

Summary
Primary antibody deficiencies (PAD) constitute the majority of all primary immunodeficiency diseases (PID) and immunoglobulin replacement forms the mainstay of therapy for many patients in this category. Secondary antibody deficiencies (SAD) represent a larger and expanding number of patients resulting from the use of a wide range of immunosuppressive therapies, in particular those targeting B cells, and may also result from renal or gastrointestinal immunoglobulin losses. While there are clear similarities between primary and secondary antibody deficiencies, there are also significant differences. This review describes a practical approach to the clinical, laboratory and radiological assessment of patients with antibody deficiency, focusing on the factors that determine whether or not immunoglobulin replacement should be used. The decision to treat is more straightforward when defined diagnostic criteria for some of the major PADs, such as common variable immunodeficiency disorders (CVID) or X‐linked agammaglobulinaemia (XLA), are fulfilled or, indeed, when there is a very low level of immunoglobulin production in association with an increased frequency of severe or recurrent infections in SAD. However, the presentation of many patients is less clear‐cut and represents a considerable challenge in terms of the decision whether or not to treat and the best way in which to assess the outcome of therapy. This decision is important, not least to improve individual quality of life and reduce the morbidity and mortality associated with recurrent infections but also to avoid inappropriate exposure to blood products and to ensure that immunoglobulin, a costly and limited resource, is used to maximal benefit.
Keywords: antibody deficiency, common variable immunodeficiency disorders (CVID), intravenous immunoglobulin (IVIg), prophylactic antibiotics, secondary antibody deficiency (SAD), subcutaneous immunoglobulin (SCIg)
OTHER ARTICLES PUBLISHED IN THIS REVIEW SERIES
Clinical challenges in the management of patients with B cell immunodeficiencies. Clinical and Experimental Immunology 2017, 188: 323–5.
The role of genomics in common variable immunodeficiency disorders. Clinical and Experimental Immunology 2017, 188: 326–32.
Progressive multi‐focal leucoencephalopathy – driven from rarity to clinical mainstream by iatrogenic immunodeficiency. Clinical and Experimental Immunology 2017, 188: 342–52.
Considerations for dosing immunoglobulin in obese patients. Clinical and Experimental Immunology 2017, 188: 353–62.
Chronic norovirus infection and common variable immunodeficiency. Clinical and Experimental Immunology 2017, 188: 363–70.
Introduction
Deficiencies in antibody production are the most frequent, clinically significant, primary immune deficiencies 1; many of them are relatively well characterized. Secondary antibody failure may develop as a consequence of various other diseases, but also as an unwanted side effect of a range of medications. Despite different pathogenesis the clinical manifestations are usually similar, including recurrent or complicated infections caused by encapsulated bacteria, predominantly of the upper and/or lower respiratory tract. Treatment approaches in such conditions are different. In secondary antibody failure, elimination of the causal mechanism is sometimes possible. In patients with mild manifestations, the ‘watch and wait approach’ in terms of immunoglobulin (Ig) replacement may be advisable, as antibiotic prophylaxis may improve the patient's health. In some cases, Ig replacement treatment is the most effective treatment, and we will focus upon how to decide when and in whom to start replacement Ig therapy.
Types of antibody failure
The situation is relatively easy in patients with well‐defined primary antibody failure conditions for which there is an internationally accepted classification scheme 2, 3. With the exception of most cases of selective IgA deficiency and transient hypogammaglobulinaemia of infancy, patients with diseases defined in the group of ‘predominantly antibody deficiencies’ 2 are advised to start Ig replacement. Similarly, this is the case for patients falling into the life‐threatening categories of severe combined immune deficiencies (SCID) and combined immune deficiencies (CID) associated with or without syndromic features (such as Wiskott–Aldrich syndrome, although not always in ataxia telangiectasia or Nijmegen breakage syndrome).
Common variable immunodeficiency disorders (CVID) and unclassified primary antibody deficiencies
From the practical viewpoint, the most significant problem with respect to the decision to initiate Ig replacement therapy (IGRT) may occur in patients with reduced, but not absent, serum levels of IgG in the range of of 4–6g/l with or without low IgA. Those with IgG levels under 4 g/l are generally more likely to require IGRT, although even here there will be exceptions. The spectrum of clinical or laboratory findings in CVID is well known to clinical immunologists, has defined diagnostic criteria ICON 4, ESID 5 and Ameratunga 6 which, generally speaking, include decreases of IgG and at least one isotype (IgA or IgM) as well as disturbed specific antibody responses after vaccination, in conjunction with severe or recurrent bacterial infections. Fulfilling CVID diagnostic criteria thus generally implies the initiation of Ig treatment 4. However, there are patients with decreased Ig levels and disturbed specific antibody responses who do not suffer from recurrent or severe respiratory infections (some may also have cytopenias, granulomas or lymphoproliferation). The benefit of starting expensive replacement Ig therapy in these patients, or in asymptomatic patients with low serum IgG alone, is more controversial 7. Over‐dependency on laboratory parameters such as serum Ig determinations can lead to confusion, as can reduced specific immune responses in the presence of mild clinical symptoms 8. The current recommendation is to follow such individuals in clinic every 6–12 months. It is important to watch for the development of bacterial infections 4, advise prompt antibiotics in such an event and to bring the clinic visit forward so that Ig therapy can be discussed. There is no clinical trial evidence that the CVID complications themselves, such as enteropathy or lymphoproliferation, respond to/are affected by replacement Ig therapy, although somewhat higher doses are required to prevent bacterial infections in such patients 9. Over time, these complications have come to the fore as a major therapeutic challenge while infections have declined, due presumably to a combination of improved Ig and antibiotic treatment 10. There is a clear need for prospective data on large multi‐centre cohorts of patients who have a limited infection burden at presentation along with decreased Ig levels and variably impaired specific antibodies, to risk stratify and inform treatment decisions.
