

Abstract
Valosin‐containing protein (VCP) or p97, a member of AAA‐ATPase protein family, has been associated with various cellular functions including endoplasmic reticulum‐associated degradation (ERAD), Golgi membrane reassembly, autophagy, DNA repair, and cell division. Recent studies identified VCP and ubiquitin proteasome system (UPS) as synthetic lethal targets in ovarian cancer. Here, we describe the preclinical activity of VCP inhibitors in ovarian cancer. Results from our studies suggest that quinazoline‐based VCP inhibitors initiate G1 cell cycle arrest, attenuate cap‐dependent translation and induce programmed cell death via the intrinsic and the extrinsic modes of apoptosis. Mechanistic studies point to the unresolved unfolded protein response (UPR) as a mechanism by which VCP inhibitors contribute to cytotoxicity. These results support an emerging concept that UPR and endoplasmic reticulum (ER) stress pathways may be targeted in ovarian cancer as a source of vulnerability. Since prolonged ER stress may result in CHOP‐mediated cell death, we tested the hypothesis that VCP inhibitors act synergistically with compounds that enhance CHOP expression. Here, we show that VCP inhibitors act synergistically with Salubrinal, an inhibitor of eIF2α dephosphorylation, by enhancing CHOP expression in ovarian cancer cell lines. Our results provide a proof‐of‐concept that VCP inhibitors can be used as a single agent and can be synergized with compounds that enhance CHOP expression to induce cell death in ovarian cancer cells.
Keywords: Ovarian cancer, DBeQ, ML240, Salubrinal, ER stress, Unfolded protein response, CB-5083, Synergy
Abbreviations
- AAA‐ATPase
ATPase associated with diverse cellular activities
- BRCA1/2
breast cancer 1/2
- PARP1
poly ADP‐ribose polymerase‐1
- DBeQ
N2,N4‐dibenzylquinazoline‐2,4‐diamine
- PBS
phosphate buffered saline
- EGF
epidermal growth factor
- TBS
tris‐buffered saline
- PERK
protein kinase R‐like endoplasmic reticulum kinase
- TCGA
the Cancer Genome Atlas
- PVDF
polyvinylidene difluoride
- IRE1α
inositol‐requiring enzyme 1 alpha
- CHOP
CCAAT/enhancer‐binding protein homologous protein
- ATF4
activating transcription factor 4
- eIF2α
eukaryotic initiation factor 2 alpha
- BiP
binding immunoglobulin protein
- Grp78
glucose regulated protein 78
- GADD34
growth arrest And DNA‐damage‐inducible 34
1. Introduction
Ovarian cancer affected 239,000 women worldwide in 2012 (Ferlay et al., 2013), and it is the leading cause of death from gynecologic cancer in the United States. Current standard‐of‐care for advanced‐stage ovarian cancer includes debulking surgery followed by adjuvant combination chemotherapy consisting of a platinum agent and a taxane agent (Chien et al., 2013). Although the combination therapy is effective and the initial response rate is over 70%, the majority of patients with advanced disease experience recurrence (Chien et al., 2013). With subsequent re‐challenge with platinum‐based chemotherapy, most will acquire resistance to chemotherapy and succumb to the disease.
Therefore, it is imperative that novel therapeutic agents be developed to treat ovarian cancer and to extend the effectiveness of platinum‐based chemotherapy. Recent advances in cancer therapeutics indicate that genetic defects in cancer can be exploited by synthetic lethality with chemical inhibitors. Synthetic lethality is a concept first reported in Drosophila genetics to describe the lethality to the fly arising from a combination of mutations in two or more genes (Nijman, 2011). This concept was extended as an approach to target cancer‐specific mutations to cause lethality in cancer cells and not in normal cells. For example, mutations in BRCA1 and BRCA2 homology recombination (HR) repair genes are common in ovarian and breast carcinomas, and cancer cells harboring these mutations are extremely sensitive to PARP1 inhibition as a result of synthetic lethality (Bryant et al., 2005; Farmer et al., 2005). Accordingly, cancer cells harboring mutations in HR repair genes can be selectively killed by PARP1 inhibitors which cause reduced toxicity to normal cells (Underhill et al., 2011).
This concept was further extended to suggest that mutations in cancer cells create new vulnerabilities that can be exploited for therapeutic benefits (Nijhawan et al., 2012). In support of this concept, several genome‐scale genetic screens with short hairpin RNAs (shRNAs) identified components of the endoplasmic reticulum (ER) stress pathway as targets of vulnerability in ovarian cancer (Cheung et al., 2011; Marcotte et al., 2012; Nijhawan et al., 2012). In particular, Valosin‐containing protein (VCP), also known as p97 AAA‐ATPase, was identified as one of the essential genes in 25 ovarian cancer cell lines compared to 75 non‐ovarian cancer cell lines (Cheung et al., 2011). Moreover, VCP was also identified as an essential gene in cyclin E1‐overexpressed cisplatin‐resistant ovarian cancer cells (Etemadmoghadam et al., 2013). With a barrel‐shaped homohexameric configuration, VCP comprises of two AAA‐ATPase domains (D1 and D2) and N‐terminal domain (Mountassif et al., 2015). Both D1 and D2 domains bind to ATP. However, the D2 domain has been shown to be primarily involved in the VCP ATPase activity. Furthermore, N‐terminal domain acts as a primary region for substrate and cofactor binding (Banerjee et al., 2016).
Several studies have associated VCP with various cellular functions including endoplasmic reticulum‐associated degradation (ERAD), Golgi membrane reassembly, autophagy and cell division (Deshaies, 2014; Meyer et al., 2002; Seguin et al., 2014). VCP is involved in the extraction of unfolded proteins from the endoplasmic reticulum. Upon extraction, these proteins undergo proteasome‐mediated degradation. Cancer cells harbor a plethora of mutations which result in increased load of unfolded proteins required for degradation. Accordingly, VCP and components of the proteasomal degradation pathway are essential for cancer cell survival and in turn present a target of vulnerability in the cancer cell that could be exploited for cancer therapy.
Several groups have recently developed specific inhibitors against VCP (Alverez et al., 2015, 2016, 2010, 2011, 2014, 2013, 2010, 2015, 2016, 2013, 2013, 2016), these efforts were reviewed and summarized elsewhere (Chapman et al., 2015). Among these specific inhibitors, DBeQ was initially developed as a reversible inhibitor of VCP/p97 that compromises protein homeostasis through impairment of both ubiquitin proteasome system (UPS) and autophagic protein clearance. DBeQ targets both D1 and D2‐ATPase domains of VCP (Fang et al., 2015). In contrast, ML240, which was derived from the scaffold of DBeQ, targets the D2‐ATPase domain (Fang et al., 2015). Another VCP inhibitor NMS‐873 is a potent noncovalent, non‐ATP‐competitive, allosteric inhibitor of VCP and activates unfolded protein response, inhibits autophagy and induces cell death (Magnaghi et al., 2013). Finally, a derivative of ML240 was further developed by Cleave Biosciences to produce orally bioavailable active compound CB‐5083 that shows promising preclinical activities (Anderson et al., 2015). Consequently, CB‐5083 is currently in two Phase I clinical trials (NCT02243917 and NCT02223598) sponsored by Cleave Biosciences.
Since VCP is identified as an essential gene in ovarian cancer cell lineages and also in Cyclin E1‐overexpressed cisplatin‐resistant ovarian cancer cell lines, we tested the potential cytotoxic activities of quinazoline‐based VCP inhibitors, DBeQ, ML240, and its clinical lead CB‐5083 in ovarian cancer cell lines. Moreover, we also determine the potential synergistic drug interactions between VCP inhibitors and Salubrinal, an agent that enhances the expression of CHOP (also known as DDIT3, DNA Damage‐Inducible Transcript 3).
2. Materials and methods
2.1. Reagents
DBeQ and ML240 were provided by Dr. Frank Schoenen at the University of Kansas (Chou et al., 2013). Salubrinal (CML0951) and tunicamycin (11089‐65‐9) were purchased Sigma–Aldrich. CB‐5083 (S8101) was purchased from Selleckchem. DBeQ, ML240, and CB‐5083 were dissolved in dimethyl sulfoxide (DMSO) at the concentration of 50 mM. Salubrinal was dissolved at the concentration of 20 mM in DMSO, and tunicamycin was dissolved at the concentration of 10 mM in DMSO. Dissolved compounds were aliquoted and stored at −80 °C. Stocks were thawed at room temperature and dissolved in appropriate media at selected concentrations immediately before use.
2.2. Cell lines and cell culture
Cancer cell lines OVCAR10, SKOV3, OVCAR5, and RMG1 were cultured in MCDB 105 (Sigma–Aldrich, M6395), medium 199 (Sigma–Aldrich, M5017), 5% fetal bovine serum (Sigma–Aldrich, F0926) and 1% streptomycin/penicillin (Sigma–Aldrich, P0781). OVCAR8 and OVSAHO cells were maintained in RPMI (Sigma–Aldrich, R8758), 10% FBS and 1% streptomycin/penicillin. All cell lines were kept in humidified incubator at 37 °C with 5% CO2. All cell lines were subjected to cell line identity confirmation.
2.3. Sulforhodamine B (SRB) assays and drug synergy studies
SRB assays were performed as previously described (Vichai and Kirtikara, 2006) with the following modifications. Briefly, 3000 and 5000 cells in 200 μl/well were plated in 96‐well plate format and cultured in appropriate growth media overnight. Next day, the cells were treated with different concentrations of DBeQ, ML240, Salubrinal and in combination for 72 h unless otherwise specified. After 72 h, cells were fixed with 10% trichloroacetic acid (Sigma–Aldrich, T6399) at 4 °C overnight. Fixed cells were then washed with running water and stained with Sulforhodamine B dye (Sigma–Aldrich‐230162) at room temperature for 45 min. Excess stain was discarded, and cells were washed with 1% acetic acid solution (Sigma–Aldrich, BP2401). Stained cells were dissolved in 10 mM TBS pH 10 and fluorescence measurements were taken at Ex 488 nm and Em 585 nm using a plate reader. Dose response curves were fitted and GI50 values were determined using GraphPad Prism four parameters. All dose–response curves were constrained with 100% on top and greater than 0% on bottom. Synergy was determined by calculating the combination indexes (CIs) obtained from the fluorescence measurements. Combination indexes were calculated based on dividing the expected effect by the observed effect. N represents the total number of CIs determined from 16 different drug combinations in duplicates that produced 20–80% effect from three independent experiments.
