

Significance
Antibiotic resistance is an ever-increasing problem; for certain pathogens, few treatment options remain. Vaccines and therapeutic antibodies represent important alternatives to antibiotics, yet despite extensive effort no approved vaccine is available for Staphylococcus aureus. Although wall carbohydrates are ideal immunotherapeutic targets due to their abundance and high level of conservation, their poor immunogenicity compared with conventional protein targets complicates the production of effective antibodies. The approach presented here fuses the high-affinity binding domains from bacteriophage lysins and autolysins that recognize specific cell wall carbohydrate epitopes to IgG Fc, creating effective therapeutic antibodies, or lysibodies. This approach is generalizable, allowing production of antibodies to poorly immunogenic carbohydrate epitopes of many Gram-positive pathogens. Lysibodies thus represent a broad class of anti-infectives.
Keywords: immunotherapy, antibody, vaccine, monoclonal, peptidoglycan hydrolase
Abstract
The cell wall of Gram-positive bacteria contains abundant surface-exposed carbohydrate molecules that are highly conserved within and often across species. The potential therapeutic usefulness of high-affinity antibodies to cell wall carbohydrates is unquestioned, however obtaining such antibodies is challenging due to the poor overall immunogenicity of these bacterial targets. Autolysins and phage lysins are peptidoglycan hydrolases, enzymes that have evolved over a billion years to degrade bacterial cell wall. Such wall hydrolases are modular enzymes, composed of discrete domains for high-affinity binding to cell wall carbohydrates and cleavage activity. In this study, we demonstrate that binding domains from autolysins and lysins can be fused to the Fc region of human IgG, creating a fully functional homodimer (or “lysibody”) with high-affinity binding and specificity for carbohydrate determinants on the bacterial surface. Furthermore, we demonstrate that this process is reproducible with three different binding domains specific to methicillin-resistant Staphylococcus aureus (MRSA). Cell-bound lysibodies induced the fixation of complement on the bacterial surface, promoted phagocytosis by macrophages and neutrophils, and protected mice from MRSA infection in two model systems. The lysibody approach could be used to target a range of difficult-to-treat pathogenic bacteria, given that cell wall hydrolases are ubiquitous in nature.
The rise of multidrug-resistant bacteria has created a need for alternatives to conventional antibiotics. One important approach is the use of therapeutic antibodies, which have recently become a mainstay in areas such as cancer therapy and inflammation and are now increasingly being developed to treat infectious diseases (1–3). Most of the latter antibodies developed thus far target virulence factors that are either secreted or bound to the bacterial surface; however, creation of opsonic antibodies to the carbohydrate components exposed on the bacterial surface remains an important yet elusive goal of immunotherapy. Carbohydrates are a major component of the Gram-positive bacterial cell wall (up to 60% in dry weight) and are invariant within and often across bacterial species. Cell wall carbohydrates are often surface-exposed and required for proper cell wall function (4–8). Although carbohydrates are attractive targets for the development of therapeutic antibodies, their structures are poor immunogens because they are T cell-independent antigens. Thus, they elicit an immune response characterized by the production of low-affinity IgMs, the absence of class-switched antibodies and memory, and a short half-life (9–11). One approach that can promote effective immunity to carbohydrates is the creation of protein–carbohydrate conjugates; recent advances in glycobiology and chemical synthesis of carbohydrates have increased the scope of this method (11). This approach, though complex and expensive, has enabled the development of effective vaccines against certain carbohydrates such as capsular polysaccharides, however capsules are often variable and require the production of a polyvalent vaccine for effective protection (12). As such, proteins represent the major class of molecular targets for antibody therapies, and attempts to target carbohydrates have been less successful (11).
Although high-affinity antibodies to cell wall carbohydrates are rare, cell wall hydrolases, which are ubiquitous in nature, can bind with very high affinity (13–15). For example, bacteria produce cell wall hydrolases (autolysins) to separate daughter cells following division and facilitate peptidoglycan turnover (16), and bacteriophages produce wall hydrolases (lysins) to release progeny from infected bacterial hosts (14, 15). Both autolysins and lysins have discrete cell wall carbohydrate binding domains required for proper function. By creating IgG Fc fusions with binding domains from different cell wall hydrolases, we produced “lysibodies” with specificity toward bacterial cell wall carbohydrate epitopes.
As a proof-of-concept, we produced lysibodies specific for the cell wall of Staphylococcus aureus. S. aureus is a leading cause of skin and soft tissue infection as well as a diverse array of severe invasive infections, making it one of the major causes of pathogen-related death in the United States (17–20). The rise in antibiotic resistance is a major concern that is not adequately addressed by the anti-infective development pipeline. In particular, methicillin-resistant S. aureus (MRSA) is now prevalent in both the hospital and community settings, representing an enormous public health burden worldwide (17, 21–23). Vaccines and therapeutic antibodies represent a prominent alternative to antibiotics; however, to date, none has successfully reached regulatory approval for S. aureus (24–27). By combining the S. aureus-specific carbohydrate binding activity of selected autolysins and lysins with the Fc effector functions of human antibodies, we created three lysibodies specific to S. aureus. These lysibodies recognized a range of clinically important staphylococcal strains, fixed complement on the bacterial surface, induced phagocytosis by macrophages and neutrophils, and protected mice from challenge with MRSA in two model systems.
Results
Lysibody Construction and Production.
IgG antibodies are composed of two heavy chains and two light chains, stabilized by disulfide bridges and noncovalent interactions. Each antibody can be functionally divided into two Fab fragments, which bind to target epitopes, a hinge region, and an Fc fragment. The Fc fragment binds to a diversity of Fc receptors, including FcRn and type I and II FcγRs, and thus determines half-life and mediates effector functions that lead to the elimination of pathogens (28). Lysibodies were produced by fusing a human IgG1 Fc with a cell wall binding domain of staphylococcal autolysin or phage lysin. Lysibodies contain a leader sequence to promote secretion and a hexahistidine tag for purification. Cysteine 220 of human IgG1 heavy chain, which in the native molecule forms a disulfide bridge with the light chain, was mutated to glycine, as a light chain is not present in lysibodies; cysteines required for the formation of disulfide bridges between the two heavy chains were retained. Thus, the final lysibody structure is a two-chain homodimer (Fig. 1A).
Fig. 1.

Lysibody construction, dimerization, formation of disulfide bridges, and binding to target organisms. (A) Schematic representations of lysibody structure. BD, binding domain; Cat, catalytic domain. (B) Structure of the expression vectors for lysibodies and controls. L, leader sequence; 6H, hexahistidine tag. (C) Purified lysibodies were run on 10% SDS/PAGE with or without the reducing agent BME and analyzed by Western blot using anti-human IgG1 Fc antibody. A duplicate gel was stained with Coomassie blue. (D) Binding of AtlA lysibody to S. aureus Wood 46 (protein A negative) was determined by deconvolution immunofluorescence microscopy. Maximum intensity projections are presented. Anti-human IgG Fc Alexa Fluor 594 conjugate, red; wheat germ agglutinin (WGA), green; DAPI, blue (scale bar, 1 μm). (E) Binding of C-terminal Fc fusion lysibodies to S. aureus Wood 46 was determined by immunofluorescence microscopy, using anti-human IgG Fc Alexa Fluor 594 conjugate (scale bar, 2 μm). Experiments were repeated three times. ScFab, single chain fragment antigen-binding.
The AtlA lysibody was created by replacing the VH and CH1 domains of human IgG1 heavy chain with the R1–R2 binding repeats of the major staphylococcal autolysin AtlA (16, 29). AtlA binds S. aureus lipoteichoic acid (LTA) on the cell surface (30), which is essential for S. aureus survival (31). AtlA binding repeats R1–R2 bind with high affinity at ∼108 binding sites per staphylococcal cell, predominantly in the vicinity of the division ring (29, 32). As control, we produced a construct with a single chain fragment variable (Fv) specific for chicken ubiquitin in place of the AtlA binding repeats, termed “ChUb construct” (Fig. 1B).
A slightly different approach was used to produce lysibodies containing binding domains from phage lysins. Unlike autolysins, the cell wall binding domain of most phage lysins is found at the C terminus of the molecule (33). Placing such domains at the N terminus of an antibody Fc region (directly replacing the Fab) results in an unnatural orientation for the lysin binding domain, potentially interfering with its function. As there are numerous examples of functional antibodies with C-terminal fusions (34), we created lysibodies using the binding domains of the S. aureus-specific lysins ClyS (35) and PlySs2 (36) fused to the C terminus of human IgG1 Fc region (Fig. 1B). As controls we also produced a lysibody containing the binding domain of the Bacillus anthracis-specific lysin PlyG (37) and a construct containing no binding domain whatsoever (“Fc only”; Fig. 1B). We used I-TASSER (38) to perform structural predictions on lysibody monomers. Each predication showed an extended structure with clear domain delineation for lysibodies, resembling the control ChUb construct—that is, a typical antibody with a single chain Fv (Fig. S1). Formation of Fc dimers would likely further stabilize these predicted structures in the correct domain organization.
Fig. S1.

Structural predictions for lysibody monomers. Protein sequences for the monomeric form of different lysibodies were analyzed by the I-TASSER server. The structures with the highest confidence rate are presented. The human IgG1 Fc region (including hinge) is colored cyan, the binding domain or single chain Fv (where applicable) is colored magenta, the hexahistidine tag is colored yellow, and linker regions are gray.
Lysibodies were produced in mammalian cells to allow glycosylation of the Fc fragment, required for effector functions. Like typical single-chain antibodies, lysibodies formed the expected dimers that were stabilized by disulfide bridges, as determined by SDS/PAGE and Western blot analysis (Fig. 1C). Elimination of disulfide bridges with β-mercaptoethanol (BME) resulted in products with one-half the molecular mass. The binding avidity of lysibodies is predicted to be significantly higher than that of the original phage lysin or autolysin as a result of dimerization.
We used fluorescence microscopy to test the binding of lysibodies to the cell wall of S. aureus. Initially, we used the protein A negative S. aureus strain Wood 46 to avoid nonspecific binding of protein A to the Fc region of lysibodies. Although protein A needs to be addressed when studying lysibody binding in vitro using purified reagents, it is less likely to inhibit lysibody activity in the blood environment (39); human serum contains abundant nonspecific antibodies that saturate protein A, as shown in Fig. S2. AtlA lysibody showed extensive labeling of the staphylococcal cell wall with some preference for the septa (Fig. 1D), similar to previous observations (29, 32). No signal was detected with the ChUb construct or the PBS control. Similarly, C-terminal fusion ClyS and PlySs2 lysibodies bound S. aureus, whereas the Fc-only construct showed no binding (Fig. 1E). The anthrax-specific PlyG lysibody showed a very slight cross-reactivity with S. aureus.
Fig. S2.

