

Abstract
Chemoresistance is a major challenge in lung cancer treatment. Recent findings have revealed that autophagic mechanism contributes significantly to immunosuppressive related chemoresistance. For that reason, targeting autophagy‐related immunosuppression is an important approach to reverse tumor drug resistance. In this study, we report for the first time that autophagy inhibition triggers upregulation of CD4+, Foxp3+ tumor infiltrating lymphocytes in late metastatic lung cancer tissues. Furthermore, autophagy blockage induces chemosensitization to carboplatin, immune activation and cell cycle arrest. This induction correlated with reduction in expression of drug resistance genes MDR1, MRP1, ABCG2 and ABCC2 along with decreased expression of PD‐L1 which is associated with severe dysfunction of tumor specific CD8+ T cells. Furthermore, experiments revealed that co‐treatment of carboplatin and autophagy inhibitor chloroquine increased lung tissue infiltration by CD4+, FoxP3+ lymphocytes and antigen‐specific immune activation. Subsequent ex vivo experiments showed the activation of carboplatin related TRAIL‐dependent apoptosis through caspase 8 and a synergistic role of miR‐155 in lung tissue infiltration by CD4+, and FoxP3+ lymphocytes. Overall, our results indicate that autophagy blockage increases lung cancer chemosensitivity to carboplatin, but also reveal that miR‐155 functions as a novel immune system activator by promoting TILs infiltration. These results indicate that targeting of autophagy can prevent cancer related immunosuppression and elucidate immune cell infiltration in tumor microenvironment thus representing a potential therapeutic strategy to inhibit lung cancer progression and metastasis.
Keywords: Lung cancer, Chemoresistance, Tumor infiltrating lymphocytes, miR‐155, TRAIL
Abbreviations
- 3‐MA
3‐methyladenine
- CQ
chloroquine
- Foxp3
forkhead box P3
- MDR1
multidrug resistance protein
- MRP1
multidrug resistance‐associated protein 1
- ABCG2
ATP‐binding cassette sub‐family G member 2
- ABCC2
ATP‐binding cassette sub‐family C member 2
- PD‐L1
programmed death‐ligand 1
- TILs
tumor infiltrating lymphocytes
- miR‐155
microRNA‐155
- TRAIL
TNF‐related apoptosis‐inducing ligand
- NSCLC
non‐small cell lung cancer
- LC3
microtubule‐associated protein 1 light chain 3
- NF‐κB
nuclear factor of kappa light polypeptide gene enhancer in B‐cells
- TRAIL
tumor necrosis factor‐related apoptosis‐inducing ligand
- DR4
death receptor 4
- DR5
death receptor 5
- STAT3
signal transducer and activator of transcription 3
1. Introduction
Lung cancer remains the leading cause of cancer‐related deaths and despite extensive research efforts, the survival rate of lung cancer patients, even with complete surgical resection, remains significantly low (Beckett et al., 2014; Tanoue et al., 2015). Increasing evidence shows that autophagy is responsible for cancer cell resistance to platinum‐based chemotherapy and tumor propagation (Yang et al., 2015; Zhou et al., 2012). Autophagy is reported to play a crucial role in lung cancer related immune suppression by promoting tumor's ability to avoid immune detection (Lorin et al., 2013; Viry et al., 2014). Enhanced autophagy is usually observed in advanced stages of metastatic tumorigenesis and is implicated in immunosuppression, with low levels of T cell infiltration in neoplastic tissues and is associated with increased chemoresistance (Bronietzki et al., 2015). For that reason activation of tumor‐infiltrating lymphocytes (TILs) within the tumor microenvironment is a crucial step to inhibit tumor propagation (deLeeuw et al., 2012; Lo Presti et al., 2014). These cytotoxic T lymphocytes (mainly CD4+, CD8+, FoxP3+) play a major role in antitumor immune responses and influence chemotherapy response (Cochaud et al., 2013; Li et al., 2014). Combinational immunotherapy with a platinum‐based regimen has emerged as standard therapy for patients with advanced stage disease and carboplatin is frequently used as a chemotherapeutic agent against lung adenocarcinoma (Ferraldeschi et al., 2007; Heigener and Reck, 2015). However, cancer cell resistance to carboplatin is one of the obstacles against successful chemotherapy (Giovannetti et al., 2012). Recent studies reveal that programmed cell death‐1 (PD‐1) which is expressed on the surface of T‐cells upon activation play a key role in chemoresistance by suppressing the immune response during lung tumorigenesis (Sznol, 2014; Tykodi, 2014). Specifically, PD‐1 as a cell surface receptor present on T cells and pro‐B cells binds its ligand PD‐L1 and PD‐L2 and triggers a net immunosuppressive effect that allows tumor cells to evade immune detection and destruction (Meng et al., 2015). In addition, miRNAs have been show to play a key role in lung cancer chemoresistance by affecting expression both PD‐1/PD‐L1 and autophagic flux through the post‐transcriptional regulation of T cell gene expression (Gong et al., 2009, 2015, 2015). Accumulating evidence suggests that these miRNAs can modulate several molecular mechanisms involved in tumor progression, autophagy activation, and metastasis and many studies have focused on the relationship between PD‐L1 and immune escape (Ohaegbulam et al., 2015; Reiss et al., 2014; Wang et al., 2013). A similar mechanism of tumor resistance includes inhibition of TRAIL binding to death receptors DR4 and DR5 which is caspase‐8‐dependent process and triggers cell apoptosis (Lemke et al., 2014). In this study we report for the first time that autophagy blockage promotes upregulation of Foxp3+ CD4+ tumor infiltrating lymphocytes in late metastatic lung cancer tissues, followed by carboplatin related TRAIL‐dependent apoptosis through caspase 8. Experimental data reveal a synergistic role of tumor suppressor miR‐155 in lung tissue infiltration by CD4+ and FoxP3+ lymphocytes and indicate that autophagy inhibition increases lung cancer chemosensitivity to carboplatin, but also show miR‐155 to function as a novel immune system activator by promoting TILs infiltration. These critical molecular mechanisms influence the development of chemoresistance in lung tumorigenesis and are an important issue for developing novel and effective therapeutic strategies.
2. Materials and methods
2.1. Ethics statement
The study was approved by the local Ethics Committee on human experimentation of the AHEPA Hospital, Medical School, Aristotle University of Thessaloniki and informed consent was obtained from each patient before the surgical procedure.
2.2. Reagents
Carboplatin was purchased from Sigma (Sigma, Germany, Europe). Dulbecco's modified eagle medium (DMEM) and fetal bovine serum (FBS) were obtained from Invitrogen (Invitrogen, Paisley, UK). DMSO (Dimethyl sulfoxide) and Tris–EDTA (Tris–Ethylene Diamine Tetraacetic Acid) were purchased from Applichem (Applichem, Darmstadt, Germany). DMEM‐Hepes, PBS (Phosphate Buffer Saline), FBS (fetal bovine serum), Trypsin–EDTA, and DMSO reagents were purchased from Invitrogen (Invitrogen, Darmstadt, Germany). The stock solution was filtered through a 0.22 μΜ syringe filter, then aliquoted and stored in the dark at room temperature.
2.3. Lung cancer tissue samples
Lung cancer tissues were obtained from AHEPA Hospital, Medical School, Aristotle University of Thessaloniki (Thessaloniki, Greece). Tissue samples were collected in the operating room immediately after surgery with non‐tumor tissues sent to Pathology for diagnosis by a certified pathologist. Biopsy isolation was performed by a pathologist using the core needle biopsy technique. For each patient, a frozen tumor sample (stored at 80 °C) and a paraffin‐embedded tissue specimen were available. A written consent was obtained from 96 lung cancer (NSCLC) patients, 37–88 years of age, prior to surgery, from each patient voluntarily involved in the usage of tissues solely for research purposes (Supplementary Table S1). Patients had read and understood the patient information document provided, and the aims and methods of this study had been fully explained to them. Patients involved had also given written informed consent to authors of this manuscript for publication of these data. The study methodologies were approved by the local Ethics Committee. The clinical investigation was conducted according to the guidelines expressed in the Declaration of Helsinki.
2.4. Ex vivo tissue culture
Lung tumor samples from consenting (chemoresistant/chemonaive) cancer patients at the AHEPA Hospital Oncology unit were collected, transferred to research lab in cold media, and processed in a sterile tissue culture hood. The culture protocol is modified based on previous studies (Leithner et al., 2014; Zarogoulidis et al., 2015). Briefly, the culture medium consists of Dulbecco's modified Eagles's medium supplemented with 5% fetal calf serum (FCS), 10 mM HEPES, 0.5 μg/ml Hydrocortisone, 1% MEM vitamins solution, 5 μg/ml insulin, 100 IU/ml penicillin, 100 μg/ml streptomycin, and 15 μg/ml gentamicin. The tissue is placed in a sterile glass dish where all necrotic and visible connective tissue is removed. Tissue samples are cut in 0.5–1.0 cm3 slices via a cryotome and placed in a 6‐well tissue culture dish with 2 ml of growth media. The dish is placed in a 37 °C, 5% CO2 incubator for 24 h. After 1 day in culture, the tissue slices were subjected to immunohistochemical analysis.
