

Abstract
Glycosylations of 4,6-tethered glucosazide donors with a panel of model acceptors revealed the effect of acceptor nucleophilicity on the stereoselectivity of these donors. The differences in reactivity among the donors were evaluated in competitive glycosylation reactions, and their relative reactivities were found to be reflected in the stereoselectivity in glycosylations with a set of fluorinated alcohols as well as carbohydrate acceptors. We found that the 2-azido-2-deoxy moiety is more β-directing than its C-2-O-benzyl counterpart, as a consequence of increased destabilization of anomeric charge development by the electron-withdrawing azide. Additional disarming groups further decreased the α-selectivity of the studied donors, whereas substitution of the 4,6-benzylidene acetal with a 4,6-di-tert-butyl silylidene led to a slight increase in α-selectivity. The C-2-dinitropyridone group was also explored as an alternative for the nonparticipating azide group, but this protecting group significantly increased β-selectivity. All studied donors exhibited the same acceptor-dependent selectivity trend, and good α-selectivity could be obtained with the weakest acceptors and most reactive donors.
Introduction
Glucosamine is a key constituent in numerous important oligosaccharides and glycoconjugates, where it can be either α- or β-linked.1−5 Whereas the former type of linkage can be reliably installed through the use of neighboring-group participation of a C-2-amide- or carbamate-based protecting group, the latter type continues to present a synthetic challenge.6−8 A thorough understanding of the glycosylation mechanism and the influence of both reaction partners and reaction conditions on glycosylation stereoselectivity is needed to enable reliable and predictable glycosylation reactions. The in-depth research conducted on conformationally restricted benzylidene mannose and glucose donors has provided important insight into the glycosylation mechanisms of this type of 1,2-cis-selective donor.9−17 To construct 1,2-cis linkages of glucosamine donors, the C-2-amino group is most commonly masked as the nonparticpating azide.18,19 Notably, benzylidene glucosazides have not been systematically investigated with respect to the stereoselectivity of glycosylations in which they are employed. The extrapolation of the stereoselectivity of benzylidene glucose donors to their glucosazide counterparts suggests that benzylidene or analogously protected glucosazides might represent an attractive class of 1,2-cis-selective glucosamine donor synthons.20,21
Recently, we advocated the use of a comprehensive set of partially fluorinated ethanols, of gradually decreasing nucleophilicity, that can be used to map how the stereoselectivity of a given glycosylation system is dependent on the nucleophilicity of the acceptor.22 The stereoselectivity of the benzylidene glucose donor system has proved to be greatly affected by the reactivity of the nucleophile.23−27 In light of the demand for 1,2-cis-selective glucosaminylations but also with the aim in mind of furthering the understanding of the stereoelectronic effects exerted by the azido group, we set out to systematically evaluate a series of glucosazide donors in a set of glycosylation reactions involving the toolset of partially fluorinated ethanols and a selection of carbohydrate acceptors. As we describe herein, changes in the structure and reactivity of the donor can be effectively mapped using our panel of model acceptors, and we observed a clear reactivity–selectivity relationship for the stereoselectivity of the glycosylations of all donors studied. Differences among the donors and the stereochemical variation in the glycosylation outcome can be explained on the basis of competition experiments and the characterization of the reactive intermediates involved.
Results and Discussion
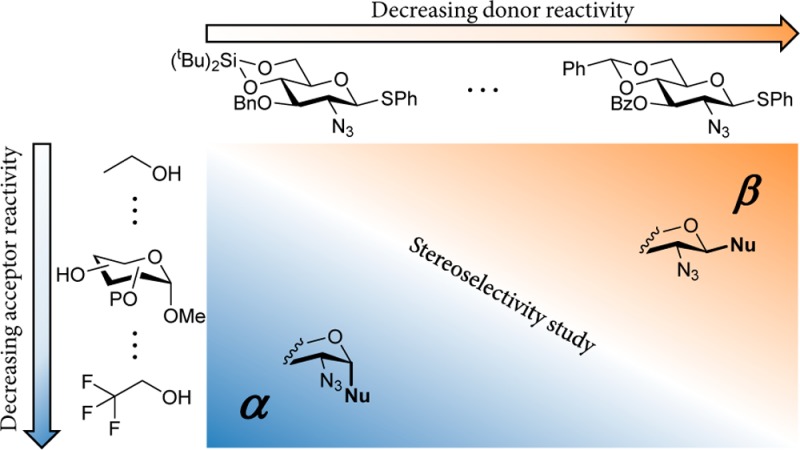
The set of (partially) fluorinated ethanol acceptors that we recently employed to relate the glycosylation stereoselectivity to the acceptor nucleophilicity is depicted in Figure 1 (compounds 6–11). Glycosylating these acceptors with benzylidene mannose, benzylidene glucose, and mannuronic acid donors, as well as fucosazide donors bearing various protecting groups, established the dependence of the stereoselectivity of the glycosylations with these donors on the nucleophilicity of the acceptor.22,28 For benzylidene-protected glucose donor 1, the gradual decrease in acceptor nucleophilicity going from ethanol to monofluoroethanol (MFE), difluoroethanol (DFE), trifluoroethanol (TFE), and hexafluoroisopropanol (HFIP) led to a gradual shift of the stereoselecivity from high β-selectivity to exclusive α-selectivity (see Table 3 below). Here, we present our results from investigating the set of conformationally restricted glucosamine donors depicted in Figure 1 (1–5). Variation in the structure of these donors is found in the cyclic protecting groups (benzylidene vs silylidene), in the functionality at the C-3-OH (benzyl vs benzoyl), and in the nature of the C-2-N-protecting group [azide vs the dinitropyridone (DNPY) group]. The DNPY group is introduced here as a nonparticipating N-protecting group.29,30 The reactivity and selectivity of the set of glucosamine donors are related to the corresponding properties of the well-studied benzylidene glucose donor 1.9,22
Figure 1.

Glucose-configured donors 1–5 and model acceptors 6–11 used in this study.
Table 3. Glycosylations of Donors 1–5 with Model Acceptors 6–11 and Carbohydrate Acceptors 25–29.
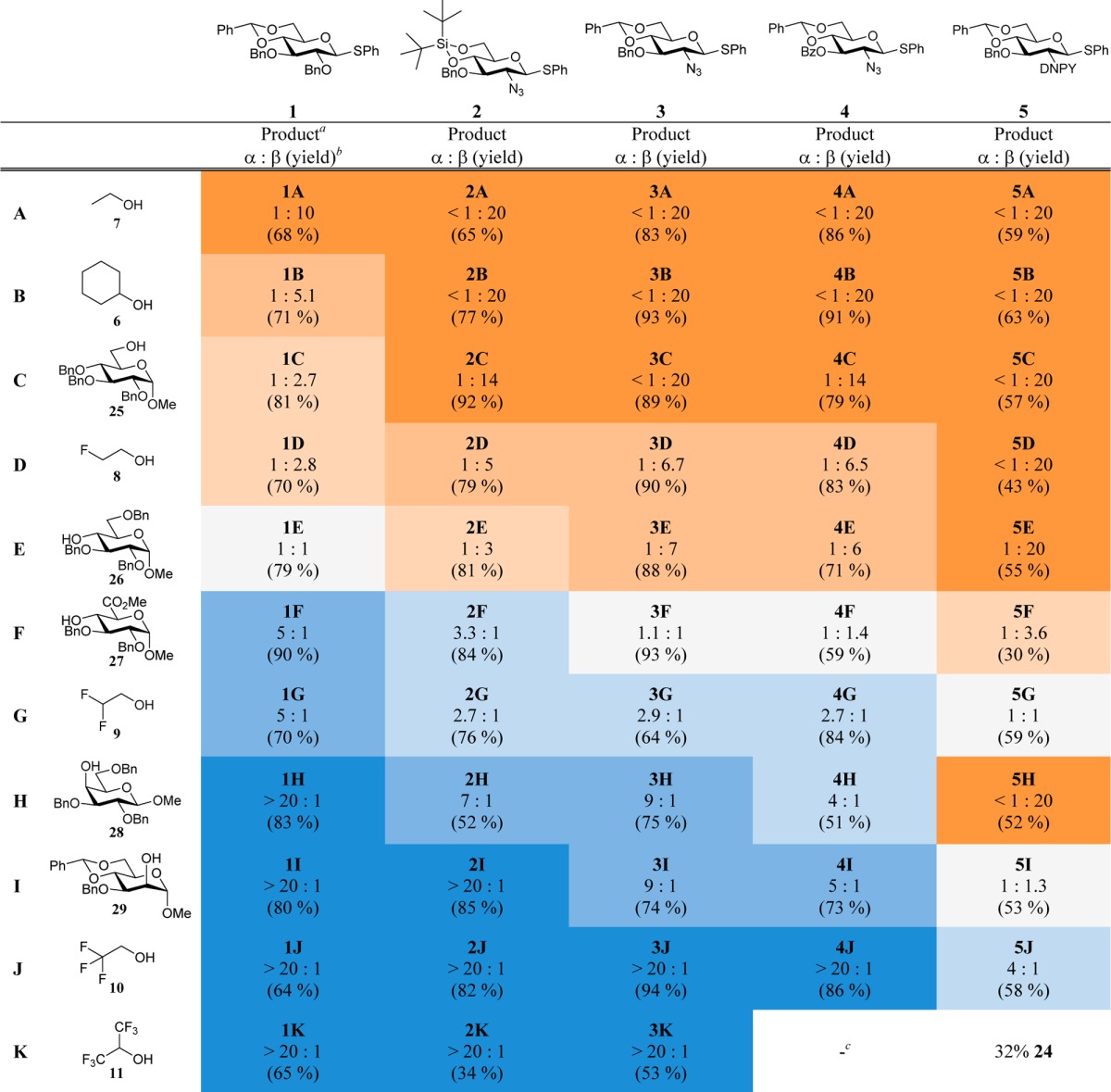
Glycosylation results for donor 1 reported previously by van der Vorm et al.22
Ratio and yield of isolated product after column chromatography; anomers were not separated.
Only the hydrolyzed donor was found.
Synthesis
We prepared benzylidene-protected glucosazide donors 3(31) and 4(32) with an O-benzyl and an O-benzoyl, respectively, at C-3, as well as silylidene-protected donor 2, from common building block 12(33) as depicted in Scheme 1. Hydrolysis of all acetyl esters and the trichloroacetamide was followed by a diazotransfer to install the desired C-2-azide.34 Subsequent introduction of the di-tert-butylsilylidene (DTBS) and the benzylidene acetal gave intermediates 13(31) and 14, respectively. Benzylation of 14 and 13 and benzoylation of 13 gave the target donor compounds 2, 3, and 4, respectively. Donor 5 was prepared in two steps from thioglucoside 15(35) by exchange of the phthaloyl group for the DNPY functionality. To this end, compound 15 was treated with ethylenediamine to give amine 16, which was treated with DNPY reagent 18(30,36) to furnish the target donor.
Scheme 1. Preparation of Donors 2–5.

Reagents and conditions: (a) (i) K2CO3, EtOH, H2O; (ii) CuSO4·5H2O, imidazole-1-sulfonyl azide hydrochloride;34 (b) di-tert-butylsilyl bis(trifluoromethanesulfonate), pyridine, 14 [71% (three steps)]; (c) PhCH(OMe)2, p-TsOH·H2O, 13 [78% (three steps)]; (d) BnBr, NaH, DMF, 2 (80%), 3 (89%); (e) BzCl, DMAP, pyridine, DCM (90%); (f) ethylenediamine, EtOH (88%); (g) 18, AcOH/pyridine (1:16, v/v) (98%); (h) K2CO3, NMP (85%); (i) HNO3, H2SO4 (60%).
Observation of Anomeric Triflates
With these five donors
in hand, we investigated the formation of potential covalent reactive
intermediates in low-temperature NMR studies.37 The donors were treated with the diphenyl sulfoxide/triflic anhydride
(Ph2SO/Tf2O)38 combination
of reagents in deuterated dichloromethane. Figure 2 shows the results of these studies, and Table 1 summarizes the anomeric
chemical shifts of the observed triflates and the temperatures at
which decomposition starts (Tdecomp) [see
also Supporting Information (SI)]. Activation
of reference donor 1 led to the formation of two species:
In addition to the anomeric triflate 19,9 the oxosulfonium triflate 19α* (6.68
ppm, 3.6 Hz) was also formed, as was confirmed by the activation of
a sample containing additional Ph2SO (see SI). Donors 2 and 3 were cleanly
converted to their anomeric α-triflates 20 and 21, respectively, by treatment with the activation couple
at −80 °C. Activation of donor 4 proceeded
more slowly, and an increase of the temperature from −80 to
−35 °C was required for complete activation. Donor 5 proved difficult to study by low-temperature NMR spectroscopy
because of significant line broadening in the resonance sets for both
the donor and the products formed upon activation. Complete activation
of the thioglycoside could only be achieved at −40 °C,
but at this temperature, decomposition of the reactive intermediates
also set in. Two anomeric signals can be discriminated in the spectrum
of the activated DNPY donor 5 (Figure 2), and we assign these as the intermediate
triflate (6.06 ppm) and oxosulfonium triflate (6.54 ppm). Unfortunately,
complete characterization was hampered by the severe line broadening.39 The reactive intermediates formed all decomposed
to give the glucal product 24. The formation of the glucal
double bond is relatively fast, as the proton at C-2 is readily eliminated
to provide the enol ether double bond that is conjugated to the DNPY
aromatic ring.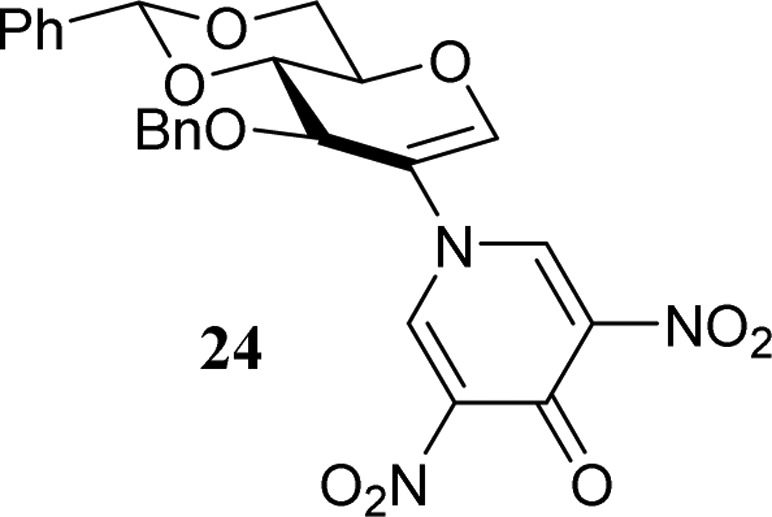
Figure 2.

1H NMR spectra of activated donors 1–5 showing their respective anomeric triflates 19–23.
Table 1. Anomeric Triflates Observed.
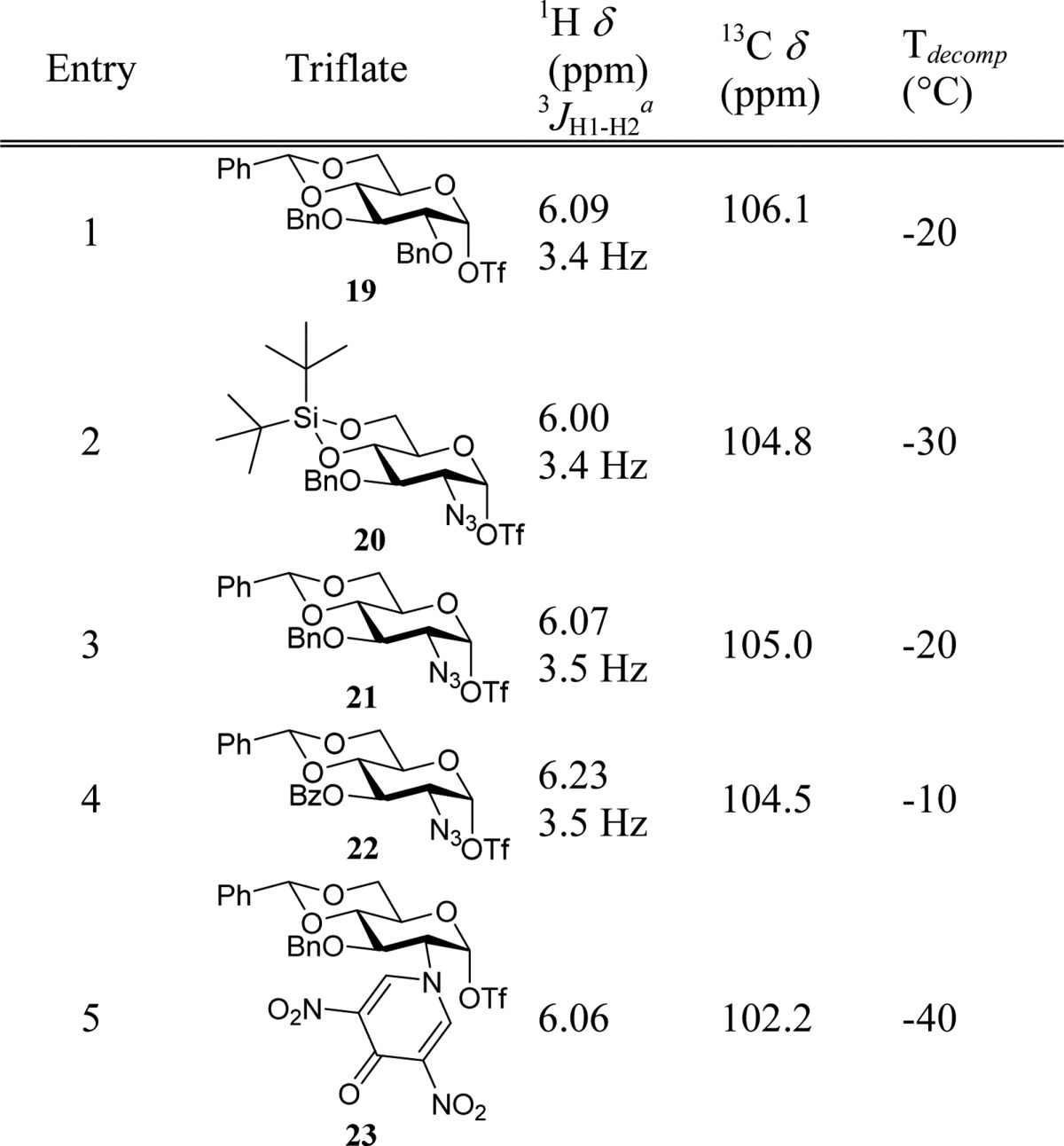
Values determined at −40 °C.
Competitive Glycosylations and Relative Reactivities
To investigate the reactivities
of donors 1–5, a series of competitive
glycosylations were performed between
the different thioglycosides.40−44 In these competition experiments, we used an in situ activation
protocol, employing N-iodosuccinimide (NIS)/trifluoromethanesulfonic
acid (TfOH) as the activator and 2,3,4-tri-O-benzyl-α-O-methyl glucose (25) as the acceptor, as is
commonly done to determine the reactivities of thioglycoside donors.45,46 It should be noted, however, that the reactivity of the thiophenyl
donor does not directly compare with the reactivity of an intermediate
triflate in the glycosylation, although it does provide an indication
of the relative disarming or arming nature of the protecting groups
present on the different donors. It is apparent from Table 2 that the azide has a profound
effect on the reactivity of donor 3, as it is completely
outcompeted by the C-2-O-benzyl donor 1.46 Silylidene donor 2 is
more reactive than donor 3, and the disaccharide products
derived from donors 2 and 3 are formed in
a 6:1 ratio. C-3-O-Benzyl donor 3 in
turn outcompetes benzoylated donor 4 slightly, as a result
of the electron-withdrawing nature of the benzoate, giving a 1.6:1
ratio of the addition products 3C and 4C.47−49 DNPY-protected donor 5 is the least reactive of the
set of donors, as it did not provide any disaccharide product in the
competition experiment with donor 4.
Table 2. Competitive Donor Activations.
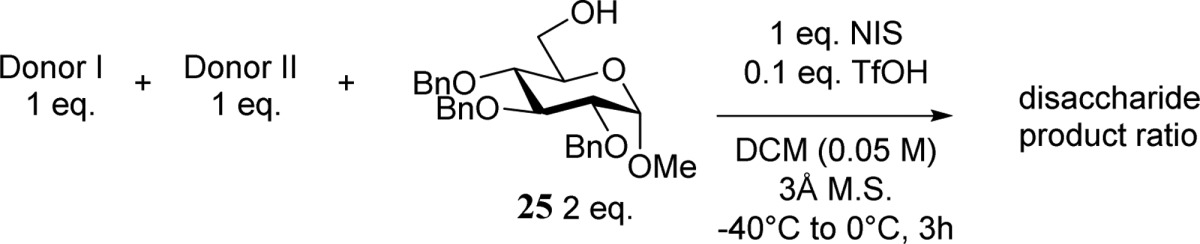
| entry | donor I | donor II | productsa | yieldb (%) |
|---|---|---|---|---|
| 1 | 1 | 2 | 1C/2C, 14:1 | 65 |
| 2 | 1 | 3 | 1C/3C, 1:0 | 80 |
| 3 | 2 | 3 | 2C/3C, 6:1 | 37 |
| 4 | 3 | 4 | 3C/4C, 1.6:1 | 39 |
| 5c | 4 | 5 | 4C/5C, 1:0 | 64 |
Determined by 1H NMR spectroscopy of the isolated disaccharide.
The disaccharide fraction was quantified after isolation by size-exclusion chromatography and related to the limiting amount of NIS (see Experimental Section).
The combined donor concentration was 0.1 M, triflic acid was added at −20 °C, the reaction mixture was heated to +15 °C overnight, and then the reaction was quenched (Et3N).
Glycosylations
With the reactivities of these five donors established, the series of glycosylations with model acceptors 6–11 and carbohydrate acceptors 25–2950−52 was undertaken using the Ph2SO/Tf2O preactivation procedure. Table 3 list all glycosylations ordered by acceptor and donor reactivities. A clear relationship between the acceptor nucleophilicity and the stereochemical outcome of the glycosylation reactions of all studied glucosamine donors was observed, in line with the results previously obtained with donor 1. Upon comparison of the outcomes of the coupling reactions of glucosazide 3 with the results obtained using C-2-O-benzyl donor 1, it becomes apparent that the latter donor reacts with higher α-selectivity. Donor 2, bearing the DTBS group, overall provides slightly more of the α-linked products than its benzylidene counterpart 3. The stereoselectivity of the condensations of donor 4, bearing an additional electron-withdrawing protecting group (i.e., the C-3-O-benzoyl), is very similar to the stereoselectivity observed with C-3-O-benzyl donor 3. Finally, donor 5, carrying the strongly electron-withdrawing DNPY group, is the most β-selective of the series of donors listed in Table 3.53
The selectivities of glycosylations with carbohydrate acceptors were also found to vary in a nucleophilicity-dependent fashion. The primary perbenzylated acceptor 25 reacted similarly to ethanol 7 to give primarily the β-linked products for all glucosamine donors studied. Secondary carbohydrate acceptors that were less nucleophilic showed variations in selectivity with the proportion of α-product increasing with decreasing acceptor reactivity. In line with our previous studies, the nucleophilicities of the secondary equatorial carbohydrate alcohols fall somewhere between the reactivities of MFE and DFE, with the reactivities of the axial hydroxyls approaching the reactivity of TFE. The differences in the reactivities of the donors are reflected in the stereoselectivities of both the glycosylations that involve the model acceptors and the glycosylations with the carbohydrate acceptors. A recurring trend is apparent for all acceptors, with the most reactive donor 1 providing the most α-linked productand the least reactive donor 5 giving the least α-linked product.