S. aureus protein A is saturated in normal human serum. Overnight cultures of S. aureus strains RN4220 (protein A positive) and Wood 46 (protein A negative) were diluted 1:100 in BHI, grown to log phase, and fixed. Cells were attached to poly-l-lysin–coated slides and blocked with PBS containing 1% BSA for 10 min. Each slide was further blocked for 1 h with 10 μL human sera, diluted in PBS containing 1% BSA to the stated concentration. The slides were then incubated with Rhodamine red-conjugated normal human IgG (nonspecific) to test binding to free protein A on the bacterial surface. Slides were processed in two batches, using sera from healthy subjects chosen at random (scale bar, 2 μm).
Target Binding Range of Lysibodies.
We evaluated the binding of lysibodies to various methicillin-resistant, vancomycin-intermediate, and vancomycin-resistant S. aureus strains (MRSA, VISA, and VRSA, respectively) using fluorescence microscopy. To avoid possible nonspecific interaction of the Fc portion of lysibodies with staphylococcal protein A, we first blocked protein A with goat and human serum and used AtlA lysibody or ChUb construct that was directly labeled with Rhodamine red. To determine the binding range of ClyS and PlySs2, we used fusion proteins of the lysin binding domain and green fluorescent protein (GFP). The binding domains of AtlA, ClyS, and PlySs2 bound all S. aureus clinical isolates tested, although some variability in fluorescent signal was observed; controls did not bind any of the strains tested (Figs. S3 and S4).
Fig. S3.
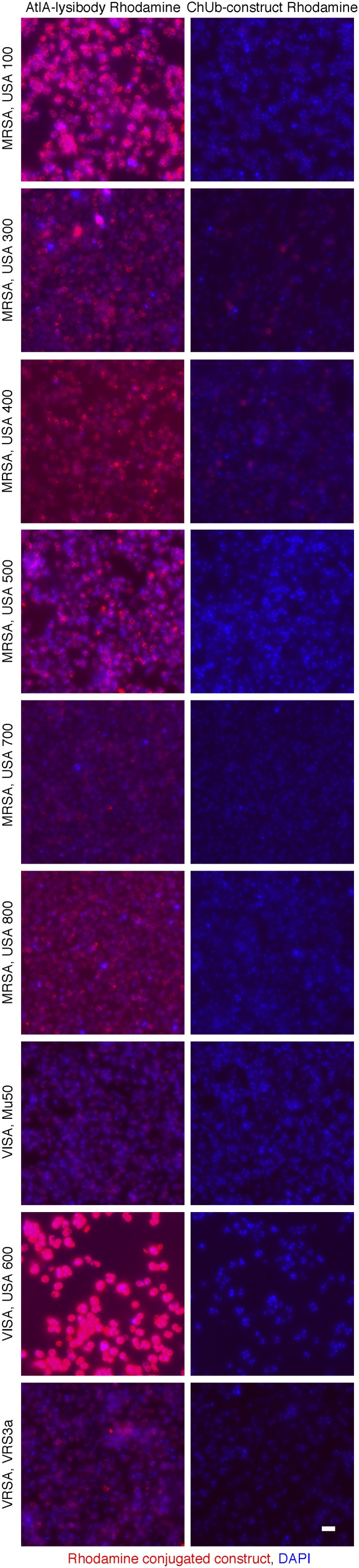
AtlA lysibody binds to a range of clinically important S. aureus strains. S. aureus strains were fixed and attached to a microscope cover glass. Protein A was blocked with goat and human serum, and the bacteria were incubated with Rhodamine red-conjugated AtlA lysibody or ChUb construct and then visualized by fluorescence microscopy. The brightness level of Mu50 and VRS3a (AtlA lysibody and ChUb construct panels) was enhanced to show binding (scale bar, 2 μm). MRSA, methicillin-resistant S. aureus; VISA, vancomycin intermediate S. aureus; VRSA, vancomycin-resistant S. aureus.
Fig. S4.

ClyS and PlySs2 binding domains specifically bind to a range of clinically important S. aureus strains. Bacterial cells were fixed, attached to a microscope cover glass, and blocked. Bacteria were incubated with purified ClyS-BD GFP fusion, PlySS2-BD GFP fusion, or GFP alone. Slides were imaged by phase-contrast and fluorescence microscopy (scale bar, 2 μm).
We further characterized the binding range of lysibodies to various staphylococcal species and other bacteria. AtlA lysibody, ClyS lysibody, and PlySs2 lysibody bound Staphylococcus epidermidis, Staphylococcus simulans, Staphylococcus hyicus, and Staphylococcus sciuri, in addition to S. aureus (Figs. S5 and S6). For AtlA lysibody and ClyS lysibody, slight to no signal was observed for nonstaphylococcal species (AtlA lysibody bound weakly to Bacillus cereus and Enterococcus faecalis, exclusively at the septum). PlySs2 lysibody, on the other hand, displayed a broad range of host binding, including staphylococci, E. faecalis, Enterococcus faecium, Streptococcus pyogenes, and Streptococcus agalactiae, consistent with the wider lytic activity range of the PlySs2 lysin (36). PlyG lysibody specifically bound B. anthracis (Fig. S6).
Fig. S5.

Binding range of AtlA lysibody. The following strains were evaluated for binding of lysibodies using fluorescence microscopy: S. aureus protein A negative Wood 46, S. epidermidis ATCC 12228S, S. simulans TNK3, S. hyicus HER1048, S. sciuri subsp. sciuri K1, B. cereus T, E. faecalis V12, S. pyogenes SF370, and E. coli DH5α. Bacterial cells were fixed, attached to a microscope cover glass, and blocked. Cells were incubated with AtlA lysibody or ChUb construct and subsequently with anti-human IgG Fc Alexa Fluor 594 conjugate. DNA was visualized using DAPI (scale bar, 2 μm).
Fig. S6.

Binding range of ClyS lysibody, PlySs2 lysibody, and PlyG lysibody. The following strains were evaluated for binding of lysibodies using fluorescence microscopy: S. aureus protein A negative Wood 46, S. epidermidis ATCC 12228S, S. simulans TNK3, S. hyicus HER1048, S. sciuri subsp. sciuri K1, B. anthracis ΔSterne, B. cereus T, E. faecalis V12, E. faecium EFSK-2, S. pyogenes SF370, and S. agalactiae 090R. Bacterial cells were fixed, attached to a microscope cover glass, and blocked. Cells were incubated with ClyS lysibody, PlySs2 lysibody, or PlyG lysibody and subsequently with anti-human IgG Fc Alexa Fluor 594 conjugate.
Lysibodies Induce Phagocytosis of S. aureus by Macrophages.
Killing of S. aureus by phagocytes is an important immunological mechanism controlling staphylococcal infections. To determine the extent of lysibody-induced phagocytosis, cells of the GFP-expressing S. aureus strain Newman/pCN57 were incubated with a monolayer of macrophages for an hour in the presence of various lysibodies. Macrophages were washed and analyzed by microscopy (Fig. 2A and Fig. S7) and flow cytometry (Fig. 2 B–D) to determine the extent of phagocytosis. We first gated on the macrophage population and then determined the percentage of highly fluorescent macrophages, indicating a substantial staphylococcal load (Fig. 2B). AtlA lysibody induced phagocytosis of staphylococci in a dose-dependent manner by both Raw 264.7 cell line macrophages and primary peritoneal murine macrophages, whereas the ChUb construct and the nonspecific monoclonal antibody 1K8 had no effect (Fig. 2C). Similarly, ClyS and PlySs2 lysibodies induced phagocytosis of S. aureus in a dose-dependent manner, whereas the B. anthracis-specific PlyG lysibody had only a minimal effect at the highest concentration, possibly due to the low cross-reactivity with S. aureus carbohydrate (Fig. 1E and 2D). To determine whether staphylococci are internalized or merely attached to the surface of macrophages, we analyzed samples treated with ClyS lysibody or PlyG lysibody using deconvolution fluorescence microscopy, which allows 3D evaluation of the cells (Fig. S7). This analysis showed a much higher bacterial load in macrophages in the presence of ClyS lysibody (Fig. S7A). Examination of sequential Z sections allowed a clear distinction between intracellular and surface-attached staphylococci (Fig. S7B). Analysis of a large population of cells showed that in the presence of ClyS lysibody the staphylococcal load per macrophage increased dramatically compared with PlyG lysibody (Fig. S7C). Roughly 75% of the staphylococci observed were inside macrophages, whereas 25% were associated with the surface or partially internalized (Fig. S7D). We next determined whether lysibodies lead to the killing of phagocytosed staphylococci. Log phase S. aureus strain Newman cells were mixed with various lysibodies and incubated with Raw 264.7 macrophages in suspension for 3 h, at a 1:1 ratio of staphylococci to macrophages. Macrophages were then lysed and the number of bacterial colony-forming units (cfus) was determined through serial dilution and plating (Fig. 2E). Under the conditions tested, AtlA ClyS and PlySs2 lysibodies led to killing of 65–80% of staphylococci compared with the PBS control. The relative killing efficiency ratios were ClyS lysibody > AtlA lysibody > PlySs2 lysibody, consistent with the results obtained in flow cytometry assays. The ChUb construct and PlyG lysibody controls did not lead to significant killing of staphylococci.
Fig. 2.

AtlA lysibody induces phagocytosis of S. aureus by macrophages. Adherent macrophages were incubated for 1 h with fluorescent S. aureus Newman/pCN57 (GFP) in the presence of various lysibodies. The cells were washed, fixed, and analyzed by microscopy and flow cytometry. (A) A representative Raw 264.7 macrophage containing fluorescent staphylococci following ClyS lysibody treatment. Staphylococci, green; WGA Alexa Fluor 594 conjugate, red. The image is presented as a maximum intensity projection (scale bar, 2 μm). Also see Fig. S7. (B) Gating scheme for flow cytometry analysis: gating on macrophages using forward and side scatter (Left), followed by determination of the percentage of highly fluorescent macrophages (Right). Black, AtlA lysibody; gray, PBS. (C) Effect of lysibody dose on phagocytosis using the N-terminal fusions AtlA lysibody and ChUb construct and 1K8 nonspecific humanized monoclonal. (D) Effect of lysibody dose on phagocytosis using C-terminal fusions: ClyS lysibody, PlySs2 lysibody, and PlyG lysibody. Experiments were performed in triplicates and repeated three times. (E) Percent killing of S. aureus Newman by Raw 264.7 macrophages in the presence of 10 µg of various lysibodies, compared with PBS control. Experiments were performed in triplicates, with two technical repeats for each biological repeat; P values were calculated using t test. FSC, forward scatter; SSC, side scatter.
Fig. S7.

