

Abstract
We used KRAS mutations to investigate the clinical relevance of circulating tumor DNA (ctDNA) measurements in patients with advanced pancreatic cancer. Fifty‐three blood samples were collected from 14 prospectively recruited patients prior to chemotherapy (gemcitabine or FOLFIRINOX) and subsequently every month during treatment. Samples were processed by density centrifugation and plasma DNA isolation. A Peptide–nucleic acid–clamp PCR was then used to detect KRAS mutations (present in >90% of pancreatic cancers) as a surrogate marker for ctDNA. Plasma samples from 29 healthy individuals were analyzed as a reference group. Results were compared to conventional monitoring measures and survival data. Median follow‐up time was 3.7 months (range 0.6–12.9 months).
Ten (71%) patients had a positive KRAS status in the plasma samples obtained prior to chemotherapy, indicating the presence of ctDNA. Among the patients who were ctDNA‐positive before chemotherapy, nine (90%) experienced disease progression during follow‐up, compared to one (25%) of four ctDNA‐negative patients (P = 0.01). The pre‐therapy ctDNA level was a statistically significant predictor of both progression‐free and overall survival (P = 0.014 and 0.010, respectively). Of the 14 patients, ten had ≥2 follow‐up samples; in several of these patients, the ctDNA level changed substantially during the course of chemotherapy. Changes in ctDNA levels corresponded both with radiological follow‐up data and CA19‐9 levels for several patients.
This pilot study supports the hypothesis that ctDNA may be used as a marker for monitoring treatment efficacy and disease progression in pancreatic cancer patients. Recruitment of more patients is ongoing to corroborate these findings.
Keywords: Circulating tumor DNA, ctDNA, Cell‐free DNA, cfDNA, KRAS, Pancreatic cancer, Liquid biopsy
Highlights
Plasma KRAS mutational status is a surrogate marker for ctDNA in pancreatic cancer.
ctDNA detected before or after chemotherapy predicts shorter survival.
ctDNA levels before chemotherapy are an independent prognostic factor.
ctDNA monitoring corresponds with radiological follow‐up data and CA19‐9 levels.
Abbreviations
- ctDNA
circulating cell-free tumor DNA
- CTC
circulating tumor cells
- PNA clamp PCR
peptide-nucleic acid clamp polymerase chain reaction
- CT
computed tomography
- MRI
magnetic resonance imaging
1. Introduction
Pancreatic cancer is the fourth leading cause of cancer‐related death in Western countries. Approximately 90% of patients who present with advanced pancreatic cancer survive <1 year, even when treated with chemotherapy. However, modern polychemotherapy is likely to extend the survival (Conroy et al., 2011; Von Hoff et al., 2013; Goldstein et al., 2015). The high death rate is explained by the advanced disease stage at the time of diagnosis, poor response to chemotherapy and de novo or acquired drug resistance (Li et al., 2004; Torre et al., 2015). Hence, informative biomarkers to identify early disease and predict progression and thus improve tumor monitoring are needed.
There is evidence that the presence of circulating tumor cells (CTCs) is associated with poor disease outcome in pancreatic cancer patients, as well as in patients suffering from other adenocarcinomas (e.g. (Bednarz‐Knoll et al., 2011; Tjensvoll et al., 2014)). Further evidence suggests that detection and genetic characterization of circulating cell‐free tumor DNA (ctDNA) might provide another, more easily accessible, source for prognostic and predictive information in several cancers (Punnoose et al., 2012; Dawson et al., 2013b; Bidard et al., 2014). ctDNA is extracellular DNA that may originate from apoptotic and necrotic tumor cells in the primary tumor, metastatic lesions or in circulation (CTCs). Thus, it is hypothesized that the ctDNA pool is expected to reflect the total tumor burden in cancer patients. Furthermore, there are indications that tumor‐derived exosomes or microvesicles may contain DNA, and it is likely that this DNA is also represented in the overall ctDNA pool (Balaj et al., 2011; Thakur et al., 2014).
Different methodological approaches have been applied for the detection of ctDNA, by identification of tumor‐specific mutations in various cancers, such as the amplification refractory mutation system (ARMS) (Spindler et al., 2012), digital PCR (Diehl et al., 2008; Taly et al., 2013; Bettegowda et al., 2014; Kinugasa et al., 2015), and different next generation sequencing approaches (Forshew et al., 2012; Dawson et al., 2013b; Bettegowda et al., 2014; Newman et al., 2014; Sausen et al., 2015; Zill et al., 2015). In pancreatic cancer, more than 90% of the tumors harbor mutations in the KRAS gene (Almoguera et al., 1988), thus, it may be a potential surrogate marker for ctDNA detection in plasma from these patients. Castells and colleagues were the first to detect circulating mutant KRAS genes in plasma from patients with pancreatic ductal adenocarcinoma in 1999 (Castells et al., 1999). They detected KRAS mutated ctDNA in 12/44 (27%) patients, and demonstrated that these patients experienced a significantly shorter survival time. Later, several other studies have also demonstrated the feasibility of detection and prognostic relevance of circulating KRAS mutations in pancreatic cancer patients (Chen et al., 2010; Dabritz et al., 2012; Kinugasa et al., 2015). Recent studies in other types of cancers also support the assumption that detection and genetic characterization of ctDNA may provide prognostic, predictive, and therapeutic information and thus could potentially be used as “liquid biopsies” in different cancer types (Diehl et al., 2008; Dawson et al., 2013a; Bettegowda et al., 2014; Thierry et al., 2014).
The aims of this study were to investigate whether KRAS mutations present in plasma‐derived cell‐free DNA from patients with advanced pancreatic cancer could be detected by our high‐fidelity polymerase‐based peptide–nucleic acid (PNA)‐clamp PCR assay (Gilje et al., 2008; Oltedal et al., 2010) and to explore the clinical relevance of ctDNA measurements in this group of patients with regard to predicting outcome and monitoring the effects of treatment.
2. Materials and methods
2.1. Patients and samples
A total of 53 blood plasma samples were collected from 14 patients with locally advanced (n = 2) or metastatic (n = 12) pancreatic cancer who were recruited at Stavanger University Hospital from September 2012 until June 2014. The median age was 64 years. Most patients had primary tumor localization in the head (n = 9) of the pancreas, grade II–III disease (n = 8) and Eastern Cooperative Oncology Group (ECOG) performance status score of 1–2 (n = 8). A partially age‐matched control group of 29 healthy volunteers was also recruited. The project was approved by the Regional Committee for Medical and Health Research Ethics (REK‐Vest 2011/475), and all participants provided written informed consent.
