

Abstract
Appropriate bone mass is maintained by bone-forming osteoblast and bone-resorbing osteoclasts. Mesenchymal stem cell (MSC) lineage cells control osteoclastogenesis via expression of RANKL and OPG (receptor activator of nuclear factor κB ligand and osteoprotegerin), which promote and inhibit bone resorption, respectively. Protein crosslinking enzymes transglutaminase 2 (TG2) and Factor XIII-A (FXIII-A) have been linked to activity of myeloid and MSC lineage cells; however, in vivo evidence has been lacking to support their function. In this study, we show in mice that TG2 and FXIII-A control monocyte-macrophage cell differentiation into osteoclasts as well as RANKL production in MSCs and in adipocytes. Long bones of mice lacking TG2 and FXIII-A transglutaminases, show compromised biomechanical properties and trabecular bone loss in axial and appendicular skeleton. This was caused by increased osteoclastogenesis, a cellular phenotype that persists in vitro. The increased potential of TG2 and FXIII-A deficient monocytes to form osteoclasts was reversed by chemical inhibition of TG activity, which revealed the presence of TG1 in osteoclasts and assigned different roles for the TGs as regulators of osteoclastogenesis. TG2- and FXIII-A-deficient mice had normal osteoblast activity, but increased bone marrow adipogenesis, MSCs lacking TG2 and FXIII-A showed high adipogenic potential and significantly increased RANKL expression as well as upregulated TG1 expression. Chemical inhibition of TG activity in the null cells further increased adipogenic potential and RANKL production. Altered differentiation of TG2 and FXIII-A null MSCs was associated with plasma fibronectin (FN) assembly defect in cultures and FN retention in serum and marrow in vivo instead of assembly into bone. Our findings provide new functions for TG2, FXIII-A and TG1 in bone cells and identify them as novel regulators of bone mass, plasma FN homeostasis, RANKL production and myeloid and MSC cell differentiation.
Maintenance of postnatal bone mass is tightly regulated by bone remodeling, which is orchestrated by reciprocal activity of bone-resorbing osteoclasts and bone-forming osteoblasts.1 Increased resorption over formation leads to bone loss (osteopenia) and ultimately osteoporosis, which is first observed in the trabecular bone that is surrounded by bone marrow. Bone marrow functions as a maturation site for mesenchymal stem cells (MSCs) that give rise to osteoblasts.2 It also serves as a maturation and homing site for hematopoietic stem cells that give rise to lymphoid and myeloid lineage cells including monocytes that can mature into osteoclasts under appropriate signals.3, 4 It can be considered that the bone and the bone marrow form a continuous organ system.2 Osteoclastogenesis is regulated by many factors, the most notable being the RANKL/OPG system (receptor activator of nuclear factor κB ligand/osteoprotegerin) which promote and inhibit osteoclastogenesis, respectively.4, 5, 6 These cytokines are secreted by bone marrow MSCs (bmMSCs), preosteoblasts, osteoblasts, osteocytes and preadipocytes and adipocytes,4, 7, 8, 9 whose activity and differentiation can be in turn be regulated by extracellular matrix (ECM).10 Bone marrow matrix (stroma) contains several extracellular molecules, including types I, III and IV collagens, and laminin and fibronectin (FN).10, 11
Transglutaminases are a family of enzymes that are able to modify glutamine residues of their specific substrate proteins. Best known modification is a Ca2+-dependent acyl-transfer reaction between polypeptide-bound glutamine (Q) residues and lysine (K) residues (or primary amines), which results in formation of a γ-glutamyl-ɛ-lysyl bond (an isopeptide crosslink/bond). This enzymatic reaction is exclusively performed by TGs and takes place mostly at the cell surface and/or in ECM compartments.12, 13, 14, 15, 16 Crosslinking can stabilize molecular structures, increase protein network formation and stiffness as well as change protein solubility and stability and enhance cell adhesion.14 TGs can also have functions that do not require their enzymatic activity.17, 18 The transglutaminase enzyme family currently contains eight genes encoding active enzymes: Factor XIII-A and TG1-7 of which TG2 and FXIII-A are found in monocyte-macrophage lineage cells and number of cells of mesenchymal origin osteoblasts as well as in bone tissue.13, 15, 19, 20, 21, 22, 23, 24 FN, a ubiquitous ECM glycoprotein required for cell adhesion, proliferation, survival and differentiation of many cell types25, 26, 27 including bmMSCs, and osteoblasts,28, 29 is a well-known substrate for TGs, particularly for FXIII-A.30, 31, 32 Postnatally, FN exists in two main forms which arise from alternate splicing of one gene: as the soluble, circulating plasma FN (pFN) produced by hepatocytes in liver, and as cellular FN (cFN) synthesized by certain tissue-resident cells.25, 33 Both plasma and cell-derived FNs undergo assembly into fibrillar network which adhesion-dependent cells bind to, and migrate, proliferate and differentiate on.26, 33 We have recently demonstrated that pFN, but not cFN, acts as a TG substrate and requires TG activity for its assembly in osteoblast and adipocyte cultures, where this pFN matrix subsequently regulates cell proliferation and differentiation and ECM deposition.23, 31 pFN is a major component of many tissues34 including bone where 90% of FN matrix is plasma-derived.34 FN deletion in liver decreases the biomechanical properties of bone.35
Despite the reported in vitro importance of TG activity for osteoblast function, neither TG2 nor FXIII-A-null mice show an overt bone phenotype.36, 37 Moreover, a recent study shows that mice deficient in both enzymes have normal bone deposition and mineralization; however in this study, maintenance of bone mass or remodeling activity of bone cells was not examined.38 Here, we also have generated a mouse lacking both TG2 and FXIII-A (Tgm2−/−;F13a1−/−). We report that the combined absence of the two produces a major bone phenotype and causes altered activity of both osteoclasts and bmMSCs. Tgm2−/−;F13a1−/− mice show severe trabecular bone loss attributable to increased osteoclastogenesis and bone resorption in vivo and bmMSC differentiation towards adipogenesis with increased RANKL production, which further contributes to a bone marrow microenvironment that is highly skewed towards bone-resorption. Both cellular phenotypes persists in vitro demonstrating that cellular TG2 and FXIII-A in the monocyte-macrophage lineage are negative regulators of osteoclastogenesis and inhibitors of adipogenesis. Interestingly, normal and Tgm2−/−;F13a1−/− osteoclasts show expression of transglutaminase 1 (TG1) whose chemical inhibition blocks osteoclastogenesis, which identifies TG1 as a promoter of bone resorption. TG1 expression is also seen in Tgm2−/−;F13a1−/− deficient bmMSC where it is significantly upregulated. Chemical inhibition of TG activity in normal and Tgm2−/−;F13a1−/− bmMSCs dramatically increases adipogenic potential of the MSCs, but affects RANKL expression only in the absence of TG2 and FXIII-A. Furthermore, TG2 and FXIII-A deficiency changes pFN homeostasis and its accumulation from blood to bone marrow and bone in vivo. It also decreases pFN assembly into matrix increases its degradation in MSC culture which is associated with increased adipogenesis. The present study is the first to report on the role for the three TGs in bone homeostasis in mice.