2.4. Clonogenic assay
500 cells/well were plated in 6‐well plates using the appropriate media. Cells were allowed to incubate overnight. Next day, the culture media was replaced with media containing appropriate compounds, and the cells were further incubated for 48 h. Following the compound treatment for 48 h, media was gently aspirated, the wells were gently washed twice with PBS pH 7.0, and the media was replaced with regular growth media. Cells were then allowed to grow in the incubator for 8–10 additional days while replacing media every 48 h. Once the colonies were optimal, media was aspirated, and 1 ml of crystal violet solution (0.5% crystal violet dye, 50% methanol, and 50% deionized distilled H20) was added to each well. The cells were stained with crystal violet solution for 30 min and then washed with water. The plates were then air‐dried, and pictures were taken using the Bio‐Rad Imager System. Colonies were counted using BioRad Quantity One version 4.6.9 software.
2.5. Puromycin incorporation assay
Puromycin incorporation was performed as previously described (Schmidt et al., 2009), with the following modification. 0.5 × 106 cells in 2 ml/well were plated in 6‐well plates and cultured in appropriate growth media overnight. Next day, growth media were substituted with media containing different concentrations of DBeQ, ML240, Salubrinal or appropriate combinations. The plates were placed in humidified incubator at 37 °C with 5% CO2 for 5.5 h, after which 2 μl of 1 mM Puromycin Stock (Goldbio, P‐600) was added to each well and incubated for additional 30 min. Cells were collected, and equal proteins were subjected to the Western blot analysis according to the established protocol. Blots were incubated with antibodies against puromycin and β‐actin.
2.6. Cellular Thermal Shift Assay (CETSA)
CETSA was performed as previously described (Jafari et al., 2014), with the following modifications. Briefly, cells were plated in cell culture dish using appropriate growth media and allowed to incubate overnight. Next day, regular growth media was replaced with culture media containing variable concentrations of DBeQ and ML240, for 2 h. Cells were trypsinized, and cell pellets were collected following centrifugation. Cell pellets were dissolved in PBS pH 7.0 and subjected to heat treatment at 57 °C. Following heat treatment, protein was precipitated using freeze‐thaw in liquid nitrogen. Cells were then subjected to centrifugation at 20,000×g, and soluble fractions were analyzed using Western blot. Blots were incubated with antibodies against VCP and β‐actin.
2.7. Transient siRNA knockdown
0.5 × 106 cells/well were plated in a 6‐well plate using appropriate growth media without antibiotics and allowed to incubate overnight. Next day, siRNAs were transfected using Oligofectamine reagent (Life Technologies, 12252‐011) according to the manufacturer's protocol. VCP siRNAs were purchased from IDT. GADD34 siRNA were purchased from Santa Cruz Biotechnology (sc‐37414). Cells were collected 48 h after transfection and equal proteins were subjected to Western blot analysis to check for transfection efficiency.
2.8. Caspase 3 activity assay
0.5 × 106 cells/well were plated in a 6‐well plate using the appropriate culture media and allowed to incubate overnight. Next day, cells were treated with culture media containing appropriate compounds. Cells were collected at different time points following the treatment using a cell scraper. Cells were lysed in caspase buffer: 20 mM PIPES, 100 mM NaCl, 1 mM EDTA, 0.1% (w/v) CHAPS, 10% sucrose, 10 mM DTT pH 7.2. Protein concentration was determined using BCA Assay. 20 μg protein was combined with 2 μl of 2 mM DEVD‐Afc (kindly provided by Dr. Wen‐Xing Ding's laboratory at the University of Kansas Medical Center) in 96‐well flat‐bottom plates. 200 μl/well caspase buffer was added. The plate was covered and incubated at 37 °C. After 2 h, fluorescence measurements were taken using a plate reader at Excitation 400 nm and Emission 510 nm. Measurements were analyzed using GraphPad Prism.
2.9. Violin plot
For the violin plot, RNA sequencing datasets (RNAseqV2 level3) from high‐grade serous ovarian cancer were downloaded from the TCGA Research Network Data Portal (http://tcga_data.nci.nih.gov/tcga). The clinical data associated with these samples was downloaded from cBioPortal (http://www.cbioportal.org/data_sets.jsp). The sensitive samples (55 samples) were defined as being disease‐free for over 24 months while the resistant samples (95 samples) were defined as being disease‐free for less than 12 months. From the data, normalized expression was extracted for the gene VCP and plotted in R with the R package ‘caroline’ using the plot function ‘volins’ (version 0.7.6. https://CRAN.R‐project.org/package.org/package=caroline). A bin‐width of h = 1650 for the kernel density estimation was used for the plots. Welch's two sample t‐test was used to calculate the p‐value.
2.10. Cell cycle analysis
1 × 106 cells were plated in a 10‐cm cell culture dishes with the appropriate culture media and were incubated overnight. Next day, cells were incubated with culture media containing the appropriate compounds for 18 h. Cells were then trypsinized, and cell pellets were collected in a 15 ml centrifuge tube. Pellets were washed with ice‐cold PBS pH 7.0 twice. Cells were then fixed using equal volumes of ice‐cold PBS and 95% ice‐cold ethanol for 1 h. Fixed cells were washed with cold PBS twice. Finally, the pellets were resuspended in 0.1% sodium citrate solution containing RNase A and PI for 30 min at 37 °C. Cell cycle profile for each treatment condition was then analyzed using flow cytometer based on previously established protocols.
2.11. Annexin V apoptotic assay
0.3 × 106 cells/well were plated in a 6‐well plate using the appropriate culture media and were allowed to incubate overnight. Cells were then incubated with culture media containing DBeQ or ML240 for 6 h. After 6 h, cells were trypsinized and centrifuged at 1500 rpm for 5 min. Cell pellets were resuspended in Annexin V Binding Buffer (BioLegend, 640914). Suspended cells underwent the assay according to the manufacturer's protocol.
2.12. Western blot and antibodies
Cells were collected at the end of treatments for Western blot analysis using a cell scraper. Proteins were extracted in 2× Laemmli Buffer (Bio‐Rad, 161‐0737) containing protease inhibitor (Roche, 05892970001) and phosphatase inhibitor (Fisher, 78440). Equal volumes of samples were loaded in SDS‐PAGE and electroblotted onto PVDF membranes. Electroblotted membranes were incubated in 5% nonfat dry milk in TBS‐Tween for 1 h at room temperature and were incubated with appropriate primary antibodies prepared in 3% Bovine Serum Albumin in TBS‐Tween overnight at 4 °C. Membranes were then washed with TBS‐Tween and incubated with appropriate horseradish peroxidase (HRP)‐conjugated secondary antibodies for 1 h at room temperature followed by the visualization of bands using the Super Signal West Femto (Pierce, 34096).
Primary antibodies used for the Western blot analysis included IRE1α (cst‐3294S), phospho‐IRE1α (Novus, nb100‐2323), CHOP (cst‐5554S), ATF4 (cst‐11815S), phospho‐eIF2α (Abcam, ab32157), VCP (sc‐20799), PARP (cst‐9542), Caspase 3 (cst‐9665P), Cleaved Caspase 3 (cst‐9661S), Caspase 9 (cst‐9508P), Caspase 8 (sc‐81656), puromycin (EMD Millipore, MABE343), β‐actin (Sigma–Aldrich, A1978), GADD34 (sc‐8327), Bip/Grp78 (cst‐3177S), p21 (sc‐397), p27 (cst‐3686S), Cyclin D1 (sc‐20044), and Cyclin E (sc‐247). Secondary antibodies included HPR‐linked anti‐rabbit IgG (cst‐7074) and HPR‐linked anti‐mouse IgG (cst‐7076). Densitometry analyses were performed using ImageJ, and graphs were plotted using GraphPad Prism.
2.13. Kaplan–Meier plots
KMplot was used to analyze the clinical association between VCP expression and outcome of patients with ovarian cancer (Gyorffy et al., 2012). The Affymetrix ID 208648_at corresponding to VCP was used in the analysis. The latest version of dataset (2015 version) was used in the analysis for both progression‐free survival (PFS) and overall survival (OS). For the PFS, a total of 1306 was included in the analysis. The median cutoff value is 778 (range 9–4128). For the OS, a total of 1582 was included in the analysis. The median cutoff value is 823 (range 9–6440). The data set includes 1144 serous and 36 endometrioid subtypes of epithelial ovarian cancer. The remaining cases are not annotated.
3. Results
3.1. DBeQ and ML240 are effective in reducing cell viability in ovarian cancer cells
We used Sulforhodamine B (SRB) assay to determine the extent of cytotoxicity induced by DBeQ and ML240 in four ovarian cancer cell lines; namely OVCAR10, OVCAR8, SKOV3, and OVCAR5. Our results indicate that the GI50 for DBeQ ranges from 2.5 ± 0.62 μM to 4.5 ± 1.3 μM; while for ML240 it ranges from 0.97 ± 0.27 μM to 2.03 ± 0.53 μM (Figures 1A–C and S1A–D) in ovarian cancer cell lines. These results suggest that VCP inhibitors are cytotoxic to ovarian cancer cells. ML240 displays higher levels of cytotoxicity compared to DBeQ in all the tested cell lines.
Figure 1.
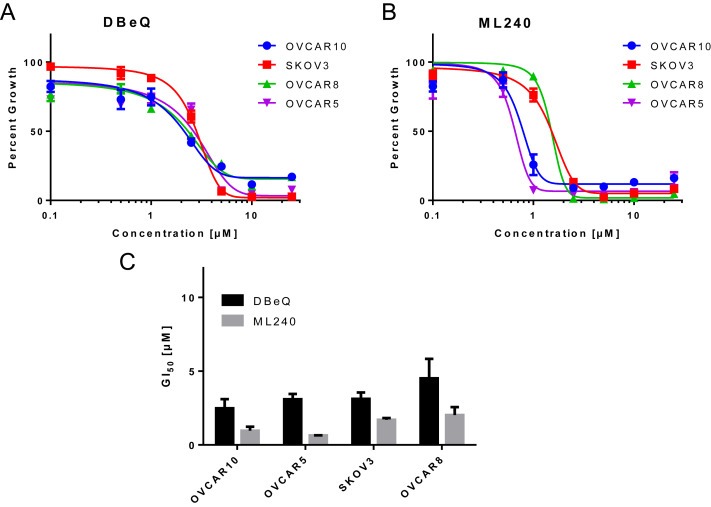
DBeQ and ML240 show increased cytotoxicity towards ovarian cancer cells . (A and B) OVCAR10, SKOV3, OVCAR8 and OVCAR5 cells were treated with vehicle (DMSO) as well as different concentrations of DBeQ and ML240 ranging up to 25 μM for 72 h in 96‐well plates. Dose response curves were generated using GraphPad Prism using four parameters nonlinear regression (curve fit). Curves were constrained on top (100%) and bottom (>0%). (C) The GI50 for DBeQ and ML240 were calculated as Mean ± SEM from 3 individual experiments for all ovarian cancer cells. Each experiment was run in triplicate for all cell lines.