2.5. Micro‐RNA transfection
MiR‐155 mimics and their appropriate negative control (NC) were purchased from Applied Biosystems (Applied Biosystems, USA) and transfected into ex vivo cultured cells at a final concentration of 50 nmol/L by using Lipofectamine 2000 (Invitrogen, Germany) according to manufacturer's instructions.
2.6. RT‐PCR
Total RNA was isolated from LC/LC‐DR treated cells using TRIZOL reagent (Invitrogen, Carlsbad, CA, USA), according to the manufacturer's instructions, with RNase‐free DNase. For quantification of mature miRNA levels, RNA was first reverse‐transcribed with miRNA‐specific primers using TaqMan MicroRNA Reverse Transcription Kit (Applied Biosystems) and qRT‐PCR was performed using a Tapman microRNA assay kit (ABI). U6 snRNA was used as control. To detect MDR1, MRP1, ABCG2, ABCC2 and PD‐L1, mRNA levels, specified primer sequences were employed (Supplementary Table S2). GAPDH was used an internal control. Each experiment was performed in triplicate.
2.7. Western blot analysis
Ex vivo cultured cells were first plated in 6‐well plates (6 × 105 cells/well). After 24 h treatment with chloroquine (20 μΜ), and carboplatin (25 μΜ), cells were subjected to western blot to determine the protein levels. Cells were washed twice with ice‐cold PBS and lysed in 100 μL of ice‐cold lysis buffer per 1 × 106 cells. The lysates were incubated on ice for 10 min, vortexed for 45 s and maintained on ice for another 10 min. Following centrifugation at 14,000 g and 4 °C for a period of 15 min, the supernatant was collected and proteins were quantified by the Bradford method. Lysate proteins dissolved in 6× Laemmli sample buffer were separated (30 μg/lane) using SDS–polyacrylamide gel electrophoresis (10% acrylamide) and electro‐transferred to PVDF (polyvinylidene difluoride) membranes. After blocking with 5% non‐fat milk in TBST buffer (50 mM Tris, 150 mM NaCl, 0.05% Tween 20, pH 7.6), the membranes were incubated for 90 min with the appropriate dilution of the primary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) in TBST plus 1% non‐fat milk. After washing, the membranes were incubated with the appropriate dilution of the horse radish peroxidase‐conjugated secondary antibody in TBST plus 1% non‐fat milk. Finally, the blots were developed by ECL (enhanced chemiluminescence) and digitized through a BioSpectrum 500 Imaging System. GAPDH was used as an internal control in all experiments.
2.8. siRNA transfection
Human ATG7 siRNA was purchased from Santa Cruz Biotechnology (Santa Cruz, California, USA) along with control siRNA. All siRNA transfections were performed with Lipofectamine 2000 Transfection Reagent (Invitrogen, Berlin, Germany) according to manufacturer instructions. Ex vivo LC‐DR cells were transfected with 100 nM siRNA for 48 h, and then treated with carboplatin (25 μΜ) for 24 h. Protein and mRNA levels were assessed by western blot analysis and RT‐PCR, respectively.
2.9. Cell proliferation assay
For detection of cell proliferation the Cell Counting Kit‐8 (CCK‐8) assay was used to monitor cell growth and the number of LC/LC‐DR viable cells was assessed by measurement of absorbance at 450 nm by FlUOstar OPTIMA (BMG LABTECH, Offenburg, Germany).
2.10. Colony formation assay
Following treatment, LC/LC‐DR cells were counted and seeded in 12‐well plates (in triplicate) at a density of 100 cells per well. The plates were incubated at 37 °C and 5% CO2 in a humidified incubator. Fresh culture medium was replaced every 2 days. After 7 days of culture, the cells were stained with crystal violet, and the numbers of colonies were counted. The rate of colony formation was calculated using the following equation: colony formation rate = (number of colonies/number of seeded cells) × 100%.
2.11. Transmission electron microscopy
Ex vivo cultured LC‐DR cells were collected and fixed in 2% paraformaldehyde, 0.1% glutaraldehyde in 0.1 M sodium cacodylate for 2 h, postfixed with 1% OsO4 for 1.5 h, and washed and stained for 1 h in 3% aqueous uranyl acetate. The samples were then washed again, dehydrated with graded alcohol and embedded in Epon‐Araldite resin (Canemco, Quebec, Canada). Ultra‐thin sections were generated using an UCT‐EMFCS Leica microtome (Leica, Germany), counterstained with 0.3% lead citrate and examined on a Philips electron microscope. The magnification of image is indicated at the bottom of micrograph images; a minimum of 20 representative cells from at least three grids were evaluated for the appearance of autophagosomes.
2.12. Acridine orange staining
As a marker of autophagy, the volume of the cellular acidic compartment was visualized by acridine orange staining. LC‐DR cells were seeded in six‐well tissue culture dishes and treated as described above for the cell viability study. Twenty‐four hours following treatment, cells were incubated with medium containing 0.1 mg/ml acridine orange (Invitrogen, A3568) for 15 min; the acridine orange was then removed, cells were washed once with PBS, fresh media was added, and fluorescent micrographs were taken using an Olympus inverted fluorescence microscope. Again, all images presented are at the same magnification. The number of cells with increased acidic vesicular organelles was determined by counting at least three representative fields per treatment condition; a minimum of three replicate experiments were conducted.
2.13. Immunohistochemistry
Human tissue sample from patients with lung cancer were immediately collected after surgery subjected to ex vivo culture and then treated with chloroquine or carboplatin (1 h, 50 μΜ) in room temperature. Then tissues were treated with anti‐human cd4, cd8, foxp3 and pd‐l1 monoclonal antibodies (Santa Cruz Biotechnology, Santa Cruz, CA). Staining with mouse IgG1 isotype was used as the negative control. Images were obtained through a Carl Zeiss microscope using image analysis software (Carl Zeiss, Berlin, Germany). The number of positive stained cells relative to the total number of cells in the tissue sections and the intensity of the positive immunosignals were quantified with Aperio ImageScope software (Vista, CA).
2.14. Immunofluorescence analysis
Ex vivo cultured cells were treated with 3‐MA (20 μΜ), and carboplatin (25 μΜ), for 24 h. Next, cells were fixed in 4% formaldehyde for 15 min at room temperature prior to cell permeabilization with 0.1% Triton X‐100 (4 °C, 10 min). Cells were saturated with PBS containing 2% BSA for 1 h at room temperature and processed for immunofluorescence with PD‐L1 antibody, respectively, at 4 °C overnight. Then, they were incubated with Alexa Fluor 488‐conjugated anti‐rabbit IgG antibody (1:100). Between all incubation steps, cells were washed three times for three minutes with PBS containing 0.2% BSA. Fluorescence signals were analyzed using a Carl Zeiss fluorescent microscope at a 100× magnification, with excitation and emission wavelengths of 488 nm and 520 nm, respectively, using image analysis software.
2.15. TILs isolation
Peripheral blood mononuclear cells (PBMC) from lung cancer patients were isolated by Ficoll density gradient centrifugation. Fresh tumor tissues were used for the isolation of TIL and non‐tumor‐infiltrating lymphocytes (NIL). In brief, fresh tumor tissues were washed three times in RPMI 1640 before being cut into small pieces. The specimen were then collected in RPMI 1640 containing 1 mg/ml collagenase IV (Sigma–Aldrich, St. Louis, MO, USA) and 10 mg/ml DNase I (Roche, Basel, Switzerland) and mechanically dissociated by using the gentle MACS Dissociator (Miltenyi Biotec, Auburn, CA, USA). Dissociated cell suspensions were further incubated 1 h at 37 °C under continuous rotation and filtered through 70 μm cell strainers to obtain cell suspensions. Cells were gently minced and passed through 70‐μm cell strainers to obtain cell suspensions. The cell suspensions were then used for FACS analysis.
2.16. Transwell migration assay
LC/LC‐DR cell invasion was evaluated using a matrigel invasion chamber. The migration assay was conducted in a 24‐well transwell cell culture apparatus fitted with multiporous polycarbonate membrane insert (8‐μm pore size) (Corning). In brief, LC/LC‐DR cells were collected and resuspended in serum free media at a density of 1 × 105 cells/ml. The top chamber of transwell was loaded with 100 μl of cell suspension and the lower chamber was filled with 0.5 ml of media supplemented with 10% FBS as a chemo attractant. After incubation at 37 °C in 5% CO2 for 24 h, the filters were removed, rinsed two times with PBS, fixed with methanol and stained with 0.5% crystal violet for 20 min. Cells on the upper side of the filter were wiped off with cotton swabs. The cells that migrated on the lower side of the filter were determined by counting specified cross‐sectional fields on the filter with a phase‐contrast microscope. The experiments were performed in triplicates.
