155/2014/709828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sperka-Gottlieb CD, Hermetter A, Paltauf F, Daum G. Lipid topology and physical properties of the outer mitochondrial membrane of the yeast, Saccharomyces cerevisiae. Biochim Biophys Acta. 1988;946:227–234. doi: 10.1016/0005-2736(88)90397-5. [DOI] [PubMed] [Google Scholar]
- 25.Cornell RB. Membrane lipid compositional sensing by the inducible amphipathic helix of CCT. Biochim Biophys Acta. 2016;1861:847–861. doi: 10.1016/j.bbalip.2015.12.022. [DOI] [PubMed] [Google Scholar]
- 26.Jo E, McLaurin J, Yip CM, St George-Hyslop P, Fraser PE. alpha-Synuclein membrane interactions and lipid specificity. J Biol Chem. 2000;275:34328–34334. doi: 10.1074/jbc.M004345200. [DOI] [PubMed] [Google Scholar]
- 27.McGillivray DJ, et al. Molecular-scale structural and functional characterization of sparsely tethered bilayer lipid membranes. Biointerphases. 2007;2:21–33. doi: 10.1116/1.2709308. [DOI] [PubMed] [Google Scholar]
- 28.Johnson JE, Cornell RB. Amphitropic proteins: Regulation by reversible membrane interactions (review) Mol Membr Biol. 1999;16:217–235. doi: 10.1080/096876899294544. [DOI] [PubMed] [Google Scholar]
- 29.Shenoy S, et al. Membrane association of the PTEN tumor suppressor: Molecular details of the protein-membrane complex from SPR binding studies and neutron reflection. PLoS One. 2012;7:e32591. doi: 10.1371/journal.pone.0032591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Valincius G, Meškauskas T, Ivanauskas F. Electrochemical impedance spectroscopy of tethered bilayer membranes. Langmuir. 2012;28:977–990. doi: 10.1021/la204054g. [DOI] [PubMed] [Google Scholar]
- 31.Sokolov VS, Kuz’min VG. Measurement of differences in the surface potentials of bilayer membranes according to the second harmonic of a capacitance current. Biofizika. 1980;25:170–172. [PubMed] [Google Scholar]
- 32.Peterson U, et al. Origin of membrane dipole potential: Contribution of the phospholipid fatty acid chains. Chem Phys Lipids. 2002;117:19–27. doi: 10.1016/s0009-3084(02)00013-0. [DOI] [PubMed] [Google Scholar]
- 33.Rostovtseva TK, Kazemi N, Weinrich M, Bezrukov SM. Voltage gating of VDAC is regulated by nonlamellar lipids of mitochondrial membranes. J Biol Chem. 2006;281:37496–37506. doi: 10.1074/jbc.M602548200. [DOI] [PubMed] [Google Scholar]
- 34.Pfefferkorn CM, et al. Depth of α-synuclein in a bilayer determined by fluorescence, neutron reflectometry, and computation. Biophys J. 2012;102:613–621. doi: 10.1016/j.bpj.2011.12.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rondelli V, et al. Amyloid-β peptides in interaction with raft-mime model membranes: A neutron reflectivity insight. Sci Rep. 2016;6:20997. doi: 10.1038/srep20997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nanda H, et al. Electrostatic interactions and binding orientation of HIV-1 matrix studied by neutron reflectivity. Biophys J. 2010;99:2516–2524. doi: 10.1016/j.bpj.2010.07.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Datta SAK, et al. HIV-1 Gag extension: Conformational changes require simultaneous interaction with membrane and nucleic acid. J Mol Biol. 2011;406:205–214. doi: 10.1016/j.jmb.2010.11.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nogales E, Wolf SG, Downing KH. Structure of the alpha beta tubulin dimer by electron crystallography. Nature. 1998;391:199–203. doi: 10.1038/34465. [DOI] [PubMed] [Google Scholar]
- 39.Nettles JH, et al. The binding mode of epothilone A on alpha,beta-tubulin by electron crystallography. Science. 2004;305:866–869. doi: 10.1126/science.1099190. [DOI] [PubMed] [Google Scholar]
- 40.Freedman H, Luchko T, Luduena RF, Tuszynski JA. Molecular dynamics modeling of tubulin C-terminal tail interactions with the microtubule surface. Proteins. 2011;79:2968–2982. doi: 10.1002/prot.23155. [DOI] [PubMed] [Google Scholar]
