

Abstract
The tumor suppressor p53 plays a key role in malignant transformation and tumor development. However, the frequency of p53 mutations within individual types of cancer is different, suggesting the existence of other mechanisms attenuating p53 tumor suppressor activity. Changes in upstream regulators of p53 such as MDM2 amplification and overexpression, expression of viral oncoproteins, estrogen receptor signaling, or changes in p53 transcriptional target genes were previously described in wild‐type p53 tumors. We identified a novel pathway responsible for attenuation of p53 activity in human cancers. We demonstrate that AGR2, which is overexpressed in a variety of human cancers and provides a poor prognosis, up‐regulates DUSP10 which subsequently inhibits p38 MAPK and prevents p53 activation by phosphorylation. Analysis of human breast cancers reveals that AGR2 specifically provides a poor prognosis in ER+ breast cancers with wild‐type p53 but not ER‐ or mutant p53 breast cancers, and analysis of independent data sets show that DUSP10 levels also have prognostic significance in this specific sub‐group of patients. These data not only reveal a novel pro‐oncogenic signaling pathway mediating resistance to DNA damaging agents in human tumors, but also has implications for designing alternative strategies for modulation of wild‐type p53 activity in cancer therapy.
Keywords: AGR2, p53, p38 MAPK, DUSP10, Drug resistance, Breast cancer
Highlights
The transcription factor p53 is the most intensively studied tumor suppressor.
AGR2‐dependent signaling pathway blocks wt p53 transcriptional activity.
p53 activity is impaired via DUSP10 up‐regulation causing inhibition of p38 MAPK.
AGR2 signaling towards p53 was demonstrated both in vitro and in vivo.
1. Introduction
Wild‐type (wt) p53 is a sequence‐specific DNA‐binding protein and stress‐activated transcription factor. Depending on cell type and stress stimuli, p53 plays multiple roles in many cellular processes, including cell cycle arrest, apoptosis, differentiation and senescence (Lane, 1992). The main role of p53 is the maintenance of genome stability and integrity, therefore p53 inactivation represents a crucial step in tumor development and progression. The role of p53 as a tumor suppressor is reflected by its high rate of mutation in human tumors. Mutations in p53 act as useful markers for the follow‐up of minimal residual disease, for comparison between primary and recurrent tumors, for tracing the origin of distant metastases and to determine tumor clonality (Olivier et al., 2010). Moreover, mutations in p53 have been consistently associated with poor prognosis in many cancers including breast, colorectal, head and neck, leukemia and others (Petitjean et al., 2007).
In cancers where wt p53 is present, the p53 network is usually altered by other genetic or epigenetic events that compromise the p53 response. The most studied is p53‐inducible E3 ubiquitin ligase MDM2 that promotes p53 proteasomal degradation through an ubiquitin‐dependent pathway (Haupt et al., 1997). Although MDM2 protein overexpression has also been found in the absence of gene amplification, it generally results from gene amplification observed in ∼7% of all human cancers (Forslund et al., 2008). Another RING finger ubiquitin ligase involved in regulation of p53 is MDMX (MDM4), which regulates MDM2 activity (Wade et al., 2013). The causative viruses of human cancer possess several distinct mechanisms to inactivate p53 functions and signaling by the alterations of post‐transcriptional modifications, localization, binding partners, turnover, and transcriptional activity (Sato and Tsurumi, 2013). Additional mechanisms resulting in attenuation of wt p53 activity are mutations of its upstream activators e.g. ATM or CHK2 (Banin et al., 1998; Hublarova et al., 2010). A strong relationship of estrogen signaling with p53 has also been documented. Activated estrogen receptors (ER) were shown to antagonize p53 function either directly through the recruitment of co‐repressors (Konduri et al., 2010), or via independent inhibition of genes responsible for execution of the p53‐triggered apoptotic program in response to DNA damage (Bailey et al., 2012).
In 1998, co‐expression of the AGR2 (anterior gradient‐2) gene and ER was detected in mammary gland cancer cells (Thompson and Weigel, 1998). Later, it was shown that AGR2 expression is directly stimulated by ER signaling (Salmans et al., 2013) and correlates with poor outcome of patients with ER‐positive breast cancer (Hrstka et al., 2010; Innes et al., 2006), which emphasizes the importance of estrogen receptors in the regulation of AGR2 expression. From a functional perspective, AGR2 was shown to promote tumor metastasis, cell survival, cell proliferation and resistance to therapy (Hrstka et al., 2013; Ramachandran et al., 2008; Wang et al., 2008). In addition to breast cancer, AGR2 was also detected in many other adenocarcinomas (Brychtova et al., 2011). Proteomic analysis of Barrett's epithelium, where alterations in TP53 gene function are observed at high frequency, identified AGR2 protein as over‐produced in pre‐neoplastic cells. Subsequent data revealed that AGR2 functions as a pro‐oncogenic survival factor that inhibits the p53 response to UV radiation (Pohler et al., 2004). However the mechanism by which AGR2 attenuates p53 activity is unknown.
In our work, we identify a novel signaling pathway where AGR2 exerts its inhibitory effect on p53 through attenuation of p38 mitogen‐activated protein kinase (p38 MAPK) activity. The anti‐cancer activity of many chemotherapy drugs relies on the induction of DNA damage and followed cellular response triggered by p53. Thus, the discovery of novel p53‐inhibitory targets may significantly contribute to drug development programs. Our data demonstrate an important role for AGR2 as a pro‐survival factor responsible for enhanced resistance to DNA damaging agents, proposing AGR2 as a target for the development of future treatments to improve anti‐cancer therapy.
2. Experimental procedures
2.1. Cell cultures and treatments
The following AGR2‐positive cell lines were used: MCF‐7 (breast cancer, ER‐positive, wt p53) and A549 (lung cancer, ER‐negative, wt p53). AGR2‐negative cell lines included in the study were H1299 (lung cancer, ER‐negative, p53 null) and ARN8 cells (clone derived from A375 cells, malignant melanoma, ER‐negative, wt p53, stably transfected with a p53 dependent β‐galactosidase reporter construct) (Blaydes and Hupp, 1998). All cell lines were cultured in glucose‐rich D‐MEM supplemented with 10% fetal bovine serum at 37 °C in a humidified atmosphere of 5% CO2. The Flp‐In™ System (Invitrogen) was used to generate H1299‐LZ4 cells containing a single integrated Flp Recombination Target (FRT) site. The coding sequence of the human AGR2 gene was stably inserted into this site using Flp recombinase mediated site‐specific DNA recombination to give H1299‐LZ4‐AGR2 cell line. To establish the knockout of all 4 copies of AGR2 in A549 cells (A549‐AGR2‐KO), TALEN (transcription activator‐like effector nucleases) pair was constructed to target AGR2 coding sequence at position 12–63 from start codon as described previously (Kim et al., 2013). TALEN expression plasmids bearing AGR2 targeting sequences were further delivered into A549 cells. Transfected cells were diluted and plated at an average density of 0.5 cells per well into a 96‐well plate. Colonies were immunochemically tested for AGR2 expression. The AGR2 non‐expressing clone denoted as A549‐AGR2‐KO was used for further experiments.