High-resolution microscopy of S. aureus Newman/pCN57 phagocytosis by Raw 267.4 macrophages. S. aureus Newman/pCN57 (expressing GFP) was added to tissue culture wells containing adherent Raw 264.7 macrophages, supplemented with 5 μg ClyS lysibody or 5 μg PlyG lysibody. Following 1 h of incubation at 37 °C, the wells were washed and macrophages were fixed. Cells were further stained with WGA Alexa Fluor 594 conjugate and imaged using deconvolution microscopy. Images are presented as maximum intensity projections. (A) Representative images of cells treated with ClyS lysibody or PlyG lysibody (scale bar, 5 μm). (B) Serial z sections at 0.6-μm intervals of a single macrophage containing fluorescent staphylococci treated with ClyS lysibody (scale bar, 5 μm). (C) The number of staphylococci in each macrophage was quantified by analyzing the image stack as presented above. The aggregate results from over 100 macrophages per condition are presented. (D) The percentage of intracellular and extracellular bacteria was determined using a similar method; partially phagocytosed bacteria were treated as extracellular.
Lysibodies Fix Complement on the Surface of S. aureus.
Complement deposition on the surface of pathogens is an important mechanism leading to their efficient removal by phagocytes. We used fluorescence microscopy to determine whether lysibodies can induce fixation of complement fragment C3b on the surface of S. aureus. Protein A negative strain Wood 46 was used to prevent a nonspecific fluorescent signal. Human serum that was preadsorbed on S. aureus to remove possible antibodies specific to this organism was used as a source of complement. AtlA, ClyS, and PlySs2 lysibodies induced complement deposition on the surface of the cells, although PlySs2 lysibody was less potent than the other two lysibodies (Fig. 3). Nonspecific controls (ChUb construct, 1K8 monoclonal antibody, PlyG lysibody, and Fc alone) did not induce complement deposition. For AtlA lysibody we also determined the extent of complement deposition on strain Newman/pCN57 using microscopy (Fig. S8A) and flow cytometry (Fig. S8B); protein A was blocked subsequent to complement deposition to prevent nonspecific fluorescent signal. This analysis demonstrated that induction of complement deposition is dose-dependent.
Fig. 3.

Lysibodies induce deposition of complement on the surface of S. aureus cells. S. aureus Wood 46 cells (protein A negative) were attached to poly-l-lysine–coated coverslips and incubated with lysibodies. The cells were then incubated with human complement, washed, fixed, and blocked. Complement was detected using rabbit anti-C3, followed by Alexa Fluor 594 conjugate; DNA was stained with DAPI. Images were obtained using deconvolution microscopy and are presented as maximum intensity projections. Experiments were repeated twice (scale bar, 1 μm).
Fig. S8.

AtlA lysibody induces deposition of complement on the surface of S. aureus cells. (A) S. aureus Newman/pCN57 (GFP) cells were attached to a poly-l-lysine–coated coverslips and incubated with AtlA lysibody or ChUb construct and then with S. aureus-adsorbed human complement. The cells were washed, fixed, and blocked (protein A was blocked with heat-inactivated serum). Complement was detected using rabbit anti-C3, followed by Alexa Fluor 594 conjugate. Deconvolution microscopy images are presented as maximum intensity projections (scale bar, 2 μm). (B) S. aureus Newman/pCN57 (GFP) cells were incubated with various concentrations of AtlA lysibody or ChUb construct, washed, and incubated with S. aureus-adsorbed human complement. The cells were then washed, fixed, and blocked (protein A was blocked with heat-inactivated serum). Complement was detected using rabbit anti-C3, followed by Alexa Fluor 594 conjugate. For flow cytometry analysis, initial gating was done on GFP-expressing cells, and then the C3 fluorescence in the red channel was evaluated.
Lysibodies Induce S. aureus Phagocytosis by Neutrophils.
Neutrophils are the first line of defense against S. aureus infection (40). Recognition of Fc by FcγRs and recognition of surface-attached C3b by C3 receptors are necessary for efficient phagocytosis by neutrophils. We evaluated the phagocytosis of fluorescent S. aureus by human neutrophils using fluorescence microscopy (Fig. 4A and Fig. S9) and flow cytometry (Fig. 4 B–E and Fig. S10). We used FITC-labeled staphylococci for increased sensitivity, as neutrophils typically phagocytosed fewer staphylococci per cell compared with macrophages. S. aureus-specific lysibodies (AtlA, ClyS, and PlySs2) induced phagocytosis of S. aureus USA300 by differentiated HL-60 neutrophils in a complement-dependent manner, whereas control constructs had no effect (Fig. 4C). Induction of phagocytosis was dose-dependent in all cases (Fig. 4D). AtlA lysibody and ClyS lysibody also induced phagocytosis of S. aureus in a complement-dependent manner using peripheral blood human polymorphonuclear cells, however PlySs2 lysibody was less effective with these cells (Fig. 4E). Similar results were obtained with S. aureus Wood 46 and USA600 (Fig. S10).
Fig. 4.

Lysibodies induce phagocytosis of S. aureus by neutrophils. HL-60 neutrophils (A–D) and human polymorphonuclear cells (E) were incubated with FITC-labeled S. aureus strain USA300 in the presence of lysibodies and S. aureus-adsorbed human complement. (A) A representative image of HL-60 neutrophils incubated with FITC-labeled S. aureus USA300 and AtlA lysibody; a maximum intensity projection is presented (scale bar, 2 μm). Also see Fig. S9. (B) Gating scheme for flow cytometry analysis: gating on neutrophils using forward and side scatter (Left) and determination of the percentage of fluorescent neutrophils (Right). Black, AtlA lysibody; gray, PBS. (C) Phagocytosis of S. aureus by HL-60 neutrophils using 5 µg AtlA lysibody, ClyS lysibody, PlySs2 lysibody, or controls in the presence or absence of complement. (D) Effect of lysibody dose on S. aureus phagocytosis by HL-60 neutrophils. (E) Phagocytosis of S. aureus by human polymorphonuclear cells using 5 µg AtlA lysibody, ClyS lysibody, PlySs2 lysibody, or controls in the presence or absence of complement. All experiments were done in triplicates and repeated two to four times. Statistical significance analysis using the t test was performed for the relevant samples. *P < 0.05, **P < 0.01, ***P < 0.001. Also see Fig. S10 (Wood 46 and USA600). FSC, forward scatter; PMNs, polymorphonuclear leukocytes; SSC, side scatter.
Fig. S9.

Comparison of the level of phagocytosis as determined by flow cytometry and high-resolution microscopy. HL-60 neutrophils were incubated with FITC-labeled S. aureus strain Wood 46 in the presence of lysibodies and S. aureus-adsorbed human complement. Cells were fixed, and each sample was divided for analysis by flow cytometry and deconvolution fluoresce microscopy. For microscopy, the cells were further stained with WGA Alexa Fluor 594 conjugate. (A) Representative images of cells treated with AtlA lysibody, ChUb construct, nonspecific 1K8 monoclonal, or PBS alone. Images are presented as maximum intensity projections (scale bar, 5 μm). White arrows represent internalized staphylococci, and yellow arrows represent staphylococci attached to the surface of phagocytes. (B) An example of the technique used to determine whether bacteria are intracellular or extracellular. The Z sections at 0.6-μm intervals are presented; intracellular bacteria are denoted with white arrows, and extracellular bacteria are denoted with yellow arrows (scale bar, 5 μm). (C) At least 150 neutrophils for each treatment group were evaluated by high-resolution fluorescence microscopy for the presence of intracellular and extracellular staphylococci. The results are presented alongside the flow cytometry results obtained from the same sample. (D) For each treatment group, the number of intracellular and extracellular bacteria observed by high-resolution fluorescence microscopy per 100 neutrophils is presented.
Fig. S10.

Lysibodies induce phagocytosis of S. aureus USA600 and Wood 46 by neutrophils. HL-60 neutrophils (A and B) and human polymorphonuclear cells (C) were incubated with FITC-labeled S. aureus strains in the presence of lysibodies and S. aureus-adsorbed human complement. (A) Phagocytosis of S. aureus by HL-60 neutrophils using 5 µg lysibody in the presence or absence of complement. (B) Effect of lysibody dose on S. aureus phagocytosis by HL-60 neutrophils. (C) Phagocytosis of S. aureus by human polymorphonuclear cells using 5 µg lysibody in the presence or absence of complement. All experiments were done in triplicates and repeated two to four times. Statistical significance analysis using the t test was performed for the relevant samples. P values are designated as follows: *P < 0.05, **P < 0.01, ***P < 0.001.
To rule out the possibility that the flow cytometry results represent bacteria attached to the surface of neutrophils rather than phagocytosed bacteria, we analyzed samples treated with AtlA lysibody or controls by both flow cytometry and deconvolution fluorescence microscopy for 3D evaluation of the cells (Fig. S9). The percentage of neutrophils containing intracellular bacteria using this method closely resembled the percentage obtained using flow cytometry. Furthermore, only a minority of staphylococci were observed attached to the surface of neutrophils, and these were often associated with phagocytic cups.
Lysibodies Protect Mice from S. aureus Infection.
Two infection models were used to test protection of mice from S. aureus infection. In a kidney abscess model, 1 mg ClyS lysibody, the B. anthracis-specific PlyG lysibody, and PBS control were each injected intraperitoneally to BALB/c female mice. Twenty-four hours later, the mice were challenged intraperitoneally with a sublethal dose of 2.5 × 106 cfu of the methicillin-resistant, VISA strain USA600. Four days later, surviving mice were euthanized, and the bacterial load in the kidneys was determined through homogenization, serial dilution, and plating. Mice treated with ClyS lysibody had a significantly reduced bacterial load compared with mice treated with PlyG lysibody or PBS (Fig. 5A).
Fig. 5.