Mechanistic Discussion
Two major trends become apparent from the table of glycosylations (Table 3). First, with decreasing acceptor nucleophilicity, the α/β ratio increases. Second, decreasing donor reactivity corresponds to a decrease in the α/β ratio. These trends also emerged during our previous studies involving benzylidene glucose, mannose, mannuronic acid, and fucosazide donors.22,28 The reactive intermediates that can play a role in the glycosylations of the conformationally restricted glucosamine donors and the reaction trajectories of the incoming nucleophiles are presented in Figure 3. Previous studies by the group of Crich have indicated that substitutions on the benzylidene glucosyl triflate 19 proceed in an SN2-like manner. In those mechanistic studies, which involved the determination of kinetic isotope effects and cation-clock methodology, isopropanol was used as an acceptor.12,14 In the kinetic scenario that was proposed, the relatively stable α-trilfate (observed by low-temperature NMR spectroscopy) is in equilibrium with its more reactive β-counterpart. In both species, the triflate can be displaced by alcohols if they are nucleophilic enough. The higher β-selectivity that is seen for the glucosazide and DNPY-glucosamine donors in comparison to donor 1 can be explained by the stronger electron-withdrawing effect of the azide with respect to the benzyl ether. This leads to a more stable covalent α-triflate and favors an associative displacement mechanism. A similar effect was observed by the group of Crich in glycosylations of the analogous 2-deoxy-2-fluoro benzylidene glucosides.54 The DNPY group is even more electron-withdrawing, leading to a further increase in β-selectivity through associative displacement. However, an SN2-like reaction pathway is less likely for the weaker nucleophiles, such as TFE and HFIP. The high α-selectivity for these acceptors can be explained perhaps more precisely by considering the involvement of more electrophilic intermediates such as the glycosyl oxocarbenium ion. The benzylidene and silylidene protecting groups restrict the conformational space that the donor pyranosides can adopt, and the intermediate oxocarbenium ion likely adopt a 3E/3H4-like conformation.55,56 Nucleophiles attack this envelope/half-chair conformer preferentially from the bottom face to lead to the α-linked prodcuts through a chairlike transition state.57 The more reactive donors more readily dissociate to form an oxocarbenium ion, and this accounts for the increased α-selectivity for these donors. Donor 2, bearing the silylidene group is the most reactive of the studied glucosamine donors. It also is slightly more flexible than the benzylidene restricted donors, and these two factors allow the activated donor to more readily form a flattened oxocarbenium ion-like intermediate. Consequently, it is the most α-selective of the studied glucosamine donors. Finally, it is notable that the C-3-O-benzoyl-protected glucosazide 4 reacts in a slightly more β-selective fashion than its C-3-O-benzyl counterpart 3. In light of the discussion above, this makes sense, as the electron-withdrawing benzoyl stabilizes the anomeric α-triflate. It contrasts, however, with the behavior of acyl groups at the C-3 position of benzylidene mannosyl donors. The 1,2-cis selectivity generally observed for these donors can be completely changed to selectively give the α-linked products by installing a C-3-O-acyl group in the donor.58,59 The difference between the benzylidene mannose and benzylidene glucose series can be found in the different geometries that the oxocarbenium ions adopts. For the benzylidene mannose system, a B2,5-like structure is one of the lower-energy oxocarbenium ion conformers.12,55,56 In this constellation, the C-3-benzoate can fold over to the electron-depleted anomeric center to provide stabilization, without a major skeletal rearrangement. For the benzylidene glucose, on the other hand, a B2,5-like structure such as 32 is significantly less favorable because this puts the C-2-azide in a flagpole position. Given the selectivities observed for this donor, influences arising from this boat conformation do not play a significant role here.
Figure 3.

Reactive intermediates and reaction pathways for the 4,6-tethered glucosazide donors.
Conclusions
A set of model acceptors of gradually changing nucleophilicity has been used to investigate how the stereochemistry of glycosylations involving 4,6-tethered glucosamine donors relates to the nucleophilicity of the acceptor. The set of acceptors was complemented by a suite of carbohydrate alcohols to translate the results obtained with the model acceptors to a more relevant glycosylation setting. Four glucosamine donors were probed that differed in the type of tether spanning the C-4 and C-6 alcohols, the nature of the protecting group at the C-3-OH, and the nature of the amino functionality at C-2. Similarly to the previously described benzylidene glucose donor 1, the stereoselectivity of the studied glucosamine donors show a strong correlation to the nucleophilicity of the acceptor, with strong nucleophiles providing completely β-selective condensations and weak nucleophiles selectively leading to the formation of the α-linked products. Benzylidene glucosazide donors are less α-selective than their C-2-O-benzyl congeners, because of the increased electron-withdrawing power of the azide, which retards the formation of an oxocarbenium ion species and favors a more associative mechanistic pathway. We have also introduced a novel protecting group for the C-2-amino group: the dinitropyridone functionality.29,30,36 Although this group is easily installed and removed from the C-2-amine, its strongly electron-withdrawing character limits its use. In the 4,6-benzylidene glucosamine donor studied here, it disarms the donor glycoside to the extent that it turns into a suboptimal glycosyl donor. A major incentive for the reported study was the good to excellent α-selectivity that has previously been reported for benzylidene glucose donor 1. Unfortunately, installation of a 4,6-benzylidene on the analogous glucosazide donors does not provide a reliable donor to affect 1,2-cis-selective glycosylations. Only with relatively poor nucleophiles are useful stereoselectivities obtained. Changing the benzylidene for a silylidene group, however, turns the donor into a more reactive glycosylating agent showing improved α-selectivity. This donor, attractive because of its fully orthogonal protecting group scheme, might find application in the future assembly of oligosaccharides featuring α-glucosamines. Finally, we note that this study provides another illustration of the application of the toolset of partially fluorinated ethanols to efficiently map the reactivity–selectivity relationship of a class of donor glycosides. Implementation of this methodology to investigate novel donor systems will provide broader insights into the different mechanistic pathways at play during glycosylations and eventually generate a complete picture of how to tune both reaction partners to achieve stereoselective glycosylation reactions in a predictable manner.
Experimental Section
General Experimental Procedures
All chemicals were of commercial grade and were used as received unless stated otherwise. Dichloromethane (DCM) was stored over activated 4 Å molecular sieves for at least 24 h before use. Trifluoromethanesulfonic anhydride (Tf2O) was distilled over P2O5 and stored at −20 °C under a nitrogen atmosphere. Triethylamine (Et3N) was distilled over CaH2 and stored over KOH pellets. Overnight temperature control was achieved with an FT902 immersion cooler (Julabo). Flash column chromatography was performed on silica gel 60 Å (0.04–0.063 mm, Screening Devices B.V.). Size-exclusion chromatography was performed on Sephadex (LH-20, GE Healthcare Life Sciences) by isocratic elution with DCM/MeOH (1:1, v/v). Thin-layer chromatography (TLC) analysis was conducted on TLC silica gel 60 (Kieselgel 60 F254, Merck) with UV detection (254 nm) and by spraying with 20% sulfuric acid in ethanol or by spraying with a solution of (NH4)6Mo7O24·H2O (25 g/L) and (NH4)4Ce(SO4)4·2H2O (10 g/L) in 10% aqueous sulfuric acid followed by charring at ±250 °C. High-resolution mass spectroscopy (HRMS) was performed on a Thermo Finnigan LTQ Orbitrap mass spectrometer equipped with an electrospray ion source in positive-ion mode (source voltage 3.5 kV, sheath-gas flow rate 10, capillary temperature 275 °C) with a resolution of R = 60.000 at m/z 400 (mass range of 150–4000). 1H and 13C NMR spectra were recorded on Bruker AV-400, Bruker DMX-400, and Bruker AV-500 NMR instruments. Chemical shifts (δ) are given in parts per million (ppm) relative to tetramethylsilane as an internal standard or the residual signal of the deuterated solvent. Coupling constants (J) are given in hertz (Hz) . All presented 13C-attached proton test (APT) spectra are proton-decoupled. NMR peak assignments were made using correlation spectroscopy (COSY) and heteronuclear single-quantum coherence (HSQC). If necessary, additional nuclear Overhauser enhancement spectroscopy (NOESY), heteronuclear multiple-bond correlation (HMBC), and gated HMBC (HMBC-GATED) experiments were used to further elucidate the structure. The anomeric product ratios were based on careful analysis of the crude reaction mixture and the purified reaction product by integration of representative 1H NMR signals. IR spectra were recorded on a Shimadzu FTIR-8300 IR spectrometer and are reported in wavenumbers (cm–1). Specific rotations were measured on a Propol automatic polarimeter or an Anton-Paar MCP-100 modular circular polarimeter at 589 nm unless otherwise stated.
General Procedure for Tf2O/Ph2SO-Mediated Glycosylations
Donor (0.1 mmol), Ph2SO (26 mg, 0.13 mmol, 1.3 equiv), and TTBP60 (62 mg, 0.25 mmol, 2.5 equiv) were coevaporated twice with dry toluene and dissolved in dry DCM (2 mL, 0.05 M donor). Activated 3 Å molecular sieves (rods, 1/16 in. in size) were added, and the reaction mixture was stirred for 1 h at room temperature under a nitrogen atmosphere. The solution was cooled to −78 °C, and Tf2O (22 μL, 0.13 mmol, 1.3 equiv) was added. The reaction mixture was allowed to warm to −60 °C (donors 1, 2, and 3), −45 °C (donor 5), and −35 °C (donor 4) and then recooled to −78 °C, after which the acceptor (0.2 mmol, 2 equiv) in DCM (0.4 mL, 0.5 M) was added. The reaction mixture was allowed to warm to −40 °C in approximately 90 min and stirred for an additional 0–18 h depending on the acceptor. The reaction was quenched with Et3N (0.1 mL, 0.72 mmol, 5.5 equiv) at −40 °C, and the mixture was diluted with DCM. The solution was transferred to a separatory funnel, water was added, the layers were separated, and the water phase was extracted once more with DCM. The combined organic layers were dried over MgSO4, filtered, and concentrated in vacuo. Purification by silica gel flash column chromatography and, when needed, sephadex LH-20 size-exclusion chromatography yielded the glycosylation product as a mixture of anomers.
General Procedure for NIS/TfOH-Mediated Competition Experiments
Donor I (0.1 mmol, 1 equiv), donor II (0.1 mmol, 1 equiv), and acceptor 25 (0.2 mmol, 2 equiv) were together coevaporated with dry toluene (2×). Dry DCM (4 mL, donor concentration of 0.05 M), a Teflon stirring bar, and 3 Å activated molecular sieves (rods, 1/16 in. in size) were added, and the mixture was stirred under a nitrogen atmosphere for 1 h at room temperature. The mixture was cooled to −40 °C, NIS (0.1 mmol, 1 equiv) was added. TfOH (50 μL of a freshly prepared 0.2 M stock solution in dry DCM, 0.1 equiv) was added, and the mixture was allowed to warm to 0 °C in 3 h. Et3N (0.1 mL) was added, and the mixture was diluted with EtOAc, washed with saturated aqueous NaS2O3 and brine, dried over Na2SO4, and concentrated in vacuo. Size-exclusion chromatography (Sephadex LH-20, 1:1 DCM/MeOH) enabled isolation of the disaccharide products and the monosaccharide remainders, which were both analyzed by NMR spectroscopy. The yield of the disaccharide fraction was determined. For the competition between donors 4 and 5, a 0.1 M concentration and a starting temperature of −20 °C were used, and the mixture was allowed to warm to +15 °C in 18 h.
General Procedure for Low-Temperature NMR Experiments
A mixture of donor (30 μmol) and Ph2SO (39 μmol) was coevaporated with dry toluene twice. [For the activation of donor 1, TTBP (75 μmol) was also added.] Under a nitrogen atmosphere, CD2Cl2 (0.6 mL) was added, and the mixture was transferred to a nitrogen-flushed NMR tube that was then closed with an NMR tube septum. The NMR magnet was cooled to −80 °C, locked, and shimmed and the sample was measured prior to activation. In a long narrow cold bath (EtOH, −85 °C) the sample was treated with Tf2O (39 μmol), shaken thrice, and cooled again after each shaking. The cold sample was wiped dry and quickly inserted back in the cold magnet. The first 1H NMR spectrum was immediately recorded. The sample was then reshimmed, and spectra were recorded in 10 °C intervals with at least 5 min of equilibration time for every temperature.
Phenyl 2-Azido-4,6-O-benzylidene-2-deoxy-1-thio-β-d-glucopyranoside (13)
To a suspension of thioglycoside 12(33) (27.14 g, 50 mmol, 1 equiv) in EtOH (200 mL) were added K2CO3 (41.5 g, 300 mmol, 6 equiv) and 20 mL of H2O, and the mixture was refluxed overnight. The flask was cooled to room temperature, and to the crude free amine61 was added the diazo transfer reagent imidazole-1-sulfonyl azide hydrochloride34 (13.10 g, 62.5 mmol, 1.25 equiv) in three equal portions, followed by a catalytic amount of CuSO4·5 H2O (125 mg, 0.5 mmol, 0.01 equiv). After the solution had been stirred for 5 h, it was filtered and reduced to one-fourth its volume in vacuo. H2O (150 mL) and 1 M aqueous HCl (150 mL) were added to obtain an acidic (pH ∼3) solution, which was extracted with EtOAc (3 × 120 mL). The combined organic layers were washed with saturated aqueous NaHCO3 (150 mL) and brine (150 mL), dried with MgSO4, and concentrated in vacuo to obtain the crude azide phenyl 2-azido-2-deoxy-1-thio-β-d-glucopyranoside.62 The crude azide (≤50 mmol) was coevaporated with toluene twice and subsequently dissolved in DMF (50 mL) and MeCN (200 mL) to which benzaldehyde dimethyl acetal (15 mL, 100 mmol, 2 equiv) and p-TsOH·H2O (950 mg, 5 mmol, 0.1 equiv) had been added. The reaction mixture was heated at 60 °C overnight and then heated at 60 °C for an additional 5 h under reduced pressure (300 mbar) to reduce the volume to one-third. The reaction was quenched by the addition of triethylamine (1 mL), and the reaction mixture was diluted with EtOAc (350 mL) and washed with H2O (2 × 100 mL), saturated aqueous NaHCO3 (1 × 100 mL), and brine (1 × 100 mL). The organic layer was dried (MgSO4) and concentrated in vacuo. The crude mixture was purified by percipitation from hot EtOAc (100 mL)/heptane (300 mL) through the addition of petroleum ether (500 mL) as the mixture was stirred and slowly cooled to 0 °C to obtain the title compound as a white powder (11.38 g, 29.5 mmol, 59%). The mother liquors were purified by flash column chromatography (8:1 to 4:1 pentane/EtOAc) to obtain an additional batch of white solid product (3.8 g, 9.6 mmol, total yield = 39.1 mmol, 78%, three steps). A purified sample could be recrystallized from either hot MeOH or EtOAc/petroleum ether to obtain white cotton-like needles. Rf 0.50 (6:1 pentane/EtOAc). Spectroscopic data were in accord with those reported previously.311H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.64–7.52 (m, 2H, CHarom), 7.50–7.43 (m, 2H, CHarom), 7.42–7.32 (m, 6H, CHarom), 5.53 (s, 1H, CHPh), 4.54 (d, 1H, J = 10.1 Hz, H-1), 4.38 (dd, 1H, J = 10.5, 4.6 Hz, H-6), 3.85–3.70 (m, 2H, H-3, H-6), 3.52–3.40 (m, 2H, H-4, H-5), 3.35 (dd, 1H, J = 10.2, 9.0 Hz, H-6), 2.75 (bs, 1H, 3-OH). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 136.8 (Cq CHPh), 133.8 (CHarom), 130.9 (Cq SPh), 129.6, 129.3, 128.8, 128.5, 126.4 (CHarom), 102.1 (CHPh), 86.9 (C-1), 80.3 (C-4), 74.2 (C-3), 70.4 (C-5), 68.5 (C-6), 65.2 (C-2). HRMS: [M + H]+ calcd for C19H20N3O4S 386.11690, found 386.11708.
Phenyl 2-Azido-2-deoxy-4,6-O-di-tert-butylsilylidene-1-thio-β-d-glucopyranoside (14)
Crude triol phenyl 2-azido-2-deoxy-1-thio-β-d-glucopyranoside (synthesized as described for compound 13) (≤10 mmol) was dissolved in pyridine (15 mL) and cooled to 0 °C. Di-tert-butylsilyl bis(trifluoromethanesulfonate) (3.6 mL, 11 mmol, 1.1 equiv) was slowly added, and the mixture was stirred for 1 h before the reaction was quenched with MeOH. The reaction mixture was diluted with 200 mL of Et2O and washed with 1 M aqueous HCl (3 × 60 mL), saturated aqueous NaHCO3 (60 mL), and brine (60 mL). The organic layer was dried with Na2SO4, filtered, and concentrated in vacuo. Purification by flash column chromatography (1–10% Et2O/pentane) afforded the silylidene-protected title compound as a colorless oil (3.10 g, 7.1 mmol, 71% over three steps). Rf 0.18 (19:1 pentane/Et2O). [α]D23 = −42.6° (c = 1.0, CHCl3). IR (neat): 652, 733, 824, 1072, 1092, 1155, 1277, 1474, 2112, 2859, 2884, 2934, 2963, 3449. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC) δ 7.57–7.51 (m, 2H, CHarom), 7.36–7.31 (m, 3H, CHarom), 4.49 (d, 1H, J = 10.2 Hz, H-1), 4.21 (dd, 1H, J = 10.2, 5.1 Hz, H-6), 3.89 (t, 1H, J = 10.2 Hz, H-6), 3.64 (t, 1H, J = 9.1 Hz, H-4), 3.56 (td, 1H, J = 9.0, 1.2 Hz, H-3), 3.40 (ddd, 1H, J = 10.1, 9.3, 5.1 Hz, H-5), 3.31 (dd, 1H, J = 10.2, 9.1 Hz, H-2), 2.92 (d, 1H, J = 1.6 Hz, 3-OH), 1.04 (s, 9H, CH3tBu), 0.97 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 133.7 (CHarom), 131.2 (Cq), 129.2, 128.7 (CHarom), 86.8 (C-1), 77.4 (C-3), 76.6 (C-4), 74.4 (C-5), 66.0 (C-6), 64.4 (C-2), 27.5, 27.0 (CH3tBu), 22.8, 20.0 (CqtBu). HRMS: [M – N2 + H]+ calcd for C20H32NO4SSi 410.18213, found 410.18220.
Phenyl 2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-1-thio-β-d-glucopyranoside (2)
Compound 14 (1.4 g, 3.2 mmol) was dissolved in DMF (15 mL) and cooled to 0 °C. Benzyl bromide (421 μL, 3.52 mmol, 1.1 equiv) and NaH (60% dispersion in mineral oil, 166 mg, 4.16 mmol, 1.3 equiv) were added, and the reaction mixture was stirred for 2 h at 0 °C and then for 1 h at room temperature. The reaction was quenched with MeOH, and H2O (100 mL) was added. The water phase was extracted three times with 30 mL of Et2O, and the combined organic layers were washed with brine (2×), dried with Na2SO4, and concentrated under reduced pressure. Purification by flash column chromatography (1%–8% Et2O/pentane) yielded compound 2 as a colorless oil (1.35 g, 2.56 mmol, 80%). Additional impurities as observed by 1H NMR spectroscopy originating from the previous crude steps could be removed by size-exclusion chromatography (Sephadex LH-20, 1:1 DCM/MeOH). Rf 0.51 (19:1 pentane/Et2O). [α]D23 = −85.0° (c = 1.0, CHCl3). IR (neat): 654, 694, 746, 766, 826, 1059, 1078, 1099, 1159, 1474, 2110, 2859, 2884, 2934, 2963. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.55–7.48 (m, 2H, CHarom), 7.43–7.37 (m, 2H, CHarom), 7.36–7.27 (m, 6H, CHarom), 4.99 (d, 1H, J = 10.7 Hz, CHH Bn), 4.81 (d, 1H, J = 10.7 Hz, CHH Bn), 4.41 (d, 1H, J = 10.2 Hz, H-1), 4.21 (dd, 1H, J = 10.3, 5.1 Hz, H-6), 3.90 (t, 1H, J = 10.2 Hz, H-6), 3.87 (dd, 1H, J = 9.5, 8.7 Hz, H-4), 3.48–3.38 (m, 2H, H-3, H-5), 3.28 (dd, 1H, J = 10.2, 9.2 Hz, H-2), 1.07 (s, 9H, CH3tBu), 1.01 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 137.9 (Cq Bn), 133.9 (CHarom), 130.9 (Cq SPh), 129.1, 128.7, 128.5, 128.5, 128.1 (CHarom), 86.4 (C-1), 84.2 (C-3), 77.8 (C-4), 75.7 (CH2 Bn), 74.7 (C-5), 66.2 (C-6), 64.2 (C-2), 27.5, 27.1 (CH3tBu), 22.7, 20.0 (CH3tBu). HRMS: [M + H]+ calcd for C27H38N3O4SSi 528.23468, found 528.23451; [M – N2 + H]+ calcd for C27H38NO4SSi 500.22853, found 500.22839.
Phenyl 2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-1-thio-β-d-glucopyranoside (3)
Compound 13 (4.36 g, 11.3 mmol) was coevaporated once with dry toluene and then dissolved in DMF (50 mL) and cooled to 0 °C. Benzyl bromide (1.9 mL, 15.8 mmol, 1.4 equiv) and NaH (60% dispersion in mineral oil, 900 mg, 22.6 mmol, 2 equiv) were added in succession, and the reaction mixture was stirred at room temperature for 4.5 h. MeOH (5 mL) was slowly added, and the reaction mixture was diluted with EtOAc (150 mL) and washed with H2O (2 × 60 mL) and brine (50 mL). The organic layer was dried (MgSO4), filtered, and concentrated in vacuo. The crude product was purified by crystallization (10 mL of hot EtOAc, addition of 100 mL of petroleum ether) to yield the title compound as a white cotton-like solid (4.79 g, 10.1 mmol, 89%). Rf 0.71 (8:1 pentane/EtOAc). Spectroscopic data were in accord with those reported previously.311H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.56 (ddt, 2H, J = 5.0, 3.4, 1.5 Hz, CHarom), 7.47 (dd, 2H, J = 7.5, 2.3 Hz, CHarom), 7.42–7.26 (m, 11H, CHarom), 5.57 (s, 1H, CHPh), 4.91 (d, 1H, J = 10.9 Hz, CHH Bn), 4.78 (d, 1H, J = 10.9 Hz, CHH Bn), 4.49 (d, 1H, J = 10.2 Hz, H-1), 4.39 (dd, 1H, J = 10.6, 5.0 Hz, H-6), 3.79 (t, 1H, J = 10.3 Hz, H-6), 3.71–3.59 (m, 2H, H-3, H-4), 3.52–3.42 (m, 1H, H-5), 3.41–3.32 (m, 1H, H-2). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 137.6, 137.1 (Cq), 134.0 (CHarom), 130.6 (Cq SPh), 129.2, 129.2, 128.8, 128.5, 128.4, 128.4, 128.1, 126.0 (CHarom), 101.3 (CHPh), 86.5 (C-1), 81.3, 81.0 (C-3, C-4), 75.3 (CH2 Bn), 70.5 (C-5), 68.5 (C-6), 64.6 (C-2). HRMS: [M + H]+ calcd for C26H26N3O4S 476.16385, found 476.16375.
Phenyl 2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-1-thio-β-d-glucopyranoside (4)
To a 0 °C solution of compound 13 (1.34 g, 3.48 mmol) in DCM (17 mL) and pyridine (1.4 mL, 34.8 mmol, 5 equiv) were added benzoyl chloride (0.61 mL, 5.22 mmol, 1.5 equiv) and DMAP (42 mg, 0.35 mmol, 0.1 equiv). The reaction mixture was allowed to stir overnight, after which H2O and DCM were added. The organic layer was separated and washed with saturated aqueous NaHCO3 and brine. The organic layer was dried with MgSO4 and concentrated in vacuo. Flash column chromatography (19:1 to 8:1 pentane/EtOAc) afforded the title compound as a white solid (1.54 g, 3.15 mmol, 90%). The product could be recrystallized from EtOAc and petroleum ether to yield a fluffy white solid. Rf 0.53 (8:1 pentane/EtOAc). Spectroscopic data were in accord with those reported previously.631H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.05 (d, 2H, J = 7.3 Hz, CHarom), 7.59 (dd, 2H, J = 6.5, 3.1 Hz, CHarom), 7.53 (t, 1H, J = 7.4 Hz, CHarom), 7.44–7.33 (m, 7H, CHarom), 7.29–7.23 (m, 3H, CHarom), 5.52 (t, 1H, J = 9.6 Hz, H-3), 5.46 (s, 1H, CHPh), 4.69 (d, 1H, J = 10.1 Hz, H-1), 4.38 (dd, 1H, J = 10.5, 4.9 Hz, H-6), 3.79 (t, 1H, J = 10.2 Hz, H-6), 3.71 (t, 1H, J = 9.5 Hz, H-4), 3.62–3.53 (m, 2H, H-2, H-5). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 165.3 (C=O Bz), 136.7 (Cq), 133.7, 133.4 (CHarom), 130.8 (Cq), 129.9, 129.3 (CHarom), 129.2 (Cq), 129.1, 128.8, 128.5, 128.2, 126.1 (CHarom), 101.3 (CHPh), 87.1 (C-1), 78.4 (C-4), 73.5 (C-3), 70.7 (C-5), 68.3 (C-6), 63.9 (C-2). HRMS: [M + H]+ calcd for C26H24N3O5S 490.14312, found 490.14305.