In some patients CVID is observed to develop over time, most frequently from IgA deficiency 11, 12. In these patients, the decline in IgG appears to be a gradual process, usually over months to years, and is not necessarily accompanied by clinical manifestations of immunodeficiency at the onset of the decline. When to test immunize such patients is difficult, but should be performed at a time most likely to assist clinical decision‐making regarding Ig replacement therapy (IGRT), and certainly if clinical features associated with PID or bacterial infections become a feature. It is important to follow the levels of specific antibodies which may be transient (memory phenotype Table 1) or absent.
Table 1.
Classification of pneumococcal serotype failure to vaccination with (PPV) adapted from 13
| Phenotype | Memory | Mild | Moderate | Severe |
|---|---|---|---|---|
|
Adults
(6–65 years) |
Initially > 70% of serotypes > 0·35 µg/ml but response lost in 6 months | Multiple serotypes < 0·35 µg/ml or inability to increase > 70% of serotypes by > twofold | < 70% of serotypes > 0·35 µg/ml but protective titre to 3 or more serotypes | Protective titres to no more than 2 serotypes and titre if present is low (< 0·35 µg/ml) |
|
Children
(2–5 years) |
Initially > 50% of serotypes > 0·35 µg/ml but response lost in 6 months | Multiple serotypes < 0·35 µg/ml or inability to increase > 70% of serotypes by > twofold | < 50% of serotypes > 0·35 µg/ml but protective titre to 3 or more serotypes | Protective titres to no more than 2 serotypes and titre if present is low (< 0·35 µg/ml) |
Mild phenotype assumes pre‐vaccination titres are less than threshold levels (< 0·35 µg/ml) and there may be a response to a second Pneumovax II booster. All phenotypes assume a history of infection. (PPV = pneumococcal polysaccharide vaccine.
Specific antibody deficiency (SPAD)
Even patients with normal serum Ig levels may have disturbed specific antibody responses, in association with a recurrent predominantly sinopulmonary pattern of infection – a condition called specific antibody deficiency (SPAD) 14. IGRT can prevent bacterial infections and is probably more beneficial than prophylactic antibiotics, although there are no reliable data for this rare condition; some of these patients may progress to develop classical primary immunodeficiencies (PIDs) and should be followed carefully. Monitoring of IGRT is complicated, as endogenous IgG levels are normal and it may be therefore be helpful to monitor specific antibody levels.
Secondary antibody deficiencies (SAD) 15
Compared to primary immune deficiencies, the situation in secondary Ig deficiencies is even more complicated. Early studies in the 1980s and 1990s showed that Ig replacement is effective in selected patients with plateau phase myeloma 16 or chronic lymphocytic leukaemia 17, although the data are old now, and new trials in patients receiving more modern anti‐malignancy therapies are required 18, 19.
SAD has a wide range of causes, including B cell lymphoproliferative disease, protein loss via the gut and renal tract, abnormal lymphatic circulation, increased catabolism and drugs (summarized in Table 2). The increased use of B cell targeted therapies such as rituximab, especially where it is used as maintenance and/or combination therapy, will result in more patients with SAD 50, 51. The growth in therapies in this area is not restricted to rituximab and includes chimeric antigen receptor‐expressing T cell therapy (CART) 26, 27, atacicept, which is a humanized TACI receptor fusion protein (transmembrane activator and calcium modulator and cyclophilin–ligand interactor: TACI) 40, 41, and imatinib (a selective tyrosine kinase inhibitor) 39. Prospective information regarding ibrutinib (a Btk inhibitor) and idelalisib (a phosphoinositide 3‐kinase delta inhibitor) in terms of Ig levels and infections will also be needed. Many of these drug targets are well known to immunologists, as mutations in the corresponding genes have been shown to cause PID. This explosion in new therapies and improved survival is likely to herald a major growth in secondary antibody deficiencies. More conventional immunosuppressive drugs may also cause significant antibody deficiencies, and there is new evidence that selected patients receiving therapy for solid organ transplantation benefit from IGRT 52.
Table 2.