Patient blood samples (9 mL in EDTA tubes) were drawn by venous puncture prior to treatment and subsequently every month during treatment. All patients were treated either with gemcitabine (n = 6) or the FOLFIRINOX chemotherapy regimen (n = 8) according to national Norwegian guidelines (Helsedirektoratet, 2015). Treatment was administered until disease progression, unacceptable toxicity, patient refusal, or treatment pause needed for patient. Every 4 weeks, data on performance status (ECOG status), use of analgesics, and patient weight were recorded, and routine laboratory tests were conducted, including blood counts and serum chemistry. Concurrently, several biomarkers, including carbohydrate antigen 19‐9 (CA19‐9), cancer antigen 125 (CA125), and carcinoembryonic antigen (CEA), were determined routinely.
Standard disease evaluation by imaging, according to RECIST 1.1 criteria (Eisenhauer et al., 2009), was applied to define treatment response. Anti‐tumor activity was assessed by computed tomography (CT) or magnetic resonance imaging (MRI) of the abdomen, chest X‐ray (or chest CT scan or MRI in case of thoracic target lesions), and other examinations, as clinically indicated to assess target and non‐target lesions. These examinations were primarily performed at baseline (screening) and every 8 weeks (earlier whenever disease progression was suspected) using the same method for each assessment.
2.2. Isolation of circulating nucleic acids from plasma
Blood samples were processed by density centrifugation using Lymphoprep™ (Axis Shield) density gradient media according to the manufacturer's instructions. Total cell‐free DNA was then isolated from 1 mL (from the first five patients only) and 4 mL (diluted 1:1 with 0.9% NaCl) of plasma using the QIAamp Circulating Nucleic Acid kit (Qiagen), as described by the manufacturer. ctDNA was eluted in 50 μl of Buffer AE, and all samples were stored at −80 °C until further analysis.
2.3. Detection of ctDNA by KRAS mutation‐based PNA clamp PCR method
The PNA‐clamp method is based on binding of a PNA to wild‐type DNA to prevent its amplification during PCR, while mutated DNA is amplified. Clamp PCR was performed in our Mx3000P real‐time PCR instrument (Stratagene/Agilent) as previously described (Gilje et al., 2008). In brief, 5 μl of plasma DNA was analyzed per reaction, and each sample was analyzed in duplicate. Moreover, all samples were analyzed with and without the PNA‐clamp added; the −PNA reaction reflected the total cell‐free DNA content in each sample. Plasma DNA from a healthy individual was used as a negative control, while the positive control was a 1:100 dilution of DNA from the colorectal carcinoma cell line LS174T (heterozygous GGT > GAT codon 12 KRAS mutation [c.35G > A]) in Caco2 DNA (KRAS wt). Positive and negative controls were included in every run. The KRAS measure ΔΔCq was computed as ΔΔCq = ΔCqwt,min − ΔCqsample where ΔCq = Cq(+PNA) − Cq(−PNA) and ΔCqwt,min denoted the lowest ΔCq measured for the wild‐type control in any run (lowest ΔCq = 11.27). We defined samples with ΔΔCq > 0 to be positive for KRAS mutated ctDNA. Reactions producing no amplification signal (no Cq values) in both replicates were scored as negative, and their ΔΔCq was set to −10.26, which was the lowest ΔΔCq value observed in this data set. Moreover, if a sample had only one replicate with an amplification signal, this Cq value was used in the further analyses. The sensitivity of the method for mutated KRAS was previously estimated to be 1:104 (Gilje et al., 2008). All ctDNA analyses were performed by two persons (S.O. and K.S.O.B.) who were blinded to patient identity, disease status, and treatment. ctDNA results were compared with conventional biochemical (CA19‐9, CA‐125, and CEA) and radiological monitoring measures.
2.4. Statistical analysis
All statistical analyses were performed with IBM SPSS version 22.0 (www.spss.com). P values of <0.05 were considered significant. Fisher's exact test was used to test for relationships between KRAS status before initiation of chemotherapy and various clinicopathological parameters. Missing data were automatically excluded from the analyses.
Kaplan–Meier estimates of clinical outcomes were determined, and differences between groups were assessed by the Mantel–Cox log‐rank test. The primary endpoints were progression‐free survival and overall survival. Progression‐free survival was defined as the elapsed time between the date of inclusion and the date of progression, or death due to any cause if the patient died before evidence of progression was obtained. Disease progression was defined by radiological imaging according to RECIST 1.1. Overall survival was defined as the elapsed time between the inclusion date and death due to any cause.
Cox univariate regression analyses were used to investigate the effects of multiple variables (initial ctDNA status [B1], ctDNA status 4 weeks after initiation of treatment [B2], age, tumor size, lymph node status, tumor grade, primary tumor localization, ECOG performance status, CA19‐9 status, CEA status, CA‐125 status, and prior anti‐cancer surgery) on progression‐free and overall survival. Univariate analyses were performed by analyzing the ctDNA status both as a categorical and as a continuous variable. Multiple Cox regression modeling was limited to initial ctDNA status (B1), tumor size, lymph node status, and tumor grade, as there were few cases with a complete data set. Multivariate analyses were performed using both the forward and backward stepwise selection of covariates. Correlation tests were performed between ctDNA and CA19‐9 levels by the non‐parametric Spearmans's rank correlation coefficient test.
3. Results
3.1. ctDNA measurements before and after initiation of chemotherapy
Plasma samples from 29 healthy individuals were analyzed to define a cut‐off for plasma KRAS mutational status and, thereby, the presence of ctDNA. According to this cut‐off, none of the normal control plasma samples were positive for KRAS mutations.
Analyses of the 14 patient plasma samples obtained before initiation of chemotherapy revealed that ten (71%) patients had a positive KRAS mutation status, indicating the presence of ctDNA. Further analyses also demonstrated that the ctDNA level before initiation of treatment was significantly higher in patients who experienced disease progression compared to patients with stable disease (P = 0.024) at follow‐up (Figure 1). Plasma samples were obtained 4 weeks after initiation of chemotherapy from nine of the 14 patients included in this study. Five of these nine patients (55%) had a positive ctDNA status.
Figure 1.
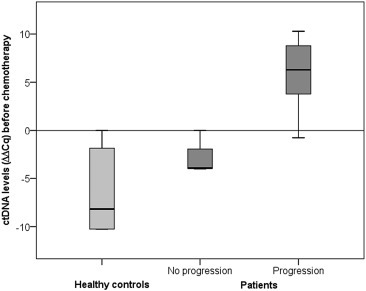
Boxplot showing ctDNA levels before initiation of chemotherapy in 14 advanced pancreatic cancer patients with and without progression during follow‐up (P = 0.024), compared to healthy controls (n = 29).