25 pmol AGR2 specific silencing RNA and 25 pmol untargeted siRNA as a control (Dharmacon, Thermo Fisher Scientific, Waltham, MA, USA) per million cells were used for transfection by AMAXA system (Lonza Group Ltd., Basel, Switzerland). Unless stated otherwise, 32 h post‐transfection the cells were exposed to 10 μM cisplatin or 0.5 μM doxorubicin (both Adriablastica, Ebewe, Unterach, Austria). After 16 h cells were harvested and lysed in NP‐40 lysis buffer [150 mM NaCl, 1% NP‐40, 50 mM TrisCl pH 8.0, 50 mM NaF, 5 mM EDTA pH 8.0 supplemented with 100× diluted protease and phosphatase inhibitors (Sigma–Aldrich Inc., St. Louis, MO, USA)].
2.2. Determination of protein expression
Immunoblotting was carried out as described previously (Hublarova et al., 2010). The membranes were probed overnight with in‐house specific anti‐p53 (DO‐1), (Vojtesek et al., 1992) anti‐p53‐S392 (FP392) (Blaydes and Hupp, 1998), anti‐MDM2 (Ab2A9) (Chen et al., 1993), anti‐p53‐S15 and anti‐p38 MAPK antibody (both Cell Signaling Technology Inc., Danvers, USA), anti‐actin AC‐40 (Sigma–Aldrich Inc., St. Louis, MO, USA), anti‐DUSP10 (Abcam, Cambridge, UK), anti‐p21WAF−1 (Ab‐1) and anti‐PARP‐1 (Ab‐2) (both Merck Millipore, Darmstadt, Germany) monoclonal antibodies, AGR2 was detected by our in‐house rabbit polyclonal antibody K47 (Hrstka et al., 2010). p38 MAPK activity was measured using p38 MAP Kinase Assay Kit (Cell Signaling Technology Inc., Danvers, USA) according to the manufacturer's instructions. Briefly, cells were transiently transfected with siRNA and subsequently exposed to cisplatin or doxorubicin. Phosphorylated (active) p38 MAP kinase was immunoprecipitated from whole cell lysates. The phosphorylation of ATF‐2 fusion protein on Thr71 caused by selectively immunoprecipitated phosphorylated p38 MAP kinase in the presence of ATP was determined. In parallel, β‐actin and AGR2 protein levels were determined to check correct protein loading and AGR2 silencing efficiency respectively. Relative fold changes in protein levels were determined using TotalLab software (Nonlinear dynamics, Newcastle upon Tyne, UK).
2.3. β‐galactosidase colorimetric assay
ARN8 cells were transfected and treated as described above and harvested using 200 μl 0.25 M TrisCl pH 7.5, cell suspensions were sonicated 3× for 5 s on ice and then centrifuged 30 min/14,000 RPM/4 °C. 150 μg of total protein was added to substrate mixture (0.001 M MgCl2, 0.045 M β‐mercaptoethanol, 0.264 mg o‐nitrophenyl‐β‐d‐galactopyranoside, 0.1 M sodium phosphate pH 7.5) in total volume of 300 μl per reaction. The reactions were incubated 30 min at 37 °C and then were stopped by 500 μl of 1 M Na2CO3, absorbance was measured at 420 nm. All samples were measured in triplicate.
2.4. Reverse transcription and quantitative PCR
Total cellular RNA was extracted by TRI‐Reagent (MRC, Cincinnati, OH, USA). cDNA synthesis was carried out using M‐MLV reverse transcriptase (Invitrogen, Carlsbad, CA, USA). Triplicate samples were subjected to quantitative PCR analysis using TaqMan Array Human MAPK pathways plates containing 92 assays to MAPK associated genes and 4 endogenous control genes (Applied Biosystems, Foster City, CA, USA). Data were analyzed using StatMiner (Integromics, Madrid, Spain). Quantitative PCR analysis using TaqMan for 18S rRNA (Applied Biosystems, Foster City, CA, USA) and SYBR Green (Sigma–Aldrich, St Louis, MO, USA) for AGR2 was performed as described previously (Hrstka et al., 2013).
2.5. Cellular viability and proliferation
MCF‐7 and A549 cells transfected with siRNA and H1299 cells transfected with corresponding plasmid were seeded into 96‐well plates using 10,000 cells per well. After 24 h incubation, doxorubicin and cisplatin were applied for an additional 48 h. 20 μl MTT reagent (2.5 mg/ml in PBS) was added after incubation with drugs and cells were incubated 3 h at 37 °C, followed by addition of 100 μl 10% SDS at room temperature overnight to dissolve violet formazan. Finally, absorbance at 595 nm was measured. Each sample was measured in five technical replicates.
In colony formation assays, one million H1299 cells were transfected with 1 μg of the construct bearing either AGR2 or p53 coding sequence. Co‐transfection by both plasmids was conducted with 0.5 μg of each construct. In total, 500 transfected cells were seeded per well of six‐well plates, 24 h later the media was changed with fresh media or media containing cisplatin or doxorubicin. Two weeks later, the cells were washed twice with PBS, fixed for 30 min in methanol and then visualized by Giemsa–Romanowski staining and quantified using TotalLab (Nonlinear dynamics, Newcastle upon Tyne, UK).
Cell proliferation and cytotoxicity of H1299‐LZ4 or H1299‐LZ4‐AGR2 cells (mock or p53 transfected) were performed on E‐plates using RTCA‐DP instrument (Roche Diagnostics GmbH, Mannheim, Germany). After transfection cells were seeded into proliferation 16‐well plates at 5,000 cells per well. After 12 h post‐seeding low doses of cisplatin and doxorubicin were applied in quadruplicate within the same experiment and proliferation was measured. Cell index was monitored every 15 min during the experiment.
2.6. Clinical samples and processing
Our hospital‐based study included 115 primary breast cancer tissue samples obtained from female patients diagnosed at Masaryk Memorial Cancer Institute (MMCI) in 2002–2005 (age 29–79 years). The study was approved by the local ethics committee of the MMCI and informed consent was obtained from each patient. All selected patients received anthracycline‐containing adjuvant chemotherapy regimens. Samples were collected within 20 min of surgical removal and immediately evaluated by a pathologist according to standardized hospital protocol. Portions of tissues were used for preparation of formalin fixed paraffin embedded tissue blocks and separate tumor pieces of ∼3 × 3 × 8 mm were stored in RNA later for 3–5 days at 4 °C and then frozen at −80 °C. RNA purification as well as determination of p53 status was carried out as described previously (Dobes et al., 2014). The main clinicopathological variables including ER and progesterone receptor (PR) status were extracted from pathological records obtained from the MMCI database. AGR2 immunohistochemical staining was performed on 4 μm thick freshly cut tissue sections. Sections were deparaffinized in xylene and rehydrated into PBS through a graded ethanol series. Endogenous peroxidase activity was quenched in 3% hydrogen peroxide in PBS for 15 min. Antigen retrieval was performed in citrate buffer pH 6.0 at 94 °C for 20 min. The sections were incubated overnight at 4 °C with anti‐AGR2 antibody (HPA007912, Sigma–Aldrich, St. Louis, MO, USA). We used the semiquantitative H‐score with a dynamic range of 0 (no staining) to 300 (diffuse intense staining), where intensity of staining is recorded on a scale of 0–3 (0 = none, 1 = weak, 2 = moderate, 3 = strong). The H‐score is calculated as the percentages of cells stained at each intensity level multiplied by that intensity level and then summed. A score <50 was considered as a negative (AGR2‐).