3.2. VCP inhibitors effectively interact and inhibit cellular VCP
We next compared the mean GI50 from several ovarian cancer cell lines with relative VCP protein expression. A moderate positive correlation was observed between the mean GI50 and relative protein expression with both DBeQ (r = 0.59) and ML240 (r = 0.45) (Figure S2A, S2C and S2D). These data are consistent with previous report indicating a positive correlation between VCP expression and GI50 for CB‐5083 (Anderson et al., 2015). Since VCP protein expression correlated with GI50 for VCP inhibitors in ovarian cancer cell lines, we tested the extent to which VCP knockdown enhances the effect of VCP inhibitors. Our results indicate 50% (siVCP‐1), 80% (siVCP‐2), and 85% (siVCP‐1 + 2) knockdown efficiency in three independent transient transfections (Figures 2A and S2B). Following the transient knockdown, cells were plated for cell viability studies. Our results show a significant reduction in GI50 for DBeQ and ML240 following VCP knockdown (Figure 2B–D). Moreover, VCP inhibitors display greater reduction of GI50 with siVCP‐2 and siVCP‐1 + 2, consistent with higher VCP knockdown efficiencies (Figure 2A–C). These results suggest that VCP is a target of DBeQ and ML240. Furthermore, we examined cell growth following transient knockdown of VCP. Our results show a significant reduction in cell growth with siVCP‐2 and siVCP‐1 + 2 (Figure 2E). These results provided the first functional validation of the previous studies identifying VCP as an essential gene in ovarian cancer survival (Cheung et al., 2011; Etemadmoghadam et al., 2013).
Figure 2.
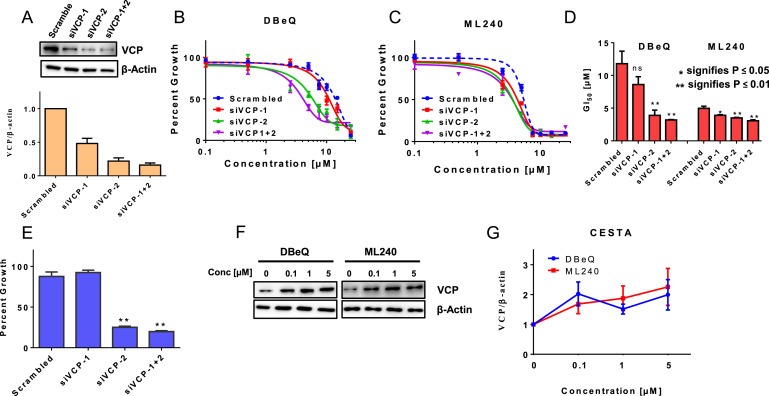
DBeQ and ML240 bind to cellular VCP/p97 . (A) SKOV3 cells were transfected with two different siRNAs targeting VCP (siVCP‐1 and siVCP‐2), and one non‐targeting scrambled small interfering RNA. Whole cell lysates following transient transfection were used to visualize the knockdown efficiencies by Western blot analysis. Densitometry analysis was performed using ImageJ to observe the relative VCP expression following three independent siRNA transfections. SKOV3 cells transfected with scramble or VCP siRNAs were treated with different concentrations of (B) DBeQ and (C) ML240 up to 25 μM for 6 h in 96‐well format and were allowed to recover under normal media for 6 additional days. Cell viability was measured using SRB Assay. Dose response curves were generated using GraphPad Prism using four parameters nonlinear regression (curve fit). Curves were constrained on top (100%) and bottom (>0%). (D) Values represent Mean ± SEM GI50 measurements following VCP inhibition based on three individual experiments. p‐Values were calculated based on the student's t‐test. (E) Values represent Percent growth ± SEM from three independent experiments following transient knockdown of VCP followed by 6 additional days of recovery in normal media. (F) SKOV3 cells were treated with DBeQ or ML240 at concentrations between 0.1 and 5 μM for 2 h. Cells were subjected to heat‐shock treatment at 57 °C. Soluble fractions were then subjected to Western blot analysis. (G) Values represent the relative thermal stability of VCP at 57 °C at different concentrations of DBeQ or ML240 in three replicates.
Cellular Thermal Shift Assay (CETSA) has been used as an effective tool to assess the binding of compounds to intended cellular targets. The assay analyzes the changes in the in vitro thermal stability of candidate cellular proteins by compounds of interest (Jafari et al., 2014). Initially, we used different temperatures following the incubation of DBeQ and ML240 for heat treatment and determined that 57 °C destabilized VCP (data not shown). Next, we evaluated the thermal stability of VCP with different concentrations of DBeQ and ML240 at 57 °C. Here, we show a shift in the thermal stability of VCP at 57 °C following 2‐hours incubation of cells with DBeQ and ML240 at concentrations ranging between 0.1 μM and 5 μM, indicating the target engagement (Figure 2F and G).
3.3. VCP inhibitors cause G1 cell cycle arrest
Given the well‐established role of VCP in cell cycle (Cao et al., 2003; Zhang et al., 1999), we performed cell cycle analysis to observe any changes in cell cycle distribution following the treatment with VCP inhibitors. We observed an increase in G1 and a decrease in S and G2/M phases with 5 μM DBeQ as well as an increase sub G0 phase with 10 μM DBeQ (Figure 3A). Similarly, we saw a reduction in S phase and an increase in sub G0 phase with ML240 treatment (Figure 3B). Furthermore, CB‐5083 treatment increased G1 and reduced S phase (Figure 3C). These results suggest that VCP inhibitors cause G1 cell cycle arrest followed by cell death. Next, we analyzed the expression of several cell cycle regulators that are substrates of the ubiquitin proteasome system (UPS) following the treatment with VCP inhibitors. We observed variable accumulation of p21, p27, Cyclin D1, and Cyclin E with DBeQ, ML240, and CB‐5083 treatments (Figure 3D). Overall, our results indicate that inhibition of VCP results in increased accumulation of cell cycle regulators that are substrates of UPS.
Figure 3.
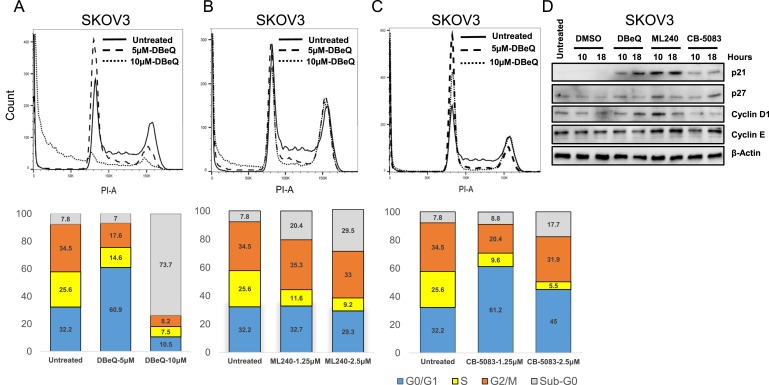
Treatment with VCP inhibitors causes G1 arrest. (A–C) PI staining was performed on SKOV3 cells treated with DBeQ [5 μM and 10 μM], ML240 [1.25 μM and 2.5 μM] and CB‐5083 [1.25 μM and 2.5 μM] for 18 h. Bar graphs represent cells in each stage of the cell cycle following VCP inhibitor treatment for 18 h (D) SKOV3 cells were incubated with DMSO (vehicle), 5 μM DBeQ, 1.25 μM ML240 or 1.25 μM CB‐5083 for 10 or 18 h. Whole cell lysates were analyzed using Western blot.
3.4. VCP inhibitors induce cell death via the apoptotic pathway
Previous studies have shown that VCP inhibitors induce the activation of caspases and apoptosis in non‐ovarian cancer cell lines (Anderson et al., 2015, 2011, 2013, 2013, 2015). We, therefore, analyzed the extent of apoptosis following the treatment with DBeQ or ML240 in ovarian cancer cells using Annexin V staining. We incubated SKOV3 cells with DBeQ [10 μM] or ML240 [5 μM] for 6 h followed by Annexin V and DAPI staining. Our results show a significant increase in Annexin V and DAPI positive cells following DBeQ and ML240 treatment (Figure 4A). Activation of procaspases is one the hallmarks of caspase‐mediated apoptotic cell death. Here, we used immunoblotting to determine PARP cleavage and activation of initiator caspases as well as effector caspases. Our results indicate the PARP cleavage at 6‐hour time point with DBeQ [10 μM] and ML240 [5 μM] treatment, which is consistent with the Annexin V‐DAPI staining (Figure 4B). We also observed the cleavage of Caspase 9 and Caspase 8 following the treatment with VCP inhibitors. Caspase 9 activation was observed at a much earlier time point (6 h) while Caspase 8 activation was observed only at 24 h following DBeQ and ML240 treatment (Figure 4B).
Figure 4.
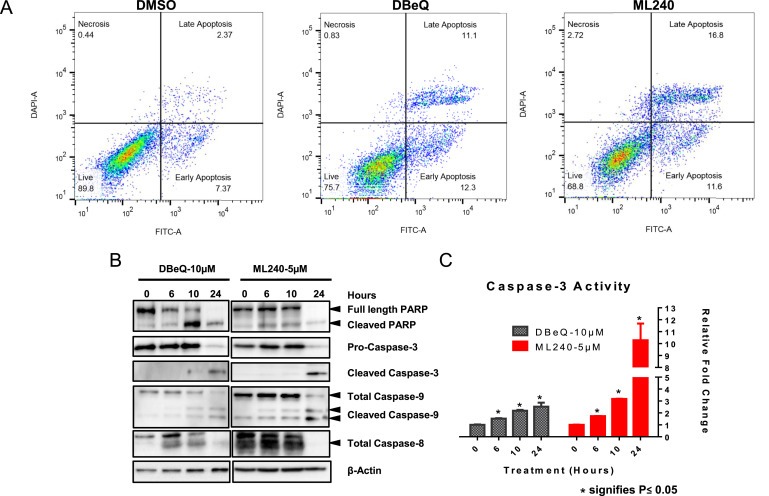
Incubation with DBeQ and ML240 induces caspase‐mediated apoptosis . (A) Annexin‐V/DAPI staining was performed following 6 h of DMSO (vehicle), 10 μM DBeQ or 5 μM ML240 treatment using SKOV3 cells. (B) OVCAR10 cells were treated with 10 μM DBeQ or 5 μM ML240 for 0, 6, 10 and 24 h. Whole cell lysates were analyzed using Western blot. (C) OVCAR10 cells were treated with 10 μM DBeQ or 5 μM ML240 for 0, 6, 10 and 24 h. The values indicate fluorescence taken from three separate samples. The caspase activity is represented as fold change of untreated group and calculated as Mean ± SEM. p‐Values were calculated based on the student's t‐test.
