

Abstract
Background: Chemotherapy options in advanced urothelial carcinoma (UC) remain limited. Here we evaluated the peptide‐based alkylating agent melphalan‐flufenamide (mel‐flufen) for UC.
Methods: UC cell lines J82, RT4, TCCsup and 5637 were treated with mel‐flufen, alone or combined with cisplatin, gemcitabine, dasatinib or bestatin. Cell viability (MTT assay), intracellular drug accumulation (liquid chromatography) apoptosis induction (apoptotic cell nuclei morphology, western blot analysis of PARP‐1/caspase‐9 cleavage and Bak/Bax activation) were evaluated. Kinome alterations were characterized by PathScan array and phospho‐Src validated by western blotting. Aminopeptidase N (ANPEP) expression was evaluated in UC clinical specimens in relation to patient outcome.
Results: In J82, RT4, TCCsup and 5637 UC cells, mel‐flufen amplified the intracellular loading of melphalan in part via aminopeptidase N (ANPEP), resulting in increased cytotoxicity compared to melphalan alone. Mel‐flufen induced apoptosis seen as activation of Bak/Bax, cleavage of caspase‐9/PARP‐1 and induction of apoptotic cell nuclei morphology. Combining mel‐flufen with cisplatin or gemcitabine in J82 cells resulted in additive cytotoxic effects and for gemcitabine also increased apoptosis induction. Profiling of mel‐flufen‐induced kinome alterations in J82 cells revealed that mel‐flufen alone did not inhibit Src phosphorylation. Accordingly, the Src inhibitor dasatinib sensitized for mel‐flufen cytotoxicity. Immunohistochemical analysis of the putative mel‐flufen biomarker ANPEP demonstrated prominent expression levels in tumours from 82 of 83 cystectomy patients. Significantly longer median overall survival was found in patients with high ANPEP expression (P = 0.02).
Conclusion: Mel‐flufen alone or in combination with cisplatin, gemcitabine or Src inhibition holds promise as a novel treatment for UC.
Keywords: Urothelial carcinoma, Melphalan‐flufenamide, Aminopeptidase N, Apoptosis, Src, Cisplatin, Gemcitabine, Dasatinib
Highlights
Mel‐flufen caused cytotoxicity and potentiated cisplatin/gemcitabine in urothelial cancer (UC) cells.
Targeting of Src by dasatinib sensitized for mel‐flufen.
Aminopeptidase N controlled mel‐flufen effect in UC cells.
Aminopeptidase N expression correlated with survival in UC patients.
Mel‐flufen holds promise as novel treatment for UC.
Abbreviations
- Akt
protein kinase B
- ALK
anaplastic lymphoma kinase
- ANPEP
aminopeptidase N
- Bak
Bcl‐2 homologous antagonist killer
- Bax
Bcl‐2‐associated X protein
- BSC
best supportive care
- CT
chemotherapy
- DSB
double strand breaks
- GC
gemcitabine cisplatin
- mel‐flufen
Lmelphalanyl‐p‐l‐fluorophenylalanine ethyl ester
- MM
multiple myeloma
- MVAC
methotrexate, vinblastine, doxorubicin, cisplatin
- PARP‐1
poly‐ADP‐ribose polymerase 1
- RTK
receptor tyrosine kinase
- UC
urothelial carcinoma
1. Introduction
Cancer of the urinary bladder is annually diagnosed in nearly half a million cases worldwide with urothelial carcinoma (UC) representing 90% of all cases (Ferlay et al., 2014; Vishnu et al., 2011). For patients with metastatic disease first line chemotherapy regimens consist of either gemcitabine or cisplatin (GC) doublet or methotrexate, vinblastine, doxorubicin, and cisplatin (MVAC) applied in combination (Sternberg et al., 1989; von der Maase et al., 2005). The efficacy of GC and MVAC is comparable but the toxicity is in favour of the GC regimen (von der Maase et al., 2005). Although the majority of the UC patients respond to primary treatment, relapse and progression are inevitable. During the last decades several small phase II trials have evaluated numerous cytotoxic drugs and regimes as second line treatment for metastatic UC yet their response rates are lower than first line treatment (Bambury and Rosenberg, 2013; Bellmunt et al., 2009). Following a positive randomised phase III trial with vinflunine, which confirmed an overall survival (OS) gain of 2.6 months over best supportive care (BSC) only, the European Medicines Agency granted market authorisation in 2009 (Bellmunt et al., 2009). Even though vinflunine is the recommended second line option, the overall survival benefit remains modest. Clearly, a significant unmet medical need is prevailing in metastatic UC when it comes to medical systemic treatment.