3.2. Survival analyses
With a median follow‐up time of 3.7 months (range 0.6–12.9 months), disease progression was encountered in 11 (78.5%) patients, and ten patients died. Nine (90%) of the ten patients with a positive ctDNA status before initiation of treatment experienced disease progression, compared to one (25%) patient with progression among the four ctDNA‐negative patients (Fisher's exact test, P = 0.01). Kaplan–Meier survival analyses indicated that patients with a positive ctDNA status before or after initiation of chemotherapy had shorter progression‐free survival (P = 0.064 and P = 0.071, respectively); although this difference did not meet statistical significance (Figure 2). A trend towards reduced overall survival among patients with a positive ctDNA status before chemotherapy initiation (P = 0.066) was also observed.
Figure 2.
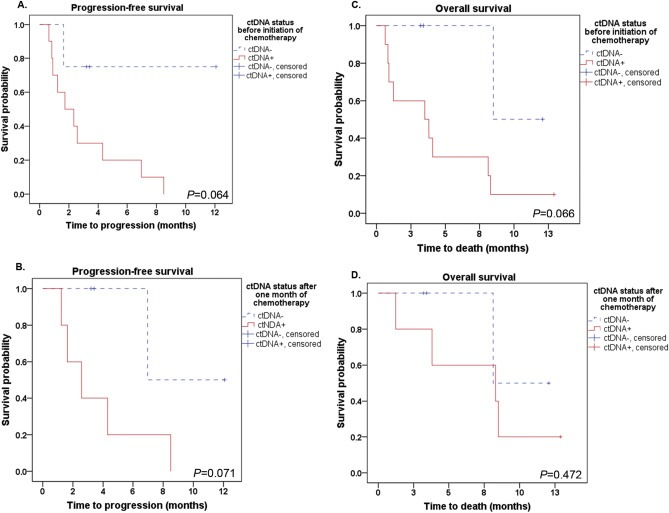
Kaplan–Meier analyses indicated a trend towards reduced progression‐free survival both before (A) and after (B) initiation of chemotherapy in pancreatic cancer patients with a positive ctDNA status (ΔΔCq > 0). Shorter overall survival was also indicated for patients who were ctDNA‐positive before chemotherapy initiation (C), while there was no association after one month of treatment (D).
Univariate and multiple Cox regression were performed to estimate the prognostic impact of ctDNA status, as well as of other clinicopathological parameters. Univariate Cox regression demonstrated that the relative level (continuous variable) of ctDNA in the plasma samples obtained before initiation of chemotherapy was a significant risk factor for both reduced progression‐free (HR = 1.287, P = 0.014) and overall (HR = 1.433, P = 0.010) survival (Table 1). None of the other covariates were significant predictors of survival. By backward and forward stepwise multiple Cox regression, it was further demonstrated that ctDNA levels before initiation of chemotherapy were an independent prognostic factor for both progression‐free (HR = 1.48, P = 0.020) and overall (HR = 1.31, P = 0.047) survival.
Table 1.
Univariate Cox regression.
| Parameter | Progression‐free survival | Overall survival | ||
|---|---|---|---|---|
| Hazard ratio | P values | Hazard ratio | P values | |
| ctDNA KRAS B1 status (pos. vs. neg.) | 5.648 | 0.100 | 5.864 | 0.099 |
| ctDNA KRAS B1 levels (continuous variable) | 1.287 | 0.014 | 1.433 | 0.010 |
| ctDNA KRAS B2 status (pos. vs. neg.) | 5.840 | 0.110 | 2.203 | 0.483 |
| ctDNA KRAS B2 levels (continuous variable) | 1.273 | 0.207 | 1.238 | 0.379 |
| Age (continuous variable) | 0.965 | 0.449 | 0.951 | 0.302 |
| Tumor size (cT2 vs. cT4) | 0.514 | 0.367 | 0.561 | 0.459 |
| Lymph node status (cN1 vs. cN2 and cN3) | 0.588 | 0.511 | 0.276 | 0.229 |
| Grade (I and II vs. III) | 1.159 | 0.849 | 0.966 | 0.966 |
| Primary tumor localization(head and body vs. tail and >1 location) | 2.933 | 0.132 | 3.772 | 0.086 |
| ECOG PS (0 vs. 1 and 2) | 0.297 | 0.134 | 0.347 | 0.194 |
| CA19‐9 status (pos. vs. neg.) | 9.352 | 0.071 | 9.352 | 0.071 |
| CEA status (pos. vs. neg.) | 2.379 | 0.277 | 1.829 | 0.455 |
| CA‐125 status (pos. vs. neg.) | 5.116 | 0.124 | 3.932 | 0.200 |
| Prior anti‐cancer surgery (yes vs. no) | 3.622 | 0.225 | 2.297 | 0.433 |
Boldface indicates significant P values.
3.3. Monitoring of ctDNA levels during the course of chemotherapy
We performed longitudinal monitoring of the disease course for ten of the 14 included patients. The remaining four patients either died within a month after study entry (n = 3) or provided only one plasma sample (n = 1). In total, 39 serial plasma samples that originated from these ten patients were analyzed; 15 (38.5%) samples showed the presence of ctDNA. Of the patients with ≥2 follow‐up samples collected, several had ctDNA levels that changed substantially during the course of chemotherapy. For at least three patients, changes in the ctDNA level also corresponded to radiological follow‐up data and to changes in CA19‐9 levels obtained every 4 weeks (Figure 3). For patient 8, ctDNA levels were significantly correlated with CA19‐9 measurements (Spearman's rho = 0.786, P = 0.021). There was also a correlation with CA19‐9 levels when all data, from all sampling time points, were included in the analysis (Spearman's rho = 0.395, P = 0.005). However, a comparison between ctDNA levels and radiological follow‐up data indicated that ctDNA measurements may reveal disease progression at an earlier stage for some patients compared to conventional monitoring methods. For example, the ctDNA levels of patient 8 had already increased 1 month before progression was diagnosed by radiological measurements (Figure 3A). ctDNA levels also seemed to reveal disease progression two months earlier than radiological imaging for patient 11 (Figure 3C). However, in patient 9 a positive ctDNA status was observed at the same time as progression was confirmed by imaging (Figure 3B).
Figure 3.