Causes of secondary antibody deficiency
| B cell lymphoproliferative disease | Protein loss | Lymphatic circulation | Increased catabolism | Drug‐related |
|---|---|---|---|---|
| Chronic lymphocytic leukaemia (CLL) 20 | Renal loss Nephrotic syndrome | Intestinal lymphangiectasia | Myotonic dystrophy 21, 22 |
Therapies targeting B cells Rituximab 23, 24, 25 CD19‐targeted chimeric antigen receptor T cells (CART) 26, 27 |
|
Non‐Hodgkins lymphoma, Hodgkins lymphoma, Diffuse large B cell lymphoma Follicular lymphoma Mantle cell lymphoma Marginal zone lymphoma Burkitt's lymphoma 28 |
Gastrointestinal loss Crohn's disease Ulcerative colitis Intestinal lymphangectasia Turner's syndrome Noonan's syndrome Klippel–Trenauny syndrome Hennekam syndrome Coeliac disease Enteric infection Menetrier's disease 29, 30, 31 |
Proteus syndrome 32
Yellow nail syndrome Noonan's syndrome |
Mycophenolate 33 Cyclophosphamide 25
Corticosteroids 34 Sulphasalazine Gold D‐penicillamine Chlorpromazine Methotrexate Clozapine 28, 35, 36, 37, 38 Imatinib 39 Atacicept 40, 41 Fludarabine and other chemotherapy |
|
|
Multiple myeloma Smouldering myeloma Monoclonal gammopathy of undetermined significance (MGUS) 42, 43, 44, 45 |
Chylothorax 46, 47 |
Anti‐epileptic medication Phenytoin Carbamazepine Sodium valproate Lamotrigine 48, 49 |
The main categories underlying secondary antibody deficiency (SAD) are shown, which include causes related to decreased production as well as increased loss or catabolism. The individual causes are very numerous and patients may therefore present with SAD from a broad range of clinical specialities, which would include Haematology, Oncology, Medicine, Rheumatology, Nephrology, primary care and others. The specialities with the highest levels of SAD are those where the disease itself (e.g. CLL, myeloma) and/or its treatment are associated with antibody deficiency. Similarly, combination and/or prolonged immunosuppressive therapy as required, for example, in transplantation, granulomatosis with polyangiitis (GPA) or neuromyelitis optica (NMO) will have a higher rate of SAD. Not shown in the Table are the transient causes of SAD, which occur in relation to interventions where intravenous fluid or blood may be needed in relation to surgery or intensive‐care settings 28.
The situation regarding the IGRT decision is different in patients where Ig deficiency is caused by conditions where it may be possible to address the cause such as in protein‐losing states or potentially in the treatment of haematological malignancy (Table 2). In SAD, given the limited evidence for many situations when antibody deficiency and impaired specific antibody production are associated with recurrent severe infection, a pragmatic approach has been suggested to assess response to prophylactic antibiotics, and if this fails then a trial of IGRT may be appropriate 15. The use of IGRT in protein‐losing states is problematic and, again, antibiotic prophylaxis would be indicated ahead of IGRT. If IGRT is required, the pharmacokinetics of the subcutaneous route may be preferable, as it avoids the peak levels and longer cycle duration of intravenous Ig (IVIg) 53, 54, 55. This would reduce infections with improved overall antibody levels afforded by more frequent dosing and limiting the higher rates of loss associated with the IgG peak levels following IVIg.
Patient assessment
Patient assessment involves gathering clinical, radiological and laboratory information, and begins with the clinical history focusing on the infection history. Obtaining a high level of detail at this stage informs treatment decisions and documents the baseline to enable objective measurements of the outcome of subsequent interventions. Such information includes the onset of infections, site, duration, seasonality, frequency and severity, including the requirement for oral antibiotics, intravenous antibiotics or hospital admission. It is also helpful to know the type(s) of pathogen, and it may be necessary to seek this information from primary care or other medical records for verification. The frequency, type, duration and route of antibiotic (or anti‐viral and anti‐fungal) used and whether there has been a requirement for second courses of antibiotics to clear infection should be sought. This should clarify whether the infections are persistent or recurrent. Information regarding smoking history (in pack‐years) and any family history of immunodeficiency or excessive infections is also important.
Antibody deficiency is characterized by sinopulmonary infection and hence careful attention to acute infections and wellbeing between infections is needed, as well as the presence of chronic symptoms of sinusitis or post‐nasal drip and chronic daily sputum production suggestive of bronchiectasis. Does sputum production return to zero between infections? If so, the target for optimal therapy should be zero daily sputum production. If this is not the case, details on volume of sputum over 24 h, colour and microbiology outside and during exacerbation are helpful. Baseline high resolution computed tomography (HRCT) imaging of the chest (and potentially sinuses) is usually undertaken to determine the presence and degree or absence of bronchiectasis and/or interstitial lung disease (ILD). This will inform the decision to treat with IGRT and antibiotics as well as the target IgG trough levels as this may need to be higher in bronchiectasis. The optimal treatment of ILD in PAD is not clear as this may represent a systemic lymphoproliferative inflammatory process 56. The natural history of an exacerbation is important (e.g. are exacerbations preceded by upper airway viral infections and do these always progress and require antibiotics), as this may help guide the patient to prompt use of antibiotics. Other baseline information includes the number of visits to the general practitioner (GP) per year, days lost from school or work, exercise capacity (distance on the level and flights of stairs) and how the patient would score themselves on a visual analogue scale (VAS) from 0 to 10 (0 being terrible and 10 being brilliant). This basic score is obtained alongside other clinical information at each clinic visit to help assess and optimize ongoing therapy. Some occupations such as nursery nursing or teaching young children carry a higher exposure to infection and impact the overall infection history. If it is decided to commence IGRT, a period of 12 months is helpful to assess the effect of therapy, as this removes seasonal infection bias and permits comparison to a 12‐month period before treatment.