The activity of Caspase 3 was determined by the Caspase 3 activity assay. Our result indicates a significant increase in Caspase 3 activity with DBeQ [10 μM] and ML240 [5 μM] at 6‐hour time point (Figure 4C). We observed a 2.5‐fold increase in Caspase 3 activity at 24 h with DBeQ and a 10‐fold increase with ML240 (Figure 4C). Overall, our data is consistent with previous studies indicating caspase‐mediated apoptosis by various VCP inhibitors (Anderson et al., 2015, 2011, 2013, 2013). Furthermore, we show that ML240 treatment results in a similar mode of cell death with higher Caspase 3 activity compared to DBeQ.
3.5. VCP inhibitors trigger the unfolded protein response (UPR)
Inhibition of VCP can impede the extraction of unfolded proteins from the ER and contribute to the unfolded protein response (UPR) (Sano and Reed, 2013). UPR is executed via changes in three transmembrane ER proteins: IRE1α, PERK, and ATF6α (Oslowski and Urano, 2011). Previous studies have shown activation of the PERK arm with DBeQ treatment (Auner et al., 2013; Chou et al., 2011). Recently, CB‐5083 treatment has been shown to activate all three arms of UPR (Anderson et al., 2015). Similarly, other VCP inhibitors, such as NMS‐873, also induce UPR in various cancer cell lines (Magnaghi et al., 2013). We decided to perform a detailed analysis comparing the effects of VCP inhibitors in the UPR pathway in ovarian cancer cells. SKOV3 cells were treated with DBeQ [10 μM], ML240 [5 μM] and CB‐5083 [2.5 μM] up to 48 h. Tunicamycin [2 μM], an inhibitor of N‐acetylglucosamine transferase, was used as a positive control to observe the activation of UPR. Our results indicate an increase in phospho‐IRE1α by 3 h with VCP inhibitors marking the activation of IRE1α branch (Figure 5A–C). We also observed an increase in the expression of CHOP and ATF4 between 3 and 12 h marking the activation of PERK arm (Figure 5A and C). We were unable to determine the status of the ATF6α. This lack of immunoreactivity may be due to low basal expression of ATF6α in SKOV3 cells. Tunicamycin treatment showed a similar increase in the expression of phospho‐IRE1α, CHOP, and ATF4 (Figure 5D). Phosphorylation of PERK upon ER stress results in the phosphorylation of eIF2α at serine 51, which has been shown to decrease cap‐dependent protein translation (Harding et al., 1999). Decrease in cap‐dependent protein translation reduces the protein load and allow cells to resolve ER stress (Rutkowski and Kaufman, 2004). To observe changes in cap‐dependent protein translation, we treated SKOV3 cells with different concentrations of DBeQ and ML240 up to 10 μM for 5.5 h and pulsed the cells with 1 μM puromycin for 30 min. Incorporated puromycin was visualized using monoclonal puromycin antibody. Results show a marked decrease in puromycin incorporation following the treatment with VCP inhibitors (Figure 5E). Collectively, these results suggest that VCP inhibitors trigger the unfolded protein response through IRE1α and PERK. Similarly, increased PERK phosphorylation and polyubiquitination of proteins following CB‐5083 treatment have been reported (Anderson et al., 2015). In line with CB‐5083, treatment with DBeQ and ML240 showed enhanced accumulation of poly‐ubiquitinated proteins (Figure S3).
Figure 5.
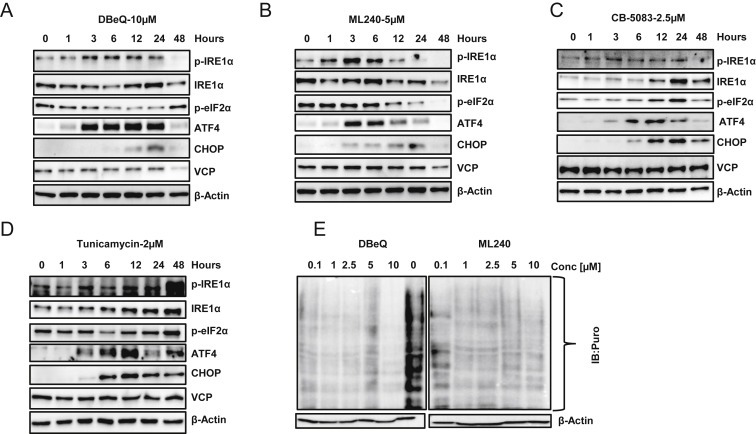
Incubation with VCP inhibitors results in activation of Unfolded Protein Response (UPR) . SKOV3 cells were incubated with (A) 10 μM DBeQ, (B) 5 μM ML240, (C) 2.5 μM CB‐5083 and (D) 2 μM Tunicamycin (positive control) for 0, 1, 3, 6, 12, 24 and 48 h. Whole cell lysates were subjected to Western blot analysis and probed for UPR‐related proteins. (E) SKOV3 cells were treated with different concentrations of DBeQ or ML240 for 6 h and pulsed with puromycin for 30 min. Whole cell lysates were analyzed for incorporated puromycin by Western blot. The results for both DBeQ and ML240 were obtained from the same blot that was later spliced together to remove the molecular weight marker. DMSO (0 μM) sample served as the control for both drugs.
3.6. VCP inhibitors synergize with Salubrinal
ER stress response is an adaptive, pro‐survival mechanism. However, prolonged ER stress activates pro‐apoptotic genes that contribute to cell death. ER stress induces CHOP expression through PERK‐mediated pathway (Harding et al., 2000), and CHOP induces the expression of pro‐apoptotic gene (PUMA and DR5) and decreases the expression of anti‐apoptotic gene (BCL2) (Cazanave et al., 2010; Ghosh et al., 2012; McCullough et al., 2001; Yamaguchi and Wang, 2004). Therefore, we reasoned that VCP inhibitors might act synergistically with compounds that induce CHOP expression. Salubrinal, a selective inhibitor of eIF2α dephosphorylation, was first identified as a compound that reduced ER stress‐induced apoptosis (Boyce et al., 2005). Since then, subsequent studies have shown that Salubrinal enhances cell death via the induction of CHOP expression (Koizumi et al., 2012; Teng et al., 2014). To assess the potential synergistic effect between VCP inhibitors and Salubrinal, we investigated four high‐grade serous ovarian cancer cell types namely; OVCAR10, OVCAR5, OVCAR8 and OVSAHO and two clear cell serous ovarian cancer cell types namely; RMG1 and SKOV3. These cells were treated with different concentrations of VCP inhibitors (DBeQ, ML240 and CB‐5083) and Salubrinal for 72 h. In this study, we used varying concentrations below the GI50 for DBeQ, ML240 and Salubrinal. For CB‐5083, we used different concentrations all below the pharmacologically achievable dose (Anderson et al., 2015). Our results show synergistic effect between DBeQ and Salubrinal as well as CB‐5083 and Salubrinal in all the tested ovarian cancer cell lines (Figures 6A and B, S4). Similar effects were observed with ML240 and Salubrinal combinations performed on OVSAHO and OVCAR8 cell lines (Figure S5A).
Figure 6.
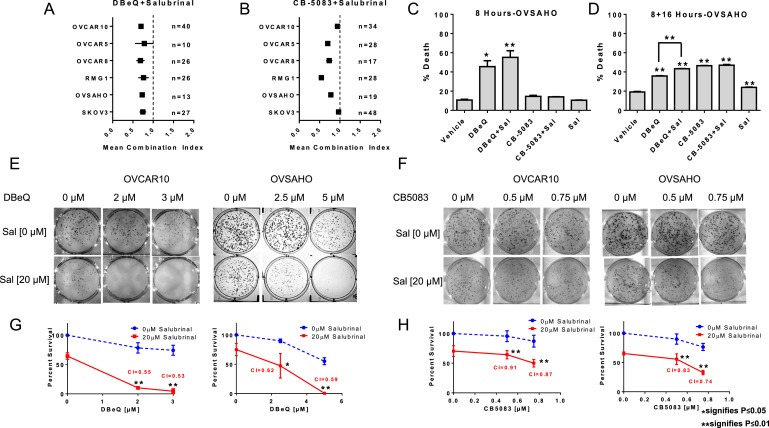
VCP inhibitors synergize with Salubrinal . Several high serous ovarian cancer cell types namely; OVCAR10, OVCAR5, OVSAHO, and OVCAR8 as well as clear cell ovarian cancer cell types; SKOV3 and RMG1 were treated with different combinations of (A) DBeQ plus Salubrinal and (B) CB‐5083 plus Salubrinal for 72 h in 96‐well format. The error bars in the figures represent 95% confidence interval of the mean combination index (CI) from 3 individual experiments with several combinations of drugs that resulted in growth inhibition between 20% and 80%. N represents the total number of CIs determined from 16 drug combinations in duplicates that produced 20–80% effect from three independent experiments. CI values less than 1 indicate synergy while values equal to or more than 1 represent additive and antagonistic effect respectively. OVSAHO cells were incubated with DBeQ [15 μM], CB‐5083 [5 μM], Salubrinal [20 μM] and combinations for (C) 8 h and (D) 8 h with drug followed by 16 h in regular media. Cells were then subjected to Annexin V/PI staining and percent cell death was calculated by adding the cells in Q1, Q2 and Q4 quadrants following flow cytometry. Percent death ± SEM were plotted using GraphPad Prism and p‐values were calculated using the student's t‐test. Representative clonogenic assay performed in OVCAR10 and OVSAHO cell lines with (E) vehicle (DMSO), DBeQ, Salubrinal, a combination of DBeQ plus Salubrinal and (F) vehicle (DMSO), CB‐5083, Salubrinal, a combination of CB‐5083 plus Salubrinal treated for 48 h in 6‐well plates. (G–H) Percent survival based on the number of colonies formed from 3 individual experiments. Combination indexes (CIs) were calculated based number of colonies formed from 3 individual experiments. p‐Values were calculated using the student's t‐test.