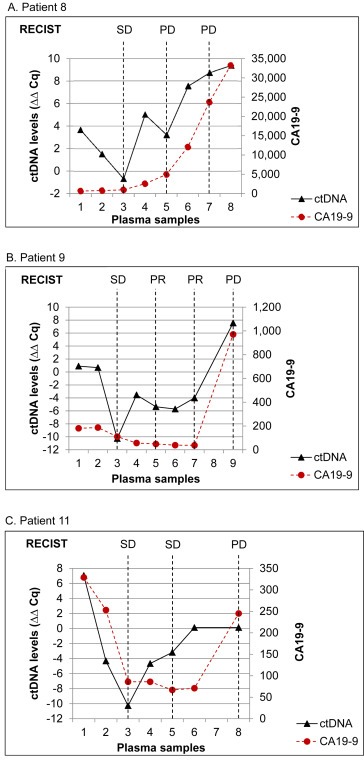
Disease monitoring by measurement of ctDNA and CA19‐9 levels in three pancreatic cancer patients. Plasma sample 1 was obtained before initiation of treatment, while plasma samples 2–9 were obtained every month during treatment with gemcitabine or FOLFIRINOX. Disease evaluation by imaging, according to RECIST 1.1 criteria, was also performed at baseline and every 8 weeks. SD, stable disease; PR, partial response; PD, progressive disease.
4. Discussion
Analysis of plasma ctDNA is emerging as a tool with several potential clinical applications. An increasing number of studies suggest that ctDNA detection and characterization may be used for treatment guidance and early detection of disease progression, as well as for monitoring treatment response and the presence of resistance mutations against targeted therapies (Misale et al., 2012; Dawson et al., 2013b; Bettegowda et al., 2014; De Mattos‐Arruda et al., 2014; Lebofsky et al., 2015).
In this pilot study, we used KRAS PNA‐clamp PCR to establish KRAS mutational status as a surrogate marker for ctDNA detection in plasma samples from patients with advanced pancreatic cancer. We detected KRAS mutated ctDNA in 71% of the patients before initiation of chemotherapy and in 56% of the patients after start of treatment. This positivity rate is higher than that described in other studies, where KRAS mutated ctDNA detection rates prior to treatment were 0% (Marchese et al., 2006), 27% (Castells et al., 1999), 33% (Chen et al., 2010), 47% (Maire et al., 2002), and 62.6% (Kinugasa et al., 2015). There may be several explanations for this discrepancy: a) we included the highest rate of metastatic patients in our study, b) sensitivity may vary among the methods used for analyses (restriction fragment length polymorphism [RFLP] PCR, Sanger sequencing, allele‐specific PCR, and digital PCR), and c) there may be differences in the amount of ctDNA used in the different assays (Castells et al., 1999; Maire et al., 2002; Marchese et al., 2006; Chen et al., 2010; Kinugasa et al., 2015).
Although 90% of patients with pancreatic cancer harbor primary tumor mutations in codons 12 and 13 of the KRAS gene, KRAS mutations in plasma are detected at rate lower than 90% for several reasons. Some patients may not have any ctDNA, or the ctDNA may come exclusively from KRAS wild‐type tumor cells in a genetically heterogeneous primary tumor (Oltedal et al., 2011; Rothe et al., 2014; Thierry et al., 2014). Therefore, parallel detection of other mutations (e.g., in KRAS codon 61, p16/CDKN2A, TP53, SMAD4 etc.) may further increase the number of ctDNA‐positive patients. The detection rate is also affected by differences in methodology. To avoid false positives among the healthy controls, we established a diagnostic threshold level for the PNA‐clamp PCR method, based on the highest measurement in an age‐matched healthy control group. A lower threshold would have increased the sensitivity of the method, but at the expense of specificity. We previously demonstrated that DNA polymerization errors during PCR lower the sensitivity of PNA‐clamp PCR (Gilje et al., 2008). By using a high‐fidelity polymerase, we increased the analytical sensitivity to 1:104 for high‐quality cellular DNA. However, the highly fragmented nature of plasma DNA can reduce the sensitivity of PNA‐clamp PCR, due to limited PCR primer binding and potentially reduced polymerase fidelity (Newman et al., 2014).
Detection of ctDNA in the plasma of pancreatic cancer patients has been shown to correlate both with primary tumor analyses and the presence of metastases, as well as survival (Castells et al., 1999; Chen et al., 2010; Kinugasa et al., 2015). Our pilot study implies that pancreatic cancer patients with KRAS mutated ctDNA in plasma before or after initiation of chemotherapy have a shorter survival time. This has also been demonstrated in previous studies (Castells et al., 1999; Chen et al., 2010; Kinugasa et al., 2015). In concordance with our data, Castells and colleagues reported shorter survival times in patients with K‐ras gene mutations in plasma prior to therapy in their study of 44 pancreatic cancer patients (P < 0.005) (Castells et al., 1999). Furthermore, Chen et al. reported a median survival of 3.9 months for patients with KRAS mutated ctDNA versus 10.2 months for the ctDNA‐negative patients (P < 0.001), and KRAS mutated ctDNA was an independent prognostic factor for survival (hazard ratio 7.39) in their study of 91 pancreatic cancer patients (Chen et al., 2010).
We wanted to explore whether changes in ctDNA levels might be useful for measurements of treatment response, as well as for early detection of recurrent disease. Continuous treatment monitoring may reveal the response to a certain systemic treatment or indicate that there should be a change in the treatment regimen due to tumor cell resistance. This is becoming more relevant in pancreatic cancer as treatment regimens are improving, with alternative treatment choices and longer survival. We therefore performed longitudinal monitoring of the disease course in our study. Our preliminary findings suggest that for some pancreatic cancer patients, ctDNA analyses may reveal response to therapy and disease progression at the same time or potentially earlier than radiologically measured tumor volumes (Figure 3). ctDNA analyses as a promising monitoring tool has also been explored for several cancers in previous studies (Diehl et al., 2013, 2013, 2008, 2015, 2014, 2015). In a study by Newman and colleagues, radiological follow‐up imaging in non‐small cell lung cancer patients was less precise in predicting treatment response and disease progression than ctDNA analyses for several patients (Newman et al., 2014). Furthermore, Diehl and colleagues described a 99.9% decrease in ctDNA levels during treatment of colorectal patients, whereas the tumor volume (composed of live and dead neoplastic cells in addition to stromal cells) decreased only slightly (Diehl et al., 2008). In pancreatic cancer patients, measurement of treatment efficacy by imaging is particularly challenging, as the tumor is often surrounded by a dense fibrotic tissue, making radiological (CT and PET scan) detection difficult (Rothenberg et al., 1996; Duffy et al., 2010). Thus, therapy decisions in this patient group are based not only on imaging, but also on evaluation of the CA19‐9 level, which is currently the most common and extensively used blood biomarker in pancreatic cancer management despite having several limitations (Duffy et al., 2010; Ballehaninna and Chamberlain, 2012). These limitations include the possibility of a misleading increase in serum samples (Dabritz et al., 2012), failure to identify early/small tumors and precancerous lesions in high‐risk patients (Ballehaninna and Chamberlain, 2012), and detection of elevated levels in only a subset of patients with resectable pancreatic cancer (Dabritz et al., 2012). All of these factors severely limit the universal applicability of serum CA19‐9 levels in pancreatic cancer management (Ballehaninna and Chamberlain, 2012). Our data suggest, however, that frequent monitoring of ctDNA levels in patients with advanced pancreatic cancer during the disease course may be a promising predictor of both responses to therapy and disease progression. Although our patient cohort is small there are also indications that the ctDNA level correlates with CA19‐9 data.