Results
TG2 and FXIII-A jointly regulate skeletal growth, bone strength and mass
Mice lacking both TG2 and FXIII-A enzymes (Tgm2−/−;F13a1−/−) were born with no obvious developmental or skeletal defects based on birth weight, and on radiographic and histological assessment of bone at birth (data not shown). By the time Tgm2−/−;F13a1−/− reached 4 weeks of age, roughly half (51.5%) of the male mice had died (Supplementary Figure S1a) – death was seemingly attributable to hemorrhaging caused by blood vessel rupture in various tissues (females were not used in this study). Although mice were normal in size at 1 month age, the surviving Tgm2−/−;F13a1−/− mice were smaller in size at 3 month age as apparent from weight/tibia measurements of 1- versus 3-month-old mice (Supplementary Figure S1b-c) and X-rays taken at those same time points (Supplementary Figure S1d). The smaller size was observed at 3 and 6 months but size was normal at 12 months age as per body weight (Supplementary Figure S1b) demonstrating that the mild growth defect was transient. This minor, but significant growth delay is likely not due to compromised nutrition intake because dentition is intact (Supplementary Figure S1d), and no behavioral alterations due to stress was observed (data not shown). Furthermore, normal eating is supported by normal serum calcium and phosphate levels (calcium levels were 9.0±0.186 and 8.8±0.071 mg/ml in WT and double-null mice, respectively, and phosphate levels 7.7±0.315 and 8.0±0.463, respectively) and unaltered bone formation/synthesis (described below, Figure 4b), these levels/parameters would be expected to be decreased under nutritional deprivation.39
Figure 4.

Increased bone marrow adiposity and RANKL production in 3-month-old Tgm2−/−;F13a1−/− mice. (a) Histomorphometric quantification of osteoblasts (N.Ob/T.Ar) and osteocytes (N.Ocy/T.Ar) (relative to trabecular area) show no significant change in 3-month-old Tgm2−/−;F13a1−/− mice. n=6. (b) Calcein double-labeling and its quantification shows no change in mineral apposition rate (MAR). n=6. Images are representative of six mice. (c) ALP staining (pink) shows active osteoblasts around trabeculae and reveals increased adipocity of the bone marrow (black arrow heads pointing to adipocytes that appear in white) in the in Tgm2−/−;F13a1−/− mice. Images are representative of three mice. (d) Quantification of adipocyte number in serial sections of bone marrow confirms the significant increase. n=3. (e and f) bmMSCs isolated from WT and Tgm2−/−;F13a1−/− mouse bone marrow show increased potential for adipogenesis under adipogenic conditions as assessed and quantified by Oil Red staining at day 13 of culture. Images are representative of three separate experiments. n=3 in Oil Red quantification. (g). RT-PCR analysis of bmMSC gene expression. The adipocyte marker, Pparg, is increased in Tgm2−/−;F13a1−/− cells grown in adipogenic media. Rankl production is dramatically increased in TG2 and FXIII-A deficient bmMSCs. Conversely Opg production is decreased. Csf1 production does not show change in the absence of the two TGs. Representative data from three separate experiments. P-values are as follows: *P<0.05, ***P<0.001, NS; not significant. Scale bar, 20 μm (b); and 200 μm (c)
Examining bone mineral density (BMD) at 1-, 3-, 6- and 12-month-old mice showed significantly lower BMD in 3-month-old Tgm2−/−;F13a1−/− double-null mice compared with WT controls and the double null at other ages (1-, 6- and 12-month-old mice) supporting transient phenotype (Supplementary Table 1 and Supplementary Figure S1e). Mineralization was intact and no unmineralized osteoid (sign of osteomalacia) was seen (Figure 1e) suggesting normal bone formation and mineralization mechanisms as reported before for TG2 and FXIII-A double null.38 The individual Tgm2−/− and F13a1−/− knockout mice showed no decrease in BMD at 3 months age (Supplementary Table 1).
Figure 1.

Trabecular bone loss in 3-month-old Tgm2−/−;F13a1−/− mice. (a) Micro-computed tomography (μCT) images of proximal tibia and lumbar vertebra trabecular bone showing bone loss in appendicular and axial skeleton of the double-null mouse. (b) No trabecular bone loss was observed in Tgm2−/− or F13a1 −/− mice. (c) Quantified μCT parameters of trabecular bone confirms dramatic and significant bone loss, that is, decreased mineralized bone volume over tissue volume (BV/TV), trabecular number (Tr.N.), trabecular thickness (Tr.Th.) and increased trabecular spacing (Tr.Sp.) in Tgm2−/−;F13a1−/− mice. μCT data derives from five mice and images are representative of five mice. (d) Three-point bending test results obtained using a MACH-1 micro-mechanical testing system of the right femur from double-null and wild-type mice. Bones of Tgm2−/−;F13a1−/− mice require less ultimate force to induce fracture. n=4–6. (e) Histology and von Kossa staining (black) of tibial trabecular bone and cartilaginous growth plates of WT and Tgm2−/−;F13a1−/− mice showing bone loss (osteopenia). Mineralization is normal and no unmineralized osteoid (osteomalacia) is seen. Cartilage and growth plate appears normal. Images are representative of three mice. (f) Cortical thickness was not decreased (C.Th). n=5. P-values are as follows: *P<0.05, **P<0.01. Scale bars, 500 μm (a, b and f); 200 μm (e)
Analysis of quantity of bone, that is, bone mass parameters in 3-month-old mice revealed severe osteopenia in Tgm2−/−;F13a1−/− mice (Figure 1). Quantitative μCT of trabecular bone from both appendicular and axial skeletal sites – from proximal tibia and lumbar vertebra – revealed dramatic trabecular bone loss in Tgm2−/−;F13a1−/− mice compared with WT controls (Figure 1a). Individual Tgm2−/− and F13a1−/− showed no trabecular bone loss at 3 month age (Figure 1b). Quantification of structural parameters from μCT data of tibial trabecular bone showed a significant decrease in trabecular bone quantity (Figure 1c). Mineralized bone volume (over tissue volume) (BV/TV), number of trabeculae (Tr.N.) and trabecular thickness (Tr.Th) were significantly decreased, and conversely, trabecular spacing (Tr.Sp.) increased (Figure 1c) in Tgm2−/−;F13a1−/− mice compared with their WT controls. Bone biomechanical quality was significantly reduced in Tgm2−/−;F13a1−/− mice compared with WT controls as analyzed by the femoral three-point bending test at the 1 and 3 month time points (Figure 1d). Bone loss in double null mice was also evident in histological analyses which showed a significant loss of bone trabeculae immediately below the cartilaginous growth plate, which itself showed no overt changes (Figure 1e). Cortical thickness (C.Th.) was not significantly altered (Figure 1f). In support of the transient phenotype, no bone loss was apparent in 1-month-old Tgm2−/−;F13a1−/− mice as per to μCT analysis (Supplementary Figure 2). Only mineralized bone volume over tissue volume (BV/TV) showed significant decrease, which may explains the decreased biomechanical strength at this age (Supplementary Figure 2).
Numerical and statistical comparison of some of the bone parameters of Tgm2−/−;F13a1−/− to individual Tgm2−/− and F13a1−/− knockouts and their controls showed consistently lower values in the double null (Supplementary Table 1-3) although ANOVA analysis between the six mouse groups showed significance only between double null and its mixed-background control.
Deletion TG2 and FXIII-A causes increased osteoclastogenesis in vivo and in vitro
To determine the cellular basis of the bone loss in Tgm2−/−;F13a1−/− mice, we analyzed bone resorption parameters in vivo. Serum measurements of the bone resorption marker (RatLapsTM) of 1-, 3-, 6- and 12-month-old mice showed significant increase in resorption in Tgm2−/−;F13a1−/− mice up to 6 month age (Figure 2a)(Supplementary Table 4 shows resorption marker levels in all six mouse groups at 3 months age). Similarly, serum osteocalcin (OCN), the majority of which has been recently described to arise from osteoclastic resorption activity,40 was significantly higher at 3-month-old Tgm2−/−;F13a1−/− mice (Figure 2b). Staining of trabecular bone for the osteoclast marker TRAP and quantification of osteoclast numbers per trabecular area (N.Oc/T.Ar.) and bone perimeter (N.Oc/Tr.Pm.) demonstrated significantly increased osteoclast numbers (twofold) in Tgm2−/−;F13a1−/− mice compared with WT mice (Figure 2c and d). Levels of bone marrow monocytes, precursor cells to osteoclasts, were unaltered as per to flow cytometric analysis indicating that only osteoclastogenesis was increased (Supplementary Figure S3). Analysis of the serum RANKL levels and RANKL/OPG ratio revealed a significantly increased production of RANKL and its dominance over OPG in Tgm2−/−;F13a1−/− mice (Figure 2e and f).
