

Significance
In Gram-negative bacteria, most lipoproteins synthesized in the inner membrane (IM) are trafficked to the outer membrane (OM). The Lol pathway is the trafficking paradigm: LolCDE releases lipoproteins from the IM; LolA shuttles them between membranes to LolB in the OM. Several OM lipoproteins are essential for viability. In apparent concordance, the Lol proteins are each essential in wild-type cells. However, we show that Escherichia coli grows well without LolA and LolB in the absence of one nonessential substrate and appropriately engineered stress responses, revealing that LolAB do not perform truly essential mechanistic roles in trafficking. Rather, LolAB are needed to prevent toxic lipoprotein mislocalization. Our findings change the prevailing paradigm and reveal the existence of an alternate trafficking route.
Keywords: outer membrane, lipoprotein trafficking, Lol pathway, Cpx response, NlpE
Abstract
The outer membrane (OM) of Gram-negative bacteria is a permeability barrier and an intrinsic antibiotic resistance factor. Lipoproteins are OM components that function in cell wall synthesis, diverse secretion systems, and antibiotic efflux pumps. Moreover, each of the essential OM machines that assemble the barrier requires one or more lipoproteins. This dependence is thought to explain the essentiality of the periplasmic chaperone LolA and its OM receptor LolB that traffic lipoproteins to the OM. However, we show that in strains lacking substrates that are toxic when mislocalized, both LolA and LolB can be completely bypassed by activating an envelope stress response without compromising trafficking of essential lipoproteins. We identify the Cpx stress response as a monitor of lipoprotein trafficking tasked with protecting the cell from mislocalized lipoproteins. Moreover, our findings reveal that an alternate trafficking pathway exists that can, under certain conditions, bypass the functions of LolA and LolB, implying that these proteins do not perform any truly essential mechanistic steps in lipoprotein trafficking. Instead, these proteins’ key function is to prevent lethal accumulation of mislocalized lipoproteins.
The outer membrane (OM) blocks entry of many currently available antibiotics, preventing their clinical use in treating Gram-negative infections amid rising rates of resistance to effective drugs (1, 2). Understanding the pathways that assemble the OM permeability barrier continues to be a key goal toward uncovering novel therapeutics (3). The OM is an essential organelle that consists of a phospholipid and lipopolysaccharide (LPS) asymmetric bilayer with resident transmembrane β-barrel proteins (termed OMPs) and lipid-anchored lipoproteins (4). Recent progress has identified the essential multiprotein machines responsible for transporting and assembling most of these components into a contiguous barrier: the Bam complex folds OMPs (5); the Lpt system delivers LPS to the cells surface (6); and the Lol pathway traffics lipoproteins to the OM (7). Notably, each of the assembly machines requires at least one OM lipoprotein component for function. For example, BamD and LptE are each essential for cell viability in Escherichia coli. In addition, OM lipoproteins are essential cofactors for peptidoglycan cell wall synthesis in many Gram-negative bacteria (8, 9), and are important virulence factors in pathogens by serving as components in protein and polysaccharide secretion systems, motility structures, and antibiotic efflux pumps (10–13). Consequently, the trafficking pathway that delivers these lipoproteins to the OM is both the linchpin of all essential OM assembly processes and a fundamental contributor to pathogenicity.
Nascent lipoproteins are directed for secretion through the Sec translocase via an N-terminal signal peptide that includes a characteristic lipobox motif (7). Following translocation, an invariant cysteine in the lipobox is diacylated and this modification permits type II signal peptidase to subsequently cleave the signal peptide (Fig. 1A) (14, 15). The modified cysteine becomes the first amino acid (+1) and is additionally acylated at its amino terminus (Fig. 1A) (16). The mature triacylated lipoprotein is now anchored into the periplasmic leaflet of the inner membrane (IM) bilayer. Most E. coli lipoproteins are targeted to the OM. Whether a lipoprotein is retained in the IM or trafficked to the OM is determined by the identity of the +2 amino acid: an aspartate causes retention, whereas most other residues allow OM targeting (17, 18). The extreme hydrophobicity of lipoprotein acyl chains poses a challenge for crossing the aqueous periplasm en route to the OM. A five-protein LolABCDE pathway traffics lipoproteins to the OM in E. coli and other Gram-negative bacteria (Fig. 1A) (7). An IM ATP-binding cassette (ABC) transporter formed by a LolCDE complex first extracts OM-targeted lipoproteins from the IM bilayer (19). LolA receives lipoproteins from LolCDE and shuttles them across the periplasm while shielding their lipophilic moieties (20). LolA delivers cargo lipoproteins to LolB, itself an OM lipoprotein, which catalyzes their insertion into the OM bilayer (21).
Fig. 1.

Depleting LolB causes toxicity because of mislocalized Lpp and OsmB. (A) The OM lipoprotein biogenesis pathway. Secreted lipoproteins are acylated by the diacylglyceryltransferase Lgt at an invariant cysteine. This modification allows type II signal peptidase Lsp to release the prolipoprotein form it signal sequence (SS). The newly formed NH2 group of the +1 cysteine is then acylated by Lnt and the mature lipoprotein enters the Lol pathway. Globomycin specifically inhibits Lsp activity (45). Compound 2 inhibits LolC and LolE function (44). (B) Loss of either Lpp or its cell wall-binding K58 residue increases cell viability when LolB is depleted. (C) Inactivation of both Lpp and the Rcs-regulated OM lipoprotein OsmB significantly improves viability during LolB depletion. (D) OsmB is responsible for the lethal toxicity during LolB depletion. pBBR1MCS is the vector control for pOsmB. B–D show growth of serially diluted cultures on indicated agar media.
Each of the Lol proteins is considered essential because each is required to deliver essential lipoproteins, such as BamD and LptE, to the OM. Both BamD and LptE are highly conserved throughout all Gram-negative bacteria, as are the IM Lol proteins and LolA (22, 23). However, no LolB homolog is apparent among α- and ε-proteobacterial classes of the major Gram-negative phylum (24). Hence, organisms such as Caulobacter crescentus and Helicobacter pylori produce lipoproteins with essential functions in the OM, but appear to lack a complete Lol pathway to deliver them to the OM. This apparent paradox prompted us to scrutinize the essentiality of LolB for lipoprotein trafficking in E. coli. Here, we show that LolB—and, surprisingly, LolA—are not essential for lipoprotein trafficking. Rather, we demonstrate that the function of both proteins is to prevent toxic accumulation of OM-targeted lipoproteins in the IM. Removing two lipoprotein substrates and manipulating an envelope stress response entirely bypasses the need for LolA and LolB, but not LolCDE. Our findings reveal the existence of an alternate trafficking pathway that receives substrates from the IM LolCDE complex and delivers them to the OM.
Results
Abundant LolB Is Required to Prevent Mislocalization of an Abundant Substrate.
To probe the essentiality of LolB in E. coli, we constructed a plasmid-based LolB depletion system where the native lolB locus was deleted and ectopic LolB expression was controlled from an arabinose-inducible promoter on a multicopy plasmid (pLolB). Cellular levels of LolB are depleted when cells are grown without the arabinose inducer and growth of otherwise wild-type cells is strictly inducer-dependent (Fig. 1B). We investigated whether these cells died directly because of a lack of LolB or because of some indirect resultant toxicity. Depleting LolB levels would compromise the Lol pathway and cause the accumulation of mislocalized lipoproteins in the IM. The most abundant protein in E. coli (∼106 molecules) is Lpp (25), an OM lipoprotein that forms a covalent linkage to the peptidoglycan cell wall via its C-terminal K58 residue (26). Intentionally mislocalizing Lpp to the IM by mutating its +2 residue is lethal, but this lethality can be suppressed by deleting K58 to prevent cell wall attachment (27). We found that either Δlpp or lpp(ΔK58) mutations modestly increased viability during LolB-depletion, confirming that a mislocalized lipoprotein contributes to the lethal toxicity caused by depleting LolB (Fig. 1B and Fig. S1). It seemed possible that mislocalization of other lipoproteins may similarly account for poor viability in LolB-depleted conditions.
Fig. S1.

Preventing Lpp cell wall cross-linking and loss of Rcs-regulated OsmB permits robust growth in LolB limiting conditions. Mutants producing an Lpp mutant lacking the K58 residue that attaches the lipoprotein to the cell wall (Δ) or the wild-type Lpp (+) were tested for viability during LolB depletion during growth in the absence of the arabinose inducer.
LolB-Depletion Causes a Toxic Activation of the Rcs Stress Response.