Genetic heterogeneity within the primary tumor, as well as between the primary tumor and metastatic lesions, has been described previously (Yachida et al., 2010; Oltedal et al., 2011; Diaz et al., 2012; Gerlinger et al., 2012; Rothe et al., 2014), and it is the main mechanism through which treatment resistance arises (Haber and Velculescu, 2014). Although many studies have reported high concordance between the primary tumor and ctDNA analyses, most studies have reported some discrepancies between the two DNA sources (Bettegowda et al., 2014; De Mattos‐Arruda et al., 2014; Thierry et al., 2014; Kinugasa et al., 2015). This could be due to genetic heterogeneity within the primary tumor, or because genetically different metastases or CTCs are contributing to the ctDNA pool. If all tumor deposits in the body are represented in the ctDNA pool, we would expect the challenges posed by intra‐tumoral genetic heterogeneity to be surmounted by ctDNA assessment. Hence, ctDNA is a promising tool for monitoring therapy response, and it is believed that monitoring of somatic genetic alterations in ctDNA during the course of therapy will reflect the dynamics of the tumor cell population in response to cancer treatment (e.g., see (Dawson et al., 2013b; De Mattos‐Arruda et al., 2014)). However, because ctDNA analysis is supposed to primarily reflect dying (necrotic or apoptotic) tumor cells rather than potential residual disease; these analyses may only complement the primary tumor and CTC analyses. This was suggested by Dawson et al., 2013, 2013 where increasing levels of both ctDNA and CTCs were found to be of prognostic value (Dawson et al., 2013b). Bettegowda et al., 2014 also suggested that ctDNA and CTCs analyses are complementary, as they found that ctDNA was often present in patients without detectable CTCs (Bettegowda et al., 2014).
5. Conclusion
This pilot study supports the hypothesis that ctDNA may be used as a marker for monitoring treatment efficacy and disease progression in patients with advanced pancreatic cancer. Our data also imply that patients with a positive ctDNA status prior to or after initiation of chemotherapy have shorter survival. Moreover, ctDNA levels before initiation of chemotherapy were an independent prognostic factor for prediction of survival in this pilot study. However, these data need further investigation in a larger cohort. Expanding the study to include several mutations could potentially provide additional prognostic information and increase ctDNA detection. It would also be of interest to investigate whether measurement of ctDNA could reveal early pancreatic cancer, as well as provide prognostic information in patients with localized disease.
Author contributions
KT participated in coordinating the study, carried out ctDNA purification, performed statistical analyses, contributed to result interpretation, and drafted the manuscript. ML contributed to result interpretation and manuscript preparation. TB participated in the study design, in coordinating the study and contributed to sample collection and manuscript preparation. SO did most of the PNA‐clamp PCR and participated in result interpretation and manuscript preparation. KSOB was a Bachelor student on the project who carried out ctDNA purification and some of the PNA‐clamp PCR and contributed to manuscript preparation. BG participated in sample collection, interpretation of the results, and manuscript preparation. JAS contributed to patient recruitment and manuscript preparation. MJ gave scientific advice and contributed to manuscript preparation. ON participated in the study design and interpretation of the results, coordinated the study, gave statistical advice and contributed to manuscript preparation. RS was the group leader and contributed in the study design, to patient recruitment, interpretation of the results, and manuscript preparation. All authors have read and approved the final manuscript.
Conflict of interest
All authors declare that they have no competing interests.
Acknowledgments
This project was supported by the Folke Hermansen Foundation, the Western Norway Regional Health Authority, and the Norwegian Cancer Society. The project is part of the strategic program called “Personalized medicine‐biomarkers and clinical studies”, which is supported by the Western Norway Regional Health Authority.
Tjensvoll Kjersti, Lapin Morten, Buhl Tove, Oltedal Satu, Steen-Ottosen Berry Katrine, Gilje Bjørnar, Søreide Jon Arne, Javle Millind, Nordgård Oddmund, Smaaland Rune, (2016), Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.11.012.