Mel‐flufen (L‐melphalanyl‐p‐l‐fluorophenylalanine ethyl ester) is a recently constructed alkylating dipeptide with melphalan as the active moiety (Gullbo et al., 2003a). Via a targeted uptake and intracellular hydrolysis by aminopeptidases, mel‐flufen offers a way to obtain significantly higher intracellular loading of melphalan resulting in improved cytotoxicity as compared to melphalan (Gullbo et al., 2003b). Mel‐flufen has been shown to have cytotoxic potential in both solid tumour cells and in multiple myeloma (MM) and in the later a phase I/II clinical trial is running (NCT01897714) (Berglund et al., 2015; Chauhan et al., 2013; Wickstrom et al., 2007). In vitro studies of mel‐flufen demonstrated that aminopeptidases, including aminopeptidase N (ANPEP or CD13), are in part regulating the tumour cell specific release of melphalan (Wickstrom et al., 2010). Interestingly, ANPEP expression has previously been described to regulate tumour cell motility and extracellular matrix degradation. With respect to urinary bladder, ANPEP expression has been found in stroma cells of the superficial lamina propria, in the muscularis propria and in blood vessels (Goo et al., 2005). An altered expression of ANPEP in cells juxtapositioned to the superficial lamina propria has been demonstrated in UC, indicative of a cancer‐associated stromal component (Liu et al., 2012). The prognostic value of tumour ANPEP expression in UC patients treated by cystectomy remains scant. However, ANPEP overexpression has in lung‐ and ovarian cancer been associated with metastasis and poor prognosis (Surowiak et al., 2006, 2001, 2006, 2011). Yet in prostate cancer and gastric carcinoma a significant better outcome for those patients with high tumour ANPEP expression was shown (Kawamura et al., 2007; Sorensen et al., 2013). In this study we evaluated and characterised in vitro cytotoxic effects of mel‐flufen in UC alone or combined with either cisplatin, gemcitabine or Src inhibition. In addition, the expression patterns in UC specimens of the putative predictive biomarker, ANPEP were also analysed.
2. Materials and methods
2.1. Cell lines, cell culture, and chemicals
The UC cell lines J82 (ATCC® HTB‐1™), TCC‐SUP (ATCC® HTB‐5™), 5637 (ATCC® HTB‐9™), and RT4 (ATCC® HTB‐2™) were obtained from American Type Culture Collection (ATCC, Manassas, VA) (Fogh et al., 1977; Nayak et al., 1977; O'Toole et al., 1978; Rigby and Franks, 1970). The cell lines were verified and authenticated by ATCC using short tandem repeat profiling and were maintained as monolayer in RPMI‐1640 medium (Invitrogen, Carlsbad, CA, USA) supplemented with fetal calf serum (10%) and glutamine (2 mM) (both from Invitrogen, Carlsbad, CA, USA). Mel‐flufen was obtained from Oncopeptides AB (Stockholm, Sweden). Melphalan (Alkeran®), cisplatin (Cisplatin Hospira), and gemcitabine (Gemzar®) were obtained from Apoteket AB, Sweden. Mel‐flufen, melphalan were prepared in DMSO, bestatin (Sigma–Aldrich, St. Louis, MO, USA) and dasatinib (Cell Signalling Technology, Danvers, MA, USA) stock solutions were made in DMSO with further dilution in culture media upon use.
2.2. Cell viability assay
Mel‐flufen and melphalan cytotoxicity was examined using either fluorometric microculture cytotoxicity assay (FMCA) or 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyl‐tetrazolium salt bromide (MTT) assay in a 96‐well format, in which 5000 cells/well were seeded in triplicates and the next day treated with drugs, either for 1 h or during 72 h of continuous exposure. For FMCA, cell viability was recorded using fluorescein diacetate (Sigma–Aldrich) incubation for 4 h followed by measurement of the hydrolysis to fluorescein at 485/520 nm. For the MTT assay, the MTT reagent (0.5 mg/ml, Sigma Aldrich) was applied for 4 h during which metabolically active cells converted it to formazan crystals which were solubilised by addition of stop solution (10% SDS in 0.01 M HCl). The resulting absorbance was measured by spectrophotometer at 570 nm. In both FMCA and MTT the absorbance in treated samples was compared to untreated for which viability was set to 100%. Data is presented as mean ± SD from three independent experiments.
For dasatinib and mel‐flufen combination experiments, dasatinib (20 nM) was added for 16 h prior to 1 h pulse treatment with mel‐flufen and 48 h post incubation with both agents. For the cisplatin and gemcitabine combination treatments the drugs were used for 1 h with 48 h or 72 h post incubation respectively. The cytotoxicity after combination treatments were scored by counting cells after 0.4% Trypan blue staining. Data is given as % viable cells relative to untreated control in three biological replicates.
2.3. Intracellular concentration assessment of mel‐flufen, melphalan and de‐ethyl‐mel‐flufen
The intracellular concentrations of mel‐flufen, de‐esterified mel‐flufen and melphalan after mel‐flufen (1 μM) and melphalan (10 μM) treatment, were examined in J82 cells at indicated time points with or without bestatin (5 μM, 1 h pre‐incubation) as previously described (Chauhan et al., 2013; Wickstrom et al., 2010) using the analytical platform from OnTarget Chemistry AB, Uppsala, Sweden. For each treatment 2.5 × 106 J82 cells/ml were resuspended in 6 ml of complete pre‐warmed RPMI media and 1 ml cell suspension taken out at indicated time points. The biotransformation was stopped by adding pre‐cooled PBS, and after washing proteins was precipitated by adding ethanol/acetonitrile (1:1, v/v) to the cell pellet. Drug concentrations were assessed in 25 μl of a sample aliquot mixed with 75 μl of an internal standard solution (1 μg/ml of fluorescein diluted 1:1 in acetonitrile: ethanol) using reversed phase gradient liquid chromatography. The drug associated spectra were recorded using positive electro spray ionisation and drug concentrations estimated using standard curves for mel‐flufen, de‐esterified mel‐flufen, and melphalan, respectively. The amount of drug was determined after correction for loading differences. From the resulting curves, the area corresponding to 0–60 min of treatment was used to calculate the AUC from three biological replicates.