Next, we analyzed the short‐term effect of VCP inhibitors in combination with Salubrinal. We treated OVSAHO cells with vehicle (DMSO), DBeQ, CB‐5083 as well as combinations DBeQ + Salubrinal and CB‐5083 + Salubrinal. After treatments, cell death was analyzed using Annexin V/PI assay. With 8 h of treatment, we observed a mean cell death of 45.56% with DBeQ and 55.23% with DBeQ + Salubrinal (Figure 6C). At the same time point, we only observed a modest increase in cell death with CB‐5083 treatment. Hence, we decided to evaluate cell death with 8 h of treatment followed by 16 h in regular media. At 8 + 16 h, our results indicate only a slight increase in cell death following Salubrinal treatment alone, while DBeQ, CB‐5083 as well as their combinations with Salubrinal all resulted in a significant increase in cell death (Figure 6D). Together, our results indicate increased cytotoxicity with long‐term (72 h) as well as short‐term (8 h and 8 + 16 h) treatment following the combination of VCP inhibitors and Salubrinal.
To further validate the results from the synergistic and cell death studies, we performed clonogenic assays in two high‐grade serous ovarian cancer cell lines: OVCAR10 and OVSAHO. Clonogenic assays show a significant reduction in colony formation with the combination of DBeQ and Salubrinal as well as CB‐5083 and Salubrinal further strengthening the combinational effect achieved with VCP inhibitors and Salubrinal. Furthermore, siRNA‐mediated knockdown of GADD34 in OVSAHO cells resulted in enhanced Caspase 3 activation. We observed a significant increase in Caspase 3 activity only in GADD34 knockdown cells following DBeQ treatment (Figure S5B and S5C). Overall, our results provide an in vitro proof‐of‐principle that inhibiting GADD34 in ovarian cancer could be one of the therapeutic strategies when developing combination therapies for VCP inhibitors.
3.7. VCP inhibitors and Salubrinal combination result in enhanced integrated stress response (ISR)
To further dissect the mechanism behind the synergistic effect observed with DBeQ and Salubrinal, we treated OVCAR10 cells with sub‐lethal dose of DBeQ [3 μM], sub‐lethal dose of Salubrinal [20 μM] and the combination of DBeQ and Salubrinal up to 48 h. Increased expression of ATF4 and CHOP was observed within 3–6 h in all three treatment conditions (Figure 6A–C). We observed decreased Grp78 expression by 24 h with DBeQ and Salubrinal alone which indicate a decrease in integrated stress response (ISR) (Figure 7A and B). However, an enhanced expression of Grp78 was observed with the combination at 24 h indicating an increase in ISR (Figure 7C). Puromycin labeling which is indicative of nascent protein translation showed a decrease in overall protein translation with DBeQ and Salubrinal combination compared to DBeQ and Salubrinal alone, further confirming enhanced IRS with the combination (Figure S6A). We also observed activation of Caspase 3 (cleaved form) at 48 h with DBeQ and Salubrinal combination suggesting that increased ISR signal triggered apoptosis (Figure 7C). Similar results were observed in OVSAHO cells (data not shown). Similarly, when lysates from different treatment conditions were analyzed, we saw enhanced CHOP expression with DBeQ and Salubrinal combination (Figure S6B). Taken together, these data suggest that GADD34 inhibition enhances the integrated stress response (ISR) induced by VCP inhibitors resulting in enhanced CHOP expression that triggers apoptosis in ovarian cancer cells.
Figure 7.
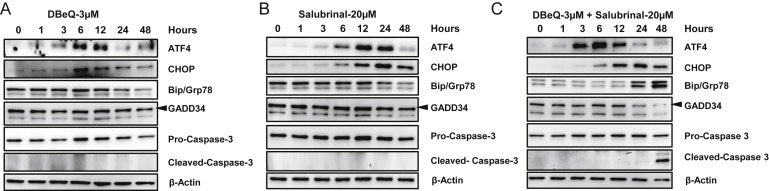
Enhanced ISR following DBeQ and Salubrinal combination results in caspase‐mediated apoptosis. OVCAR10 cells were incubated with (A) sub‐lethal dose of DBeQ [3 μM], (B) sub‐lethal dose of Salubrinal [20 μM], (C) combination of DBeQ [3 μM] and Salubrinal [20 μM] for 1, 3, 6, 12, 24 and 48 h. Whole cell lysates were subjected to Western blot analysis and probed with the above antibodies.
3.8. Lower VCP expression in ovarian cancer is associated with poor outcome in patients with ovarian cancer
Since elevated expression of VCP is associated with poor prognosis in various cancer types (Tsujimoto et al., 2004, 2003, 2004, 2003), we determined the association between VCP expression and clinical outcome in patients with ovarian cancer. Surprisingly, Kaplan–Meier analyses of 1648 ovarian tumor samples, provided by the online KM plotter (Gyorffy et al., 2012), indicate that lower expression of VCP is associated with shorter progression‐free survival (PFS) and overall survival (OS) in ovarian cancer (Figure 8A and B). To further understand this puzzling clinical outcome association, we analyzed the ovarian carcinoma RNA sequencing datasets from the Cancer Genome Atlas (Cancer Genome Atlas Research Network, 2011). We compared VCP gene expression in tumor samples collected from high‐grade serous ovarian cancer patients that were either sensitive or resistant to the platinum‐based chemotherapy. Our results show a significant reduction of VCP expression in tumor samples that were collected from patients with resistance to chemotherapy (p‐value = 0.01532) (Figure 8C). Taken together, these results indicate that ovarian carcinomas with lower expression of VCP are associated with poor response to platinum‐based chemotherapy, poor progression‐free survival, and poor overall survival. Although these results are in contrast to previously reported studies in other cancer types, the result is highly relevant to ovarian cancer because our results point to the clinical potential of using VCP inhibitors in the setting of platinum resistance given that these tumors have lower levels of VCP expression and are likely more sensitive to VCP inhibitors.
Figure 8.
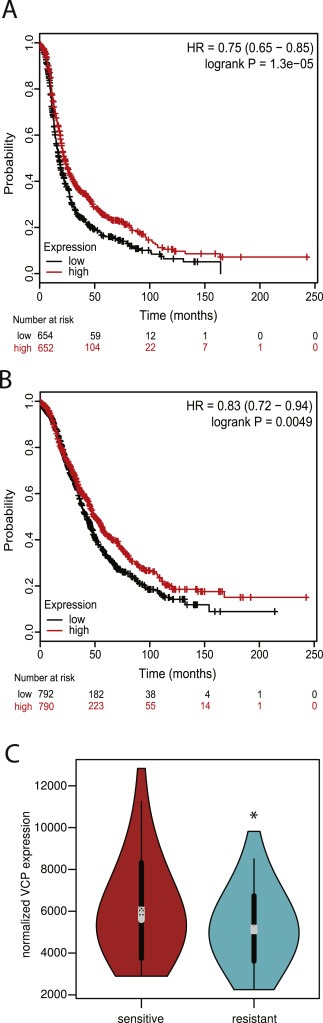
Association between VCP expression and clinical outcome. (A) Lower expression of VCP in ovarian carcinomas is associated with early disease recurrence (progression‐free survival). (B) Lower expression of VCP in ovarian carcinomas is associated with poor overall survival. (C) RNA sequencing dataset representing high‐grade serous ovarian cancer patient samples that are sensitive (n = 55) and resistant (n = 95) to established chemotherapy was downloaded from the TCGA Research Network Data Portal. VCP gene expression between the two datasets was compared using violin plot. p‐Value = 0.01532, Welch's two sample t‐test.
4. Discussion
In this study, we provided evidence that ovarian cancer cells are sensitive to VCP inhibitors. In particular, cells with lower levels of cellular VCP are more sensitive to the inhibitors. These results are consistent with prior studies indicating a strong inverse correlation between VCP expression and sensitivity to VCP inhibitors (Anderson et al., 2015). In addition, we provided evidence that VCP inhibitors interact with VCP and confer thermal stability to VCP. Collectively, these results demonstrate drug–target interactions and further support the view that lower levels of cellular VCP require a lower amount of compounds to inhibit the activity, and that on‐target inhibition of VCP by these agents contributes to cytotoxicity. These results are potentially significant given that VCP expression is significantly lower in high‐grade serous carcinomas collected from patients with chemotherapy‐resistant ovarian cancer.
To define the molecular mechanisms associated with cytotoxicity induced by VCP inhibitors, we determined the proportion of cells in different phases of cell cycle following drug treatment. We observed that VCP inhibitors induced G1 arrest, as evident by a decrease in S phase proportion following 18 h of drug treatment. Subsequent to G1 arrest, we observed an increase in sub‐G1 population and a concomitant decrease in G1 population, suggesting that these compounds induce cell death in the G1 population. Similar results were reported by Magnaghi et al. with VCP inhibitor NMS‐873 (Magnaghi et al., 2013). VCP/p97 has been shown to be involved in ubiquitin‐mediated degradation of cell cycle regulator Cyclin E (Dai and Li, 2001). We showed that treatment with VCP inhibitors resulted in the accumulation of Cyclin E, Cyclin D1, p21, and p27. Since we used a p53 null cell line, the accumulation of p21 was independent of p53. We observed activation of Caspase 8 and Caspase 9, and increased the activity of Caspase 3, indicating that both extrinsic and intrinsic apoptotic pathways are activated by VCP inhibitors. Similar results were reported by various groups using the same or structurally different VCP inhibitors (Anderson et al., 2015; Chou et al., 2011; Magnaghi et al., 2013). These results are also consistent with a recent report indicating that VCP inhibitor, CB‐5083, induces cell death via DR5‐and CHOP‐dependent pathway (Anderson et al., 2015).
ML240 has been shown to bind selectively to the D2 domain and inhibit the D2‐ATPase activity of VCP; while DBeQ inhibits the activity in both D1 and D2 domains (Chou et al., 2014). Similarly, CB‐5083 which was derived from the scaffold of ML240 is a specific inhibitor of D2 domain of VCP (Zhou et al., 2015). Our results indicate that inhibition of D2‐ATPase is sufficient to cause the cytotoxic effect in ovarian cancer cells. In addition, specific inhibition of D2‐ATPase domain of VCP with ML240 may be attributed to the lower GI50 values across all the tested cell lines.