Figure 2.

Osteoclastogenesis, bone resorption and RANKL production are increased in Tgm2−/−;F13a1−/− mice. (a) Analysis of the bone resorption marker (serum COL I telopeptide fragments) (RatLapsTM) shows a significant increase in Tgm2−/−;F13a1−/− mice up to 6 months age compared with WT mice. n=5–7. (b) Serum osteocalcin levels are elevated in 3-month-old Tgm2−/−;F13a1−/− mice compared with WT mice of same age. n=6. (c) The presence of osteoclasts was increased in lumbar trabecular bone (Tb) in 3-month-old Tgm2−/−;F13a1−/− mice compared with WT controls as evident by tartrate-resistant acid phosphatase (TRAP, pink) staining of histology sections (counterstained with hematoxylin). BM; bone marrow. Images are representative of six mice. (d) Quantification of osteoclast numbers relative to trabecular area (N.Oc./T.Ar) shows a significant, twofold increase in 3-month-old mice Tgm2−/−;F13a1−/− mice. (e and f) Analysis of the serum RANKL levels and RANKL/OPG ratio shows twofold increase in RANKL production and significant skewing towards RANKL dominance in 3-month-old mice Tgm2−/−;F13a1−/− mice. n=6. P-values are as follows: *P<0.05, ** P<0.01, ***P<0.001. Scale bar, 40 μm
To determine whether the increased osteoclastogenesis was caused solely by an increased RANKL levels, we isolated monocytes from the bone marrow of 2-month-old Tgm2−/−;F13a1−/− and WT mice (this age is optimal for isolation), and examined their osteoclastogenesis and resorption potential in vitro. A 6-day exposure of monocytes to M-CSF (monocyte colony-stimulating factor) and RANKL resulted in increased osteoclastogenesis from Tgm2−/−;F13a1−/− monocytes compared with WT monocytes (Figure 3a). Quantification of osteoclast number demonstrated a twofold increase (Figure 3b) and significantly increased TRAP levels in the culture media (Figure 3c). Functional analysis of resorption potency of the osteoclast cultures demonstrated a visible and significant increase in the area of resorption pits after exposure to a bone-mimicking osteological surface (Figure 3d). Data indicated that TG2 and FXIII-A are inhibitors of osteoclast formation.
Figure 3.

Monocytes from Tgm2−/−;F13a1−/− mice show increased differentiation into osteoclasts in vitro – chemical inhibition of TG activity, however, blocks osteoclastogenesis due to presence of TG1. (a) Bone marrow monocytes isolated from 2-month-old mice were differentiated into osteoclasts with M-CSF and RANKL for 6 days. Tartrate-resistant acid phosphatase (TRAP) staining of the cultures showing increased osteoclast numbers and increased size in the combined absence of TG2 and FXIII-A. Images are representative of three separate experiments. (b) Osteoclast counts from the cultures show a twofold increase in osteoclast numbers from Tgm2−/−;F13a1−/− monocytes. n=3 (three separate experiments). (c) Quantification of TRAP staining from media shows significant increase in Tgm2−/−;F13a1−/− cultures. Triplicate analysis from three separate experiments is presented. (d) Tgm2−/−;F13a1−/− osteoclasts show dramatically increased resorption activity as visualized by resorption pit assay on mineral coated plates. Images are representative of three experiments. Quantification of the resorbed area (%) shows significantly increased, threefold osteoclast activity of Tgm2−/−;F13a1−/− osteoclasts. n=3. (e) Chemical inhibition of TG activity with NC9 in WT and Tgm2−/−;F13a1−/− cultures, however, shows complete attenuation of osteclastogenesis suggesting presence of other TGs. Triplicate analysis from three separate experiments in presented. (f) RT-PCR analyses of Tgm1 expression during osteoclastogenesis shows increase upon M-CSF stimulation in both WT and Tgm2−/−;F13a1−/− cells. Representative data from three separate experiments is shown. (g) qRT-PCR analysis shows that Tgm1 is not upregulated in Tgm2−/−;F13a1−/− osteoclasts. n=3. P-values are as follows: ***P<0.001. NS; not significant. Scale bars, 200 μm
Chemical inhibition of TG activity inhibits osteoclastogenesis – presence of TG1 in osteoclasts
To examine if chemical inhibition of TG activity would result the same cellular behavior as the absence of TG2 and FXIII-A, we treated monocytes during osteoclastogenesis with NC9 which is a broad spectrum, irreversible TG inhibitor.41 Surprisingly, treatment of WT and Tgm2−/−;F13a1−/− monocytes with the inhibitor resulted in complete blockage of osteoclast formation in both, indicating that TG activity from another enzyme may be present and responsible for osteoclastogenesis (Figure 3e). Indeed, RT- and qRT-PCR screening of expression of TG family members showed strong expression of Tgm1 (TG1) in both Tgm2−/−;F13a1−/− and WT osteoclasts (Supplementary Figure S4) where it was upregulated during osteoclastogenesis in response to M-CSF (Figure 3f). Tgm1 was not upregulated in the absence of TG2 and FXIII-A and showed same level expression as WT (Figure 3g) suggesting that it does not compensate for the absence of TG2 and/or FXIII-A, but functions to promote osteoclastogenesis as opposed to TG2 and FXIII-A which inhibit the process.
TG2 and FXIII-A deficiency increases bone marrow adipogenesis and RANKL production by bmMSCs
To continue our examination of bone cell function as well as to determine the source for skewed RANKL/OPG balance towards RANKL production in bone marrow, we analyzed osteoblast function and numbers in vivo. Histomorphometric analysis of 3-month-old mice showed no significant change in osteoblast (N.Ob/T.Ar) or osteocyte numbers (N.Ocy/T.Ar) in Tgm2−/−;F13a1−/− versus WT mice (Figure 4a). Assessment of dynamic bone formation by serial calcein injections in the 3-month-old mice showed no change in mineral apposition rate (MAR) in WT versus Tgm2−/−;F13a1−/− mice (2.09±0.12 μm/day and 2.12±0.25 μm/day, respectively) (Figure 4b). Histological analysis of ALP (alkaline phosphatase levels/positive cells) showed similar ALP staining around bone trabeculae in Tgm2−/−;F13a1−/− and WT bone (Figure 4c). As apparent from the histological images and cell counts of serial bone marrow sections (Figure 4c and d) Tgm2−/−;F13a1−/− bone marrow showed a dramatically and significantly increased number of adipocytes. Cultures of bmMSCs isolated from 2-month-old mice (this age is optimal for isolation) Tgm2−/−;F13a1−/− mice showed significantly increased potential to differentiate into adipocytes under adipogenic conditions as visualized and quantified by Oil Red staining (Figure 4e and f). Moreover, mRNA expression analysis showed higher Pparg2 production by Tgm2−/−;F13a1−/− cells upon treatment with adipogenic media (Figure 4g). Most striking was the very high and significantly increased RANKL production in Tgm2−/−;F13a1−/− cells compared with WT cells (Figure 4g and Supplementary Figure S4a). Opg production was decreased in Tgm2−/−;F13a1−/− cells but this was not significant (Figure 4g and Supplementary Figure S5a). mRNA expression of M-CSF (Csf1) was not altered (Figure 4g).