Several stress responsive signal transduction systems use OM lipoproteins as stress sensors. The Rcs two-component phosphorelay uses the OM lipoprotein RcsF to detect defects throughout the envelope (28–30). Stress conditions are thought to enable RcsF to reach across the periplasm to promote activation of the RcsC IM histidine kinase, which then phosphorylates the RcsB response regulator, allowing it to control expression of Rcs regulon genes (28, 30, 31). Mislocalizing RcsF to the IM by mutating its +2 residue causes strong activation of the Rcs response even in the absence of stress (32). We hypothesized that RcsF may mislocalize during LolB-depleted growth and inappropriately activate the Rcs response. Indeed, Δlpp ΔrcsF cells exhibited markedly increased viability during LolB-depletion compared with Δlpp cells (Fig. 1C). Moreover, we observed similarly robust viability in Δlpp ΔrcsB cells that still produce the RcsF lipoprotein but cannot induce the regulon (Fig. 1C). Clearly, mislocalized RcsF is not itself toxic during LolB-depletion; rather, the resultant activation of the Rcs regulon is toxic.
We sought to identify the regulon members responsible for toxicity by screening knockout alleles of candidate RcsB-regulated genes (29). We found that toxicity was completely dependent upon induction of the osmB gene, encoding a small OM-targeted lipoprotein whose cellular function remains unclear. We observed the same robust viability in Δlpp ΔosmB cells during LolB-depletion as in Δlpp ΔrcsB or Δlpp ΔrcsF cells (Fig. 1C). Moreover, expressing OsmB in trans restored the toxicity of LolB depletion to Δlpp ΔosmB cells (Fig. 1D). The basis for OsmB toxicity remains to be fully elucidated.
Our results identified two sources of lethal toxicities that are caused by lowered LolB levels. Mitigating these toxicities by deleting lpp and rcsF, rcsB, or osmB significantly improved viability during LolB-depleted conditions. However, we were unable to delete lolB outright in these strains; they require leaky expression from pLolB. To more tightly regulate LolB production, we constructed single-copy LolB-depletion strain by placing an arabinose-inducible lolB at an ectopic chromosomal locus (λlolB) and then deleting the native lolB gene. Using this tightly regulated, single-copy LolB-depletion system, cells grown without inducer exhibited poor viability even when both lpp and rcsF were deleted, confirming that these cells continue to require LolB function (Fig. S2, lane 1).
Fig. S2.
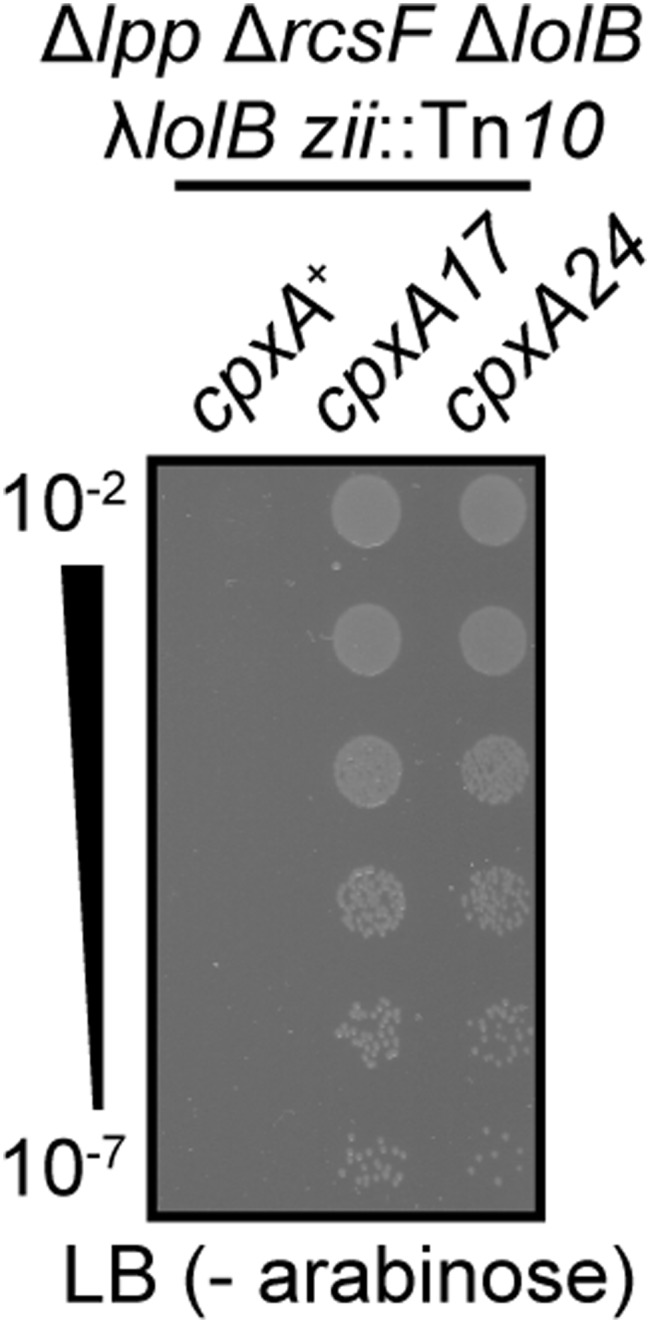
Activating CpxA is sufficient for permitting inducer-independent growth of single-copy LolB depletion ΔrcsF Δlpp cells. Serially diluted saturated cultures were plated onto media lacking the arabinose inducer.
The Cpx Response Monitors Lipoprotein Trafficking and Protects Against Defects.
A second stress response that employs an OM lipoprotein sensor is the Cpx two-component system. Adhesion of E. coli to abiotic surfaces is thought to allow the OM lipoprotein NlpE to reach across the periplasm to activate the CpxA IM histidine kinase, thereby increasing phosphorylation of the response regulator CpxR and enabling it to induce the Cpx regulon (33, 34). We examined the contribution of the Cpx response to growth during LolB-depleted conditions. The absence of CpxR severely reduced viability of Δlpp ΔrcsB cells during LolB-depletion (Fig. 2A). Hence, rather than being harmful, the Cpx response actually protects cells against the consequences of reduced LolB levels. Mislocalizing NlpE to the IM by mutating its +2 residue induces the Cpx response, likely by facilitating CpxA interaction (35). We hypothesized that NlpE may mislocalize to IM when LolB is depleted and thus activate the Cpx response. Indeed, we found that Δlpp ΔrcsB ΔnlpE cells exhibited the same reduced viability during LolB-depletion as Δlpp ΔrcsB ΔcpxR cells (Fig. 2A). Hence, activation of the Cpx response relies entirely on signaling that originates from NlpE. Strikingly, these data identify the Cpx system as a watchdog of lipoprotein trafficking: by monitoring the biogenesis of NlpE, the Cpx system can mount a protective response when the trafficking pathway is impaired.
Fig. 2.

The Cpx response protects the cell from impaired lipoprotein trafficking. (A) The Cpx response monitors trafficking efficiency via the sensor NlpE and is required for survival during LolB-depletion. (B) Activating mutations in CpxA allow cells to grow without LolB. Images show growth of serially diluted cultures on indicated agar media.
Activating the Cpx Response Bypasses Essentiality of LolB.
We took advantage of the λlolB system to more rigorously test the essentiality of LolB by searching for spontaneous mutants that could suppress the inducer-dependent growth phenotype. Two such mutant strains were isolated from media lacking inducer (Fig. 2B) and both carry suppressor mutations in cpxA. The first mutation was a 12-bp duplication that caused an insertion of VLML after amino acid 27, near the periplasmic sensor domain of the protein. CpxA sensor domain mutations commonly induce the Cpx response (36). Indeed, the second suppressor mutation was a 32-amino acid sensor domain deletion corresponding to the well-characterized activating allele cpxA24 (36, 37). We tested whether activating cpxA mutations were sufficient for suppression by introducing cpxA24 and a previously characterized activating mutation, cpxA17 (A188E) (36, 37), into the Δlpp ΔrcsF ΔlolB λlolB strain. Both cpxA mutations enabled growth in the absence of inducer, confirming that activating CpxA permits growth despite extremely low levels of LolB (Fig. S2).
We hypothesized that the cpxA suppressor mutations might allow inducer-independent growth because they bypass the essentiality of lolB outright. We introduced cpxA24 into Δlpp ΔrcsB cells that expressed lolB only from its native locus. We then used genetic linkage analysis to quantify how readily we could introduce a ΔlolB::cam deletion by cotransduction while selecting for a nearby ΔychN::kan allele. Astonishingly, lolB could be deleted from cpxA24 Δlpp ΔrcsB cells as readily as from cells complemented with lolB in trans (carrying pLolB) (Table 1). Notably, this finding indicates that there is no selective pressure to maintain lolB in cpxA24 Δlpp ΔrcsB cells. Clearly, lolB is no longer essential in cpxA24 Δlpp ΔrcsB cells. Moreover, we could also readily delete lolB in Δlpp ΔrcsB cells carrying plasmid pLD404 that overexpresses NlpE (33), underscoring that activation of the Cpx response allows for complete bypass of LolB. To date we have been unable to identify any single Cpx regulon member that is required for total LolB independence.