Lysibodies protect mice from MRSA infection in kidney abscess and bacteremia models. (A) Five-week-old female BALB/C mice were injected i.p. with 1 mg of the S. aureus-specific ClyS lysibody, B. anthracis-specific PlyG lysibody, or PBS. One day later, the mice were injected i.p. with 2.5 × 106 S. aureus USA600 (methicillin-resistant, vancomycin-intermediate) in 5% mucin. Mouse viability was monitored daily for 4 d, at which time the mice were euthanized, and the bacterial load per gram in the kidneys was determined through homogenization, serial dilution, and plating. Aggregate data from four experiments are presented (n = 10 in each group). Statistical significance was determined using two-tailed Mann–Whitney test. (B) Five-week-old female BALB/C mice were injected i.p. with 0.3 mg AtlA lysibody or PBS (n = 17 in each group). One day later, mice were injected i.p. with 2 × 106 S. aureus MW2 (USA400, methicillin-resistant) in 5% mucin. Mouse viability was monitored for 8 d. Data represent aggregate results from four experiments. Statistical significance was determined using the Gehan–Breslow–Wilcoxon test.
To test protection from bacteremia, 0.3 mg AtlA lysibody and PBS control were each injected intraperitoneally to female BALB/c mice. Twenty-four hours later, the mice were challenged intraperitoneally with 2 × 106 cfu of the MRSA strain MW2 (USA400). Mouse viability was monitored for 8 d, at which time surviving mice were euthanized. AtlA lysibody-treated mice had significantly improved survival rates compared with control mice (Fig. 5B).
Discussion
In this study, we report the development of a solution to a long-standing problem in immunology—how to create high-affinity opsonic antibodies to invariant and abundant bacterial cell wall carbohydrates. We demonstrate that binding domains from cell wall hydrolases can direct the Fc portion of an antibody to bacterial wall carbohydrates, enabling the fusion homodimer to function like a normal antibody: efficiently binding, opsonizing, inducing complement fixation, promoting phagocytosis of bacteria by macrophages and neutrophils, and protecting animals in infection models. As proof of concept, we produced three different lysibodies specific for S. aureus, using the binding domains from either the major staphylococcal autolysin AtlA, or two phage lysins—ClyS and PlySs2.
The major advantage of lysibodies compared with typical monoclonal antibodies is the ability to bind abundant carbohydrate targets on the bacterial wall that are highly conserved and thus unlikely to mutate to avoid binding. Many surface carbohydrates are critical for proper cell wall function (7). In S. aureus, the membrane-bound carbohydrate LTA is essential (31), and although mutants lacking wall teichoic acid (WTA) are viable, they have impaired pathogenicity and are less able to colonize the host (41). When using a binding domain derived from the pathogen’s own autolysin, as in the case of the AtlA lysibody, mutations that would prevent lysibody binding would necessarily also disturb the action of the native autolysin, interfering with cell division. Resistance to lysibodies containing a binding domain from a phage lysin is also unlikely; lysins have evolved over a billion years to bind wall targets that cannot easily mutate, as a phage that does not produce a functional lysin would be trapped inside the infected host and thus lost from the population (42). Supporting this, resistance to phage lysins was not observed following selection procedures that readily produce antibiotic resistance (37, 43). Another advantage of targeting wall carbohydrates is that they are often conserved across species, resulting in a broad range of target organisms. For example, AtlA lysibody and ClyS lysibody bound all strains of S. aureus tested as well as several coagulase-negative staphylococci, whereas PlySs2 lysibody bound streptococci and enterococci in addition to staphylococci. The protein targets of monoclonal antibodies, on the other hand, are often variable even at the species level and may not always be expressed (44–47). This may limit the strain coverage of certain monoclonal antibodies and could also result in the selection of escape mutants during the course of treatment.
A potential drawback associated with frequent/repeated lysibody treatment could be that part of the lysibody molecule is foreign to humans and may therefore elicit an immune response, the effects of which are currently unknown. However, reduction of binding domain immunogenicity could be achieved through reduction of T effector epitopes and introduction of Treg epitopes (48). Furthermore, development of antilysibody antibodies in a treated patient would likely only occur after the infecting organism has already been eliminated. The intended clinical use of lysibodies is to treat life-threatening infections, either alone or in combination with conventional therapy to help boost the opsonic response to the infecting organism. For many individuals, such life-threatening infections are a once-in-a-lifetime event for a given pathogen, and thus, development of antilysibody antibodies would be irrelevant for these patients. In cases of repeated life-threatening episodes, however, a lysibody with a different binding domain could be used; as we have shown here, development of multiple distinct lysibodies to a single organism is quite possible.
There is an urgent clinical need to create new treatment options to staphylococcal infections (17). Other than antibiotics, to which staphylococci show increased resistance, no other anti-infective is currently available. Hospitalized patients and those undergoing immunosuppressive therapy are particularly vulnerable, as highly virulent drug-resistant bacteria have become endemic in many healthcare facilities (19). Recent technological advances in the production of recombinant antibodies have made the routine use of these molecules in the clinic increasingly feasible. Therapeutic antibodies and vaccines are now aggressively being pursued as an alternative treatment for antibiotic-resistant bacterial pathogens, as indicated by the number of such agents reaching advanced stages of clinical trials (1–3). To date, all attempts to produce an effective S. aureus vaccine or therapeutic antibodies have failed to protect patients in clinical trials despite showing promise in preclinical studies. Although the reason for this failure is still unclear, it has been proposed that targeting only a single surface antigen may not have been sufficient to confer protective immunity, due to the vast array of redundant virulence factors S. aureus possesses. Staphylococcal protein A has also been proposed as a factor preventing the development of effective immunity, by nonspecifically activating B cells, leading to the deletion of potentially protective B-cell clones (49). Many current vaccine development efforts are targeting multiple factors in an attempt to overcome some of these limitations (50). In this context, lysibodies could provide a unique solution. By targeting carbohydrate epitopes that are highly abundant, conserved, and indispensible, lysibodies may lead to a better outcome compared with previous single-target therapeutic antibodies.
In conclusion, our approach opens an avenue for the development of therapeutic antibodies, using binding domains that were optimized through evolution. Lysibodies could be produced for a range of additional Gram-positive pathogenic bacteria, given the wealth of autolysins and phage lysins found in nature (15). Lysibodies therefore represent a broad class of anti-infectives that resolve the long-standing problem of effectively targeting bacterial surface carbohydrates with antibodies. More broadly, our results strongly suggest that proteins or protein domains with high-affinity binding for the surface of a bacterium, virus, or parasite may be Fc-modified to produce a functional opsonic antibody.
Methods
Ethics Statement.
Procedures involving human subjects were approved by the Rockefeller University Institutional Review Board (IRB number VFI-0790). Informed consent was obtained from all subjects. Mouse work was approved by and mice were cared for in accordance with the Rockefeller University Institutional Animal Care and Use Committee (protocol 14691H) (51).
Additional Methods.
A list of primers used in this study is provided in Table S1. A list of strains used in this study is provided in Table S2. Additional methods are provided in SI Methods.
Table S1.
Primers used in this study
| Primer name | Orientation | Sequence |
| abVec_MCS_5_AgeI_SalI | 5′ | CCGGTAGCGGCCGCCTGCAGGGATCCTCTAGAGATATCCAGCTGAAG |
| abVec_MCS_3_AgeI_SalI | 3′ | TCGACTTCAGCTGGATATCTCTAGAGGATCCCTGCAGGCGGCCGCTA |
| H6GS_5_AgeI_NotI | 5′ | CCGGTCATCATCATCATCATCATGGAGGAGGAGGAAGCGGAGGAGGAAGC |
| H6GS_3_AgeI_NotI | 3′ | GGCCGCTTCCTCCTCCGCTTCCTCCTCCTCCATGATGATGATGATGATGA |
| AtlA_R1R2_5_NotI | 5′ | CCCGCGGCCGCATGACAACTACCCCTACTACACCATCAAAACC |
| AtlA_R1R2_3_PstI | 3′ | GGGCTGCAGAGCTGTTTTTGGTTGTGCTACTGC |
| hIgG1_Fc_5_PstI | 5′ | CCCCTGCAGCCCAAATCTGGTGACAAAACTC |
| hIgG1_Fc_3_HinDIII | 3′ | CTTAAGCTTTCATTTACCCGGAGACAGGG |
| hIgG1_Fc_5_NotI | 5′ | CAAGCGGCCGCCCCAAATCTGGTGACAAAACTC |
| hIgG1_Fc_3_PstI | 3′ | CACCTGCAGTTTACCCGGAGACAGGGAG |
| New_MCS_5_PstI_SalI | 5′ | GGGGGGATCCGGGTCTAGAGGAAGATCTGGAGGAGGAGGGGATATCAAG |
| New_MCS_3_PstI_SalI | 3′ | TCGACTTGATATCCCCTCCTCCTCCAGATCTTCCTCTAGACCCGGATCCCCCCTGCA |
| ClyS-BD_5_SalI | 5′ | CCGGTCGACCATGAATAAGATCACAAATAAAGTTAAACCACC |
| ClyS-BD_3_HinDIII | 3′ | CCCAAGCTTTTAAAACACTTCTTTCACAATCAATCTCTC |
| PlySs2-BD_5_SalI | 5′ | GAGGTCGACCACACCGCCTGGCACGGTCGCACAG |
| PlySs2-BD_3_HinDIII | 3′ | GAGAAGCTTTTATTTAAATGTACCCCAAGCATTG |
| ChickUbi_5_NotI | 5′ | GGGGCGGCCGCATGGCCGAGGTGCAGCTGTTGGAG |
| ChickUbi_3_PstI | 3′ | CCCCTGCAGTCGTTTGATTTCCACCTTGGTCCCTTG |
| PlyG-BD_5_SalI | 5′ | GGGGTCGACTCATGTGGCGACTACTTCACC |
| PlyG-BD_3_HinDIII | 3′ | CCCAAGCTTTTATTTAACTTCATACCACCAACC |
| hIgG1_Fc-only_5_NotI | 5′ | CCCGCGGCCGCCCCAAATCTGGTGACAAAACTC |
| ClyS-BD_5_XbaI | 5′ | CCGTCTAGAATGAATAAGATCACAAATAAAGTTAAACCACC |
| ClyS-BD_3_PstI | 3′ | GCGCTGCAGTTAAAACACTTCTTTCACAATCAATCTCTC |
| H6_GFP_5_EcoRI | 5′ | CGCGAATTCATGAGTAAAGGAGAACTTCATCATCATCATCATCATTCCTCCGCCATGAGTAAAGGAGAAGAACTTTTC |
| GFP_3_KpnI | 3′ | GAGGGTACCTTTGTATAGTTCATCCATGCC |