Upregulation of TG1 in the absence of TG2 and FXIII-A bmMSCs – all three regulate RANKL production
Interestingly, both WT and Tgm2−/−;F13a1−/− bmMSCs also showed Tgm1 expression (Supplementary Figure S5b) which was significantly higher in the knockout cells (Figure 5a). Inhibition of TG activity by NC9 in both WT and Tgm2−/−;F13a1−/− bmMSC cultures under adipogenic conditions, significantly increased adipogenic transcription factor Pparg2 expression (Figure 5b and c) suggesting that TG1 can regulate bmMSC fate and that its production is activated in the absence of TG2 and FXIII-A. Interestingly, NC9 upregulated RANKL mRNA significantly only in Tgm2−/−;F13a1−/− bmMSC but not in WT cells, suggesting that TG2 and FXIII-A proteins, but not their enzymatic activity, regulates TG1 which then negatively regulates RANKL mRNA production.
Figure 5.
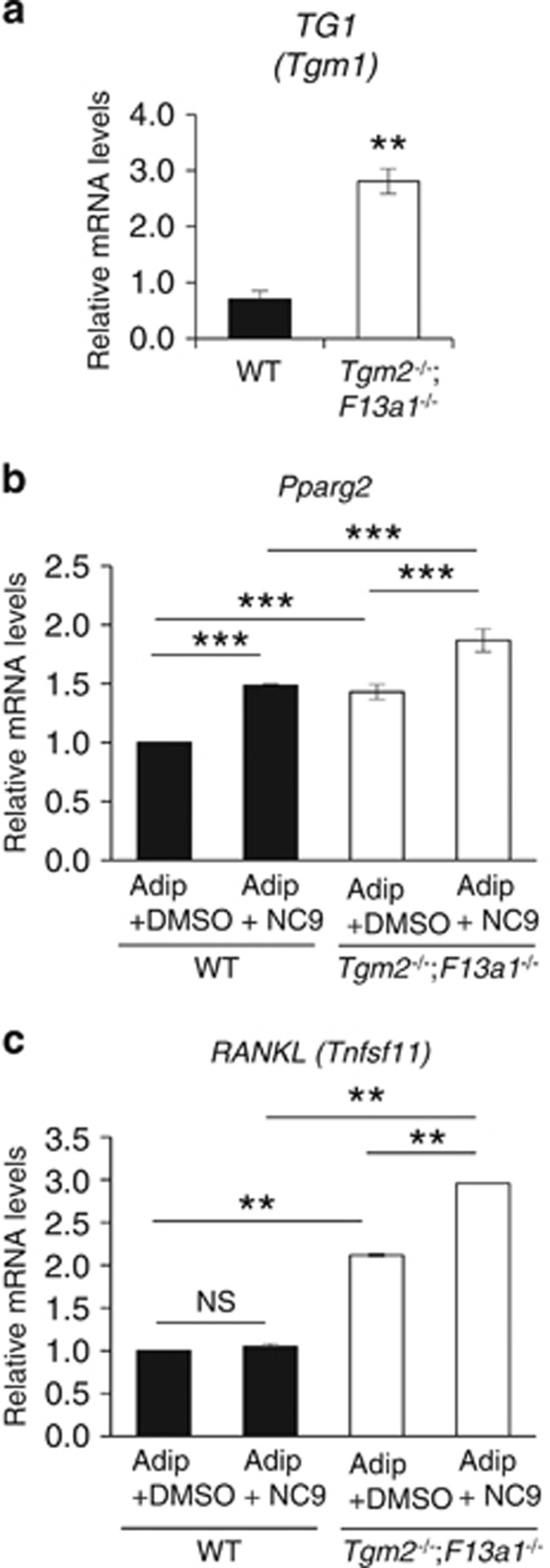
Tgm1 is upregulated in Tgm2−/−;F13a1−/− bmMSCs and its chemical inhibition promotes PPARγ and RANKL production. (a) qRT-PCR analyses of Tgm1 in bmMSCs shows significant upregulation in double null cells isolated from 2-month-old mice. Data is from three separate experiments. (b) PPARγ (Pparg) and (c) RANKL (Tnfsf11) expression in bmMSC after adipogenic treatment and NC9 increased adipogenic potential of and increased RANKL production in Tgm2−/−;F13a1−/− cells which both are further increased by TG inhibitor NC9 in double null cells. P-values are as follows: **P<0.01, ***P<0.001. NS; not significant
TG2 and FXIII-A regulate pFN assembly and homeostasis in liver-plasma-bone axis
To examine which biochemical conditions in the bone marrow may be contributing to the microenvironment that switches MSC differentiation towards adipogenesis in Tgm2−/−;F13a1−/− mice we first confirmed that bone marrow in these mice is indeed devoid of TG activity (Supplementary Figure S6a). We also considered that FN – which we have shown before to be the main TG transamidation substrate in osteoblast and adipocyte cell cultures23, 31 – can regulate the differentiation of the two cell types.31, 35, 42 Thus, we examined FN homeostasis in WT and Tgm2−/−;F13a1−/− mouse bones and bone marrow. Sequential extraction of bone with the chaotropic agent Guanidium-HCl (G1 and G2), mineral dissolving EDTA and DOC/SDS-detergents (Figure 6a and b) showed that significantly more FN was present in the G1 and G2 extracts, and less in the last DOC/SDS extracts, strongly suggesting that FN in 3-month-old Tgm2−/−;F13a1−/− bone has increased solubility. The increased FN in the G1-extract was not attributable to increased EDA-FN secretion by cells in the bone marrow – while EDA-FN was produced in Tgm2−/−;F13a1−/− osteoblasts, EDA-FN, was not externalized or found in the bone extracts (G1-extract) nor bone marrow (Supplementary Figure S6b-c). However, total FN levels were clearly increased in bone marrow (Figure 6c) suggesting that this represented pFN. Immunohistological analysis of bone sections showed more intense total FN staining in the osteoid layer and in the bone marrow space of Tgm2−/−;F13a1−/− mice compared with WT mice. Cells adhering to this FN layer appeared morphologically larger and rounder (Figure 6d).
Figure 6.

Tgm2−/−;F13a1−/− mice show alterations in FN matrix solubility in bone, and increased FN levels in bone marrow. (a and b) Sequential bone protein extraction with Guanidium-HCl (G1), EDTA, a repeat of Guanidium-HCl (G2) and DOC/SDS, followed by western blot analysis using anti-FN antibody shows higher levels of FN in G1 and G2 extracts and decreased levels in the DOC/SDS extract in 3-month-old Tgm2−/−;F13a1−/− mice compared with WT controls of same age. In particular, the high-molecular weight (HMW) FN is absent in Tgm2−/−;F13a1−/− mouse bone. Quantification of FN levels in G1-, G2- and DOC/SDS extracts shows that the FN changes are significant. (c) Western blot analysis of FN levels in bone marrow flushes shows its increase. (d) Histological staining of bone sections for total FN (antibody used does not distinguish between cellular and plasma FN) (pink) shows more intense FN staining in the osteoid (Os) layer of double-null mice. This was accompanied by a rounder morphology of osteoblasts. Images are representative of three mice. All data is collected from 3-month-old mice. P-values are as follows: *P<0.05, **P<0.01. Scale bar, 200 μm
Analysis of pFN levels in WT and Tgm2−/−;F13a1−/− mouse serum showed a dramatic and significant increase indicative of defective integration into tissues (Figure 7a). Serum FN levels were significantly higher in 1-, 3- and 6-month-old mice, but not in 12-month-old mice (Supplementary Figure S7), which correlates with the other parameters, particularly the resorption marker (Figure 3a). Cellular (EDA-FN) levels were not increased in serum (Figure 7b). Hepatocytes isolated from WT and Tgm2−/−;F13a1−/− showed no altered FN production (Figure 7c) further supporting the concept of a tissue-integration defect. Intraperitoneal injections of Oyster488-labeled pFN into WT and Tgm2−/−;F13a1−/− mice for 3 consecutive days (Figure 7d) followed by fluorometric detection in serum, bone marrow and bone (G1-extract), showed that the double-null mice had significantly higher levels of pFN in serum and bone marrow compared with WT mice, and a conversely decreased amount in bone (Figure 7e). Furthermore, as the marrow space also showed a significant decrease in TG activity (Supplementary Figure S6a) pFN matrix in this milieu is likely not appropriately crosslinked and/or assembled into fibrillar networks. Indeed, Tgm2−/−;F13a1−/− bmMSCs had decreased ability to assemble exogenously added biotinylated pFN into DOC-soluble fibrils (Figure 8a). pFN was more prone to fragmentation indicating that non-crosslinked bpFN and/or bpFN matrix was biochemically labile (Figure 8a). Tgm2−/−;F13a1−/− bmMSCs did not assemble bpFN into rounded nodules like in WT cells (Figure 8c). This decreased pFN matrix assembly was not linked to bmMSC proliferation as seen from the results of MTT assay and Ki67 staining of the bmMSC cultures (Figure 8 b and c).