Phenyl 2-Amino-3-O-benzyl-4,6-O-benzylidene-2-deoxy-1-thio-β-d-glucopyranoside (16)
Fully protected glycoside 15(35) (9.11 g, 15.7 mmol) was dissolved in 160 mL of EtOH and heated to reflux, after which ethylene diamine (52 mL, 785 mmol, 50 equiv) was added in three portions and reflux was maintained overnight. The reaction mixture was concentrated under reduced pressure and mixed with toluene (100 mL) and 45 g of silica gel, and the mixture was evaporated to dryness. Column chromatography (8:2 to 2:1 pentane/EtOAc) gave the free amine as a white solid (6.19 g, 13.76 mmol, 88%) that could be recrystallized in EtOAc/petroleum ether. Rf 0.40 (2:1 pentane/EtOAc). mp 136.1–137.5 °C. [α]D20 = −33.5° (c = 0.57, CHCl3). IR (thin film): 698, 748, 1026, 1069, 1123, 1371, 1452, 1583, 2870, 3030, 3059. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.56–7.44 (m, 4H, CHarom), 7.42–7.24 (m, 11H, CHarom), 5.59 (s, 1H, CHPh), 4.99 (d, 1H, J = 11.3 Hz, CHH Bn), 4.68 (d, 1H, J = 11.2 Hz, CHH Bn), 4.58 (d, 1H, J = 9.9 Hz, H-1), 4.38 (dd, 1H, J = 10.5, 5.0 Hz, H-6), 3.81 (t, 1H, J = 10.3 Hz, H-6), 3.72 (t, 1H, J = 9.2 Hz, H-4), 3.59 (t, 1H, J = 9.0 Hz, H-3), 3.52 (td, 1H, J = 9.7, 4.9 Hz, H-5), 2.91 (t, 1H, J = 9.4 Hz, H-2), 1.75 (bs, 2H, NH2). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 138.2, 137.4 (Cq), 133.0 (CHarom), 131.8 (Cq SPh), 129.1, 128.6, 128.4, 128.3, 128.3, 128.0, 126.0 (CHarom), 101.3 (CHPh), 89.6 (C-1), 82.2, 82.2 (C3, C-4), 75.1 (CH2 Bn), 70.7 (C-5), 68.8 (C-6), 55.5 (C-2). HRMS: [M + H]+ calcd for C26H28NO4S 450.17336, found 450.17238.
1-(4-Nitrophenyl)-4-pyridone (17)
Following the procedure of You and Twieg,64 4-hydroxypyridine (14.3 g, 150 mmol), 4-chloronitrobenzene (22.9 g, 145 mmol), and K2CO3 (20.7 g, 150 mmol) were suspended in N-methyl-2-pyrrolidone (110 mL) and heated at 150 °C for 2 h. The hot solution was then poured directly onto ice and allowed to precipitate until all of the ice had melted. The suspension was then filtered and washed four times with cold H2O. The resulting solid was dried under a vacuum at 100 °C until dry. Yield: 26.6 g, 123 mmol, 85%. IR (neat): 606, 692, 741, 752, 843, 1015, 1111, 1198, 1285, 1339, 1495, 1514, 1582, 1638, 3071. 1H NMR (DMSO, 400 MHz, H–H COSY, HSQC): δ 8.38 (d, 2H, J = 9.1 Hz), 8.14 (d, 2H, J = 7.8 Hz), 7.86 (d, 2H, J = 9.1 Hz), 6.29 (d, 2H, J = 7.8 Hz). 13C-APT NMR (DMSO, 101 MHz, HSQC): δ 177.6, 147.1, 145.9, 139.2, 125.3, 123.2, 118.3. HRMS: [M + H]+ calcd for C11H9N2O3 217.06077, found 217.06074.
3,5-Dinitro-1-(4-nitrophenyl)-4-pyridone (18)
In a modification of the procedure of Matsumura et al.,30 an ice-cooled three-neck flask equipped with a condenser was charged with 120 mL of H2SO4 (30% SO3), and then 120 mL of fuming 99% HNO3 was slowly added. To this cold mixture was added pyridone 17 (21.6 g, 100 mmol) in small portions. When addition was complete, the mixture was slowly brought to 130 °C and stirred for 40 h. The mixture was allowed to cool and then poured over ice, stirred for 3 h, filtered, and washed three times with cold water. Yield: 18.4 g, 60 mmol, 60%. Purity (NMR): 90%. Tetranitro [3,5-dinitro-1-(2,4-dinitrophenyl)-4-pyridone 1H NMR (DMSO, 400 MHz): δ 9.42 (s, 2H), 9.05 (d, 1H, J = 2.6 Hz), 8.87 (dd, 1H, J = 8.8, 2.6 Hz), 8.32 (d, 1H, J = 8.7 Hz)] and dinitro [3-nitro-1-(4-nitrophenyl)-4-pyridone 1H NMR (DMSO, 400 MHz): δ 9.18 (d, 1H, J = 2.5 Hz), 8.43 (d, 2H, J = 9.0 Hz), 8.26 (dd, 1H, J = 7.8, 2.5 Hz), 7.99 (d, 2H, J = 9.1 Hz), 6.68 (d, 1H, J = 7.9 Hz)] impurities were present (ratios varied slightly upon repetition). IR (neat): 717, 768, 789, 853, 910, 1141, 1261, 1306, 1350, 1449, 1514, 1591, 1672, 3076. 1H NMR (DMSO, 400 MHz): δ 9.38 (s, 1H), 8.47 (d, 1H, J = 9.0 Hz), 8.05 (d, 1H, J = 9.1 Hz). 13C-APT NMR (DMSO, 101 MHz): δ 159.3, 147.6, 145.5, 142.1, 141.6, 125.7, 125.1. HRMS: [M + H]+ calcd for C11H7N4O7 307.03093, found 307.03123.
Phenyl 2-(3,5-Dinitro-4-pyridone)-3-O-benzyl-4,6-O-benzylidene-2-deoxy-1-thio-β-d-glucopyranoside (5)
Free amine 16 (3.6 g, 8 mmol) and reagent 18 (2.7 g, 8.8 mmol, 1.1 equiv) were dissolved in pyridine (48 mL) and AcOH (4 mL) and left to stir for 30 min. The mixture was diluted with EtOAc and washed with 1 M aqueous HCl (5×) and saturated aqueous NaHCO3 (1×). The organic layer was dried (MgSO4), filtered, and concentrated under reduced pressure. Column chromatography: DCM until all of the nitroanaline had been removed, followed by 1% to 5% acetone in DCM. Yield: 4.84 g, 7.8 mmol (98%) as a yellow solid. Rf 0.21 (DCM), [α]D20 = 10.5° (c = 0.5, CHCl3). IR (thin film): 604, 696, 746, 989, 1055, 1094, 1211, 1300, 1329, 1516, 1674, 2856, 2926, 3034, 3059. 1H NMR (acetone-d6, 400 MHz, H–H COSY, HSQC): δ 8.74 (s, 2H, CH pyridone), 7.63–7.54 (m, 2H, CHarom), 7.51–7.39 (m, 5H, CHarom), 7.39–7.31 (m, 3H, CHarom), 7.21–7.14 (m, 3H, CHarom), 7.14–7.07 (m, 2H, CHarom), 5.84 (s, 1H, CHPh), 5.73 (d, 1H, J = 10.4 Hz, H-1), 4.84 (d, 1H, J = 12.1 Hz, CHH Bn), 4.62 (d, 1H, J = 12.1 Hz, CHH Bn), 4.55–4.47 (m, 1H, H-3), 4.44–4.39 (m, 1H, H-6), 4.39 (t, 1H, J = 8.9 Hz, H-2), 4.06–3.91 (m, 3H, H-4, H-5, H-6). 13C-APT NMR (acetone-d6, 101 MHz, HSQC): δ 159.9 (C=O pyridone), 143.1 (Cq NO2), 138.5, 137.8 (Cq), 133.4 (CHarom), 131.7 (Cq SPh), 130.3, 129.7, 129.5, 129.2, 129.0, 129.0, 127.0 (CHarom), 102.0 (CHPh), 85.9 (C-1), 83.0 (C-4), 77.0 (C-3), 74.7 (CH2 Bn), 71.6 (C-2), 70.9 (C-5), 68.8 (C-6). HRMS: [M + H]+ calcd for C31H28N3O9S 618.15408, found 618.15375.
Trifluoromethanesulfonyl 2,3-Di-O-benzyl-4,6-O-benzylidene-α-d-glucopyranoside (19).9
1H NMR (CD2Cl2, T = 213 K, 400 MHz, H–H COSY, HSQC): δ 6.08 (d, 1H, J = 3.5 Hz, H-1), 5.59 (s, 1H, CHPh), 4.89 (d, 1H, J = 11.0 Hz, CHH Bn), 4.85–4.69 (m, 3H, CHH Bn, CH2 Bn), 4.29 (dd, 1H, J = 10.3, 4.8 Hz, H-6), 4.09–3.94 (m, 2H, H-3, H-5), 3.86–3.70 (m, 3H, H-2, H-4, H-6). 13C-APT NMR (CD2Cl2, T = 213 K, 101 MHz, HSQC): δ 106.1 (C-1), 100.8 (CHPh), 79.6 (C-4), 77.0 (C-3), 76.3 (C-2), 75.0, 74.1 (CH2 Bn), 67.4 (C-6), 65.8 (C-5).
Trifluoromethanesulfonyl 2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-α-d-glucopyranoside (20)
1H NMR (CD2Cl2, T = 243 K, 400 MHz, H–H COSY, HSQC): δ 6.08 (d, 1H, J = 3.5 Hz, H-1), 5.64 (s, 1H, CHPh), 4.98 (d, 1H, J = 10.6 Hz, CHH Bn), 4.78 (d, 1H, J = 10.6 Hz, CHH Bn), 4.32 (dd, 1H, J = 10.4, 4.9 Hz, H-6), 4.11–4.00 (m, 2H, H-3, H-5), 3.94–3.86 (m, 2H, H-2, H-4), 3.82 (t, 1H, J = 10.3 Hz, H-6). 13C-APT NMR (CD2Cl2, T = 243 K, 101 MHz, HSQC): δ 137.2, 136.7 (Cq), 130.5, 128.4, 128.4, 125.9 (CHarom), 105.0 (C-1), 101.3 (CHPh), 80.6 (C-4), 76.4 (C-3), 75.3 (CH2 Bn), 67.6 (C-6), 66.2 (C-5), 61.4 (C-2).
Trifluoromethanesulfonyl 2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-α-d-glucopyranoside (21)
1H NMR (CD2Cl2, T = 243 K, 400 MHz, H–H COSY, HSQC): δ 6.23 (d, 1H, J = 3.5 Hz, H-1), 5.80 (t, 1H, J = 10.0 Hz, H-3), 5.54 (s, 1H, CHPh), 4.36 (dd, 1H, J = 10.4, 4.9 Hz, H-6), 4.21 (td, 1H, J = 9.9, 4.9 Hz, H-5), 4.12 (dd, 1H, J = 10.2, 3.5 Hz, H-2), 3.98 (t, 1H, J = 9.8 Hz, H-4), 3.86 (t, 1H, J = 10.3 Hz, H-6). 13C-APT NMR (CD2Cl2, T = 243 K, 101 MHz, HSQC): δ 104.5 (C-1), 101.8 (CHPh), 77.5 (C-4), 69.3 (C-3), 67.6 (C-6), 66.4 (C-5), 60.9 (C-2).
Trifluoromethanesulfonyl 2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α-d-glucopyranoside (22)
1H NMR (CD2Cl2, T = 233 K, 400 MHz, H–H COSY, HSQC, HMBC): δ 6.00 (d, 1H, J = 3.4 Hz, H-1), 5.08 (d, 1H, J = 10.1 Hz, CHH Bn), 4.81 (d, 1H, J = 10.2 Hz, CHH Bn), 4.15–4.06 (m, 2H, H-4, H-6), 3.95–3.84 (m, 3H, H-3, H-5, H-6), 3.79 (dd, 1H, J = 10.1, 3.4 Hz, H-2), 1.07 (s, 9H, CH3tBu), 1.00 (s, 9H, CH3tBu). 13C-APT NMR (CD2Cl2, T = 233 K, 101 MHz, HSQC, HMBC): δ 118.9 (q, J = 317.6 Hz, CF3), 104.8 (C-1), 78.8 (C-3), 76.9 (C-4), 75.7 (CH2 Bn), 70.0 (C-5), 65.3 (C-6), 60.6 (C-2), 27.0, 26.4 (CH3tBu), 22.5, 19.7 (CqtBu). 13C-HMBC NMR (CD2Cl2, 101 MHz): δ 104.8 (JC1–H1 = 187 Hz, C-1).
3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-d-glucal (24)
Off-white solid. Rf 0.20 (7:3 pentane/EtOAc). [α]D23 = +85.9° (c = 0.32, DCM). IR (thin film): 698, 720, 753, 1007, 1059, 1095, 1192, 1247, 1304, 1351, 1516, 1679, 2880, 2924, 3072. 1H NMR (acetone-d6, 500 MHz, H–H COSY, HSQC): δ 8.72 (s, 2H, CH pyridone), 7.62–7.53 (m, 2H, CHarom), 7.49–7.37 (m, 4H, CHarom, H-1), 7.29–7.14 (m, 5H, CHarom), 5.88 (s, 1H, CHPh), 4.93–4.88 (m, 2H, CHH Bn, H-3), 4.68 (d, 1H, J = 11.8 Hz, CHH Bn), 4.46 (dd, 1H, J = 10.5, 5.2 Hz, H-6), 4.37 (dd, 1H, J = 10.4, 6.9 Hz, H-4), 4.30 (td, 1H, J = 10.2, 5.1 Hz, H-5), 4.03 (t, 1H, J = 10.3 Hz, H-6). 13C-APT NMR (acetone-d6, 101 MHz, HSQC): δ 160.0 (C=O pyridone), 149.3 (C-1), 144.7 (CH pyridone), 142.8 (Cq NO2), 138.4 (Cq Bn, Ph), 129.8, 129.2, 129.1, 129.0, 128.8, 127.0 (CHarom), 122.1 (C-2), 101.9 (CHPh), 80.2 (C-4), 74.7 (CH2 Bn), 74.6 (C-3), 70.7 (C-5), 68.2 (C-6). HRMS: [M + H]+ calcd for C25H22N3O9 508.13506, found 508.13465.
Ethyl 2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-β-d-glucopyranoside (2A)
Donor 2 and ethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (0% to 5% Et2O in pentane) to yield glycosylation product 2A (30 mg, 65 μmol, 65%, α/β < 1:20) as a colorless oil. Rf 0.35 (5% Et2O in pentane). [α]D23 = −69.6° (c = 0.5, CHCl3). IR (neat): 652, 768, 827, 962, 1082, 1161, 1474, 2112, 2859, 2932. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.46–7.40 (m, 2H, CHarom), 7.39–7.27 (m, 3H, CHarom), 4.99 (d, 1H, J = 11.0 Hz, CHH Bn), 4.81 (d, 1H, J = 10.9 Hz, CHH Bn), 4.31 (dd, 1H, J = 7.7, 1.7 Hz, H-1), 4.16 (dd, 1H, J = 10.3, 5.0 Hz, H-6), 3.98–3.87 (m, 3H, CHH—CH3 Et, H-4, H-6), 3.61 (dq, 1H, J = 9.5, 7.1 Hz, CHH—CH3 Et), 3.41–3.28 (m, 3H, H-2, H-3, H-5), 1.26 (t, 3H, J = 7.1 Hz, CH3 Et), 1.08 (s, 9H, CH3tBu), 1.01 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 138.3 (Cq), 128.5, 128.4, 128.0 (CHarom), 102.1 (C-1), 82.4 (C-3), 78.1 (C-4), 75.4 (CH2 Bn), 70.5 (C-5), 66.4 (C-6), 66.1 (CH2 Et), 65.6 (C-2), 27.6, 27.2 (CH3tBu), 22.8, 20.1 (CqtBu), 15.2 (CH3 Et). HRMS: [M – N2 + H]+ calcd for C23H38NO5Si 436.25138, found 436.25132.
Cyclohexyl 2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-β-d-glucopyranoside (2B)
Donor 2 and cyclohexanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (4:1 to 0:1 pentane/toluene) to yield glycosylation product 2B (40 mg, 77 μmol, 77%, α/β < 1:20) as a colorless oil. Rf 0.43 (5% Et2O in pentane). [α]D20 = −44.3° (c = 1.0, CHCl3). IR (thin film): 696, 768, 827, 961, 1080, 1163, 1364, 2112, 2859, 2934. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.45–7.40 (m, 2H, CHarom), 7.38–7.27 (m, 3H, CHarom), 4.97 (d, 1H, J = 11.1 Hz, CHH Bn), 4.81 (d, 1H, J = 11.1 Hz, CHH Bn), 4.42 (d, 1H, J = 7.8 Hz, H-1), 4.15 (dd, 1H, J = 10.3, 5.0 Hz, H-6), 3.99–3.89 (m, 2H, H-3, H-6), 3.64 (tt, 2H, J = 9.2, 3.8 Hz, CH Cy), 3.40–3.24 (m, 3H, H-2, H-4, H-5), 1.96–1.83 (m, 2H, CH2 Cy), 1.80–1.71 (m, 2H, CH2 Cy), 1.55–1.48 (m, 1H, CH2 Cy), 1.47–1.37 (m, 2H, CH2 Cy), 1.34–1.20 (m, 3H, CH2 Cy), 1.08 (s, 9H, tBu), 1.01 (s, 9H, tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 138.4 (Cq), 128.5, 128.3, 127.9 (CHarom), 100.7 (C-1), 82.3 (C-4), 78.3 (CH Cy), 78.0 (C-3), 75.4 (CH2 Bn), 70.5 (C-5), 66.4 (C-6), 65.8 (C-2), 33.6, 31.7 (CH2 Cy), 27.6, 27.2 (CH3tBu), 25.6 (CH2 Cy), 24.1, 23.9 (Cq tBu), 22.8, 20.1 (CH2 Cy). HRMS: [M – N2 + H]+ calcd for C27H44NO5Si 490.29833, found 490.29811.
Methyl 6-O-(2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α/β-d-glucopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (2C)
Donor 2 and acceptor 25 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (1:0 to 9:1 pentane/EtOAc) to yield glycosylation product 2C (81 mg, 92 μmol, 92%, α/β = 1:14) as a white solid. Rf 0.42 (4:1 pentane/EtOAc). [α]D23 = −18.6° (c = 1.0, CHCl3). IR (thin film): 654, 969, 735, 827, 962, 1028, 1070, 1161, 1362, 1454, 2112, 2859, 2931. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 7.45–7.39 (m, 2H, CHarom), 7.38–7.25 (m, 18H, CHarom), 4.99 (d, 1H, J = 11.0 Hz, CHH Bn), 4.98 (d, 1H, J = 10.9 Hz, CHH Bn), 4.94 (d, 1H, J = 11.1 Hz, CHH Bn), 4.85–4.76 (m, 3H, CHH Bn, 2 × CHH Bn), 4.66 (d, 1H, J = 11.1 Hz, CHH Bn), 4.64 (d, 1H, J = 12.1 Hz, CHH Bn), 4.60 (d, 1H, J = 3.6 Hz, H-1), 4.17 (d, 1H, J = 7.9 Hz, H-1′), 4.15–4.10 (m, 1H, H-6′), 4.05–3.96 (m, 2H, H-3, H-6), 3.96–3.87 (m, 2H, H-4′, H-6′), 3.76 (ddd, 1H, J = 9.9, 4.2, 1.7 Hz, H-5), 3.70 (dd, 1H, J = 10.7, 4.2 Hz, H-6), 3.59 (t, 1H, J = 9.5 Hz, H-4), 3.54 (dd, 1H, J = 9.6, 3.5 Hz, H-2), 3.40 (dd, 1H, J = 9.7, 7.9 Hz, H-2′), 3.37–3.26 (m, 5H, CH3 Ome, H-3′, H-5′), 1.07 (s, 9H, CH3tBu), 1.01 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 138.9, 138.6, 138.2, 138.1 (Cq), 128.6, 128.5, 128.5, 128.4, 128.3, 128.1, 128.0, 128.0, 127.9, 127.8, 127.7 (CHarom), 102.2 (C-1′), 98.3 (C-1), 82.5 (C-3′), 82.2 (C-3), 79.9 (C-2), 77.9 (C-4′), 77.7 (C-4), 75.9, 75.4, 75.0, 73.6 (CH2 Bn), 70.6 (C-5′), 69.7 (C-5), 68.6 (C-6), 66.3 (C-6′), 65.6 (C-2′), 55.3 (OMe), 27.5, 27.1 (CH3tBu), 22.8, 20.1 (CqtBu). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 400 MHz): δ 4.87 (d, 1H, J = 3.6 Hz, H-1′), 4.52 (d, 1H, J = 3.4 Hz, H-1). 13C-APT NMR (CDCl3, 101 MHz): δ 98.1, 98.0, 68.3. HRMS: [M + NH4]+ calcd for C49H67N4O10Si 899.46210, found 899.46246.
2-Fluoroethyl 2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α/β-d-glucopyranoside (2D)
Donor 2 and 2-fluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:0 to 0:1 pentane/toluene to 2% Et2O in toluene) to yield glycosylation product 2D (37.8 mg, 79 μmol, 79%, α/β = 1:5.5) as a colorless oil. Rf 0.20 (toluene). Data reported for a 1.00:0.18 mixture of anomers: IR (neat): 654, 768, 827, 962, 1080, 1161, 1472, 2112, 2859, 2932. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.45–7.40 (m, 2.36H, CHarom), 7.39–7.27 (m, 3.54H, CHarom), 5.06 (d, 0.18H, J = 10.7 Hz, CHH Bnα), 4.99 (d, 1H, J = 10.9 Hz, CHH Bnβ), 4.86 (d, 0.18H, J = 3.6 Hz, H-1α), 4.82 (d, 0.18H, J = 10.6 Hz, CHH Bnα), 4.82 (d, 1H, J = 10.9 Hz, CHH Bnβ), 4.71–4.61 (m, 1.18H, CHHFα, CHHFβ), 4.58–4.47 (m, 1.18H, CHHFα, CHHFβ), 4.37 (d, 1H, J = 7.6 Hz, H-1β), 4.17 (dd, 1H, J = 10.3, 5.1 Hz, H-6β), 4.12–3.78 (m, 5.26H, CH2—CH2Fα, CH2—CH2Fβ, H-3α, H-4α, H-4β, H-5α, H-6α, H-6α, H-6β), 3.44–3.29 (m, 3.18H, H-2α, H-2β, H-3β, H-5β), 1.09 (s, 1.62H, CH3tBuα), 1.08 (s, 9H, CH3tBuβ), 1.03 (s, 1.62H CH3tBuα), 1.01 (s, 9H, CH3tBuβ). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 138.3 (Cq,α), 138.2 (Cq,β), 128.6 (CHarom Bnα), 128.5 (CHarom Bnβ), 128.5 (CHarom Bnα), 128.4 (CHarom Bnβ), 128.0 (CHarom Bnα,β), 102.4 (C-1β), 98.3 (C-1α), 82.7 (d, J = 170.0 Hz, CH2Fβ), 82.4 (d, J = 170.6 Hz, CH2Fα), 82.3 (C-3β), 79.3, 79.0 (C-3α, C-4α), 78.0 (C-4β), 75.6 (CH2 Bnα), 75.5 (CH2 Bnβ), 70.6 (C-5β), 69.0 (d, J = 20.3 Hz, CH2—CH2Fβ), 67.3 (d, J = 20.1 Hz, CH2—CH2Fα), 66.8 (C-5α), 66.7 (C-6α), 66.3 (C-6β), 65.5 (C-2β), 62.5 (C-2α), 27.5, 27.1 (CH3tBuα,β), 23.1 (CqtBuα), 22.8 (CqtBuβ), 20.1 (CqtBuα), 20.1 (CqtBuβ). HRMS: [M – N2 + H]+ calcd for C23H37FNO5Si 454.24195, found 454.24188.
Methyl 4-O-(2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α/β-d-glucopyranosyl)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (2E)
Donor 2 and acceptor 26 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (1:0 to 9:1 pentane/EtOAc) to yield glycosylation product 2E (72 mg, 82 μmol, 82%, α/β = 1:3) as a colorless oil. Rf 0.23 and 0.41 (9:1 pentane/EtOAc). IR (thin film): 654, 696, 735, 768, 827, 962, 1090, 1159, 1271, 1362, 1454, 2110, 2859, 2932. Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 7.44–7.39 (m, 2H, CHarom), 7.39–7.21 (m, 18H, CHarom), 4.98 (d, 1H, J = 10.8 Hz, CHH Bn), 4.83–4.74 (m, 4H, CHH Bn, CH2 Bn, CHH Bn), 4.68 (d, 1H, J = 11.9 Hz, CHH Bn), 4.62 (d, 1H, J = 12.2 Hz, CHH Bn) 4.59 (d, 1H, J = 3.6 Hz, H-1), 4.44 (d, 1H, J = 11.9 Hz, CHH Bn), 4.23 (d, 1H, J = 8.0 Hz, H-1′), 3.97 (dd, 1H, J = 10.6, 3.0 Hz, H-6), 3.94–3.73 (m, 5H, H-3, H-4, H-4′, H-5, H-6′), 3.71–3.66 (m, 1H, H-6), 3.55–3.47 (m, 2H, H-2, H-6′), 3.38 (s, 3H, CH3 OMe), 3.27–3.21 (m, 1H, H-2′), 3.20–3.14 (m, 1H, H-3′), 3.06 (td, 1H, J = 9.9, 5.1 Hz, H-5′), 1.06 (s, 9H, CH3tBu), 0.97 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 139.4, 138.4, 138.1, 137.9 (Cq), 128.5, 128.5, 128.5, 128.4, 128.4, 128.2, 128.1, 128.0, 128.0, 127.9, 127.4, 127.3 (CHarom), 101.0 (C-1′), 98.4 (C-1), 82.6 (C-3′), 80.2 (C-3), 79.2 (C-2), 78.1 (C-4′), 77.0 (C-4), 75.3, 75.3, 73.6, 73.6 (CH2 Bn), 70.2 (C-5′), 69.7 (C-5), 68.3 (C-6), 66.2 (C-6′), 66.1 (C-2′), 55.4 (OMe), 27.6, 27.1 (CH3tBu), 22.7, 20.0 (Cq tBu). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.67 (d, 1H, J = 4.0 Hz, H-1), 5.11 (d, 1H, J = 10.6 Hz, CHH Bn), 5.06 (d, 1H, J = 10.6 Hz, CHH Bn), 4.87 (d, 1H, J = 10.6 Hz, CHH Bn), 4.79 (d, 1H, J = 10.6 Hz, CHH Bn), 4.75 (d, 1H, J = 12.0 Hz, CHH Bn), 4.09 (t, 1H, J = 9.0 Hz, H-3), 3.56 (dd, 1H, J = 9.6, 3.5 Hz, H-2), 3.38 (s, CH3 OMe), 3.21 (dd, 1H, J = 10.2, 4.0 Hz, H-2′), 1.06 (s, 9H, CH3tBu), 1.04 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz): δ 138.8, 138.3, 138.2, 138.0, 128.6, 128.4, 128.3, 128.1, 128.0, 128.0, 128.0, 127.9, 127.7, 127.6, 127.4, 127.3, 97.9, 97.7 (C-1, C-1′), 82.1 (C-3), 80.6 (C-2), 79.1, 79.0, 75.6, 75.1, 73.7, 73.4, 69.6, 69.2, 67.5, 66.5, 62.3 (C-2′), 55.4, 27.6, 27.2, 22.8, 20.1. HRMS: [M + NH4]+ calcd for C49H67N4O10Si 899.46210, found 899.46246.