2.4. Assessment of apoptotic cell nuclei morphology
Apoptotic cell nuclei morphology was examined in ethanol fixed cells in which DAPI containing mounting media (Vector Labs, Burlingame, CA, USA) was used to stain the DNA of the nuclei of the cells. The resulting DAPI‐stained cell nuclei were examined using the FL‐1 channel on a fluorescent microscope (Zeiss Axioplan 2, Zeiss, and Thornwood, NY, USA). The number of cells with chromatin condensation and/or fragmentation of nuclei in a total number of 200 cells were recorded as % of apoptotic cells. Data shown is the mean ± SD of three biological replicates.
2.5. Assessment of pro‐apoptotic conformational changes of Bak and Bax
Bak and Bax activation was measured by flow cytometry using antibodies recognizing N‐terminal conformational changes previously shown to be associated with pro‐apoptotic function of either protein (Griffiths et al., 1999; Mandic et al., 2002). J82 cells were after treatment fixed in 4% paraformaldehyde solution in PBS for 15 min and stained with antibodies against Bak (AM03, clone TC100; Oncogene Research Products, CA, USA), 1:50, Bax (clone 6A7; BD Biosciences Pharmingen, San Diego, CA, USA), 1:250 in 100 μl of PBS containing digitonin (100 μg/ml) as permeabilising agent for 1 h. A secondary Alexa 488 labelled anti‐mouse antibody was applied for 45 min after which Bak or Bax associated IFL signal was recorded in the FL‐1 channel of the FACS Calibur flow cytometer (BD Bioscience). Cell Quest software (BD Bioscience) was used to process the data. The accurate staining with Bak or Bax antibodies was verified using an isotype IgG1 negative control antibody (data not shown). The fold change, in Bak or Bax associated median fluorescence intensity, was compared to untreated cells and mean ± SD from three replicates are presented.
2.6. Cell cycle distribution
Cell cycle distribution were analysed in ethanol‐fixed J82 cells stained with PI staining as previously described (Darzynkiewicz and Huang, 2004). The ModFit LT (Verity Software House, Topsham, ME, USA) program was applied on the data to allow cell distribution into the different phases.
2.7. Western blot analysis
Protein extracts were made from cells using RIPA buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 0.5% Igepal, 5 mM EDTA, and 0.1% SDS) with protease and phosphatase inhibitor cocktail tablets (Roche Diagnostics AB, Stockholm, Sweden). Protein concentration was assayed by BCA Protein Assay Reagent (Interchim, Montlucon Cedex, France) and 50 μg of total protein were loaded onto either Bis Tris 4–12% or Tris Acetate 3–8% gels (NuPAGE, Invitrogen). The gels were electrophoresed (150V, 90 min) in MOPS or Tris‐acetate g buffer (NuPAGE, Invitrogen) and proteins transferred to nitrocellulose membranes (Optitran BA‐885, Whatman, GE Healthcare, Pittsburgh, PA, USA) at 30V for 90 min in transfer buffer (NuPAGE, Invitrogen) containing 10% methanol. Membranes were blocked in Odyssey buffer (Li‐Cor Biosciences, Bad Homburg, Germany) diluted in tris‐buffered saline (TBS) 1:1 for 60 min. The primary antibodies (diluted in blocking buffer and incubated overnight) were: PARP‐1 (#H250), caspase‐9 (#7150) (both from Santa Cruz Biotechnology, Santa Cruz, CA, USA), phospho‐Src (Y416) (Cell Signalling, Danvers, MA, USA), and γH2AX (Abcam, Cambridge, MA, USA). GAPDH (Trevigen, Gaithersburg, MD, USA) or β‐tubulin (Sigma Aldrich) was used as loading controls. Signals from the primary antibodies were visualized by adding IR‐Dye‐linked secondary antibodies (LI‐COR Biosciences, Bad Homburg, Germany) followed by recording the signals on the Odyssey platform. The amount of cleaved PARP‐1 relative to full length PARP‐1 was quantified by densitometry and after correcting for loading differences.
2.8. PathScan RTK signalling antibody array analysis
For kinome profiling the PathScan RTK signalling antibody array (#7949, Cell Signalling Technology) was used in which protein lysate from J82 cells (2 × 106) were assayed according to the manufacturer's instructions. Detection of antibody signals was made by a biotin‐streptavidin‐conjugated antibody on the Odyssey® Sa Infrared Imaging System (LI‐COR). Signal from each spot was recorded and average intensity after background subtraction was taken and further processed. Only kinases giving signal above negative controls in untreated J82 cells and which showed consistent signalling (differing <20% between the two duplicated spotted kinases), were analysed further in which sample normalisation was made using the positive controls on each slide. Treatment induced kinase signal was calculated relative to untreated J82 cells which was set to an arbitrary value of 100.
2.9. Immunohistochemical analysis of ANPEP expression in tumour samples from UC patients
ANPEP immunohistochemical analysis was carried out on tumour specimens from 83 patients treated by cystectomy, as previously described (Shah et al., 2014). Prior to immunohistochemistry, endogenous peroxidase activity was quenched with 0.5% peroxidase solution in TBS for 30 min. The tumour sections were blocked for 30 min at 20 °C in TBS supplemented with 5% bovine milk and 1% bovine serum albumin followed by incubation with ANPEP antibody 1:200 overnight. The ANPEP antibody used was produced by immunising rabbits with a peptide directed against the ANPEP sequence: aa 627–640: C‐TGYYRVNYDEENWR from which monospecific IgG (concentration 0.48 mg/ml) was isolated. To visualise the primary antibody binding, a biotinylated anti‐rabbit IgG secondary antibody (Vector Labs) was applied (30 min, 1:200) followed by avidin‐biotin peroxidase complex solution (30 min) and 3,3‐diaminobenzidine solution (6 min). Breast and ovarian tumours served as positive controls. Staining was evaluated by a pathologist who was uninformed of the patients' outcomes. In a defined area of the specimen, the proportion of positively stained tumour cells was stratified into 5 groups (0%, 1%–25%, 26%–50%, 51%–75%, and 76%–100%) to obtain the score value and the intensity was divided into 4 groups (0, negative; 1, weak; 2, moderate; and 3, strong). The non‐malignant cells provided background reference. The product of the score and intensity was dichotomised into high or low and, used to correlate the staining with the outcome data. To enhance the clarity of the micrographs, Adobe Photoshop CS5E v 12.0.4 × 64 (Adobe Systems Inc, San Jose, CA) was applied.