Figure 7.

Alterations in plasma FN homeostasis in 3-month-old Tgm2−/−;F13a1−/−. (a) Plasma (pFN) levels are significantly and dramatically increased in 3-month-old double-null mouse serum. n=6. (b) Cellular FN levels show no significant changes between the serum of Tgm2−/−;F13a1−/− and WT mice. n=6. (c) Hepatocytes isolated from 3-month-old Tgm2−/−;F13a1−/− and WT mice produce and secrete FN at similar levels as seen by Western blot analysis of FN in culture media. Albumin was used as control protein for media analysis, and actin blotting shows the cellular density of the samples where media was collected from. (CM; conditioned media, CE; cell extract). Western blots are representative of three separate experiments. (d) The Oyster488-labeled FN (Oy488pFN) injection regime. Mice (n=3)(3 month old) were injected intraperitoneally on 3 consecutive days with 1 mg of labeled FN. On day 5, mice were sacrificed and tissue fluids and tissues were collected. (e) Fluorometric detection of Oy488pFN in serum, bone marrow and the bone G1-extract. A significant increase in Oy488pFN retention was seen in serum and bone marrow in 3-month-old Tgm2−/−;F13a1−/− mice compared with control WT mice of same age. Conversely, a decrease was seen in Oy488FN incorporation in bone G1-extract. (f) Schematic diagram representing pFN homeostasis in the blood-bone marrow-bone axis and its defect in Tgm2−/−;F13a1−/− mice where its accumulation is decreased in bone likely contributing to higher levels in the marrow compartment and in blood. P-values are as follows: *P<0.05, ***P<0.001
Figure 8.

Plasma FN matrix assembly defect and its increased degradation in Tgm2−/−;F13a1−/− bmMSC cultures. (a). BMMSCs isolated from 2-month-old mice were grown with biotinylated plasma FN (bpFN) and assembly of DOC-soluble matrix was examined by Western blotting. A decrease is seen in DOC-soluble bpFN matrix in Tgm2−/−;F13a1−/− mouse cultures. A smaller pFN fragment is also seen in Tgm2−/−;F13a1−/− cultures indicative of increased degradation. Western blot image is representative of two separate cell culture experiments. (b) Double-null bmMSCs show no altered proliferation under regular media or adipogenic media as per MTT assay. Triplicate analysis from two separate experiments is presented. (c) Immunofluoresence analysis of bpFN assembly (green), cell density (DAPI, blue) proliferation (Ki67+DAPI)(pink) around bpFN. WT bmMSCs assemble bpFN into clear rounded patches that are associated with cell nodules. Tgm2−/−;F13a1−/− bpFN matrix lacks this rounded shape and nodular configuration of the cells. Cell proliferation appears normal. Immunofluoresence images are representative of two separate cell culture experiments. NS, not significant. Scale bars, 10 μm
Discussion
In vitro work on transglutaminases TG2 and FXIII-A has strongly suggested them to have a role in bone formation; however, the respective individual knockout mice have not shown a bone phenotype possibly due to compensation from each other.43 Nevertheless, a recent report suggests that their double deletion neither results in overt changes to skeleton nor bone deposition defect.38 In this study we have also generated a mouse lacking TG2 and FXIII-A. Our evidence supports these previous in vivo findings showing no phenotype in osteoblasts; however, we provide new evidence via additional skeletal phenotyping that TG2 and FXIII-A do have a synergistic function in the maintenance of bone mass. TG2 and FXIII-A regulate osteoclastogenesis, RANKL expression and bone marrow adipogenesis suggesting a role in myeloid lineage and MSC cell differentiation in mice. The parameters revealing this phenotype were not examined previously. This is the first report to identify TGs in the regulation of bone remodeling and in balancing bone cell activity via RANKL.
The presence of TG2 and FXIII-A in monocytes and macrophages is well documented,15, 20, 44, 45 and a recent report shows the relevance of FXIII-A in osteoclast function in arthritis where FXIII-A deficiency is protective of osteoclast-mediated bone erosion.46 In our study, we show that absence of TG2 and FXIII-A result in dramatic 'increase' in osteoclastogeneis in vivo – a phenotype that persists in vitro. An unexpected finding was that monocytes and osteoclasts express TG1, which is increased during osteoclastogenesis both in normal mice and in the absence of TG2 and FXIII-A. Blocking of TG activity with a chemical inhibitor, NC9,41 in both WT and double null monocytes/osteoclast cultures inhibits osteoclastogenesis suggesting that TG1 is required for osteoclast formation. Thus, our data supports the findings of Rahgu et al.46 who showed blocking of osteoclastogenesis by cystamine as TG inhibitor. Our data reveals a role for TG1 in osteoclasts and a regulatory circuitry between the three TGs. TG1 is a keratinocyte TG responsible for the formation of cornified cell envelope that acts as protective skin barrier. Mutations in TGM1 cause lamellar ichthyosis.14 No skeletal phenotype has been reported for Tgm1 deficient mice that die soon after birth.47 TG1 expression has not been previously documented in any bone cells.
Although TG2 and FXIII-A have been implicated in osteoblast function and activity in vitro,28, 31, 48, 49 the double-null mouse phenotype reported before38 and our work here strongly suggests that they do not regulate osteoblast activity per se but regulate the function of their precursor cells in the postnatal skeleton. This is supported by the increased adipogenesis in double null bone marrow and by the enhanced potential of TG2 and FXIII-A double null bmMSCs to differentiate to adipocytes as well as by our previous findings showing enhanced adipogenesis in both FXIII-A as well as TG2-deficient mouse embryonic fibroblasts.23, 24 The absence of osteoblast phenotype may at first appear contradictory to the in vitro data where us and others demonstrated a role for TG2 and FXIII-A in matrix deposition and cell adhesion in human (HOS, HOBs and MG-63 cells)49 and mouse osteoblasts MC3T3-E1 and primary cells).21 However, the absence of bone deposition phenotype does not exclude the possibility that the function of TG2 and FXIII-A in osteoblasts could be to enhance adhesion of osteoblasts as well as to provide quality to the bone and strengthen the organic bone matrix via orchestrating or assisting assembly of ECM proteins, such as pFN fibrillogenesis, as shown in our study. Indeed, quality and quantity of pFN network in bone may affect total matrix quality via downstream effects on lysyl oxidase crosslinking activity,50 BMP1 activation51 and subsequent collagen fibril assembly/maturation. This role for TGs in osteoblasts is currently under investigation.