Table 1.
LolB is dispensable in Δlpp ΔrcsB cpxA24 cells
| Recipient strain | ΔlolB::cam cotransduction frequency (%)* |
| [pLolB]† | 49 |
| Δlpp | 0 |
| ΔrcsB | 0 |
| Δlpp ΔrcsB | 0 |
| Δlpp ΔrcsB cpxA24 | 46 |
P1vir lysates of a ΔychN::kan ΔlolB::cam strain were used to transduce the indicated recipient strains. To determine cotransduction frequency, Kanr transductants were first selected, and then 300 of these transductants were replica plated to determine the number of Kanr Camr transductants.
Linkage analysis was performed in media supplemented with arabinose.
Essential Lipoproteins Are Efficiently Trafficked in the Absence of LolB.
Despite an incomplete Lol pathway, essential lipoproteins must be reaching the OM to support viability of cpxA24 Δlpp ΔrcsB cells that lack lolB. The Bam machine, responsible for the essential process of folding OMPs into the OM, is a five-member complex that relies on four OM lipoproteins (5, 38) (Fig. 3A): BamD is an essential lipoprotein component (39); the BamBCE lipoproteins are not individually essential, but do play important roles for efficient OMP folding (5). For example, loss of BamB results in lowered OMP levels, whereas loss of both BamB and BamE is lethal (40). The final component, BamA, is itself a β-barrel OMP.
Fig. 3.

The Bam machine is fully formed and fully function in cells that lack LolB. (A) Schematic of the five protein OM Bam machine. Solid lines indicate direct protein–protein interactions (38, 56, 57). Bam lipoproteins are in black, essential proteins are in bold. (B) The absence of LolB does not reduce the levels of OMPs assembled by the Bam machine. BamA and the mature, folded LptDOX are essential Bam substrates. LamB and OmpA are nonessential OMPs. OMPs and Bam lipoproteins were detected by immunoblotting of whole-cell samples from the indicated strains, probed with antisera raised against each protein. (C) The BamCDE lipoproteins copurify with His6-BamA equally well in the presence or absence of LolB. Input samples and Ni-NTA-purified eluate samples were subjected to immunoblotting using antisera specific for each Bam protein.
We exploited the Bam machine’s extensive reliance on OM lipoproteins to gauge the efficiency of lipoprotein trafficking in cells lacking LolB by assessing OMP levels. We observed that two nonessential OMP substrates, LamB and OmpA, were completely unaffected by loss of lolB (Fig. 3B). The Bam machine has two essential OMP substrates: the LPS insertase LptD and BamA. LptD is the cell’s most complicated OMP substrate: its folding requires both assistance from an additional essential OM lipoprotein (LptE) and disulfide bond rearrangement to achieve the oxidized, functional form (LptDOX) (41, 42). We found that cpxA24 Δlpp ΔrcsB cells lacking lolB produced the same levels of both BamA and LptDOX as lolB+ controls (Fig. 3B). These data suggested OMP folding was not compromised in cells lacking LolB.
We also investigated Bam complex composition to assess the efficiency of Bam lipoprotein trafficking to the OM when LolB is absent. We constructed ΔbamA strains that were complemented with plasmid-expressed His6-tagged BamA. The tagged BamA was affinity purified from whole cells and we then measured copurification of the BamCDE lipoproteins. Remarkably, we observed no difference in Bam machine composition between lolB+ and ΔlolB cells in the cpxA24 Δlpp ΔrcsB background (Fig. 3C). This finding was particularly striking for BamC because this lipoprotein can only be copurified with BamA indirectly, through an interaction with another lipoprotein (BamD). Clearly, our data shows that BamCDE lipoproteins are efficiently delivered to the OM in cpxA24 Δlpp ΔrcsB cells that lack LolB. Although we do not have antisera to detect BamB, it’s absence causes significant reductions in OMP levels (43). Because we did not observe any such OMP defects above, our data are consistent with BamB being present in Bam machines of cpxA24 Δlpp ΔrcsB ΔlolB cells lacking LolB. Collectively, our findings demonstrate that in cells lacking LolB, the Bam complex remains fully formed and fully functional for OMP assembly, despite the absence of a recognized pathway to traffic four of its five components to the OM.
LolA Is also Nonessential for Trafficking but LolCDE Are Required.
Clearly, cells that lack LolB can deliver essential lipoproteins to the OM via some alternate LolB-independent pathway. We wondered if other Lol proteins were similarly nonessential, or if they participated in the alternate pathway and remained essential. We again used genetic linkage analysis to test lolA essentiality in cpxA24 Δlpp ΔrcsB cells. Remarkably, lolA could be readily deleted from Δlpp ΔrcsB cells in a cpxA24-dependent manner (Table 2). Moreover, we could construct a cpxA24 Δlpp ΔrcsB ΔlolA ΔlolB strain that lack both LolA and LolB and only produces the IM LolCDE complex. However, we are unable to delete lolCDE in any background. Indeed, the cpxA24 Δlpp ΔrcsB background is highly sensitive to depletion of LolCDE (Fig. S3). Accordingly, we also observed that cpxA24 Δlpp ΔrcsB cells remained sensitive to “compound 2,” a pyrazole inhibitor of LolC and LolE (Table S1) (44). The minimum inhibitory concentration (MIC) of compound 2 was significantly lower in the ΔlolA and ΔlolB derivatives of cpxA24 Δlpp ΔrcsB. Indeed, the ΔlolA and ΔlolB derivatives are more sensitive to a range of antibiotics (Fig. S4), suggesting increased cell permeability. We do not yet understand the basis for this permeability. The cpxA24 Δlpp ΔrcsB strain and its ΔlolA/B/AB derivatives cells also remained sensitive to the Lsp signal peptidase inhibitor globomycin (45) (Table S1). Lsp activity requires that substrate prelipoproteins are first diacylated (14). The sensitivity of cpxA24 Δlpp ΔrcsB cells to globomycin demonstrates that Lsp activity remains essential and we hence infer that diacylation still occurs in these cells to allow for Lsp activity.
Table 2.
LolA and LolB are dispensable in Δlpp ΔrcsB cpxA24 cells
| Recipient strain | ΔlolA::kan cotransduction frequency (%)* |
| [pLolA]† | 89 |
| Δlpp | 0 |
| ΔrcsB | 0 |
| Δlpp ΔrcsB | 0 |
| Δlpp ΔrcsB cpxA24 | 86 |
| Δlpp ΔrcsB cpxA24 ΔlolB::cam | 91 |
P1vir lysates of a zca-1230::Tn10 ΔlolA::kan strain were used to transduce the indicated recipient strains. To determine cotransduction frequency, Tetr transductants were first selected, and then 300 of these transductants were replica plated to determine the number of Tetr Kanr transductants.
Linkage analysis was performed in media supplemented with the arabinose inducer.
Fig. S3.

lolCDE remain essential in Δlpp ΔrcsB cpxA24 cells. Strains were constructed carrying pLolCDE, encoding arabinose-inducible lolCDE. ΔlolCDE::cam was then introduced by transduction on media supplemented with 0.2% arabinose. Serial dilutions of the strains were plated onto LB media (− arabinose) or LB supplemented with 0.2% arabinose (+ arabinose).
Table S1.
Δlpp ΔrcsB cpxA24 cells are sensitive to Lsp and LolCDE inhibitors
| Strain | MIC compound 2 (µg/mL) | MIC globomycin (µM) |
| Δlpp ΔrcsB | 64 | >20 |
| Δlpp ΔrcsB cpxA24 | 64 | >20 |
| Δlpp ΔrcsB cpxA24 ΔlolA | 2 | 20 |
| Δlpp ΔrcsB cpxA24 ΔlolB | 0.25 | 10 |
| Δlpp ΔrcsB cpxA24 ΔlolA ΔlolB | 1 | 20 |
Fig. S4.

ΔlolA and ΔlolB derivatives of cpxA24 Δlpp ΔrcsB exhibit increased antibiotic sensitivity. Strain sensitivity to indicated antibiotics was determine by measuring the diameter of growth inhibition around an antibiotic disk, as described in SI Materials and Methods. Zone of clearance denotes the diameter of clearance minus the diameter of the disk (6 mm).