2.10. Statistical analyses
Student's t‐test was used to compare mean values of two groups. Clinical characteristics of the 83 patients were evaluated using the Pearson 2‐sided chi‐square test. Survival probabilities were calculated with Kaplan–Meier method (Breslow test) and Cox proportional hazard regression model, respectively. Survival was assessed from the date of cystectomy to the date of the event analysed (relapse or death). Statistical assessment was performed by replacing the tumour category (pT), lymph node status (pN) and the World Health Organisation grade after cystectomy values of 0 with the corresponding value (≠0) from the diagnostic transurethral resection of the bladder performed prior to the cystectomy. P < 0.05 was considered statistically significant. SPSS version 22 (IBM, Somers, NY) was used for all statistical analyses.
3. Results
3.1. Mel‐flufen induces cytotoxicity in UC cells
The efficacy of alkylating peptide mel‐flufen in UC was studied in the UC cell lines J82, RT4, 5637, and TCC‐SUP. At 72 h mel‐flufen caused significant alteration of cell morphology in all the UC cell lines while treatment with melphalan had less effect (Figure 1A). Cell viability analysis also revealed significantly decreased viability after mel‐flufen treatment in all the cell lines, yet to a different degree (Figure 1B). Thus, at 1 μM mel‐flufen, the J82, TCCSup, and 5637 cell lines, showed less than 30% viability, whereas in RT4 cells a higher concentration, 5 μM, caused a similar decrease (Figure 1B). Accordingly, the cell kill effect ratio of melphalan and mel‐flufen demonstrated a 15–100‐fold higher effect at 1 or 5 μM respectively, in favour of mel‐flufen. Less difference was observed in RT4, the cell line most refractory to melphalan, in which mel‐flufen only showed a 4‐fold higher cell killing effect at 5 μM (Supplementary Figure S1).
Figure 1.
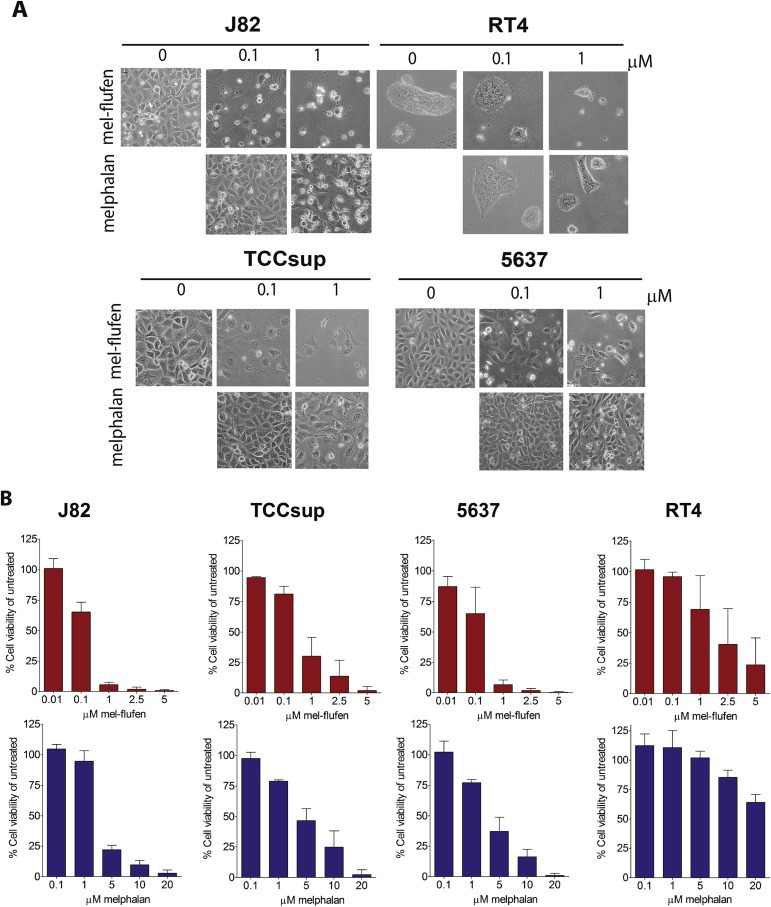
Mel‐flufen induces cytotoxicity in UC cells. J82, RT4, 5637 and TCC‐SUP UC cells were treated with mel‐flufen or melphalan for 72 h. (A) Photos showing cell morphology upon treatment. (B) Cell viability after 72 h of continuous treatments with either mel‐flufen (upper panel) or melphalan (lower panel). Data shown is the mean ± SD from 3 experiments. The ratio between mel‐flufen and melphalan for the cell lines is presented in Supplementary Figure. S1.