2.7. Ex vivo chemosensitivity assay
A portion of a patient's solid tumor, about the size of a core biopsy, was mechanically disaggregated and established in primary culture. Cellular viability was determined by Trypan blue dye exclusion test. The cell suspension was than diluted to 0.8–1.0 × 106/ml in culture medium: RPMI‐1640 (Sigma–Aldrich Inc., St. Louis, MO, USA) supplemented with 15% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 300 mg/ml glutamine, 0.3 U/ml R‐Humulin, 10 μg/ml holo‐Transferrin and 5.6 μg/ml Amphotericin‐B. Ex vivo sensitivity of isolated tumor cells to anti‐cancer drugs was determined according to Mosmann's protocol (Mosmann, 1983) with several modifications. Briefly, six final concentrations of each tested agent (including doxorubicin which was tested preferentially in case of a lower number of cells) were tested in triplicate, negative control (wells with culture medium only) and positive controls (wells with cell suspension without drug) were used as well. Tumor cells were incubated for 72 h at 37 °C/5% CO2 and analyzed by MTT assay. The measured values of the absorbance were converted into the program Chemorezist 1.0 (Regner et al., 2000) and the dose response curves exported for each agent tested. The EC50 value was derived from the average response curve and the samples evaluated as resistant: EC50 > 1/16 × maximal tested concentration of the drug (cmax); partially sensitive: EC50 = 1/16–1/32 × cmax; sensitive: EC50 < 1/32 × cmax.
3. Results
3.1. AGR2‐dependent inhibition of p53 activity
The tumor suppressor wt p53 is critically important for the cellular response to DNA damage, including that caused by chemotherapy. We investigated the potential links between AGR2, chemotherapy, and p53 activation, based on the previous observations linking AGR2 with resistance to therapy. First, we analyzed the effect of cisplatin and doxorubicin treatment on AGR2 protein level in A549 and MCF‐7 cell lines (Figure 1). Although induction of AGR2 expression was observed predominantly in MCF‐7, probably due to a different cellular content, these results encouraged us to assess the effect of AGR2 on p53 in tumor cells exposed to DNA damaging drugs. To generalize the effects of AGR2, two different cancer cell lines A549 and MCF‐7 (both expressing AGR2 and wt p53) were studied. Both cell lines were pretreated with siRNA against AGR2 for 32 h and then exposed for 16 h to cisplatin or doxorubicin. Treatment with these drugs induced p53 levels, along with Ser15 and Ser392 phosphorylation, especially in response to doxorubicin treatment. Simultaneously, the level of p53 downstream regulated genes MDM2 and CDKN1A (encoding p21WAF−1) were elevated, indicating the induction of p53 transcriptional activity (Figure 1, Figure S1). However, AGR2‐depleted cells showed clearly increased p53 level in comparison with cells pretreated by control siRNA (in A549: 1.5× for untreated cells, 1.2× in response to cisplatin, 2.3× in response to doxorubicin; in MCF‐7: 2× for untreated cells, 2.1× in response to cisplatin and 1.7× in response to doxorubicin). Similar effects were observed also for phosphorylation at Ser15 and Ser392, as well as in the levels of p53 target genes, which were higher in cells pretreated with AGR2 specific siRNA, indicating enhanced transcriptional activity of p53 in response to AGR2 depletion (Figure 1). More than two‐fold increase in the level of MDM2 has been observed in AGR2 silenced A549 cells exposed to both cisplatin and doxorubicin in comparison to cells pretreated with control siRNA, while p21WAF−1 was induced by doxorubicin only, but again 1.2× more after AGR2 silencing. In MCF‐7 cells the effect was clear predominantly in response to cisplatin treatment, where 3.3× for MDM2 and 1.6× stronger induction for p21WAF−1 was observed after AGR2 depletion (Figure S1).
Figure 1.
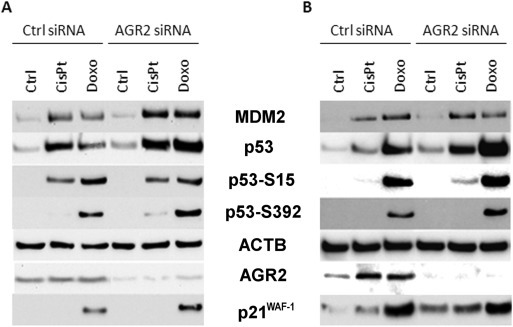
The effect of AGR2 level on p53 and its downstream regulated proteins. The figure shows representative Western blots of A549 cells (A) and MCF‐7 cells (B) transfected either with control or AGR2 specific siRNA and treated with cisplatin or doxorubicin. The levels of p53, p53 phosphorylation at Ser15 and Ser392, MDM2 and p21WAF−1 protein were determined. Beta‐actin (ACTB) was used as a loading control, AGR2 expression was determined to confirm siRNA efficacy. Biologically independent experiments were conducted at least 3 times with very similar results. The changes in the protein levels corresponding to this figure are graphically quantified in Figure S1.
To demonstrate that AGR2 attenuates the transcriptional activity of p53, ARN8 cells carrying p53‐responsive β‐galactosidase reporter were used. These cells were transiently transfected with AGR2 expressing plasmid and subsequently exposed to doxorubicin and cisplatin. AGR2 transfection clearly decreased transactivation of the p53‐responsive β‐galactosidase reporter in comparison with mock transfected cells (Figure 2). Taken together, our data show that AGR2 reduces endogenous p53 levels, post‐translational modification at Ser15 and Ser392 and transcriptional activity in response to both cisplatin and doxorubicin.
Figure 2.
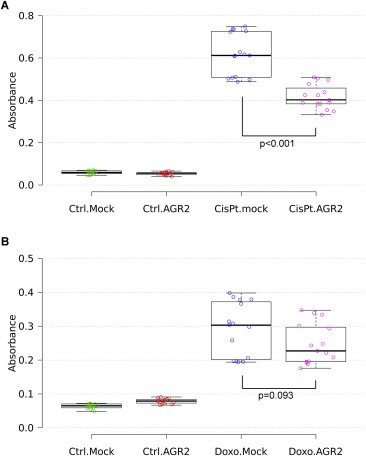
AGR2 blocks p53 transactivation. ARN8 cells bearing β‐galactosidase reporter construct were transiently transfected with either empty pcDNA3‐GW plasmid or carrying the coding sequence for AGR2 (2 μg of plasmid per million cells using Amaxa nucleofector). Mock or AGR2 transfected cells were treated with either cisplatin (A) or doxorubicin (B). Determination of p53 transcriptional activity of p53 was conducted using β‐galactosidase colorimetric assay in three independent biological replicates, each performed in quintuplicate. Statistical significance of differences in β‐galactosidase activity was calculated using One‐way Anova test.