Methyl (Methyl 4-O-[2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α/β-d-glucopyranosyl]-2,3-di-O-benzyl-α-d-glucopyranosyl uronate) (2F)
Donor 2 and acceptor 27 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (1:0 to 9:1 pentane/EtOAc) to yield glycosylation product 2F (69 mg, 84 μmol, 84%, α/β = 3.3:1) as a white solid. Rf 0.36 and 0.39 (9:1 pentane/EtOAc). IR (thin film): 654, 696, 735, 827, 1042, 1144, 1387, 1751, 2108, 2859, 2934. Data for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 7.43–7.38 (m, 2H, CHarom), 7.37–7.25 (m, 13H, CHarom), 5.45 (d, 1H, J = 4.1 Hz, H-1′), 5.07–5.02 (m, 2H, 2 × CHH Bn), 4.90 (d, 1H, J = 10.6 Hz, CHH Bn), 4.84–4.78 (m, 1H, CHH Bn), 4.75 (d, 1H, J = 12.0 Hz, CHH Bn), 4.59 (d, 1H, J = 12.2 Hz, CHH Bn), 4.57 (d, 1H, J = 3.5 Hz, H-1), 4.21–4.17 (m, 1H, H-5), 4.09–4.01 (m, 3H, H-3, H-4, H-6′), 3.91–3.85 (m, 1H, H-4′), 3.83–3.73 (m, 5H, CH3 CO2Me, H-3′, H-6′), 3.62 (td, 1H, J = 10.1, 5.0 Hz, H-5′), 3.58–3.53 (m, 1H, H-2), 3.41 (s, 3H, CH3 OMe), 3.23 (dd, 1H, J = 10.2, 4.1 Hz, H-2′), 1.07 (s, 9H, CH3tBu), 1.05 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 169.2 (C=O CO2Me), 138.7, 138.2, 137.8 (Cq), 128.7, 128.5, 128.5, 128.5, 128.3, 128.2, 128.0, 127.7, 127.6 (CHarom), 98.5, 98.4 (C-1, C-1′), 81.0 (C-3), 79.9 (C-2), 79.0, 79.0 (C-3′, C-4′), 76.2 (C-4), 75.5, 75.4, 73.6 (CH2 Bn), 70.2 (C-5), 67.0 (C-5′), 66.4 (C-6′), 62.4 (C-2′), 55.9 (OMe), 52.9 (CO2Me), 27.6, 27.2 (CH3tBu), 22.9, 20.0 (CqtBu). Diagnostic peaks for the β-anomer: 1H NMR (CDCl3, 400 MHz): δ 4.98 (d, 1H, J = 10.9 Hz, CHH Bn), 4.39 (d, 1H, J = 7.7 Hz, H-1′), 4.02–3.96 (m, 1H), 3.82 (s, 3H, CH3 CO2Me), 3.52 (dd, 1H, J = 9.5, 3.6 Hz, H-2), 1.05 (s, 9H, CH3tBu), 0.97 (s, 9H, CH3, tBu). 13C-APT NMR (CDCl3, 101 MHz): δ 170.2, 139.1, 138.1, 138.1, 128.6, 128.5, 128.4, 128.3, 128.3, 128.1, 128.0, 127.5, 127.3, 101.9 (C-1′), 98.9 (C-1), 82.5, 79.6, 79.4, 78.8, 78.0, 75.4, 73.9, 70.4, 69.9, 66.1, 55.9, 52.8, 27.5, 27.1, 22.8, 20.0. HRMS: [M + NH4]+ calcd for C43H61N4O11Si 837.41006, found 837.41042.
2,2-Difluoroethyl 2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α/β-d-glucopyranoside (2G)
Donor 2 and 2,2-difluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:0 to 0:1 pentane/toluene to 2% Et2O in toluene) to yield glycosylation product 2G (38.1 mg, 76 μmol, 76%, α/β = 2.7:1) in two fractions (24.3 mg of α only, 13.8 mg of α/β = 0.3:1) as white solids. Rf 0.43 β, 0.31 α (toluene). IR (neat): 654, 766, 826, 1070, 1474, 2108, 2860, 2934. Data for the α-anomer: [α]D23 = +35.6° (c = 0.86, CHCl3). 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.45–7.40 (m, 2H, CHarom), 7.39–7.27 (m, 3H, CHarom), 5.95 (tt, 1H, J = 55.2, 4.1 Hz, CHF2), 5.06 (d, 1H, J = 10.6 Hz, CHH Bn), 4.85 (d, 1H, J = 3.6 Hz, H-1), 4.82 (d, 1H, J = 10.7 Hz, CHH Bn), 4.13–4.08 (m, 1H, H-6), 3.98–3.92 (m, 1H, H-3/4), 3.92–3.72 (m, 5H, CH2—CHF2, H-3/4, H-5, H-6), 3.35 (dd, 1H, J = 10.1, 3.6 Hz, H-2), 1.09 (s, 9H, CH3tBu), 1.03 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 138.1 (Cq), 128.6, 128.5, 128.1 (CHarom), 113.8 (t, J = 241.6 Hz, CHF2), 98.7 (C-1), 79.0, 78.9 (C-3, C-4), 75.7 (CH2 Bn), 67.3 (t, J = 28.6 Hz, CH2—CHF2), 67.1 (C-5), 66.6 (C-6), 62.4 (C-2), 27.5, 27.1 (CH3tBu), 22.8, 20.1 (CqtBu). Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.44–7.39 (m, 2H, CHarom), 7.38–7.29 (m, 3H, CHarom), 5.92 (tdd, 1H, J = 55.3, 5.1, 3.4 Hz, CHF2), 4.99 (d, 1H, J = 10.9 Hz, CHH Bn), 4.81 (d, 1H, J = 11.0 Hz, CHH Bn), 4.35 (s, 1H, J = 7.7 Hz, H-1), 4.17 (dd, 1H, J = 10.3, 5.0 Hz, H-6), 4.02–3.74 (m, 4H, CH2—CHF2, H-4, H-6), 3.42–3.30 (m, 3H, H-2, H-3, H-5), 1.09 (s, 9H, CH3tBu), 1.01 (s, 9H, CH3, tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 138.1 (Cq), 128.5, 128.4, 128.1 (CHarom), 114.1 (t, J = 241.4 Hz, CHF2), 102.5 (C-1), 82.2 (C-3), 77.9 (C-4), 75.5 (CH2 Bn), 70.7 (C-5), 68.8 (dd, J = 29.3, 28.8 Hz, CH2—CHF2), 66.2 (C-6), 65.4 (C-2), 27.5, 27.1 (CH3tBu), 22.8, 20.1 (CqtBu). HRMS: [M – N2 + H]+ calcd for C23H36F2NO5Si 472.23253, found 472.23239.
Methyl 4-O-(2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α/β-d-glucopyranosyl)-2,3,6-tri-O-benzyl-β-d-galactopyranoside (2H)
Donor 2 and acceptor 28 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (1:0 to 9:1 pentane/EtOAc) to yield glycosylation product 2H (46 mg, 52 μmol, 52%, α/β = 7:1) as a colorless oil. Rf 0.33 and 0.51 (9:1 pentane/EtOAc). IR (thin film): 652, 696, 735, 826, 1001, 1036, 1206, 1364, 1454, 2108, 2859, 2932. Data for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 7.46–7.41 (m, 2H, CHarom), 7.41–7.23 (m, 18H, CHarom), 5.10 (d, 1H, J = 10.3 Hz, CHH Bn), 4.89–4.81 (m, 3H, CHH Bn, 2 × CHH Bn), 4.79 (d, 1H, J = 3.7 Hz, H-1′), 4.72 (d, 1H, J = 10.6 Hz, CHH Bn), 4.68 (d, 1H, J = 13.0 Hz, CHH Bn), 4.58–4.44 (m, 3H, CH2 Bn, H-5′), 4.23 (d, 1H, J = 7.6 Hz, H-1), 4.07–3.99 (m, 2H, H-4, H-6), 3.99–3.88 (m, 3H, H-3′, H-4′, H-6′), 3.76 (t, 1H, J = 10.1 Hz, H-6′), 3.67–3.58 (m, 2H, H-2, H-6), 3.56 (s, 3H, CH3 OMe), 3.48 (dd, 1H, J = 8.9, 5.5 Hz, H-5), 3.38 (dd, 1H, J = 10.0, 2.9 Hz, H-3), 3.33 (dd, 1H, J = 9.7, 3.7 Hz, H-2′), 1.06 (s, 9H, CH3tBu), 1.02 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 138.8, 138.4, 138.2, 137.7 (Cq), 128.6, 128.6, 128.5, 128.5, 128.5, 128.3, 128.2, 127.9, 127.8 (CHarom), 105.1 (C-1), 99.2 (C-1′), 79.9, 79.9 (C-2, C-3′), 79.6, 79.4 (C-3, C-4′), 75.7, 75.6 (CH2 Bn), 75.0 (C-4), 73.6 (CH2 Bn), 72.9 (C-5), 72.6 (CH2 Bn), 67.1, 67.0 (C-6, C-6′), 66.9 (C-5′), 63.2 (C-2′), 57.5 (OMe), 27.5, 27.3 (CH3tBu), 22.7, 20.2 (CqtBu). Diagnostic peaks for the β-anomer: 1H NMR (CDCl3, 400 MHz): δ 4.94 (d, 0.14H, J = 11.1 Hz, CHH Bn), 4.27 (d, 0.14H, J = 7.7 Hz, H-1), 3.22–3.16 (m, 0.28H), 3.20–3.09 (m, 2H). 13C-APT NMR (CDCl3, 101 MHz): δ 105.1 (C-1), 102.0 (C-1′), 82.3, 78.0, 75.4, 75.3, 73.7, 73.7, 73.5, 73.4, 70.4, 69.6, 66.4, 65.6, 57.3, 27.6, 27.2, 22.8, 20.1. HRMS: [M + NH4]+ calcd for C49H67N4O10Si 899.46210, found 899.46243.
Methyl 2-O-(2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α-d-glucopyranosyl)-3-O-benzyl-4,6-O-benzylidene-α-d-mannopyranoside (2I)
Donor 2 and acceptor 29 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (1:0 to 9:1 pentane/EtOAc) to yield glycosylation product 2I (67 mg, 85 μmol, 85%, α/β > 20:1) as a white solid. Rf 0.54 (9:1 pentane/EtOAc). [α]D20 = +44.3° (c = 1.34, CHCl3). IR (thin film): 696, 827, 937, 1040, 1088, 1130, 1364, 2108, 2859, 2957. Data for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 7.54–7.47 (m, 2H, CHarom), 7.46–7.41 (m, 2H, CHarom), 7.41–7.22 (m, 11H, CHarom), 5.65 (s, 1H, CHPh), 5.23 (d, 1H, J = 3.6 Hz, H-1′), 5.09 (d, 1H, J = 10.6 Hz, CHH Bn), 4.89–4.82 (m, 2H, CHH Bn, CHH Bn), 4.74–4.65 (m, 2H, CHH Bn, H-1), 4.31–4.21 (m, 2H, H-4, H-6), 4.11–3.92 (m, 5H, H-2, H-3, H-3′, H-4′, H-6′), 3.92–3.76 (m, 4H, H-5, H-5′, H-6, H-6′), 3.36 (s, 3H, CH3 OMe), 3.27 (dd, 1H, J = 10.0, 3.7 Hz, H-2′), 1.09 (s, 9H), 1.05 (s, 9H). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 138.6, 138.4, 137.7 (Cq), 129.0, 128.5, 128.5, 128.4, 128.3, 128.3, 128.0, 127.6, 127.5, 127.4, 126.2, 126.1 (CHarom), 101.7 (CHPh), 101.0 (C-1), 99.4 (C-1′), 79.3 (C-4), 79.1, 78.9 (C-3′, C-4′), 76.0, 75.6 (C-2, C-3), 75.6, 73.0 (CH2 Bn), 69.0 (C-6), 67.2 (C-5′), 66.6 (C-6′), 64.1 (C-5), 62.6 (C-2′), 55.2 (CH3 OMe), 27.5, 27.2 (CH3tBu), 22.8, 20.2 (CqtBu). Diagnostic peaks for the β-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.60 (s, 1H, CHPh), 4.98 (d, 1H, J = 11.3 Hz, CHH Bn), 4.39 (d, 1H, J = 8.2 Hz, H-1′), 3.57–3.48 (m, 1H). 13C-APT NMR (CDCl3, 101 MHz): δ 101.6, 99.6, 81.8, 78.5, 77.8, 76.4, 75.2, 74.4, 72.3, 70.9, 65.7, 55.1, 27.5, 27.1. HRMS: [M + NH4]+ calcd for C42H59N4O10Si 807.39950, found 807.39931.
2,2,2-Trifluoroethyl 2-Azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α-d-glucopyranoside (2J)
Donor 2 and 2,2,2-trifluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 30 min at −40 °C) and purified by flash column chromatography (1:0 to 0:1 pentane/toluene to 2% Et2O in toluene) to yield glycosylation product 2J (42.4 mg, 82 μmol, 82%, α/β > 20:1) as a colorless oil. Rf 0.36 (toluene). [α]D23 = +32.6° (c = 1.0, CHCl3). IR (neat): 654, 766, 826, 1036, 1082, 1159, 1279, 1472, 2108, 2859, 2930. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.45–7.39 (m, 2H, CHarom), 7.38–7.27 (m, 3H, CHarom), 5.07 (d, 1H, J = 10.6 Hz, CHH Bn), 4.88 (d, 1H, J = 3.6 Hz, H-1), 4.82 (d, 1H, J = 10.6 Hz, CHH Bn), 4.14–4.07 (m, 1H, H-6), 4.03–3.93 (m, 3H, CH2—CF3, H-4), 3.92–3.80 (m, 3H, H-3, H-5, H-6), 3.36 (dd, 1H, J = 10.0, 3.6 Hz, H-2), 1.09 (s, 9H, CH3tBu), 1.03 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 138.1 (Cq), 128.6, 128.5, 128.1 (CHarom), δ 123.5 (q, J = 278.4 Hz, CF3), 98.8 (C-1), 78.9 (C-3), 78.8 (C-4), 75.7 (CH2 Bn), 67.4 (C-5), 66.5 (C-6), 65.2 (q, J = 35.4 Hz, CH2—CF3), 62.2 (C-2), 27.5, 27.1 (CH3tBu), 22.8, 20.1 (CqtBu). HRMS: [M – N2 + H]+ calcd for C23H35F3NO5Si 490.22311, found 490.22292.
1,1,1,3,3,3-Hexafluoro-2-propyl 2-azido-3-O-benzyl-2-deoxy-4,6-O-di-tert-butylsilylidene-α-d-glucopyranoside (2K)
Donor 2 and 1,1,1,3,3,3-hexafluoro-2-propanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 72 h at −40 °C) and purified by flash column chromatography (4:1 to 0:1 pentane/toluene) to yield glycosylation product 2K (20 mg, 34 μmol, 34%, α/β > 20:1) as a white solid. Rf 0.38 (9:1 pentane/Et2O). [α]D20 = +31.2° (c = 0.50, CHCl3). IR (thin film): 689, 827, 1030, 1098, 1221, 1288, 1368, 2112, 2860, 2934. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC, NOESY): δ 7.45–7.28 (m, 5H, CHarom), 5.12–5.04 (m, 2H, CHH Bn, H-1), 4.83 (d, 1H, J = 10.5 Hz, CHH Bn), 4.40 (hept, 1H, J = 5.7 Hz, CH HFIP), 4.09 (dd, 1H, J = 9.4, 4.0 Hz, H-6), 4.03–3.91 (m, 2H, H-4, H-5), 3.91–3.83 (m, 2H, H-3, H-6), 3.42 (dd, 1H, J = 10.2, 3.8 Hz, H-2), 1.08 (s, 9H, CH3tBu), 1.03 (s, 9H, CH3tBu). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 138.0 (Cq), 128.6, 128.5, 128.2 (CHarom), 100.4 (C-1), 78.7 (C-3), 78.4 (C-4), 75.8 (CH2 Bn), 73.3 (p, J = 33.2 Hz), 68.1 (C-5), 66.1 (C-6), 61.9 (C-2), 27.5, 27.0 (CH3tBu), 22.8, 20.1 (CqtBu). HRMS: [M – N2 + H]+ calcd for C24H34F6NO5Si 558.21050, found 558.21009.
Ethyl 2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-β-d-glucopyranoside (3A)
Donor 3 and ethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:0 to 0:1 hexane/toluene to 5% EtOAc in toluene) to yield glycosylation product 3A (34.3 mg, 83 μmol, 83%, α/β < 1:20) as a white solid. Rf 0.58 (9:1 toluene/EtOAc). [α]D23 = −79.6° (c = 0.69, CHCl3). IR (neat): 692, 993, 1098, 1186, 1267, 1365, 1452, 2111, 2878, 2979. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.50–7.46 (m, 2H, CHarom), 7.41–7.28 (m, 8H, CHarom), 5.57 (s, 1H, CHPh), 4.91 (d, 1H, J = 11.2 Hz, CHH Bn), 4.79 (d, 1H, J = 11.3 Hz, CHH Bn), 4.37 (d, 1H, J = 8.2 Hz, H-1), 4.34 (dd, 1H, J = 10.6, 5.0 Hz, H-6), 3.96 (dq, 1H, J = 9.7, 7.1 Hz, CHH Et), 3.80 (t, 1H, J = 10.3 Hz, H-6), 3.70 (t, 1H, J = 9.0 Hz, H-4), 3.66 (dq, 1H, J = 9.7, 7.2 Hz, CHH Et), 3.54 (t, 1H, J = 9.3 Hz, H-3), 3.44 (dd, 1H, J = 9.5, 8.0 Hz, H-2), 3.39 (td, 2H, J = 9.8, 5.0 Hz, H-5), 1.29 (t, 3H, J = 7.1 Hz, CH3 Et). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 138.0, 137.2 (Cq), 129.2, 128.5, 128.4, 128.3, 128.0, 126.1 (CHarom), 102.5 (C-1), 101.4 (CHPh), 81.7 (C-4), 79.1 (C-3), 75.0 (CH2 Bn), 68.7 (C-6), 66.3 (CH2 Et), 66.3, 66.2 (C-2, C-5), 15.2 (CH3 Et). 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 102.5 (JC1,H1 = 161 Hz, C-1). HRMS: [M + NH4]+ calcd for C22H29N4O5 429.21325, found 429.21321.
Cyclohexyl 2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-β-d-glucopyranoside (3B)
Donor 3 and cyclohexanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:1 to 0:1 hexane/toluene to 5% EtOAc in toluene) to yield glycosylation product 3B (43 mg, 93 μmol, 93%, α/β < 1:20) as a white solid. Rf 0.23 (toluene). [α]D23 = −60.5° (c = 0.86, DCM). IR (neat): 696, 748, 998, 1092, 1275, 1365, 1452, 2108, 2858, 2933. Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.50–7.44 (m, 2H, CHarom), 7.42–7.27 (m, 8H, CHarom), 5.56 (s, 1H, CHPh), 4.90 (d, 1H, J = 11.4 Hz, CHH Bn), 4.79 (d, 1H, J = 11.4 Hz, CHH Bn), 4.47 (d, 1H, J = 7.8 Hz, H-1), 4.32 (dd, 1H, J = 10.5, 5.0 Hz, H-6), 3.79 (t, 1H, J = 10.3 Hz, H-6), 3.74–3.64 (m, 2H, H-4, CH Cyc), 3.50 (t, 1H, J = 9.2 Hz, H-3), 3.44 (dd, 1H, J = 9.6, 7.8 Hz, H-2), 3.36 (td, 1H, J = 9.7, 5.0 Hz, H-5), 1.99–1.87 (m, 2H, CH2 Cyc), 1.82–1.72 (m, 2H, J = 15.2, 4.4 Hz, CH2 Cyc), 1.56–1.37 (m, 3H, CH2 Cyc), 1.36–1.20 (m, 3H, CH2 Cyc). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 138.0, 137.3 (Cq), 129.1, 128.5, 128.4, 128.3, 127.9, 126.1 (CHarom), 101.4 (CHPh), 101.0 (C-1), 81.6 (C-4), 79.0 (C-3), 78.5 (CH Cyc), 75.0 (CH2 Bn), 68.8 (C-6), 66.5 (C-2), 66.3 (C-5), 33.6, 31.8, 25.6, 24.1, 23.9 (CH2 Cyc). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.59 (s, 0.04H, CHPh), 5.03 (d, 0.04H, J = 3.7 Hz, H-1), 4.12 (t, 0.04H, J = 9.5 Hz, H-3), 4.00 (td, 0.04H, J = 9.9, 4.8 Hz, H-5), 3.28 (dd, 0.04H, J = 10.0, 3.7 Hz, H-2). 13C-APT NMR (CDCl3, 101 MHz): δ 101.4 (CHPh), 97.1 (C-1), 76.0 (C-3), 63.8 (C-2), 62.9 (C-5). HRMS: [M + NH4]+ calcd for C26H35N4O5 483.26020, found 483.25991.
Methyl 6-O-(2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-β-d-glucopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (3C)
Donor 3 and acceptor 25 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 4:1 pentane/EtOAc) to yield glycosylation product 3C (73.7 mg, 89 μmol, 89%, α/β < 1:20) as a white solid. Rf 0.42 (4:1 pentane/EtOAc). Spectroscopic data were in accord with those reported previously.38 [α]D23 = −32.2° (c = 1.0, CHCl3). IR (neat): 696, 737, 999, 1028, 1070, 1090, 1277, 1362, 1497, 2108, 2876, 2926. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 7.47 (dd, 2H, J = 7.3, 2.5 Hz, CHarom), 7.42–7.25 (m, 23H, CHarom), 5.55 (s, 1H, CHPh), 4.99 (d, 1H, J = 10.9 Hz, CHH 3-OBn), 4.95 (d, 1H, J = 11.2 Hz, CHH 4-OBn), 4.91 (d, 1H, J = 11.2 Hz, CHH 3′-OBn), 4.85–4.76 (m, 3H, CHH 3-OBn, CHH 2-OBn, CHH 3′-OBn), 4.70–4.63 (m, 2H, CHH 4-OBn, CHH 2-OBn), 4.61 (d, 1H, J = 3.6 Hz, H-1), 4.30 (dd, 1H, J = 10.5, 5.0 Hz, H-6′), 4.23 (d, 1H, J = 7.9 Hz, H-1′), 4.07 (d, 1H, J = 8.9 Hz, H-6), 4.00 (t, 1H, J = 9.3 Hz, H-3), 3.81–3.72 (m, 3H, H-5, H-6, H-6′), 3.69 (t, 1H, J = 9.1 Hz, H-4′), 3.60 (t, 1H, J = 9.3 Hz, H-4), 3.59–3.46 (m, 3H, H-2, H-2′, H-3′), 3.37 (s, 3H, CH3 OMe), 3.36–3.29 (m, 1H, H-5′). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 138.8, 138.5, 138.2, 137.8, 137.2 (Cq), 129.2, 128.6, 128.5, 128.5, 128.4, 128.3, 128.3, 128.1, 128.1, 128.0, 127.9, 127.9, 127.7, 126.1 (CHarom), 102.4 (C-1′), 101.4 (CHPh), 98.4 (C-1), 82.2 (C-3), 81.5 (C-4′), 79.8 (C-2), 79.3 (C-3′), 77.6 (C-4), 75.9 (CH2 3-OBn), 75.0, 75.0 (CH2 3′-OBn, 4-OBn), 73.6 (CH2 2-OBn), 69.6 (C-5), 68.7, 68.6 (C-6, C-6′), 66.3 (C-5′), 66.1 (C-2′), 55.4 (OMe). HRMS: [M + NH4]+ calcd for C48H55N4O10 847.39127, found 847.39224.