- 41.Zhang R, Alushin GM, Brown A, Nogales E. Mechanistic origin of microtubule dynamic instability and its modulation by EB proteins. Cell. 2015;162:849–859. doi: 10.1016/j.cell.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang L, Rozek A, Hancock REW. Interaction of cationic antimicrobial peptides with model membranes. J Biol Chem. 2001;276:35714–35722. doi: 10.1074/jbc.M104925200. [DOI] [PubMed] [Google Scholar]
- 43.Michaud-Agrawal N, Denning EJ, Woolf TB, Beckstein O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J Comput Chem. 2011;32:2319–2327. doi: 10.1002/jcc.21787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu P, Agrafiotis DK, Theobald DL. Fast determination of the optimal rotational matrix for macromolecular superpositions. J Comput Chem. 2010;31:1561–1563. doi: 10.1002/jcc.21439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Theobald DL. Rapid calculation of RMSDs using a quaternion-based characteristic polynomial. Acta Crystallogr A. 2005;61:478–480. doi: 10.1107/S0108767305015266. [DOI] [PubMed] [Google Scholar]
- 46.Coletta A, et al. Low-complexity regions within protein sequences have position-dependent roles. BMC Syst Biol. 2010;4:43. doi: 10.1186/1752-0509-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mosior M, McLaughlin S. Electrostatics and reduction of dimensionality produce apparent cooperativity when basic peptides bind to acidic lipids in membranes. Biochim Biophys Acta. 1992;1105:185–187. doi: 10.1016/0005-2736(92)90178-o. [DOI] [PubMed] [Google Scholar]
- 48.Strandberg E, Tiltak D, Ehni S, Wadhwani P, Ulrich AS. Lipid shape is a key factor for membrane interactions of amphipathic helical peptides. Biochim Biophys Acta. 2012;1818:1764–1776. doi: 10.1016/j.bbamem.2012.02.027. [DOI] [PubMed] [Google Scholar]
- 49.Zinser E, et al. Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J Bacteriol. 1991;173:2026–2034. doi: 10.1128/jb.173.6.2026-2034.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pecqueur L, et al. A designed ankyrin repeat protein selected to bind to tubulin caps the microtubule plus end. Proc Natl Acad Sci USA. 2012;109:12011–12016. doi: 10.1073/pnas.1204129109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sirajuddin M, Rice LM, Vale RD. Regulation of microtubule motors by tubulin isotypes and post-translational modifications. Nat Cell Biol. 2014;16:335–344. doi: 10.1038/ncb2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Minoura I, et al. Overexpression, purification, and functional analysis of recombinant human tubulin dimer. FEBS Lett. 2013;587:3450–3455. doi: 10.1016/j.febslet.2013.08.032. [DOI] [PubMed] [Google Scholar]
- 53.Vemu A, et al. Structure and dynamics of single-isoform recombinant neuronal human tubulin. J Biol Chem. 2016;291:12907–12915. doi: 10.1074/jbc.C116.731133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sheldon KL, Gurnev PA, Bezrukov SM, Sackett DL. Tubulin tail sequences and post-translational modifications regulate closure of mitochondrial voltage-dependent anion channel (VDAC) J Biol Chem. 2015;290:26784–26789. doi: 10.1074/jbc.M115.678854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rakovska B, et al. Structure and function of the membrane anchoring self-assembled monolayers. Langmuir. 2015;31:846–857. doi: 10.1021/la503715b. [DOI] [PubMed] [Google Scholar]
- 56.Rostovtseva TK, Gurnev PA, Chen MY, Bezrukov SM. Membrane lipid composition regulates tubulin interaction with mitochondrial voltage-dependent anion channel. J Biol Chem. 2012;287:29589. doi: 10.1074/jbc.M112.378778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kirby BJ, et al. Phase-sensitive specular neutron reflectometry for imaging the nanometer scale composition depth profile of thin-film materials. Curr Opin Colloid Interface Sci. 2012;17:44–53. [Google Scholar]