3.2. Identification of AGR2‐dependent mechanism responsible for inhibition of p53
To identify downstream effectors of AGR2 signaling pathway, we screened mRNA levels of 92 key genes known to be involved in MAPK signaling in both A549 and MCF‐7 cells pretreated either with control siRNA or AGR2 specific siRNA and then exposed to cisplatin or doxorubicin (Table S1). Based on the criteria that changes in gene expression must be of the same nature in response to both cisplatin and doxorubicin and that the extent of these changes should be at least 20%, we found only one gene, Dual Specificity Phosphatase 10 (DUSP10) also known as MAP Kinase Phosphatase 5 (MKP5). DUSP10 mRNA levels were reduced in both MCF‐7 and A549 in response to AGR2 depletion (Figure 3A and B). The involvement of AGR2 in maintaining DUSP10 levels was then confirmed in biologically independent experiments using RT‐qPCR (data not shown) and at the protein level. Decreased DUSP10 protein levels were seen predominantly in control MCF‐7 cells exposed only to AGR2 siRNA in comparison with A549 cells showing only negligible reduction of DUSP10. However, the effect was more pronounced in response to both cisplatin and doxorubicin and a clear decrease in DUSP10 expression was observed in both cell lines (Figure 3C and D).
Figure 3.
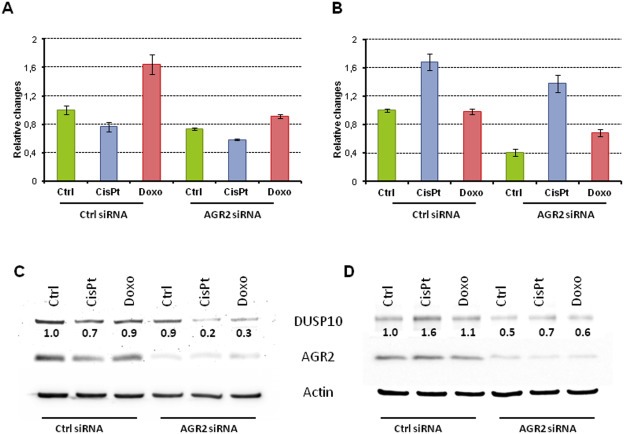
AGR2 influences DUSP10 expression. Inhibition of AGR2 decreases DUSP10 mRNA in both A549 (A) and MCF‐7 (B) cells. Protein levels show similar trends in both A549 (C) and MCF‐7 (D) cells. For this representative figure, the relative fold changes in DUSP10 expression in relation to untreated cells with endogenous AGR2 expression are indicated under corresponding bands.
To investigate the link between DUSP10 and AGR2 in relation to p53 activity, A549, A549‐AGR2‐KO (AGR2 homozygous knockout) and MCF‐7 cells were transfected with DUSP10 expression plasmid and exposed to either cisplatin or doxorubicin (Figure 4). In response to these drugs two to three times lower induction of p53 was observed in DUSP10 transfected A549 and MCF‐7 cells compared to their mock transfected counterparts (Figure 4, Figure S2). A similar effect was observed for p53 phosphorylation, as well as expression of p21WAF−1 and MDM2. Interestingly, the introduction of DUSP10 into A549‐AGR2‐KO cells also attenuated p53 activity, when compared with mock transfected A549‐AGR2‐KO cells, but to a lesser extent (e.g. decrease in total p53 level 1.2× for cisplatin and 1.4× for doxorubicin treatment) than in parental A549 cells with endogenous AGR2 expression (Figure 4, Figure S3 – highlighted in blue), indicating potential role of AGR2 in DUSP10 mediated attenuation of p53. A synergistic effect of AGR2 and DUSP10 is also supported by the finding that the level of p53 is higher in A549‐AGR2‐KO cells than in parental AGR2‐positive cells, in both mock and DUSP10 transfected cells. Similar results were obtained for posttranslational modifications and expression of the p53 downstream targets p21WAF−1 and MDM2. The results from this experiment correspond with the findings shown in Figure 1 and similar trends are observed for all three cellular models used, providing internal validation of the data.
Figure 4.
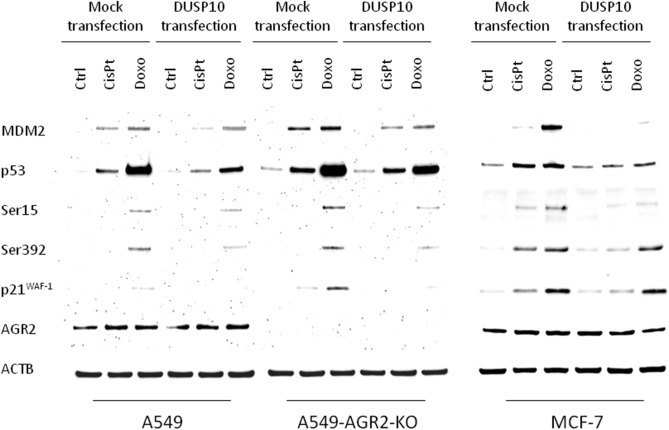
DUSP10 expression directly influences p53. A549, A549‐AGR2‐KO and MCF‐7 cells were either mock or pcDNA3‐GW‐ DUSP10 transfected to determine the total level of p53, phosphorylation at Ser15 and Ser392, as well as the expression of p53 downstream regulated proteins p21WAF−1 and MDM2. ACTB normalized protein levels are shown graphically in Figures S2 and S3.
Dual specificity protein phosphatases inactivate their target kinases by dephosphorylating both phosphoserine/threonine and phosphotyrosine residues. DUSP10 shows substrate selectivity towards the stress activated MAP kinases, predominately p38 MAPK and c‐Jun amino‐terminal kinase (JNK) (Owens and Keyse, 2007). Given that p38 MAPK phosphorylates p53 at Ser15 and Ser392 (Lim et al., 2007), we studied p38 MAPK as a potential effector of the AGR2‐dependent inhibitory pathway towards p53. p38 MAPK protein levels were immunochemically determined in both AGR2 expressing and AGR2‐siRNA treated cells (Figure 5). Since p38 MAPK protein levels do not necessarily reflect catalytic activity, we determined p38 MAPK activity using a nonradioactive IP‐kinase assay in the same lysates. This assay clearly demonstrated increased phosphorylation of ATF‐2 fusion protein by immobilized p38MAPK in response to both cisplatin and doxorubicin treatment. This effect was significantly enhanced when AGR2 was silenced (Figure 5). Increased catalytic activity even in the absence of increased p38MAPK protein levels after AGR2 silencing supports a role of AGR2 on DUSP10 expression, which in turn leads to inhibition of p38 MAPK activity. Taken together these data indicate a novel AGR2‐triggered signaling pathway that attenuates p53 activity in response to DNA damage (Figure 6).
Figure 5.