It is also very important to obtain a careful history of previous and ongoing diseases and treatments, particularly those associated with SAD (Table 1). It is likely that there will be increasing referrals from a range of specialities (including haematology, oncology, transplantation, rheumatology) where combinations of the underlying disease or treatment result in secondary antibody deficiency. Improved treatments using agents which target B cells (such as rituximab), especially when used in combination with cytotoxic agents (e.g. fludarabine), or as maintenance therapy, will increase further the proportion of patients developing antibody deficiency over time 15.
Laboratory assessment
The laboratory assessment encompasses serum levels of Igs (IgG, IgA and IgM) and specific antibodies to tetanus, haemophilus and pneumococcus, although other specific antibodies may be used. The aim is to assess both the quantity (Ig levels) and the quality (responses to conjugated and unconjugated vaccines) of humoral immunity. Baseline blood tests include a full blood count (FBC), renal and liver function tests, including albumin in particular and C‐reactive protein (CRP). Flow cytometric determination of lymphocyte phenotypes [T, B and natural killer (NK) cells] to determine if B cell numbers are normal and if there is any evidence of T or NK cell abnormality. The analysis of class‐switched memory B cells (CSMB) does not generally affect the decision for IGRT, but may be helpful in patients without a clear diagnosis. Haematological malignancy should be excluded.
The determination of vaccine responses form an important part of the assessment and the results carry greater weight than the absolute level of IgG, especially if there is only a mild to moderate reduction in IgG. In general, the more severe the reduction in IgG the greater the likelihood of impaired or non‐durable responses to test vaccination.
In practice, there is a hierarchy of responses to vaccination, with tetanus being the most likely to result in so‐called ‘protective’ post‐vaccination levels, followed by haemophilus and then polysaccharide pneumococcal vaccination (PPV). Both tetanus toxoid and conjugated haemophilus behave as protein vaccines, while PPV is a pure polysaccharide vaccine, as is 23‐valent Pneumovax II. It is important to distinguish between protein and carbohydrate responses, which rely upon different immune pathways. Protective levels are not known for PID patients; each laboratory must determine their own ‘normal post‐immunization response levels’ in a large group of healthy individuals in order to advise clinicians. There are several assays for carbohydrate responses; interpretation is complicated 4 and the reader is referred to their local laboratory.
Some patients may be referred who have already been commenced on IGRT where the full clinical and laboratory information of the initial assessment may not be available or complete. The question then arises as to whether Ig therapy is still needed. In this situation, again the clinical and historical infection history and impact of Ig are important. While it is not possible to perform a complete assessment of antibody function while on IGRT, some test information is helpful. As most Ig products contain minimal amounts of IgA and IgM, if these are present in normal concentrations this reflects endogenous production. It is possible to undertake flow cytometry and, if B cells are normal in number, to determine if CSMB cells are also normal using the EUROclass panel. It may also be possible to assess vaccination responses to PPV for IgA and IgM responses and potentially to assess polysaccharide responses to antigens where the antibodies in Ig preparations are low, such as Typhi V or tick‐born encephalitis 7. The vaccination response in treated patients can be also measured by determination of specific antibody‐forming cells by enzyme‐linked immunospot (ELISPOT) assay 57. If there is a low infection burden and the clinical and laboratory evidence is supportive of intact endogenous antibody production, a period of Ig washout of 3–6 months may be undertaken, to allow full reassessment of endogenous antibody quantity and quality to help determine if future IGRT is required.
Baseline lung function with gas exchange should be obtained, and a high‐resolution computed tomography (HRCT) scan of the chest if this has not been performed recently. This is to establish if there is any existing end organ damage, such as bronchiectasis or evidence of interstitial lung disease. It is also helpful to establish if there is splenomegaly and any lymphadenopathy. The testing described is not exhaustive, and will vary with clinical practice and health‐care setting.
Conclusions
When and in whom to initiate IGRT requires a combination of laboratory, radiological and clinical information to determine the risk of future infections, morbidity and mortality for each individual patient. These individual decisions involve experience in assessment of all information and should be made in close liaison with the patient, with a balance of risks and benefits discussed.
This is much more straightforward when there is clear evidence of antibody deficiency, impaired vaccine responses and a significant infection burden and potentially end organ damage such as bronchiectasis or when patients fulfil criteria for well‐defined PIDs, as this generally implies the initiation of IGRT. In those without such findings, it is reasonable to take a ‘watch and wait’ approach with regular review and the added safety of prompt access to antibiotics. The likelihood of requiring IGRT is related broadly inversely to IgG levels (particularly with levels below 4 g/l), but there will always be exceptions (including SPAD) and individual assessment is key. Some patients may benefit from prophylactic antibiotics and will require assessment of the efficacy of the chosen antibiotic regimen (Table 3).
Table 3.