| PlySs2-BD_5_XbaI | 5′ | GAGTCTAGAACACCGCCTGGCACGGTCGCACAG |
| PlySs2-BD_3_PstI | 3′ | GGGCTGCAGTTATTTAAATGTACCCCAAGCATTG |
| GFP_3_PstI | 3′ | CGCCTGCAGTTATTTGTATAGTTCATCCATGCCATGTG |
Restriction sites are underlined.
Table S2.
Bacterial strains used in this study
| Organism | Source |
| B. anthracis, ΔSterne | Ref. 37 |
| B. cereus, T | Ref. 37 |
| Bacillus subtilis, SL4 | Fischetti Laboratory Bacteria Collection, The Rockefeller University |
| E. faecalis, V12 | Fischetti Laboratory Bacteria Collection, The Rockefeller University |
| E. faecium, EFSK-2 | Alexander Tomasz, The Rockefeller University, New York, NY |
| E. coli, DH5α | Invitrogen |
| S. aureus, NRS105, Wood 46, (MSSA, protein A negative) | NARSA |
| S. aureus, Newman (MSSA) | Ref. 35 |
| S. aureus, NRS623, RN4220/pCN57 (MSSA, constitutive GFP expression) | NARSA |
| S. aureus, Newman/pCN57 (MSSA, constitutive GFP expression) | This work |
| S. aureus, NRS382, USA100 (MRSA) | NARSA |
| S. aureus, NRS383, USA200 (MRSA) | NARSA |
| S. aureus, NRS384, USA300 (MRSA) | NARSA |
| S. aureus, MW2, USA400 (MRSA) | Ref. 35 |
| S. aureus, NRS385, USA500 (MRSA) | NARSA |
| S. aureus, NRS22, USA600, HIP07930 (VISA) | NARSA |
| S. aureus, NRS386, USA700 (MRSA) | NARSA |
| S. aureus, NRS387, USA800 (MRSA) | NARSA |
| S. aureus, NRS1, Mu50, (VISA) | NARSA |
| S. aureus, VRS2, HIP11983 (VRSA) | NARSA |
| S. aureus, VRS3a, HIP13170 (VRSA) | NARSA |
| S. epidermidis, ATCC 12228 | ATCC |
| S. hyicus, HER1048 | Hans Wolfgang Ackermann, Université Laval, Québec, Canada |
| S. sciuri subsp. sciuri, K1 | Ref. 35 |
| S. simulans, TNK3 | Ref. 35 |
| S. agalactiae 090R | Fischetti Laboratory Bacteria Collection, The Rockefeller University |
| S. pyogenes, SF370 | Fischetti Laboratory Bacteria Collection, The Rockefeller University |
ATCC, American Type Culture Collection; MRSA, methicillin-resistant S. aureus; MSSA, methicillin-sensitive S. aureus; NARSA, Network on Antimicrobial Resistance in S. aureus; VISA, vancomycin-intermediate S. aureus; VRSA, vancomycin-resistant S. aureus.
SI Methods
Cell Lines, Bacteria, and Media.
The 293T cells were obtained from Michel Nussenzweig at the Rockefeller University and were grown in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% heat-inactivated FBS (Sigma) and 2 mM sodium pyruvate (Sigma). The Raw 264.7 murine macrophage cell line was obtained from ATCC (ATCC Number TIB-71) and grown in minimum essential media (MEM; Gibco, Life technologies), containing 1 mM sodium pyruvate and 10% heat inactivated FBS. HL-60 cells were obtained from ATCC (ATCC Number CCL241) and propagated in RPMI 1640 (Gibco, Life Technologies), containing GlutaMax (Gibco, Life Technologies), 10% heat-inactivated FBS, penicillin, and streptomycin. Differentiation of HL-60 was performed using a similar medium, lacking antibiotics, and supplemented with 100 mM N,N-dimethylformamide (DMF, ≥99.8% purity, Sigma), in accordance with established procedures (52). All mammalian tissue cultures were incubated at 37 °C, 5% CO2. All cell lines were tested for mycoplasma at Memorial Sloan Kettering Cancer Center Antibody and Bioresource Core Facility, using the MycoAlert PLUS Assay (Lonza), and found to be clear of mycoplasma.
Bacterial strains used in this work are denoted in Table S2. S. aureus strain Newman/pCN57 was created by transforming strain Newman with plasmid pCN57, which expresses GFP from the strong PblaZ promoter. Escherichia coli strains were grown in LB medium. Staphylococci, enterococci, and bacilli were grown in brain heart infusion (BHI) broth (BD). Streptococci were grown in Todd–Hewitt medium (Difco) supplemented with 1% yeast extract (Fisher Scientific). Bacterial strains were grown at 37 °C with shaking except for streptococci and enterococci, which were grown stationary at 37 °C.
Reagents.
Dulbecco’s PBS (DPBS) without calcium chloride and magnesium chloride was from Gibco, and DPBS/Modified with calcium chloride and magnesium chloride was from HyClone. Goat serum was from Sigma. Goat anti-human IgG (gamma chain specific) alkaline phosphatase antibody (Sigma, A3187) was used at 1:5,000 dilution for Western blots. Goat anti-human IgG, Fcγ fragment-specific, DyLight 594 conjugate (Jackson ImmunoResearch, 109–515-008) was used at 1:1,000 dilution. Polyclonal rabbit anti-human C3c Complement (Dako, A0062, 9.6 g/L) was used at 1:500 dilution for microscopy and 1:2,000 dilution for flow cytometry. Goat anti-rabbit IgG Alexa Fluor 594 conjugate highly cross-adsorbed (Life Technologies, A11037) was used at 1:1,000 dilution for microscopy and 1:2,000 for flow cytometry. WGA Alexa Fluor 488 and Alexa Fluor 594 conjugates (Molecular Probes) were used at 5 μg/mL, DAPI (Sigma) was used at 1 μg/mL, and Fluorescein isothiocyanate (FITC) isomer 1 (Sigma) and NHS-Rhodamine red (Thermo Scientific) were used according to manufacturer instructions. Other reagents were from Sigma, unless otherwise noted.
Construction of AtlA Lysibody and ChUb Construct Expression Vectors.
The multicloning site of the mammalian expression vector AbVec-hIgG1 (53), GenBank ID FJ475055, was replaced with a new multicloning site containing the following restriction sites in order: AgeI, NotI, PstI, BamHI, XbaI, EcoRV, PvuII, SalI. This was done by aligning primers abVec_MCS_5_AgeI_SalI and abVec_MCS_3_AgeI_SalI (Table S1) and inserting the resulting double-stranded DNA fragment between the AgeI and SalI sites of AbVec-hIgG1, yielding pAR323.
pAR401_AtlA lysibody was derived from pAR323 through the insertion of the following DNA fragments: A DNA fragment encoding amino acids TGHHHHHHGGGGSGGGSGR, created by aligning primers H6GS_5_AgeI_NotI and H6GS_3_AgeI_NotI, was inserted between sites AgeI and NotI. A DNA fragment encoding the R1–R2 binding domains of S. aureus Newman AtlA was amplified using primers AtlA_R1R2_5_NotI and AtlA_R1R2_3_PstI, and the resulting PCR product was inserted into the NotI and PstI sites. The Fc region of human IgG1, encompassing the hinge region and constant domains 2 and 3, was amplified using primers hIgG1_Fc_5_PstI and hIgG1_Fc_3_HinDIII and inserted between the PstI and HinDIII sites, thereby replacing the original human IgG1 Fc fragment found on this plasmid. Nucleotide changes encoded on primer hIgG1_Fc_5_PstI resulted in a change of the original N-terminal–most cysteine encoded on this fragment, which normally forms a disulfide bridge with the light chain to a glycine.
To construct the control plasmid pAR444_ChUb construct, a single-chain Fv fragment specific for chicken ubiquitin that was obtained from the domain antibody phage library (54) was amplified using primers ChickUbi_5_NotI and ChickUbi_3_PstI and inserted into the NotI and PstI sites of pAR401_AtlA lysibody, thereby replacing the AtlA binding repeats R1–R2. The Fc-alone construct was created by amplifying the human IgG1 Fc region using primers hIgG1_Fc-only_5_NotI and hIgG1_Fc_3_HinDIII and inserting the resulting PCR product between the NotI and HinDIII sites of pAR401_AtlA lysibody, yielding pAR450_Fc-only.
For the creation of C-terminal fusion lysibodies, the multicloning site of pAR323 was further modified by replacing the region between the PstI and SalI restriction sites with a DNA fragment obtained by the alignment of primers New_MCS_5_PstI_SalI and New_MCS_3_PstI_SalI, which contains the following restriction sites: PstI, BamHI, XbaI, BglII, EcoRV, SalI. A DNA fragment encoding a hexahistidine tag and a short glycine/serine linker was created by aligning primers H6GS_5_AgeI_NotI and H6GS_3_AgeI_NotI and was inserted between the AgeI and NotI restriction sites. The hinge region and constant domains CH2 and CH3 of human IgG1 were amplified using primers hIgG1_Fc_5_NotI and hIgG1_Fc_3_PstI and were inserted into the NotI and PstI sites; a cysteine in the hinge region, which normally forms the disulfide bond with the light chain, was changed to a glycine.
Various phage lysin binding domains were cloned into the SalI and HinDIII sites of the resulting plasmid as follows: The ClyS binding domain was amplified with primers ClyS-BD_5_SalI and ClyS-BD_3_HinDIII, yielding pAR422_ClyS lysibody. The PlySs2 binding domain was amplified with primers PlySs2-BD_5_SalI and PlySs2-BD_3_HinDIII, yielding pAR423_PlySs2 lysibody. The binding domain of PlyG was amplified with primers PlyG-BD_5_SalI and PlyG-BD_3_HinDIII, yielding pAR465_PlyG lysibody.
Construction of Lysin Binding Domains and GFP Fusion Proteins.
A fusion of the binding domain from the ClyS lysin to GFP was produced by inserting the following PCR products into pBAD24: The ClyS binding domain was amplified using primers ClyS-BD_5_XbaI and ClyS-BD_3_PstI and was inserted between the XbaI and PstI sites. GFP_mut2 was amplified using primers H6_GFP_5_EcoRI and GFP_3_KpnI (a hexahistidine tag was added on the 5′ primer) and inserted between the EcoRI and KpnI sites, yielding pAR160_GFP_ClyS-BD (55).
The binding domain of PlySs2 was amplified using primers PlySs2-BD_5_XbaI and PlySs2-BD_3_PstI and the resulting PCR product was inserted into the XbaI and PstI sites of pAR159 (a pBAD24-based plasmid, containing a hexahistidine-tagged GFP) (56), yielding pAR517_GFP_PlySs2-BD.
A control construct containing only a hexahistidine-tagged GFP was produced by amplifying the GFP_mut2 gene using primers H6_GFP_5_EcoRI and GFP_3_PstI and inserting the resulting PCR product into the EcoRI and PstI sites of pBAD24, yielding pAR518_GFP.
Expression and Purification of Lysibodies.
Lysibodies and similar constructs were produced in 293T cells using polyethylenimine (PEI) transient transfection method. For a 1 L reaction, 2 mg of the expression vector DNA and 500 μg helper vector were mixed in 10 mL optimem medium (Gibco, Life Technologies). We then mixed in 10 mL optimem medium containing 2.5 mg PEI, and the reaction was left at room temperature for 15 min. The mix was then added to 1 L DMEM containing 2 mM pyruvate and 10 mL Nutridoma-SP (Roche). Alternatively, 1 L FreeStyle 293 Expression Medium (Life Technologies) was used. The 293T cells were grown in 15-cm tissue culture plates to 70–80% confluence, washed with DPBS/modified containing calcium and magnesium (HyClone), and incubated with 20 mL transfection mix per plate for 6 d.
