

Abstract
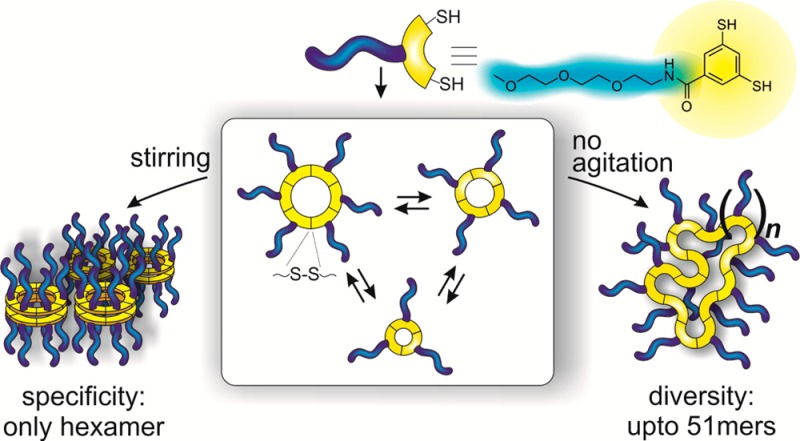
With the advent of reversible covalent chemistry the study of the interplay between covalent bond formation and noncovalent interactions has become increasingly relevant. Here we report that the interplay between reversible disulfide chemistry and self-assembly can give rise either to molecular diversity, i.e., the emergence of a unprecedentedly large range of macrocycles or to molecular specificity, i.e., the autocatalytic emergence of a single species. The two phenomena are the result of two different modes of self-assembly, demonstrating that control over self-assembly pathways can enable control over covalent bond formation.
Introduction
Biological systems function by virtue of a complex and concurrent interplay of covalent bond formation and noncovalent assembly processes. For example, the noncovalent assembly of protein complexes affects the ability of the protein to catalyze covalent chemical reactions.1 On the other hand, covalent histone modifications control the noncovalent binding of a DNA strand within the chromatin complex.2 In chemistry, traditionally the processes of covalent and noncovalent bond formation occur sequentially: for example, first the potential host and guest molecules are synthesized by covalent chemical means and subsequently, host–guest binding or self-assembly is investigated. In many cases such separation of covalent and noncovalent processes cannot be avoided as the conditions required for organic synthesis are often incompatible with those required for noncovalent interactions. However, with the advent of reversible covalent chemistry,3−5 the development of systems featuring concurrent covalent and noncovalent chemistries has become possible. Dynamic covalent systems constructed from different building blocks tend to lead to diverse mixtures of products which continuously exchange building blocks via reversible covalent bond formation (dynamic combinatorial libraries, DCLs).6−8 Noncovalent interactions9 can then be utilized to channel the building blocks into specific DCL members that optimally engage in molecular recognition. This effect has been exploited for the dynamic combinatorial discovery of synthetic receptors10−17 and ligands for biomolecules18,19 by exposing the system to a corresponding template.
The theory of template-induced amplification of specific DCL members is well-established.20−25 More recently, also molecular recognition processes that take place in the absence of added templates,26 occurring between library members have been explored, leading to interlocked structures,27−31 self-replicating molecules32−38 and self-assembling materials.39−41 In these systems, noncovalent interactions within (for interlocked structures) or between (for self-replicating and self-assembling materials) specific library members shift the equilibrium toward molecules that engage most efficiently in noncovalent interactions. Such behavior is relevant from the perspectives of the origin-of-life42−44 and the de novo synthesis of life,45−49 as it leads to the spontaneous and often autocatalytic emergence of specific molecules from complex mixtures, where these molecules had acquired information and are able to pass this information on to the next generation during self-replication. Self-assembly phenomena are also intimately linked with materials science, as supramolecular objects based on molecules containing dynamic covalent bonds undergoing spontaneous self-assembly can be regarded as self-synthesizing.34,50−52 Despite the considerable interest in dynamic combinatorial self-assembly, the number of such systems is limited, particularly when it comes to amphiphile self-assembly.39,50,53−55 Moreover, in contrast to the well-established theory of template-induced amplification,20−25 the theoretical understanding on how the selection of different modes of self-assembly relates to covalent selection remains underdeveloped. We now report how two different self-assembly pathways induce dramatically different responses in the behavior of libraries made from the same building block. One pathway leads to a diverse set of unprecedentedly large macrocycles, while a second pathway leads to the autocatalytic formation of one specific macrocycle.
Results and Discussion
In order to explore self-assembly in dynamic combinatorial chemistry, we designed amphiphilic building block 1 (Scheme 1), containing a short polar oligo(ethylene oxide) chain connected to a nonpolar aromatic ring functionalized with two thiol groups for reversible covalent disulfide chemistry. Under slightly basic conditions dithiols are (partially) deprotonated to give thiolates which are oxidized slowly by atmospheric oxygen (or faster using sodium perborate) to disulfides. Thiolates also react with disulfides in a reversible manner, which enables exchange, and thus, dynamic covalent chemistry between the disulfides.
Scheme 1. Dynamic Combinatorial Chemistry of Building Block 1.
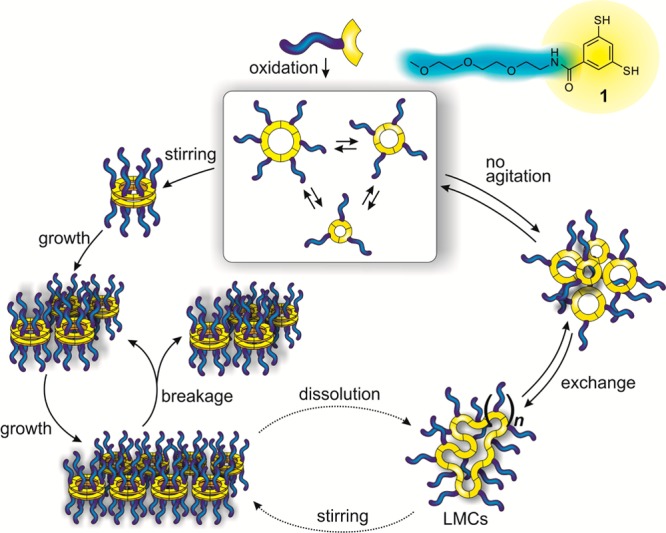
The oxidation of building block 1 in a 9:1 mixture of aqueous borate buffer (pH = 8.2) and dimethylformamide (DMF) in the absence of mechanical agitation yielded a DCL which consisted mainly of cyclic trimers and tetramers, but featured a considerable amount (30 mol %) of larger macrocyclic species (LMCs) as well, from cyclic 7mer to cyclic 44mer (Figure 1A). The identity of the observed species was confirmed by LC–MS analysis, as shown in Section 6 of the Supporting Information (SI).56 Although we could not exclude that some of the large oligomers are present in form of interlocked species, the relative simplicity of the UPLC chromatograms suggests that they are monocyclic (as for one given macrocycle size, numerous interlocked species with different hydrophobicity can be formed, which would substantially complicate the chromatograms). In general, the occurrence of such large macrocycles under relatively dilute conditions (6.0 mM in 1) is unprecedented as the production of a large number of small macrocycles is usually preferred over producing a small number of larger entities for entropic reasons,41 (although enthalpic effects due to differences in interfacial energy also cannot be excluded).