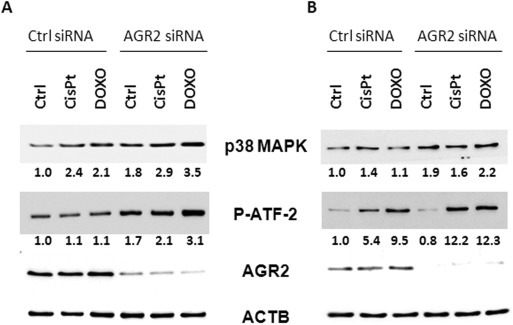
Inhibition of AGR2 increases p38 MAPK activity. A549 (A) and MCF‐7 cells (B) were transfected either with control or AGR2 specific siRNA and then exposed to cisplatin or doxorubicin. Changes in p38 MAPK activity directly correspond to fold changes in degree of phosphorylation of ATF‐2 fusion protein. Relative fold changes in amounts of phosphorylated ATF‐2 fusion protein as well as p38MAPK are indicated under corresponding bands. These numbers were determined after normalization to ACTB in relation to untreated cells with unaffected endogenous AGR2 expression. This is a representative figure of two independent determinations of p38 MAPK expression and activity.
Figure 6.
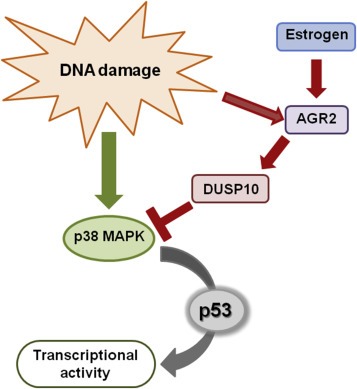
Schematic representation of AGR2 signaling pathway. In response to DNA damage and other stimuli, AGR2 expression is elevated and induces DUSP10 expression. This process results in p38 MAPK inhibition and impaired p53 activity.
3.3. AGR2 mediates drug resistance in vitro and in vivo
The identification of an AGR2‐dependent signaling pathway responsible for attenuation of wt p53 transcriptional activity led us to investigate whether AGR2 expression may contribute to increased cell survival in response to chemotherapy, whose efficiency can greatly depend on the activity of p53. We used the MTT assay in A549 and MCF‐7 cells either transfected with siRNA against AGR2 or control siRNA and then exposed to either cisplatin or doxorubicin. Increased drug sensitivity was observed in AGR2‐depleted cells (Figure S4). A similar trend was observed for the ability of AGR2 to block p53 functions and to rescue development of colonies (Figure S5). Briefly, clonogenic assays with H1299 cells (AGR2 and p53 negative) transfected either with TP53 or co‐transfected with TP53 and AGR2 together demonstrated increased colony development in cells with both TP53 and AGR2 in comparison with H1299 cells expressing p53 only.
The pro‐survival potential of AGR2 was also determined using real time monitoring of cell viability and proliferation kinetics using xCELLigence system. The proliferation of H1299‐LZ4 cells (AGR2 negative) and H1299‐LZ4‐AGR2 (stably expressing AGR2) after transient transfection by expression vector coding p53 was monitored. The rate of proliferation in untreated H1299 cells was very similar for both AGR2‐negative and ‐positive H1299 cells. Dramatic changes in proliferation were observed after introduction of wt p53, since H1299‐LZ4‐AGR2 cells were able to partially attenuate p53 activity and keep cellular proliferation, albeit with lower intensity (Figure S6A – black curve). When the cells were exposed to cisplatin and doxorubicin, we observed similar trends for both drugs (Figure S6B, C). AGR2 expressing cells showed increased proliferation rates compared to H1299‐LZ4 cells, highlighting AGR2's pro‐proliferative and pro‐survival potential. In the presence of drugs, AGR2 rescued cells transfected by wt p53 even with increased efficiency compared to untreated cells, demonstrating that AGR2's inhibitory effect towards p53 is enhanced in response to genotoxic stress.
AGR2 expression in ex vivo cultured cells from breast cancer patients was determined to analyze the ability of AGR2 to mediate resistance to cytotoxic agents in primary cancers. This clinical study focused on chemosensitivity prediction of ex vivo cultivated malignant breast cancer cells was conducted at MMCI from August 2003 to September 2005 (Michalova et al., 2008; Poprach et al., 2008). In total 31 successfully evaluated breast cancer samples enabled us to determine AGR2 expression in relation to doxorubicin sensitivity. We found significantly higher AGR2 mRNA level in tumor samples evaluated as doxorubicin insensitive (p = 0.022, Mann–Whitney U Test), supporting the role of AGR2 in resistance to DNA damaging drugs (Figure S7).
The effect of AGR2 expression determined by immunohistochemical staining on outcome of breast cancer patients treated by anthracyclines was evaluated in a cohort of 115 consecutive patients. No significant effect of AGR2 level on patients' outcome was found (p = 0.313, Log Rank test) (Figure S8A). Taking into account that AGR2 exerts its oncogenic functions via wt p53 inhibition, we focused only on tumors with wt p53 (80 cases) from the cohort of 115 patients, but again did not find a significant difference in overall survival with respect to AGR2 (p = 0.159, Log Rank test) (Figure S8B). Since ER status represents an important positive prognostic factor and was shown to regulate AGR2 expression, only wt p53 and concurrently ER‐positive cases were selected. In this group of 67 patients, we found significantly worse overall survival of patients with AGR2 over‐expressing tumors (p = 0.043, Log Rank test) (Figure 7).
Figure 7.
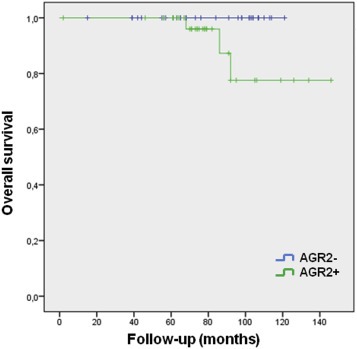
Kaplan–Meier plots in relation to AGR2 expression. Overall survival of 67 patients with both positive ER and wt p53 selected from group of 115 consecutive breast cancer patients who received anthracyclines in adjuvant treatment.
To support our clinical results, the patients' outcome was analyzed in relation to expression of AGR2 and DUSP10 using the online survival analysis software Kaplan–Meier Plotter (http://kmplot.com/analysis/index.php?p=service) to assess the prognostic value of selected biomarkers using transcriptomic data in breast cancer patients (Gyorffy et al., 2010). Following our selection criteria involving wt p53 and ER‐positivity (n = 234) we found both AGR2 (p = 0.034) and DUSP10 (p = 0.002) to have significant impact on patients' relapse free survival (Figure S9). On the other hand and similarly to our clinical data (Figure S8), the evaluation of AGR2 and DUSP10 expression in the whole cohort of 3554 breast tumors showed even a reverse trend for AGR2 and an order of magnitude less significance for DUSP10 (Figure S10).
4. Discussion
Unremitting efforts in p53 research over the past 35 years clearly illustrate that pathways driven by p53 or controlling p53 represent promising targets of newly developed anti‐cancer therapeutics. The regulation of wt p53 is predominantly accomplished through two main mechanisms, comprising regulation of p53 stability by a series of distinct E3 ligases (Lee and Gu, 2010) and modulation of transcriptional activity via a myriad of posttranslational modifications including, but probably not limited to, phosphorylation, acetylation, methylation, glycosylation, ubiquitination, neddylation, sumoylation and poly‐ribosylation (Kruse and Gu, 2009). This implies that p53 is under tight regulation by a large number of both negative and positive signals. Interestingly, many p53 negative regulators are in parallel p53 target genes forming auto‐regulatory negative feedback loops with p53 (Harris and Levine, 2005). Overexpression and/or amplification of these negative regulators is proposed to attenuate p53 functions and promote tumorigenesis and indeed have been observed frequently in tumors (Jiang et al., 2015; Yu et al., 2014).