References
- Almoguera, C. , Shibata, D. , Forrester, K. , Martin, J. , Arnheim, N. , Perucho, M. , 1988. Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 53, 549–554. [DOI] [PubMed] [Google Scholar]
- Balaj, L. , Lessard, R. , Dai, L. , Cho, Y.J. , Pomeroy, S.L. , Breakefield, X.O. , Skog, J. , 2011. Tumour microvesicles contain retrotransposon elements and amplified oncogene sequences. Nat. Commun. 2, 180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballehaninna, U.K. , Chamberlain, R.S. , 2012. The clinical utility of serum CA 19-9 in the diagnosis, prognosis and management of pancreatic adenocarcinoma: an evidence based appraisal. J. Gastrointest. Oncol. 3, 105–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bednarz-Knoll, N. , Alix-Panabieres, C. , Pantel, K. , 2011. Clinical relevance and biology of circulating tumor cells. Breast Cancer Res. 13, 228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettegowda, C. , Sausen, M. , Leary, R.J. , Kinde, I. , Wang, Y. , Agrawal, N. , Bartlett, B.R. , Wang, H. , Luber, B. , Alani, R.M. , Antonarakis, E.S. , Azad, N.S. , Bardelli, A. , Brem, H. , Cameron, J.L. , Lee, C.C. , Fecher, L.A. , Gallia, G.L. , Gibbs, P. , Le, D. , Giuntoli, R.L. , Goggins, M. , Hogarty, M.D. , Holdhoff, M. , Hong, S.M. , Jiao, Y. , Juhl, H.H. , Kim, J.J. , Siravegna, G. , Laheru, D.A. , Lauricella, C. , Lim, M. , Lipson, E.J. , Marie, S.K. , Netto, G.J. , Oliner, K.S. , Olivi, A. , Olsson, L. , Riggins, G.J. , Sartore-Bianchi, A. , Schmidt, K. , Shih, L.,M. , Oba-Shinjo, S.M. , Siena, S. , Theodorescu, D. , Tie, J. , Harkins, T.T. , Veronese, S. , Wang, T.L. , Weingart, J.D. , Wolfgang, C.L. , Wood, L.D. , Xing, D. , Hruban, R.H. , Wu, J. , Allen, P.J. , Schmidt, C.M. , Choti, M.A. , Velculescu, V.E. , Kinzler, K.W. , Vogelstein, B. , Papadopoulos, N. , Diaz, L.A. , 2014. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci. Transl Med. 6, 224ra224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidard, F.C. , Madic, J. , Mariani, P. , Piperno-Neumann, S. , Rampanou, A. , Servois, V. , Cassoux, N. , Desjardins, L. , Milder, M. , Vaucher, I. , Pierga, J.Y. , Lebofsky, R. , Stern, M.H. , Lantz, O. , 2014. Detection rate and prognostic value of circulating tumor cells and circulating tumor DNA in metastatic uveal melanoma. Int. J. Cancer. 134, 1207–1213. [DOI] [PubMed] [Google Scholar]
- Castells, A. , Puig, P. , Mora, J. , Boadas, J. , Boix, L. , Urgell, E. , Sole, M. , Capella, G. , Lluis, F. , Fernandez-Cruz, L. , Navarro, S. , Farre, A. , 1999. K-ras mutations in DNA extracted from the plasma of patients with pancreatic carcinoma: diagnostic utility and prognostic significance. J. Clin. Oncol. 17, 578–584. [DOI] [PubMed] [Google Scholar]
- Chen, H. , Tu, H. , Meng, Z.Q. , Chen, Z. , Wang, P. , Liu, L.M. , 2010. K-ras mutational status predicts poor prognosis in unresectable pancreatic cancer. Eur. J. Surg. Oncol. 36, 657–662. [DOI] [PubMed] [Google Scholar]
- Conroy, T. , Desseigne, F. , Ychou, M. , Bouche, O. , Guimbaud, R. , Becouarn, Y. , Adenis, A. , Raoul, J.L. , Gourgou-Bourgade, S. , De La Fouchardiere, C. , Bennouna, J. , Bachet, J.B. , Khemissa-Akouz, F. , Pere-Verge, D. , Delbaldo, C. , Assenat, E. , Chauffert, B. , Michel, P. , Montoto-Grillot, C. , Ducreux, M. , 2011. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 364, 1817–1825. [DOI] [PubMed] [Google Scholar]
- Dabritz, J. , Preston, R. , Hanfler, J. , Oettle, H. , 2012. K-ras mutations in the plasma correspond to computed tomographic findings in patients with pancreatic cancer. Pancreas. 41, 323–325. [DOI] [PubMed] [Google Scholar]
- Dawson, S.J. , Rosenfeld, N. , Caldas, C. , 2013. Circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 369, 93–94. [DOI] [PubMed] [Google Scholar]
- Dawson, S.J. , Tsui, D.W. , Murtaza, M. , Biggs, H. , Rueda, O.M. , Chin, S.F. , Dunning, M.J. , Gale, D. , Forshew, T. , Mahler-Araujo, B. , Rajan, S. , Humphray, S. , Becq, J. , Halsall, D. , Wallis, M. , Bentley, D. , Caldas, C. , Rosenfeld, N. , 2013. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N. Engl. J. Med. 368, 1199–1209. [DOI] [PubMed] [Google Scholar]
- De Mattos-Arruda, L. , Weigelt, B. , Cortes, J. , Won, H.H. , Ng, C.K. , Nuciforo, P. , Bidard, F.C. , Aura, C. , Saura, C. , Peg, V. , Piscuoglio, S. , Oliveira, M. , Smolders, Y. , Patel, P. , Norton, L. , Tabernero, J. , Berger, M.F. , Seoane, J. , Reis-Filho, J.S. , 2014. Capturing intra-tumor genetic heterogeneity by de novo mutation profiling of circulating cell-free tumor DNA: a proof-of-principle. Ann. Oncol. 25, 1729–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz, L.A. , Williams, R.T. , Wu, J. , Kinde, I. , Hecht, J.R. , Berlin, J. , Allen, B. , Bozic, I. , Reiter, J.G. , Nowak, M.A. , Kinzler, K.W. , Oliner, K.S. , Vogelstein, B. , 2012. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature. 486, 537–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl, F. , Schmidt, K. , Choti, M.A. , Romans, K. , Goodman, S. , Li, M. , Thornton, K. , Agrawal, N. , Sokoll, L. , Szabo, S.A. , Kinzler, K.W. , Vogelstein, B. , Diaz, L.A. , 2008. Circulating mutant DNA to assess tumor dynamics. Nat. Med. 14, 985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy, M.J. , Sturgeon, C. , Lamerz, R. , Haglund, C. , Holubec, V.L. , Klapdor, R. , Nicolini, A. , Topolcan, O. , Heinemann, V. , 2010. Tumor markers in pancreatic cancer: a European Group on Tumor Markers (EGTM) status report. Ann. Oncol. 21, 441–447. [DOI] [PubMed] [Google Scholar]