2-Fluoroethyl 2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranoside (3D)
Donor 3 and 2-fluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:0 to 0:1 pentane/toluene to 5% EtOAc in toluene) to yield glycosylation product 3D (38.5 mg, 90 μmol, 90%, α/β = 1:6.7) as a white solid. Rf 0.40 (19:1 toluene/EtOAc). IR (neat): 696, 748, 996, 1028, 1072, 1091, 1174, 1276, 1368, 1454, 2108, 2873, 2917. Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.50–7.46 (m, 2H, CHarom), 7.41–7.36 (m, 5H, CHarom), 7.36–7.25 (m, 3H, CHarom), 5.57 (s, 1H, CHPh), 4.91 (d, 1H, J = 11.2 Hz, CHH Bn), 4.79 (d, 1H, J = 11.3 Hz, CHH Bn), 4.69–4.64 (m, 1H, CHHF), 4.55 (dt, 1H, J = 4.6, 2.9 Hz, CHHF), 4.42 (d, 1H, J = 7.9 Hz, H-1), 4.34 (dd, 1H, J = 10.5, 5.0 Hz, H-6), 4.11 (ddd, 0.5H, J = 12.2, 4.8, 2.9 Hz, CHH—CFH2), 4.03 (ddd, 0.5H, J = 12.2, 4.7, 3.0 Hz, CHH—CFH2), 3.92 (ddd, 0.5H, J = 12.2, 5.9, 3.2 Hz, CHH—CFH2), 3.86 (ddd, 0.5H, J = 12.2, 6.0, 3.3 Hz, CHH—CFH2), 3.80 (t, 1H, J = 10.3 Hz, H-6), 3.71 (t, 1H, J = 9.2 Hz, H-4), 3.56 (t, 1H, J = 9.2 Hz, H-3), 3.48 (dd, 1H, J = 9.5, 7.9 Hz, H-2), 3.39 (td, 1H, J = 9.7, 4.9 Hz, H-5). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 137.8, 137.1 (Cq), 129.2, 128.5, 128.4, 128.3, 128.0, 126.1 (CHarom), 102.7 (C-1), 101.4 (CHPh), 82.6 (d, J = 170.1 Hz, CFH2), 81.5 (C-4), 79.0 (C-3), 75.1 (CH2 Bn), 69.3 (d, J = 20.1 Hz, CH2—CFH2), 68.6 (C-6), 66.3 (C-5), 66.1 (C-2). 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 102.7 (JC1,H1 = 162 Hz, C-1). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 5.58 (s, 0.15H, CHPh), 4.96 (d, 0.15H, J = 10.9 Hz, CHH Bn), 4.95 (d, 0.15H, J = 3.7 Hz, H-1), 4.81 (d, 0.15H, J = 11.0 Hz, CHH Bn), 4.29 (dd, 0.15H, J = 10.2, 4.9 Hz, H-6). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 101.5 (CHPh), 98.8 (C-1), 82.8 (C-4), 76.2 (C-3), 75.2 (CH2 Bn), 68.9 (C-6), 63.0, 62.9 (C-2, C-5). 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 98.8 (JC1,H1 = 172 Hz, C-1). HRMS: [M + NH4]+ calcd for C22H28FN4O5 447.20382, found 447.20355.
Methyl 4-O-(2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranosyl)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (3E)
Donor 3 and acceptor 26 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 4:1 pentane/EtOAc) to yield glycosylation product 3E (73.3 mg, 88 μmol, 88%, α/β = 1:7) as a white solid. Rf 0.51 α, 0.43 β (4:1 pentane/EtOAc). IR (neat): 696, 737, 1049, 1092, 1362, 1454, 2110, 2868. Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, TOCSY): δ 7.68–7.60 (m, 2H, CHarom), 7.52–7.18 (m, 23H, CHarom), 5.47 (s, 1H, CHPh), 4.89 (d, 1H, J = 11.2 Hz, CHH Bn), 4.87 (d, 1H, J = 10.9 Hz, CHH Bn), 4.81 (d, 1H, J = 10.9 Hz, CHH Bn), 4.78 (d, 1H, J = 12.2 Hz, CHH Bn), 4.75 (d, 1H, J = 11.2 Hz, CHH Bn), 4.71 (d, 1H, J = 12.0 Hz, CHH Bn), 4.63 (d, 1H, J = 12.1 Hz, CHH Bn), 4.60 (d, 1H, J = 3.7 Hz, H-1), 4.41 (d, 1H, J = 12.0 Hz, CHH Bn), 4.19 (d, 1H, J = 7.6 Hz, H-1′), 4.11 (dd, 1H, J = 10.6, 5.0 Hz, H-6′), 4.00–3.90 (m, 2H, H-4, H-6), 3.85 (t, 1H, J = 9.3 Hz, H-3), 3.75 (dt, 1H, J = 9.8, 2.4 Hz, H-5), 3.69 (dd, 1H, J = 10.8, 1.9 Hz, H-6), 3.56 (t, 1H, J = 9.0 Hz, H-4′), 3.51 (dd, 1H, J = 9.5, 3.7 Hz, H-2), 3.45–3.38 (m, 4H, H-6′, CH3 OMe), 3.36–3.27 (m, 2H, H-2′, H-3′), 3.00 (td, 1H, J = 9.8, 5.0 Hz, H-5′). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 139.3, 138.3, 137.8, 137.8, 137.3 (Cq), 131.1, 129.4, 128.6, 128.4, 128.3, 128.2, 128.2, 128.1, 128.1, 127.9, 127.9, 127.6, 126.0, 124.8 (CHarom), 101.3, 101.2 (CHPh, C-1′), 98.4 (C-1), 81.7 (C-4′), 80.1 (C-3), 79.2 (C-3′), 79.0 (C-2), 76.9 (C-4), 75.4, 74.7, 73.6, 73.5 (CH2 Bn), 69.7 (C-5), 68.6 (C-6′), 68.0 (C-6), 66.6 (C-2′), 65.8 (C-5′), 55.4 (OMe). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.71 (d, 1H, J = 4.0 Hz, H-1′), 5.53 (s, 1H, CHPh), 5.11 (d, 1H, J = 10.7 Hz, CHH Bn), 4.95 (d, 1H, J = 10.9 Hz, CHH Bn). 13C-APT NMR (CDCl3, 101 MHz): δ 98.1, 97.8, 82.7, 82.1, 80.5, 76.2, 75.1, 73.3, 73.0, 69.4, 69.1, 68.7, 63.4, 62.9. HRMS: [M + Na]+ calcd for C48H51N3O10Na 852.34667, found 852.34668.
Methyl (Methyl 4-O-[2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranosyl]-2,3-di-O-benzyl-α-d-glucopyranosyl uronate) (3F)
Donor 3 and acceptor 27 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 4:1 pentane/EtOAc) to yield glycosylation product 3F (71.8 mg, 93 μmol, 93%, α/β = 1.1:1) as a white solid. Rf 0.54 (4:1 pentane/EtOAc). Spectroscopic data were in accord with those reported previously.21 IR (neat): 696, 735, 914, 989, 1028, 1045, 1090, 1267, 1369, 1454, 1749, 2108, 2870, 2916. Data reported for a 1:1 mixture of anomers: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 7.48–7.41 (m, 4H, CHarom), 7.41–7.24 (m, 36H, CHarom), 5.53 (s, 1H, CHPhα), 5.51 (d, 1H, J = 3.9 Hz, H-1′α), 5.47 (s, 1H, CHPhβ), 5.04 (d, 1H, J = 10.5 Hz, CHH Bn), 4.94 (d, 1H, J = 11.0 Hz, CHH Bn), 4.91–4.82 (m, 4H, 2 × CHH Bn, 2 × CHH Bn), 4.81–4.72 (m, 4H, 2 × CHH Bn, 2 × CHH Bn), 4.64–4.58 (m, 2H, 2 × CHH Bn), 4.57 (d, 2H, J = 3.5 Hz, H-1α,β), 4.43 (d, 1H, J = 8.1 Hz, H-1′β), 4.26 (dd, 1H, J = 10.3, 4.8 Hz, H-6′α), 4.24–4.19 (m, 2H, H-5α, H-5β), 4.09–3.99 (m, 4H, H-3β, H-4α, H-4β, H-6′β), 3.97 (t, 1H, J = 9.5 Hz, H-3′α), 3.89 (t, 1H, J = 9.2 Hz, H-3α), 3.82 (s, 3H, CH3 CO2Me), 3.81 (s, 3H, CH3 CO2Me), 3.72–3.56 (m, 4H, H-2β, H-4′α, H-4′β, H-6′α), 3.56–3.46 (m, 3H, H-2α, H-3′β, H-5′α), 3.46–3.38 (m, 7H, 2 × CH3 OMe, H-6′β), 3.36–3.29 (m, 2H, H-2′α, H-2′β), 3.26 (td, 1H, J = 9.7, 5.0 Hz, H-5′β). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 170.0, 170.0 (C=O CO2Me), 139.1, 138.5, 138.0, 137.9, 137.9, 137.8, 137.4, 137.2 (Cq), 129.2, 129.1, 128.7, 128.6, 128.5, 128.5, 128.4, 128.3, 128.3, 128.3, 128.3, 128.2, 128.1, 128.0, 128.0, 127.8, 127.7, 127.5, 127.4, 126.1, 126.1 (CHarom), 102.3 (C-1′β), 101.4 (CHPhβ), 101.3 (CHPhα), 98.9, 98.6 (C-1α, C-1β), 98.5 (C-1′ α), 82.4 (C-4′α), 81.6 (C-4′β), 81.1 (C-3β), 79.6 (C-2β, C-4β), 79.5 (C-3α), 79.4 (C-3′β), 78.7 (C-2α), 76.3 (C-3′α), 75.6 (CH2 Bn), 75.5 (C-4α), 75.1, 75.0, 73.9, 73.7 (CH2 Bn), 70.0, 69.9 (C-5α, C-5β), 68.5, 68.5 (C-6α, C-6β), 66.7 (C-2′β), 66.2 (C-5′β), 63.0 (C-5′α), 62.8 (C-2′α), 55.9, 55.9 (OMe), 53.0, 52.8 (CO2Me). HRMS: [M + NH4]+ calcd for C42H49N4O11 785.33923, found 785.34007.
2,2-Difluoroethyl 2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranoside (3G)
Donor 3 and 2,2-difluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:0 to 0:1 pentane/toluene to 5% EtOAc in toluene) to yield glycosylation product 3G (28.8 mg, 64 μmol, 64%, α/β = 2.9:1) as a white solid. Rf 0.15 and 0.18 (toluene). IR (neat): 698, 747, 998, 1070, 1093, 1372, 1454, 2109, 2867, 2934. Data reported for a 1:0.35 mixture of anomers: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.52–7.45 (m, 2.70H, CHarom), 7.43–7.26 (m, 10.80H, CHarom), 5.95 (tt, 1H, J = 55.2, 4.2 Hz, CF2Hα), 5.94 (tt, 0.35H, J = 55.3, 3.8 Hz, CF2Hβ), 5.58 (s, 1H, CHPhα), 5.57 (s, 0.35H, CHPhβ), 4.96 (d, 1H, J = 10.9 Hz, CHH Bnα), 4.93 (d, 1H, J = 3.9 Hz, H-1α), 4.92 (d, 0.35H, J = 11.3 Hz, CHH Bnβ), 4.80 (d, 1H, J = 11.0 Hz, CHH Bnα), 4.79 (d, 0.35H, J = 11.3 Hz, CHH Bnβ), 4.40 (d, 0.35H, J = 7.9 Hz, H-1β), 4.34 (dd, 0.35H, J = 10.5, 5.0 Hz, H-6β), 4.29 (dd, 1H, J = 10.2, 4.8 Hz, H-6α), 4.08 (t, 1H, J = 9.5 Hz, H-3α), 4.02–3.67 (m, 6.4H, H-4α, H-4β, H-5α, H-6α, H-6β, CH2—CF2Hα, CH2—CF2Hβ), 3.56 (t, 0.35H, J = 9.2 Hz, H-3β), 3.50–3.35 (m, 1.70H, H-2α, H-2β, H-5β). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 137.8, 137.8, 137.1, 137.1 (Cq), 129.4, 129.3, 129.1, 128.6, 128.5, 128.5, 128.4, 128.1, 126.1 (CHarom), 114.0 (t, J = 241.5 Hz, CF2Hβ), 113.8 (t, J = 241.6 Hz, CF2Hα), 102.8 (C-1β), 101.6 (CHPhα), 101.5 (CHPhβ), 99.3 (C-1α), 82.6 (C-4α), 81.4 (C-4β), 78.9 (C-3β), 76.0 (C-3α), 75.3 (CH2 Bnα), 75.1 (CH2 Bn), 69.0 (t, J = 29.0 Hz, CH2—CF2Hβ), 68.8 (C-6), 68.5 (C-6), 67.5 (t, J = 28.7 Hz, CH2—CF2Hα), 66.4 (C-5β), 66.0 (C-2β), 63.3 (C-5α), 62.9 (C-2α). HRMS: [M + H]+ calcd for C22H24F2N3O5 448.16785, found 448.16761.
Methyl 4-O-(2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranosyl)-2,3,6-tri-O-benzyl-β-d-galactopyranoside (3H)
Donor 3 and acceptor 28 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 4:1 pentane/EtOAc) to yield glycosylation product 3H (62.2 mg, 75 μmol, 75%, α/β = 9:1) as a white solid. Rf 0.52 (4:1 pentane/EtOAc). IR (neat): 696, 735, 995, 1030, 1072, 1090, 1368, 1454, 1497, 2106, 2862, 2920. Data for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 7.50–7.46 (m, 2H, CHarom), 7.42–7.25 (m, 23H, CHarom), 5.51 (s, 1H, CHPh), 4.98 (d, 1H, J = 10.7 Hz, CHH 3′-OBn), 4.90 (d, 1H, J = 11.0 Hz, CHH 2-OBn), 4.88 (d, 1H, J = 3.7 Hz, H-1′), 4.84–4.76 (m, 3H, CHH 2-OBn, CHH 3′-OBn, CHH 3-OBn), 4.69 (d, 1H, J = 12.4 Hz, CHH 3-OBn), 4.59–4.51 (m, 2H, CH2 6-OBn), 4.30 (td, 1H, J = 10.1, 4.9 Hz, H-5′), 4.25 (d, 1H, J = 7.6 Hz, H-1), 4.14–4.07 (m, 2H, H-3′, H-4), 4.03 (t, 1H, J = 8.9 Hz, H-6), 3.80 (dd, 1H, J = 10.2, 4.9 Hz, H-6′), 3.70–3.60 (m, 3H, H-2, H-4′, H-6), 3.55 (s, 3H, CH3 OMe), 3.54–3.48 (m, 2H, H-5, H-6′), 3.44–3.36 (m, 2H, H-2′, H-3). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 138.7, 138.3, 138.1, 137.7, 137.6 (Cq), 129.0, 128.6, 128.5, 128.5, 128.4, 128.3, 128.3, 128.3, 128.2, 128.0, 127.7, 127.6, 126.1 (CHarom), 105.3 (C-1), 101.2 (CHPh), 99.4 (C-1′), 83.1 (C-4′), 80.1 (C-3), 78.9 (C-2), 77.0 (C-3′), 75.3 (CH2 3′-OBn), 75.1 (CH2 2-OBn), 74.7 (C-4), 73.6 (CH2 6-OBn), 73.0 (CH2 3-OBn), 72.9 (C-5), 68.9 (C-6′), 67.1 (C-6), 63.8 (C-2′), 62.9 (C-5′), 57.4 (OMe). Diagnostic peaks for the β-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.54 (s, 1H, CHPh), 3.92 (dd, 1H, J = 9.7, 7.7 Hz, H-2), 3.76–3.71 (m, 1H, H-6), 3.22 (td, 1H, J = 9.7, 4.8 Hz, H-5′). 13C-APT NMR (CDCl3, 101 MHz): δ 105.1 (C-1), 102.6 (C-1′), 101.4, 81.5, 81.3, 79.5, 79.0, 74.0, 73.4, 69.4, 68.6, 66.3, 66.0, 57.4. HRMS: [M + NH4]+ calcd for C48H55N4O10 847.39127, found 847.39206.
Methyl 2-O-(2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranosyl)-3-O-benzyl-4,6-O-benzylidene-α-d-mannopyranoside (3I)
Donor 3 and acceptor 29 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 4:1 pentane/EtOAc) to yield glycosylation product 3I (54.7 mg, 74 μmol, 74%, α/β = 9:1) as a white solid. Rf 0.74 (7/2 pentane/EtOAc). IR (neat): 696, 746, 997, 1036, 1074, 1090, 1128, 1371, 1454, 2106, 2862, 2922. Data for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 7.54–7.47 (m, 4H, CHarom), 7.44–7.25 (m, 16H, CHarom), 5.66 (s, 1H, CHPh), 5.60 (s, 1H, CHPh′), 5.39 (d, 1H, J = 3.7 Hz, H-1′), 5.00 (d, 1H, J = 11.0 Hz, CHH Bn), 4.90 (d, 1H, J = 12.2 Hz, CHH Bn), 4.84 (d, 1H, J = 10.9 Hz, CHH Bn), 4.73–4.66 (m, 2H, H-1, CHH Bn), 4.34–4.24 (m, 3H, H-4, H-6, H-6′), 4.17 (dd, 1H, J = 10.2, 9.0 Hz, H-3′), 4.09 (dd, 1H, J = 3.1, 1.7 Hz, H-2), 4.00 (dd, 1H, J = 9.9, 3.1 Hz, H-3), 3.95–3.86 (m, 2H, H-5′, H-6), 3.83–3.70 (m, 3H, H-4′, H-5, H-6′), 3.36 (s, 3H, CH3 OMe), 3.32 (dd, 1H, J = 10.2, 3.8 Hz, H-2′). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 138.7, 138.0, 137.7, 137.2 (Cq), 129.2, 129.0, 128.6, 128.5, 128.4, 128.3, 128.2, 128.0, 127.6, 127.4, 126.2, 126.0 (CHarom), 101.7 (CHPh), 101.5 (CHPh′), 101.0 (C-1), 99.8 (C-1′), 82.9 (C-4′), 79.4 (C-4), 75.9 (C-3), 75.6 (C-3′), 75.5 (C-2), 75.3, 73.3 (CH2 Bn), 69.0, 68.9 (C-6, C-6′), 64.1 (C-5), 63.3 (C-5′), 63.0 (C-2′), 55.0 (OMe). Diagnostic peaks for the β-anomer: 1H NMR (CDCl3, 400 MHz): δ 4.43 (d, 0.1H, J = 8.0 Hz, H-1′), 3.62 (dd, 0.1H, J = 9.6, 8.0 Hz, H-2′), 3.51 (t, 0.1H, J = 9.3 Hz, H-4). 13C-APT NMR (CDCl3, 101 MHz): δ 102.0 (C-1′), 78.5 (C-4), 66.4 (C-2′). HRMS: [M + NH4]+ calcd for C41H47N4O10 755.32867, found 755.32921.
2,2,2-Trifluoroethyl 2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-α-d-glucopyranoside (3J)
Donor 3 and 2,2,2-trifluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 1 h at −40 °C) and purified by flash column chromatography (1:0 to 0:1 pentane/toluene to 5% EtOAc in toluene) to yield glycosylation product 3J (44 mg, 94 μmol, 94%, α/β > 20:1) as a colorless oil. Rf 0.24 (toluene). [α]D23 = +25.9° (c = 0.88, DCM). IR (neat): 697, 747, 1001, 1034, 1090, 1165, 1279, 1373, 2108, 2865, 2934. Data for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.51–7.48 (m, 2H, CHarom), 7.42–7.28 (m, 8H, CHarom), 5.58 (s, 1H, CHPh), 4.99–4.94 (m, 2H, CHH Bn, H-1), 4.80 (d, 1H, J = 10.9 Hz, CHH Bn), 4.29 (dd, 1H, J = 10.2, 4.8 Hz, H-6), 4.10 (dd, 1H, J = 10.0, 9.0 Hz, H-3), 3.98 (qd, 2H, J = 8.5, 3.1 Hz, CH2—CF3), 3.91 (td, 1H, J = 9.9, 4.8 Hz, H-5), 3.79–3.70 (m, 2H, H-4, H-6), 3.43 (dd, 1H, J = 10.0, 3.7 Hz, H-2). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 137.8, 137.1 (Cq), 129.3, 128.6, 128.5, 128.3, 128.1, 126.1 (CHarom), 123.5 (q, J = 278.5 Hz), 101.6 (CHPh), 99.4 (C-1), 82.5 (C-4), 75.9 (C-3), 75.3 (CH2 Bn), 68.7 (C-6), 65.4 (q, J = 35.4 Hz), 63.5 (C-5), 62.7 (C-2). 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 102.5 (JC1,H1 = 173 Hz, C-1). Diagnostic peaks for the β-anomer: 1H NMR (CDCl3, 400 MHz): δ 4.44 (d, 0.03H, J = 7.9 Hz, H-1), 4.34 (dd, 0.03H, J = 10.8, 5.3 Hz, H-6), 3.56 (t, 0.03H, J = 9.2 Hz), 3.48 (dd, 0.03H, J = 10.0, 7.9 Hz, H-2). 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 102.4 (JC–H = 150 Hz, C-1). HRMS: [M + NH4]+ calcd for C22H26F3N4O5 483.18498, found 483.18463.
1,1,1,3,3,3-Hexafluoro-2-propyl 2-Azido-3-O-benzyl-4,6-O-benzylidene-2-deoxy-α-d-glucopyranoside (3K)
Donor 3 and 1,1,1,3,3,3-hexafluoroisopropanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 72 h at −40 °C) and purified by flash column chromatography (1:0 to 9:1 pentane/EtOAc) to yield glycosylation product 3K (28.1 mg, 53 μmol, 53%, α/β > 20:1) as a colorless oil. [α]D23 = +25.8° (c = 0.5, CHCl3). IR (neat): 689, 748, 999, 1092, 1196, 1219, 1287, 1368, 2108, 2868, 2928. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 7.54–7.45 (m, 2H, CHarom), 7.44–7.27 (m, 8H, CHarom), 5.59 (s, 1H, CHPh), 5.13 (d, 1H, J = 3.9 Hz, H-1), 4.99 (d, 1H, J = 10.8 Hz, CHH Bn), 4.82 (d, 1H, J = 10.8 Hz, CHH Bn), 4.40 (hept, 1H, J = 5.9 Hz, CH HFIP), 4.26 (dd, 1H, J = 10.3, 4.9 Hz, H-6), 4.10 (dd, 1H, J = 10.0, 9.1 Hz, H-3), 3.98 (td, 1H, J = 10.0, 4.9 Hz, H-5), 3.80–3.73 (m, 2H, H-4, H-6), 3.51 (dd, 1H, J = 10.1, 3.9 Hz, H-2). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 137.6, 136.9 (Cq), 129.3, 128.6, 128.5, 128.4, 128.2, 126.1 (CHarom), 120.9 (q, J = 283 Hz, CF3), 101.6 (CHPh), 101.0 (C-1), 82.1 (C-4), 75.9 (C-3), 75.5 (CH2 Bn), 74.0, 73.7 (hept, J = 32.8 Hz, CH HFIP), 68.3 (C-6), 64.2 (C-5), 62.5 (C-2). HRMS: [M + H]+ calcd for C23H21F6N3O5 534.14582, found 534.14569.
Ethyl 2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-β-d-glucopyranoside (4A)
Donor 4 and ethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:1:0 to 0:1:0 to 0:19:1 pentane/toluene/EtOAc) to yield glycosylation product 4A (36 mg, 85 μmol, 85%, α/β < 1:20) as a white solid. Rf 0.44 (19:1 toluene/EtOAc). [α]D23 = −53.7° (c = 1.04, DCM). IR (thin film): 709, 1001, 1026, 1070, 1094, 1180, 1263, 1375, 1726, 2110, 2872. Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.11–8.05 (m, 2H, CHarom), 7.60–7.55 (m, 1H, CHarom), 7.48–7.43 (m, 2H, CHarom), 7.40–7.35 (m, 2H, CHarom), 7.32–7.27 (m, 3H, CHarom), 5.50 (s, 1H, CHPh), 5.40 (t, 1H, J = 9.8 Hz, H-3), 4.59 (d, 1H, J = 7.9 Hz, H-1), 4.38 (dd, 1H, J = 10.6, 5.0 Hz, H-6), 4.02 (dq, 1H, J = 9.5, 7.1 Hz, CHH Et), 3.83 (t, 1H, J = 10.3 Hz, H-6), 3.80–3.67 (m, 2H, H-4, CHH Et), 3.64 (dd, 1H, J = 10.0, 7.9 Hz, H-2), 3.56 (td, 1H, J = 9.7, 5.0 Hz, H-5), 1.32 (t, 3H, J = 7.1 Hz, CH3 Et). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 165.5 (C=O Bz), 136.9 (Cq), 133.4, 130.1 (CHarom), 129.7 (Cq), 129.2, 128.6, 128.3, 126.2 (CHarom), 102.8 (C-1), 101.6 (CHPh), 79.0 (C-4), 71.8 (C-3), 68.7 (C-6), 66.6, 66.6 (C-5, CH2 Et), 65.0 (C-2), 15.2 (CH3 Et). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 5.88 (t, 0.03H, J = 9.9 Hz, H-3), 5.53 (s, 0.03H, CHPh), 5.06 (d, 0.03H, J = 3.6 Hz, H-1), 4.32 (dd, 0.03H, J = 10.4, 4.9 Hz, H-6), 3.32 (dd, 0.03H, J = 10.3, 3.6 Hz, H-2). HRMS: [M + NH4]+ calcd for C22H27N4O6 443.19251, found 443.19234.