Recent comprehensive proteomic technologies provide the opportunity to discover novel, clinically relevant oncogenic pathways that would have been over‐looked by cancer gene screens of the past. One of these recently discovered oncogenes related to p53 is AGR2. AGR2 induces metastasis, acts as a survival factor mediating drug resistance and has a direct involvement in malignant transformation (Brychtova et al., 2011). The ability of AGR2 to inhibit p53 represents another key role of AGR2 in human malignancies.
AGR2 expression has been described in a range of human malignancies including breast tumors, where the regulation of AGR2 expression is best described. Breast cancer encompasses a group of heterogeneous diseases where chemotherapy represents a standard treatment option predominantly for high risk patients, who benefit significantly from the addition of chemotherapy to endocrine treatment (Albain et al., 2010; Paik et al., 2006). p53 plays an important role in sensitivity to chemotherapy, as illustrated by many studies linking TP53 mutations to drug resistance (Dobes et al., 2014; Chrisanthar et al., 2011). TP53 mutations have especially been linked to resistance towards DNA damaging agents such as anthracyclines and mitomycin in breast cancer. Although alterations in TP53 are observed at a relatively high frequency, it is reasoned that many other pathways, mainly in tumors with wt p53, may contribute to inhibition of this tumor suppressor. This is consistent with the fact that 70–80% of breast carcinomas are ER‐positive and most of these express wt p53 (Olivier et al., 2006). One explanation could be that ER‐dependent signaling, including AGR2 upregulation, actively participates in attenuation of p53 activity and in this way contributes to the malignant phenotype in the early stages of breast cancer development. Conversely, in advanced breast tumors where endocrine resistance is usually developed, p53 is almost always mutated (Bouchalova et al., 2014; Yamashita et al., 2006). This is consistent with numerous studies showing crosstalk between p53 and ER resulting in repression of p53 activity (Bailey et al., 2012; Konduri et al., 2010; Liu et al., 2009; Sayeed et al., 2007). We and others have demonstrated that AGR2 expression is directly regulated by estrogens and is associated with poor outcomes for ER‐positive breast cancer patients (Hrstka et al., 2010; Innes et al., 2006; Thompson and Weigel, 1998). These findings indicate that ER‐dependent induction of AGR2 may contribute to attenuation of p53 activity. Interestingly, tamoxifen induces AGR2 both in vivo and in vitro (Hengel et al., 2011; Hrstka et al., 2010), which is in accordance with observations that not only estrogen signaling via ER but also tamoxifen treatment may inhibit p53 activity (Bailey et al., 2012). On the other hand, to generalize inhibitory effect of AGR2 towards p53 in response to DNA damage, we used A549 cells to confirm that AGR2 blocks p53 transcriptional activity also independently of ER signaling, albeit with varying involvement of other corresponding signaling pathways with respect to cellular context.
Based on the critical role of p53 in carcinogenesis and response to genotoxic therapies, clarification and detailed description of the mechanism by which AGR2 exerts its p53 inhibitory activity may have impact on therapeutic strategies for the treatment of cancer with respect to AGR2 and p53 status. We found that AGR2 up‐regulates DUSP10 mRNA, whose product belongs to a subset of protein tyrosine phosphatases responsible for dephosphorylation of threonine and tyrosine residues on MAPKs (Jeffrey et al., 2007), while DUSP10 exhibits increased substrate selectivity for p38 MAPK (Theodosiou et al., 1999). With this in mind, it is reasonable to assume that AGR2 induces DUSP10 expression that subsequently inhibits p38 MAPK, well known to be involved in regulation of p53 via post‐translational modifications enabling stabilization and activation of p53. Taken together, we have discovered a novel mechanism that contributes to AGR2‐mediated oncogenic and anti‐therapeutic effects through attenuating p53 transcriptional activity in response to DNA damage.
In summary, DNA damaging drugs are frequently used for the treatment of many human cancers, including breast, however resistance is often observed. Identification of this novel mechanism, through which AGR2 inhibits p53, may lead to development of useful diagnostic and therapeutic approaches focused on activation of p53 tumor suppressor. In accordance, AGR2 was shown to contribute to cell death resistance induced by the commonly used DNA damaging agents both in vitro and in mouse xenografts (Hengel et al., 2011; Ramachandran et al., 2008; Simpson et al., 2010). Our study extends these findings by showing a significant association of elevated AGR2 mRNA levels with resistance to doxorubicin treatment in ex vivo cultured human breast cancer primocultures, as well as significant association of induced AGR2 protein levels with worse overall survival of wt p53, ER‐positive breast cancer patients. Although it remains important to comprehensively determine whether AGR2 could be a potential drug target for reactivation of the p53 pathway in cancer cells, our findings clearly indicate that AGR2 could be useful as a prognostic marker in ER‐positive wt p53 breast tumors and potentially also as a druggable molecular target (Arumugam et al., 2015).
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Acknowledgments
The work was supported by the project MEYS – NPS I – LO1413, MH CZ – DRO (MMCI, 00209805), IGA NT/13794‐4/2012 and GACR 13‐00956S.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.12.003.
Hrstka Roman, Bouchalova Pavla, Michalova Eva, Matoulkova Eva, Muller Petr, Coates Philip J., Vojtesek Borivoj, (2016), AGR2 oncoprotein inhibits p38 MAPK and p53 activation through a DUSP10‐mediated regulatory pathway, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.12.003.