Our data support a model in which an alternate trafficking pathway receives substrate lipoproteins from LolCDE at the IM but delivers them to the OM via a LolA- and LolB-independent mechanism. In fact, the LolAB pathway appears to compete for substrates with the alternate pathway. We noted that the cpxA24 Δlpp ΔrcsB strain lacking lolA grow equally well as its lolA+ parent (Fig. 4A). In contrast, the cpxA24 Δlpp ΔrcsB strain that lacks lolB exhibit a consistent growth defect compared with its lolB+ parent (Fig. 4A). Part of this growth defect might be attributable to polar effects of the knockout lolB allele on the essential ispE gene that lies downstream. However, we found that additionally deleting lolA improved growth of ΔlolB cells (Fig. 4A), suggesting that LolA production in the absence of LolB might be detrimental. We tested this hypothesis directly by introducing a plasmid encoding an arabinose-inducible lolA (pLolA) into cpxA24 Δlpp ΔrcsB ΔlolB cells. Inducing LolA overproduction was extremely toxic in cells lacking lolB but was well tolerated in all lolB+ control strains (Fig. 4B). It appears likely that overproducing LolA when LolB is absent titrates lipoproteins into dead-end LolA-bound complexes that cannot be inserted into the OM.
Fig. 4.

LolA is toxic in cells lacking LolB. (A) Δlpp ΔrcsB cpxA24 cells lacking lolB grow to a lower final culture density than isogenic lolB+ cells; the additional loss of lolA increases the final culture density of Δlpp ΔrcsB cpxA24 ΔlolB cells. (B) Overproduction of LolA causes lethal toxicity in Δlpp ΔrcsB cpxA24 cells that lack lolB. pBAD18 is the vector control for pLolA. Serial dilutions of saturated cultures were plated onto indicated solid medium.
Discussion
The current model of lipoprotein trafficking posits that after extraction from the IM by LolCDE, all lipoproteins transit the periplasm and reach the OM via LolA and LolB, respectively. Our findings change this paradigm. We reveal that an undiscovered, LolAB-independent, route for lipoprotein trafficking to the OM exists and is capable of supporting cell viability under certain conditions. We conclude that LolA and LolB do not perform any truly essential mechanistic steps and so are not absolutely required for lipoprotein trafficking. In contrast, LolCDE play a critical role and are fundamentally essential. The as yet undiscovered system must provide a redundant means by which to bringing lipoproteins across the periplasm and anchor them into the OM. Our findings suggest that the LolAB-dependent pathway is critically important to prevent mislocalization of OM-destined lipoproteins in the IM. Lowering LolA or LolB levels is acutely toxic for the cell not because it deprives the OM of essential lipoproteins, but because it allows two lipoprotein substrates—Lpp and OsmB—to accumulate in the IM to toxic levels. Lpp toxicity is clearly caused by the covalent attachment of the protein to the cell wall from the IM. OsmB contains glycine-zipper domains that feature in several pore-forming proteins (46). We suspect that mislocalized OsmB might form ion-permeable pores through the IM; this is a lethal event in E. coli because IM integrity is required to maintain the membrane potential that powers cellular energy generation. LolA and LolB are critically important in wild-type cells to protect the IM from damage that can be inflicted by lipoproteins that are en route to the OM.
Extensive biochemical evidence has established that LolA cannot insert lipoproteins into membranes but that it can receive lipoprotein substrates from LolCDE (19, 20, 47–49). On the other hand, analogous studies of LolB show that it can insert lipoproteins into membranes but it cannot receive them from LolCDE; its source for lipoprotein substrates is LolA (21, 48, 49). The fact that the established trafficking pathway relies on both LolA and LolB, together, is consistent with our essentiality study. Under conditions where LolB is no longer essential, we observe that LolA is likewise nonessential. Indeed, when LolB is missing, overproducing LolA is acutely toxic, likely because of titration of lipoproteins into dead-end LolA-bound products that cannot be inserted into the OM. Such titration would deprive an alternate trafficking pathway of essential substrates to support viability. Moreover, this finding suggests that the alternate pathway is not only independent of LolA, but that it cannot use LolA.
It has long been noted that conservation of LolB is poor in α- and ε-proteobacteria (22, 24). It is possible that organisms of these classes produce a structural or functional homolog of LolB that cannot be readily identified. Alternatively, LolA might be sufficient in such cells. Curiously, genome-scale transposon mutagenesis studies of the α-proteobacterium C. crescentus and the ε-proteobacteria H. pylori hint that their lolA homologs might not be essential (50, 51). It is tempting to speculate that the LolAB-independent trafficking pathway in E. coli may be conserved in these species.
As much as our findings disprove the essentiality of LolA and LolB for lipoprotein trafficking, they underscore the fundamental requirement for the LolCDE proteins. This fact is perhaps not surprising: lipoprotein acylation remains essential in our LolAB-bypass system and the cell requires a mechanism to extract OM-destined lipoproteins from the IM as a prerequisite for their subsequent trafficking. It is remarkable that a second, LolAB-independent, trafficking pathway appears to be compatible with LolCDE so that it can receive lipoprotein substrates. Moreover, this alternate pathway has sufficient capacity to not affect the efficiency with which the four lipoproteins of the Bam machine are brought to the OM.
The σE stress response monitors—among other cell envelope processes—biogenesis of one class of OM proteins, the β-barrel OMPs, and can protect the cell against defects in the OMP assembly pathway (52, 53). A comparable protective response for the lipoprotein class of OM proteins has not previously been identified. Our findings reveal that the Cpx system fulfills this role. When lipoprotein trafficking defects occur during LolB depletion, both the Rcs and the Cpx stress-signaling pathways are induced by mislocalization of their respective lipoprotein sensors RcsF and NlpE, but the consequences of the resultant signaling through the two pathways are very different. Whereas Rcs signaling is acutely toxic and lethal to the cell, Cpx signaling significantly improves cell viability. This fact leads us to conclude that Cpx (and not Rcs) is dedicated to alleviating stress caused by lipoprotein trafficking defects. CpxA monitors the efficiency of lipoprotein trafficking through its interaction with NlpE, revealing a new physiological role for this lipoprotein. When trafficking is impaired, mislocalized NlpE accumulates in the IM and is sensed by CpxA, activating a protective response that significantly improves cell viability. Activating the Cpx system is required to bypass LolAB completely. This requirement seems consistent with our demonstration that LolAB act to protect the IM. Indeed, one of the long-established roles of the Cpx system is to maintain IM homeostasis (54). We do not yet know if Cpx activation is required to combat the toxicity caused by other mislocalized lipoproteins or if it is required to increase production of components of the LolAB-independent pathway. It may be that Cpx activation contributes in both a positive and a negative fashion. Indeed, attempts to identify a single, responsible Cpx regulon member have so far been unsuccessful. Additional studies are needed to understand the functional roles of this envelope stress response.
Materials and Methods
Strains and Growth Conditions.
Strains and plasmids used in this study are provided in Tables S2 and S3, respectively. See Table S4 for oligonucleotides used. Detailed descriptions of strain and plasmid constructions are provided in SI Materials and Methods.
Table S2.