3.2. Mel‐flufen treatment results in intracellular loading of melphalan in UC cells
Increased intracellular accumulation of melphalan has been shown upon mel‐flufen treatment of tumour cells in vitro (Chauhan et al., 2013; Wickstrom et al., 2010) and here mel‐flufen, the de‐esterified form, and the parental compound melphalan, were examined in J82 cells after mel‐flufen (1 μM) or melphalan (10 μM) treatment (Figure 2A,B). Assessment of mel‐flufen and the de‐esterified form of mel‐flufen in J82 cells showed a clear accumulation of both these molecules (Figure 2A) which is in marked contrast to results in breast‐, lung‐, cervical carcinoma, and neuroblastoma (Wickstrom et al., 2010). In line with previous data, mel‐flufen treatment caused a gradual increase in melphalan accumulation with a concomitant decrease in mel‐flufen (Figure 2A). Importantly, even if a 10‐fold lower concentration of mel‐flufen (1 μM) was applied, the concentrations of melphalan measured were still higher than with melphalan (10 μM) (Figure 2A). Accordingly the Area Under the Curve (AUC) ratio of melphalan in mel‐flufen and melphalan treated J82 cells revealed a 1.7‐fold difference and taking into consideration the 10‐fold difference in concentration applied, this translates to an almost 20‐fold difference in favour of mel‐flufen (Figure 2B). Melphalan, the active component of mel‐flufen, acts by causing DNA cross‐links, which, in s‐phase of the cell cycle, inhibits replication function resulting in DNA double strand breaks (DSB) (Lee et al., 2010). Indeed, increased DNA DSB formation measured as γH2AX was found at 24 h after mel‐flufen treatment of J82 cells as compared to melphalan (Figure 2C).
Figure 2.
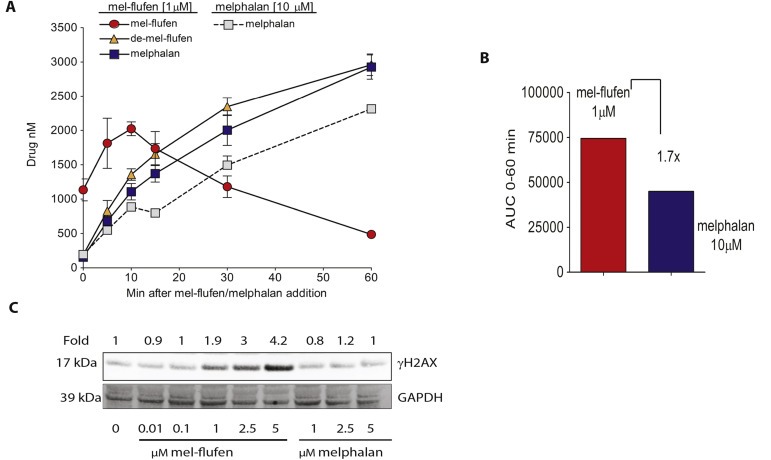
Mel‐flufen treatment of UC cells results in increased loading of melphalan. J82 cells were treated with mel‐flufen (1 μM) or melphalan (10 μM). The intracellular concentration of mel‐flufen, de‐esterified mel‐flufen or melphalan was measured using HPLC‐MS/MS. (A) Solid line: intracellular concentration of mel‐flufen, de‐esterified mel‐flufen or melphalan after mel‐flufen treatment. Dotted line: intracellular concentration of melphalan after melphalan treatment. (B) AUC 0–60 min after mel‐flufen (1 μM) or melphalan (10 μM) treatment. Please note the 10‐fold difference in concentration and the 1.7 fold higher loading of melphalan after mel‐flufen treatment. (A–B) is based on 3 biological replicates. (C) γH2AX was examined in J82 cells after 1 h treatment of mel‐flufen or melphalan and 24 h post incubation in which GAPDH served as loading control.
3.3. Increased apoptotic signalling in UC cells after mel‐flufen treatment involves activation of Bak and Bax
Analysis of apoptosis induction, in response to mel‐flufen or melphalan treatment in the UC cells, revealed a higher amount of cells with apoptotic cell nuclei after mel‐flufen treatment than after treatment with equimolar doses of melphalan (Figure 3A). Hence, approximately 18 and 20% of the J82 cells had nuclei with clear apoptotic features at 1 and 5 μM mel‐flufen respectively (Figure 3A, red bars). In response to melphalan 5 μM the percentage of cells with apoptotic nuclei was only 10%, showing a higher potency of mel‐flufen relative to melphalan (Figure 3A, grey bars).
Figure 3.