In addition, we provided evidence that VCP inhibitors can be combined with agents that modify ER stress to enhance cytotoxic activities of VCP inhibitors (Figure 9). Our results indicate that VCP inhibitors activate ER stress pathway and that cytotoxicity of VCP inhibitors can be enhanced by Salubrinal, a GADD34 inhibitor that prolongs the phosphorylation of eIF2α. Although the transient phosphorylation of eIF2α is a critical adaptive response to ER stress, resulting in a decrease in cap‐dependent translation to lower protein load, sustained phosphorylation of eIF2α, in the presence of upstream ER stress, could induce CHOP and CHOP‐dependent apoptosis (Marciniak et al., 2004). Consistent with this hypothesis, we observed drug synergies between VCP inhibitors and Salubrinal. It is important to note that recent studies by Parzych et al. reported that Guanabenz, a GADD34 inhibitor, decreases cell death induced by DBeQ (Parzych et al., 2015). Parzych et al. suggested that inadequate regulation of protein translation contributes to cell death by DBeQ and NMS‐873. This conclusion is based on the fact that Guanabenz and cycloheximide attenuated protein synthesis and cell death induced by VCP inhibitors. These authors also pointed out that cycloheximide reduces the expression of CHOP. Therefore, it is possible that inhibition of CHOP expression rather than attenuation of global protein translation by cycloheximide contributes to the attenuation of cell death by VCP inhibitors. Our assertion is supported by Anderson et al. who reported that knockdown of CHOP was sufficient to blunt cell death induced by VCP inhibitor CB‐5083 (Anderson et al., 2015). Furthermore, caution must be exercised not to extrapolate transient cytoprotection conferred by agents that inhibit protein synthesis to the long‐term cytoprotection by these agents against VCP inhibitors. This caution is prompted by our observation that GADD34 inhibitor Salubrinal enhances cell death induced by VCP inhibitors in long‐term cell viability assays. Furthermore, supplementary data provided by Magnaghi et al. indicates that actinomycin D, which attenuates protein synthesis through transcriptional inhibition, enhances the cytotoxic effect of VCP knockdown by two different siRNAs (Magnaghi et al., 2013). Therefore, a complete block on protein synthesis may not confer the long‐term cytoprotection against VCP inhibition.
Figure 9.
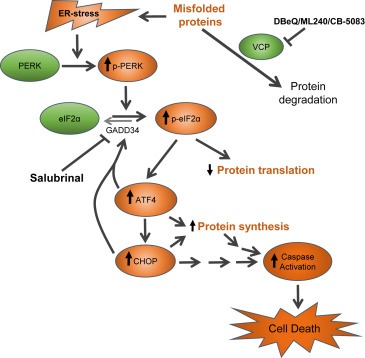
A proposed mechanism of action by the combination of VCP inhibitors and ER stress‐prolonging agent Salubrinal . VCP facilitates the removal of unfolded proteins. When VCP is inhibited by DBeQ, ML240, and CB‐5083, increased accumulation of unfolded proteins induces ER stress and activates PERK pathway. PERK phosphorylates eIF2α and attenuates cap‐dependent translation, which lower protein load to restore protein homeostasis. ER stress also activates the sequential induction of ATF4 and CHOP transcription factors which together transactivate GADD34 and increase protein synthesis. The presence of sustained ER stress (either by continuous exposure to ER stress activating agents) or unresolved ER stress (due to inhibition of GADD34 by Salubrinal) can enhance or prolong the expression of CHOP and contribute to caspase‐mediated apoptosis. ER stress and unfolded protein response induced by VCP inhibitors are shown in orange color.
It is likely that GADD34 inhibitors will contribute to stronger attenuation of cap‐dependent translation by maintaining the phosphorylation of eIF2α, given that without these inhibitors the downstream transcription factors ATF4 and CHOP will transactivate GADD34 to enhance dephosphorylation of eIF2α and to restart protein translation (Han et al., 2013). Therefore, the transient effect of GADD34 inhibitors is a stronger attenuation of cap‐dependent translation. However, CHOP expression is induced under these conditions, and CHOP has been shown to contribute to cell death induced by VCP inhibitor CB‐5083 (Anderson et al., 2015). Therefore, although stronger attenuation of protein translation produced by GADD34 inhibitors might afford transient cytoprotection by reducing the protein load, unabated CHOP expression induced by GADD34 inhibitors may enhance cell death by VCP inhibitors under the prolonged exposure.
In our studies, we found that Salubrinal, another GADD34 inhibitor, enhances the effect of DBeQ and CB‐5083 in several ovarian cancer cell lines. Unlike Parzych et al. studies which determined the cytotoxic effect at 8 h of treatment with one combined dose, our studies determined both the short‐term and the long‐term effect of the several combined doses for VCP inhibitors and Salubrinal. We observed synergistic activities of the combined drugs across several concentrations in both short‐term and long‐term assays in several ovarian cancer cell lines. Therefore, the differences in the outcome of these two studies may be attributable to the differences in cell models, the exposure time, and the differences in off‐target effects of Salubrinal and guanabenz.
Prolonged ER stress and subsequent cell death is an important mechanism of action of drugs that target the ubiquitin proteasome system (UPS). Bortezomib was the first compound successful in targeting the UPS in clinical settings; however, it has not been very effective against solid tumors (Huang et al., 2014). The failure to elicit similar effect has been attributed to the lack of prolonged suppression of proteasome in vivo in solid tumor (Deshaies, 2014). VCP inhibitors provide an exciting avenue for the development of novel chemotherapy in all tumors with enhanced unfolded protein load. In addition, identifying compounds that prolong the ER stress and enhance the cytotoxic effect of VCP inhibitors may be useful as a combination treatment strategy in solid tumors. Our results provide a proof‐of‐principle that VCP inhibitors and the ER stress pathways could be targeted in combination with the ER stress‐prolonging agent Salubrinal for enhanced cytotoxicity. Moreover, transient knockdown of GADD34 by RNAi also enhances the Caspase 3 activity induced by VCP inhibitor DBeQ (Figure S5C). Collectively, these results support the role of targeting GADD34 to enhance the cytotoxic effect of VCP inhibitors.
Recently, several groups have identified the pathway involved in protein homeostasis as a target of vulnerability in ovarian cancer. For example, Cheung et al. used 102 cancer cell lines and performed genome‐scale synthetic lethal screen with short hairpin RNAs (shRNAs) (Cheung et al., 2011). In this study, VCP was identified as one of the 22 putative essential genes in ovarian cancer cells. In addition, in their follow‐up studies, Hahn and Bowtell groups identified VCP as one of the essential genes in Cyclin E1‐overexpressing, cisplatin‐resistant ovarian cancer cells (Etemadmoghadam et al., 2013). Finally, Marcotte and Brown et al. identified several components of protein homeostasis, such as SNRPD‐1, PSMD1, PSMA1, PSMB2, RPS17, and EIF3B, as essential genes in several cancer cell lines (Marcotte et al., 2012). Therefore, results from our study indicating that VCP/p97, a component of protein homeostasis, can be targeted by DBeQ, ML240, and CB‐5083 and that these agents induce ER stress and apoptosis in ovarian cancer cells are highly relevant for advancing VCP inhibitors as potential therapeutics for ovarian cancer.
Given that pharmacodynamics and pharmacokinetics of CB‐5083, which is orally bioavailable, are already demonstrated, and that CB‐5083 is well‐tolerated in mice, it would be important to further develop this agent for additional preclinical and clinical studies. In addition, since VCP inhibitors induce ER stress, attenuate protein translation, increase ubiquitylation of proteins, it would be important to determine the similarity and differences in the mechanism of cell death induced by these agents compared to proteasome inhibitors, such as bortezomib. In particular, bortezomib and Hsp90 inhibitors also cause proteotoxic stress, and their mechanisms of action include lowering BRCA1 expression and enhancing sensitivity to PARP inhibitors and cisplatin (Johnson et al., 2013; Neri et al., 2011; Stecklein et al., 2012). Therefore, it would be important to determine the extent to which VCP inhibitors enhances sensitivity to conventional and emerging chemotherapeutics.
Disclosure of potential conflicts of interest
F.J.S. has a financial interest in Cleave Biosciences. Other authors have no financial conflict of interest to disclose.
Author's contributions
Conception and design: Jeremy Chien, Frank Schoenen, and Prabhakar Bastola.
Development of methodology: Jeremy Chien, Prabhakar Bastola.
Acquisition of data: Prabhakar Bastola.
Additional: Lisa Neums performed VCP expression and violin plots, and Jeremy Chien performed Kaplan–Meier analyses.
Analysis and interpretation of data: Jeremy Chien and Prabhakar Bastola.
Writing, review, and revision of the manuscript: Jeremy Chien, Prabhakar Bastola, Lisa Neums, and Frank Schoenen.
Administration, technical, or material support: Jeremy Chien.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
The study is funded by the University of Kansas Endowment Association, the University of Kansas Cancer Center Support Grant (P30‐CA168524), the Cancer Center Cancer Biology program, the American Cancer Society Research Scholar (125618‐RSG‐14‐067‐01‐TBE), and the Department of Defense Ovarian Cancer Research Program under award number (W81XWH‐10‐1‐0386). Views and opinions of, and endorsements by the author(s) do not reflect those of the US Army or the Department of Defense. We acknowledge the Flow Cytometry Core Laboratory, which is sponsored, in part, by the NIH/NIGMS COBRE grant P30 GM103326.
1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.09.005.
Bastola Prabhakar, Neums Lisa, Schoenen Frank J., Chien Jeremy, (2016), VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death and synergistically kill ovarian cancer cells in combination with Salubrinal, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.09.005.