2.17. Cell cycle analysis
Flow cytometric analysis was performed to define the cell cycle distribution for chloroquine treated (20 μΜ), carboplatin treated (25 μΜ) and control drug resistant LC‐DR cells for 24 h. In brief, cells grown in 100 mm culture dishes were harvested, resuspended in cold PBS, and fixed with 9 volumes of 70% ethanol. Cells were then stained for total DNA content with a solution containing 50 μg/ml propidium iodide, 50 μg/ml RNaseA, 0.1% Triton X‐100 and 0.1 mmol/L EDTA in PBS for 30 min at 37 °C. Cell cycle distribution was analyzed with a FACScan flow cytometer (BD Biosciences, San Jose, CA).
2.18. Statistical analysis
The results are expressed as mean ± SD from at least three separate experiments performed in triplicate. The differences between groups were determined with a two‐tailed Student's t‐test or ANOVA using GraphPad software. The results are presented as represent the mean ± SD of at least three independent experiments. Differences were considered statistically significant at P < 0.05. Statistically significant data are indicated by asterisks (*P < 0.05, **P < 0.01).
3. Results
3.1. Autophagy inhibition augments carboplatin activity in ex vivo metastatic lung cancer cells
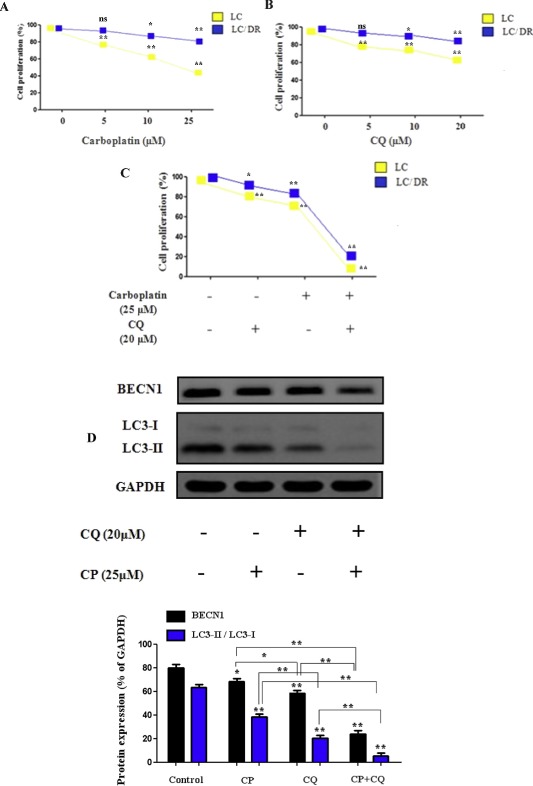
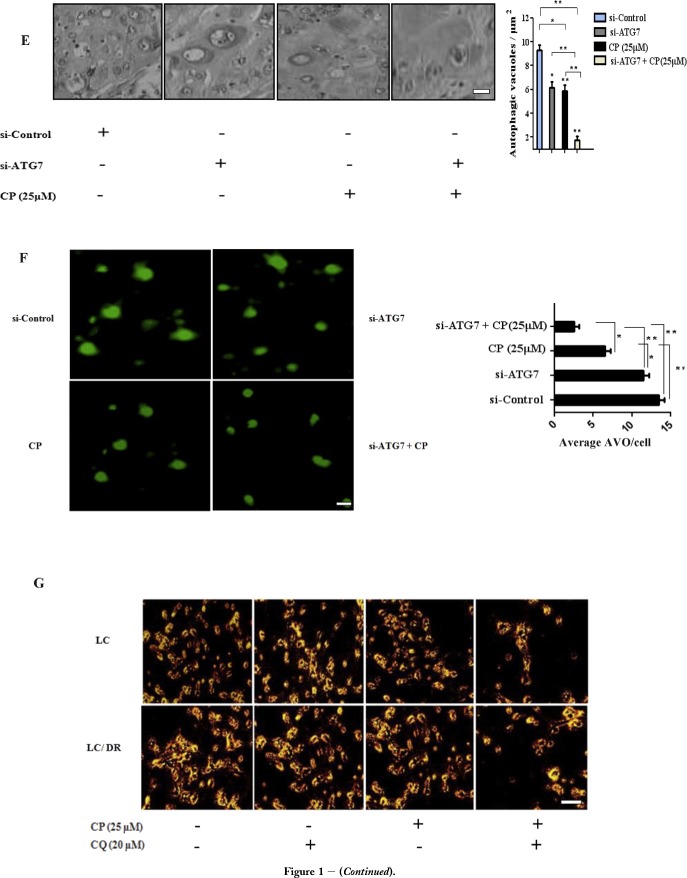
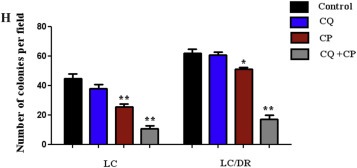
To investigate the mechanism of autophagy inhibition in chemosensitization of ex vivo cancer cells, we analyzed the synergistic effects of autophagy blockage and carboplatin, treatment in lung tumor tissues. Results showed that carboplatin or chloroquine alone induced limited inhibition of proliferation in drug‐resistant LC‐DR cells compared with its effects in no resistant cells (LC) (Figure 1A and B). Co‐treatment with chloroquine had significant inhibitory effects on the proliferation of both LC/LC‐DR cells and interacted synergistically with carboplatin to induce cell death (Figure 1C). In addition, autophagy inhibition alone had minor inhibitory effects on the proliferation of drug resistant lung cancer LC/DR cells, indicating that blocking autophagy promotes sensitization of cells to carboplatin (Figure 1B and C, Supplementary Tables S3–S5). Next, we evaluated the levels of Beclin expression and LC3‐I to LC3‐II conversion which are key markers of autophagic flux in drug resistant LC/DR cells. Co‐treatment induced downregulation in LC3‐II accumulation followed by reduction in Beclin 1 expression indicating decrease formation of autophagosomes (Figure 1D). Furthermore, the effect of co‐treatment was more evident in non resistant LC cells (Supplementary Fig. 1). To verify these results we performed autophagy inhibition using siATG7 in drug resistant LC‐DR cells. Transmission electron microscopy (TEM) and acridine orange staining was used to check the formation of autophagosomes in carboplatin‐treated cells (Figure 1E and F). Untreated drug resistant LC/DR cells exhibited increased accumulation of large autophagic vacuoles with typical double‐layer membrane containing organelle remnants, whereas only a few vacuoles were observed in siATG7 transfected/carboplatin treated cells (Figure 1E). Furthermore, to determine the long‐term inhibitory effects of autophagy blockage on the proliferation of lung cancer cells, a colony formation assay was performed. The results showed that autophagy inhibition alone distinctly inhibited the colony growth of LC (Figure 1F) but not drug resistant LC‐DR cells, however significantly limited the LC‐DR colony number when combined with carboplatin (Figure 1 G and H). Equivalent to decreased autophagosome, apoptosis was markedly enhanced in chloroquine plus carboplatin‐treated group, compared with treatment with carboplatin alone, chloroquine alone and control (data in results Section 3.5). Overall, these results indicate that autophagy blockage in combination with carboplatin could downregulate the drug resistant mechanism in chemoresistant cancer cells and promote carboplatin related apoptosis.
Figure 1.
Autophagy inhibition chemosensitizes ex vivo lung cancer cells to carboplatin induced apoptosis. (A–C) The CCK8 cell proliferation assay was performed to determine the proliferation of LC/LC‐DR cells after 24 h treatment with autophagy inhibitor chloroquine (20 μΜ) and carboplatin (25 μΜ). Data represent the mean ± SD of three independent experiments (D) The conversion from LCE‐I to LCE‐II, as well as Beclin‐1 expression was detected by western blot. The densitometric analysis of LC3II/LC3I protein ratio and Beclin 1 expression was calculated from the average of three experiments and normalized by GAPDH. (E) TEM images depicting ultrastructures of autophagosomes in cells transfected with siATG7 and then treated with carboplatin (25 μΜ) for 24 h. The number of autophagic vacuoles (indicated by arrows) under TEM was calculated by continuous counts in two fields under high resolution (Scale bar, 100 μm). (F) Acridine orange (AO) staining of autophagic vesicles in transfected LC‐DR cells treated with carboplatin (25 μΜ) for 24 h. AO images were taken 24 h post treatment using an inverted fluorescence microscope. Average number of autophagic vacuoles (AVOs) per cell was counted for each concentration used and is presented in the graph. Data represent the mean ± SD of three separate experiments (Scale bar, 10 μm). (G) Representative images of colony formation assay of LC/LC‐DR cells (magnification ×200). (H) Quantitative analysis of the colony formation rates. Chloroquine in combination with carboplatin significantly inhibited the colony‐forming ability of LC/LC‐DR cells following 24 h treatment (Scale bar, 20 μm). Data represent the mean ± SD of three separate experiments. (*P < 0.05; **P < 0.01).