Figure 1.
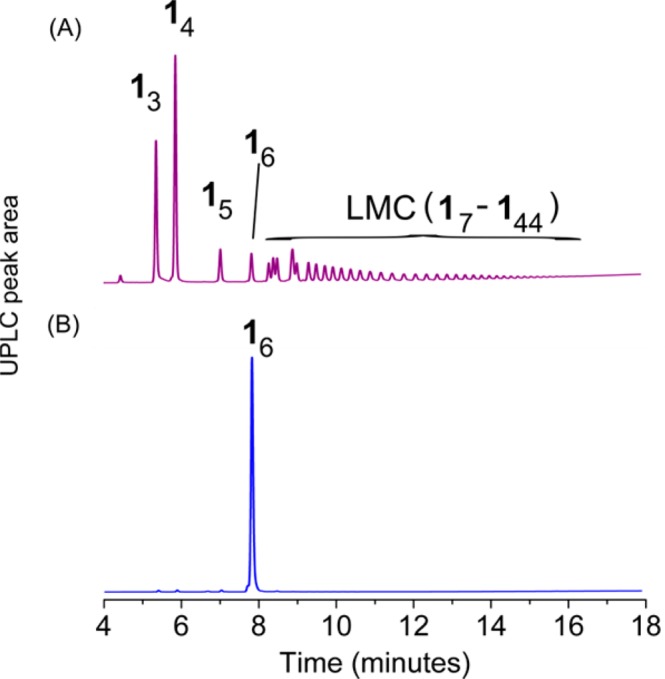
UPLC analyses of DCLs made from 6.0 mM building block 1 in a 9:1 mixture of aqueous borate buffer (50 mM, pH 8.2) and DMF (A) without agitation and (B) stirred at 1200 rpm.
In order to rationalize this unusual behavior, we performed a series of experiments to gain more insight into the self-assembly properties of the oligomers formed from 1. First, the effect of cosolvents was investigated. Thus, we prepared DCLs from 1, with identical building block concentration (6.0 mM) but with increasing amounts of DMF as a cosolvent (from 10 to 90% V/V) and investigated the composition of the DCLs with UPLC. The libraries were prepared by oxidizing the monomer with sodium perborate to 85% in 30 min in a solvent mixture of DMF and aqueous borate buffer. The composition of the DCLs was monitored for up to 2 months, but remained essentially unchanged after 40 days. For detailed experimental information, see the SI Section 5.
Trimers and tetramers were always the main components in the DCLs. However, the overall LMC content as well as the maximal detected macrocycle size decreased upon increasing DMF content (Figure 2A), giving rise finally to DCLs consisting exclusively of trimers and tetramers at high DMF concentrations. Thus, the formation of LMCs appears to be inhibited by the presence of the organic cosolvent, suggesting a role for hydrophobic interactions in LMC formation. We observed that in libraries with a DMF content less than 10% V/V and building block concentrations higher than 1 mM, occasionally, phase separation occurred. The composition of the separated phase was, however, similar to that of the solution. For further information see the SI (Figures S104–106). Together, these results suggested that the LMCs are formed upon aggregation of trimers and tetramers under the given conditions.
Figure 2.
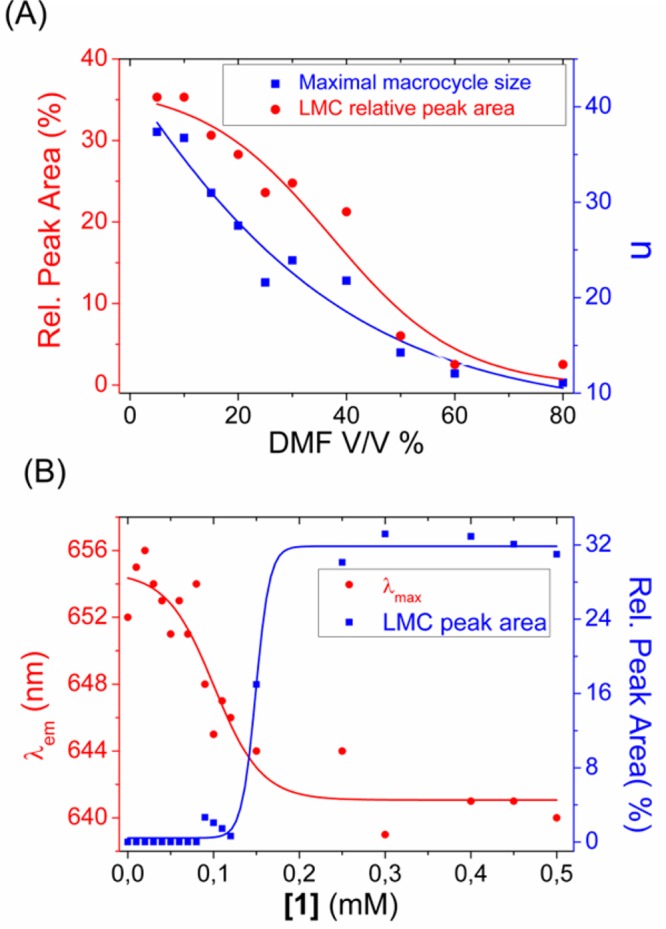
(A) LMC percentage (left axis) and maximal detected LMC size (n, right axis) of DCLs prepared from 1 (6.0 mM) in 50 mM borate buffer (pH = 8.2) with different amounts of DMF as a cosolvent. (B) Fluorescence emission maximum (left axis, λexc = 553 nm) and LMC content (right axis) of solutions containing 230 nM Nile Red and a DCL prepared from 1 (50 mM borate buffer, pH = 8.2, without cosolvent), at different building block concentrations. Lines are drawn to guide the eye.