FN is a well-established TG substrate32, 52, 53 although the exact in vivo physiological role of its transglutaminylation and crosslinking has remained unclear. Accumulating evidence suggests though that particularly FXIIII-A promotes FN fibrillogenesis in vitro30 and we have shown that the main crosslinking/transamidation substrate of FXIII-A is liver (serum)-derived pFN in cell cultures using inhibitor NC9 and FXIII-A deficient mouse embryonic fibroblasts.23, 31 Here we provide first in vivo evidence on the role of TG2 and FXIII-A in whole-body pFN homeostasis by showing solubility changes of FN matrix in the double null mouse as well as decreased integration of injected pFN into bone tissue and its pooling into serum and bone marrow fluid. In cultures, TG2- and FXIII-A-deficient bmMSCs form poorly the cell clustering nodules of pFN. This likely promotes bmMSC differentiation to adipocytes as FN is a regulator of preadipocyte stage jointly with Pref-1/Dlk-1 (refs 24, 42), which we have previously shown to be absent from TG2-deficient mouse embryonic fibroblasts.24 Although we did not dissect the mechanism linking TGs, adipogenesis and RANKL, recent advances suggest that the link may involve at least canonical Wnt-pathway, which inhibits adipogenesis, suppresses RANKL production,54 and which is regulated by TG255, 56 as well as FN.57 TG2 was also recently shown to correlate with RANKL production in human periodontal ligament cells as part of the engaged inflammatory response in periodontitis.58 Links between MSC differentiation towards adipogenesis with increased RANKL expression also exists.59 The mechanisms how TGs regulate osteoclastogenesis may involve c-Src and regulation of cytoskeletal dynamics.56 It is likely that these functions involve both transamidation (pFN crosslinking) and non-transamidating functions.
In summary, our data links TG2 and FXIII-A for the first time to maintenance of bone mass and marrow stem cell fate as well as coupling of bone cells in vivo which identifies them as potential targets to modulate RANKL levels in osteoporosis.
Materials and Methods
More in Supplementary Materials and Methods.
Mice
Generation of individual Tgm2−/− and F13a1−/− knockout mice were described previously.36, 37 All mice were maintained in a pathogen-free animal facility and kept on a diurnal cycle and fed a normal chow diet. The experiments described in this study were approved by the Animal Care Committee of McGill University. Crossbreeding of the two genotypes was done using heterozygote female F13a1−/+ (on CBA/CaJ background) and knockout male Tgm2−/− (on C57BL/6) mice to avoid the spontaneous miscarriages that occur for F13a1−/− mice.60 Despite numerous attempts, we failed to generate double null mice from the Tgm2+/−;F13a1+/− breeding which could have generated matching wild-type controls. Mating Tgm2−/−;F13a1−/− males with Tgm2−/−;F13a1+/− females resulted in Tgm2−/−;F13a1−/− mice that we used in our experiments. Since this breeding scheme does not generate littermate wild-type mice, mixed-background wild-type controls were generated by parallel breeding by crossing C57BL/6 and CBA/CaJ strains. Genotyping was done as described previously.36, 37 Genomic DNA was extracted from tail biopsies using Genomic DNA Extraction Kit (Life Technologies Inc., Burlington, ON, Canada). The weight, BMD and resorption parameters of all genotypes was monitored up to 1 year (n=6–17). For analysis of individual knockouts, age and gender matched littermate controls were used.
Serum markers
Mice were fasted for 6 h, anesthetized with isoflurane, and whole blood was collected by cardiac puncture into serum collection tubes. Bone resorption levels were assessed by measuring C-terminal telopeptides of type I collagen in mouse serum using the RatLapsTM Kit (Immunodiagnostics Systems, Gaithersburg, MD, USA). Total mouse osteocalcin levels were measured with kits from Biomedical Technologies (Ward Hill, MA, USA). OPG and RANKL levels were measured with kits from Abcam (Toronto, ON, Canada), total serum FN and serum cellular FN were measured with kits from MyBioSource, (San Diego, CA, USA). Calcium and phosphate were measured using a kit from Sekisui Diagnostics (Lexington, MA, USA).
Micro-Computed tomography (μCT)
Tibias and vertebrae of 3-month-old mice (n=5) were dissected and fixed in 3.7% formaldehyde for 24 h at room temperature and washed with 70% ethanol. μCT was performed on trabecular and cortical bone, with quantification done for the tibia samples, using a SkyScan 1072 (Aartselaar, Antwerp, Belgium) instrument. Mineralized bone volume, tissue volume, and trabecular and cortical bone parameters were recorded. SkyScan software was used for three-dimensional reconstructions.
Osteoclast cultures, TRAP staining and resorption assay
Bone marrow monocytes were isolated and cultured from 6–8-week-old mice as follows. Mice were sacrificed, and long bones were dissected and kept in ice-cold PBS. Bone marrow was collected by centrifugation.61 Marrow was resuspended in complete alpha MEM (Invitrogen), supplemented with 10% FBS, penicillin and streptomycin, 1 mM sodium pyruvate and 1 mM l-glutamine, and then passed through 70 μm cell strainer. After centrifugation, the cell pellet was resuspended in red blood cell (RBC) lysis buffer (Sigma-Aldrich, Oakville, ON, Canada) for 10 minute on ice. RBC lysis buffer was then neutralized with complete medium. Cells were pelleted and washed twice with complete medium. Cell counts were done, and 15 × 106 cells were cultured with 25 ng/ml M-CSF for 24 h in a humidified 37°C incubator with 5% CO2. The following day, non-adherent cells were collected and centrifuged. Cells were plated at 50 000 cells/cm2 and supplemented with 50 ng/ml M-CSF and 50 ng/ml RANKL (Peprotech Inc., Rocky Hill, NJ, USA). Cells were treated every two days, and TRAP staining was done at day 5. For TRAP staining, cultured medium was removed and cells were washed with PBS and fixed in 3.7% formaldehyde for 10 min at room temperature. TRAP staining was done according to the manufacturer's instructions (Sigma) and quantified using a colorimetric kit (Corning Life Sciences, Corning, NY, USA). Resorption assays were conducted using Corning Osteo Assay plates (Corning Life Sciences) having a bone biomimetic synthetic surface coating. Cells were plated into the plates at 50 000 cells/cm2 and were cultured for further 5 days in the presence of M-CSF and RANKL as described above. Resorption pits were visualized by light microscopy after von Kossa staining and quantified as per to instructions of plate manufacturer.
Bone marrow mesenchymal stem cell isolation and culture
Bone marrow was collected from 6–8-week-old mice by removing the bone marrow from long bones via a centrifugation step. Bone marrow cells were resuspended in MesenCult Complete Media with stimulatory supplement (StemCell Technologies, Vancouver, BC, Canada). Cell suspensions were passed through a 70 μm cell strainer and pelleted by centrifugation. Cells were resuspended in media and plated at 1 500 000 cells/cm2 in tissue culture plates. After the cells had reached 80% confluency, adipogenic differentiation was initiated by adding MesenCult Media with adipogenic supplements (StemCell Technologies) or MesenCult Complete Media for the control cultures. Media was changed every 3 days, and the experiment was terminated on day 13. Lipid were visualized and quantified by Oil red staining as before.23
Hepatocyte isolation
Hepatocytes were isolated from mouse livers following a previously published protocol.62 Briefly, 3-month-old mice were anesthetized with pentobarbital and liver perfusion was done through the heart and through the portal vein using perfusion medium and using a peristaltic pump. Collagenase-containing solution was perfused for 10 min, and the liver was dissected and transferred into a petri dish containing perfusion medium I (as per Li et al.62). The liver capsule was disrupted with tweezers and hepatocytes were released by gentle shaking. Cells were passed through a 70-μm cell strainer and centrifuged, then washed with fresh perfusion medium II (as in Li et al.62) and resuspended in William E medium (Sigma). Hepatocytes were plated at 0.4 × 106 cells/ml in culture plates coated with type I collagen (Sigma), and plates were placed at 37 °C incubator with 5% CO2 for 6–8 h to form a monolayer. Unattached cells were removed and fresh William E media was added. Hepatocytes were cultured up to 4 days, after which culture media was collected and cells were extracted for analysis.