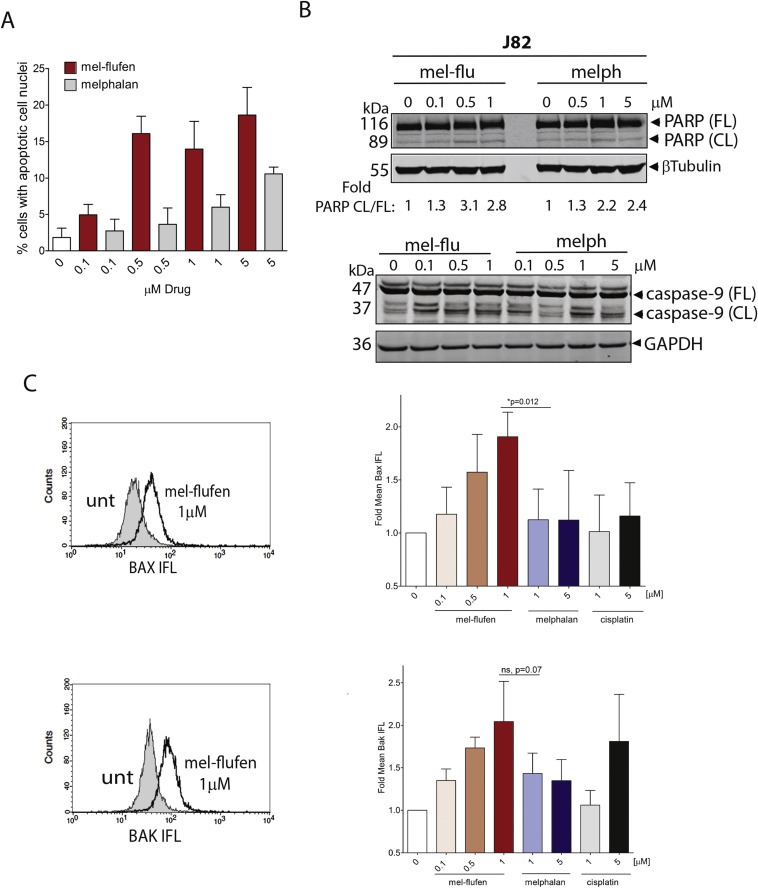
Mel‐flufen induces apoptosis in UC cell lines. (A) J82 cells were treated with mel‐flufen (red bars) or melphalan (grey bars) for 1 h and post incubated for 48 h. The nuclear morphology of the cells was assessed by DAPI‐staining and subsequent visualization in the FL‐1 channel of a Fluorescence microscope. The percentage of cells showing apoptotic nuclear morphology of 200 cells examined is given. Results shown are the mean ± SD from three biological replicates. Statistical difference between mel‐flufen and melphalan were reached at doses 0.5 μM, 1 μM and 5 μM (P = 0.0009, P = 0.0087 and P = 0.002 respectively). (B) Apoptosis was confirmed in J82 cells by analysing PARP‐1 and caspase‐9 cleavage by western blot treated as in (A) β‐tubulin and GAPDH was used as loading control for PARP‐1 and caspase‐9 analyses respectively. For PARP, the fold ratio CL/FL was determined by densitometry and is presented. (C) Bak or Bax activation in J82 cells was analysed by flow cytometry after 1 h treatment with mel‐flufen or melphalan, or after 24 h with cisplatin. Left: histogram showing Bak or Bax activation as a shift to the right in the diagram. Filled: untreated; unfilled: mel‐flufen treated. Right: Fold Bax or Bak mean IFL is presented relative to untreated cells. Data shown are the mean ± SD from three biological replicates. Upper panel: Bax, t‐test mel‐flufen 1 μM vs. melphalan 1 μM P = 0.012. Lower panel: Bak, t‐test mel‐flufen 1 μM vs. melphalan 1 μM P = 0.07.
Consistent with these findings, mel‐flufen treatment of J82 cells also resulted in higher amount of cleaved PARP‐1, a marker of caspase activity than did melphalan at equimolar concentrations (Figure 3B, upper panel). Activation of apoptosis in response to mel‐flufen was also verified by increased caspase‐9 cleavage (Figure 3B, lower panel). The superiority of mel‐flufen over melphalan in causing apoptotic nuclear morphology, at equimolar concentrations, was also confirmed in the other UC cell lines tested (TCCSup, RT4, and 5637) (Supplementary Figure S2, A–C, left panels). Moreover, mel‐flufen also caused PARP‐1 and caspase‐9 cleavage in 5637 as well as PARP‐1 cleavage in TCCSup (Supplementary Figure S2, A–B, right panels). In contrast, only a minor PARP‐1 and caspase‐9 cleavage was evident after either mel‐flufen, melphalan or cisplatin treatment in RT4 cells (data not shown). Thus the decreased cell viability and increased apoptotic nuclear cell morphology in response to mel‐flufen likely is executed by other pathways in RT4 cells.
Activation of the Bcl‐2 proteins Bak and Bax is central in DNA‐damage induced apoptosis after DNA damaging agents (Griffiths et al., 1999; Mandic et al., 2002). Such activation can be measured with antibodies recognizing the N‐terminal activity related conformational changes by flow cytometry. This approach was used to analyse Bak and Bax activation after mel‐flufen, melphalan or cisplatin treatment in J82 cells (Figure 3C). A dose dependent activation of either Bak or Bax was evident upon mel‐flufen treatment resulting in a 2‐fold activation at 1 μM, a dose which caused clear PARP‐1 cleavage and induction of apoptotic nuclear morphology (Figure 3C). Melphalan and cisplatin treatment did not activate Bax in these cells albeit activation of Bak was evident (Figure 3C). Thus, mel‐flufen caused a significantly higher activation of Bax as compared to melphalan at 1 μM (Figure 3C, upper panel, P = 0.012), whereas for Bak the difference in activation was non‐significant (Figure 3C, lower panel, P = 0.07). All in all these results show that mel‐flufen caused more prominent Bax activation in UC cells than did melphalan or cisplatin (Figure 3C).
Mel‐flufen‐ and melphalan‐induced cell cycle effects were also analysed in J82 cells (Supplementary Figure S3). Mel‐flufen (0.5 or 1 μM) caused a pronounced S‐phase accumulation whereas melphalan only caused increase in G2/M‐fraction.