In order to demonstrate that the formation of LMCs is a consequence of the aggregation of trimers and tetramers, we investigated the system using a Nile Red fluorescence assay. Nile Red is a solvatochromic dye, featuring low fluorescence intensity in aqueous solution due to aggregation, but when incorporated into hydrophobic microenvironments it shows a significant fluorescence increase and a characteristic blue shift of the emission maximum, as a result of encapsulation of the dye molecules by the hydrophobic microenvironment and consequent disaggregation.57 In a nonstirred, oxidized DCL of 1 (featuring trimers, tetramers and LMCs) in aqueous buffer, Nile Red showed significantly higher fluorescence intensity compared to that in buffer, whereas in the absence of Nile Red, neither the DCL, nor the buffer showed fluorescence (see SI Figure S105). This indicated that in aqueous buffer, the DCL contained aggregates providing a hydrophobic microenvironment to the dye. In order to estimate the critical aggregation concentration (CAC), fully oxidized DCLs with an increasing (0.01–1 mM) building block concentration (containing 230 nM Nile Red) were prepared in aqueous buffer and the shift of the fluorescence emission maximum was monitored. As shown in Figure 2B, a sharp decrease in the fluorescence emission maximum was detected between 0.05 and 0.15 mM overall building block concentration, indicating that aggregation starts taking place in this concentration range. UPLC analyses of the samples showed that at low concentrations, only trimers and tetramers were present, but the LMC content showed a sharp increase in approximately the same concentration range where the Nile Red fluorescence intensity decreased (0.1–0.2 mM), indicating that the formation of LMCs and aggregation are correlated.
We attempted to gain insight into the nanoscale structure of the aggregates. In fresh samples, light microscopy showed the formation of spherical droplets with diameters between 5 and 30 μm, which form larger, needle-like aggregates upon aging (see SI Figure S108). Similarly, confocal fluorescence microscopy (Nile Red staining) of a freshly prepared DCL containing LMCs showed the presence of spherical aggregates with diameters between 5 and 10 μm, confirming that the trimer/tetramer aggregates and the LMCs self-assemble into microscale objects (SI Figure S109). We also analyzed the samples by transmission electron microscopy (TEM) searching for smaller nanosize assemblies, but failed to detect any.
We interpret these results as follows: Upon oxidation and exchange, the cyclic trimer and tetramer form first, as observed from previous investigations of DCLs prepared from building blocks bearing the same dithiol core.34,41,44,58 These two amphiphilic species, featuring a hydrophobic macrocycle core and hydrophilic tri(ethylene oxide) chains, are capable of self-assembling into supramolecular aggregates upon exceeding a critical aggregation concentration,59,60 as shown by the Nile Red fluorescence assay. The high local concentration of disulfides in these aggregates exceeds the effective molarity for ring closing of the smaller macrocycles, allowing LMCs to be formed. As this process takes place mainly in a separated microphase, the overall changes in the noncovalent interactions between the macrocycles upon transitioning from trimers and tetramers to LMCs is probably small (the overall hydrophobic interactions between trimers and tetramers are probably comparable to those between LMCs). Thus, in general, the formation of these LMCs at the covalent level is enabled by the hydrophobicity-driven self-assembly of smaller oligomers at the noncovalent level. Upon addition of organic cosolvents, such as DMF, the solvent environment becomes less polar and aggregation of trimers and tetramers occur to a lesser extent, resulting in a decreasing LMC content and size in DCLs with higher cosolvent content.
In sharp contrast to the molecular diversity observed in the nonagitated DCL prepared from 1, in a stirred library the cyclic hexamer (16) emerges exclusively, as shown in Figure 1B. The hexamer assembles as two-dimensional aggregates (vide infra) that separate from the solution as a solid precipitate, which enabled its easy isolation by simple centrifugation and freeze-drying in 52% yield (for a detailed procedure, see SI Section 10). We suspected that the phase separation of the hexamer is driven by hydrophobic interactions. In order to prove this hypothesis, we assessed whether the hexamers would disassemble again upon exposing them to organic (co)solvents. An isolated sample of the hexamer was dissolved in a mixture of water and acetonitrile (MeCN:H2O 2:1 with 0.1% TFA) at a concentration of 0.13 mM (Figure 3A) and the composition of the sample was monitored with UPLC. After 7 days, only 7% of the DCL was present in the form of hexamers and the rest was converted to trimers, tetramers and pentamers (Figure 3B), whereas in the DCL formed after 12 days, only 4% of hexamers were present and 29% of the library consisted of LMCs (Figure 3C). Similar results were obtained when a sample of isolated hexamer was dissolved in pure MeOH (c = 0.44 mM): in this case, predominantly trimers and tetramers were detected at equilibrium (Figure 3D). The amount of trimers and tetramers increased parallelly (to 60 and 20 mol %, respectively), alongside with the decrease in the amount of hexamers to 4 mol %. In this case, however, no LMCs were detectable at any stage of the process, which is in line with our previous observations concerning the role of hydrophobic interactions in the formation of larger oligomers. It is worth noting that under conditions which favor the formation of LMCs (i.e., in at least partially aqueous environment), the tetramer is favored over the trimer (see Figure 1A and Figure 3C), whereas under conditions where LMCs are not present, the trimer is the favored species (Figure 3D), which might explain that upon the dissolution of the hexamer, the trimer emerges first (Figure 3B).
Figure 3.
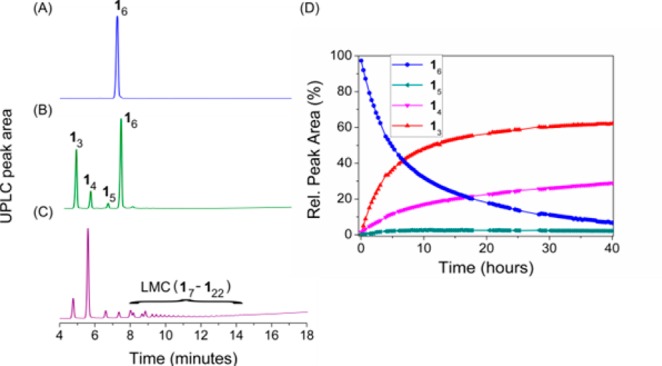
UPLC chromatogram of the isolated hexamer dissolved in MeCN:H2O 2:1 (0.1 V/V % TFA) after (A) 0 days, (B) 7 days and (C) 12 days. (D) Temporal evolution of a DCL prepared by dissolving the hexamer of 1 in MeOH (0.44 mM).