- Eisenhauer, E.A. , Therasse, P. , Bogaerts, J. , Schwartz, L.H. , Sargent, D. , Ford, R. , Dancey, J. , Arbuck, S. , Gwyther, S. , Mooney, M. , Rubinstein, L. , Shankar, L. , Dodd, L. , Kaplan, R. , Lacombe, D. , Verweij, J. , 2009. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer. 45, 228–247. [DOI] [PubMed] [Google Scholar]
- Forshew, T. , Murtaza, M. , Parkinson, C. , Gale, D. , Tsui, D.W. , Kaper, F. , Dawson, S.J. , Piskorz, A.M. , Jimenez-Linan, M. , Bentley, D. , Hadfield, J. , May, A.P. , Caldas, C. , Brenton, J.D. , Rosenfeld, N. , 2012. Noninvasive identification and monitoring of cancer mutations by targeted deep sequencing of plasma DNA. Sci. Transl Med. 4, 136–168. [DOI] [PubMed] [Google Scholar]
- Gerlinger, M. , Rowan, A.J. , Horswell, S. , Larkin, J. , Endesfelder, D. , Gronroos, E. , Martinez, P. , Matthews, N. , Stewart, A. , Tarpey, P. , Varela, I. , Phillimore, B. , Begum, S. , Mcdonald, N.Q. , Butler, A. , Jones, D. , Raine, K. , Latimer, C. , Santos, C.R. , Nohadani, M. , Eklund, A.C. , Spencer-Dene, B. , Clark, G. , Pickering, L. , Stamp, G. , Gore, M. , Szallasi, Z. , Downward, J. , Futreal, P.A. , Swanton, C. , 2012. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilje, B. , Heikkila, R. , Oltedal, S. , Tjensvoll, K. , Nordgard, O. , 2008. High-fidelity DNA polymerase enhances the sensitivity of a peptide nucleic acid clamp PCR assay for K-ras mutations. J. Mol. Diagn. 10, 325–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein, D. , El-Maraghi, R.H. , Hammel, P. , Heinemann, V. , Kunzmann, V. , Sastre, J. , Scheithauer, W. , Siena, S. , Tabernero, J. , Teixeira, L. , Tortora, G. , Van Laethem, J.L. , Young, R. , Penenberg, D.N. , Lu, B. , Romano, A. , Von Hoff, D.D. , 2015. nab-Paclitaxel plus gemcitabine for metastatic pancreatic cancer: long-term survival from a phase III trial. J. Natl. Cancer Inst. 107, [DOI] [PubMed] [Google Scholar]
- Haber, D.A. , Velculescu, V.E. , 2014. Blood-based analyses of cancer: circulating tumor cells and circulating tumor DNA. Cancer Discov. 4, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helsedirektoratet, 2015. Nasjonalt handlingsprogram med retningslinjer for diagnostikk, behandling og oppfølging av pancreaskreft. [Google Scholar]
- Kidess, E. , Heirich, K. , Wiggin, M. , Vysotskaia, V. , Visser, B.C. , Marziali, A. , Wiedenmann, B. , Norton, J.A. , Lee, M. , Jeffrey, S.S. , Poultsides, G.A. , 2015. Mutation profiling of tumor DNA from plasma and tumor tissue of colorectal cancer patients with a novel, high-sensitivity multiplexed mutation detection platform. Oncotarget. 6, 2549–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinugasa, H. , Nouso, K. , Miyahara, K. , Morimoto, Y. , Dohi, C. , Tsutsumi, K. , Kato, H. , Matsubara, T. , Okada, H. , Yamamoto, K. , 2015. Detection of K-ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer. 121, 2271–2280. [DOI] [PubMed] [Google Scholar]
- Lebofsky, R. , Decraene, C. , Bernard, V. , Kamal, M. , Blin, A. , Leroy, Q. , Rio Frio, T. , Pierron, G. , Callens, C. , Bieche, I. , Saliou, A. , Madic, J. , Rouleau, E. , Bidard, F.C. , Lantz, O. , Stern, M.H. , Le Tourneau, C. , Pierga, J.Y. , 2015. Circulating tumor DNA as a non-invasive substitute to metastasis biopsy for tumor genotyping and personalized medicine in a prospective trial across all tumor types. Mol. Oncol. 9, 783–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, D. , Xie, K. , Wolff, R. , Abbruzzese, J.L. , 2004. Pancreatic cancer. Lancet. 363, 1049–1057. [DOI] [PubMed] [Google Scholar]
- Maire, F. , Micard, S. , Hammel, P. , Voitot, H. , Levy, P. , Cugnenc, P.H. , Ruszniewski, P. , Puig, P.L. , 2002. Differential diagnosis between chronic pancreatitis and pancreatic cancer: value of the detection of KRAS2 mutations in circulating DNA. Br. J. Cancer. 87, 551–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchese, R. , Muleti, A. , Pasqualetti, P. , Bucci, B. , Stigliano, A. , Brunetti, E. , De Angelis, M. , Mazzoni, G. , Tocchi, A. , Brozzetti, S. , 2006. Low correspondence between K-ras mutations in pancreatic cancer tissue and detection of K-ras mutations in circulating DNA. Pancreas. 32, 171–177. [DOI] [PubMed] [Google Scholar]
- Misale, S. , Yaeger, R. , Hobor, S. , Scala, E. , Janakiraman, M. , Liska, D. , Valtorta, E. , Schiavo, R. , Buscarino, M. , Siravegna, G. , Bencardino, K. , Cercek, A. , Chen, C.T. , Veronese, S. , Zanon, C. , Sartore-Bianchi, A. , Gambacorta, M. , Gallicchio, M. , Vakiani, E. , Boscaro, V. , Medico, E. , Weiser, M. , Siena, S. , Di Nicolantonio, F. , Solit, D. , Bardelli, A. , 2012. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature. 486, 532–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman, A.M. , Bratman, S.V. , To, J. , Wynne, J.F. , Eclov, N.C. , Modlin, L.A. , Liu, C.L. , Neal, J.W. , Wakelee, H.A. , Merritt, R.E. , Shrager, J.B. , Loo, B.W. , Alizadeh, A.A. , Diehn, M. , 2014. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat. Med. 20, 548–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltedal, S. , Gilje, B. , Korner, H. , Aasprong, O.G. , Tjensvoll, K. , Heikkila, R. , Smaaland, R. , Nordgard, O. , 2010. Detection of occult metastases in sentinel lymph nodes from colon cancer patients by K-ras mutation peptide nucleic acid clamp PCR. Ann. Surg. 251, 1087–1091. [DOI] [PubMed] [Google Scholar]