Graded antibiotic regimens
| Antibiotic regimen | Dosing schedule | Additional options | Emergency plan | Example |
|---|---|---|---|---|
| Intermittent antibiotics | None | Attend GP with symptoms | n.a. | |
| None | Early use of home back‐up antibiotics | Co‐amoxyclav 625 mg tds for 2 weeks held at home | ||
| Prophylactic antibiotics during the winter months with home rescue during the summer | Low‐dose and full‐dose options, e.g. Azithromycin 250–500 mg 3 days/week | Early use of home back‐up antibiotics |
Azithromycin 3 days/week plus back‐up Co‐amoxyclav for 2 weeks held at home |
|
| Ongoing prophylaxis | Prophylactic antibiotics | Low‐dose and full‐dose options, e.g. Azithromycin 250–500 mg 3 days/week | Early use of home back‐up antibiotics |
Azithromycin 3 days/week plus back‐up Co‐amoxyclav for 2 weeks held at home |
| Rotating prophylactic antibiotics | Early use of home back‐up antibiotics | |||
| Prophylactic antibiotics | Nebulized antibiotics | Early use of home back‐up antibiotics |
Azithromycin 3 days/week plus back‐up Co‐amoxyclav for 2 weeks held at home |
|
| Prophylactic antibiotics | Intermittent planned IVAB | Early use of home back‐up antibiotics |
Azithromycin 3 days/week plus back‐up Co‐amoxyclav for 2 weeks held at home |
Antibiotic prescribing should take into account the previous culture and sensitivity results as well as any allergies, tolerance and the likelihood of pseudomonas or macrolide‐resistant Haemophilus influenzae. If there has been no response to a back‐up course of antibiotics and a different second course of antibiotics, there should be a review and consideration for intravenous antibiotic (IVAB) treatment. Prophylactic and back‐up antibiotics should be different classes (e.g. macrolide and penicillin) and not an increase in dose of the existing prophylactic regimen. Monitoring and additional patient information may be needed, such as electrocardiogram (ECG) and hearing alterations for those on long‐term macrolides. There are many potential antibiotic options and the examples are illustrative, with individual decisions being made on clinical grounds. Nebulized antibiotics and intermittent IVAB are used mainly with severe bronchiectasis and pseudomonas colonization. GP = general practitioner; n.a. = not applicable; tds = three times a day.
The algorithm in Fig. 1 aims to balance evidence of antibody deficiency and the current burden of ongoing infection and end organ damage, such as bronchiectasis, and the prognosis of the individual patient. Additional important features that need to be taken into consideration are the non‐infectious complications of some primary antibody deficiencies, such as CVID, which include autoimmune cytopenias, lymphoproliferation, enteropathy or granulomata. In practice, when the situation is less clear cut, a period of observation with support and a clear plan of action for infections may be necessary to help inform decision‐making for the patient and clinician.
Figure 1.
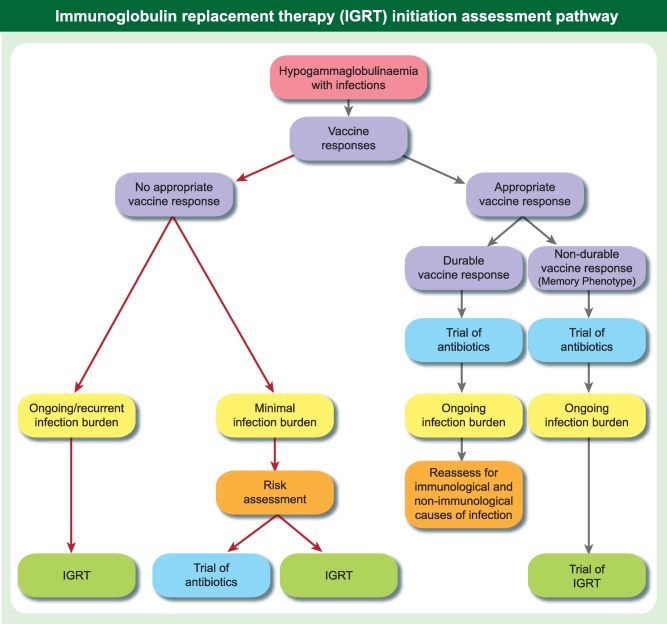
Immunoglobulin replacement therapy (IGRT) initiation assessment pathway. The Figure shows an outline assessment pathway for patients presenting with hypogammaglobulinaemia with a requirement to integrate clinical and laboratory findings and, where appropriate, to determine the impact of a trial of antibiotics. It is important to be able to reassess a decision if there is a change in the clinical or laboratory picture and to evaluate the effect of introduction of antibiotics or IGRT. Infection burden is a multi‐dimensional concept, which is difficult to condense into a number, but as a guide for adults a minimal infection burden would represent viral infections not requiring antibiotics and two or less 1‐week courses of oral antibiotics in 12 months. The pathway is a simplified outline of a complex area where clinical experience and judgement is required for each individual case to optimize care.
It is essential to be able to modify decisions. In patients who have a persistent significant infection burden despite prophylactic antibiotics in the context of antibody deficiency, a 12‐month initial trial of IGRT is justified with careful documentation of infections before and after therapy, to determine the degree of benefit. The trial must be undertaken with the patient's informed consent, to ensure that there is agreement in terms of the degree of improvement over baseline that will support ongoing IGRT.
The numbers of patients with SAD is likely to continue to increase, and interventions in this heterogeneous group may differ from PAD. In some settings the antibody deficiency may be reversible, and in others consideration will need to be given to additional infection risks not associated with antibody deficiency, such as neutropenia. A pragmatic approach of a trial of prophylactic antibiotics (Table 3) ahead of consideration of IGRT in those with reduction in IgG, disturbed vaccine responses and a significant infection burden has been suggested 15.