Strains used in this study
| Strain | Genotype | Source |
| MC4100 | F− {[araD139]B/r Δ(argF-lac)169 λ− e14− flhD5301 Δ(fruK-yeiR)725(fruA25) relA1 rpsL150 rbsR22 thi-1 Δ(fimB-fimE)632(::IS1) deoC1} | (67) |
| NR754 | MC4100 Ara+ | (68) |
| MG2150 | NR754/pLolB | Present study |
| MG2162 | NR754 ΔlolB/pLolB | Present study |
| MG2164 | NR754 ΔlolB Δlpp::kan/pLolB | Present study |
| MG2165 | NR754 ΔlolB ΔrcsB::kan/pLolB | Present study |
| MG2166 | NR754 ΔlolB ΔrcsF::kan/pLolB | Present study |
| MG2180 | NR754 ΔlolB Δlpp/pLolB | Present study |
| MG2200 | NR754 ΔlolB Δlpp ΔrcsB::kan/pLolB | Present study |
| MG2201 | NR754 ΔlolB Δlpp ΔrcsF::kan/pLolB | Present study |
| MG2222 | NR754 ΔlolB lpp(ΔK58) ΔynhG::kan ΔrcsF/pLolB | Present study |
| MG2221 | NR754 ΔlolB lpp(ΔK58) ΔynhG::kan ΔrcsB/pLolB | Present study |
| MG2246 | NR754 ∆(λatt-lom)::bla-ParaBAD::lolB ∆lpp ∆rcsF ∆lolB::kan | Present study |
| MG2268 | NR754 ∆(λatt-lom)::bla-ParaBAD::lolB ∆lpp ∆rcsF ∆lolB::kan cpxA(VLML) | Present study |
| MG2302 | NR754 nadA::Tn10 ∆lpp ∆rcsF ∆lolB::kan cpxA(VLML) | Present study |
| MG2382 | NR754 ∆(λatt-lom)::bla-ParaBAD::lolB ∆lpp ∆rcsF ∆lolB::kan cpxA24 | Present study |
| MG2448 | NR754 ∆(λatt-lom)::bla-ParaBAD::lolB ∆lpp ∆rcsF ∆lolB::kan zii::Tn10 cpxA+ | Present study |
| MG2449 | NR754 ∆(λatt-lom)::bla-ParaBAD::lolB ∆lpp ∆rcsF ∆lolB::kan zii::Tn10 cpxA17 | Present study |
| MG2450 | NR754 ∆(λatt-lom)::bla-ParaBAD::lolB ∆lpp ∆rcsF ∆lolB::kan zii::Tn10 cpxA24 | Present study |
| MG2579 | NR754 Δlpp | Present study |
| MG2592 | NR754 Δlpp ΔrcsB | Present study |
| MG2612 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 | Present study |
| MG2795 | NR754 ΔychN::kan ΔlolB::cam/pLolB | Present study |
| MG3056 | NR754 Δlpp ΔrcsB ΔbamA zii::Tn10 cpxA+ / pZS21::His6-BamA | Present study |
| MG3057 | NR754 Δlpp ΔrcsB ΔbamA zii::Tn10 cpxA24 / pZS21::His6-BamA | Present study |
| MG3058 | NR754 Δlpp ΔrcsB ΔbamA zii::Tn10 cpxA24 ΔlolB::cam / pZS21::His6-BamA | Present study |
| MG3114 | NR754 ΔlolB ΔrcsF::kan ΔcpxR::spec/pLolB | Present study |
| MG3117 | NR754 ΔlolB Δlpp ΔosmB::kan/pLolB | Present study |
| MG3137 | NR754 ΔlolB ΔosmB::kan/pLolB | Present study |
| MG3139 | NR754 zca-1230::Tn10 ΔlolA::kan/pBAD33LolA | Present study |
| MG3187 | NR754 ΔlolB Δlpp ΔosmB::kan/pLolB pBBR1MCS | Present study |
| MG3188 | NR754 ΔlolB Δlpp ΔosmB::kan/pLolB pOsmB | Present study |
| MG3262 | NR754 ΔlolB lpp(ΔK58) ΔynhG::kan/pLolB | Present study |
| MG3275 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 ΔlolB | Present study |
| MG3323 | NR754 ΔlolB ΔrcsF::kan ΔnlpE::spec/pLolB | Present study |
| MG3324 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 ΔlolA | Present study |
| MG3348 | NR754 Δlpp ΔrcsB cpxA+ bla | Present study |
| MG3349 | NR754 Δlpp ΔrcsB cpxA24 bla | Present study |
| MG3350 | NR754 Δlpp ΔrcsB cpxA24 bla ΔlolB::cam | Present study |
| MG3395 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 ΔlolA ΔlolB | Present study |
| MG3438 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA+/pLolCDE | Present study |
| MG3439 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24/pLolCDE | Present study |
| MG3452 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA+/pLolA | Present study |
| MG3453 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24/pLolA | Present study |
| MG3454 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 ΔlolA/pLolA | Present study |
| MG3455 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 ΔlolB/pLolA | Present study |
| MG3470 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA+/pBAD18 | Present study |
| MG3471 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24/pBAD18 | Present study |
| MG3472 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 ΔlolA/pBAD18 | Present study |
| MG3473 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 ΔlolB/pBAD18 | Present study |
| MG3487 | NR754 ΔlolB lpp(ΔK58) ΔynhG::kan ΔosmB/pLolB | Present study |
| MG3488 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 ΔlolA ΔlolB/pBAD18 | Present study |
| MG3489 | NR754 Δlpp ΔrcsB zii::Tn10 cpxA24 ΔlolA ΔlolB/pLolA | Present study |
| MG3495 | NR754/pLolA | Present study |
| MG3496 | NR754 ΔlolB Δlpp ΔrcsB ΔosmB::kan/pLolB pBBR1MCS | Present study |
| MG3497 | NR754 ΔlolB Δlpp ΔrcsB ΔosmB::kan/pLolB pOsmB | Present study |
| MG3498 | NR754 ΔlolB Δlpp ΔrcsF ΔosmB::kan/pLolB pBBR1MCS | Present study |
| MG3499 | NR754 ΔlolB Δlpp ΔrcsF ΔosmB::kan/pLolB pOsmB | Present study |
Table S3.
Plasmids used in this study
| Name | Description | Source |
| pBAD18 | Ampr cloning vector | (63) |
| pLolA | lolA cloned into pBAD18, arabinose-inducible expression | T.J.S. laboratory strain collection |
| pLolB | lolB cloned into pBAD18, arabinose-inducible expression | Present study |
| pLolCDE | lolCDE cloned into pBAD18, arabinose-inducible expression | Present study |
| pBAD33 | Camr cloning vector | (63) |
| pBAD33::LolA | lolA cloned into pBAD33, arabinose-inducible expression | Present study |
| pBBR1MCS | Camr cloning vector | (64) |
| pOsmB | osmB cloned into pBBR1MCS, lac promoter induced | Present study |
| pZS21::His6-BamA | Constitutive expression of His6-tagged BamA | (40) |
Table S4.
Oligonucleotides used in this study
| Name | Sequence (5′-3′) |
| lolB_18_NheI_F | ttagctagcagggttataactgcaacgtatctcaag |
| lolB_18_SphI_R | actgcatgctcattatttcactatccagttatccatttttaac |
| lolCDE_pBAD18_NheI | acggctagccgtgtctttgctacagcaacc |
| lolCDE_pBAD18_SphI | ttggcatgccttgttttaatgtactgcctttactgg |
| lolB-Kan_F | ctagcattaagggttataactgcaacgtatctcaaggacttgtcatcactgtgtaggctggagctgcttc |
| lolB-Kan_R | gtaatgtataaaaacagattaagttttgccggagagggccactgtgtccgcatatgaatatcctccttagttcctattc |
| lolB_screen_F_-400 | tgctcttcaacgctaagataaagtt |
| lolB_screen_R_+1100 | gttagtatttcaccaacgccttc |
| lolCDE_kan1_F | gattgatttacgggggcttttcagattagccctgacgatcacttacagttcagacgtttagtgtaggctggagctgcttc |
| lolCDE_kan1_R | ctgcaactgccgaccgctatcaaacacgccaagcgcaatttttgttccaccaatatcaaacatatgaatatcctccttagttcctattc |
| lolCDE_screen_F | ctgacgatcacttacagttcagacg |
| lolCDE_screen_R | ccaagcgcaatttttgttcc |
| lolB_int_F | gtgcgcaatcttaatcagtatcag |
| lolB_int_R | gtgtcataaccaccataaacaacct |
| osmB_F_XhoI | agactcgagcaggagagagtattatgtttgtaacgagc |
| osmB_R_SpeI | cctactagtttatttaccgacctggtgaccaataac |
| lolA_SacI_F | actgagctccgggagtgacgtaatttgagg |
| lolA_SphI_R | tgagcatgcacagattgctcactcaggtgc |
Description of the Suppressor Selection.
A detailed description of suppressor selection is provided in SI Materials and Methods. Briefly, cultures of a Δlpp ΔrcsF ΔlolB::kan λlolB (MG2246) strain were plated onto media lacking arabinose to select for mutants capable of inducer-independent growth. Suppressor mutant strains were transduced with nadA::Tn10 to test whether the nearby bla-marked (AmpR) λlolB construct could be removed. The absence of lolB in the resultant AmpS strain was confirmed by Illumina whole-genome sequencing and with intra-lolB diagnostic PCR. Whole-genome sequencing identified suppressor mutations in cpxA.
Affinity Copurification of Bam Lipoprotein with His6-BamA.
Composition of the Bam complex assessed as previously described (55) and is detailed in SI Materials and Methods. Briefly, equivalent numbers of cells were taken from cultures, lysed in BugBuster (EMD Millipore) supplemented with 5 µg/mL lysozyme, 250 U of benzonase, and 1 mM phenylmethylsulfonyl fluoride (PMSF). Debris was removed following 20-min centrifugation at 20,000 × g and lysates were incubated with Ni-NTA resin for 16 h at 4 °C. Resins were washed and proteins eluted from resin by washing with 200 mM imidazole.