3.2. Chloroquine/carboplatin treatment downregulates expression of drug resistance genes and restores metastatic lung cancer cells sensitivity to carboplatin
In order to further investigate autophagy promotion of chemoresistance we treated LC‐DR cells with autophagy inhibitor chloroquine and then assessed the expression of drug resistance genes MDR1, MRP1, ABCG2 and ABCC2. We observed that cell resistance associated gene expression was highest in untreated cells as compared to both chloroquine and carboplatin treated cells (Figure 2A). The protein expression of MDR1 and ABCC2 was also found to be significantly higher in LC‐DR control cells compared with ABCG2 and ABCC2 expression levels. Treatment with chloroquine markedly reduced MDR1, MRP1 and ABCG2 protein levels, followed by mRNA reduction of MRP1 and ABCC2 levels (Figure 2A and B). Furthermore, chloroquine/carboplatin treatment significantly decreased both protein and mRNA expression of each drug resistance marker in both cell lines (Figure 2B, Supplementary Fig. S2). Next, we evaluated the effect of co‐treatment on the metastatic phenotype of LC and LC‐DR cells by transwell migration assay. Data reveal a significant reduction in cell migration and metastatic potential of LC and LC‐DR cells in chloroquine plus carboplatin‐treated group compared with untreated cells (Figure 2C). This can be explained by the fact that ex vivo cancer cells (even the non resistant control cells) exhibit some “mild” type of chemoresistance. In addition, carboplatin treated cells displayed lower migration rate compared to chloroquine treated cells (Figure 2D). Overall, these data show that chloroquine/carboplatin treatment could decrease expression of drug resistance genes, by inhibiting metastatic potential and re‐establishing lung cancer cells sensitivity to carboplatin.
Figure 2.
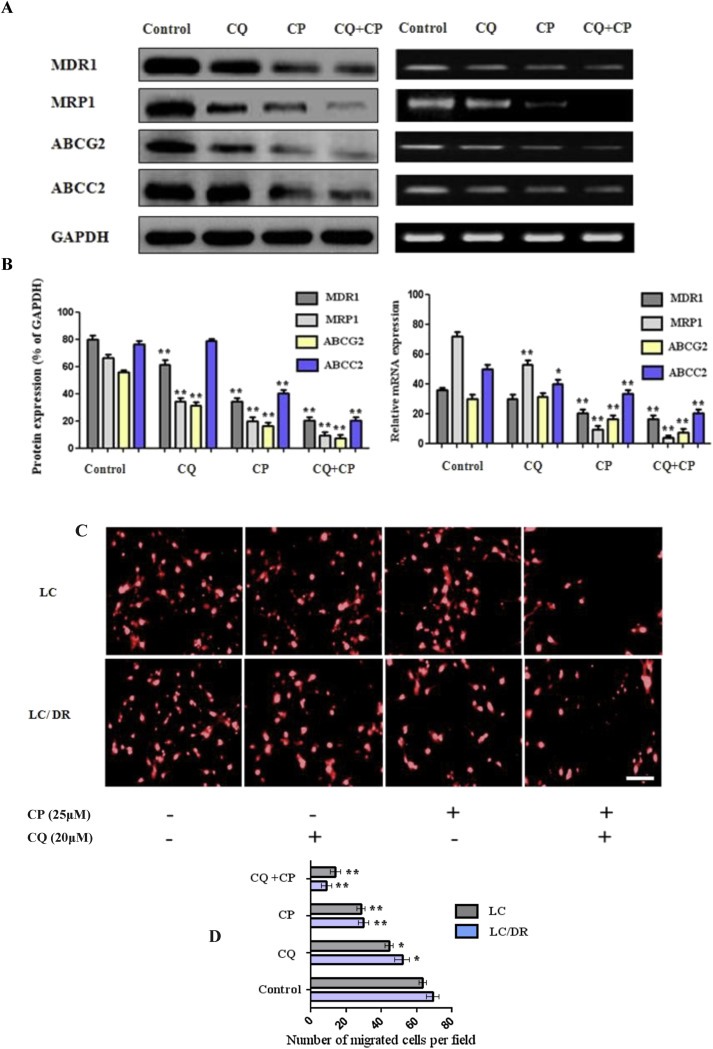
Co‐treatment of chloroquine and carboplatin inhibits cellular migration and downregulates expression of drug resistance genes. (A–B) The protein and mRNA expression levels of MDR1, MRP1, ABCG2 and ABCC2 were assayed by western blot and RT‐PCR, respectively. LC/LC‐DR cells were treated with autophagy inhibitor chloroquine (20 μΜ) or carboplatin (25 μΜ) for 24 h. (C–D) Effect of chloroquine or carboplatin on cell migration using transwell migration assay. Ex vivo cultured cells treated for 24 h with autophagy inhibitor chloroquine (20 μΜ) or carboplatin (25 μΜ) and subjected to migration assay to determine cellular migration (magnification ×200) (Scale bar, 10 μm). Data represent the mean ± SD of three independent experiments. (*P < 0.05; **P < 0.01). GAPDH was used as a loading control for western blot analysis. Densitometric analysis of each protein level was calculated from the average of three experiments. Each value was expressed as the ratio of the measured protein to GAPDH level (p < 0.001). For the RT‐PCR GAPDH was used as an internal control in the mRNA analysis experiments.
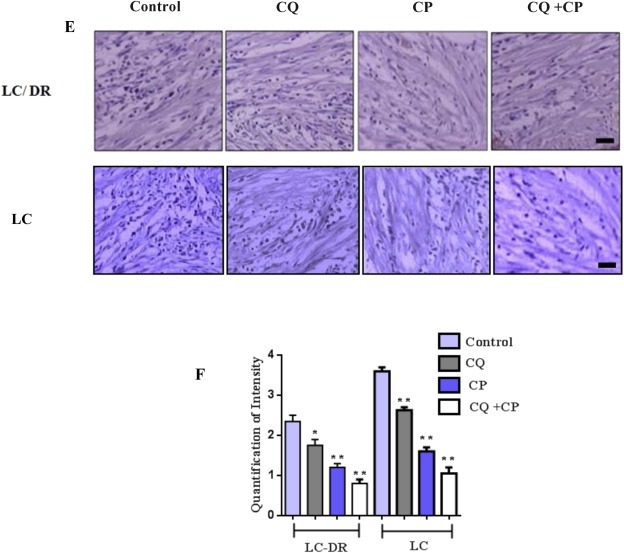
3.3. Combination blockage increases lung tissue infiltration by CD4+, FoxP3+ and regulates expression of stage specific FoxP3+ TILs
Previous studies have shown that chemoresistance is influenced by lymphocyte infiltration in the tumor microenvironment and that the presence of tumor‐infiltrating lymphocytes (TILs) affects clinical outcome (Guthrie et al., 2013; Templeton et al., 2014). However, the role of autophagy in lung tissue infiltration remains unknown. To investigate this, the expression levels of CD4+, CD8+ and FoxP3 TILs from lung tissue samples were evaluated. Results show that the number of CD4+ TILs was significantly depleted in late metastatic stages (III and IV) than in early stages (I, II) of lung cancer samples (Figure 3A). Furthermore, the number of CD8+ TILs correlated with tumor stage progression and was downregulated following tumor propagation (Figure 3B and Supplementary Table S6). In addition, CD4+ and CD8+ TILs levels were decreased in patients with stage III and stage IV lung cancer (Figure 3A and B). Elevated FoxP3+ TILs numbers were observed only in tumor stages I and II expect late metastatic grade IV (Figure 3C) but were depleted in severe lung cancer patients. FoxP3+ TILs were only present in early stages and CD4+/Foxp3+ plus CD8+/Foxp3+ numbers were highly expressed in the majority of patients (Supplementary Table S6). In addition the cytokine levels of Th1+/CD4+ cells was evaluated in metastatic and non metastatic patients (Supplementary Fig. S3). To further evaluate TIL response to chemotherapy, lung cancer biopsies (stage IV) were treated with autophagy inhibitor chloroquine along with carboplatin and subjected to immunohistochemical analysis. Results show the number of CD4+ and FoxP3+ TILs significantly increased in chloroquine plus carboplatin treated group compared with chloroquine‐treated alone or control cells. In contrast, no significant differences in the number CD8+ TILs were observed between control, chloroquine or combination group (Figure 3D). Overall, these results indicate that inhibition of autophagy in the presence of carboplatin could elucidate lymphocyte infiltration in the tumor microenvironment and promote CD4+, FoxP3+ TILs upregulation by controlling expression of stage specific TILs.
Figure 3.

Inhibition of autophagy in the presence of carboplatin elucidates lymphocyte infiltration in patients during progressive lung carcinogenesis. Expression levels of (A) CD4, (B) CD8 and (C) FoxP3 tumor‐infiltrating lymphocytes in lung cancer stage specific (I–IV) samples. (D) Representative immunohistochemical images of TILs in lung cancer specimens. Immunohistochemical staining for CD4, CD8 and Foxp3 in lung biopsies from control subjects and biopsies treated with chloroquine and carboplatin. Sections of human lung biopsies were obtained from patients with lung cancer and following chloroquine/carboplatin treatment (50 μΜ, 1 h). Human CD4, CD8 and Foxp3, were detected using anti‐human monoclonal antibodies. Staining with mouse IgG1 isotype was used as the negative control. (E) Quantification of immunohistochemistry by the intensity of the positive immunosignals in the tissue sections are showed in histograms (*P < 0.05, **P < 0.01). Images were obtained through a Carl Zeiss microscope using image analysis software (Scale bar, 50 μm). Stained tumor cells are shown at a final magnification (×400).