The majority of referrals to immunology clinics for antibody deficiency occur in the context of an existing infection history, with some referrals requesting clarification of abnormal results, especially when early detection of antibody deficiency follows screening 28, 58, 59. A decision to ‘watch and wait’ must be open to review if the situation changes; if, for example, it becomes clear that initially protective vaccine responses are non‐durable and decline over a period of 6–12 months to below protective levels or infections recur, Ig therapy may be reconsidered.
An improved understanding of the limits of current IGRT strategies in terms of preventing subclinical infection which may be viral will be important for decisions in those with more modest reductions in IgG 60. For patients already receiving IGRT it is hoped the that Burden of Infection in Primary Antibody Deficiency (BIPAD) study, which samples patients and controls every 2 weeks for 12 months, will help to define the types, frequency and duration of infections in the upper airway of these patients.
Increasing use of molecular diagnostics will probably refine further the risk profiles of patients presenting with antibody deficiency and feed into therapeutic decision‐making. The use of screening for antibody deficiency will result in patients presenting earlier with a much shorter diagnostic delay, and hence a much more limited infection history and less end organ damage 28, 58, 59. Discussions with the patient with a hitherto limited infection burden may thus be modified regarding the risks and benefits of a significant intervention such as IGRT. Equally the numbers of patients in whom diagnostic criteria are not met will increase, and the practical decision of whom to treat and when will need ongoing review.
Disclosure
S. J. has received support from CSL Behring, Baxter, Biotest, BPL, LFB, Shire, Grifols and Octapharma for projects, advisory boards, meetings and clinical trials. J. L. obtained fees for lectures from Baxter, Biotest and CSL Behring and obtained fees for consulting from Octapharma, Baxter and Baxalta. H. C. has received support from CSL Behring, Baxter Healthcare, Biotest, BPL, LFB, Shire, Grifols and Octapharma for research projects, advisory boards and educational projects in the past but none currently.
Author contributions
S. J., H. C. and J. L. designed and wrote the paper.
References
- 1. Gathmann B, Grimbacher B, Beauté J et al The European internet‐based patient and research database for primary immunodeficiencies: results 2006–2008. Clin Exp Immunol 2009; 157:3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Picard C, Al‐Herz W, Bousfiha A et al Primary immunodeficiency diseases: an update on the classification from the international union of immunological societies expert committee for primary immunodeficiency 2015. J Clin Immunol 2015; 35:696–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jolles S. The variable in common variable immunodeficiency: a disease of complex phenotypes. J Allergy Clin Immunol Pract 2013; 1:545–56. [DOI] [PubMed] [Google Scholar]
- 4. Bonilla FA, Barlan I, Chapel H et al International consensus document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract 2016; 4:38–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Conley ME, Notarangelo LD, Etzioni A. Diagnostic criteria for primary immunodeficiencies. Representing PAGID (Pan‐American Group for Immunodeficiency) and ESID (European Society for Immunodeficiencies). Clin Immunol 1999; 93:190–7. [DOI] [PubMed] [Google Scholar]
- 6. Ameratunga R, Woon ST, Gillis D, Koopmans W, Steele R. New diagnostic criteria for CVID. Expert Rev Clin Immunol 2014; 10:183–6. [DOI] [PubMed] [Google Scholar]
- 7. Wolf HM, Thon V, Litzman J, Eibl MM. Detection of impaired IgG antibody formation facilitates the decision on early immunoglobulin replacement in hypogammaglobulinemic patients. Front Immunol 2015; 6:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Driessen GJ, Dalm VA, van Hagen PM et al Common variable immunodeficiency and idiopathic primary hypogammaglobulinemia: two different conditions within the same disease spectrum. Haematologica 2013; 98:1617–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lucas M, Lee M, Lortan J, Lopez‐Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol 2010; 125:1354–60 e4 . [DOI] [PubMed] [Google Scholar]
- 10. Resnick ES, Moshier EL, Godbold JH, Cunningham‐Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood 2012; 119:1650–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Espanol T, Catala M, Hernandez M, Caragol I, Bertran JM. Development of a common variable immunodeficiency in IgA‐deficient patients. Clin Immunol Immunopathol 1996; 80:333–5. [DOI] [PubMed] [Google Scholar]
- 12. Aghamohammadi A, Mohammadi J, Parvaneh N et al Progression of selective IgA deficiency to common variable immunodeficiency. Int Arch Allergy Immunol 2008; 147:87–92. [DOI] [PubMed] [Google Scholar]
- 13. Orange JS, Ballow M, Stiehm ER et al Use and interpretation of diagnostic vaccination in primary immunodeficiency: a working group report of the basic and clinical immunology interest section of the American Academy of Allergy, Asthma and Immunology. J Allergy Clin Immunol 2012; 130:S1–24. [DOI] [PubMed] [Google Scholar]