References
- Albain, K.S. , Barlow, W.E. , Shak, S. , Hortobagyi, G.N. , Livingston, R.B. , Yeh, I.T. , Ravdin, P. , Bugarini, R. , Baehner, F.L. , Davidson, N.E. , Sledge, G.W. , Winer, E.P. , Hudis, C. , Ingle, J.N. , Perez, E.A. , Pritchard, K.I. , Shepherd, L. , Gralow, J.R. , Yoshizawa, C. , Allred, D.C. , Osborne, C.K. , Hayes, D.F. , 2010. Prognostic and predictive value of the 21-gene recurrence score assay in postmenopausal women with node-positive, oestrogen-receptor-positive breast cancer on chemotherapy: a retrospective analysis of a randomised trial. Lancet Oncol. 11, 55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arumugam, T. , Deng, D. , Bover, L. , Wang, H. , Logsdon, C.D. , Ramachandran, V. , 2015. New blocking antibodies against novel AGR2-C4.4A pathway reduce growth and metastasis of pancreatic tumors and increase survival in Mice. Mol. Cancer Ther. 14, 941–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey, S.T. , Shin, H. , Westerling, T. , Liu, X.S. , Brown, M. , 2012. Estrogen receptor prevents p53-dependent apoptosis in breast cancer. Proc. Natl. Acad. Sci. U. S. A. 109, 18060–18065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banin, S. , Moyal, L. , Shieh, S. , Taya, Y. , Anderson, C.W. , Chessa, L. , Smorodinsky, N.I. , Prives, C. , Reiss, Y. , Shiloh, Y. , Ziv, Y. , 1998. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 281, 1674–1677. [DOI] [PubMed] [Google Scholar]
- Blaydes, J.P. , Hupp, T.R. , 1998. DNA damage triggers DRB-resistant phosphorylation of human p53 at the CK2 site. Oncogene. 17, 1045–1052. [DOI] [PubMed] [Google Scholar]
- Bouchalova, P. , Nenutil, R. , Muller, P. , Hrstka, R. , Appleyard, M.V. , Murray, K. , Jordan, L.B. , Purdie, C.A. , Quinlan, P. , Thompson, A.M. , Vojtesek, B. , Coates, P.J. , 2014. Mutant p53 accumulation in human breast cancer is not an intrinsic property or dependent on structural or functional disruption but is regulated by exogenous stress and receptor status. J. Pathol. 233, 238–246. [DOI] [PubMed] [Google Scholar]
- Brychtova, V. , Vojtesek, B. , Hrstka, R. , 2011. Anterior gradient 2: a novel player in tumor cell biology. Cancer Lett. 304, 1–7. [DOI] [PubMed] [Google Scholar]
- Chen, J. , Marechal, V. , Levine, A.J. , 1993. Mapping of the p53 and mdm-2 interaction domains. Mol. Cell Biol. 13, 4107–4114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrisanthar, R. , Knappskog, S. , Lokkevik, E. , Anker, G. , Ostenstad, B. , Lundgren, S. , Risberg, T. , Mjaaland, I. , Skjonsberg, G. , Aas, T. , Schlichting, E. , Fjosne, H.E. , Nysted, A. , Lillehaug, J.R. , Lonning, P.E. , 2011. Predictive and prognostic impact of TP53 mutations and MDM2 promoter genotype in primary breast cancer patients treated with epirubicin or paclitaxel. PLoS One. 6, e19249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobes, P. , Podhorec, J. , Coufal, O. , Jureckova, A. , Petrakova, K. , Vojtesek, B. , Hrstka, R. , 2014. Influence of mutation type on prognostic and predictive values of TP53 status in primary breast cancer patients. Oncol. Rep. 32, 1695–1702. [DOI] [PubMed] [Google Scholar]
- Forslund, A. , Zeng, Z. , Qin, L.X. , Rosenberg, S. , Ndubuisi, M. , Pincas, H. , Gerald, W. , Notterman, D.A. , Barany, F. , Paty, P.B. , 2008. MDM2 gene amplification is correlated to tumor progression but not to the presence of SNP309 or TP53 mutational status in primary colorectal cancers. Mol. Cancer Res. 6, 205–211. [DOI] [PubMed] [Google Scholar]
- Gyorffy, B. , Lanczky, A. , Eklund, A.C. , Denkert, C. , Budczies, J. , Li, Q. , Szallasi, Z. , 2010. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731. [DOI] [PubMed] [Google Scholar]
- Harris, S.L. , Levine, A.J. , 2005. The p53 pathway: positive and negative feedback loops. Oncogene. 24, 2899–2908. [DOI] [PubMed] [Google Scholar]
- Haupt, Y. , Maya, R. , Kazaz, A. , Oren, M. , 1997. Mdm2 promotes the rapid degradation of p53. Nature. 387, 296–299. [DOI] [PubMed] [Google Scholar]
- Hengel, S.M. , Murray, E. , Langdon, S. , Hayward, L. , O'Donoghue, J. , Panchaud, A. , Hupp, T. , Goodlett, D.R. , 2011. Data-independent proteomic screen identifies novel tamoxifen agonist that mediates drug resistance. J. Proteome Res. 10, 4567–4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrstka, R. , Nenutil, R. , Fourtouna, A. , Maslon, M.M. , Naughton, C. , Langdon, S. , Murray, E. , Larionov, A. , Petrakova, K. , Muller, P. , Dixon, M.J. , Hupp, T.R. , Vojtesek, B. , 2010. The pro-metastatic protein anterior gradient-2 predicts poor prognosis in tamoxifen-treated breast cancers. Oncogene. 29, 4838–4847. [DOI] [PubMed] [Google Scholar]
- Hrstka, R. , Brychtova, V. , Fabian, P. , Vojtesek, B. , Svoboda, M. , 2013. AGR2 predicts tamoxifen resistance in postmenopausal breast cancer patients. Dis. Markers. 35, 207–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hublarova, P. , Greplova, K. , Holcakova, J. , Vojtesek, B. , Hrstka, R. , 2010. Switching p53-dependent growth arrest to apoptosis via the inhibition of DNA damage-activated kinases. Cell. Mol. Biol. Lett. 15, 473–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innes, H.E. , Liu, D. , Barraclough, R. , Davies, M.P. , O'Neill, P.A. , Platt-Higgins, A. , de Silva Rudland, S. , Sibson, D.R. , Rudland, P.S. , 2006. Significance of the metastasis-inducing protein AGR2 for outcome in hormonally treated breast cancer patients. Br. J. Cancer. 94, 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey, K.L. , Camps, M. , Rommel, C. , Mackay, C.R. , 2007. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat. Rev. Drug Discov. 6, 391–403. [DOI] [PubMed] [Google Scholar]
- Jiang, L. , Kon, N. , Li, T. , Wang, S.J. , Su, T. , Hibshoosh, H. , Baer, R. , Gu, W. , 2015. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 520, 57–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, Y. , Kweon, J. , Kim, A. , Chon, J.K. , Yoo, J.Y. , Kim, H.J. , Kim, S. , Lee, C. , Jeong, E. , Chung, E. , Kim, D. , Lee, M.S. , Go, E.M. , Song, H.J. , Kim, H. , Cho, N. , Bang, D. , Kim, J.S. , 2013. A library of TAL effector nucleases spanning the human genome. Nat. Biotechnol. 31, 251–258. [DOI] [PubMed] [Google Scholar]
- Konduri, S.D. , Medisetty, R. , Liu, W. , Kaipparettu, B.A. , Srivastava, P. , Brauch, H. , Fritz, P. , Swetzig, W.M. , Gardner, A.E. , Khan, S.A. , Das, G.M. , 2010. Mechanisms of estrogen receptor antagonism toward p53 and its implications in breast cancer therapeutic response and stem cell regulation. Proc. Natl. Acad. Sci. U. S. A. 107, 15081–15086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse, J.P. , Gu, W. , 2009. Modes of p53 regulation. Cell. 137, 609–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, D.P. , 1992. Cancer. p53, guardian of the genome. Nature. 358, 15–16. [DOI] [PubMed] [Google Scholar]