Cyclohexyl 2-Azido-3-O-benozyl-4,6-O-benzylidene-2-deoxy-β-d-glucopyranoside (4B)
Donor 4 and cyclohexanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:1:0 to 0:1:0 to 0:19:1 pentane/toluene/EtOAc) to yield glycosylation product 4B (43.6 mg, 91 μmol, 91%, α/β < 1:20) as a white solid. Rf 0.18 (toluene). [α]D23 = −41.2° (c = 0.87, DCM). IR (thin film): 613, 708, 999, 1096, 1263, 1730, 2110, 2859, 2934. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.11–8.05 (m, 2H, CHarom), 7.61–7.54 (m, 1H, CHarom), 7.49–7.42 (m, 2H, CHarom), 7.41–7.35 (m, 2H, CHarom), 7.31–7.27 (m, 3H, CHarom), 5.50 (s, 1H, CHPh), 5.38 (t, 1H, J = 9.9 Hz, H-3), 4.70 (d, 1H, J = 7.9 Hz, H-1), 4.37 (dd, 1H, J = 10.6, 5.0 Hz, H-6), 3.89–3.71 (m, 3H, H-4, H-6, CHO Cyc), 3.64 (dd, 1H, J = 10.1, 7.9 Hz, H-2), 3.55 (td, 1H, J = 9.8, 5.0 Hz, H-5), 2.04–1.90 (m, 2H, 2 × CHH Cyc), 1.85–1.73 (m, 2H, 2 × CHH Cyc), 1.58–1.40 (m, 3H, CHH Cyc, 2 × CHH Cyc), 1.39–1.20 (m, 3H, 3 × CHH Cyc). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 165.5 (C = O Bz), 136.9 (Cq), 133.4, 130.0 (CHarom), 129.7 (Cq), 129.2, 128.5, 128.3, 126.2 (CHarom), 101.6 (CHPh), 101.2 (C-1), 79.0 (C-4), 78.8 (CH Cyc), 71.8 (C-3), 68.7 (C-6), 66.6 (C-5), 65.2 (C-2), 33.7, 31.7, 25.6, 24.1, 23.9 (CH2 Cyc). HRMS: [M + NH4]+ calcd for C26H33N4O6 497.23946, found 497.23932.
Methyl 6-O-(2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (4C)
Donor 4 and acceptor 25 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 3/1 pentane/EtOAc) to yield glycosylation product 4C (67 mg, 79 μmol, 79%, α/β = 1:14) as a white solid. Rf 0.24 (4:1 pentane/EtOAc). [α]D20 = −17.5° (c = 1.34, CHCl3). IR (thin film): 696, 748, 1002, 1028, 1068, 1092, 1263, 1313, 1369, 1452, 1730, 2110, 2872, 2918. Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 8.09–8.04 (m, 2H, CHarom), 7.60–7.53 (m, 1H, CHarom), 7.48–7.41 (m, 2H, CHarom), 7.40–7.24 (m, 20H, CHarom), 5.47 (s, 1H, CHPh), 5.42 (t, 1H, J = 9.8 Hz, H-3′), 5.00 (d, 1H, J = 10.9 Hz, CHH Bn), 4.95 (d, 1H, J = 11.1 Hz, CHH Bn), 4.84 (d, 1H, J = 11.0 Hz, CHH Bn), 4.80 (d, 1H, J = 12.2 Hz, CHH Bn), 4.68–4.63 (m, 2H, 2 × CHH Bn), 4.61 (d, 1H, J = 3.5 Hz, H-1), 4.43 (d, 1H, J = 8.0 Hz, H-1′), 4.33 (dd, 1H, J = 10.5, 5.0 Hz, H-6′), 4.12 (d, 1H, J = 9.1 Hz, H-6), 4.02 (t, 1H, J = 9.3 Hz, H-3), 3.84–3.72 (m, 4H, H-4′, H-5, H-6, H-6′), 3.69 (dd, 1H, J = 9.9, 8.0 Hz, H-2′), 3.57 (t, 1H, J = 9.2 Hz, H-4), 3.54 (dd, 1H, J = 9.7, 3.6 Hz, H-2), 3.49 (td, 1H, J = 9.8, 5.0 Hz, H-5′), 3.39 (s, 3H, CH3 OMe). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 165.4 (C=O), 138.7, 138.3, 138.2, 136.7 (CHarom), 133.5, 130.0 (CHarom), 129.4 (Cq Bz), 129.1, 128.6, 128.6, 128.5, 128.5, 128.3, 128.3, 128.1, 128.0, 128.0, 127.9, 127.9, 127.7, 126.1 (CHarom), 102.6 (C-1′), 101.5 (CHPh), 98.3 (C-1), 82.1 (C-3), 79.8 (C-2), 78.7 (C-4′), 77.7 (C-4), 75.8, 75.0, 73.6 (CH2 Bn), 71.9 (C-3′), 69.7 (C-5), 68.9 (C-6), 68.5 (C-6′), 66.6 (C-5′), 65.2 (C-2′), 55.4 (CH3 OMe). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.79 (t, 0.07H, J = 9.9 Hz, H-3′), 5.50 (s, 0.07H, CHPh), 5.08 (d, 0.07H, J = 3.5 Hz, H-1′), 4.24 (dd, 0.07H, J = 10.3, 4.8 Hz, H-6′), 3.41 (s, 0.21H, CH3 OMe). 13C-APT NMR (CDCl3, 101 MHz): δ 101.7 (CHPh), 99.3 (C-1′), 98.1 (C-1), 82.1, 80.0, 79.7, 77.6, 75.8, 75.2, 70.0, 69.4, 67.0, 62.8, 62.1, 55.4. HRMS: [M + NH4]+ calcd for C48H53N4O11 861.37053, found 861.37064.
2-Fluoroethyl 2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranoside (4D)
Donor 4 and 2-fluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:1:0 to 0:1:0 to 0:16:1 pentane/toluene/EtOAc) to yield glycosylation product 4D (36.6 mg, 83 μmol, 83%, α/β = 1:6.5) as a white solid. Rf 0.41 (19:1 toluene/EtOAc). IR (thin film): 700, 748, 879, 1001, 1026, 1070, 1093, 1179, 1261, 1722, 2108, 2868. Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.10–8.05 (m, 2H, CHarom), 7.61–7.54 (m, 1H, CHarom), 7.49–7.43 (m, 2H, CHarom), 7.41–7.36 (m, 2H, CHarom), 7.33–7.27 (m, 3H, CHarom), 5.50 (s, 1H, CHPh), 5.41 (t, 1H, J = 9.8 Hz, H-3), 4.69 (ddt, 1H, J = 4.6, 3.2, 1.8 Hz, CHH—CH2F), 4.65 (d, 1H, J = 7.9 Hz, H-1), 4.58 (ddt, 1H, J = 4.5, 3.3, 1.8 Hz, CHH—CH2F), 4.38 (dd, 1H, J = 10.5, 4.9 Hz, H-6), 4.14 (dddd, 1H, J = 30.3, 11.9, 4.6, 3.1 Hz, CHHF), 3.95 (dddd, 1H, J = 27.1, 12.1, 5.5, 3.4 Hz, CHHF), 3.83 (t, 1H, J = 10.3 Hz, H-6), 3.79 (t, 1H, J = 9.5 Hz, H-4), 3.69 (dd, 1H, J = 10.0, 7.9 Hz, H-2), 3.57 (td, 1H, J = 9.7, 5.0 Hz, H-5). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 165.4 (C=O), 136.8 (Cq Ph), 133.5, 130.0 (CHarom), 129.5 (Cq Bz), 129.2, 128.6, 128.5, 128.3, 126.3, 126.2 (CHarom), 103.0 (C-1), 101.6 (CHPh), 82.5 (d, J = 170.5 Hz, CH2F), 78.8 (C-4), 71.7 (C-3), 69.5 (d, J = 20.2 Hz, CH2—CH2F), 68.5 (C-6), 66.7 (C-5), 64.9 (C-2). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.89 (t, 0.17H, J = 9.9 Hz, H-3), 5.53 (s, 0.17H, CHPh), 5.12 (d, 0.17H, J = 3.6 Hz, H-1), 4.33 (dd, 0.17H, J = 10.4, 5.0 Hz, H-6), 3.38 (dd, 0.17H, J = 10.4, 3.6 Hz, H-2). 13C-APT NMR (CDCl3, 101 MHz): δ 101.8, 99.5 (C-1), 82.4 (d, J = 170.8 Hz), 79.6, 68.9, 67.8 (d, J = 20.2 Hz), 63.1, 61.8. HRMS: [M + NH4]+ calcd for C22H26FN4O6 461.18309, found 461.18292.
Methyl 4-O-(2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranosyl)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (4E)
Donor 4 and acceptor 26 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 4:1 pentane/EtOAc) to yield glycosylation product 4E (60 mg, 71 μmol, 71%, α/β = 1:6) as a white solid. Rf 0.67 (4:1 pentane/EtOAc). IR (thin film): 696, 733, 914, 999, 1026, 1045, 1090, 1177, 1263, 1314, 1366, 1452, 1730, 2108, 2866, 2899. Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 8.10–8.04 (m, 2H, CHarom), 7.61–7.53 (m, 1H, CHarom), 7.49–7.24 (m, 22H, CHarom), 5.40 (s, 1H, CHPh), 5.19 (t, 1H, J = 9.8 Hz, H-3′), 4.90 (d, 1H, J = 10.8 Hz, CHH Bn), 4.83 (d, 1H, J = 10.8 Hz, CHH Bn), 4.81–4.73 (m, 2H, 2 × CHH Bn), 4.67–4.60 (m, 2H, CHH Bn, H-1), 4.42 (d, 1H, J = 12.0 Hz, CHH Bn), 4.31 (d, 1H, J = 8.0 Hz, H-1′), 4.17 (dd, 1H, J = 10.6, 5.0 Hz, H-6′), 4.08–3.98 (t, 1H, J = 9.4 Hz, H-4), 3.96 (dd, 1H, J = 10.8, 2.4 Hz, H-6), 3.86 (t, 1H, J = 9.3 Hz, H-3), 3.79–3.74 (m, 1H, H-5), 3.71 (dd, 1H, J = 10.7, 1.7 Hz, H-6), 3.61 (t, 1H, J = 9.5 Hz, H-4′), 3.54 (dd, 1H, J = 9.6, 3.7 Hz, H-2), 3.52–3.45 (m, 2H, H-2′, H-6′), 3.39 (s, 3H, CH3 OMe), 3.08 (td, 1H, J = 9.7, 5.0 Hz, H-5′). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 165.3 (C=O), 139.3, 138.3, 137.6, 136.9 (Cq), 133.4, 129.9 (CHarom), 128.9 (Cq Bz), 128.6, 128.5, 128.5, 128.3, 128.3, 128.2, 127.9, 127.8, 126.2 (CHarom), 101.5, 101.4 (CHPh, C-1′), 98.4 (C-1), 80.1 (C-3), 79.1 (C-2), 78.9 (C-4′), 76.8 (C-4), 75.6, 73.7, 73.6 (CH2 Bn), 72.0 (C-3′), 69.7 (C-5), 68.6 (C-6′), 67.8 (C-6), 66.1 (C-5′), 65.6 (C-2′), 55.5 (OMe). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.91 (d, 0.17H, J = 3.9 Hz, H-1′), 5.80 (t, 0.17H, J = 10.0 Hz, H-3′), 5.47 (s, 0.17H, CHPh), 5.15 (d, 0.17H, J = 10.7 Hz, CHH Bn), 4.74 (d, 0.17H, J = 12.0 Hz, CHH Bn), 4.09 (t, 0.17H, J = 9.2 Hz), 3.29 (dd, 0.17H, J = 10.5, 3.9 Hz, H-2′). 13C-APT NMR (CDCl3, 101 MHz): δ 165.6, 138.6, 138.1, 137.9, 137.0, 133.4, 130.0, 129.6, 129.0, 128.6, 128.6, 128.5, 128.4, 128.2, 128.1, 127.6, 127.6, 127.4, 101.6 (CHPh), 98.6 (C-1′), 97.7 (C-1), 82.1, 80.7, 79.5, 75.0, 73.6, 73.3, 72.8, 69.5, 69.3, 68.9, 68.7, 63.7, 61.9, 55.5. HRMS: [M + NH4]+ calcd for C48H53N4O11 861.37053, found 861.37081.
Methyl (Methyl 4-O-[2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranosyl]-2,3-di-O-benzyl-α-d-glucopyranosyl uronate) (4F)
Donor 4 and acceptor 27 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 4:1 pentane/EtOAc) to yield glycosylation product 4F (46 mg, 59 μmol, 59%, α/β = 1:1.4) as a white solid. Rf 0.56 (4:1 pentane/EtOAc). IR (thin film): 698, 750, 991, 1026, 1047, 1092, 1178, 1263, 1452, 1730, 2110, 2868, 2938. Data reported for a 0.7:1 mixture of anomers: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 8.09–8.03 (m, 3.4H, CHarom), 7.61–7.52 (m, 1.7H, CHarom), 7.49–7.25 (m, 28.9H, CHarom), 5.76 (dd, 0.7H, J = 9.5, 10.3 Hz, H-3′α), 5.70 (d, 0.7H, J = 3.9 Hz, H-1′α), 5.47 (s, 0.7H, CHPhα), 5.41 (s, 1H, CHPhβ), 5.36 (t, 1H, J = 9.8 Hz, H-3′β), 5.10 (d, 0.7H, J = 10.6 Hz, CHH Bnα), 4.93 (d, 1H, J = 10.9 Hz, CHH Bnβ), 4.87 (d, 1H, J = 10.9 Hz, CHH Bnβ), 4.83 (d, 0.7H, J = 10.6 Hz, CHH Bnα), 4.79 (d, 1H, J = 13.6 Hz, CHH Bnβ), 4.76 (d, 0.7H, J = 13.6 Hz, CHH Bnα), 4.66–4.62 (m, 2H, CHH Bnβ, H-1′β), 4.61–4.58 (m, 2.4H, CHH Bnα, H-1α, H-1β), 4.31 (dd, 0.7H, J = 10.0, 4.6 Hz, H-6′α), 4.28 (d, 0.7H, J = 9.7 Hz, H-5α), 4.23 (d, 1H, J = 9.9 Hz, H-5β), 4.19–4.06 (m, 3.4H, H-3α, H-4α, H-4β, H-6′β), 3.91 (t, 1H, J = 9.2 Hz, H-3), 3.85 (s, 2.1H, CH3 CO2Meα), 3.83 (s, 3H, CH3 CO2Meβ), 3.79–3.59 (m, 3.8H, H-2α, H-4′α, H-4′β, H-5′α, H-6′α), 3.59–3.44 (m, 4H, H-2β, H-2′β, H-5′β, H-6′β), 3.43 (s, 3H, CH3 OMeβ), 3.42 (s, 2.1H, CH3 OMeα), 3.31 (dd, 1H, J = 10.4, 3.9 Hz, H-2′α). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 170.2, 169.9 (C=O CO2Me), 165.5, 165.3 (C=O Bz), 139.1, 138.4, 138.0, 137.7, 137.0, 136.8 (Cq Bn, CHPh), 133.4, 133.4, 130.0, 130.0 (CHarom), 129.5, 129.5 (Cq Bz), 129.1, 129.1, 128.7, 128.6, 128.5, 128.5, 128.5, 128.3, 128.3, 128.2, 128.1, 127.8, 127.7, 127.6, 127.5, 126.2, 126.2 (CHarom), 102.3 (C-1′β), 101.7 (CHPhα), 101.5 (CHPhβ), 99.0 (C-1′α), 98.8 (C-1β), 98.5 (C-1α), 81.1 (C-3α), 79.9 (C-2α), 79.5, 79.5 (C-3β, C-4β), 79.2 (C-4′α), 78.9, 78.7 (C-2β, C-4′β), 75.7, 75.4 (CH2 Bn), 75.2 (C-4α), 73.9, 73.6 (CH2 Bn), 71.9 (C-3′β), 69.8, 69.8 (C-5α, C-5β), 69.6 (C-3′α), 68.5, 68.4 (C-6′α, C-6′β), 66.5 (C-5′β), 65.6 (C-2′β), 63.2 (C-5′α), 61.7 (C-2′α), 56.0, 56.0 (CH3 OMe), 53.1, 53.0 (CH3 CO2Me). HRMS: [M + NH4]+ calcd for C42H47N4O12 799.31850, found 799.31869.
2,2-Difluoroethyl 2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranoside (4G)
Donor 4 and 2,2-difluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations and purified by flash column chromatography (1:1:0 to 0:1:0 to 0:19:1 pentane/toluene/EtOAc) to yield glycosylation product 4G (38.6 mg, 84 μmol, 84%, α/β = 2.7:1) as a white solid. Rf 0.49 (19:1 toluene/EtOAc). IR (thin film): 709, 997, 1026, 1069, 1094, 1265, 1726, 2108, 2870. Data for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.11–8.05 (m, 2H, CHarom), 7.63–7.53 (m, 1H, CHarom), 7.50–7.36 (m, 4H, CHarom), 7.34–7.27 (m, 3H, CHarom), 6.02 (tt, 1H, J = 55.1, 4.2 Hz, CHF2), 5.91–5.81 (m, 1H, H-3), 5.53 (s, 1H, CHPh), 5.11 (d, 1H, J = 3.6 Hz, H-1), 4.33 (dd, 1H, J = 10.4, 4.9 Hz, H-6), 4.07 (ddd, 1H, J = 14.8, 6.4, 3.7 Hz, H-5), 3.99–3.77 (m, 4H, H-4, H-6), 3.42 (dd, 1H, J = 10.4, 3.6 Hz, H-2). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 165.5 (C=O Bz), 136.8 (Cq), 133.5, 130.1 (CHarom), 129.5 (Cq Bz), 128.5, 128.3, 126.2 (CHarom), 113.7 (t, J = 241.7 Hz, CHF2), 101.8 (CHPh), 99.8 (C-1), 79.4 (C-4), 69.3 (C-3), 68.7 (C-6), 67.6 (t, J = 29.0 Hz, CH2—CHF2), 63.5 (C-5), 61.8 (C-2). Data for the β-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.97 (tdd, 0.37H, J = 55.2, 4.8, 3.5 Hz, CHF2), 5.51 (s, 0.37H, CHPh), 5.42 (t, 0.37H, J = 9.8 Hz, H-3), 4.63 (d, 0.37H, J = 7.9 Hz, H-1), 4.38 (dd, 0.37H, J = 10.5, 5.0 Hz, H-6), 4.12–3.99 (m, 0.37H, CHH—CHF2), 3.98–3.76 (m, 1.11H, CHH—CHF2, H-4, H-6), 3.69 (dd, 0.37H, J = 10.0, 7.9 Hz, H-2), 3.58 (td, 0.37H, J = 9.8, 5.0 Hz, H-5). 13C-APT NMR (CDCl3, 101 MHz): δ 165.4, 136.7, 133.5, 130.0, 129.4, 129.2, 128.6, 126.2, 113.8 (t, J = 241.5 Hz), 103.1, 101.7, 78.7 (C-4), 71.5 (C-3), 69.1 (t, J = 29.0 Hz, CH2—CHF2), 68.4 (C-6), 66.8 (C-5), 64.9 (C-2). HRMS: [M + H]+ calcd for C22H22F2N3O6 462.14712, found 462.14699.
Methyl 4-O-(2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranosyl)-2,3,6-tri-O-benzyl-β-d-galactopyranoside (4H)
Donor 4 and acceptor 28 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 4:1 pentane/EtOAc) to yield glycosylation product 4H (43 mg, 52 μmol, 52%, α/β = 4:1) as a white solid. Rf 0.36 (4:1 pentane/EtOAc). IR (thin film): 698, 737, 997, 1072, 1094, 1265, 1452, 1730, 2106, 2862, 2930. Data for the α-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 8.12–8.05 (m, 2H, CHarom), 7.57 (t, 1H, J = 7.4 Hz, CHarom), 7.45 (t, 2H, J = 7.7 Hz, CHarom), 7.42–7.20 (m, 20H, CHarom), 5.85 (t, 1H, J = 9.9 Hz, H-3′), 5.44 (s, 1H, CHPh), 5.07 (d, 1H, J = 3.9 Hz, H-1′), 4.93 (d, 1H, J = 11.0 Hz, CHH Bn), 4.84 (d, 1H, J = 10.9 Hz, CHH Bn), 4.79 (d, 1H, J = 12.4 Hz, CHH Bn), 4.74 (d, 1H, J = 12.4 Hz, CHH Bn), 4.55 (s, 2H, CH2 Bn), 4.46 (td, 1H, J = 9.9, 4.9 Hz, H-5′), 4.26 (d, 1H, J = 7.6 Hz, H-1), 4.17 (d, 1H, J = 3.1 Hz, H-4), 3.93 (t, 1H, J = 9.1 Hz, H-6), 3.85 (dd, 1H, J = 10.2, 5.0 Hz, H-6′), 3.81–3.70 (m, 2H, H-2, H-4′), 3.64 (dd, 2H, J = 9.1, 5.4 Hz, H-6), 3.57–3.50 (m, 5H, CH3 OMe, H-5, H-6′), 3.47 (dd, 1H, J = 10.4, 3.9 Hz, H-2′), 3.42 (dd, 1H, J = 10.0, 3.0 Hz, H-3). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 165.3 (C=O Bz), 138.7, 138.3, 137.6, 137.2 (Cq), 133.3, 130.0 (CHarom), 129.8 (Cq), 128.9, 128.7, 128.6, 128.6, 128.5, 128.5, 128.4, 128.4, 128.3, 128.2, 128.2, 128.1, 127.9, 127.7, 127.6, 127.6, 126.3, 126.2 (CHarom), 105.2 (C-1), 101.4 (CHPh), 99.4 (C-1′), 80.0 (C-4′), 79.8 (C-3), 79.0 (C-2), 75.2 (CH2 Bn), 74.4 (C-4), 73.6, 73.2 (CH2 Bn), 72.6 (C-5), 70.2 (C-3′), 68.8 (C-6′), 67.1 (C-6), 62.8, 62.7 (C-2′, C-5′), 57.0 (OMe). Diagnostic peaks for the β-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.47 (s, 0.25H, CHPh), 5.34 (t, 0.25H, J = 9.8 Hz, H-3′), 4.30 (d, 0.25H, J = 7.7 Hz, H-1′), 4.09 (d, 0.25H, J = 2.7 Hz, H-4). 13C-APT NMR (CDCl3, 101 MHz): δ 105.1 (C-1), 102.7 (C-1′), 101.5 (CHPh), 81.2, 79.6, 78.9, 75.3, 74.6, 73.9, 73.6, 73.3, 71.7, 69.4, 68.5, 66.2, 65.0, 57.3. HRMS: [M + NH4]+ calcd for C48H53N4O11 861.37053, found 861.37067.