As the hexamer emerged as the sole product from a mechanically agitated DCL, we suspected it was capable of self-replication (i.e., catalyzing its own formation driven by nanoscale self-assembly). Thus, we monitored the change in the concentrations of the library members in time. Building block 1 was dissolved to a concentration of 2.0 mM in aqueous borate buffer and the library was stirred at 1200 rpm. The relative amount of the hexamer showed sigmoidal growth (Figure 4A): a lag phase (corresponding to a slow nucleation event) was followed by a rapid increase in the concentration of the hexamer, whereupon all DCL material was converted into hexamer. Similar behavior was observed at building block concentrations close to the CAC of the LMCs, i.e., at 0.05–0.5 mM, whereas the lag phase increased at decreasing stirring rates (see SI Figures S124 and S125, respectively). In order to prove the autocatalytic nature of the formation of the hexamer, we performed seeding experiments: a preoxidized, nonagitated DCL prepared of 1 (6.0 mM) in a 9:1 mixture of borate buffer (50 mM) and DMF, was seeded with 5 and 10 mol % (with respect to the overall building block concentration) preformed hexamer, respectively. Even in a nonstirred sample, an immediate and sharp increase of the hexamer concentration was observed compared to the nonseeded control (Figure 4B), indicating that the formation of the hexamer is autocatalytic. Additionally, we observed that in the presence of increasing amounts of organic cosolvents both the replication rate and the final hexamer content decreased, in line with our previous observations on cosolvent effects (see Figure 2A and SI Figure S123).
Figure 4.
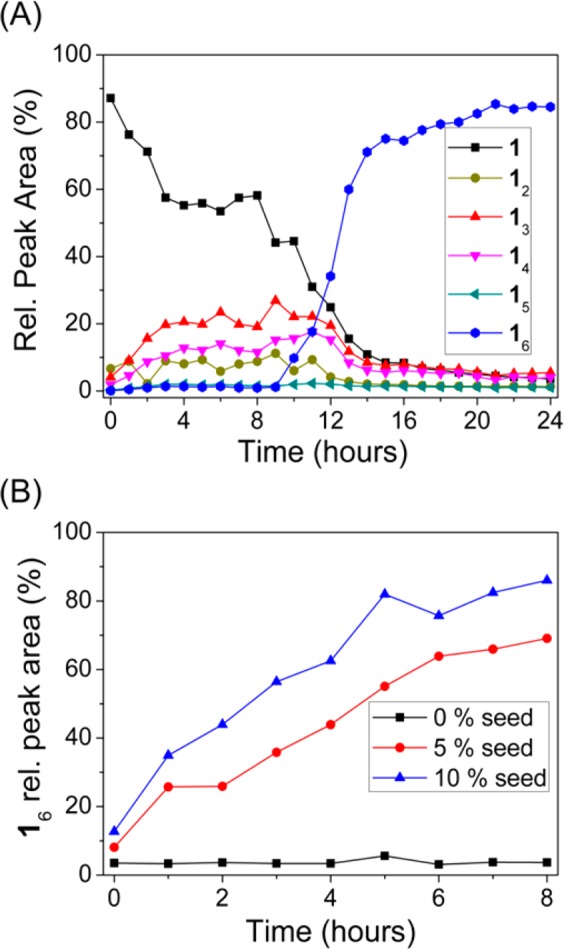
(A) Change of the product distribution with time in a DCL prepared of building block 1, showing the characteristic sigmoidal growth of the hexamer. (B) Change of the relative concentration of the hexamer of 1 in a DCL prepared from 1 (6.0 mM) in a 9:1 mixture of aqueous borate buffer (50 mM, pH = 8.2) and DMF without seeding (squares) and upon seeding with 5.0% (circles) and with 10% (triangles) preformed hexamer seed at t = 0 min.
On the basis of previous examples,34,36,39,41,51,58 we expected the self-replicating species to self-assemble into well-defined nanoscale structures. As powder X-ray diffraction experiments conducted on the isolated hexamer did not deliver sufficiently informative data (see SI Figure S122), we proceeded to study the process of self-replication with cryo-TEM. A DCL at 6.0 mM building block concentration was prepared and preoxidized to 80% with sodium perborate. Stirring was continued and samples were taken at various time points in order to monitor the dynamics of aggregate formation in parallel with UPLC and cryo-TEM. The results are shown in Figure 5. At the beginning of the monitoring process (at 3 mol % hexamer content), no nanoscale assemblies were observed (Figure 5A), in line with the previous observation that LMCs form microscale aggregates which are too large to be observed with TEM. However, after 2 h (4 mol % hexamer content), long, sharp-edged nanoribbons (length: 400–600 nm, width: 15–30 nm) were observed, which laterally associated into bundles (Figure 5B). As the self-replication continued (16 mol % hexamer content) the ribbons became more elongated (600–800 nm) and more abundant (Figure 5C). At 18 mol % hexamer content the single nanoribbons were not observed anymore, and the bundles (30–40 nm wide) became the prevalent nanoscale objects (Figure 5D). At a later stage of replication (65 mol % hexamer content) the bundles grew several micrometers long and up to 80 nm wide (Figure 5E). In aged samples (72 h after the onset of the replication) with 100 mol % hexamer content, the elongated bundles gave way to irregular platelets, with a size of ca. 100–150 nm in both directions (Figure 5F). AFM measurements showed similar results, confirming the presence of nanoribbons and -platelets with a constant height of 2–3 nm during the entire self-replication process (Figure 6 and SI Figure S128). Confocal fluorescence microscopy (Nile Red staining) also indicated the presence of nanoribbons with lengths of 5–10 μm (see SI Figure S127).
Figure 5.

Cryogenic (A–D) and negative stain (E,F) TEM images of a stirred DCL made from preoxidized (80%, NaBO3) building block 1 (6.0 mM) in various stages of the self-replication process at (A) 3%; t = 0 h (B) 4%; t = 2 h, (C) 16%; t = 3.5 h, (D) 18%; t = 20 h, (E) 65%; t = 43 h, (F) 100% hexamer content; t = 72 h.
Figure 6.