For N-terminal fusion lysibodies and monoclonal antibodies that do not contain a hexahistidine tag, medium from transfection plates was spun down, filtered, and proteins were precipitated with 60% ammonium sulfate at 4 °C overnight. Samples were centrifuged at 6,000 rpm using a Sorvall RC-5B centrifuge equipped with a GS-3 rotor. The protein pellet was suspended in 25 mL PBS containing two tablets of Complete protease inhibitor mixture tablet (Roche) and dialyzed against PBS using a membrane with 12–14 kDa cutoff (Spectrum Laboratories) for 24 h at 4 °C with three buffer changes. The dialyzed mix was centrifuged to remove precipitates and mixed end-over-end with protein G Sepharose beads (GE Healthcare) at 4 °C overnight. The protein G beads were loaded on a column and washed with 20 column volumes of PBS. The construct was eluted with 3 mL of a solution composed of 0.1 M glycine, pH 2.7, three times, and each eluted fraction was immediately neutralized with 450 μL of a solution composed of 1 M Tris, pH 9.0. Positive fractions were concentrated using an Amicon ultrafiltration device with a 10 kDa molecular mass cutoff membrane. The buffer was changed to DPBS/modified through three cycles of volume reduction and dilution in DPBS/modified. Protein concentration was determined according to absorbance at 280 nm, using a ND-1000 spectrophotometer (Nanodrop). The final product was stored in aliquots at –80 °C.
Supernatants of C-terminal fusion lysibodies were filtered through a 0.22-μm filter (Millipore) and loaded on a NiNTA column, calibrated with MCAC buffer (30 mM Tris, pH 7.4, 0.5 M NaCl, 10% glycerol). The column was washed thoroughly with MCAC buffer and MCAC containing 20 mM imidazole. Lysibodies were eluted with MCAC containing 150 mM imidazole, and positive fractions were processed as described above. Purification of GFP binding domain fusion protein was done using metal affinity chromatography as previously described (56).
Fluorescence Microscopy: Binding of Constructs to the Bacterial Surface.
Bacteria were fixed for 15 min at room temperature and 30 min on ice, using 2.6% paraformaldehyde, 0.012% glutaraldehyde, and 30 mM phosphate buffer, pH 7.4. Fixed cells were washed with PBS and attached to poly-l-lysine–coated cover glass. The cells were washed and blocked for 15 min with 10% normal goat serum. For bacteria not expressing protein A, lysibody was diluted to 2 μg/mL in PBS containing 2% BSA and 1% gelatin, and fluorescent conjugates were diluted 1:1,000. Bacteria were incubated with each for 1 h at room temperature. Microscopy using binding domain–GFP fusions was done in a similar manner, using PBS containing 2% BSA and 1% gelatin as blocking agent and dilution buffer. Microscopy studies using lysibodies and clinical S. aureus strains that express protein A were performed by blocking fixed cells with PBS containing 2% BSA and 1% gelatin, 10% goat serum, and 20% human serum sequentially. Lysibodies were conjugated to Rhodamine red according to manufacturer instructions (Thermo scientific), diluted to 5 μg/mL in PBS containing 2% BSA and 1% gelatin, and incubated with the cells for 1 h at room temperature. Slides were mounted in 50% glycerol and 0.1% p-phenylenediamine in PBS, pH 8. Phase microscopy was performed using a Nikon Eclipse E400 microscope, equipped with a Nikon 100×/1.25 oil immersion lens, and a Retiga EXi fast 1394 camera (QImaging). QCapture Pro version 5.1.1.14 software (QImaging) was used for image capture and processing. Deconvolution microscopy was performed using a DeltaVision image restoration microscope (Applied Precision/Olympus) equipped with CoolSnap QE cooled CCD camera (Photometrics). An Olympus 100×/1.40 N.A., UPLS Apo oil immersion objective was used in conjunction with a 1.5× optovar. The z stacks were taken at 0.15-μm intervals. Images were deconvolved using the SoftWoRx software (Applied Precision/DeltaVision) and corrected for chromatic aberrations.
Raw 264.7 Phagocytosis Assay.
S. aureus Newman/pCN57 was streaked on a BHI plate containing 10 μg/mL erythromycin and was grown at 37 °C for 1 d and then at 25 °C for an additional day. S. aureus cells from several separate colonies were scraped off the plate, washed once in PBS, and resuspended to a final OD600 of 0.3. Raw 264.7 macrophages were seeded at 5 × 105 cells per well of a 24-well plate 2 d before the experiment. The wells were washed with 1 mL PBS, and supplemented with 300 μL MEM medium without serum, constructs at different concentrations and 30 μL bacteria (roughly 1 × 107 cells); the plates were incubated at 37 °C 5% CO2 for 1 h. The wells were washed three times with PBS to remove extracellular bacteria, and the cells were fixed using 1 mL per well of 1% paraformaldehyde in PBS for 1 h at 4 °C. Each well was then washed with 1 mL PBS, and the cells were scraped off the plate in 200 μL PBS, using a 10 μL disposable inoculation loop. The cell suspension was transferred to a U-bottomed 96-well plate and analyzed using a C6 flow cytometer (BD-accuri) with the CFlow software. Forward and side scatter were used to gate the macrophage population. Macrophages displaying elevated fluorescence in the green channel were denoted as positive for phagocytosis of S. aureus.
For microscopy experiments, samples were processed in a similar manner using glass-bottomed wells. The cells were further stained with WGA Alexa Fluor 594 and analyzed by deconvolution microscopy to verify the presence of fluorescent S. aureus within macrophages.
S. aureus Killing by Macrophages.
S. aureus cultures were diluted 1:100 from an overnight culture and grown to an OD600 of 0.5. Cells were harvested, washed in DPBS, resuspended in DPBS to an OD600 of 0.3 (∼108 cells per mL), and further adjusted to 107 cells per mL. To each well of a U-bottom 96-well plate, 10 μg of the relevant lysibody were added in a final volume of 90 μL HBSS containing 0.1% gelatin, and then 10 μL of bacteria were added. The plate was placed on a shaker at 200 rpm at 4 °C for 1 h. Raw 264.7 macrophages were grown to semiconfluence, the medium was replaced with DPBS, and the cells were removed from the plate using a cell scraper. The cells were centrifuged and suspended in HBSS to a final concentration of 106 cells per mL. We added 100 μL of macrophages to each well of the 96-well plate, resulting in a 200 μL final volume, and the plate was placed on a shaker at 200 rpm at 37 °C for 3 h. The cfu quantification was performed by removing 20 μL of the mixture into 180 μL 0.2% saponin. Following 5 min of incubation, the mix was serially diluted 1:10 in distilled water and plated on BHI plates. Experiments were done in triplicates, with two technical repeats for each biological repeat.
Phagocytosis Assay: Murine Peritoneal Macrophage.
We intraperitoneally injected 5–6-wk-old female BALB/c mice with 1 mL of Brewer thioglycollate medium modified (BD). Mice were euthanized after 4–5 d, and the peritoneal cavity was washed with DPBS without calcium and magnesium to obtain macrophages. Macrophages were washed, and 5 × 105 cells were added to each well of a 24-well plate and incubated for 1 h at 37 °C 5% CO2. The wells were washed three times with DPBS to remove nonadherent cells and supplemented with 1 mL MEM medium without serum. Phagocytosis assays were performed as described for the Raw 264.7 cells; however, detachment of adherent cells following fixation was done using 250 μL 0.25% trypsin in PBS, pH 7.2, 0.1% EDTA, for 30 min at 37 °C, followed by gentle pipetting with a 1-mL pipette tip.
Preparation of Human Complement, Adsorbed on S. aureus.
Blood was obtained from a healthy human donor by venipuncture and was immediately placed on ice for 2 h until clotting occurred. Clots were removed by centrifugation, and the serum was passed through a 0.22-μm filter. EDTA was added to a final concentration of 25 mM to prevent complement activation, and the serum was adsorbed on S. aureus Newman/pCN57. For each milliliter of serum adsorbed, a washed pellet from 10 mL overnight culture and a washed pellet from 10 mL late logarithmic stage culture were used, to account for possible variability of surface epitopes between growth stages. The serum was rotated end-over-end with S. aureus cells for 30 min at 4 °C, centrifuged to remove cells, and filtered through a 0.22-μm filter. The serum was then dialyzed against 1.5 L of a solution composed of 5 mM Hepes containing 0.9% NaCl, pH 7.4, using a membrane with a cutoff limit of 12–14 kDa, for 16 h with two buffer changes. The serum was then frozen in liquid nitrogen as single-use aliquots and stored at –80 °C until use.
Complement Fixation: Sample Preparation for Microscopy.
S. aureus Wood 46 (protein A negative) was grown on a BHI plate for 1 d at 37 °C and then 1 d at 25 °C. Several separate colonies were scraped off the plates and suspended in PBS to a final OD600 of 1.0. The cells were attached to acid-washed poly-l-lysine–coated cover slides. Lysibodies and other constructs were diluted to a final concentration of 1 mg/mL in DPBS containing calcium and magnesium (HyClone), and 10 μL were added to each slide and incubated at room temperature for 1 h. The cells were washed three times with PBS and once with DGHB (5 mM Hepes, 71 mM NaCl, 0.15 mM CaCl2, 0.5 mM MgCl2, 2.5% glucose, 0.1% gelatin, pH 7.4) and incubated for 20 min at 37 °C with 30 μL DGHB containing 0.5% human serum that was adsorbed on S. aureus. The cells were washed thoroughly with PBS and fixed with 50 μL 2.6% paraformaldehyde in PBS for 1 h at 4 °C. The cells were then washed again with PBS and blocked with PBS containing 2% BSA and 1% gelatin overnight at 4 °C. The slides were washed with PBS and incubated with 10 μL rabbit anti-C3 diluted 1:500, followed by three PBS washes, and then incubated with 10 μL goat anti-rabbit Alexa Fluor 594 conjugate diluted 1:1,000 and 1 μg/mL DAPI; incubation steps were 1 h at room temperature each. The slides were mounted and imaged as described above.
For microscopy on Newman/pCN57 (protein A positive, expressing cytoplasmic GFP), the cells were blocked following fixation using PBS containing 2% BSA and 1% gelatin, followed by heat-inactivated goat and human sera.
Complement Fixation: Sample Preparation for Flow Cytometry.