SI Materials and Methods
Bacterial Strains and Growth Conditions.
Strains and plasmids used in this study are listed in Tables S2 and S3, respectively. Strains were grown in lysogeny broth (LB, 10 g/L NaCl) under aeration at 37 °C unless otherwise noted. Where appropriate, LB media were supplemented with kanamycin (Kan, 25 µg/mL), ampicillin (Amp, 25–125 µg/mL), tetracycline (Tet, 20 µg/mL), and arabinose [Ara, 0.2% (vol/vol)]. Antibiotics targeting lipoproteins biogenesis globomycin and compound 2 were sourced from Terry Roemer, Merck Research Laboratories, Kenilworth, NJ. Kanamycin deletion–insertion mutations of lpp, rcsB, rcsF, osmB were obtained from the Keio collection (58). The lpp(Δ58) allele linked to ΔynhG::kan has been previously reported (59). The zca-1230::Tn10 allele has also been previously described (60). Alleles were routinely moved by via P1vir transduction. cpxA alleles were moved via cotransduction with zii::Tn10 (37) or cpxPQ-bla (61). Keio alleles were cured of their Kanr cassette using pCP20 (62).
Plasmid Construction.
Oligonucleotides used in this study are listed in Table S4. The pLolB plasmid that expresses LolB from an arabinose-inducible promoter was constructed by PCR amplifying the lolB gene using oligonucleotides lolB_18_NheI_F and lolB_18_SphI_R, and cloning the product into NheI-SphI digested pBAD18 vector (63).
The pLolCDE plasmid that expresses LolC, LolD, and LolE from an arabinose-inducible promoter was constructed by PCR amplifying the lolCDE operon with oligonucleotides lolCDE_pBAD18_NheI and lolCDE_pBAD18_SphI, and cloning the product into NheI-SphI digested pBAD18 vector.
The pOsmB plasmid that expresses OsmB from a lactose-inducible promoter was constructed by PCR amplifying osmB with oligonucleotides osmB_F_XhoI and osmB_R_SpeI, and cloning the product into XhoI-SpeI–digested pBBR1MCS vector (64).
The pBAD33::LolA plasmid that expresses LolA from an arabinose-inducible promoter was constructed by PCR amplifying the lolA gene using oligonucleotides lolA_SacI_F and lolA_SphI_R, and cloning the product into SacI-NheI sites of pBAD33 (63).
Construction of Knockout lol Alleles.
Deletion–insertion alleles of lolB and lolCDE were constructed using λRed allelic replacement (62). Recombineering strain DY378 was first transformed with complementing pLolB or pLolCDE plasmids. Expression of λRed recombinase was induced in the resultant strains, as previously described (65). Induced cells were electroporated with PCR amplified pKD3 (for Kanr mutations) or pKD4 (for Camr mutations) with oligonucleotides lolB-Kan_F and lolB-Kan_R for lolB, or with oligonucleotides lolCDE_kan1_F and lolCDE_kan1_R for the lolCDE operon. Mutations were confirmed by arabinose-dependent growth of the resulting strains and by diagnostic PCR with oligonucleotides flanking the lolB locus (lolB_screen_F_-400 and lolB_screen_R_+1100) or the lolCDE operon (lolCDE_screen_F and lolCDE_screen_R).
Construction of Single-Copy Arabinose-Inducible lolB Strain.
To construct an ectopic, arabinose-inducible chromosomal lolB construct, we used λInCh method (66) to integrate a fragment of pLolB into the native λatt. This construct could complement a deletion of the native lolB when cells were grown in the presence of arabinose. We moved the λatt(ParaBAD::lolB-bla) construct into indicated strains by P1vir transduction and selecting for Ampr. This construct is abbreviated as λlolB in the main text and herein.
Selection of Spontaneous lolB Bypass Suppressor Mutations.
We selected for lolB bypass suppressors by plating 0.1 mL of overnight culture of strain MG2246 (Table S2) onto LB agar lacking arabinose. Plates were incubated overnight at 37 °C. We collected colonies and confirmed their ability to grow in the absence of arabinose. To determine whether suppressor mutants could tolerate complete loss of lolB, we introduced nadA::Tn10 by P1vir transduction. The nadA locus is genetically tightly linked to the Ampr-marked lolB construct at λatt (λlolB, the sole source of lolB in the strain). If mutant strains maintained an absolute requirement for lolB, Tetr nadA::Tn10 transductants would all be Ampr. The genetic linkage between nadA and λatt loci is ∼80%. Hence, if mutant strains bypassed the need for lolB entirely, we expected that 80% of Tetr nadA::Tn10 transductants would also be Amps, having lost the λlolB construct because of cotransduction of the wild-type λatt locus. We identified two MG2246-derived spontaneous mutant strains (designated MG2268 and MG2382) that could readily lose the λlolB construct following nadA::Tn10 transduction. The absence of any lolB in the nadA::Tn10 derivative of the suppressor strain MG2268 (designated MG2302) was confirmed by a diagnostic PCR using intralolB oligonucleotides lolB_int_F and lolB_int_R which failed to amplify a product.
Whole-genome Illumina sequencing of MG2302 confirmed the absence of lolB in the strain and identified a 12-nt insertion that in cpxA the introduced VLML after amino acid 27 of CpxA. PCR amplification and Sanger sequencing of the cpxA locus in MG2382 found a deletion mutation that corresponded to the well-characterized cpxA24 allele. Transduction of the cpxA mutant loci into the MG2268 parent strain allowed the strain to grow without arabinose and become AmpS following nadA::Tn10 transduction. Both phenotypes confirmed the cpxA mutations were sufficient for lolB bypass.
Efficiency of Plating Assays.
To assess viability of strains on growth media with and without arabinose, serial 10-fold dilutions of saturated overnight cultures were prepared in 96-well plates. A microplate replicator was used to replica plate ∼3 µL of each dilution onto indicated media. Plates were incubated overnight at 37 °C.
MIC Assay.
Saturated overnight cultures were diluted with LB broth to a A600nm of 1.0. Cultures were then diluted 1:1,000 into fresh LB broth, and 98 µL of diluted culture was added to each well of a 96-well plate. Serial dilutions of indicated antibiotics (2 µL) were added to each well. Plates were incubated at 37 °C overnight. Culture density was determined by measuring A600nm and the MIC was determined by identifying the lowest concentration of antibiotic required for A600nm measurements that were equivalent to fresh LB broth.
Genetic Linkage Assays.
To assess lolAB gene essentiality, knockout alleles were introduced into recipient strains indirectly, via cotransduction with a nearby marker. lolB essentiality was determined by transducing recipient cells with a P1vir lysate of strain MG2795 where ΔychN::kan is linked to lolB::cam. Kanr transductants were selected and then screened for Camr to determine cotransduction frequency. lolA essentiality was determined by transducing recipient cells with a P1vir lysate of strain MG3139 where zca-1230::Tn10 is linked to ΔlolA::kan. Tetr transductants were selected and then screened for Kanr to determine cotransduction frequency. cpxA24 recipients (MG3349) and cpxA+ control (MG3348) were marked with bla near the cpxA locus.
Growth Curve Analysis.
Saturated overnight cultures were diluted 1:100 into fresh LB broth and 2 mL was aliquoted into wells of a 24-well microtiter plate (Costar 3526). Plates were sealed with breathable film (Breathe Easy sealing membrane, Sigma-Aldrich). Plates were grown for 7 h at 37 °C with agitation in a BioTek H1 plate reader, recording A600nm in 10-min intervals.
Antibiotics Disk Assay.
A 0.1-mL aliquot of saturated overnight culture was used to inoculate 3 mL of molten LB top agar [agar at 0.75% (wt/vol)]. The mixture was poured onto a prewarmed LB plate and allowed to set. Antibiotic BBL Sensi-Discs (BD) were placed on top of the overlay and plates were incubated upright overnight at 37 °C. Zones of clearance were measured as the diameter of growth inhibition around the antibiotic disk minus the diameter of the disk (6 mm).
Immunoblotting.
Logarithmic-phase cultures were standardized by absorbance at 600 nm (OD600nm) measurement and aliquots were collected, pelleted, and resuspended in SDS/PAGE sample buffer. LptDOX samples were prepared under nonreducing conditions, where SDS/PAGE sample buffer lacked β-mercaptoethanol. SDS/PAGE was used to resolve proteins, which were then transferred onto nitrocellulose membranes. Immunoblotting was performed overnight with polyclonal rabbit antisera: anti-LptD (1:5,000), anti-BamA (1:30,000), anti-BamC (1:30,000), anti-BamD (1:30,000), anti-BamE (1:30,000), anti-LamB (1:30,000). Blots were subsequently incubated with anti-rabbit antibodies conjugated to HRP for 2 h. Blots were developed by enhanced chemiluminescence (Amersham) and visualized by exposure to X-ray film.