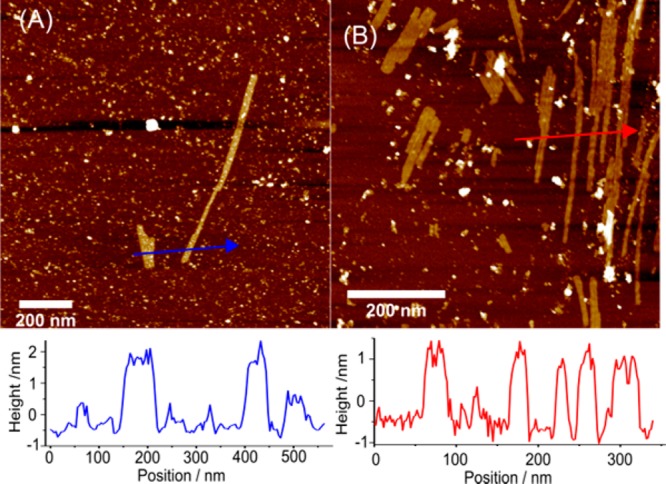
AFM images of a stirred DCL made from preoxidized (80%, NaBO3) building block 1 (6.0 mM) at (A) 54% (B) 100% conversion to hexamer (5 months old sample).
These results show that the self-replication of the hexamers is concomitant with formation of 2-dimensional nanoscale assemblies. Based on the results of AFM and TEM measurements, we hypothesize the following steps of self-assembly: Initially, the hexamer molecules pack into fibers, featuring the hydrophobic aromatic rings at their core, which is surrounded by the hydrophilic oligo(ethylene oxide) units (Scheme 1). This hydrophobicity-driven arrangement might be further stabilized by clustering of the side chains, which is pronounced for methyl-terminated oligo(ethylene oxide) chains.61,62 Exponential self-replication in this system is possibly a consequence of a fiber breakage-elongation mechanism, established previously for peptide-based replicators.36 Yet, unlike in the previous systems, where the fibers were observed to elongate only longitudinally, the fibers assembled from the hexamer are capable of stabilizing themselves by lateral association as well (Figure 5B), forming nanoribbons. The reason for the different self-assembly behavior might arise from the different structure of the side chains: whereas in the case of the previously reported peptide replicators, β-sheet interactions between the peptide side chains contribute significantly to the stabilization of the fibers, the interaction strength between the oligo(ethylene oxide) side chains is considerably smaller compared to the energy gain resulting from the hydrophobically driven association of the aromatic cores. The observation that the edges of the nanoribbons are remarkably straight in the TEM and AFM images suggests that fibers act as precursors in the formation of nanoribbons.
The fact that elongated structures are produced during early stages of the assembly process suggests that assembly at the extremities of the ribbons is initially faster than growth from the flanks of the structures and also faster than their lateral association. However, as mechanical agitation is continued and a considerable amount of the smaller macrocycles are consumed lateral association becomes the main assembly pathway, giving rise to wider and shorter platelets. This transformation of nanoribbons to platelets presumably represent a transition from a kinetically preferred assembly to a thermodynamically preferred one. Note that the height of the assemblies is constant throughout the entire replication process, i.e., the association of the nanoribbons proceeds only in one dimension.
We also considered the possibility that Na+ ions might act as templates by forming crown-ether like chelates upon interactions with the oligo(ethylene oxide) side chains of neighboring molecules of the hexamer. However, the replication rate shows no readily interpretable dependence on the concentration of Na+ in the 0–50 mM range (see SI Figure S126).
We were also interested to what extent the molecular structure of the monomer affects the dual assembly modes described above. Thus, we synthesized two analogues of 1 containing one ethylene oxide unit less (2) and more (3) than 1. Analogue 2 oxidized only very slowly when exposed to air in the absence of stirring, possibly due to its low solubility in aqueous borate buffer. However, quick oxidation with perborate in the absence of agitation gave rise to a DCL containing mainly trimers and tetramers but also a significant amount of LMCs (Figure 7A). Fluorescence microscopy (see SI Figure S112) showed spherical aggregates which were similar to those observed for 1, whereas with TEM no nanoscale structures were detectable (see SI Figure S111). Addition of large amounts of cosolvent led to the disappearance of LMCs, analogously to 1. When stirred, the cyclic tetramer emerged as the only product (Figure 7B). Fluorescence microscopy and TEM showed the presence of nanoribbons (see SI Figures S131–132). Similarly to the hexamer formed from 1, this tetramer could also be easily isolated in 76% yield. Compared to the hexamer of 1, the tetramer of 2 showed a weak autocatalytic effect; more precise analysis of the seeding process was hampered by analytical difficulties related to the poor solubility of the tetramer of 2 (see SI Figures S133–134). Assembly of 2 occurs for a smaller macrocycle size than for 1 which can be rationalized based on the fact that building block 2 is more hydrophobic than 1. Therefore, fewer units of 1 are required to generate a sufficient hydrophobic driving force to enable self-assembly.41
Figure 7.
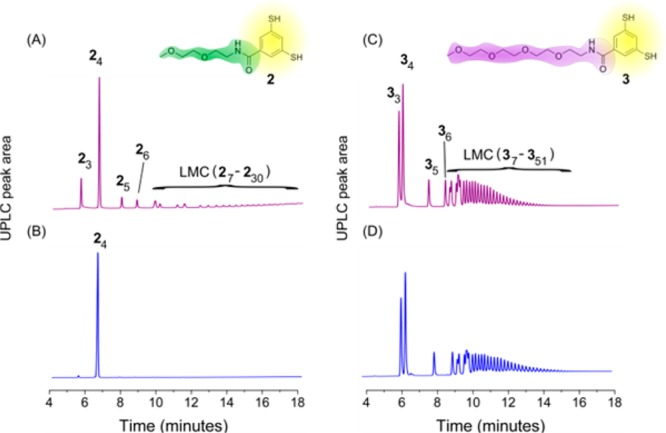
UPLC traces of DCLs prepared from (A) 2, quickly oxidized with NaBO3 and left unagitated (B) 2, stirred for 3 days (C) 3, unagitated after 7 days (D) 3, stirred for 7 days.
In sharp contrast, 3 does not show preference for any specific macrocycle and, regardless of mechanical agitation, gives rise to LMCs up to 55mer upon oxidation (Figure 7C and D). The mechanism of the formation of the LMCs for 3 resembles that for 1, as shown by cosolvent addition and fluorescence experiments (see SI Figures S113–S117). TEM shows no detectable aggregates (see SI Figure S118), whereas in fluorescence microscopy, spherical aggregates similar to those observed in case of 1, were detectable (see SI Figure S119). Dynamic light scattering indicated the presence of aggregates with a diameter of ca. 2 μm (see SI Figure S120).