S. aureus Newman/pCN57 was grown on a BHI plate for 1 day at 37 °C and for another day at 25 °C. Several separate colonies were scraped off the plates and suspended in PBS to a final OD600 of 1.0. We mixed 30 μL bacteria with lysibodies or controls at various concentrations, and the final volume was adjusted to 200 μL with PBS. The cells were rotated at 4 °C for 2 h and washed with 0.5 mL saline and then 100 μL GVB (gelatin veronal buffer; Sigma). The cells were suspended in 300 μL 3% human complement (adsorbed on S. aureus) in GVB, and the tubes were rotated at 37 °C for 15 min. The samples were then immediately placed on ice, and EDTA was added to a final concentration of 20 mM to stop complement fixation. The samples were washed twice with PBS and fixed with 250 μL 2.6% paraformaldehyde in PBS (phosphate adjusted to 40 mM, pH 7.4) for 1 h at 4 °C. The samples were then washed twice with PBS, and the pellet was blocked with 100 μL PBS containing 2% BSA and 1% gelatin for 20 min. We added 100 μL 10% heat-inactivated goat serum for an additional 20 min. Then, 10 μL heat-inactivated human serum was added to each sample for an additional 20 min, to block protein A. The cells were then washed with PBS, and each tube was suspended in 100 μL rabbit anti-C3 antibody diluted 1:2,000 in PBS containing 2% BSA and 1% gelatin and rotated for 1 h at room temperature. The cells were washed with PBS, suspended in 100 μL goat anti-rabbit Alexa Fluor 594 conjugate diluted 1:2,000 in PBS containing 2% BSA and 1% gelatin, and the tubes were rotated for 1 h at room temperature. The cells were then washed with PBS and resuspended in 200 μL PBS. Samples were analyzed using a BD-Accuri C6 flow cytometer and the CFlow and FlowJo softwares. Unlabeled and monolabeled samples were used to calibrate compensation values. Gating was done on GFP-positive cells to exclude non-S. auereus particles, and C3b signal distribution was determined.
Preparation of Human Peripheral Polymorphonuclear Leukocytes, HL-60 Neutrophils, and FITC-Labeled S. aureus for Phagocytosis Assays.
HL-60 neutrophils were propagated and differentiated as described by Romero-Steiner et al. (52). Primary human neutrophils were isolated from the blood of healthy volunteers collected in tubes containing acid citrate dextrose (ACD). For each 9 mL of blood, 4.5 mL of 6% dextran (molecular mass ∼100,000 kDa) and 0.9% NaCl were added. The tubes were left stationary at room temperature for 30 min to allow red blood cells to settle, and the top layer was collected. The cells were centrifuged, and residual red blood cells were lysed for 30 s in 0.2% NaCl, and the solution was supplemented by an equal volume of 1.6% NaCl. The cells were centrifuged, and the process was repeated two more times. The cells were then suspended in 10 mL DPBS (without calcium and magnesium), and 3 mL Ficoll–Hypaque solution (density, 1.077 g/L) was layered at the bottom of the tube. The cells were centrifuged at 900 g for 20 min at 20 °C, and the cell pellet was washed once with DPBS without calcium and magnesium and suspended in HBSS containing 0.1% gelatin at a final concentration of 1 × 108 cells per mL.
For phagocytosis assays, S. aureus strains were grown on BHI plates for 1 d at 37 °C and for another day at 25 °C. Several separate colonies were scraped off the plates and resuspended in PBS to a final OD600 of 1.0 and fixed for 1 h at 4 °C with 2.6% paraformaldehyde in PBS (phosphate concentration adjusted to 40 mM, pH 7.4). Bacterial cells were labeled with FITC (Sigma) according to manufacturer instructions and washed thoroughly. Cells were suspended in PBS containing 14% glycerol and frozen in single-use aliquots at –80 °C.
Phagocytosis of S. aureus by HL-60 Neutrophils, and Human Peripheral Polymorphonuclear Leukocytes.
Neutrophils were adjusted to 1 × 108 cells per mL, and FITC-labeled S. aureus cells were adjusted to 5 × 107 cells per mL in HBSS (Gibco 14025) containing 0.1% gelatin. Phagocytosis assay was modified from Nanra et al. (57). To each well of a round-bottomed 96-well plate were added 60 μL HBSS containing 0.1% gelatin, 10 μL lysibody or control diluted in HBSS containing 0.1% gelatin, and 10 μL diluted bacteria. The plate was shaken at 200 rpm, 4 °C for 1 h. Then, 10 μL of 5% S. aureus-adsorbed complement and 10 μL HL-60 cells or primary human neutrophils adjusted to 1 × 108 cells per mL in HBSS containing 0.1% gelatin were added to each well, and the plate was shaken at 200 rpm at 37 °C for 1 h. Cells were then fixed in 2.6% paraformaldehyde, 30 mM sodium phosphate, pH 7.4, for 1 h at 4 °C. The cells were washed, suspended in 200 μL PBS, and analyzed using a C6 flow cytometer (BD-accuri). Forward and side scatter were used to gate the neutrophil population, and cells displaying an increase in fluorescence were denoted as positive for phagocytosis of S. aureus. For microscopy experiments, samples were processed in a similar manner and stained with WGA Alexa Fluor 594. Cells were then mounted on microscope slides and analyzed by deconvolution microscopy to verify the presence of fluorescent S. aureus within neutrophils.
Mouse Kidney Abscess and Bacteremia Models.
The mouse kidney abscess model was modified from Daniel et al. (35). Five-wk-old BALB/c female mice (Jackson Laboratories) were each injected intraperitoneally with 1 mg lysibody or control, in a total volume of 500 μL. S. aureus strain USA600 (MRSA, VISA) was diluted 1:100 in BHI from an overnight culture and grown at 37 °C with shaking at 200 rpm to an OD600 of 0.5. Cells were harvested, washed with saline, and suspended in saline to an OD600 of 1.0. Bacteria were diluted in saline and adjusted to 2.5 × 106 cfu per mouse (injection volume, 500 μL) in saline 5% hog gastric mucin (Sigma). Injection of mice was performed 24 h following injection of lysibodies. Actual injected cfu was determined through plating. Mouse viability was monitored every 24 h, and after 4 d, surviving mice were euthanized. Both kidneys were dissected and ground in 1 mL 0.5% saponin. Samples were serially diluted and streaked on BHI plates for quantification of bacterial load.
Bacteremia models were performed in a similar manner with the following changes: Each mouse was injected intraperitoneally with 0.3 mg lysibody or PBS and 24 h later with 2 × 106 cfu of a mouse-adapted MW2 USA400 S. aureus strain in saline containing 5% hog gastric mucin (35). Mouse viability was monitored for 8 d, at which point surviving mice were euthanized.
In all experiments, animals were of the same age, sex, and genetic background and were randomly assigned to experimental groups. Sample size was chosen to provide adequate statistical power. Researchers were not blinded to the experimental group identity.
Statistical Analysis.
Significance for all in vitro phagocytosis assays was determined using two-tailed Student’s t test. Bacterial loads in the renal abscess model were analyzed using the two-tailed Mann–Whitney test for statistical significance. For statistical analysis, mice that succumbed to infection were assigned a kidney load value equivalent to the live mouse with the highest load in the experimental group. Statistical significance for mouse bacteremia models was determined using the Gehan–Breslow–Wilcoxon test. Data analysis was done using Prism version 5.0c (GraphPad Software).
Data Availability.
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Acknowledgments
We thank Svetlana Mazel and members of the Rockefeller University Flow Cytometry Resource Center for their help and advice; Alison North of the Rockefeller University Bio-Imaging Resource Center for advice regarding fluorescent microscopy; Yevgeniy Mayr for his help with the characterization of certain constructs; and Adam Vigil, Michael Wittekind, Ray Schuch, and Jimmy Rotolo for suggestions and useful discussions, as well as the gift of 1K8 antibody. This work was supported in part by funds from ContraFect Corp and Rockefeller University.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1619249114/-/DCSupplemental.
References
- 1.Casadevall A, Dadachova E, Pirofski LA. Passive antibody therapy for infectious diseases. Nat Rev Microbiol. 2004;2:695–703. doi: 10.1038/nrmicro974. [DOI] [PubMed] [Google Scholar]
- 2.Bebbington C, Yarranton G. Antibodies for the treatment of bacterial infections: Current experience and future prospects. Curr Opin Biotechnol. 2008;19:613–619. doi: 10.1016/j.copbio.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 3.DiGiandomenico A, Sellman BR. Antibacterial monoclonal antibodies: The next generation? Curr Opin Microbiol. 2015;27:78–85. doi: 10.1016/j.mib.2015.07.014. [DOI] [PubMed] [Google Scholar]
- 4.Weidenmaier C, Peschel A. Teichoic acids and related cell-wall glycopolymers in Gram-positive physiology and host interactions. Nat Rev Microbiol. 2008;6:276–287. doi: 10.1038/nrmicro1861. [DOI] [PubMed] [Google Scholar]
- 5.Ellwood DC. The wall content and composition of Bacillus substilis var. niger grown in a chemostat. Biochem J. 1970;118:367–373. doi: 10.1042/bj1180367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Reichmann NT, Gründling A. Location, synthesis and function of glycolipids and polyglycerolphosphate lipoteichoic acid in Gram-positive bacteria of the phylum Firmicutes. FEMS Microbiol Lett. 2011;319:97–105. doi: 10.1111/j.1574-6968.2011.02260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown S, Santa Maria JP, Jr, Walker S. Wall teichoic acids of gram-positive bacteria. Annu Rev Microbiol. 2013;67:313–336. doi: 10.1146/annurev-micro-092412-155620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matias VR, Beveridge TJ. Native cell wall organization shown by cryo-electron microscopy confirms the existence of a periplasmic space in Staphylococcus aureus. J Bacteriol. 2006;188:1011–1021. doi: 10.1128/JB.188.3.1011-1021.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mond JJ, Lees A, Snapper CM. T cell-independent antigens type 2. Annu Rev Immunol. 1995;13:655–692. doi: 10.1146/annurev.iy.13.040195.003255. [DOI] [PubMed] [Google Scholar]
- 10.Snapper CM, Mond JJ. A model for induction of T cell-independent humoral immunity in response to polysaccharide antigens. J Immunol. 1996;157:2229–2233. [PubMed] [Google Scholar]
- 11.Astronomo RD, Burton DR. Carbohydrate vaccines: Developing sweet solutions to sticky situations? Nat Rev Drug Discov. 2010;9:308–324. doi: 10.1038/nrd3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ada G, Isaacs D. Carbohydrate-protein conjugate vaccines. Clin Microbiol Infect. 2003;9:79–85. doi: 10.1046/j.1469-0691.2003.00530.x. [DOI] [PubMed] [Google Scholar]