3.4. Mel‐flufen in combination chemotherapy regimens for treatment of UC cells
The effect of combined mel‐flufen and gemcitabine or mel‐flufen and cisplatin, was analysed in J82 cells (Figure 4A–B). It was found that the mel‐flufen‐induced reduction in cell number was further potentiated by gemcitabine (Figure 4A, left panel). The combined treatment also caused cleavage of PARP‐1 which was most evident at 0.5 μM of mel‐flufen (Figure 4A, right panel) The combined effect of mel‐flufen and cisplatin was also analysed and demonstrated a more prominent cell death as compared to either agent alone (Figure 4B). Albeit this combination also resulted in PARP‐1 cleavage, the addition of mel‐flufen to cisplatin did not cause any further increase in PARP‐1 cleavage as compared to cisplatin single treatment (Figure 4B, right panel) suggesting that other mechanisms than apoptosis might be involved in the observed additive effect. Nevertheless, our data support that mel‐flufen may be used in combination with either gemcitabine or cisplatin with at least additive effects.
Figure 4.
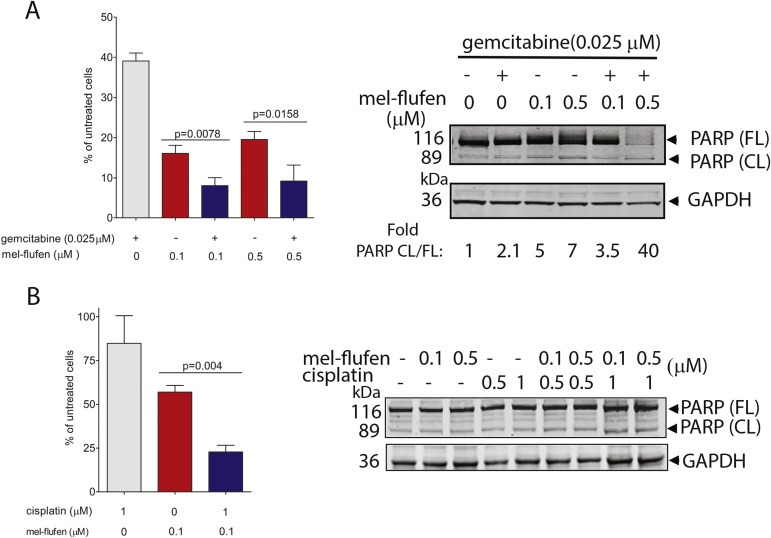
Combined mel‐flufen and gemcitabine or cisplatin treatment decreases cell survival in UC cells. (A) Left: J82 cells were treated with either mel‐flufen (0.1 or 0.5 μM), gemcitabine (0.025 μM), or a combination, during 1 h and post incubated for 72 h. The number of cells were counted using Trypan blue and is presented as % of untreated control. Data shown is the mean ± SD of three experiments. Student's t‐test was used to achieve the indicated P‐values. Right: PARP‐1 cleavage was examined in J82 cells after mel‐flufen, gemcitabine, and combined treatment. GAPDH was used as loading control. Fold ratio CL/FL PARP was determined by densitometry. (B) Left: J82 cells were treated with either mel‐flufen (0.1 μM) or cisplatin (1 μM) for 1 h and post incubated for another 48 h and counted as in (A). Right: PARP‐1 cleavage was examined in J82 cells after mel‐flufen, cisplatin, and their combination. GAPDH was used as loading control.
3.5. Profiling of mel‐flufen kinome alterations suggest Src inhibition as a candidate for potentiation of mel‐flufen response in UC
In order to further understand signalling pathways in UC cells that were altered or remained active and could drive refractoriness to mel‐flufen, profiling of the kinome was carried out at 24 h post mel‐flufen treatment in J82 cells using PathScan RTK signalling antibody array (Figure 5A, upper panel). For comparison, the kinome was also profiled in response to melphalan or cisplatin (Figure 5A, lower panel left and right respectively). Analysis revealed a 30% or more reduction in phosphorylation of 6 out of the 27 kinases/growth factor receptors analysed (Figure 5A, upper panel, green bars). Among them were PDGFR, FGFR3, c‐Kit and InsR, illustrating that mel‐flufen treatment indeed blocked survival pathways operative in UC cells. Yet several kinases and growth factors were still phosphorylated after mel‐flufen treatment and hence could contribute to survival of the UC cells upon treatment (Figure 5A, upper panel, red bars). The kinase which demonstrated the highest activity and which showed a 5‐fold increase in phosphorylation in mel‐flufen treated J82 cells was Src. Interestingly, Src phosphorylation was also evident upon treatment with melphalan and cisplatin, pointing towards a survival promoting role of this kinase in UC (Figure 5A, lower panels). Indeed Src has previously been reported in other UC cells to be phosphorylated and to be a driver of cisplatin resistance (Levitt et al., 2010). As a next step we therefore set out to validate the observed Src phosphorylation using western blotting (Figure 5B). Western blot analysis of Src phosphorylation‐status verified that neither mel‐flufen, nor melphalan, or cisplatin inhibited the high level of phosphorylated Src found in untreated J82 cells. Thus an at least 2‐fold increased level of phosphorylated Src was found as compared to untreated cells (Figure 5B, lower panel). Levitt et al. previously reported that addition of the Src kinase inhibitor dasatinib could sensitise UC cells to cisplatin (Levitt et al., 2010). Based on this finding and our results we next examined if dasatinib could sensitise UC to mel‐flufen (Figure 5C,D). We found dasatinib to clearly impair phosphorylated Src Y416 in a dose‐dependent manner, an effect that was maintained when dasatinib and mel‐flufen were combined (Figure 5C, right panel). More specifically, dasatinib sensitised UC cells for mel‐flufen induced cell death (Figure 5D). Thus, while dasatinib alone caused a 35% reduction in cell viability, the combination with mel‐flufen generated a further reduction in viability resulting in only 11% and 15% viable cells in the combined regimen using mel‐flufen concentrations of 2.5 μM and 1 μM respectively (Figure 5D, right panel). These data support that Src signalling may protect UC cells from mel‐flufen and that dasatinib is a potential sensitizer for mel‐flufen treatment.