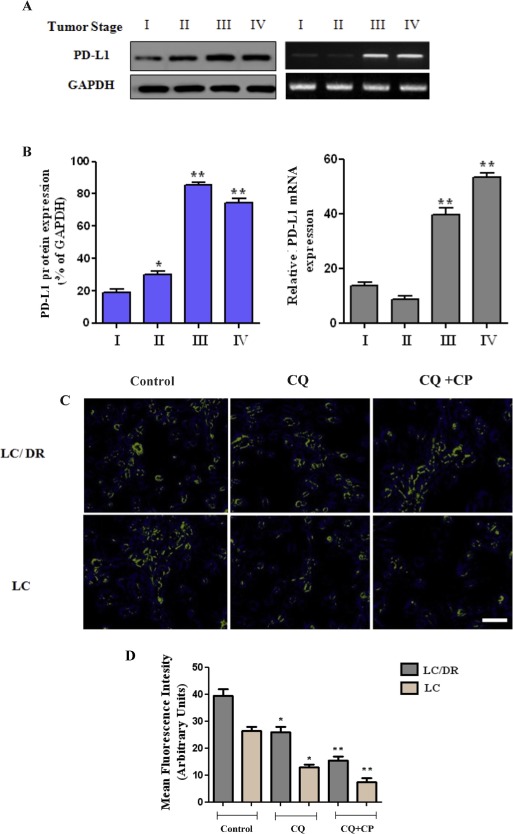
3.4. Role of autophagy inhibition on PD‐L1 expression and clinicopathological correlation with tumor progression
Recent evidence highlight the relationship between immune system regulation and lymphocyte infiltration with programmed cell death‐ligand 1 (PD‐L1), and its clinical role in lung cancer‐related immunosuppression (D'Incecco et al., 2015; Wang et al., 2015a). To address this issue, lung tissue PD‐L1 expression was analyzed by western blot, RT‐PCR, immunofluorescence and immunohistochemical analysis. As shown in Figure 4A, both stage III and stage IV tumor samples exhibit elevated PD‐L1 compared with low levels of PD‐L1 detected in stage I. Positive tumor PD‐L1 expression was identified in 14 of 21 (67%) stage III biopsies, and 27 of 32 (91%) stage IV biopsies (Supplementary Table S7). Equally, augmented PD‐L1 expression was observed in stromal cells compared to tumor lesions, however the predominant pattern of staining in the biopsies was non‐peripheral (intratumoral). To further investigate PD‐L1 response to chemotherapy, LC/LC‐DR cells were treated with autophagy inhibitor chloroquine and carboplatin and subjected to immunofluorescence (Figure 4C and D). In addition, immunohistochemical analysis was performed in lung biopsies following chloroquine and carboplatin treatment (50 μΜ, 1 h) (Figure 4E). Results show a higher PD‐L1 expression pattern in both drug resistant LC‐DR and LC cells, indicating the crucial role of PD‐L1 in regulating the chemoresistance mechanism (Figure 4E and F). Treatment with chloroquine plus carboplatin drastically reduced PD‐L1 levels in both cell types. In summary, these results indicate, the key role of autophagy on PD‐L1 modulation and its clinicopathological correlation with lung tumor progression and immunosuppression.
Figure 4.
Autophagy inhibition on PD‐L1 expression and clinicopathological correlation with tumor progression. (A–B) The protein and mRNA expression level of PD‐L1 was assayed by western blot and RT‐PCR, respectively. (C–D) LC/LC‐DR cells were treated with autophagy inhibitor chloroquine (20 μΜ) or carboplatin (25 μΜ) for 24 h and PD‐L1 expression was measured by immunofluorescence analysis (magnification ×200). (F–G) Immunohistochemical analysis was performed in lung biopsies following chloroquine and carboplatin treatment (50 μΜ) for 1 h using mouse IgG1 isotype as negative control. Images were obtained through a Carl Zeiss microscope using image analysis software (Scale bar, 50 μm) (magnification ×400). (D) Quantification of immunohistochemistry by the intensity of the positive immunosignals in the tissue sections are showed in histograms (*P < 0.05, **P < 0.01). Stained tumor cells are shown at a final magnification (×200). Data represent the mean ± SD of three independent experiments. (*P < 0.05; **P < 0.01).
3.5. Autophagy inhibition promotes carboplatin related TRAIL‐dependent apoptosis through caspase 8
In order to examine whether autophagy inhibition promotes carboplatin associated induction of TRAIL apoptosis, the expression of DR4 and DR5 receptors was evaluated following autophagy blockage. In this regard, inhibition of autophagy by chloroquine was evaluated according to apoptotic expression and release of TRAIL downstream molecules DR4 and DR5 (Figure 5A and B). Furthermore, the activity of caspase 8 and cleaved caspase 8 increased following co‐treatment, suggesting that autophagy blockage triggers caspase‐8 response during apoptosis. Inhibition of autophagy further increased the apoptotic activity and enhanced the expression of TRAIL markers DR4 and DR5 by up regulating caspase 8 activation (Figure 5A and B). Autophagy blockage along with carboplatin treatment induced a dose‐dependent apoptosis in ex vivo cancer cells. Next, we investigated whether combination treatment‐induced apoptosis relates to cell cycle regulation. The cell cycle was analyzed by flow cytometry 24 h following treatment. The results show that percentage of cells increased in G0/G1 phase and decreased in the S and G2/M phases significantly in co‐treatment group compared to single treated or the control group (Figure 5C and D). Collectively, the results demonstrate that autophagy inhibition can promote carboplatin related TRAIL‐dependent apoptosis and trigger G0/G1 cell cycle arrest.
Figure 5.
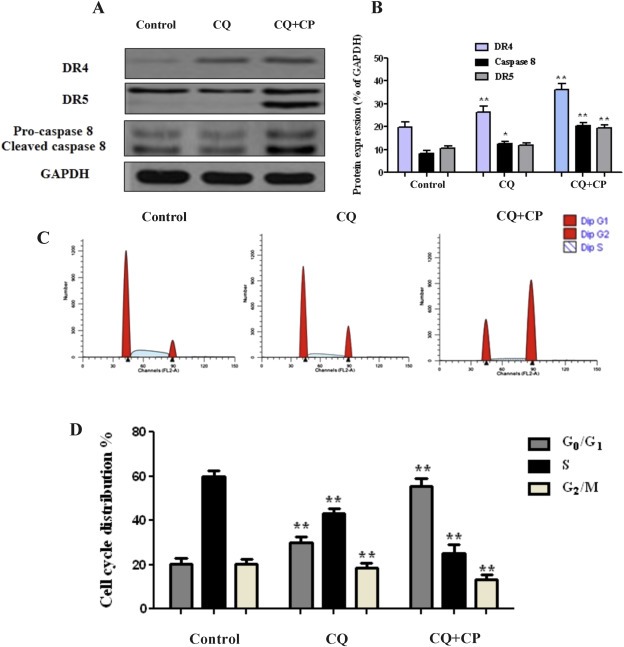
Autophagy inhibition activates carboplatin related TRAIL apoptosis. (A–B) LC‐DR cells were treated with autophagy inhibitor chloroquine (20 μΜ) or carboplatin (25 μΜ) for 24 h. Protein levels of DR4, DR5 and caspase 8 were analyzed by western blot. Ex vivo lung cultured cells were treated with chloroquine (20 μΜ) and carboplatin (25 μΜ) for 24 h and subjected to FACS analysis. Data represent the mean ± SD of three independent experiments (*P < 0.05, **P < 0.01). (C–D) Co‐treatment of chloroquine‐carboplatin induces cell cycle G0/G1 arrest in lung cancer cells. Ex vivo lung cultures were treated with chloroquine (20 μΜ) and carboplatin (25 μΜ). After 24 h, cells were collected and subjected to flow cytometric analysis of cell cycle distribution. Data show the representative of three independent experiments (*P < 0.05, **P < 0.01). GAPDH was used as a loading control for western blot analysis. Densitometric analysis of each protein level was calculated from the average of three experiments. Data represent the mean ± SD of three separate experiments. (*P < 0.05; **P < 0.01).