- 13.Loessner MJ, Kramer K, Ebel F, Scherer S. C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol Microbiol. 2002;44:335–349. doi: 10.1046/j.1365-2958.2002.02889.x. [DOI] [PubMed] [Google Scholar]
- 14.Fischetti VA. Bacteriophage endolysins: A novel anti-infective to control Gram-positive pathogens. Int J Med Microbiol. 2010;300:357–362. doi: 10.1016/j.ijmm.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pastagia M, Schuch R, Fischetti VA, Huang DB. Lysins: The arrival of pathogen-directed anti-infectives. J Med Microbiol. 2013;62:1506–1516. doi: 10.1099/jmm.0.061028-0. [DOI] [PubMed] [Google Scholar]
- 16.Biswas R, et al. Activity of the major staphylococcal autolysin Atl. FEMS Microbiol Lett. 2006;259:260–268. doi: 10.1111/j.1574-6968.2006.00281.x. [DOI] [PubMed] [Google Scholar]
- 17.Tong SY, Davis JS, Eichenberger E, Holland TL, Fowler VG., Jr Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev. 2015;28:603–661. doi: 10.1128/CMR.00134-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klevens RM, et al. Active Bacterial Core surveillance (ABCs) MRSA Investigators Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA. 2007;298:1763–1771. doi: 10.1001/jama.298.15.1763. [DOI] [PubMed] [Google Scholar]
- 19.Boucher HW, Corey GR. Epidemiology of methicillin-resistant Staphylococcus aureus. Clin Infect Dis. 2008;46:S344–S349. doi: 10.1086/533590. [DOI] [PubMed] [Google Scholar]
- 20.Centers for Disease Control and Prevention 2014 Active Bacterial Core Surveillance Report, Emerging Infections Program Network, Methicillin Resistant Staphylococcus aureus. Available at https://www.cdc.gov/abcs/reports-findings/survreports/mrsa14.html. Accessed September 27, 2016.
- 21.McCaig LF, McDonald LC, Mandal S, Jernigan DB. Staphylococcus aureus-associated skin and soft tissue infections in ambulatory care. Emerg Infect Dis. 2006;12:1715–1723. doi: 10.3201/eid1211.060190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noskin GA, et al. National trends in Staphylococcus aureus infection rates: Impact on economic burden and mortality over a 6-year period (1998-2003) Clin Infect Dis. 2007;45:1132–1140. doi: 10.1086/522186. [DOI] [PubMed] [Google Scholar]
- 23.DeLeo FR, Chambers HF. Reemergence of antibiotic-resistant Staphylococcus aureus in the genomics era. J Clin Invest. 2009;119:2464–2474. doi: 10.1172/JCI38226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fattom AI, Horwith G, Fuller S, Propst M, Naso R. Development of StaphVAX, a polysaccharide conjugate vaccine against S. aureus infection: From the lab bench to phase III clinical trials. Vaccine. 2004;22:880–887. doi: 10.1016/j.vaccine.2003.11.034. [DOI] [PubMed] [Google Scholar]
- 25.Fowler VG, et al. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery: A randomized trial. JAMA. 2013;309:1368–1378. doi: 10.1001/jama.2013.3010. [DOI] [PubMed] [Google Scholar]
- 26.Jansen KU, Girgenti DQ, Scully IL, Anderson AS. Vaccine review: “Staphyloccocus aureus vaccines: Problems and prospects”. Vaccine. 2013;31:2723–2730. doi: 10.1016/j.vaccine.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 27.Fowler VG, Jr, Proctor RA. Where does a Staphylococcus aureus vaccine stand? Clinical Microbiology and Infection. 2014;20:66–75. doi: 10.1111/1469-0691.12570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bournazos S, Ravetch JV. Fcγ receptor pathways during active and passive immunization. Immunol Rev. 2015;268:88–103. doi: 10.1111/imr.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baba T, Schneewind O. Targeting of muralytic enzymes to the cell division site of Gram-positive bacteria: Repeat domains direct autolysin to the equatorial surface ring of Staphylococcus aureus. EMBO J. 1998;17:4639–4646. doi: 10.1093/emboj/17.16.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fischer W, Rösel P, Koch HU. Effect of alanine ester substitution and other structural features of lipoteichoic acids on their inhibitory activity against autolysins of Staphylococcus aureus. J Bacteriol. 1981;146:467–475. doi: 10.1128/jb.146.2.467-475.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gründling A, Schneewind O. Synthesis of glycerol phosphate lipoteichoic acid in Staphylococcus aureus. Proc Natl Acad Sci USA. 2007;104:8478–8483. doi: 10.1073/pnas.0701821104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yamada S, et al. An autolysin ring associated with cell separation of Staphylococcus aureus. J Bacteriol. 1996;178:1565–1571. doi: 10.1128/jb.178.6.1565-1571.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson DC, et al. Endolysins as antimicrobials. Adv Virus Res. 2012;83:299–365. doi: 10.1016/B978-0-12-394438-2.00007-4. [DOI] [PubMed] [Google Scholar]
- 34.Kontermann RE. Dual targeting strategies with bispecific antibodies. MAbs. 2012;4:182–197. doi: 10.4161/mabs.4.2.19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daniel A, et al. Synergism between a novel chimeric lysin and oxacillin protects against infection by methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2010;54:1603–1612. doi: 10.1128/AAC.01625-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gilmer DB, Schmitz JE, Euler CW, Fischetti VA. Novel bacteriophage lysin with broad lytic activity protects against mixed infection by Streptococcus pyogenes and methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother. 2013;57:2743–2750. doi: 10.1128/AAC.02526-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schuch R, Nelson D, Fischetti VA. A bacteriolytic agent that detects and kills Bacillus anthracis. Nature. 2002;418:884–889. doi: 10.1038/nature01026. [DOI] [PubMed] [Google Scholar]
- 38.Yang J, et al. The I-TASSER Suite: Protein structure and function prediction. Nat Methods. 2015;12:7–8. doi: 10.1038/nmeth.3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nordenfelt P, et al. Antibody orientation at bacterial surfaces is related to invasive infection. J Exp Med. 2012;209:2367–2381. doi: 10.1084/jem.20120325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spaan AN, Surewaard BG, Nijland R, van Strijp JA. Neutrophils versus Staphylococcus aureus: A biological tug of war. Annu Rev Microbiol. 2013;67:629–650. doi: 10.1146/annurev-micro-092412-155746. [DOI] [PubMed] [Google Scholar]
- 41.Weidenmaier C, et al. Lack of wall teichoic acids in Staphylococcus aureus leads to reduced interactions with endothelial cells and to attenuated virulence in a rabbit model of endocarditis. J Infect Dis. 2005;191:1771–1777. doi: 10.1086/429692. [DOI] [PubMed] [Google Scholar]
- 42.Fischetti VA. Bacteriophage lysins as effective antibacterials. Curr Opin Microbiol. 2008;11:393–400. doi: 10.1016/j.mib.2008.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Loeffler JM, Nelson D, Fischetti VA. Rapid killing of Streptococcus pneumoniae with a bacteriophage cell wall hydrolase. Science. 2001;294:2170–2172. doi: 10.1126/science.1066869. [DOI] [PubMed] [Google Scholar]
- 44.Dreisbach A, et al. Profiling the surfacome of Staphylococcus aureus. Proteomics. 2010;10:3082–3096. doi: 10.1002/pmic.201000062. [DOI] [PubMed] [Google Scholar]
- 45.Ziebandt AK, et al. Proteomics uncovers extreme heterogeneity in the Staphylococcus aureus exoproteome due to genomic plasticity and variant gene regulation. Proteomics. 2010;10:1634–1644. doi: 10.1002/pmic.200900313. [DOI] [PubMed] [Google Scholar]
- 46.Golubchik T, et al. Within-host evolution of Staphylococcus aureus during asymptomatic carriage. PLoS One. 2013;8:e61319. doi: 10.1371/journal.pone.0061319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Laabei M, et al. Evolutionary trade-offs underlie the multi-faceted virulence of Staphylococcus aureus. PLoS Biol. 2015;13:e1002229. doi: 10.1371/journal.pbio.1002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Groot AS, Terry F, Cousens L, Martin W. Beyond humanization and de-immunization: Tolerization as a method for reducing the immunogenicity of biologics. Expert Rev Clin Pharmacol. 2013;6:651–662. doi: 10.1586/17512433.2013.835698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pozzi C, et al. Phagocyte subsets and lymphocyte clonal deletion behind ineffective immune response to Staphylococcus aureus. FEMS Microbiol Rev. 2015;39:750–763. doi: 10.1093/femsre/fuv024. [DOI] [PubMed] [Google Scholar]
- 50.Giersing BK, Dastgheyb SS, Modjarrad K, Moorthy V. Status of vaccine research and development of vaccines for Staphylococcus aureus. Vaccine. 2016;34:2962–2966. doi: 10.1016/j.vaccine.2016.03.110. [DOI] [PubMed] [Google Scholar]
- 51.American Veterinary Medical Association . AVMA Guidelines for the Euthanasia of Animals: 2013 Edition. American Veterinary Medical Association; Schaumburg, IL: 2013. [Google Scholar]
- 52.Romero-Steiner S, et al. Standardization of an opsonophagocytic assay for the measurement of functional antibody activity against Streptococcus pneumoniae using differentiated HL-60 cells. Clin Diagn Lab Immunol. 1997;4:415–422. doi: 10.1128/cdli.4.4.415-422.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Niro R, et al. Rapid generation of rotavirus-specific human monoclonal antibodies from small-intestinal mucosa. J Immunol. 2010;185:5377–5383. doi: 10.4049/jimmunol.1001587. [DOI] [PubMed] [Google Scholar]
- 54.Lee CM, Iorno N, Sierro F, Christ D. Selection of human antibody fragments by phage display. Nat Protoc. 2007;2:3001–3008. doi: 10.1038/nprot.2007.448. [DOI] [PubMed] [Google Scholar]
- 55.Dhingra N, et al. Attenuated neutrophil axis in atopic dermatitis compared to psoriasis reflects TH17 pathway differences between these diseases. J Allergy Clin Immunol. 2013;132:498–501. doi: 10.1016/j.jaci.2013.04.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Raz A, Talay SR, Fischetti VA. Cellular aspects of the distinct M protein and SfbI anchoring pathways in Streptococcus pyogenes. Mol Microbiol. 2012;84:631–647. doi: 10.1111/j.1365-2958.2012.08047.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nanra JS, et al. Heterogeneous in vivo expression of clumping factor A and capsular polysaccharide by Staphylococcus aureus: Implications for vaccine design. Vaccine. 2009;27:3276–3280. doi: 10.1016/j.vaccine.2009.01.062. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article (and its supplementary information files).