- 14. Wall LA, Dimitriades VR, Sorensen RU. Specific antibody deficiencies. Immunol Allergy Clin North Am 2015; 35:659–70. [DOI] [PubMed] [Google Scholar]
- 15. Dhalla F, Misbah SA. Secondary antibody deficiencies. Curr Opin Allergy Clin Immunol 2015; 15:505–13. [DOI] [PubMed] [Google Scholar]
- 16. Chapel HM, Lee M, Hargreaves R, Pamphilon DH, Prentice AG. Randomised trial of intravenous immunoglobulin as prophylaxis against infection in plateau‐phase multiple myeloma. The UK Group for Immunoglobulin Replacement Therapy in Multiple Myeloma. Lancet 1994; 343:1059–63. [DOI] [PubMed] [Google Scholar]
- 17. Cooperative Group for the Study of Immunoglobulin in Chronic Lymphocytic Leukemia . Intravenous immunoglobulin for the prevention of infection in chronic lymphocytic leukemia. A randomized, controlled clinical trial. Cooperative group for the study of immunoglobulin in chronic lymphocytic leukemia. N Engl J Med 1988; 319:902–7. [DOI] [PubMed] [Google Scholar]
- 18. Friman V, Winqvist O, Blimark C, Langerbeins P, Chapel H, Dhalla F. Secondary immunodeficiency in lymphoproliferative malignancies. Hematol Oncol 2016; 34:121–32. [DOI] [PubMed] [Google Scholar]
- 19. Sanchez‐Ramon S, Dhalla F, Chapel H. Challenges in the role of gammaglobulin replacement therapy and vaccination strategies for hematological malignancy. Front Immunol 2016; 7:317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hamblin AD, Hamblin TJ. The immunodeficiency of chronic lymphocytic leukaemia. Br Med Bull 2008; 87:49–62. [DOI] [PubMed] [Google Scholar]
- 21. Kaminsky P, Lesesve JF, Jonveaux P, Pruna L. IgG deficiency and expansion of CTG repeats in myotonic dystrophy. Clin Neurol Neurosurg 2011; 113:464–8. [DOI] [PubMed] [Google Scholar]
- 22. Kim J, Hayton WL, Robinson JM, Anderson CL. Kinetics of FcRn‐mediated recycling of IgG and albumin in human: pathophysiology and therapeutic implications using a simplified mechanism‐based model. Clin Immunol 2007; 122:146–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Casulo C, Maragulia J, Zelenetz AD. Incidence of hypogammaglobulinemia in patients receiving rituximab and the use of intravenous immunoglobulin for recurrent infections. Clin Lymph Myeloma Leuk 2013; 13:106–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. van Vollenhoven RF, Emery P, Bingham CO III et al Long‐term safety of rituximab in rheumatoid arthritis: 9.5‐year follow‐up of the global clinical trial programme with a focus on adverse events of interest in RA patients. Ann Rheum Dis 2013; 72:1496–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Venhoff N, Effelsberg NM, Salzer U et al Impact of rituximab on immunoglobulin concentrations and B cell numbers after cyclophosphamide treatment in patients with ANCA‐associated vasculitides. PLOS ONE 2012; 7:e37626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Davila ML, Riviere I, Wang X et al Efficacy and toxicity management of 19‐28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med 2014; 6:224ra25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maude SL, Frey N, Shaw PA et al Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014; 371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jolles S, Borrell R, Zouwail S et al Calculated globulin (CG) as a screening test for antibody deficiency. Clin Exp Immunol 2014; 177:671–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Braamskamp MJ, Dolman KM, Tabbers MM. Clinical practice. Protein‐losing enteropathy in children. Eur J Pediatr 2010; 169:1179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Duraisingham SS, Buckland MS, Grigoriadou S, Longhurst HJ. Secondary antibody deficiency. Expert Rev Clin Immunol 2014; 10:583–91. [DOI] [PubMed] [Google Scholar]
- 31. Dominguez‐Pinilla N, Benitez EM, Gonzalez‐Tome MI, Ruiz‐Contreras J, Gonzalez‐Granado LI. Invasive pneumococcal infection secondary to hypogammaglobulinemia due to Menetrier disease. Pediatr Infect Dis J 2013; 32:578. [DOI] [PubMed] [Google Scholar]
- 32. Hodge D, Misbah SA, Mueller RF, Glass EJ, Chetcuti PA. Proteus syndrome and immunodeficiency. Arch Dis Child 2000; 82:234–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boddana P, Webb LH, Unsworth J, Brealey M, Bingham C, Harper SJ. Hypogammaglobulinemia and bronchiectasis in mycophenolate mofetil‐treated renal transplant recipients: an emerging clinical phenomenon? Clin Transplant 2011; 25:417–9. [DOI] [PubMed] [Google Scholar]
- 34. Kawano T, Matsuse H, Obase Y et al Hypogammaglobulinemia in steroid‐dependent asthmatics correlates with the daily dose of oral prednisolone. Int Arch Allergy Immunol 2002; 128:240. [DOI] [PubMed] [Google Scholar]
- 35. Abe S, Suzuki T, Hori T, Baba A, Shiraishi H. Hypogammaglobulinemia during antipsychotic therapy. Psychiatry Clin Neurosci 1998; 52:115–7. [DOI] [PubMed] [Google Scholar]