3.6. Role of MiR‐155 in lung tissue infiltration by CD4+, and FoxP3+ lymphocytes
Increasing evidence accentuate on the role of specific microRNAs in triggering lung cancer associated inflammation and tissue infiltration by TILs (Liao et al., 2015; Roy and Sen, 2011). To explore this, we used miR‐155 which represents a key factor in lung carcinogenesis by regulating innate and adaptive immunity response (Gao et al., 2014; Thomsen et al., 2015). However, the role of miR‐155 expression in immune cells in solid tumor development is still unknown. In this regard, we assessed the expression levels of miR‐155 expression levels in metastatic lung cancer tissues compared to non‐metastatic samples tissues using RT‐PCR. The results showed that miR‐155 level was significantly lower in lung tissue than in the adjacent normal tissue (Figure 6A and B). To establish the correlation between miR‐155 expression and lung cancer progression, we further compared the miR‐155 levels in lung chemoresistant and chemosensitive tissues and found that the relative mean levels of miR‐155 in pulmonary tissues with metastasis (n = 47) were lower than that in those without metastasis (n = 49) (Figure 6A). Furthermore, the percentage of chemoresistant lung cancer samples with downregulated miR‐155 was higher than in chemonaive tissues (Figure 6B). These data show that miR‐155 plays a substantial role in lung cancer metastasis. Treatment of ex vivo culture tissues with chloroquine and carboplatin radically enhanced miR‐155 expression compared with chloroquine treated or control cells (Figure 6C). Furthermore, we evaluated the role of miR‐155 in lymphocyte infiltration in lung tissue samples and the effect on lymphocytes (TILs) in clinical outcome. Transfection of miR‐155 in ex vivo lung cancer tissues caused CD4+, FoxP3+ upregulation and TIL numbers were significantly increased in chloroquine plus carboplatin treated group compared with single treatment or control (Figure 6D). In addition, FoxP3+ TILs were also upregulated in both in chloroquine and carboplatin group compared with control. Overall, these results demonstrate that co‐treatment of chloroquine plus carboplatin triggers upregulation of miR‐155 expression and synergistically enhances lymphocytic infiltration in tumor microenvironment by eliciting CD4+ and FoxP3+ TILs expression.
Figure 6.
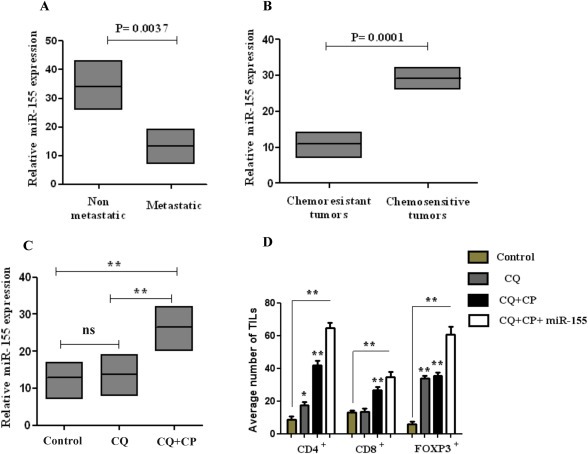
MiR‐155 is deregulated in metastatic chemoresistant lung tissues. (A–B) Relative expression of miR‐155 in metastatic lung cancer tissues in comparison with chemonaive non‐metastatic tissues. (B) miR‐155 expression was significantly lower in patients with advanced pathological stage (III–IV) and lymph node metastasis. (C) LC‐DR cells were treated with autophagy inhibitor chloroquine (20 μΜ) or carboplatin (25 μΜ) for 24 h and miR‐155 expression was evaluated. (D) Effect of miR‐155 transfected LC‐DR cells in TILs expression. MiR‐155 expression was normalized to U6 expression. Data represent the mean ± SD of three independent experiments (*P < 0.05; **P < 0.01).
4. Discussion
Numerous studies have implicated the crucial role of autophagy in cancer chemoresistance (Duffy et al., 2015). However, the exact mechanisms underlying this phenomenon are still unknown and represent a significant barrier to the improvement of the long‐term overcome of NSCLC patients (Kovarik et al., 2014; Sui et al., 2014). Our study highlights for the first time the involvement of autophagy in the metastatic mechanism that drives lung cancer immunosuppressive metastasis and inhibits T cell infiltration. Correlation of ex vivo experiments with clinical patient data show an early immunosuppressive phenotypic response mainly characterized by autophagy activation and apoptosis inhibition. Recent studies have also indicated that autophagy promoted chemoresistance through in hypoxia‐mediated induction of autophagy (Li et al., 2016; Song et al., 2009). Furthermore, latest evidence emphasizes the cyto‐protective functions of autophagy in cancer cells through miR‐152 as a autophagy‐regulating miRNA that plays a role in cisplatin resistance. Expression of miR‐152 was dramatically downregulated in the cisplatin‐resistant cell lines A2780/CP70, SKOV3/DDP compared with their respective parental cells (He et al., 2015). Likewise, autophagy is also involved in immune system regulation and immunoediting (Chen et al., 2014b). T cell immunoglobulin‐ and mucin domain‐containing molecule‐4 (TIM‐4) was found to repress tumor‐specific immunity triggered by chemotherapy‐induced tumor cell death. TIM‐4 was highly expressed on tumor‐associated myeloid cells and was activated by autophagy‐mediated degradation of ingested tumors, leading to reduced antigen presentation and impaired CTL responses (Baghdadi et al., 2013). Recent findings reveal that, autophagy inhibition re‐sensitizes paclitaxel‐resistant triple negative breast cancer cells to caspase‐dependent/chemotherapy‐induced apoptosis and can overcome drug resistance of BCR‐ABL‐positive chronic myeloid leukemia cells, respectively (Wen et al., 2015; Zeng et al., 2015). Furthermore, autophagy inhibition was shown to upregulate T cell lymphocyte expression in colorectal carcinoma. Specifically, deletion of Atg7 caused increase in the number of CD45 T cells, an infiltration of CD11c+ cells and increase in the numbers of cytotoxic CD8+ T cells and Th1 cells in the intestinal epithelium (Lévy et al., 2015). Equally in our study we showed that co‐treatment of carboplatin and autophagy inhibitor chloroquine increased lung tissue infiltration by CD4+, FoxP3+ lymphocytes and antigen‐specific inflammatory immune activation. There is ample evidence that the presence of tumor‐infiltrating T lymphocytes is associated with a favorable prognostic in cancer patients (Rahir and Moser, 2012). Tumor‐infiltrating CD8+ and FOXP3+ lymphocytes have been shown to shape tumor response in triple‐negative breast cancer and are important predictors of pathologically complete responses to neoadjuvant chemotherapy, especially in the hormone receptors (−)/HER2(+) group (Miyashita et al., 2014; Yamaguchi et al., 2012). These observations highlight the capacity of tumor‐specific T cells to reach tumor microenvironment and shape immune resistance and immunotherapy (Trivedi et al., 2014). Another important issue in tumor related chemoresistance is the expression of specific miRs that sculpture immune phenotype and promote cancer metastasis. Especially in late metastatic neoplasia stages the specific miR members regulate tumor accumulation and sculpture immune response to support extended tumorigenesis. Recently, miR‐200 was proven to act as cell‐autonomous suppressor of EMT and metastasis, by targeting PD‐L1. Moreover, ZEB1, an EMT activator and transcriptional repressor of miR‐200, relieves miR‐200 repression of PD‐L1 on tumor cells, leading to CD8+ T‐cell immunosuppression and metastasis (Chen et al., 2014a). Likewise, overexpression of tumor suppressor miR‐34a blocked PD‐L1 expression, and reduced PD‐L1 surface expression and functions as a potential immunotherapeutic target in acute myeloid leukemia by blocking PD‐L1 specific T cell apoptosis (Wang et al., 2015b). These findings also explore the role of PD‐L1 in tumor immune escape. A latest study identified miR‐197/CKS1B/STAT3‐mediated PD‐L1 network in NSCLC, independent of immunoinhibitory signals. In particular, miR‐197 was downregulated in platinum‐resistant NSCLC specimens, resulting in the promotion of chemoresistance, tumorigenicity, and pulmonary metastasis in vitro and in vivo. Mechanistic investigations reveal that a miR‐197‐mediated CKS1B/STAT3 axis exerts tumor progression regulated by various oncogenic genes (Bcl‐2, c‐Myc, and cyclin D1), and PD‐L1 is a putative biomarker of this axis (Fujita et al., 2015). Furthermore, miR‐197 mimic sensitizes PD‐L1 (high) drug‐resistant cells to chemotherapy for overcoming lung chemoresistance and combined chemotherapy with current existing pharmacological regimens may be crucial of patient survival. Moreover the cross talking roles between tumor infiltrating lymphocytes and microRNA expression via targeting autophagy remains to be elucidated in the near future. Enhanced resistance in solid tumors provides a better understanding of the pathogenesis and development of NSCLC and may be quite important implication for future therapy. Tumoral immunity is a key event for acquiring chemoresistance and many data show towards immune regulation in hypoxic tumor microenvironments. What's more, a number of studies have shown that the metastatic potential of carcinoma cells by supporting TRAIL inhibition and autophagy may be required for the maintenance of tumor potential and provide resistance to chemotherapy (Liu et al., 2013; Wang et al., 2014). This induction of TRAIL apoptosis is mainly associated with reduction in autophagy levels and proliferation inhibition by neoplastic cells and can trigger immune system activation in tumor microenvironment thus signifying a prospective therapeutic strategy to inhibit lung cancer progression and metastasis. In conclusion, our findings reveal that autophagy blockage increases lung cancer chemosensitivity to carboplatin, but also divulge miR‐155 role as an immune system activator by promoting TILs infiltration. These results indicate that targeting of autophagy can prevent cancer related immunosuppression and elucidate T cell infiltration in tumor microenvironment and thus identifying a novel molecular mechanism that regulates chemoresistance and represents a therapeutic target for the treatment of metastatic lung cancer.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Acknowledgments
This work was funded by grant from the “ΙΚΥ Fellowship – Siemens Program”.