- 58.Shekhar P, Nanda H, Lösche M, Heinrich F. Continuous distribution model for the investigation of complex molecular architectures near interfaces with scattering techniques. J Appl Phys. 2011;110:102216–10221612. doi: 10.1063/1.3661986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kienzle PA, et al. 2011. Refl1D (version 0.6.19) (Univ of Maryland, College Park, MD)
- 60.Towns J, et al. XSEDE: Accelerating scientific discovery. Comput Sci Eng. 2014;16:62–74. [Google Scholar]
- 61.Humphrey W, Dalke A, Schulten K. VMD: Visual molecular dynamics. J Mol Graph. 1996;14:27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 62.Monticelli L, et al. The MARTINI coarse-grained force field: Extension to proteins. J Chem Theory Comput. 2008;4:819–834. doi: 10.1021/ct700324x. [DOI] [PubMed] [Google Scholar]
- 63.Marrink SJ, Risselada HJ, Yefimov S, Tieleman DP, de Vries AH. The MARTINI force field: Coarse grained model for biomolecular simulations. J Phys Chem B. 2007;111:7812–7824. doi: 10.1021/jp071097f. [DOI] [PubMed] [Google Scholar]
- 64.Yesylevskyy SO, Schäfer LV, Sengupta D, Marrink SJ. Polarizable water model for the coarse-grained MARTINI force field. PLOS Comput Biol. 2010;6:e1000810. doi: 10.1371/journal.pcbi.1000810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Markosyan S, et al. Effect of confinement on DNA, solvent and counterion dynamics in a model biological nanopore. Nanoscale. 2014;6:9006–9016. doi: 10.1039/c3nr06559f. [DOI] [PubMed] [Google Scholar]
- 66.Van Der Spoel D, et al. GROMACS: Fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 67.Rzepiela AJ, et al. Reconstruction of atomistic details from coarse-grained structures. J Comput Chem. 2010;31:1333–1343. doi: 10.1002/jcc.21415. [DOI] [PubMed] [Google Scholar]
- 68.Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys J. 2009;97:50–58. doi: 10.1016/j.bpj.2009.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klauda JB, et al. Update of the CHARMM all-atom additive force field for lipids: Validation on six lipid types. J Phys Chem B. 2010;114:7830–7843. doi: 10.1021/jp101759q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Phillips JC, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Brooks BR, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yoo JJ, Aksimentiev A. Improved parametrization of Li+, Na+, K+, and Mg2+ ions for all-atom molecular dynamics simulations of nucleic acid systems. J Phys Chem Lett. 2012;3:45–50. [Google Scholar]
- 73.Shaw DE, et al. Anton, a special-purpose machine for molecular dynamics simulation. Commun ACM. 2008;51:91–97. [Google Scholar]
- 74.Shan Y, Klepeis JL, Eastwood MP, Dror RO, Shaw DE. Gaussian split Ewald: A fast Ewald mesh method for molecular simulation. J Chem Phys. 2005;122:54101. doi: 10.1063/1.1839571. [DOI] [PubMed] [Google Scholar]
- 75.Deol SS, Bond PJ, Domene C, Sansom MSP. Lipid-protein interactions of integral membrane proteins: A comparative simulation study. Biophys J. 2004;87:3737–3749. doi: 10.1529/biophysj.104.048397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Domene C, Bond PJ, Deol SS, Sansom MSP. Lipid/protein interactions and the membrane/water interfacial region. J Am Chem Soc. 2003;125:14966–14967. doi: 10.1021/ja0364874. [DOI] [PubMed] [Google Scholar]
- 77.Chothia C, Levitt M, Richardson D. Helix to helix packing in proteins. J Mol Biol. 1981;145:215–250. doi: 10.1016/0022-2836(81)90341-7. [DOI] [PubMed] [Google Scholar]
- 78.Ujwal R, et al. The crystal structure of mouse VDAC1 at 2.3 A resolution reveals mechanistic insights into metabolite gating. Proc Natl Acad Sci USA. 2008;105:17742–17747. doi: 10.1073/pnas.0809634105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]