Affinity Copurification of Bam Lipoprotein with His6-BamA.
Bam lipoprotein association with a His6-tagged BamA β-barrel was performed as described previously (55). Briefly, haploid bamA strains were constructed by deleting the chromosomal locus and expressing His6-BamA from a pZS21 plasmid (Table S3). Strains expressing an untagged BamA were also constructed to confirm the specificity of affinity purification. Strains expressing His6-BamA were grown overnight in LB broth, and then back diluted to an A600nm of 0.05 for subculture. Constructed strains were grown in 200 mL of LB and grown to an A600nm density of ∼1.0. Cells were harvested, washed, and resuspended in 1 mL of 20 mM KH2PO4 (pH 7.2), 150 mM NaCl buffer. The cell resuspension was diluted 10-fold in BugBuster (EMD Millipore) supplemented with 5 µg/mL lysozyme, 250 U of benzonase, and 1 mM PMSF. Cells were lysed by gentle agitation for 20 min at room temperature. The lysate was centrifuged for 20 min at 20,000 × g to remove nonlyzed cells. The supernatant was then incubated with 80 µL of Ni-NTA resin (Qiagen) at 4 °C for 16 h. The resin was pelleted by centrifugation and washed 5 times with 1 mL of 20 mM KH2PO4 (pH 7.2), 150 mM NaCl buffer supplemented with 20 mM imidazole. Resin-bound proteins were eluted in 0.25 mL of 20 mM KH2PO4 (pH 7.2), 150 mM NaCl buffer supplemented with 200 mM imidazole. Aliquots were mixed with SDS/PAGE sample buffer and protein content analyzed by immunoblotting.
Acknowledgments
We thank members of the T.J.S. laboratory for helpful discussions; Daniel Kahne (Harvard University) for comments on the manuscript; Terry Roemer (Merck Research Laboratories) for providing globomycin and compound 2; and Charles Cowles for constructing pLolA and the ΔlolA::kan allele. This work was supported by Grants R35 GM118024 and R01 GM34821 (to T.J.S.).
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1702248114/-/DCSupplemental.
References
- 1.Boucher HW, et al. Bad bugs, no drugs: No ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48:1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Delcour AH. Outer membrane permeability and antibiotic resistance. Biochim Biophys Acta. 2009;1794:808–816. doi: 10.1016/j.bbapap.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cox G, Wright GD. Intrinsic antibiotic resistance: Mechanisms, origins, challenges and solutions. Int J Med Microbiol. 2013;303:287–292. doi: 10.1016/j.ijmm.2013.02.009. [DOI] [PubMed] [Google Scholar]
- 4.Nikaido H. Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev. 2003;67:593–656. doi: 10.1128/MMBR.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ricci DP, Silhavy TJ. The Bam machine: A molecular cooper. Biochim Biophys Acta. 2012;1818:1067–1084. doi: 10.1016/j.bbamem.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Okuda S, Sherman DJ, Silhavy TJ, Ruiz N, Kahne D. Lipopolysaccharide transport and assembly at the outer membrane: The PEZ model. Nat Rev Microbiol. 2016;14:337–345. doi: 10.1038/nrmicro.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okuda S, Tokuda H. Lipoprotein sorting in bacteria. Annu Rev Microbiol. 2011;65:239–259. doi: 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- 8.Paradis-Bleau C, et al. Lipoprotein cofactors located in the outer membrane activate bacterial cell wall polymerases. Cell. 2010;143:1110–1120. doi: 10.1016/j.cell.2010.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Typas A, et al. Regulation of peptidoglycan synthesis by outer-membrane proteins. Cell. 2010;143:1097–1109. doi: 10.1016/j.cell.2010.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akama H, et al. Crystal structure of the drug discharge outer membrane protein, OprM, of Pseudomonas aeruginosa: Dual modes of membrane anchoring and occluded cavity end. J Biol Chem. 2004;279:52816–52819. doi: 10.1074/jbc.C400445200. [DOI] [PubMed] [Google Scholar]
- 11.Dong C, et al. Wza the translocon for E. coli capsular polysaccharides defines a new class of membrane protein. Nature. 2006;444:226–229. doi: 10.1038/nature05267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goyal P, et al. Structural and mechanistic insights into the bacterial amyloid secretion channel CsgG. Nature. 2014;516:250–253. doi: 10.1038/nature13768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kovacs-Simon A, Titball RW, Michell SL. Lipoproteins of bacterial pathogens. Infect Immun. 2011;79:548–561. doi: 10.1128/IAI.00682-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tokunaga M, Tokunaga H, Wu HC. Post-translational modification and processing of Escherichia coli prolipoprotein in vitro. Proc Natl Acad Sci USA. 1982;79:2255–2259. doi: 10.1073/pnas.79.7.2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sankaran K, Wu HC. Lipid modification of bacterial prolipoprotein. Transfer of diacylglyceryl moiety from phosphatidylglycerol. J Biol Chem. 1994;269:19701–19706. [PubMed] [Google Scholar]
- 16.Gupta SD, Gan K, Schmid MB, Wu HC. Characterization of a temperature-sensitive mutant of Salmonella typhimurium defective in apolipoprotein N-acyltransferase. J Biol Chem. 1993;268:16551–16556. [PubMed] [Google Scholar]
- 17.Yamaguchi K, Yu F, Inouye M. A single amino acid determinant of the membrane localization of lipoproteins in E. coli. Cell. 1988;53:423–432. doi: 10.1016/0092-8674(88)90162-6. [DOI] [PubMed] [Google Scholar]
- 18.Masuda K, Matsuyama S, Tokuda H. Elucidation of the function of lipoprotein-sorting signals that determine membrane localization. Proc Natl Acad Sci USA. 2002;99:7390–7395. doi: 10.1073/pnas.112085599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yakushi T, Masuda K, Narita S, Matsuyama S, Tokuda H. A new ABC transporter mediating the detachment of lipid-modified proteins from membranes. Nat Cell Biol. 2000;2:212–218. doi: 10.1038/35008635. [DOI] [PubMed] [Google Scholar]
- 20.Matsuyama S, Tajima T, Tokuda H. A novel periplasmic carrier protein involved in the sorting and transport of Escherichia coli lipoproteins destined for the outer membrane. EMBO J. 1995;14:3365–3372. doi: 10.1002/j.1460-2075.1995.tb07342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuyama Si, Yokota N, Tokuda H. A novel outer membrane lipoprotein, LolB (HemM), involved in the LolA (p20)-dependent localization of lipoproteins to the outer membrane of Escherichia coli. EMBO J. 1997;16:6947–6955. doi: 10.1093/emboj/16.23.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tokuda H, Matsuyama S. Sorting of lipoproteins to the outer membrane in E. coli. Biochim Biophys Acta. 2004;1693:5–13. doi: 10.1016/j.bbamcr.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 23.LoVullo ED, Wright LF, Isabella V, Huntley JF, Pavelka MS., Jr Revisiting the Gram-negative lipoprotein paradigm. J Bacteriol. 2015;197:1705–1715. doi: 10.1128/JB.02414-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wilson MM, Bernstein HD. Surface-exposed lipoproteins: An emerging secretion phenomenon in Gram-negative bacteria. Trends Microbiol. 2016;24:198–208. doi: 10.1016/j.tim.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li G-W, Burkhardt D, Gross C, Weissman JS. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell. 2014;157:624–635. doi: 10.1016/j.cell.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Braun V, Rehn K. Chemical characterization, spatial distribution and function of a lipoprotein (murein-lipoprotein) of the E. coli cell wall. The specific effect of trypsin on the membrane structure. Eur J Biochem. 1969;10:426–438. doi: 10.1111/j.1432-1033.1969.tb00707.x. [DOI] [PubMed] [Google Scholar]
- 27.Yakushi T, Tajima T, Matsuyama S, Tokuda H. Lethality of the covalent linkage between mislocalized major outer membrane lipoprotein and the peptidoglycan of Escherichia coli. J Bacteriol. 1997;179:2857–2862. doi: 10.1128/jb.179.9.2857-2862.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Konovalova A, Mitchell AM, Silhavy TJ. A lipoprotein/β-barrel complex monitors lipopolysaccharide integrity transducing information across the outer membrane. eLife. 2016;5:e15276. doi: 10.7554/eLife.15276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laubacher ME, Ades SE. The Rcs phosphorelay is a cell envelope stress response activated by peptidoglycan stress and contributes to intrinsic antibiotic resistance. J Bacteriol. 2008;190:2065–2074. doi: 10.1128/JB.01740-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cho S-H, et al. Detecting envelope stress by monitoring β-barrel assembly. Cell. 2014;159:1652–1664. doi: 10.1016/j.cell.2014.11.045. [DOI] [PubMed] [Google Scholar]