- 36. Snowden N, Dietch DM, Teh LS, Hilton RC, Haeney MR. Antibody deficiency associated with gold treatment: natural history and management in 22 patients. Ann Rheum Dis 1996; 55:616–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van Rossum MA, Fiselier TJ, Franssen MJ et al Dutch Juvenile Chronic Arthritis Study Group. Effects of sulfasalazine treatment on serum immunoglobulin levels in children with juvenile chronic arthritis. Scand J Rheumatol 2001; 30:25–30. [DOI] [PubMed] [Google Scholar]
- 38. Williams A, Scott DL, Greenwood A, Huskisson EC. The clinical value of measuring immunoglobulins when assessing penicillamine therapy in rheumatoid arthritis. Clin Rheumatol 1988; 7:347–53. [DOI] [PubMed] [Google Scholar]
- 39. Santachiara R, Maffei R, Martinelli S et al Development of hypogammaglobulinemia in patients treated with imatinib for chronic myeloid leukemia or gastrointestinal stromal tumor. Haematologica 2008; 93:1252–5. [DOI] [PubMed] [Google Scholar]
- 40. Ginzler EM, Wax S, Rajeswaran A et al Atacicept in combination with MMF and corticosteroids in lupus nephritis: results of a prematurely terminated trial. Arthritis Res Ther 2012; 14:R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kappos L, Hartung HP, Freedman MS et al Atacicept in multiple sclerosis (ATAMS): a randomised, placebo‐controlled, double‐blind, phase 2 trial. Lancet Neurol 2014; 13:353–63. [DOI] [PubMed] [Google Scholar]
- 42. Hargreaves RM, Lea JR, Griffiths H et al Immunological factors and risk of infection in plateau phase myeloma. J Clin Pathol 1995; 48:260–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kyle RA, Gertz MA, Witzig TE et al Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003; 78:21–33. [DOI] [PubMed] [Google Scholar]
- 44. Kyle RA, Therneau TM, Rajkumar SV et al A long‐term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346:564–9. [DOI] [PubMed] [Google Scholar]
- 45. Perez‐Persona E, Vidriales MB, Mateo G et al New criteria to identify risk of progression in monoclonal gammopathy of uncertain significance and smoldering multiple myeloma based on multiparameter flow cytometry analysis of bone marrow plasma cells. Blood 2007; 110:2586–92. [DOI] [PubMed] [Google Scholar]
- 46. McGrath EE, Blades Z, Anderson PB. Chylothorax: aetiology, diagnosis and therapeutic options. Respir Med 2010; 104:1–8. [DOI] [PubMed] [Google Scholar]
- 47. Nair SK, Petko M, Hayward MP. Aetiology and management of chylothorax in adults. Eur J Cardiothorac Surg 2007; 32:362–9. [DOI] [PubMed] [Google Scholar]
- 48. Ashrafi M, Hosseini SA, Abolmaali S et al Effect of anti‐epileptic drugs on serum immunoglobulin levels in children. Acta Neurol Belg 2010; 110:65–70. [PubMed] [Google Scholar]
- 49. Hayman G, Bansal A. Antibody deficiency associated with carbamazepine. BMJ 2002; 325:1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaplan B, Kopyltsova Y, Khokhar A, Lam F, Bonagura V. Rituximab and immune deficiency: case series and review of the literature. J Allergy Clin Immunol Pract 2014; 2:594–600. [DOI] [PubMed] [Google Scholar]
- 51. Levy R, Mahevas M, Galicier L et al Profound symptomatic hypogammaglobulinemia: a rare late complication after rituximab treatment for immune thrombocytopenia. Report of 3 cases and systematic review of the literature. Autoimmun Rev 2014; 13:1055–63. [DOI] [PubMed] [Google Scholar]
- 52. Sarmiento E, Diez P, Arraya M et al Early intravenous immunoglobulin replacement in hypogammaglobulinemic heart transplant recipients: results of a clinical trial. Transpl Infect Dis 2016; 18:832–43. [DOI] [PubMed] [Google Scholar]
- 53. Berger M, Jolles S, Orange JS, Sleasman JW. Bioavailability of IgG administered by the subcutaneous route. J Clin Immunol 2013; 33:984–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Jolles S, Orange JS, Gardulf A et al Current treatment options with immunoglobulin G for the individualization of care in patients with primary immunodeficiency disease. Clin Exp Immunol 2015; 179:146–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hlavackova E, Liska M, Jicinska H, Navratil J, Litzman J. Secondary combined immunodeficiency in pediatric patients after the fontan operation: three case reports. Int Arch Allergy Immunol 2016; 170:251–6. [DOI] [PubMed] [Google Scholar]
- 56. Jolles S, Carne E, Brouns M, El‐Shanawany T, Williams P, Marshall C, Fielding P. FDG PET‐CT imaging of therapeutic response in granulomatous lymphocytic interstitial lung disease (GLILD) in common variable immunodeficiency (CVID). Clin Exp Immunol 2017; 187:138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chovancova Z, Vlkova M, Litzman J, Lokaj J, Thon V. Antibody forming cells and plasmablasts in peripheral blood in CVID patients after vaccination. Vaccine 2011; 29:4142–50. [DOI] [PubMed] [Google Scholar]
- 58. Holding S, Jolles S. Current screening approaches for antibody deficiency. Curr Opin Allergy Clin Immunol 2015; 15:547–55. [DOI] [PubMed] [Google Scholar]
- 59. Holding S, Khan S, Sewell WA, Jolles S, Dore PC. Using calculated globulin fraction to reduce diagnostic delay in primary and secondary hypogammaglobulinaemias: results of a demonstration project. Ann Clin Biochem 2015; 52:319–26. [DOI] [PubMed] [Google Scholar]
- 60. Jolles S. Subclinical infection and dosing in primary immunodeficiencies. Clin Exp Immunol 2014; 178(Suppl.1):67–9. [DOI] [PMC free article] [PubMed] [Google Scholar]