Methyl 2-O-(2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-α/β-d-glucopyranosyl)-3-O-benzyl-4,6-O-benzylidene-α-d-mannopyranoside (4I)
Donor 4 and acceptor 29 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (19:1 to 3/1 pentane/EtOAc) to yield glycosylation product 4I (55 mg, 73 μmol, 73%, α/β = 5:1) as a white solid. Rf 0.36 (4:1 pentane/EtOAc). IR (thin film): 671, 750, 1037, 1092, 1265, 1373, 1730, 2108, 2913. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 8.15–8.04 (m, 2H, CHarom), 7.59–7.53 (m, 1H, CHarom), 7.53–7.25 (m, 17H, CHarom), 5.92 (dd, 1H, J = 10.4, 9.5 Hz, H-3′), 5.67 (s, 1H, CHPh), 5.54 (s, 1H, CHPh′), 5.51 (d, 1H, J = 3.8 Hz, H-1′), 4.94 (d, 1H, J = 12.2 Hz, CHH Bn), 4.73 (d, 1H, J = 1.5 Hz, H-1), 4.69 (d, 1H, J = 12.2 Hz, CHH Bn), 4.39 (t, 1H, J = 9.7 Hz, H-4), 4.32 (dd, 1H, J = 10.4, 4.9 Hz, H-6′), 4.27 (dd, 1H, J = 10.3, 4.7 Hz, H-6), 4.14 (dd, 1H, J = 3.1, 1.6 Hz, H-2), 4.06–3.99 (m, 2H, H-3, H-5′), 3.95 (t, 1H, J = 10.3 Hz, H-6), 3.86–3.77 (m, 3H, H-4′, H-5, H-6′), 3.38–3.33 (m, 4H, CH3 OMe, H-2′). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 165.5 (C=O Bz), 138.8, 137.7, 136.8 (Cq), 133.4, 130.1 (CHarom), 129.6 (Cq), 129.2, 128.9, 128.5, 128.4, 128.3, 128.3, 128.3, 128.3, 127.6, 127.6, 127.4, 126.2, 126.2, 126.2, 126.1 (CHarom), 101.7, 101.6 (CHPh), 100.9 (C-1), 100.1 (C-1′), 79.7 (C-4′), 79.5 (C-4), 75.9, 75.9 (C-2, C-3), 73.6 (CH2 Bn), 69.2 (C-3′), 68.9 (C-6), 68.8 (C-6′), 64.1 (C-5), 63.3 (C-5′), 61.9 (C-2′), 54.9 (OMe). Diagnostic peaks for the β-anomer: 1H NMR (CDCl3, 400 MHz): δ 5.60 (s, 0.2H, CHPh), 5.50 (s, 0.2H, CHPh), 5.40 (t, 0.2H, J = 9.8 Hz, H-3′), 4.87 (d, 0.2H, J = 1.4 Hz, H-1), 4.80 (d, 0.2H, J = 12.3 Hz, CHH Bn), 3.57 (td, 0.2H, J = 9.9, 4.3 Hz), 3.40 (s, 0.6H, CH3 OMe). 13C-APT NMR (CDCl3, 101 MHz): δ 101.8 (C-1′), 100.0 (CHPh), 99.4 (C-1), 78.7, 78.5, 76.4, 74.4, 72.4, 71.5, 68.9, 68.5, 66.9, 65.1, 64.2, 55.2. HRMS: [M + NH4]+ calcd for C41H45N4O11 769.30793, found 769.30780.
2,2,2-Trifluoroethyl 2-Azido-3-O-benzoyl-4,6-O-benzylidene-2-deoxy-α-d-glucopyranoside (4J)
Donor 4 and 2,2,2-trifluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 30 min at −40 °C) and purified by flash column chromatography (19:1 to 17:3 pentane/EtOAc) to yield glycosylation product 4J (41 mg, 86 μmol, 86%, α/β > 20:1) as a white solid. Rf 0.15 (toluene). [α]D20 = +78.9° (c = 1.03, CHCl3). IR (thin film): 702, 989, 1085, 1177, 1275, 1717, 2112, 2864. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.13–8.03 (m, 2H, CHarom), 7.60–7.53 (m, 1H, CHarom), 7.48–7.38 (m, 4H, CHarom), 7.33–7.28 (m, 3H, CHarom), 5.87 (t, 1H, J = 10.0 Hz, H-3), 5.53 (s, 1H, CHPh), 5.14 (d, 1H, J = 3.6 Hz, H-1), 4.33 (dd, 1H, J = 10.4, 4.9 Hz, H-6), 4.14–3.97 (m, 4H, CH2CF3, H-5), 3.84 (t, 1H, J = 9.5 Hz, H-4), 3.80 (t, 1H, J = 10.3 Hz, H-6), 3.44 (dd, 1H, J = 10.4, 3.6 Hz, H-2). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 165.5 (C=O), 136.7 (Cq), 133.5, 130.0 (CHarom), 129.5 (Cq Bz), 129.2, 128.5, 127.6, 126.2 (CHarom), 123.42 (q, J = 278.6 Hz), 101.8 (CHPh), 99.9 (C-1), 79.3 (C-4), 69.1 (C-3), 68.6 (C-6), 65.41 (q, J = 35.6 Hz), 63.7 (C-5), 61.6 (C-2). HRMS: [M + NH4]+ calcd for C22H24F3N4O6 497.16425, found 497.16425.
Ethyl 3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-β-d-glucopyranoside (5A)
Donor 5 and ethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 1 h at −40 °C) and purified by flash column chromatography (19:1 to 8:2 pentane/EtOAc) to yield glycosylation product 5A (32 mg, 59 μmol, 59%, α/β < 1:20) as a yellow solid alongside donor 5 (14 mg). Rf 0.60 (7:3 pentane/EtOAc). [α]D23 = +156.9° (c = 0.64, CHCl3). IR (thin film): 698, 998, 1093, 1213, 1303, 1330, 1516, 1679, 2930. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.58 (s, 2H, CH pyridone), 7.56 (dd, 2H, J = 7.4, 2.2 Hz, CHarom), 7.45–7.38 (m, 3H, CHarom), 7.06–6.97 (m, 5H, CHarom), 5.66 (s, 1H, CHPh), 5.32 (d, 1H, J = 8.3 Hz, H-1), 4.70 (d, 1H, J = 11.7 Hz, CHH Bn), 4.57 (dd, 1H, J = 10.2, 8.7 Hz, H-3), 4.53 (d, 1H, J = 11.6 Hz, CH H Bn), 4.43 (dd, 1H, J = 10.3, 4.6 Hz, H-6), 3.99–3.81 (m, 4H, CHH Et, H-4, H-5, H-6), 3.72 (dd, 1H, J = 10.3, 8.3 Hz, H-2), 3.60 (dq, 1H, J = 9.9, 7.1 Hz, CHH Et), 1.08 (t, 3H, J = 7.1 Hz, CH3 Et). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 160.6 (C=O pyridone), 141.7 (Cq NO2 pyridone), 141.4 (CH pyridone), 137.1, 136.6 (Cq), 129.4, 128.9, 128.7, 128.5, 128.5, 126.3 (CHarom), 101.9 (CHPh), 99.3 (C-1), 82.8 (C-4), 75.4 (C-3), 74.9 (CH2 Bn), 73.8 (C-2), 68.7 (C-6), 66.5 (CH2 Et), 65.7 (C-5), 15.1 (CH3 Et). HRMS: [M + H]+ calcd for C27H28N3O10 554.17692, found 554.17642.
Cyclohexyl 3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-β-d-glucopyranoside (5B)
Donor 5 and cyclohexanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 1 h at −40 °C) and purified by flash column chromatography (19:1 to 8:2 pentane/EtOAc) to yield 51 mg of glycosylation product 5B as an inseperable mixture with donor 5 (13 mg of 5, 38 mg of 5B, 63 μmol, 63%, α/β < 1:20) as a yellow solid. Rf 0.75 (7:3 pentane/EtOAc). Rf 0.55 (7:3 pentane/EtOAc). IR. (thin film): 697, 718, 749, 789, 910, 997, 1092, 1212, 1302, 1330, 1516, 1623, 1674, 2931. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.60 (s, 2H, CH pyridone), 7.56 (d, 2H, J = 4.7 Hz, CHarom), 7.48–7.33 (m, 3H, CHarom), 7.10–6.96 (m, 5H, CHarom), 5.65 (s, 1H, CHPh), 5.41 (d, 1H, J = 8.2 Hz, H-1), 4.71 (d, 1H, J = 11.7 Hz, CHH Bn), 4.66–4.50 (m, 2H, CHH Bn, H-3), 4.44 (dd, 1H, J = 10.6, 5.2 Hz, H-6), 3.98–3.80 (m, 3H, H-4, H-5, H-6), 3.79–3.61 (m, 2H, CH Cy, H-2), 1.91–1.77 (m, 1H, CH2 Cy), 1.71–1.54 (m, 2H, CH2 Cy), 1.54–1.45 (m, 1H, CH2 Cy), 1.43–0.96 (m, 6H, CH2 Cy). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 160.5 (C=O pyridone), 141.6 (Cq NO2 pyridone), 141.4 (CH pyridone), 137.1, 136.7 (Cq), 128.8, 128.6, 128.4, 126.3 (CHarom), 101.9 (CHPh), 98.1 (C-1), 82.7 (C-4), 78.8 (CH Cy), 75.6 (C-3), 74.8 (CH2 Bn), 74.0 (C-2), 68.8 (C-6), 65.7 (C-5), 33.3, 31.7, 25.3, 23.9, 23.6 (CH2 Cy). HRMS: [M + H]+ calcd for C31H34N3O10 608.22387, found 608.22352.
Methyl 6-O-(3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-β-d-glucopyranosyl)-2,3,4-tri-O-benzyl-α-d-glucopyranoside (3C)
Donor 5 and acceptor 25 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (9:1 to 7:3 pentane/EtOAc) to yield glycosylation product 5C (55 mg, 54 μmol, 54%, α/β < 1:20) as a yellow solid. Rf 0.45 (7:3 pentane/EtOAc). [α]D20 = +90.5° (c = 0.92, CHCl3). IR (thin film): 698, 1001, 1069, 1094, 1213, 1331, 1454, 1522, 1678, 2868. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 8.19 (s, 2H, CH pyridone), 7.54 (dd, 2H, J = 7.6, 2.1 Hz, CHarom), 7.45–7.38 (m, 3H, CHarom), 7.34–7.22 (m, 13H, CHarom), 7.20–7.12 (m, 5H, CHarom), 7.04 (dd, 2H, J = 6.6, 2.9 Hz, CHarom), 5.66 (s, 1H, CHPh), 4.92 (d, 1H, J = 11.0 Hz, CHH Bn), 4.85 (d, 1H, J = 8.3 Hz, H-1′), 4.83–4.66 (m, 2H, 2 × CHH Bn), 4.72 (d, 1H, J = 10.9 Hz, CHH Bn), 4.69 (d, 1H, J = 12.0 Hz, CHH Bn), 4.60 (d, 1H, J = 12.2 Hz, CHH Bn), 4.60 (d, 1H, J = 12.0 Hz, CHH Bn), 4.46 (d, 1H, J = 3.4 Hz, H-1), 4.39 (dd, 1H, J = 10.6, 5.0 Hz, H-6′), 4.34 (d, 1H, J = 11.3 Hz, CHH Bn), 4.10 (t, 1H, J = 7.9 Hz, H-3′), 4.01 (dd, 1H, J = 10.8, 1.8 Hz, H-6), 3.91 (t, 1H, J = 9.2 Hz, H-3), 3.89–3.82 (m, 2H, H-4′, H-6), 3.77–3.69 (m, 2H, H-2′, H-5), 3.65 (td, 1H, J = 9.7, 5.0 Hz, H-5′), 3.52 (dd, 1H, J = 10.8, 7.1 Hz, H-6), 3.39 (dd, 1H, J = 9.6, 3.5 Hz, H-2), 3.21 (s, 3H, CH3 OMe), 3.13 (dd, 1H, J = 9.9, 8.9 Hz, H-4). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 159.4 (C=O pyridone), 141.7 (Cq NO2 pyridone), 140.2 (CH pyridone), 138.6, 138.0, 138.0, 136.7, 135.8 (Cq), 129.5, 129.1, 129.0, 128.6, 128.5, 128.5, 128.5, 128.2, 128.1, 128.0, 128.0, 127.9, 127.7, 127.6, 126.1 (CHarom), 101.8 (CHPh), 100.1 (C-1′), 97.9 (C-1), 82.3 (C-4′), 81.6 (C-3), 79.8 (C-2), 78.2 (C-4), 75.7, 74.8, 74.3, (CH2 Bn), 74.0 (C-3′), 73.3 (CH2 Bn), 72.7 (C-2′), 70.4 (C-6), 69.3 (C-5), 68.4 (C-6′), 66.1 (C-5′), 55.1 (OMe). 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 100.1 (J = 163 Hz, C-1′). HRMS: [M + H]+ calcd for C53H54N3O15 972.35494, found 972.35546.
2-Fluoroethyl 3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-β-d-glucopyranoside (5D)
Donor 5 and 2-fluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 1 h at −40 °C) and purified by flash column chromatography (19:1 to 8:2 pentane/EtOAc) to yield glycosylation product 5D (24 mg, 43 μmol, 43%, α/β < 1:20) as a yellow solid alongside donor 5 (15.6 mg). Rf 0.42 (3/2 pentane/EtOAc). [α]D23 = +142.9° (c = 0.48, CHCl3). IR (thin film): 698, 752, 1070, 1096, 1213, 1304, 1331, 1518, 1680, 2870, 3064. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.58 (s, 2H, CH pyridone), 7.63–7.49 (m, 2H, CHarom), 7.47–7.36 (m, 3H, CHarom), 7.09–6.96 (m, 5H, CHarom), 5.66 (s, 1H, CHPh), 5.46 (d, 1H, J = 8.3 Hz, H-1), 4.71 (d, 1H, J = 11.7 Hz, CHH Bn), 4.57 (dd, 1H, J = 10.3, 8.7 Hz, H-3), 4.53 (d, 1H, J = 11.7 Hz, CHH Bn), 4.48–4.42 (m, 2H, CHHF, H-6), 4.33 (t, 1H, J = 4.1 Hz, CHHF), 4.09–3.81 (m, 5H, CH2—CH2F, H-4, H-5, H-6), 3.77 (dd, 1H, J = 10.3, 8.4 Hz, H-2). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 160.6 (C=O pyridone), 141.5, 141.5 (Cq NO2, CH pyridone), 137.0, 136.6 (Cq), 129.4, 129.0, 128.7, 128.6, 128.5, 126.3 (CHarom), 101.9 (CHPh), 99.8 (C-1), 82.7 (C-4), 82.5 (d, J = 169.4 Hz, CH2F), 75.3 (C-3), 74.9 (CH2 Bn), 73.6 (C-2), 69.5 (d, J = 19.5 Hz, CH2—CH2F), 68.6 (C-6), 65.8 (C-5). HRMS: [M + H]+ calcd for C27H27FN3O10 572.16760, found 572.16705.
Methyl 4-O-(3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-β-d-glucopyranosyl)-2,3,6-tri-O-benzyl-α-d-glucopyranoside (5E)
Donor 5 and acceptor 26 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (9:1 to 7:3 pentane/EtOAc) to yield glycosylation product 5E (54 mg, 56 μmol, 56%, α/β < 1:20) as a yellow solid. Rf 0.37 (7:3 pentane/EtOAc). [α]D23 = +83.3° (c = 0.84, CHCl3). IR (thin film): 696, 734, 997, 1028, 1039, 1092, 1209, 1302, 1327, 1454, 1522, 1682, 2862, 2900, 3030, 3065. 1H NMR (CDCl3, 500 MHz, H–H COSY, HSQC, HMBC, NOESY): δ 7.74 (s, 2H, CH pyridone), 7.57–7.53 (m, 2H, CHarom), 7.49–7.38 (m, 8H, CHarom), 7.36–7.25 (m, 13H, CHarom), 7.04–7.00 (m, 2H, CHarom), 5.57 (s, 1H, CHPh), 4.90 (d, 1H, J = 11.7 Hz, CHH Bn), 4.77 (d, 1H, J = 12.1 Hz, CHH Bn), 4.74 (d, 1H, J = 12.3 Hz, CHH Bn), 4.69 (d, 1H, J = 11.7 Hz, CHH Bn), 4.66 (d, 1H, J = 12.0 Hz, CHH Bn), 4.63 (d, 1H, J = 12.1 Hz, CHH Bn), 4.56 (d, 1H, J = 12.3 Hz, CHH Bn), 4.54 (d, 1H, J = 3.6 Hz, H-1), 4.35 (d, 1H, J = 8.2 Hz, H-1′), 4.27–4.20 (m, 2H, CHH Bn, H-6′), 3.92 (t, 1H, J = 9.5 Hz, H-4), 3.70 (t, 1H, J = 9.3 Hz, H-3), 3.67 (t, 1H, J = 9.0 Hz, H-4′), 3.58 (t, 1H, J = 10.4 Hz, H-6′), 3.53–3.45 (m, 2H, H-2, H-3′), 3.43 (dd, 1H, J = 11.4, 1.5 Hz, H-6), 3.40–3.34 (m, 1H, H-5), 3.31 (s, 2H, CH3 OMe), 3.18 (dd, 1H, J = 10.5, 8.3 Hz, H-2′), 3.04 (dd, 1H, J = 11.3, 2.6 Hz, H-6), 2.92 (td, 1H, J = 9.8, 5.1 Hz, H-5′). 13C-APT NMR (CDCl 3, 126 MHz, HSQC, HMBC): δ 159.3 (C=O pyridone), 141.8 (Cq NO2 pyridone), 139.6 (CH pyridone), 139.4, 138.1, 137.8, 136.8, 135.7 (Cq), 129.6, 129.3, 129.2, 129.0, 128.9, 128.6, 128.6, 128.5, 128.4, 128.1, 128.1, 127.8, 126.1 (CHarom), 101.8 (CHPh), 98.1 (C-1), 97.6 (C-1′), 82.4 (C-4′), 79.7 (C-2), 79.2 (C-3), 75.3 (CH2 Bn), 74.4 (C-4), 73.9, 73.6, 73.5 (CH2 Bn), 73.0 (C-3′), 72.5 (C-2′), 69.2 (C-5), 68.4 (C-6′), 68.1 (C-6), 65.6 (C-5′), 55.7 (OMe). HRMS: [M + H]+ calcd for C53H54N3O15 972.35494, found 972.35519.
Methyl (Methyl 4-O-[3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-α/β-d-glucopyranosyl]-2,3-di-O-benzyl-α-d-glucopyranosyl uronate) (5F)
Donor 5 and acceptor 27 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (9:1 to 7:3 pentane/EtOAc) to yield glycosylation product 5F (27 mg, 30 μmol, 30%, α/β = 1:3.6) as a yellow solid. Rf 0.51 (7:3 pentane/EtOAc). IR (thin film): 648, 698, 733, 789, 910, 995, 1090, 1171, 1209, 1302, 1331, 1454, 1520, 1678, 1744, 2932. Data for the β-anomer: 1H NMR (CDCl3, 500 MHz, H–H COSY, HSQC, HMBC): δ 8.06 (s, 2H, CH pyridone), 7.50 (dd, 2H, J = 7.3, 2.0 Hz, CHarom), 7.45–7.35 (m, 6H, CHarom), 7.33–7.18 (m, 9H, CHarom), 7.03 (dd, 2H, J = 6.9, 2.2 Hz, CHarom), 6.96 (dd, 1H, J = 14.5, 6.9 Hz, CHarom), 5.55 (s, 1H, CHPh), 5.17 (d, 1H, J = 8.2 Hz, H-1′), 4.92 (d, 1H, J = 11.3 Hz, CHH Bn), 4.82–4.72 (m, 3H, CHH Bn, 2 × CHH Bn), 4.61–4.55 (m, 2H, 2 × CHH Bn), 4.51 (d, 1H, J = 3.3 Hz, H-1), 4.14 (dd, 1H, J = 10.6, 4.8 Hz, H-6′), 3.97–3.88 (m, 2H, H-3′, H-4), 3.83 (d, 1H, J = 9.7 Hz, H-5), 3.82–3.73 (m, 2H, H-3, H-4′), 3.54–3.43 (m, 6H, CH3 CO2Me, H-2, H-2′, H-5′), 3.42–3.36 (m, 4H, CH3 OMe, H-6′). 13C-APT NMR (CDCl3, 126 MHz, HSQC, HMBC): δ 170.0 (C=O CO2Me), 159.5 (C=O pyridone), 141.5 (Cq NO2 pyridone), 140.4 (CH pyridone), 139.1, 137.8, 136.7, 135.7 (Cq), 129.5, 129.2, 129.2, 129.1, 129.0, 128.9, 128.7, 128.7, 128.6, 128.5, 128.4, 128.2, 128.2, 127.7, 127.7, 126.1, 126.0, 125.7 (CHarom), 101.7 (CHPh), 98.8 (C-1′), 98.3 (C-1), 82.3 (C-4′), 79.1 (C-3), 78.8 (C-2), 77.8 (C-4), 75.5, 74.2 (CH2 Bn), 74.0 (C-3′), 73.7 (CH2 Bn), 72.9 (C-2′), 68.8 (C-5), 68.2 (C-6′), 65.9 (C-5′), 56.1 (OMe), 52.9 (CO2Me). 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 98.8 (J = 167 Hz, C-1′). Diagnostic peaks for the α-anomer: 1H NMR (CDCl3, 500 MHz, H–H COSY, HSQC): δ 5.74 (d, 0.28H, J = 3.9 Hz, H-1′), 5.61 (s, 0.28H, CHPh), 5.03 (d, 0.28H, J = 12.7 Hz, CHH Bn), 4.70 (d, 0.28H, J = 12.3 Hz), 4.62 (d, 0.28H, J = 12.2 Hz, CHH Bn), 4.46 (d, 0.28H, J = 12.3 Hz, CHH Bn), 4.37 (dd, 0.28H, J = 10.5, 4.9 Hz, H-6′), 4.29 (d, 0.28H, J = 9.9 Hz, H-5), 4.08 (t, 0.28H, J = 9.4 Hz), 3.69 (dd, 0.28H, J = 10.6, 3.9 Hz, H-2′). 13C-APT NMR (CDCl3, 126 MHz, HSQC): δ 169.7, 159.2, 141.2, 101.8 (CHPh), 96.8 (C-1′), 82.6, 80.7, 79.8, 74.5, 74.4, 73.1, 72.2, 69.6, 69.5, 68.1, 63.0, 56.1, 53.3. 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 96.8 (J = 181 Hz, C-1′). HRMS: [M + H]+ calcd for C47H48N3O16 910.30291, found 910.30315.
2,2-Difluoroethyl 3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-α/β-d-glucopyranoside (5G)
Donor 5 and 2,2-difluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 1 h at −40 °C) and purified by flash column chromatography (19:1 to 8:2 pentane/EtOAc) to yield glycosylation product 5G (17.3 mg, 29 μmol of α-anomer; 17.8 mg, 30 μmol of β-anomer; α/β = 1:1; 59%) as a yellow solid alongside donor 5 (11 mg). Rf 0.12 and 0.30 (7:3 pentane/EtOAc). IR (thin film): 698, 997, 1069, 1094, 1211, 1300, 1339, 1520, 1684, 2922. Data for the α-anomer: 1H NMR (acetone-d6, 400 MHz, H–H COSY, HSQC): δ 8.91 (s, 2H, CH pyridone), 7.61–7.53 (m, 2H, CHarom), 7.48–7.37 (m, 3H, CHarom), 7.25–7.10 (m, 5H, CHarom), 6.16 (tt, 1H, J = 55.0, 3.7 Hz, CHF2), 5.83 (s, 1H, CHPh), 5.56 (d, 1H, J = 3.7 Hz, H-1), 4.91 (d, 1H, J = 11.9 Hz, CHH Bn), 4.79 (dd, 1H, J = 10.7, 3.7 Hz, H-2), 4.71–4.62 (m, 2H, CHH Bn, H-3), 4.37 (dd, 1H, J = 10.1, 4.9 Hz, H-6), 4.18–4.06 (m, 2H, CHH—CHF2, H-5), 4.03 (dd, 1H, J = 9.6, 8.6 Hz, H-4), 3.96–3.83 (m, 2H, CHH—CHF2, H-6). 13C-APT NMR (acetone-d6, 101 MHz, HSQC): δ 160.0 (C=O pyridone), 142.9 (Cq NO2 pyridone), 142.6 (CH pyridone), 138.6, 138.5 (Cq), 129.8, 129.2, 129.0, 129.0, 128.9, 127.0 (CHarom), 115.0 (t, J = 239.2 Hz, CHF2), 102.1 (CHPh), 98.8 (C-1), 83.5 (C-4), 75.0 (CH2 Bn), 74.7 (C-3), 69.9 (C-2), 68.9 (C-6), 67.93 (t, J = 27.3 Hz, CH2—CHF2), 63.9 (C-5). Data for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.67 (s, 2H, CH pyridone), 7.60–7.52 (m, 2H, CHarom), 7.46–7.37 (m, 3H, CHarom), 7.03–6.93 (m, 5H, CHarom), 5.75 (tt, 1H, J = 54.9, 3.9 Hz, CHF2), 5.65 (s, 1H, CHPh), 5.59 (d, 1H, J = 8.3 Hz, H-1), 4.76–4.64 (m, 2H, CHH Bn, H-3), 4.50 (d, 1H, J = 11.6 Hz, CHH Bn), 4.45 (dd, 1H, J = 10.4, 4.9 Hz, H-6), 4.05 (td, 1H, J = 9.7, 5.0 Hz, H-5), 4.00–3.80 (m, 4H, CH2—CHF2, H-4, H-6), 3.77 (dd, 1H, J = 10.3, 8.4 Hz, H-2). 13C-APT NMR (CDCl3, 101 MHz, HSQC): δ 160.9 (C=O pyridone), 141.7 (Cq NO2 pyridone), 141.5 (CH pyridone), 137.0, 136.7 (Cq), 129.4, 128.8, 128.6, 128.5, 128.4, 126.4 (CHarom), 113.4 (t, J = 241.5 Hz, 102.0 (CHPh), 99.9 (C-1), 82.6 (C-4), 75.5 (C-3), 75.1 (CH2 Bn), 73.7 (C-2), 68.9 (t, J = 27.8 Hz, CH2—CHF2), 68.6 (C-6), 65.8 (C-5). HRMS: [M + H]+ calcd for C27H26F2N3O10 590.15808, found 590.15741.