Fibronectin labeling and injections
Labeling was done with a similar approach as published before.35 One milligram of human pFN was labeled with Oyster-488 (LUMINARTIS, Muenster, Germany) and injected intraperitoneally into 12-wk-old mice on 3 consecutive days. Four experimental groups were used: WT labeled with pFN-Oyster-488, WT labeled with inactivated Oyster-488, double-null mice labeled with pFN-Oyster-488 and double-null mice labeled with inactivated Oyster-488 (n=3). Inactivation of Oyster-488 was done by incubating the dye for 1 h in 0.1 M Tris-buffer, pH 8.4. On day 5, mice were sacrificed, and bone marrow was collected by removing growth plates and centrifuging out soft interior tissue. This was followed by a cleaning step with PBS, and bones were used for bone protein extraction as described above. Only the G1-extract was prepared. The fluorescence signal from Oyster-488 labeled pFN in extracted material was measured with a Synergy 2 Multi-Mode plate Reader (BioTek, Winooski, VT, USA) equipped with Gen5 2.05 data analysis software (Winooski, VT, USA). The background signal from the inactivated labeling control was subtracted from the experimental group.
Statistical analyses
All data are presented as mean and error bars represent S.E.M. of mouse groups with an n of 5–11. In cell culture experiments, S.E.M. derived from either two or three independent experiments. Statistical significance between two groups was determined by two-tailed, unpaired Student's t-test using statistics software. P-values are indicated as follows: *P<0.05, **P<0.01, and ***P<0.001. One way ANOVA followed by Tukey's multiple comparison posttest was used to compare multiple groups using Origin 8. The significance level was set at *P<0.05.
Acknowledgments
We thank the staff of the McGill Centre for Bone and Periodontal Research for help with this work and Dr Alessandra Balduini from University of Pavia and Tufts University for helpful discussions. This study was supported by grants to MTK from the Canadian Institutes of Health Research (CIHR) (MOP-119403) and from the CIHR Institute of Musculoskeletal Health and Arthritis, and the CIHR Institute of Genetics. CC and AS were supported by stipends from the China Scholarship Council and the Faculty of Dentistry of McGill University. VDM received stipends from the Faculty of Dentistry at McGill University and the CIHR Osler Systems Biology Training program. MTK and MM are members of the Fonds de Recherche – Santé (FRQ-S) Network for Oral and Bone Health Research.
Footnotes
Supplementary Information accompanies this paper on Cell Death and Differentiation website (http://www.nature.com/cdd)
Edited by M Piacentini
The authors declare no conflict of interest.
Supplementary Material
References
- Siddiqui JA, Partridge NC. Physiological bone remodeling: systemic regulation and growth factor involvement. Physiology (Bethesda, Md) 2016; 31: 233–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianco P, Robey PG. Marrow stromal stem cells. J Clin Invest 2000; 105: 1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celso CL, Scadden DT. The haematopoietic stem cell niche at a glance. J Cell Sci 2011; 124: 3529–3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce BF, Xing L. Functions of RANKL/RANK/OPG in bone modeling and remodeling. Arch Biochem Biophys 2008; 473: 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998; 93: 165–176. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA 1998; 95: 3597–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeshita S, Fumoto T, Naoe Y, Ikeda K. Age-related marrow adipogenesis is linked to increased expression of RANKL. J Biol Chem 2014; 289: 16699–16710, jbc. M114. 547919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien CA, Nakashima T, Takayanagi H. Osteocyte control of osteoclastogenesis. Bone 2013; 54: 258–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittrant Y, Gorin Y, Mohan S, Wagner B, Abboud-Werner S. Colony-stimulating factor-1 (CSF-1) directly inhibits receptor activator of nuclear factor-κB ligand (RANKL) expression by osteoblasts. Endocrinology 2009; 150: 4977–4988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alford AI, Kozloff KM, Hankenson KD. Extracellular matrix networks in bone remodeling. Int J Biochem Cell Biol 2015; 65: 20–31. [DOI] [PubMed] [Google Scholar]
- Nilsson SK, Debatis ME, Dooner MS, Madri JA, Quesenberry PJ, Becker PS. Immunofluorescence characterization of key extracellular matrix proteins in murine bone marrow in situ. J Histochem Cytochem 1998; 46: 371–377. [DOI] [PubMed] [Google Scholar]
- Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 2003; 4: 140–156. [DOI] [PubMed] [Google Scholar]
- Iismaa SE, Mearns BM, Lorand L, Graham RM. Transglutaminases and disease: lessons from genetically engineered mouse models and inherited disorders. Physiol Rev 2009; 89: 991–1023. [DOI] [PubMed] [Google Scholar]
- Eckert RL, Kaartinen MT, Nurminskaya M, Belkin AM, Colak G, Johnson GV et al. Transglutaminase regulation of cell function. Physiol Rev 2014; 94: 383–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muszbek L, Bereczky Z, Bagoly Z, Komáromi I, Katona É. Factor XIII: a coagulation factor with multiple plasmatic and cellular functions. Physiol Rev 2011; 91: 931–972. [DOI] [PubMed] [Google Scholar]
- Aeschlimann D, Thomazy V. Protein crosslinking in assembly and remodelling of extracellular matrices: the role of transglutaminases. Connect Tissue Res 2000; 41: 1–27. [DOI] [PubMed] [Google Scholar]
- Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol 2000; 148: 825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanchan K, Fuxreiter M, Fesus L. Physiological, pathological, and structural implications of non-enzymatic protein-protein interactions of the multifunctional human transglutaminase 2. Cell Mol Life Sci 2015; 72: 3009–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen MT, El-Maadawy S, Rasanen NH, McKee MD. Tissue transglutaminase and its substrates in bone. J Bone Miner Res 2002; 17: 2161–2173. [DOI] [PubMed] [Google Scholar]
- Nurminskaya MV, Belkin AM. Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol 2012; 294: 1–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jallad HF, Nakano Y, Chen JL, McMillan E, Lefebvre C, Kaartinen MT. Transglutaminase activity regulates osteoblast differentiation and matrix mineralization in MC3T3-E1 osteoblast cultures. Matrix Biol 2006; 25: 135–148. [DOI] [PubMed] [Google Scholar]
- Nurminskaya M, Kaartinen MT. Transglutaminases in mineralized tissues. Front Biosci 2006; 11: 1591–1606. [DOI] [PubMed] [Google Scholar]
- Myneni VD, Hitomi K, Kaartinen MT. Factor XIII-A transglutaminase acts as a switch between preadipocyte proliferation and differentiation. Blood 2014; 124: 1344–1353. [DOI] [PubMed] [Google Scholar]
- Myneni VD, Melino G, Kaartinen MT. Transglutaminase 2—a novel inhibitor of adipogenesis. Cell Death Dis 2015; 6: e1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov R, Yamada KM. Fibronectin at a glance. J Cell Sci 2002; 115: 3861–3863. [DOI] [PubMed] [Google Scholar]
- Schwarzbauer JE, DeSimone DW. Fibronectins, their fibrillogenesis, and in vivo functions. Cold Spring Harb Perspect Biol 2011; 3: pii: a005041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George EL, Georges-Labouesse EN, Patel-King RS, Rayburn H, Hynes RO. Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 1993; 119: 1079–1091. [DOI] [PubMed] [Google Scholar]
- Al-Jallad HF, Nakano Y, Chen JLY, McMillan E, Lefebvre C, Kaartinen MT. Transglutaminase activity regulates osteoblast differentiation and matrix mineralization in MC3T3-E1 osteoblast cultures. Matrix Biol 2006; 25: 135–148. [DOI] [PubMed] [Google Scholar]