The presented data for DCLs formed from 1 support a complex self-assembly energy landscape (Scheme 2), where mechanically triggered autocatalysis allows to access specific assembly modes by lowering activation barriers. In the absence of mechanical agitation, the oxidized monomers first form trimers and tetramers, which self-assemble into less defined spherical aggregates. The corresponding part of the (simplified) energy landscape can be represented by a wide and relatively shallow energy minimum, with several local minima, corresponding to the trimer/tetramer aggregates and LMCs, whose mutual interconversion reactions feature low activation barriers. However, among the aggregates formed, also the hexamer assemblies, capable of autocatalytic growth, are present. Nevertheless, these small aggregates (primary nuclei) require a very long time to grow as the number of autocatalytic fiber ends is negligible. This implies a high energy barrier toward the formation of the thermodynamically more stable hexamer nanoribbons. Upon stirring, however, mechanical energy is administered to the system, resulting in the breakage of the primary nuclei. As a result of this process, the number of free fiber ends growth rapidly (potentially exponentially36), which allows for the autocatalytic growth to set in. The primary hexamer assemblies (fibers) serve as a template for the formation of further hexamer molecules, either at their ends (resulting in longer fibers) or at their side (leading to nanoribbons). In other words, mechanical energy supply lowers the activation barrier of hexamer formation by enabling an autocatalytic pathway. DCLs made from building block 2 follow very similar assembly paths compared to DCLs made from 1, except that now the tetramer and not the hexamer is the species that assembles into fibers and ribbons.
Scheme 2. Simplified Potential Energy Landscape of Dynamic Combinatorial Libraries of Building Block 1 in Water.
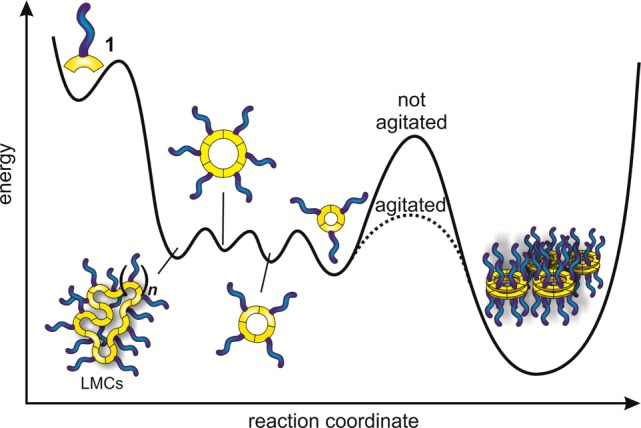
Conclusions
In conclusion we showed for the first time in the context of dynamic combinatorial chemistry that a single building block can give rise to two systems featuring remarkably different modes of self-assembly. Without agitation, self-assembly of cyclic trimers and tetramers into less-defined aggregates and subsequent disulfide exchange leads to a diverse mixture of unprecedentedly large covalent macrocycles. With agitation one specific macrocycle self-assembling into well-defined nanoribbons and -platelets forms in an autocatalytic manner, enabled again by disulfide exchange. Thus, due to the presence of dynamic covalent bonds, the difference in self-assembly modes at the noncovalent level is also reflected at the covalent level. The fact that aggregation is accompanied by a net rearrangement of disulfide bonds only in the former case is most likely a result of the higher thermodynamic stability of the hexamer assemblies (due to the close packing of hexamer units) compared to the ill-defined aggregates of trimers and tetramers. Systems that may be channeled into distinct self-assembly pathways are receiving increasing attention in nanotechnology and materials science in the last years.63−68 Extension of these systems to incorporate a dynamic covalent level, as shown now in our work, opens the way to multifaceted dynamic self-assembling systems of potential interest in the context of materials science and artificial life.
Acknowledgments
Jacob Baas is gratefully acknowledged for the PXRD measurements. Gaël Schaeffer is gratefully acknowledged for fruitful discussions. We are grateful for support from the ERC, NWO, COST Action CM1304, Marie Curie ITN ResMoSys and the Dutch Ministry of Education, Culture and Science (Gravitation Program 024.001.035).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b01814.
Synthetic procedures, NMR spectra, UPLC and LC–MS methods, methods of DCL and sample preparation, mass spectra, (cryo)-TEM and AFM measurements, additional UPLC chromatograms, fluorescence spectra, (cryo)-TEM, AFM, optical and fluorescence microscopy images, powder X-ray diffractogram of 16, additional graphs for the characterization of the self-replication of 1 and 2, as well as the self-assembly properties of the LMCs of 1–3 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Nissen P.; Hansen J.; Ban N.; Moore P. B.; Steitz T. A. Science 2000, 289, 920–930. 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- Suganuma T.; Workman J. L. Annu. Rev. Biochem. 2011, 80, 473–499. 10.1146/annurev-biochem-061809-175347. [DOI] [PubMed] [Google Scholar]
- Meguellati K.; Ladame S. Top. Curr. Chem. 2011, 322, 291–314. 10.1007/128_2011_277. [DOI] [PubMed] [Google Scholar]