References
- Alverez, C. , Arkin, M.R. , Bulfer, S.L. , Colombo, R. , Kovaliov, M. , LaPorte, M.G. , Lim, C. , Liang, M. , Moore, W.J. , Neitz, R.J. , Yan, Y. , Yue, Z. , Huryn, D.M. , Wipf, P. , 2015. Structure-activity study of bioisosteric trifluoromethyl and pentafluorosulfanyl indole inhibitors of the AAA ATPase p97. ACS Med. Chem. Lett.. 6, 1225–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alverez, C. , Bulfer, S.L. , Chakrasali, R. , Chimenti, M.S. , Deshaies, R.J. , Green, N. , Kelly, M. , LaPorte, M.G. , Lewis, T.S. , Liang, M. , Moore, W.J. , Neitz, R.J. , Peshkov, V.A. , Walters, M.A. , Zhang, F. , Arkin, M.R. , Wipf, P. , Huryn, D.M. , 2016. Allosteric indole amide inhibitors of p97: identification of a novel probe of the ubiquitin pathway. ACS Med. Chem. Lett.. 7, 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson, D.J. , Le Moigne, R. , Djakovic, S. , Kumar, B. , Rice, J. , Wong, S. , Wang, J. , Yao, B. , Valle, E. , Kiss von Soly, S. , Madriaga, A. , Soriano, F. , Menon, M.K. , Wu, Z.Y. , Kampmann, M. , Chen, Y. , Weissman, J.S. , Aftab, B.T. , Yakes, F.M. , Shawver, L. , Zhou, H.J. , Wustrow, D. , Rolfe, M. , 2015. Targeting the AAA ATPase p97 as an approach to treat cancer through disruption of protein homeostasis. Cancer Cell. 28, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auner, H.W. , Moody, A.M. , Ward, T.H. , Kraus, M. , Milan, E. , May, P. , Chaidos, A. , Driessen, C. , Cenci, S. , Dazzi, F. , Rahemtulla, A. , Apperley, J.F. , Karadimitris, A. , Dillon, N. , 2013. Combined inhibition of p97 and the proteasome causes lethal disruption of the secretory apparatus in multiple myeloma cells. PLoS One. 8, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee, S. , Bartesaghi, A. , Merk, A. , Rao, P. , Bulfer, S.L. , Yan, Y. , Green, N. , Mroczkowski, B. , Neitz, R.J. , Wipf, P. , Falconieri, V. , Deshaies, R.J. , Milne, J.L. , Huryn, D. , Arkin, M. , Subramaniam, S. , 2016. 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science. 351, 871–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce, M. , Bryant, K.F. , Jousse, C. , Long, K. , Harding, H.P. , Scheuner, D. , Kaufman, R.J. , Ma, D. , Coen, D.M. , Ron, D. , Yuan, J. , 2005. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 307, 935–939. [DOI] [PubMed] [Google Scholar]
- Bryant, H.E. , Schultz, N. , Thomas, H.D. , Parker, K.M. , Flower, D. , Lopez, E. , Kyle, S. , Meuth, M. , Curtin, N.J. , Helleday, T. , 2005. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 434, 913–917. [DOI] [PubMed] [Google Scholar]
- Bursavich, M.G. , Parker, D.P. , Willardsen, J.A. , Gao, Z.H. , Davis, T. , Ostanin, K. , Robinson, R. , Peterson, A. , Cimbora, D.M. , Zhu, J.F. , Richards, B. , 2010. 2-Anilino-4-aryl-1,3-thiazole inhibitors of Valosin-containing protein (VCP or p97). Bioorg. Med. Chem. Lett.. 20, 1677–1679. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, 2011. Integrated genomic analyses of ovarian carcinoma. Nature. 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao, K. , Nakajima, R. , Meyer, H.H. , Zheng, Y. , 2003. The AAA-ATPase Cdc48/p97 regulates spindle disassembly at the end of mitosis. Cell. 115, 355–367. [DOI] [PubMed] [Google Scholar]
- Cazanave, S.C. , Elmi, N.A. , Akazawa, Y. , Bronk, S.F. , Mott, J.L. , Gores, G.J. , 2010. CHOP and AP-1 cooperatively mediate PUMA expression during lipoapoptosis. Am. J. Physiol. Gastrointest. Liver Physiol.. 299, G236–G243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, E. , Maksim, N. , de la Cruz, F. , La Clair, J.J. , 2015. Inhibitors of the AAA+ chaperone p97. Molecules. 20, 3027–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung, H.W. , Cowley, G.S. , Weir, B.A. , Boehm, J.S. , Rusin, S. , Scott, J.A. , East, A. , Ali, L.D. , Lizotte, P.H. , Wong, T.C. , Jiang, G. , Hsiao, J. , Mermel, C.H. , Getz, G. , Barretina, J. , Gopal, S. , Tamayo, P. , Gould, J. , Tsherniak, A. , Stransky, N. , Luo, B. , Ren, Y. , Drapkin, R. , Bhatia, S.N. , Mesirov, J.P. , Garraway, L.A. , Meyerson, M. , Lander, E.S. , Root, D.E. , Hahn, W.C. , 2011. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. USA. 108, 12372–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien, J. , Kuang, R. , Landen, C. , Shridhar, V. , 2013. Platinum-sensitive recurrence in ovarian cancer: the role of tumor microenvironment. Front. Oncol.. 3, 251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, T.F. , Brown, S.J. , Minond, D. , Nordin, B.E. , Li, K. , Jones, A.C. , Chase, P. , Porubsky, P.R. , Stoltz, B.M. , Schoenen, F.J. , Patricelli, M.P. , Hodder, P. , Rosen, H. , Deshaies, R.J. , 2011. Reversible inhibitor of p97, DBeQ, impairs both ubiquitin-dependent and autophagic protein clearance pathways. Proc. Natl. Acad. Sci. USA. 108, 4834–4839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, T.F. , Bulfer, S.L. , Weihl, C.C. , Li, K. , Lis, L.G. , Walters, M.A. , Schoenen, F.J. , Lin, H.J. , Deshaies, R.J. , Arkin, M.R. , 2014. Specific inhibition of p97/VCP ATPase and kinetic analysis demonstrate interaction between D1 and D2 ATPase domains. J. Mol. Biol.. 426, 2886–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, T.F. , Li, K. , Frankowski, K.J. , Schoenen, F.J. , Deshaies, R.J. , 2013. Structure-activity relationship study reveals ML240 and ML241 as potent and selective inhibitors of p97 ATPase. ChemMedChem. 8, 297–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou, T.F. , Li, K. , Nordin, B.E. , Porubsky, P. , Frankowski, K. , Patricelli, M.P. , Aube, J. , Schoenen, F.J. , Deshaies, R. , 2010. Selective, Reversible Inhibitors of the AAA ATPase p97 Probe Reports from the NIH Molecular Libraries Program; Bethesda (MD) [PubMed] [Google Scholar]
- Dai, R.M. , Li, C.-C.H. , 2001. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat. Cell Biol.. 3, 740–744. [DOI] [PubMed] [Google Scholar]
- Deshaies, R.J. , 2014. Proteotoxic crisis, the ubiquitin-proteasome system, and cancer therapy. BMC Biol.. 12, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etemadmoghadam, D. , Weir, B.A. , Au-Yeung, G. , Alsop, K. , Mitchell, G. , George, J. , Australian ovarian Cancer study, G., Davis, S. , D'Andrea, A.D. , Simpson, K. , Hahn, W.C. , Bowtell, D.D. , 2013. Synthetic lethality between CCNE1 amplification and loss of BRCA1. Proc. Natl. Acad. Sci. USA. 110, 19489–19494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, C.J. , Gui, L. , Zhang, X. , Moen, D.R. , Li, K. , Frankowski, K.J. , Lin, H.J. , Schoenen, F.J. , Chou, T.F. , 2015. Evaluating p97 inhibitor analogues for their domain selectivity and potency against the p97-p47 complex. ChemMedChem. 10, 52–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, H. , McCabe, N. , Lord, C.J. , Tutt, A.N. , Johnson, D.A. , Richardson, T.B. , Santarosa, M. , Dillon, K.J. , Hickson, I. , Knights, C. , Martin, N.M. , Jackson, S.P. , Smith, G.C. , Ashworth, A. , 2005. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 434, 917–921. [DOI] [PubMed] [Google Scholar]
- Ferlay, J. , Soerjomataram, I. , Ervik, M. , Dikshit, R. , Eser, S. , Mathers, C. , Rebelo, M. , Parkin, D. , Forman, D. , Bray, F. , 2013. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [internet] International Agency for Research on Cancer; Lyon, France: [Google Scholar]
- Ghosh, A.P. , Klocke, B.J. , Ballestas, M.E. , Roth, K.A. , 2012. CHOP potentially co-operates with FOXO3a in neuronal cells to regulate PUMA and BIM expression in response to ER stress. PloS One. 7, e39586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui, L. , Zhang, X. , Li, K. , Frankowski, K.J. , Li, S. , Wong, D.E. , Moen, D.R. , Porubsky, P.R. , Lin, H.J. , Schoenen, F.J. , Chou, T.F. , 2016. Evaluating p97 inhibitor analogues for potency against p97-p37 and p97-Npl4-Ufd1 complexes. ChemMedChem. 11, 953–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyorffy, B. , Lanczky, A. , Szallasi, Z. , 2012. Implementing an online tool for genome-wide validation of survival-associated biomarkers in ovarian-cancer using microarray data from 1287 patients. Endocr. Relat. Cancer. 19, 197–208. [DOI] [PubMed] [Google Scholar]
- Han, J. , Back, S.H. , Hur, J. , Lin, Y.H. , Gildersleeve, R. , Shan, J. , Yuan, C.L. , Krokowski, D. , Wang, S. , Hatzoglou, M. , Kilberg, M.S. , Sartor, M.A. , Kaufman, R.J. , 2013. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat. Cell Biol.. 15, 481–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, H.P. , Novoa, I. , Zhang, Y. , Zeng, H. , Wek, R. , Schapira, M. , Ron, D. , 2000. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol. Cell. 6, 1099–1108. [DOI] [PubMed] [Google Scholar]
- Harding, H.P. , Zhang, Y. , Ron, D. , 1999. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 397, 271–274. [DOI] [PubMed] [Google Scholar]
- Huang, Z. , Wu, Y. , Zhou, X. , Xu, J. , Zhu, W. , Shu, Y. , Liu, P. , 2014. Efficacy of therapy with bortezomib in solid tumors: a review based on 32 clinical trials. Future Oncol.. 10, 1795–1807. [DOI] [PubMed] [Google Scholar]
- Jafari, R. , Almqvist, H. , Axelsson, H. , Ignatushchenko, M. , Lundback, T. , Nordlund, P. , Martinez Molina, D. , 2014. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc.. 9, 2100–2122. [DOI] [PubMed] [Google Scholar]