- Tang C-H, Yang R-S, Liou H-C, Fu W-M. Enhancement of fibronectin synthesis and fibrillogenesis by BMP-4 in cultured rat osteoblast. J Bone Miner Res 2003; 18: 502–511. [DOI] [PubMed] [Google Scholar]
- Mosher DF, Schad PE, Vann JM. Cross-linking of collagen and fibronectin by factor XIIIa. Localization of participating glutaminyl residues to a tryptic fragment of fibronectin. J Biol Chem 1980; 255: 1181–1188. [PubMed] [Google Scholar]
- Cui C, Wang S, Myneni VD, Hitomi K, Kaartinen MT. Transglutaminase activity arising from Factor XIIIA is required for stabilization and conversion of plasma fibronectin into matrix in osteoblast cultures. Bone 2014; 59: 127–138. [DOI] [PubMed] [Google Scholar]
- Corbett SA, Lee L, Wilson CL, Schwarzbauer JE. Covalent cross-linking of fibronectin to fibrin is required for maximal cell adhesion to a fibronectin-fibrin matrix. J Biol Chem 1997; 272: 24999–25005. [DOI] [PubMed] [Google Scholar]
- To WS, Midwood KS. Plasma and cellular fibronectin: distinct and independent functions during tissue repair. Fibrogenesis Tissue Repair 2011; 4: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti FA, Chauhan AK, Iaconcig A, Porro F, Baralle FE, Muro AF. A major fraction of fibronectin present in the extracellular matrix of tissues is plasma-derived. J Biol Chem 2007; 282: 28057–28062. [DOI] [PubMed] [Google Scholar]
- Bentmann A, Kawelke N, Moss D, Zentgraf H, Bala Y, Berger I et al. Circulating fibronectin affects bone matrix, whereas osteoblast fibronectin modulates osteoblast function. J Bone Miner Res 2010; 25: 706–715. [DOI] [PubMed] [Google Scholar]
- De Laurenzi V, Melino G. Gene disruption of tissue transglutaminase. Mol Cell Biol 2001; 21: 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer P, Metzner HJ, Zettlmeissl G, Li M, Smith AG, Lathe R et al. Targeted inactivation of the mouse locus encoding coagulation factor XIII-A: hemostatic abnormalities in mutant mice and characterization of the coagulation deficit. Thromb Haemost 2002; 88: 967–974. [PubMed] [Google Scholar]
- Cordell PA, Newell LM, Standeven KF, Adamson PJ, Simpson KR, Smith KA et al. Normal bone deposition occurs in mice deficient in factor XIII-A and transglutaminase 2. Matrix Biol 2015; 43: 85–96. [DOI] [PubMed] [Google Scholar]
- Bonjour JP. Calcium and phosphate: a duet of ions playing for bone health. J Am Coll Nutr 2011; 30: 438s–448s. [DOI] [PubMed] [Google Scholar]
- Lacombe J, Karsenty G, Ferron M. In vivo analysis of the contribution of bone resorption to the control of glucose metabolism in mice. Mol Metab 2013; 2: 498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Jallad HF, Myneni VD, Piercy-Kotb SA, Chabot N, Mulani A, Keillor JW et al. Plasma membrane factor XIIIA transglutaminase activity regulates osteoblast matrix secretion and deposition by affecting microtubule dynamics. PLoS One 2011; 6: 0015893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhao L, Smas C, Sul HS. Pref-1 interacts with fibronectin to inhibit adipocyte differentiation. Mol Cell Biol 2010; 30: 3480–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarantino U, Oliva F, Taurisano G, Orlandi A, Pietroni V, Candi E et al. FXIIIA and TGF-β over-expression produces normal musculo-skeletal phenotype in TG2−/− mice. Amino Acids 2009; 36: 679–684. [DOI] [PubMed] [Google Scholar]
- Szondy Z, Sarang Z, Molnar P, Nemeth T, Piacentini M, Mastroberardino PG et al. Transglutaminase 2−/− mice reveal a phagocytosis-associated crosstalk between macrophages and apoptotic cells. Proc Natl Acad Sci USA 2003; 100: 7812–7817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin K, Simpson K, Beckers C, Brown J, Vacher J, Ouwehand W et al. Use of a novel floxed mouse to characterise the cellular source of plasma coagulation FXIII-A. Lancet (London, England) 2015; 385: S39. [DOI] [PubMed] [Google Scholar]
- Raghu H, Cruz C, Rewerts CL, Frederick MD, Thornton S, Mullins ES et al. Transglutaminase factor XIII promotes arthritis through mechanisms linked to inflammation and bone erosion. Blood 2015; 125: 427–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki M, Yamashita F, Ishida-Yamamoto A, Yamada K, Kinoshita C, Fushiki S et al. Defective stratum corneum and early neonatal death in mice lacking the gene for transglutaminase 1 (keratinocyte transglutaminase). Proc Natl Acad Sci USA 1998; 95: 1044–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aeschlimann D, Mosher D, Paulsson M. Tissue transglutaminase and factor XIII in cartilage and bone remodeling. Semin Thromb Hemost 1996; 22: 437–443. [DOI] [PubMed] [Google Scholar]
- Heath DJ, Downes S, Verderio E, Griffin M. Characterization of tissue transglutaminase in human osteoblast-like cells. J Bone Miner Res 2001; 16: 1477–1485. [DOI] [PubMed] [Google Scholar]
- Fogelgren B, Polgár N, Szauter KM, Újfaludi Z, Laczkó R, Fong KSK et al. Cellular fibronectin binds to lysyl oxidase with high affinity and is critical for its proteolytic activation. J Biol Chem 2005; 280: 24690–24697. [DOI] [PubMed] [Google Scholar]
- Huang G, Zhang Y, Kim B, Ge G, Annis DS, Mosher DF et al. Fibronectin binds and enhances the activity of bone morphogenetic protein 1. J Biol Chem 2009; 284: 25879–25888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosher DF. Cross-linking of cold-insoluble globulin by fibrin-stabilizing factor. J Biol Chem 1975; 250: 6614–6621. [PubMed] [Google Scholar]
- Mosher DF, Schad PE. Cross-linking of fibronectin to collagen by blood coagulation factor XIIIa. J Clin Invest 1979; 64: 781–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spencer GJ, Utting JC, Etheridge SL, Arnett TR, Genever PG. Wnt signalling in osteoblasts regulates expression of the receptor activator of NFkappaB ligand and inhibits osteoclastogenesis in vitro. J Cell Sci 2006; 119: 1283–1296. [DOI] [PubMed] [Google Scholar]
- Deasey S, Nurminsky D, Shanmugasundaram S, Lima F, Nurminskaya M. Transglutaminase 2 as a novel activator of LRP6/beta-catenin signaling. Cell Signal 2013; 25: 2646–2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello S, Cao L, Matei D. Tissue transglutaminase regulates beta-catenin signaling through a c-Src-dependent mechanism. FASEB J 2013; 27: 3100–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielefeld KA, Amini-Nik S, Whetstone H, Poon R, Youn A, Wang J et al. Fibronectin and beta-catenin act in a regulatory loop in dermal fibroblasts to modulate cutaneous healing. J Biol Chem 2011; 286: 27687–27697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matarese G, Curro M, Isola G, Caccamo D, Vecchio M, Giunta ML et al. Transglutaminase 2 up-regulation is associated with RANKL/OPG pathway in cultured HPDL cells and THP-1-differentiated macrophages. Amino Acids 2015; 47: 2447–2455. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Fumoto T, Naoe Y, Ikeda K. Age-related marrow adipogenesis is linked to increased expression of RANKL. J Biol Chem 2014; 289: 16699–16710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koseki-Kuno S, Yamakawa M, Dickneite G, Ichinose A. Factor XIII A subunit-deficient mice developed severe uterine bleeding events and subsequent spontaneous miscarriages. Blood 2003; 102: 4410–4412. [DOI] [PubMed] [Google Scholar]
- Boraschi-Diaz I, Komarova SV. The protocol for the isolation and cryopreservation of osteoclast precursors from mouse bone marrow and spleen. Cytotechnology 201468: 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward A, Tosh D Isolation and culture of adult mouse hepatocytes. In: Ward A, Tosh D (eds). Mouse Cell Culture Methods in Molecular Biology, Humana Press, 2010, pp 185–196. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.