- Jin Y.; Yu C.; Denman R. J.; Zhang W. Chem. Soc. Rev. 2013, 42, 6634. 10.1039/c3cs60044k. [DOI] [PubMed] [Google Scholar]
- Jin Y.; Wang Q.; Taynton P.; Zhang W. Acc. Chem. Res. 2014, 47, 1575–1586. 10.1021/ar500037v. [DOI] [PubMed] [Google Scholar]
- Corbett P. T.; Leclaire J.; Vial L.; West K. R.; Wietor J.-L.; Sanders J. K. M.; Otto S. Chem. Rev. 2006, 106, 3652–3711. 10.1021/cr020452p. [DOI] [PubMed] [Google Scholar]
- Otto S. Acc. Chem. Res. 2012, 45, 2200–2210. 10.1021/ar200246j. [DOI] [PubMed] [Google Scholar]
- Cougnon F. B. L.; Sanders J. K. M. Acc. Chem. Res. 2012, 45, 2211–2221. 10.1021/ar200240m. [DOI] [PubMed] [Google Scholar]
- Furlan R. L. E.; Otto S.; Sanders J. K. M. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 4801–4804. 10.1073/pnas.022643699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto S.; Furlan R. L. E.; Sanders J. K. M. Science 2002, 297, 590–593. 10.1126/science.1072361. [DOI] [PubMed] [Google Scholar]
- Lippert A. R.; Naganawa A.; Keleshian V. L.; Bode J. W. J. Am. Chem. Soc. 2010, 132, 15790–15799. 10.1021/ja107314p. [DOI] [PubMed] [Google Scholar]
- Mahon C. S.; Fulton D. A. Chem. Sci. 2013, 4, 3661. 10.1039/c3sc51413g. [DOI] [Google Scholar]
- Hamieh S.; Saggiomo V.; Nowak P.; Mattia E.; Ludlow R. F.; Otto S. Angew. Chem., Int. Ed. 2013, 52, 12368–12372. 10.1002/anie.201305744. [DOI] [PubMed] [Google Scholar]
- Ulatowski F.; Sadowska-Kuziola A.; Jurczak J. J. Org. Chem. 2014, 79, 9762–9770. 10.1021/jo501956h. [DOI] [PubMed] [Google Scholar]
- Riddell I. A.; Ronson T. K.; Clegg J. K.; Wood C. S.; Bilbeisi R. A.; Nitschke J. R. J. Am. Chem. Soc. 2014, 136, 9491–9498. 10.1021/ja504748g. [DOI] [PubMed] [Google Scholar]
- Ratjen L.; Vantomme G.; Lehn J.-M. Chem. - Eur. J. 2015, 21, 10070–10081. 10.1002/chem.201501192. [DOI] [PubMed] [Google Scholar]
- Brachvogel R.-C.; Hampel F.; von Delius M. Nat. Commun. 2015, 6, 7129. 10.1038/ncomms8129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen D. J.; Manuguerra I.; Kjelstrup M. B.; Gothelf K. V. Angew. Chem., Int. Ed. 2014, 53, 14415–14418. 10.1002/anie.201405761. [DOI] [PubMed] [Google Scholar]
- Mondal M.; Hirsch A. K. H. Chem. Soc. Rev. 2015, 44, 2455. 10.1039/C4CS00493K. [DOI] [PubMed] [Google Scholar]
- Grote Z.; Scopelliti R.; Severin K. Angew. Chem., Int. Ed. 2003, 42, 3821–3825. 10.1002/anie.200351623. [DOI] [PubMed] [Google Scholar]
- Severin K. Chem. - Eur. J. 2004, 10, 2565–2580. 10.1002/chem.200305660. [DOI] [PubMed] [Google Scholar]
- Saur I.; Severin K. Chem. Commun. 2005, 41, 1471–1473. 10.1039/b417824f. [DOI] [PubMed] [Google Scholar]
- Corbett P. T.; Otto S.; Sanders J. K. M. Chem. - Eur. J. 2004, 10, 3139–3143. 10.1002/chem.200400300. [DOI] [PubMed] [Google Scholar]
- Corbett P. T.; Sanders J. K. M.; Otto S. J. Am. Chem. Soc. 2005, 127, 9390–9392. 10.1021/ja0509026. [DOI] [PubMed] [Google Scholar]
- Corbett P. T.; Sanders J. K. M.; Otto S. Chem. - Eur. J. 2008, 14, 2153–2166. 10.1002/chem.200701413. [DOI] [PubMed] [Google Scholar]
- Solà J.; Lafuente M.; Atcher J.; Alfonso I. Chem. Commun. 2014, 50, 4564–4566. 10.1039/c4cc00245h. [DOI] [PubMed] [Google Scholar]
- Chung M.-K.; White P. S.; Lee S. J.; Gagné M. R. Angew. Chem., Int. Ed. 2009, 48, 8683–8686. 10.1002/anie.200903478. [DOI] [PubMed] [Google Scholar]
- Cougnon F. B. L.; Au-Yeung H. Y.; Pantoş G. D.; Sanders J. K. M. J. Am. Chem. Soc. 2011, 133, 3198–3207. 10.1021/ja111407m. [DOI] [PubMed] [Google Scholar]
- Ayme J.-F.; Beves J. E.; Leigh D. A.; McBurney R. T.; Rissanen K.; Schultz D. Nat. Chem. 2012, 4, 15–20. 10.1038/nchem.1193. [DOI] [PubMed] [Google Scholar]
- Ponnuswamy N.; Cougnon F. B. L.; Clough J. M.; Pantoş G. D.; Sanders J. K. M. Science 2012, 338, 783–785. 10.1126/science.1227032. [DOI] [PubMed] [Google Scholar]
- Ponnuswamy N.; Cougnon F. B. L.; Pantoş G. D.; Sanders J. K. M. J. Am. Chem. Soc. 2014, 136, 8243–8251. 10.1021/ja4125884. [DOI] [PubMed] [Google Scholar]
- Sadownik J. W.; Philp D. Angew. Chem., Int. Ed. 2008, 47, 9965–9970. 10.1002/anie.200804223. [DOI] [PubMed] [Google Scholar]
- Rubinov B.; Wagner N.; Rapaport H.; Ashkenasy G. Angew. Chem., Int. Ed. 2009, 48, 6683–6686. 10.1002/anie.200902790. [DOI] [PubMed] [Google Scholar]
- Carnall J. M. A.; Waudby C. A.; Belenguer A. M.; Stuart M. C. A.; Peyralans J. J.-P.; Otto S. Science 2010, 327, 1502–1506. 10.1126/science.1182767. [DOI] [PubMed] [Google Scholar]
- Dadon Z.; Samiappan M.; Shahar A.; Zarivach R.; Ashkenasy G. Angew. Chem., Int. Ed. 2013, 52, 9944–9947. 10.1002/anie.201303900. [DOI] [PubMed] [Google Scholar]
- Colomb-Delsuc M.; Mattia E.; Sadownik J. W.; Otto S. Nat. Commun. 2015, 6, 7427. 10.1038/ncomms8427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak P.; Colomb-Delsuc M.; Otto S.; Li J. J. Am. Chem. Soc. 2015, 137, 10965–10969. 10.1021/jacs.5b04380. [DOI] [PubMed] [Google Scholar]