1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.08.005.
Zarogoulidis Paul, Petanidis Savvas, Domvri Kalliopi, Kioseoglou Efrosini, Anestakis Doxakis, Freitag Lutz, Zarogoulidis Konstantinos, Hohenforst-Schmidt Wolfgang, Eberhardt Wilfried, (2016), Autophagy inhibition upregulates CD4+ tumor infiltrating lymphocyte expression via miR-155 regulation and TRAIL activation, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.08.005.
References
- Baghdadi, M. , Yoneda, A. , Yamashina, T. , Nagao, H. , Komohara, Y. , Nagai, S. , Akiba, H. , Foretz, M. , Yoshiyama, H. , Kinoshita, I. , Dosaka-Akita, H. , Takeya, M. , Viollet, B. , Yagita, H. , Jinushi, M. , 2013. TIM-4 glycoprotein-mediated degradation of dying tumor cells by autophagy leads to reduced antigen presentation and increased immune tolerance. Immunity. 39, 1070–1081. [DOI] [PubMed] [Google Scholar]
- Beckett, P. , Tata, L.J. , Hubbard, R.B. , 2014. Risk factors and survival outcome for non-elective referral in non-small cell lung cancer patients–analysis based on the National Lung Cancer Audit. Lung Cancer. 83, 396–400. [DOI] [PubMed] [Google Scholar]
- Bronietzki, A.W. , Schuster, M. , Schmitz, I. , 2015. Autophagy in T-cell development, activation and differentiation. Immunol. Cell Biol.. 93, 25–34. [DOI] [PubMed] [Google Scholar]
- Chen, L. , Gibbons, D.L. , Goswami, S. , Cortez, M.A. , Ahn, Y.H. , Byers, L.A. , Zhang, X. , Yi, X. , Dwyer, D. , Lin, W. , Diao, L. , Wang, J. , Roybal, J.D. , Patel, M. , Ungewiss, C. , Peng, D. , Antonia, S. , Mediavilla-Varela, M. , Robertson, G. , Jones, S. , Suraokar, M. , Welsh, J.W. , Erez, B. , Wistuba, I.I. , Chen, L. , Peng, D. , Wang, S. , Ullrich, S.E. , Heymach, J.V. , Kurie, J.M. , Qin, F.X. , 2014. Metastasis is regulated via microRNA-200/ZEB1 axis control of tumour cell PD-L1 expression and intratumoral immunosuppression. Nat. Commun.. 5, 5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, P. , Cescon, M. , Bonaldo, P. , 2014. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy. 10, 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochaud, S. , Giustiniani, J. , Thomas, C. , Laprevotte, E. , Garbar, C. , Savoye, A.M. , Curé, H. , Mascaux, C. , Alberici, G. , Bonnefoy, N. , Eliaou, J.F. , Bensussan, A. , Bastid, J. , 2013. IL-17A is produced by breast cancer TILs and promotes chemoresistance and proliferation through ERK1/2. Sci. Rep.. 3, 3456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- deLeeuw, R.J. , Kost, S.E. , Kakal, J.A. , Nelson, B.H. , 2012. The prognostic value of FoxP3+ tumor-infiltrating lymphocytes in cancer: a critical review of the literature. Clin. Cancer Res.. 18, 3022–3029. [DOI] [PubMed] [Google Scholar]
- D'Incecco, A. , Andreozzi, M. , Ludovini, V. , Rossi, E. , Capodanno, A. , Landi, L. , Tibaldi, C. , Minuti, G. , Salvini, J. , Coppi, E. , Chella, A. , Fontanini, G. , Filice, M.E. , Tornillo, L. , Incensati, R.M. , Sani, S. , Crinò, L. , Terracciano, L. , Cappuzzo, F. , 2015. PD-1 and PD-L1 expression in molecularly selected non-small-cell lung cancer patients. Br. J. Cancer. 112, 95–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy, A. , Le, J. , Sausville, E. , Emadi, A. , 2015. Autophagy modulation: a target for cancer treatment development. Cancer Chemother. Pharmacol.. 75, 439–447. [DOI] [PubMed] [Google Scholar]
- Ferraldeschi, R. , Baka, S. , Jyoti, B. , Faivre-Finn, C. , Thatcher, N. , Lorigan, P. , 2007. Modern management of small-cell lung cancer. Drugs. 67, 2135–2152. [DOI] [PubMed] [Google Scholar]
- Fujita, Y. , Yagishita, S. , Hagiwara, K. , Yoshioka, Y. , Kosaka, N. , Takeshita, F. , Fujiwara, T. , Tsuta, K. , Nokihara, H. , Tamura, T. , Asamura, H. , Kawaishi, M. , Kuwano, K. , Ochiya, T. , 2015. The clinical relevance of the miR-197/CKS1B/STAT3-mediated PD-L1 network in chemoresistant non-small-cell lung cancer. Mol. Ther.. 23, 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, F. , Chang, J. , Wang, H. , Zhang, G. , 2014. Potential diagnostic value of miR-155 in serum from lung adenocarcinoma patients. Oncol. Rep.. 31, 351–357. [DOI] [PubMed] [Google Scholar]
- Giovannetti, E. , Toffalorio, F. , De Pas, T. , Peters, G.J. , 2012. Pharmacogenetics of conventional chemotherapy in non-small-cell lung cancer: a changing landscape?. Pharmacogenomics. 13, 1073–1086. [DOI] [PubMed] [Google Scholar]
- Gong, A.Y. , Zhou, R. , Hu, G. , Li, X. , Splinter, P.L. , O'Hara, S.P. , LaRusso, N.F. , Soukup, G.A. , Dong, H. , Chen, X.M. , 2009. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-gamma-induced B7-H1 expression in cholangiocytes. J. Immunol.. 182, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie, G.J. , Charles, K.A. , Roxburgh, C.S. , Horgan, P.G. , McMillan, D.C. , Clarke, S.J. , 2013. The systemic inflammation-based neutrophil-lymphocyte ratio: experience in patients with cancer. Crit. Rev. Oncol. Hematol.. 88, 218–230. [DOI] [PubMed] [Google Scholar]
- He, J. , Yu, J.J. , Xu, Q. , Wang, L. , Zheng, J.Z. , Liu, L.Z. , Jiang, B.H. , 2015. Downregulation of ATG14 by EGR1-MIR152 sensitizes ovarian cancer cells to cisplatin-induced apoptosis by inhibiting cyto-protective autophagy. Autophagy. 11, 373–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heigener, D. , Reck, M. , 2015. Exploring the potential of immuno-oncology-based treatment for patients with non-small cell lung cancer. Expert Rev. Anticancer Ther.. 15, 69–83. [DOI] [PubMed] [Google Scholar]
- Kovarik, M. , Hronek, M. , Zadak, Z. , 2014. Clinically relevant determinants of body composition, function and nutritional status as mortality predictors in lung cancer patients. Lung Cancer. 84, 1–6. [DOI] [PubMed] [Google Scholar]
- Leithner, K. , Wohlkoenig, C. , Stacher, E. , Lindenmann, J. , Hofmann, N.A. , Gallé, B. , Guelly, C. , Quehenberger, F. , Stiegler, P. , Smolle-Jüttner, F.M. , Philipsen, S. , Popper, H.H. , Hrzenjak, A. , Olschewski, A. , Olschewski, H. , 2014. Hypoxia increases membrane metallo-endopeptidase expression in a novel lung cancer ex vivo model – role of tumor stroma cells. BMC Cancer. 14, 40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemke, J. , von Karstedt, S. , Zinngrebe, J. , Walczak, H. , 2014. Getting TRAIL back on track for cancer therapy. Cell Death Differ.. 21, 1350–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévy, J. , Cacheux, W. , Bara, M.A. , L'Hermitte, A. , Lepage, P. , Fraudeau, M. , Trentesaux, C. , Lemarchand, J. , Durand, A. , Crain, A.M. , Marchiol, C. , Renault, G. , Dumont, F. , Letourneur, F. , Delacre, M. , Schmitt, A. , Terris, B. , Perret, C. , Chamaillard, M. , Couty, J.P. , Romagnolo, B. , 2015. Intestinal inhibition of Atg7 prevents tumour initiation through a microbiome-influenced immune response and suppresses tumour growth. Nat. Cell Biol.. 17, 1062–1073. [DOI] [PubMed] [Google Scholar]
- Li, B. , Zhu, X. , Sun, L. , Yuan, L. , Zhang, J. , Li, H. , Ye, Z. , 2014. Induction of a specific CD8+ T-cell response to cancer/testis antigens by demethylating pre-treatment against osteosarcoma. Oncotarget. 5, 10791–10802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L.Q. , Xie, W.J. , Pan, D. , Chen, H. , Zhang, L. , 2016. Inhibition of autophagy by bafilomycin A1 promotes chemosensitivity of gastric cancer cells. Tumour Biol.. 37, 653–659. [DOI] [PubMed] [Google Scholar]