- Johnson, N. , Johnson, S.F. , Yao, W. , Li, Y.C. , Choi, Y.E. , Bernhardy, A.J. , Wang, Y. , Capelletti, M. , Sarosiek, K.A. , Moreau, L.A. , Chowdhury, D. , Wickramanayake, A. , Harrell, M.I. , Liu, J.F. , D'Andrea, A.D. , Miron, A. , Swisher, E.M. , Shapiro, G.I. , 2013. Stabilization of mutant BRCA1 protein confers PARP inhibitor and platinum resistance. Proc. Natl. Acad. Sci. USA. 110, 17041–17046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi, M. , Tanjung, N.G. , Chen, A. , Dynlacht, J.R. , Garrett, J. , Yoshioka, Y. , Ogawa, K. , Teshima, T. , Yokota, H. , 2012. Administration of salubrinal enhances radiation-induced cell death of SW1353 chondrosarcoma cells. Anticancer Res.. 32, 3667–3673. [PubMed] [Google Scholar]
- Magnaghi, P. , D'Alessio, R. , Valsasina, B. , Avanzi, N. , Rizzi, S. , Asa, D. , Gasparri, F. , Cozzi, L. , Cucchi, U. , Orrenius, C. , Polucci, P. , Ballinari, D. , Perrera, C. , Leone, A. , Cervi, G. , Casale, E. , Xiao, Y. , Wong, C. , Anderson, D.J. , Galvani, A. , Donati, D. , O'Brien, T. , Jackson, P.K. , Isacchi, A. , 2013. Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat. Chem. Biol.. 9, 548–556. [DOI] [PubMed] [Google Scholar]
- Marciniak, S.J. , Yun, C.Y. , Oyadomari, S. , Novoa, I. , Zhang, Y. , Jungreis, R. , Nagata, K. , Harding, H.P. , Ron, D. , 2004. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev.. 18, 3066–3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte, R. , Brown, K.R. , Suarez, F. , Sayad, A. , Karamboulas, K. , Krzyzanowski, P.M. , Sircoulomb, F. , Medrano, M. , Fedyshyn, Y. , Koh, J.L. , van Dyk, D. , Fedyshyn, B. , Luhova, M. , Brito, G.C. , Vizeacoumar, F.J. , Vizeacoumar, F.S. , Datti, A. , Kasimer, D. , Buzina, A. , Mero, P. , Misquitta, C. , Normand, J. , Haider, M. , Ketela, T. , Wrana, J.L. , Rottapel, R. , Neel, B.G. , Moffat, J. , 2012. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov.. 2, 172–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough, K.D. , Martindale, J.L. , Klotz, L.O. , Aw, T.Y. , Holbrook, N.J. , 2001. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol. Cell. Biol.. 21, 1249–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, H.H. , Wang, Y. , Warren, G. , 2002. Direct binding of ubiquitin conjugates by the mammalian p97 adaptor complexes, p47 and Ufd1-Npl4. EMBO J.. 21, 5645–5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mountassif, D. , Fabre, L. , Zaid, Y. , Halawani, D. , Rouiller, I. , 2015. Cryo-EM of the pathogenic VCP variant R155P reveals long-range conformational changes in the D2 ATPase ring. Biochem. Biophys. Res. Commun.. 468, 636–641. [DOI] [PubMed] [Google Scholar]
- Neri, P. , Ren, L. , Gratton, K. , Stebner, E. , Johnson, J. , Klimowicz, A. , Duggan, P. , Tassone, P. , Mansoor, A. , Stewart, D.A. , Lonial, S. , Boise, L.H. , Bahlis, N.J. , 2011. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood. 118, 6368–6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijhawan, D. , Zack, T.I. , Ren, Y. , Strickland, M.R. , Lamothe, R. , Schumacher, S.E. , Tsherniak, A. , Besche, H.C. , Rosenbluh, J. , Shehata, S. , Cowley, G.S. , Weir, B.A. , Goldberg, A.L. , Mesirov, J.P. , Root, D.E. , Bhatia, S.N. , Beroukhim, R. , Hahn, W.C. , 2012. Cancer vulnerabilities unveiled by genomic loss. Cell. 150, 842–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman, S.M. , 2011. Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett.. 585, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslowski, C.M. , Urano, F. , 2011. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol.. 490, 71–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parzych, K. , Chinn, T.M. , Chen, Z. , Loaiza, S. , Porsch, F. , Valbuena, G.N. , Kleijnen, M.F. , Karadimitris, A. , Gentleman, E. , Keun, H.C. , Auner, H.W. , 2015. Inadequate fine-tuning of protein synthesis and failure of amino acid homeostasis following inhibition of the ATPase VCP/p97. Cell Death Dis.. 6, e2031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polucci, P. , Magnaghi, P. , Angiolini, M. , Asa, D. , Avanzi, N. , Badari, A. , Bertrand, J. , Casale, E. , Cauteruccio, S. , Cirla, A. , Cozzi, L. , Galvani, A. , Jackson, P.K. , Liu, Y. , Magnuson, S. , Malgesini, B. , Nuvoloni, S. , Orrenius, C. , Sirtori, F.R. , Riceputi, L. , Rizzi, S. , Trucchi, B. , O'Brien, T. , Isacchi, A. , Donati, D. , D'Alessio, R. , 2013. Alkylsulfanyl-1,2,4-triazoles, a new class of allosteric valosine containing protein inhibitors. Synthesis and structure-activity relationships. J. Med. Chem.. 56, 437–450. [DOI] [PubMed] [Google Scholar]
- Rutkowski, D.T. , Kaufman, R.J. , 2004. A trip to the ER: coping with stress. Trends Cell Biol.. 14, 20–28. [DOI] [PubMed] [Google Scholar]
- Sano, R. , Reed, J.C. , 2013. ER stress-induced cell death mechanisms. Biochim. Biophys. Acta. 1833, 3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, E.K. , Clavarino, G. , Ceppi, M. , Pierre, P. , 2009. SUnSET, a nonradioactive method to monitor protein synthesis. Nat. Methods. 6, 275–277. [DOI] [PubMed] [Google Scholar]
- Seguin, S.J. , Morelli, F.F. , Vinet, J. , Amore, D. , De Biasi, S. , Poletti, A. , Rubinsztein, D.C. , Carra, S. , 2014. Inhibition of autophagy, lysosome and VCP function impairs stress granule assembly. Cell Death Differ.. 21, 1838–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stecklein, S.R. , Kumaraswamy, E. , Behbod, F. , Wang, W. , Chaguturu, V. , Harlan-Williams, L.M. , Jensen, R.A. , 2012. BRCA1 and HSP90 cooperate in homologous and non-homologous DNA double-strand-break repair and G2/M checkpoint activation. Proc. Natl. Acad. Sci. USA. 109, 13650–13655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teng, Y. , Gao, M. , Wang, J. , Kong, Q. , Hua, H. , Luo, T. , Jiang, Y. , 2014. Inhibition of eIF2alpha dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells. Cell Death Dis.. 5, e1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto, Y. , Tomita, Y. , Hoshida, Y. , Kono, T. , Oka, T. , Yamamoto, S. , Nonomura, N. , Okuyama, A. , Aozasa, K. , 2004. Elevated expression of Valosin-containing protein (p97) is associated with poor prognosis of prostate cancer. Clin. Cancer Res.. 10, 3007–3012. [DOI] [PubMed] [Google Scholar]
- Underhill, C. , Toulmonde, M. , Bonnefoi, H. , 2011. A review of PARP inhibitors: from bench to bedside. Ann. Oncol.. 22, 268–279. [DOI] [PubMed] [Google Scholar]
- Vichai, V. , Kirtikara, K. , 2006. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat. Protoc.. 1, 1112–1116. [DOI] [PubMed] [Google Scholar]
- Wijeratne, E.M. , Gunaherath, G.M. , Chapla, V.M. , Tillotson, J. , de la Cruz, F. , Kang, M. , U'Ren, J.M. , Araujo, A.R. , Arnold, A.E. , Chapman, E. , Gunatilaka, A.A. , 2016. Oxaspirol B with p97 inhibitory activity and other oxaspirols from Lecythophora sp. FL1375 and FL1031, endolichenic fungi inhabiting Parmotrema tinctorum and Cladonia evansii . J. Nat. Prod.. 79, 340–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi, H. , Wang, H.G. , 2004. CHOP is involved in endoplasmic reticulum stress-induced apoptosis by enhancing DR5 expression in human carcinoma cells. J. Biol. Chem.. 279, 45495–45502. [DOI] [PubMed] [Google Scholar]
- Yamamoto, S. , Tomita, Y. , Hoshida, Y. , Takiguchi, S. , Fujiwara, Y. , Yasuda, T. , Yano, M. , Nakamori, S. , Sakon, M. , Monden, M. , Aozasa, K. , 2003. Expression level of Valosin-containing protein is strongly associated with progression and prognosis of gastric carcinoma. J. Clin. Oncol.. 21, 2537–2544. [DOI] [PubMed] [Google Scholar]
- Yamamoto, S. , Tomita, Y. , Hoshida, Y. , Toyosawa, S. , Inohara, H. , Kishino, M. , Kogo, M. , Nakazawa, M. , Murakami, S. , Iizuka, N. , Kidogami, S. , Monden, M. , Kubo, T. , Ijuhin, N. , Aozasa, K. , 2004. Expression level of Valosin-containing protein (VCP) as a prognostic marker for gingival squamous cell carcinoma. Ann. Oncol.. 15, 1432–1438. [DOI] [PubMed] [Google Scholar]
- Yamamoto, S. , Tomita, Y. , Nakamori, S. , Hoshida, Y. , Nagano, H. , Dono, K. , Umeshita, K. , Sakon, M. , Monden, M. , Aozasa, K. , 2003. Elevated expression of Valosin-containing protein (p97) in hepatocellular carcinoma is correlated with increased incidence of tumor recurrence. J. Clin. Oncol.. 21, 447–452. [DOI] [PubMed] [Google Scholar]
- Zhang, S.H. , Liu, J. , Kobayashi, R. , Tonks, N.K. , 1999. Identification of the cell cycle regulator VCP (p97/CDC48) as a substrate of the band 4.1-related protein-tyrosine phosphatase PTPH1. J. Biol. Chem.. 274, 17806–17812. [DOI] [PubMed] [Google Scholar]
- Zhou, H.J. , Wang, J. , Yao, B. , Wong, S. , Djakovic, S. , Kumar, B. , Rice, J. , Valle, E. , Soriano, F. , Menon, M.K. , Madriaga, A. , Kiss von Soly, S. , Kumar, A. , Parlati, F. , Yakes, F.M. , Shawver, L. , Le Moigne, R. , Anderson, D.J. , Rolfe, M. , Wustrow, D. , 2015. Discovery of a first-in-class, potent, selective, and orally bioavailable inhibitor of the p97 AAA ATPase (CB-5083). J. Med. Chem.. 58, 9480–9497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data