- Clixby G.; Twyman L. Org. Biomol. Chem. 2016, 14, 4170–4184. 10.1039/C6OB00280C. [DOI] [PubMed] [Google Scholar]
- Nguyen R.; Allouche L.; Buhler E.; Giuseppone N. Angew. Chem., Int. Ed. 2009, 48, 1093–1096. 10.1002/anie.200804602. [DOI] [PubMed] [Google Scholar]
- Li J.; Carnall J. M. A.; Stuart M. C. A.; Otto S. Angew. Chem., Int. Ed. 2011, 50, 8384–8386. 10.1002/anie.201103297. [DOI] [PubMed] [Google Scholar]
- Malakoutikhah M.; Peyralans J. J.-P.; Colomb-Delsuc M.; Fanlo-Virgós H.; Stuart M. C. A.; Otto S. J. Am. Chem. Soc. 2013, 135, 18406–18417. 10.1021/ja4067805. [DOI] [PubMed] [Google Scholar]
- Ruiz-Mirazo K.; Briones C.; de la Escosura A. Chem. Rev. 2014, 114, 285–366. 10.1021/cr2004844. [DOI] [PubMed] [Google Scholar]
- Kurihara K.; Okura Y.; Matsuo M.; Toyota T.; Suzuki K.; Sugawara T. Nat. Commun. 2015, 6, 8352. 10.1038/ncomms9352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadownik J. W.; Mattia E.; Nowak P.; Otto S. Nat. Chem. 2016, 8, 264–269. 10.1038/nchem.2419. [DOI] [PubMed] [Google Scholar]
- Szostak J. W.; Bartel D. P.; Luisi P. L. Nature 2001, 409, 387–390. 10.1038/35053176. [DOI] [PubMed] [Google Scholar]
- Luisi P. L.; Ferri F.; Stano P. Naturwissenschaften 2006, 93, 1–13. 10.1007/s00114-005-0056-z. [DOI] [PubMed] [Google Scholar]
- Hammer D. A.; Kamat N. P. FEBS Lett. 2012, 586, 2882–2890. 10.1016/j.febslet.2012.07.044. [DOI] [PubMed] [Google Scholar]
- Mann S. Acc. Chem. Res. 2012, 45, 2131–2141. 10.1021/ar200281t. [DOI] [PubMed] [Google Scholar]
- Caschera F.; Noireaux V. Curr. Opin. Chem. Biol. 2014, 22, 85–91. 10.1016/j.cbpa.2014.09.028. [DOI] [PubMed] [Google Scholar]
- Minkenberg C. B.; Florusse L.; Eelkema R.; Koper G. J. M.; van Esch J. H. J. Am. Chem. Soc. 2009, 131, 11274–11275. 10.1021/ja902808q. [DOI] [PubMed] [Google Scholar]
- Pal A.; Malakoutikhah M.; Leonetti G.; Tezcan M.; Colomb-Delsuc M.; Nguyen V. D.; van der Gucht J.; Otto S. Angew. Chem., Int. Ed. 2015, 54, 7852. 10.1002/anie.201501965. [DOI] [PubMed] [Google Scholar]
- Boekhoven J.; Hendriksen W. E.; Koper G. J. M.; Eelkema R.; van Esch J. H. Science 2015, 349, 1075–1079. 10.1126/science.aac6103. [DOI] [PubMed] [Google Scholar]
- Minkenberg C. B.; Li F.; van Rijn P.; Florusse L.; Boekhoven J.; Stuart M. C. A.; Koper G. J. M.; Eelkema R.; van Esch J. H. Angew. Chem., Int. Ed. 2011, 50, 3421–3424. 10.1002/anie.201007401. [DOI] [PubMed] [Google Scholar]
- Minkenberg C. B.; Homan B.; Boekhoven J.; Norder B.; Koper G. J. M.; Eelkema R.; van Esch J. H. Langmuir 2012, 28, 13570–13576. 10.1021/la302294r. [DOI] [PubMed] [Google Scholar]
- Kim J.; Baek K.; Shetty D.; Selvapalam N.; Yun G.; Kim N. H.; Ko Y. H.; Park K. M.; Hwang I.; Kim K. Angew. Chem., Int. Ed. 2015, 54, 2693–2697. 10.1002/anie.201411842. [DOI] [PubMed] [Google Scholar]
- Careful examination of the mass spectrometry data clearly shows that the large oligomers are cyclic and not linear. See the SI Section 6 for more detailed information.
- Greenspan P. J. Cell Biol. 1985, 100, 965–973. 10.1083/jcb.100.3.965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonetti G.; Otto S. J. Am. Chem. Soc. 2015, 137, 2067–2072. 10.1021/ja512644f. [DOI] [PubMed] [Google Scholar]
- Helttunen K.; Shahgaldian P. New J. Chem. 2010, 34, 2704–2714. 10.1039/c0nj00123f. [DOI] [Google Scholar]
- Jie K.; Zhou Y.; Yao Y.; Huang F. Chem. Soc. Rev. 2015, 44, 3568–3587. 10.1039/C4CS00390J. [DOI] [PubMed] [Google Scholar]
- Hammouda B.; Ho D. L. A.; Kline S. Macromolecules 2004, 37, 6932–6937. 10.1021/ma049623d. [DOI] [Google Scholar]
- Wang Y.; Kim Y.; Lee M. Angew. Chem., Int. Ed. 2016, 55, 13122–13126. 10.1002/anie.201607143. [DOI] [PubMed] [Google Scholar]
- Besenius P.; Portale G.; Bomans P. H. H.; Janssen H. M.; Palmans A. R. A.; Meijer E. W. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 17888–17893. 10.1073/pnas.1009592107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissman H.; Rybtchinski B. Curr. Opin. Colloid Interface Sci. 2012, 17, 330–342. 10.1016/j.cocis.2012.10.001. [DOI] [Google Scholar]
- Wang C.-W.; Sinton D.; Moffitt M. G. ACS Nano 2013, 7, 1424–1436. 10.1021/nn305197m. [DOI] [PubMed] [Google Scholar]
- Jacobs W. M.; Frenkel D. Soft Matter 2015, 11, 8930–8938. 10.1039/C5SM01841B. [DOI] [PubMed] [Google Scholar]
- Tantakitti F.; Boekhoven J.; Wang X.; Kazantsev R. V.; Yu T.; Li J.; Zhuang E.; Zandi R.; Ortony J. H.; Newcomb C. J.; Palmer L. C.; Shekhawat G. S.; de la Cruz M. O.; Schatz G. C.; Stupp S. I. Nat. Mater. 2016, 15, 469–476. 10.1038/nmat4538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y.; Zheng Y.; Xiong W.; Peng C.; Zhang Y.; Duan R.; Che Y.; Zhao J. Sci. Rep. 2016, 6, 27335. 10.1038/srep27335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.