- Oltedal, S. , Aasprong, O.G. , Moller, J.H. , Korner, H. , Gilje, B. , Tjensvoll, K. , Birkemeyer, E.M. , Heikkila, R. , Smaaland, R. , Nordgard, O. , 2011. Heterogeneous distribution of K-ras mutations in primary colon carcinomas: implications for EGFR-directed therapy. Int. J. Colorectal Dis. 26, 1271–1277. [DOI] [PubMed] [Google Scholar]
- Punnoose, E.A. , Atwal, S. , Liu, W. , Raja, R. , Fine, B.M. , Hughes, B.G. , Hicks, R.J. , Hampton, G.M. , Amler, L.C. , Pirzkall, A. , Lackner, M.R. , 2012. Evaluation of circulating tumor cells and circulating tumor DNA in non-small cell lung cancer: association with clinical endpoints in a phase II clinical trial of pertuzumab and erlotinib. Clin. Cancer Res. 18, 2391–2401. [DOI] [PubMed] [Google Scholar]
- Reinert, T. , Scholer, L.V. , Thomsen, R. , Tobiasen, H. , Vang, S. , Nordentoft, I. , Lamy, P. , Kannerup, A.S. , Mortensen, F.V. , Stribolt, K. , Hamilton-Dutoit, S. , Nielsen, H.J. , Laurberg, S. , Pallisgaard, N. , Pedersen, J.S. , Orntoft, T.F. , Andersen, C.L. , 2015. Analysis of circulating tumour DNA to monitor disease burden following colorectal cancer surgery. Gut. 10.1136/gutjnl-2014-308859 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Rothe, F. , Laes, J.F. , Lambrechts, D. , Smeets, D. , Vincent, D. , Maetens, M. , Fumagalli, D. , Michiels, S. , Drisis, S. , Moerman, C. , Detiffe, J.P. , Larsimont, D. , Awada, A. , Piccart, M. , Sotiriou, C. , Ignatiadis, M. , 2014. Plasma circulating tumor DNA as an alternative to metastatic biopsies for mutational analysis in breast cancer. Ann. Oncol. 25, 1959–1965. [DOI] [PubMed] [Google Scholar]
- Rothenberg, M.L. , Abbruzzese, J.L. , Moore, M. , Portenoy, R.K. , Robertson, J.M. , Wanebo, H.J. , 1996. A rationale for expanding the endpoints for clinical trials in advanced pancreatic carcinoma. Cancer. 78, 627–632. [DOI] [PubMed] [Google Scholar]
- Sausen, M. , Phallen, J. , Adleff, V. , Jones, S. , Leary, R.J. , Barrett, M.T. , Anagnostou, V. , Parpart-Li, S. , Murphy, D. , Kay Li, Q. , Hruban, C.A. , Scharpf, R. , White, J.R. , O'dwyer, P.J. , Allen, P.J. , Eshleman, J.R. , Thompson, C.B. , Klimstra, D.S. , Linehan, D.C. , Maitra, A. , Hruban, R.H. , Diaz, L.A. , Von Hoff, D.D. , Johansen, J.S. , Drebin, J.A. , Velculescu, V.E. , 2015. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat. Commun. 6, 7686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spindler, K.L. , Pallisgaard, N. , Vogelius, I. , Jakobsen, A. , 2012. Quantitative cell-free DNA, KRAS, and BRAF mutations in plasma from patients with metastatic colorectal cancer during treatment with cetuximab and irinotecan. Clin. Cancer Res. 18, 1177–1185. [DOI] [PubMed] [Google Scholar]
- Taly, V. , Pekin, D. , Benhaim, L. , Kotsopoulos, S.K. , Le Corre, D. , Li, X. , Atochin, I. , Link, D.R. , Griffiths, A.D. , Pallier, K. , Blons, H. , Bouche, O. , Landi, B. , Hutchison, J.B. , Laurent-Puig, P. , 2013. Multiplex picodroplet digital PCR to detect KRAS mutations in circulating DNA from the plasma of colorectal cancer patients. Clin. Chem. 59, 1722–1731. [DOI] [PubMed] [Google Scholar]
- Thakur, B.K. , Zhang, H. , Becker, A. , Matei, I. , Huang, Y. , Costa-Silva, B. , Zheng, Y. , Hoshino, A. , Brazier, H. , Xiang, J. , Williams, C. , Rodriguez-Barrueco, R. , Silva, J.M. , Zhang, W. , Hearn, S. , Elemento, O. , Paknejad, N. , Manova-Todorova, K. , Welte, K. , Bromberg, J. , Peinado, H. , Lyden, D. , 2014. Double-stranded DNA in exosomes: a novel biomarker in cancer detection. Cell Res. 24, 766–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierry, A.R. , Mouliere, F. , El Messaoudi, S. , Mollevi, C. , Lopez-Crapez, E. , Rolet, F. , Gillet, B. , Gongora, C. , Dechelotte, P. , Robert, B. , Del Rio, M. , Lamy, P.J. , Bibeau, F. , Nouaille, M. , Loriot, V. , Jarrousse, A.S. , Molina, F. , Mathonnet, M. , Pezet, D. , Ychou, M. , 2014. Clinical validation of the detection of KRAS and BRAF mutations from circulating tumor DNA. Nat. Med. 20, 430–435. [DOI] [PubMed] [Google Scholar]
- Tjensvoll, K. , Nordgard, O. , Smaaland, R. , 2014. Circulating tumor cells in pancreatic cancer patients: methods of detection and clinical implications. Int. J. Cancer. 134, 1–8. [DOI] [PubMed] [Google Scholar]
- Torre, L.A. , Bray, F. , Siegel, R.L. , Ferlay, J. , Lortet-Tieulent, J. , Jemal, A. , 2015. Global cancer statistics, 2012. CA Cancer J. Clin. 65, 87–108. [DOI] [PubMed] [Google Scholar]
- Von Hoff, D.D. , Ervin, T. , Arena, F.P. , Chiorean, E.G. , Infante, J. , Moore, M. , Seay, T. , Tjulandin, S.A. , Ma, W.W. , Saleh, M.N. , Harris, M. , Reni, M. , Dowden, S. , Laheru, D. , Bahary, N. , Ramanathan, R.K. , Tabernero, J. , Hidalgo, M. , Goldstein, D. , Van Cutsem, E. , Wei, X. , Iglesias, J. , Renschler, M.F. , 2013. Increased survival in pancreatic cancer with nab-paclitaxel plus gemcitabine. N. Engl. J. Med. 369, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachida, S. , Jones, S. , Bozic, I. , Antal, T. , Leary, R. , Fu, B. , Kamiyama, M. , Hruban, R.H. , Eshleman, J.R. , Nowak, M.A. , Velculescu, V.E. , Kinzler, K.W. , Vogelstein, B. , Iacobuzio-Donahue, C.A. , 2010. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 467, 1114–1117. (England) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zill, O.A. , Greene, C. , Sebisanovic, D. , Siew, L. , Leng, J. , Vu, M. , Hendifar, A.E. , Wang, Z. , Atreya, C.E. , Kelley, R.K. , Van Loon, K. , Ko, A.H. , Tempero, M.A. , Bivona, T.G. , Munster, P.N. , Talasaz, A. , Collisson, E. , 2015. Cell-free DNA next-generation sequencing in pancreatobiliary carcinomas. Cancer Discov. 5, 1040–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]