Methyl 4-O-(3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-β-d-glucopyranosyl)-2,3,6-tri-O-benzyl-β-d-galactopyranoside (5H)
Donor 5 and acceptor 28 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (9:1 to 7:3 pentane/EtOAc) to yield glycosylation product 5H (51 mg, 52 μmol, 52%, α/β < 1:20) as a yellow solid. Rf 0.49 (7:3 pentane/EtOAc). [α]D20 = +35.5° (c = 0.85, CHCl3). IR (thin film): 698, 750, 999, 1072, 1094, 1213, 1454, 1522, 1682, 2868. 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 8.20 (s, 2H, CH pyridone), 7.55–7.49 (m, 2H, CHarom), 7.47–7.40 (m, 3H, CHarom), 7.40–7.15 (m, 18H, CHarom), 7.07–7.00 (m, 2H, CHarom), 5.63 (s, 1H, CHPh), 5.03 (d, 1H, J = 8.3 Hz, H-1′), 4.80 (d, 1H, J = 12.2 Hz, CHH Bn), 4.64 (d, 1H, J = 10.4 Hz, CHH Bn), 4.61 (d, 1H, J = 12.3 Hz, CHH Bn), 4.57 (d, 1H, J = 12.2 Hz, CHH Bn), 4.50 (s, 2H, CH2 Bn), 4.47 (d, 1H, J = 12.3 Hz, CHH Bn), 4.29 (d, 1H, J = 10.4 Hz, CHH Bn), 4.20 (dd, 1H, J = 10.5, 5.0 Hz, H-6′), 4.13 (d, 1H, J = 7.6 Hz, H-1), 3.91 (d, 1H, J = 2.6 Hz, H-4), 3.88–3.78 (m, 2H, H-3′, H-4′), 3.74 (t, 1H, J = 10.3 Hz, H-6′), 3.70 (dd, 1H, J = 9.9, 8.4 Hz, H-2′), 3.66–3.52 (m, 2H, H-6, H-6), 3.46 (s, 4H, CH3 OMe, H-5), 3.41–3.33 (m, 2H, H-3, H-5′), 2.93 (dd, 1H, J = 9.6, 7.6 Hz, H-2). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 159.4 (Cq pyridone), 141.6 (Cq NO 2 pyridone), 140.5 (CH pyridone), 138.2, 138.0, 137.6, 136.6, 135.6 (Cq), 129.6, 129.3, 129.1, 129.1, 128.9, 128.7, 128.6, 128.6, 128.4, 128.4, 128.1, 128.0, 127.7, 127.5, 126.1 (CHarom), 104.8 (C-1), 101.8 (CHPh), 99.6 (C-1′), 82.3 (C-4′), 80.2, 80.2 (C-2, C-3), 75.4 (CH2 Bn), 74.6 (C-4), 74.5, 74.4 (CH2 Bn), 74.2 (C-3′), 73.5 (CH2 Bn), 72.5 (C-5), 72.3 (C-2′), 68.6 (C-6), 68.2 (C-6′), 66.2 (C-5′), 57.3 (OMe). 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 104.8 (J = 159 Hz, C-1), 99.6 (J = 165 Hz, C-1′). HRMS: [M + H]+ calcd for C53H54N3O15 972.35494, found 972.35542.
Methyl 2-O-(3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-α/β-d-glucopyranosyl)-3-O-benzyl-4,6-O-benzylidene-α-d-mannopyranoside (5I)
Donor 5 and acceptor 29 were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 18 h at −40 °C) and purified by flash column chromatography (9:1 to 7:3 pentane/EtOAc) to yield glycosylation product 5I (47 mg, 53 μmol, 53%, α/β = 1:1.3) as a yellow solid. Rf 0.34 and 0.49 (7:3 pentane/EtOAc). IR: (thin film): 646, 696, 731, 789, 908, 997, 1090, 1123, 1211, 1302, 1333, 1454, 1518, 1624, 1674, 2910. Data reported for a 0.8:1 mixture of anomers: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC, HMBC): δ 8.48 (s, 2H, pyridoneβ), 8.33 (s, 1.6H, pyridoneα), 7.58–7.26 (m, 22.8H, CHarom), 7.23–7.01 (m, 11.8H, CHarom), 6.99–6.94 (m, 1.6H, CHarom), 5.67 (s, 1.8H, CHPh′α, CHPh′β), 5.62 (s, 0.8H, CHPhα), 5.53 (s, 1H, CHPhβ), 5.27 (d, 0.8H, J = 3.9 Hz, H-1′α), 5.24 (d, 1H, J = 8.3 Hz, H-1′β), 4.84 (d, 1H, J = 12.1 Hz, CHH Bnβ), 4.79 (d, 0.8H, J = 12.2 Hz, CHH Bnα), 4.75 (d, 1H, J = 12.1 Hz, CHH Bnβ), 4.70 (d, 1H, J = 12.2 Hz, CHH Bnβ), 4.67 (d, 0.8H, J = 1.1 Hz, H-1α), 4.64 (d, 1H, J = 12.1 Hz, CHH Bnβ), 4.57 (d, 0.8H, J = 12.2 Hz, CHH Bnα), 4.52 (d, 0.8H, J = 11.1 Hz, CHH Bnα), 4.50 (dd, 1H, J = 10.3, 8.2 Hz, H-3′β), 4.46–4.41 (m, 1H, H-6′β), 4.33 (d, 0.8H, J = 11.1 Hz, CHH Bnα), 4.34–4.26 (m, 1.6H, H-6α, H-6′α), 4.22–4.15 (m, 2.8H, H-1β, H-2β, H-3′α), 4.10–3.96 (m, 4.4H, H-2α, H-2′α, H-2′β, H-5′α, H-6β), 3.95–3.83 (m, 8.2H, H-3α, H-3β, H-4β, H-4′α, H-4′β, H-5′β, H-6α, H-6′α, H-6′β), 3.81–3.74 (m, 1.6H, H-4α, H-5α), 3.61 (dq, 1H, J = 9.0, 4.5 Hz, H-5β), 3.50 (t, 1H, J = 10.3 Hz, H-6β), 3.38 (s, 2.4H, CH3 OMeα), 3.15 (s, 3H, CH3 OMeβ). 13C-APT NMR (CDCl3, 101 MHz, HSQC, HMBC): δ 159.9, 159.7 (C = O pyridone), 142.1 (CH pyridoneα), 141.7 (Cq NO2 pyridone), 140.9 (CH pyridoneβ), 140.8 (Cq NO2 pyridone), 138.2, 137.6, 137.4, 137.4, 136.8, 136.6, 136.5, 136.0 (Cq), 129.6, 129.4, 129.1, 129.0, 128.9, 128.8, 128.7, 128.6, 128.5, 128.4, 128.4, 128.3, 128.3, 127.8, 127.8, 127.6, 126.2, 126.1, 126.1 (CHarom), 101.9, 101.8 (CHPh′α,β), 101.6, 101.6 (CHPhα,β), 100.7 (C-1α), 99.8 (C-1′α), 99.2 (C-1β), 98.8 (C-1′β), 83.1 (C-4′α), 82.4 (C-4′β), 79.6 (C-4α), 79.1 (C-2α), 78.5 (C-4β), 76.1 (C-2β), 75.0 (C-3α), 74.5, 74.5, 74.3 (CH2 Bn), 74.2 (C-3β, C-3′β), 72.9 (C-2′β), 72.7 (CH2 Bn), 72.5 (C-3′α), 69.9 (C-2′α), 68.5, 68.5, 68.4 (C-6α,β, C-6′α,β), 66.1 (C-5′β), 63.7 (C-5β), 63.3 (C-5α), 63.1 (C-5′α), 55.1, 55.1 (OMe). 13C-HMBC-GATED NMR (CDCl3, 101 MHz): δ 99.8 (J = 176 Hz, C-1′α), 98.8 (J = 164 Hz, C-1′β). HRMS: [M + H]+ calcd for C46H46N3O15 880.29234, found 880.29252.
2,2,2-Trifluoroethyl 3-O-Benzyl-4,6-O-benzylidene-2-deoxy-2-(3,5-dinitro-4-pyridone)-α/β-d-glucopyranoside (5J)
Donor 5 and 2,2,2-trifluoroethanol were condensed using the general procedure for Tf2O/Ph2SO-mediated glycosylations (for an additional 1 h at −40 °C) and purified by flash column chromatography (19:1 to 8:2 pentane/EtOAc) to yield glycosylation product 5J (28 mg, 46 μmol of α-anomer; 7 mg, 12 μmol of β-anomer; α/β = 4:1; 58%) as a yellow solid alongside glucal 24 (4 mg) and donor 5 (13 mg). Rf 0.15 and 0.67 (7:3 pentane/EtOAc). IR (thin film): 698, 754, 1001, 1071, 1096, 1169, 1215, 1279, 1304, 1331, 1520, 1680, 2855, 2924, 3065. Data for the α-anomer: 1H NMR (acetone-d6, 400 MHz, H–H COSY, HSQC): δ 8.92 (s, 2H, CH pyridone), 7.60–7.54 (m, 2H, CHarom), 7.47–7.37 (m, 3H, CHarom), 7.24–7.13 (m, 5H, CHarom), 5.84 (s, 1H, CHPh), 5.65 (d, 1H, J = 3.7 Hz, H-1), 4.92 (d, 1H, J = 11.8 Hz, CHH Bn), 4.84 (dd, 1H, J = 10.7, 3.7 Hz, H-2), 4.73 (dd, 1H, J = 10.7, 8.4 Hz, H-3), 4.70 (d, 1H, J = 11.8 Hz, CHH Bn), 4.51–4.38 (m, 1H, CHH—CF3), 4.38 (dd, 1H, J = 10.1, 4.7 Hz, H-6), 4.30–4.17 (m, 1H, CHH—CF3), 4.12 (dd, 1H, J = 9.8, 4.7 Hz, H-5), 4.09–4.02 (m, 1H, H-4), 3.93 (t, 1H, J = 10.0 Hz, H-6). 13C-APT NMR (acetone-d6, 101 MHz, HSQC): δ 160.0 (C=O pyridone), 142.9 (Cq NO2 pyridone), 142.6 (CH pyridone), 138.6, 138.5 (Cq), 129.8, 129.2, 129.0, 129.0, 128.9, 127.0 (CHarom), 124.72 (q, J = 277.4 Hz, CF3), 102.1 (CHPh), 98.8 (C-1), 83.4 (C-4), 75.1 (CH2 Bn), 74.6 (C-3), 69.8 (C-2), 68.8 (C-6), 65.84 (q, J = 35.0 Hz, CH2—CF3), 64.1 (C-5). Diagnostic peaks for the β-anomer: 1H NMR (CDCl3, 400 MHz, H–H COSY, HSQC): δ 8.61 (s, 2H, CH pyridone), 7.56 (dd, 2H, J = 6.6, 2.9 Hz, CHarom), 7.41 (dd, 3H, J = 5.0, 1.7 Hz, CHarom), 7.00 (s, 5H, CHarom), 5.66 (s, 1H, CHPh), 5.58 (d, 1H, J = 8.3 Hz, H-1), 4.75–4.62 (m, 2H, CHH Bn, H-3), 4.52 (d, 1H, J = 11.7 Hz, CHH Bn), 4.46 (dd, 1H, J = 10.5, 4.8 Hz, H-6), 4.17–3.99 (m, 3H, CH2—CF3, H-5), 3.91–3.82 (m, 2H, H-4, H-6), 3.78 (dd, 1H, J = 10.0, 8.5 Hz, H-2). HRMS: [M + H]+ calcd for C27H25F3N3O10 608.14865, found 608.14825.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.7b00470.
NMR spectra of all new compounds, decomposition study and identification of anomeric triflates by NMR spectroscopy, 1H NMR spectra of isolated disaccharide fractions of competition experiments (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Capila I.; Linhardt R. J. Angew. Chem., Int. Ed. 2002, 41 (3), 390–412. . [DOI] [PubMed] [Google Scholar]
- Spillmann D.; Lindahl U. Curr. Opin. Struct. Biol. 1994, 4 (5), 677–682. 10.1016/S0959-440X(94)90165-1. [DOI] [Google Scholar]
- Griffiths A. J.; Davies D. B. Carbohydr. Polym. 1991, 14 (3), 241–279. 10.1016/0144-8617(91)90079-R. [DOI] [Google Scholar]
- Griffiths A. J.; Davies D. B. Carbohydr. Polym. 1991, 14 (4), 339–365. 10.1016/0144-8617(91)90001-S. [DOI] [Google Scholar]
- Boltje T. J.; Buskas T.; Boons G.-J. Nat. Chem. 2009, 1 (8), 611–622. 10.1038/nchem.399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christina A. E.; van der Marel G. A.; Codée J. D. C. In Modern Synthetic Methods in Carbohydrate Chemistry; Werz D. B., Vidal S., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2013; pp 97–124. [Google Scholar]
- Nigudkar S. S.; Demchenko A. V. Chem. Sci. 2015, 6 (5), 2687–2704. 10.1039/C5SC00280J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett C. S.Selective Glycosylations: Synthetic Methods and Catalysts; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2017. [Google Scholar]
- Crich D.; Cai W. J. Org. Chem. 1999, 64 (13), 4926–4930. 10.1021/jo990243d. [DOI] [PubMed] [Google Scholar]
- Crich D.; Sun S. J. Am. Chem. Soc. 1997, 119 (46), 11217–11223. 10.1021/ja971239r. [DOI] [Google Scholar]
- Crich D.; Sun S. J. Org. Chem. 1996, 61 (14), 4506–4507. 10.1021/jo9606517. [DOI] [PubMed] [Google Scholar]
- Huang M.; Garrett G. E.; Birlirakis N.; Bohé L.; Pratt D. A.; Crich D. Nat. Chem. 2012, 4 (8), 663–667. 10.1038/nchem.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crich D.; Chandrasekera N. S. Angew. Chem., Int. Ed. 2004, 43 (40), 5386–5389. 10.1002/anie.200453688. [DOI] [PubMed] [Google Scholar]
- Adero P. O.; Furukawa T.; Huang M.; Mukherjee D.; Retailleau P.; Bohé L.; Crich D. J. Am. Chem. Soc. 2015, 137 (32), 10336–10345. 10.1021/jacs.5b06126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.; Furukawa T.; Retailleau P.; Crich D.; Bohé L. Carbohydr. Res. 2016, 427, 21–28. 10.1016/j.carres.2016.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohé L.; Crich D. Carbohydr. Res. 2015, 403, 48–59. 10.1016/j.carres.2014.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M.; Retailleau P.; Bohé L.; Crich D. J. Am. Chem. Soc. 2012, 134 (36), 14746–14749. 10.1021/ja307266n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongat A. F. G.; Demchenko A. V. Carbohydr. Res. 2007, 342 (3–4), 374–406. 10.1016/j.carres.2006.10.021. [DOI] [PubMed] [Google Scholar]
- For the use of C2-N-C3-O-carbamate-protected glucosamines, see, for example: ref (6) andCodée J. D. C.; Ali A.; Overkleeft H. S.; van der Marel G. A. C. R. Chim. 2011, 14 (2–3), 178–193. 10.1016/j.crci.2010.05.010. [DOI] [Google Scholar]
- Cid M. B.; Alfonso F.; Martín-Lomas M. Chem. - Eur. J. 2005, 11 (3), 928–938. 10.1002/chem.200400746. [DOI] [PubMed] [Google Scholar]
- Li J.; Dai Y.; Li W.; Laval S.; Xu P.; Yu B. Asian J. Org. Chem. 2015, 4 (8), 756–762. 10.1002/ajoc.201500113. [DOI] [Google Scholar]
- van der Vorm S.; Hansen T.; Overkleeft H. S.; van der Marel G. A.; Codee J. D. C. Chem. Sci. 2017, 8 (3), 1867–1875. 10.1039/C6SC04638J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C.; Leo A.; Taft R. W. Chem. Rev. 1991, 91 (2), 165–195. 10.1021/cr00002a004. [DOI] [Google Scholar]
- Ammer J.; Mayr H. J. Phys. Org. Chem. 2013, 26 (1), 59–63. 10.1002/poc.3064. [DOI] [Google Scholar]
- Minegishi S.; Kobayashi S.; Mayr H. J. Am. Chem. Soc. 2004, 126 (16), 5174–5181. 10.1021/ja031828z. [DOI] [PubMed] [Google Scholar]
- Beaver M. G.; Woerpel K. A. J. Org. Chem. 2010, 75 (4), 1107–1118. 10.1021/jo902222a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krumper J. R.; Salamant W. A.; Woerpel K. A. J. Org. Chem. 2009, 74 (21), 8039–8050. 10.1021/jo901639b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagen B.; Ali S.; Overkleeft H. S.; van der Marel G. A.; Codée J. D. C. J. Org. Chem. 2017, 82 (2), 848–868. 10.1021/acs.joc.6b02593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura E.; Ariga M.; Tohda Y.; Kawashima T. Tetrahedron Lett. 1981, 22 (8), 757–758. 10.1016/0040-4039(81)80143-8. [DOI] [Google Scholar]
- Matsumura E.; Kobayashi H.; Nishikawa T.; Ariga M.; Tohda Y.; Kawashima T. Bull. Chem. Soc. Jpn. 1984, 57 (7), 1961–1965. 10.1246/bcsj.57.1961. [DOI] [Google Scholar]
- Lindberg J.; Öhberg L.; Garegg P. J.; Konradsson P. Tetrahedron 2002, 58 (7), 1387–1398. 10.1016/S0040-4020(01)01241-8. [DOI] [Google Scholar]
- Emmadi M.; Kulkarni S. S. J. Org. Chem. 2011, 76 (11), 4703–4709. 10.1021/jo200342v. [DOI] [PubMed] [Google Scholar]
- Coutant C.; Jacquinet J.-C. J. Chem. Soc., Perkin Trans. 1 1995, (12), 1573–1581. 10.1039/p19950001573. [DOI] [Google Scholar]
- Goddard-Borger E. D.; Stick R. V. Org. Lett. 2007, 9 (19), 3797–3800. 10.1021/ol701581g. [DOI] [PubMed] [Google Scholar]
- Chiara J. L.; García Á.; Cristóbal-Lumbroso G. J. Org. Chem. 2005, 70 (10), 4142–4151. 10.1021/jo050185y. [DOI] [PubMed] [Google Scholar]
- Trans-amination by double Michael addition–elimination with a nucleophilic amine ensures deprotection. Typical conditions included 0.2 M solution of DNPY-protected compound in pyridine at room temperature treated with amine (2 equiv) and stirred for 30 min. Extractive workup gave deprotected amines in excellent yield (>90%). Efficient nucleophilic amines are ethylenediamine (DNPY product is water-soluble), propylamine, hexylamine, and hydrazine.
- Frihed T. G.; Bols M.; Pedersen C. M. Chem. Rev. 2015, 115 (11), 4963–5013. 10.1021/cr500434x. [DOI] [PubMed] [Google Scholar]
- Codée J. D. C.; Litjens R. E. J. N.; den Heeten R.; Overkleeft H. S.; van Boom J. H.; van der Marel G. A. Org. Lett. 2003, 5 (9), 1519–1522. 10.1021/ol034312t. [DOI] [PubMed] [Google Scholar]
- The chemical shifts of the resonances in donor 5 are very sensitive to temperature, concentration, and chemical environment. It was ensured that the sample had not precipitated during the low-temperature NMR experiments at all temperatures; the cause of the line broadening is therefore attributed to the poor solvation and mobility of the donor in dichloromethane.
- Lee J.-C.; Greenberg W. A.; Wong C.-H. Nat. Protoc. 2007, 1 (6), 3143–3152. 10.1038/nprot.2006.489. [DOI] [PubMed] [Google Scholar]
- Douglas N. L.; Ley S. V.; Lücking U.; Warriner S. L. J. Chem. Soc., Perkin Trans. 1 1998, (1), 51–66. 10.1039/a705275h. [DOI] [Google Scholar]
- Zhang Z.; Ollmann I. R.; Ye X.-S.; Wischnat R.; Baasov T.; Wong C.-H. J. Am. Chem. Soc. 1999, 121 (4), 734–753. 10.1021/ja982232s. [DOI] [Google Scholar]
- Pedersen C. M.; Marinescu L. G.; Bols M. Chem. Commun. 2008, (21), 2465–2467. 10.1039/b801305e. [DOI] [PubMed] [Google Scholar]
- Kamkhachorn T.; Parameswar A. R.; Demchenko A. V. Org. Lett. 2010, 12 (13), 3078–3081. 10.1021/ol101089u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walvoort M. T. C.; de Witte W.; van Dijk J.; Dinkelaar J.; Lodder G.; Overkleeft H. S.; Codée J. D. C.; van der Marel G. A. Org. Lett. 2011, 13 (16), 4360–4363. 10.1021/ol2016862. [DOI] [PubMed] [Google Scholar]
- de Jong A.-R.; Hagen B.; van der Ark V.; Overkleeft H. S.; Codée J. D. C.; Van der Marel G. A. J. Org. Chem. 2012, 77 (1), 108–125. 10.1021/jo201586r. [DOI] [PubMed] [Google Scholar]
- Mootoo D. R.; Konradsson P.; Udodong U.; Fraser-Reid B. J. Am. Chem. Soc. 1988, 110 (16), 5583–5584. 10.1021/ja00224a060. [DOI] [Google Scholar]
- Andrews C. W.; Rodebaugh R.; Fraser-Reid B. J. Org. Chem. 1996, 61 (16), 5280–5289. 10.1021/jo9601223. [DOI] [PubMed] [Google Scholar]
- Fraser-Reid B.; López J. C. In Reactivity Tuning in Oligosaccharide Assembly; Fraser-Reid B., Cristóbal López J., Eds.; Springer: Berlin, 2011; pp 1–29. [Google Scholar]
- Balmond E. I.; Coe D. M.; Galan M. C.; McGarrigle E. M. Angew. Chem., Int. Ed. 2012, 51 (36), 9152–9155. 10.1002/anie.201204505. [DOI] [PubMed] [Google Scholar]
- Ennis S. C.; Cumpstey I.; Fairbanks A. J.; Butters T. D.; Mackeen M.; Wormald M. R. Tetrahedron 2002, 58 (46), 9403–9411. 10.1016/S0040-4020(02)01221-8. [DOI] [Google Scholar]
- Ingle A. B.; Chao C.-S.; Hung W.-C.; Mong K.-K. T. Org. Lett. 2013, 15 (20), 5290–5293. 10.1021/ol402519c. [DOI] [PubMed] [Google Scholar]
- The moderate yields obtained in glycosylations with donor 5 are attributed to the unidentified baseline (TLC) material when the reaction was activated and stirred at −40 °C. For model acceptors 6–10, the donor was activated to −50 °C, and unreacted donor was consistently recovered.
- Crich D.; Li L. J. Org. Chem. 2007, 72 (5), 1681–1690. 10.1021/jo062294y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield D. M. Carbohydr. Res. 2007, 342 (12–13), 1726–1740. 10.1016/j.carres.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Hosoya T.; Kosma P.; Rosenau T. Carbohydr. Res. 2015, 411, 64–69. 10.1016/j.carres.2015.03.010. [DOI] [PubMed] [Google Scholar]
- Smith D. M.; Woerpel K. A. Org. Biomol. Chem. 2006, 4 (7), 1195–1201. 10.1039/b600056h. [DOI] [PubMed] [Google Scholar]
- Crich D.; Cai W.; Dai Z. J. Org. Chem. 2000, 65 (5), 1291–1297. 10.1021/jo9910482. [DOI] [PubMed] [Google Scholar]
- This striking stereodirecting effect has been hypothesized to be caused by remote participation, but direct evidence for this type of participation on benzylidene mannose donors has not yet been found. Other stereoelectronic stabilizing effects might be responsible for the apparent stereoselectivity and await further study. For a discussion on remote participation, see:; a Crich D.; Hu T.; Cai F. J. Org. Chem. 2008, 73 (22), 8942–8953. 10.1021/jo801630m. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Baek J. Y.; Lee B.-Y.; Jo M. G.; Kim K. S. J. Am. Chem. Soc. 2009, 131 (48), 17705–17713. 10.1021/ja907252u. [DOI] [PubMed] [Google Scholar]; c Komarova B. S.; Orekhova M. V.; Tsvetkov Y. E.; Nifantiev N. E. Carbohydr. Res. 2014, 384, 70–86. 10.1016/j.carres.2013.11.016. [DOI] [PubMed] [Google Scholar]
- Crich D.; Smith M.; Yao Q.; Picione J. Synthesis 2001, 2001 (02), 0323–0326. 10.1055/s-2001-10798. [DOI] [Google Scholar]
- Benakli K.; Zha C.; Kerns R. J. J. Am. Chem. Soc. 2001, 123 (38), 9461–9462. 10.1021/ja0162109. [DOI] [PubMed] [Google Scholar]
- Buskas T.; Garegg P. J.; Konradsson P.; Maloisel J.-L. Tetrahedron: Asymmetry 1994, 5 (11), 2187–2194. 10.1016/S0957-4166(00)86294-1. [DOI] [Google Scholar]
- Emmadi M.; Kulkarni S. S. J. Org. Chem. 2011, 76 (11), 4703–4709. 10.1021/jo200342v. [DOI] [PubMed] [Google Scholar]
- You F.; Twieg R. J. Tetrahedron Lett. 1999, 40 (50), 8759–8762. 10.1016/S0040-4039(99)01694-9. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.