- 31.Konovalova A, Perlman DH, Cowles CE, Silhavy TJ. Transmembrane domain of surface-exposed outer membrane lipoprotein RcsF is threaded through the lumen of β-barrel proteins. Proc Natl Acad Sci USA. 2014;111:E4350–E4358. doi: 10.1073/pnas.1417138111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shiba Y, et al. Exploring the relationship between lipoprotein mislocalization and activation of the Rcs signal transduction system in Escherichia coli. Microbiology. 2012;158:1238–1248. doi: 10.1099/mic.0.056945-0. [DOI] [PubMed] [Google Scholar]
- 33.Snyder WB, Davis LJ, Danese PN, Cosma CL, Silhavy TJ. Overproduction of NlpE, a new outer membrane lipoprotein, suppresses the toxicity of periplasmic LacZ by activation of the Cpx signal transduction pathway. J Bacteriol. 1995;177:4216–4223. doi: 10.1128/jb.177.15.4216-4223.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danese PN, Silhavy TJ. CpxP, a stress-combative member of the Cpx regulon. J Bacteriol. 1998;180:831–839. doi: 10.1128/jb.180.4.831-839.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miyadai H, Tanaka-Masuda K, Matsuyama S, Tokuda H. Effects of lipoprotein overproduction on the induction of DegP (HtrA) involved in quality control in the Escherichia coli periplasm. J Biol Chem. 2004;279:39807–39813. doi: 10.1074/jbc.M406390200. [DOI] [PubMed] [Google Scholar]
- 36.Raivio TL, Silhavy TJ. Transduction of envelope stress in Escherichia coli by the Cpx two-component system. J Bacteriol. 1997;179:7724–7733. doi: 10.1128/jb.179.24.7724-7733.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cosma CL, Danese PN, Carlson JH, Silhavy TJ, Snyder WB. Mutational activation of the Cpx signal transduction pathway of Escherichia coli suppresses the toxicity conferred by certain envelope-associated stresses. Mol Microbiol. 1995;18:491–505. doi: 10.1111/j.1365-2958.1995.mmi_18030491.x. [DOI] [PubMed] [Google Scholar]
- 38.Bakelar J, Buchanan SK, Noinaj N. The structure of the β-barrel assembly machinery complex. Science. 2016;351:180–186. doi: 10.1126/science.aad3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malinverni JC, et al. YfiO stabilizes the YaeT complex and is essential for outer membrane protein assembly in Escherichia coli. Mol Microbiol. 2006;61:151–164. doi: 10.1111/j.1365-2958.2006.05211.x. [DOI] [PubMed] [Google Scholar]
- 40.Sklar JG, et al. Lipoprotein SmpA is a component of the YaeT complex that assembles outer membrane proteins in Escherichia coli. Proc Natl Acad Sci USA. 2007;104:6400–6405. doi: 10.1073/pnas.0701579104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee J, et al. Characterization of a stalled complex on the β-barrel assembly machine. Proc Natl Acad Sci USA. 2016;113:8717–8722. doi: 10.1073/pnas.1604100113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chng S-S, et al. Disulfide rearrangement triggered by translocon assembly controls lipopolysaccharide export. Science. 2012;337:1665–1668. doi: 10.1126/science.1227215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wu T, et al. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell. 2005;121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 44.Nayar AS, et al. Novel antibacterial targets and compounds revealed by a high-throughput cell wall reporter assay. J Bacteriol. 2015;197:1726–1734. doi: 10.1128/JB.02552-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogeley L, et al. Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science. 2016;351:876–880. doi: 10.1126/science.aad3747. [DOI] [PubMed] [Google Scholar]
- 46.Kim S, et al. Transmembrane glycine zippers: Physiological and pathological roles in membrane proteins. Proc Natl Acad Sci USA. 2005;102:14278–14283. doi: 10.1073/pnas.0501234102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanamaru K, Taniguchi N, Miyamoto S, Narita S, Tokuda H. Complete reconstitution of an ATP-binding cassette transporter LolCDE complex from separately isolated subunits. FEBS J. 2007;274:3034–3043. doi: 10.1111/j.1742-4658.2007.05832.x. [DOI] [PubMed] [Google Scholar]
- 48.Okuda S, Tokuda H. Model of mouth-to-mouth transfer of bacterial lipoproteins through inner membrane LolC, periplasmic LolA, and outer membrane LolB. Proc Natl Acad Sci USA. 2009;106:5877–5882. doi: 10.1073/pnas.0900896106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taniguchi N, Matsuyama S, Tokuda H. Mechanisms underlying energy-independent transfer of lipoproteins from LolA to LolB, which have similar unclosed β-barrel structures. J Biol Chem. 2005;280:34481–34488. doi: 10.1074/jbc.M507388200. [DOI] [PubMed] [Google Scholar]
- 50.Salama NR, Shepherd B, Falkow S. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol. 2004;186:7926–7935. doi: 10.1128/JB.186.23.7926-7935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Christen B, et al. The essential genome of a bacterium. Mol Syst Biol. 2011;7:528. doi: 10.1038/msb.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lima S, Guo MS, Chaba R, Gross CA, Sauer RT. Dual molecular signals mediate the bacterial response to outer-membrane stress. Science. 2013;340:837–841. doi: 10.1126/science.1235358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo MS, Gross CA. Stress-induced remodeling of the bacterial proteome. Curr Biol. 2014;24:R424–R434. doi: 10.1016/j.cub.2014.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Raivio TL. Everything old is new again: An update on current research on the Cpx envelope stress response. Biochim Biophys Acta. 2014;1843:1529–1541. doi: 10.1016/j.bbamcr.2013.10.018. [DOI] [PubMed] [Google Scholar]
- 55.Ricci DP, Hagan CL, Kahne D, Silhavy TJ. Activation of the Escherichia coli β-barrel assembly machine (Bam) is required for essential components to interact properly with substrate. Proc Natl Acad Sci USA. 2012;109:3487–3491. doi: 10.1073/pnas.1201362109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Han L, et al. Structure of the BAM complex and its implications for biogenesis of outer-membrane proteins. Nat Struct Mol Biol. 2016;23:192–196. doi: 10.1038/nsmb.3181. [DOI] [PubMed] [Google Scholar]
- 57.Gu Y, et al. Structural basis of outer membrane protein insertion by the BAM complex. Nature. 2016;531:64–69. doi: 10.1038/nature17199. [DOI] [PubMed] [Google Scholar]
- 58.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol. 2006;2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cowles CE, Li Y, Semmelhack MF, Cristea IM, Silhavy TJ. The free and bound forms of Lpp occupy distinct subcellular locations in Escherichia coli. Mol Microbiol. 2011;79:1168–1181. doi: 10.1111/j.1365-2958.2011.07539.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Singer M, et al. A collection of strains containing genetically linked alternating antibiotic resistance elements for genetic mapping of Escherichia coli. Microbiol Rev. 1989;53:1–24. doi: 10.1128/mr.53.1.1-24.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grabowicz M, Koren D, Silhavy TJ. The CpxQ sRNA negatively regulates Skp to prevent mistargeting of β-Barrel outer membrane proteins into the cytoplasmic membrane. MBio. 2016;7:e00312–e00316. doi: 10.1128/mBio.00312-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kovach ME, Phillips RW, Elzer PH, Roop RM, 2nd, Peterson KM. pBBR1MCS: A broad-host-range cloning vector. Biotechniques. 1994;16:800–802. [PubMed] [Google Scholar]
- 65.Yu D, et al. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boyd D, Weiss DS, Chen JC, Beckwith J. Towards single-copy gene expression systems making gene cloning physiologically relevant: Lambda InCh, a simple Escherichia coli plasmid-chromosome shuttle system. J Bacteriol. 2000;182:842–847. doi: 10.1128/jb.182.3.842-847.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Casadaban MJ. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976;104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- 68.Button JE, Silhavy TJ, Ruiz N. A suppressor of cell death caused by the loss of sigmaE downregulates extracytoplasmic stress responses and outer membrane vesicle production in Escherichia coli. J Bacteriol. 2007;189:1523–1530. doi: 10.1128/JB.01534-06. [DOI] [PMC free article] [PubMed] [Google Scholar]