- Liao, C. , Xiao, W. , Zhu, N. , Liu, Z. , Yang, J. , Wang, Y. , Hong, M. , 2015. Radiotherapy suppressed tumor-specific recruitment of regulator T cells via up-regulating microR-545 in Lewis lung carcinoma cells. Int. J. Clin. Exp. Pathol.. 8, 2535–2544. [PMC free article] [PubMed] [Google Scholar]
- Liu, H. , He, Z. , Simon, H.U. , 2013. Targeting autophagy as a potential therapeutic approach for melanoma therapy. Semin. Cancer Biol.. 23, 352–360. [DOI] [PubMed] [Google Scholar]
- Lo Presti, E. , Dieli, F. , Meraviglia, S. , 2014. Tumor-infiltrating γδ T lymphocytes: pathogenic role, clinical significance, and differential programing in the tumor microenvironment. Front. Immunol.. 5, 607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorin, S. , Hamaï, A. , Mehrpour, M. , Codogno, P. , 2013. Autophagy regulation and its role in cancer. Semin. Cancer Biol.. 23, 361–379. [DOI] [PubMed] [Google Scholar]
- Meng, X. , Huang, Z. , Teng, F. , Xing, L. , Yu, J. , 2015. Predictive biomarkers in PD-1/PD-L1 checkpoint blockade immunotherapy. Cancer Treat. Rev.. 41, 868–876. [DOI] [PubMed] [Google Scholar]
- Miyashita, M. , Sasano, H. , Tamaki, K. , Chan, M. , Hirakawa, H. , Suzuki, A. , Tada, H. , Watanabe, G. , Nemoto, N. , Nakagawa, S. , Ishida, T. , Ohuchi, N. , 2014. Tumor-infiltrating CD8+ and FOXP3+ lymphocytes in triple-negative breast cancer: its correlation with pathological complete response to neoadjuvant chemotherapy. Breast Cancer Res. Treat.. 148, 525–534. [DOI] [PubMed] [Google Scholar]
- Ohaegbulam, K.C. , Assal, A. , Lazar-Molnar, E. , Yao, Y. , Zang, X. , 2015. Human cancer immunotherapy with antibodies to the PD-1 and PD-L1 pathway. Trends Mol. Med.. 21, 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahir, G. , Moser, M. , 2012. Tumor microenvironment and lymphocyte infiltration. Cancer Immunol. Immunother.. 61, 751–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiss, K.A. , Forde, P.M. , Brahmer, J.R. , 2014. Harnessing the power of the immune system via blockade of PD-1 and PD-L1: a promising new anticancer strategy. Immunotherapy. 6, 459–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, S. , Sen, C.K. , 2011. MiRNA in innate immune responses: novel players in wound inflammation. Physiol. Genomics. 43, 557–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, J. , Qu, Z. , Guo, X. , Zhao, Q. , Zhao, X. , Gao, L. , Sun, K. , Shen, F. , Wu, M. , Wei, L. , 2009. Hypoxia-induced autophagy contributes to the chemoresistance of hepatocellular carcinoma cells. Autophagy. 5, 1131–1144. [DOI] [PubMed] [Google Scholar]
- Sui, X. , Kong, N. , Zhu, M. , Wang, X. , Lou, F. , Han, W. , Pan, H. , 2014. Cotargeting EGFR and autophagy signaling: a novel therapeutic strategy for non-small-cell lung cancer. Mol. Clin. Oncol.. 2, 8–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sznol, M. , 2014. Blockade of the B7-H1/PD-1 pathway as a basis for combination anticancer therapy. Cancer J.. 20, 290–295. [DOI] [PubMed] [Google Scholar]
- Tanoue, L.T. , Tanner, N.T. , Gould, M.K. , Silvestri, G.A. , 2015. Lung cancer screening. Am. J. Respir. Crit. Care Med.. 191, 19–33. [DOI] [PubMed] [Google Scholar]
- Templeton, A.J. , McNamara, M.G. , Šeruga, B. , Vera-Badillo, F.E. , Aneja, P. , Ocaña, A. , Reibowitz-Amit, L. , Sonpavde, G. , Knox, J.J. , Tran, B. , Tannock, I.F. , Amir, E. , 2014. Prognostic role of neutrophil-to-lymphocyte ratio in solid tumors: a systematic review and meta-analysis. J. Natl. Cancer Inst.. 106, dju124 [DOI] [PubMed] [Google Scholar]
- Thomsen, K.G. , Terp, M.G. , Lund, R.R. , Søkilde, R. , Elias, D. , Bak, M. , Litman, T. , Beck, H.C. , Lyng, M.B. , Ditzel, H.J. , 2015. miR-155, identified as anti-metastatic by global miRNA profiling of a metastasis model, inhibits cancer cell extravasation and colonization in vivo and causes significant signaling alterations. Oncotarget. 6, 29224–29239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi, S. , Jie, H.B. , Ferris, R.L. , 2014. Tumor antigen-specific monoclonal antibodies and induction of T-cell immunity. Semin. Oncol.. 41, 678–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tykodi, S.S. , 2014. PD-1 as an emerging therapeutic target in renal cell carcinoma: current evidence. Onco Targets Ther.. 7, 1349–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viry, E. , Paggetti, J. , Baginska, J. , Mgrditchian, T. , Berchem, G. , Moussay, E. , Janji, B. , 2014. Autophagy: an adaptive metabolic response to stress shaping the antitumor immunity. Biochem. Pharmacol.. 92, 31–42. [DOI] [PubMed] [Google Scholar]
- Wang, A. , Wang, H.Y. , Liu, Y. , Zhao, M.C. , Zhang, H.J. , Lu, Z.Y. , Fang, Y.C. , Chen, X.F. , Liu, G.T. , 2015. The prognostic value of PD-L1 expression for non-small cell lung cancer patients: a meta-analysis. Eur. J. Surg. Oncol.. 41, 450–456. [DOI] [PubMed] [Google Scholar]
- Wang, X. , Li, J. , Dong, K. , Lin, F. , Long, M. , Ouyang, Y. , Wei, J. , Chen, X. , Weng, Y. , He, T. , Zhang, H. , 2015. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal.. 27, 443–452. [DOI] [PubMed] [Google Scholar]
- Wang, F. , Lin, J. , Xu, R. , 2014. The molecular mechanisms of TRAIL resistance in cancer cells: help in designing new drugs. Curr. Pharm. Des.. 20, 6714–6722. [DOI] [PubMed] [Google Scholar]
- Wang, W. , Li, F. , Mao, Y. , Zhou, H. , Sun, J. , Li, R. , Liu, C. , Chen, W. , Hua, D. , Zhang, X. , 2013. miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Hum. Genet.. 132, 641–648. [DOI] [PubMed] [Google Scholar]
- Wen, J. , Yeo, S. , Wang, C. , Chen, S. , Sun, S. , Haas, M.A. , Tu, W. , Jin, F. , Guan, J.L. , 2015. Autophagy inhibition re-sensitizes pulse stimulation-selected paclitaxel-resistant triple negative breast cancer cells to chemotherapy-induced apoptosis. Breast Cancer Res. Treat.. 149, 619–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, R. , Tanaka, M. , Yano, A. , Tse, G.M. , Yamaguchi, M. , Koura, K. , Kanomata, N. , Kawaguchi, A. , Akiba, J. , Naito, Y. , Ohshima, K. , Yano, H. , 2012. Tumor-infiltrating lymphocytes are important pathologic predictors for neoadjuvant chemotherapy in patients with breast cancer. Hum. Pathol.. 43, 1688–1694. [DOI] [PubMed] [Google Scholar]
- Yang, H.Z. , Ma, Y. , Zhou, Y. , Xu, L.M. , Chen, X.J. , Ding, W.B. , Zou, H.B. , 2015. Autophagy contributes to the enrichment and survival of colorectal cancer stem cells under oxaliplatin treatment. Cancer Lett.. 361, 128–136. [DOI] [PubMed] [Google Scholar]
- Zarogoulidis, P. , Petanidis, S. , Kioseoglou, E. , Domvri, K. , Anestakis, D. , Zarogoulidis, K. , 2015. MiR-205 and miR-218 expression is associated with carboplatin chemoresistance and regulation of apoptosis via Mcl-1 and Survivin in lung cancer cells. Cell Signal.. 27, 1576–1588. [DOI] [PubMed] [Google Scholar]
- Zeng, X. , Zhao, H. , Li, Y. , Fan, J. , Sun, Y. , Wang, S. , Wang, Z. , Song, P. , Ju, D. , 2015. Targeting Hedgehog signaling pathway and autophagy overcomes drug resistance of BCR-ABL-positive chronic myeloid leukemia. Autophagy. 11, 355–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, S. , Zhao, L. , Kuang, M. , Zhang, B. , Liang, Z. , Yi, T. , Wei, Y. , Zhao, X. , 2012. Autophagy in tumorigenesis and cancer therapy: Dr. Jekyll or Mr. Hyde?. Cancer Lett.. 323, 115–127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data
Supplementary data