- Lee, J.T. , Gu, W. , 2010. The multiple levels of regulation by p53 ubiquitination. Cell Death Differ. 17, 86–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, Y.P. , Lim, T.T. , Chan, Y.L. , Song, A.C. , Yeo, B.H. , Vojtesek, B. , Coomber, D. , Rajagopal, G. , Lane, D. , 2007. The p53 knowledgebase: an integrated information resource for p53 research. Oncogene. 26, 1517–1521. [DOI] [PubMed] [Google Scholar]
- Liu, W. , Ip, M.M. , Podgorsak, M.B. , Das, G.M. , 2009. Disruption of estrogen receptor alpha-p53 interaction in breast tumors: a novel mechanism underlying the anti-tumor effect of radiation therapy. Breast Cancer Res. Treat. 115, 43–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalova, E. , Poprach, A. , Nemeckova, I. , Nenutil, R. , Valik, D. , Zaloudik, J. , Vyzula, R. , Vojtesek, B. , 2008. Chemosensitivity prediction in tumor cells ex vivo–difficulties and limitations of the method. Klin. Onkol. 21, 93–97. [PubMed] [Google Scholar]
- Mosmann, T. , 1983. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J. Immunol. Methods. 65, 55–63. [DOI] [PubMed] [Google Scholar]
- Olivier, M. , Langerod, A. , Carrieri, P. , Bergh, J. , Klaar, S. , Eyfjord, J. , Theillet, C. , Rodriguez, C. , Lidereau, R. , Bieche, I. , Varley, J. , Bignon, Y. , Uhrhammer, N. , Winqvist, R. , Jukkola-Vuorinen, A. , Niederacher, D. , Kato, S. , Ishioka, C. , Hainaut, P. , Borresen-Dale, A.L. , 2006. The clinical value of somatic TP53 gene mutations in 1,794 patients with breast cancer. Clin. Cancer Res. 12, 1157–1167. [DOI] [PubMed] [Google Scholar]
- Olivier, M. , Hollstein, M. , Hainaut, P. , 2010. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2, a001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owens, D.M. , Keyse, S.M. , 2007. Differential regulation of MAP kinase signalling by dual-specificity protein phosphatases. Oncogene. 26, 3203–3213. [DOI] [PubMed] [Google Scholar]
- Paik, S. , Tang, G. , Shak, S. , Kim, C. , Baker, J. , Kim, W. , Cronin, M. , Baehner, F.L. , Watson, D. , Bryant, J. , Costantino, J.P. , Geyer, C.E. , Wickerham, D.L. , Wolmark, N. , 2006. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J. Clin. Oncol. 24, 3726–3734. [DOI] [PubMed] [Google Scholar]
- Petitjean, A. , Achatz, M.I. , Borresen-Dale, A.L. , Hainaut, P. , Olivier, M. , 2007. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 26, 2157–2165. [DOI] [PubMed] [Google Scholar]
- Pohler, E. , Craig, A.L. , Cotton, J. , Lawrie, L. , Dillon, J.F. , Ross, P. , Kernohan, N. , Hupp, T.R. , 2004. The Barrett's antigen anterior gradient-2 silences the p53 transcriptional response to DNA damage. Mol. Cell. Proteomics. 3, 534–547. [DOI] [PubMed] [Google Scholar]
- Poprach, A. , Michalova, E. , Pavlik, T. , Lakomy, R. , Vyskocil, J. , Nemeccek, R. , Zaloudik, J. , Vyzula, R. , Kocak, I. , Kocakova, I. , 2008. Actual state of ex vivo chemoresistance testing of malignant tumors in Masaryk Memorial Cancer Institute Brno. Klin. Onkol. 21, 116–121. [PubMed] [Google Scholar]
- Ramachandran, V. , Arumugam, T. , Wang, H. , Logsdon, C.D. , 2008. Anterior gradient 2 is expressed and secreted during the development of pancreatic cancer and promotes cancer cell survival. Cancer Res. 68, 7811–7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regner, B. , Dusek, L. , Hajduch, M. , 2000. Software chemoresist version 1.0 – a complex tool for data analysis a management of tumor drug resistance testing. Klin. Onkol. 13, 30–32. [Google Scholar]
- Salmans, M.L. , Zhao, F. , Andersen, B. , 2013. The estrogen-regulated anterior gradient 2 (AGR2) protein in breast cancer: a potential drug target and biomarker. Breast Cancer Res. 15, 204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato, Y. , Tsurumi, T. , 2013. Genome guardian p53 and viral infections. Rev. Med. Virol. 23, 213–220. [DOI] [PubMed] [Google Scholar]
- Sayeed, A. , Konduri, S.D. , Liu, W. , Bansal, S. , Li, F. , Das, G.M. , 2007. Estrogen receptor alpha inhibits p53-mediated transcriptional repression: implications for the regulation of apoptosis. Cancer Res. 67, 7746–7755. [DOI] [PubMed] [Google Scholar]
- Simpson, N.E. , Lambert, W.M. , Watkins, R. , Giashuddin, S. , Huang, S.J. , Oxelmark, E. , Arju, R. , Hochman, T. , Goldberg, J.D. , Schneider, R.J. , Reiz, L.F. , Soares, F.A. , Logan, S.K. , Garabedian, M.J. , 2010. High levels of Hsp90 cochaperone p23 promote tumor progression and poor prognosis in breast cancer by increasing lymph node metastases and drug resistance. Cancer Res. 70, 8446–8456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosiou, A. , Smith, A. , Gillieron, C. , Arkinstall, S. , Ashworth, A. , 1999. MKP5, a new member of the MAP kinase phosphatase family, which selectively dephosphorylates stress-activated kinases. Oncogene. 18, 6981–6988. [DOI] [PubMed] [Google Scholar]
- Thompson, D.A. , Weigel, R.J. , 1998. hAG-2, the human homologue of the Xenopus laevis cement gland gene XAG-2, is coexpressed with estrogen receptor in breast cancer cell lines. Biochem. Biophys. Res. Commun. 251, 111–116. [DOI] [PubMed] [Google Scholar]
- Vojtesek, B. , Bartek, J. , Midgley, C.A. , Lane, D.P. , 1992. An immunochemical analysis of the human nuclear phosphoprotein p53. New monoclonal antibodies and epitope mapping using recombinant p53. J. Immunol. Methods. 151, 237–244. [DOI] [PubMed] [Google Scholar]
- Wade, M. , Li, Y.C. , Wahl, G.M. , 2013. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer. 13, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Z. , Hao, Y. , Lowe, A.W. , 2008. The adenocarcinoma-associated antigen, AGR2, promotes tumor growth, cell migration, and cellular transformation. Cancer Res. 68, 492–497. [DOI] [PubMed] [Google Scholar]
- Yamashita, H. , Toyama, T. , Nishio, M. , Ando, Y. , Hamaguchi, M. , Zhang, Z. , Kobayashi, S. , Fujii, Y. , Iwase, H. , 2006. p53 protein accumulation predicts resistance to endocrine therapy and decreased post-relapse survival in metastatic breast cancer. Breast Cancer Res. 8, R48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. , Yue, X. , Zhao, Y. , Li, X. , Wu, L. , Zhang, C. , Liu, Z. , Lin, K. , Xu-Monette, Z.Y. , Young, K.H. , Liu, J. , Shen, Z. , Feng, Z. , Hu, W. , 2014. LIF negatively regulates tumour-suppressor p53 through Stat3/ID1/MDM2 in colorectal cancers. Nat. Commun. 5, 5218 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data