Figure 5.

Kinome profiling of UC cells after mel‐flufen treatment reveal Src as a driver of mel‐flufen refractoriness and identifies possible combination regimen. (A) The phosphorylation status of kinases and growth factor receptors were profiled in J82 cell lysates after mel‐flufen (1 μM, 24 h), melphalan (1 μM, 24 h), or cisplatin (1 μM, 24 h) exposure using PathScan RTK signalling antibody array. The phosphorylation levels of the kinases in untreated J82 cells were arbitrary set to 100 and the values in the treated samples calculated accordingly. Red bars: kinases/growth factor receptors showing at least 1.3 fold increased phosphorylation level as compared to untreated cells; Green bars: kinases/growth factor receptors showing at least 1.3 fold decreased phosphorylation level as compared to untreated cells; Grey bars: non‐altered kinases/growth factor receptors. (B) Validation of Src phosphorylation at Y416 in J82 cells using western blot with GAPDH as loading control (upper panel). Quantification of Src phosphorylation in J82 cells after indicated treatments. The western blot values were obtained by densitometry of the bands and the PathScan RTK signalling antibody array from values obtained as in (A). (C) Left: J82 cells were treated with dasatinib for 48 h and effect on Src phosphorylation at Y416 was examined by western blot with GAPDH as loading control. Right: J82 cells were treated with 20 nM dasatinib for 16 h and thereafter, with mel‐flufen for 1 h and post incubated for 48 h. The effect on Src phosphorylation at Y416 was examined by western blot in which GAPDH was used as loading control. (D) J82 cells were treated as in (C, right) and effect on cell morphology (left) and cytotoxicity (right) examined. Left: Photos showing cell morphology after indicated treatments. Right: Cells were counted using Trypan blue and is depicted as % of untreated control. Data presented is the mean ± SD in three experiments. Student's t‐test was applied.
3.6. Mel‐flufen intracellular loading and cell death are controlled by aminopeptidases and aminopeptidase N displays increased expression in clinical UC specimens
Mel‐flufen biotransformation and cell killing effects has in other tumour types been shown to depend on aminopeptidases (Chauhan et al., 2013; Wickstrom et al., 2010). The effect of bestatin‐mediated aminopeptidase blockade on mel‐flufen effects in UC was therefore evaluated (Figure 6A). Bestatin promptly blocked mel‐flufen biotransformation and reduced melphalan accumulation with about 40% (Figure 6A, left panel). Importantly, bestatin also caused an approximately 50% reduction in mel‐flufen‐induced cell death up to a concentration of 1 μM mel‐flufen (Figure 6A, right panel), hence illustrating a critical role of aminopeptidases in mel‐flufen effects in UC. One of the aminopeptidases which bestatin is inhibiting is ANPEP, which hence constitute a putative mel‐flufen biomarker. In order to find further support for mel‐flufen as a therapeutic agent in UC we therefore examined ANPEP expression by traditional immunohistochemistry in clinical UC specimen obtained from 83 patients whose demographic characteristics previously were reported (Shah et al., 2014). Analysis of ANPEP expression in these specimens revealed that all but one case had higher ANPEP staining in tumour as compared to non‐tumour tissue (Figure 6B). Within these tumours, ANPEP expression varied but was localised mainly to the UC cells and not to the surrounding stroma (Figure 6B). The degree of ANPEP staining in the specimen varied both in intensity as well as in the extent of cells showing ANPEP positivity (Figure 6C). Thus intensity was weak in 48 cases; moderate in 26 cases; and strong in 8 cases respectively (Figure 6C, upper chart). However the ANPEP IHC score revealed that in the majority of the cases (n = 77) most of the cells were labelled (76–100%) with the others cases distributed into 0%: (n = 1); 26%–50%: (n = 1) and 51%–75%: (n = 4) (Figure 6C, lower chart). In 60 out of 83 cases (72%) ANPEP was mainly confined to the cytoplasm whereas in 2 patients (2%) only membranous staining was evident. The remaining 20 patients (24%) showed both cytoplasmatic and membranous staining. By dichotomising the ANPEP tumour expression (staining intensity x score for percentage of positive cells) of the 83 patients with UC treated by cystectomy, the survival data and ANPEP expression were analysed by the Kaplan–Meier method and Breslow test (Figure 6D). Patients with high ANPEP expression had significantly longer overall survival (OS) than patients with low ANPEP expression (P = 0.02, median OS 8.1 years vs 3.2 years, mean OS 7.9 years vs 5.7 years) (Figure 6D). When stratifying the group's disease free survival (DFS) on ANPEP, no significant difference was found (P = 0.094, median DFS not reached vs not reached, mean DFS 8.7 years vs 6.9 years) (Figure 6D). Inclusion of ANPEP in the univariate Cox proportional hazard regression model did not improve statistical significance for OS (P = 0.053) or DFS (P = 0.194). The 2‐sided Pearson chi‐square test showed significant differences in the distribution of high versus low ANPEP expression in relation to pT ≤ 1 vs ≥ 2 (P = 0.024). However, no significances were found for gender (P = 0.163), UC specific cause of death (P = 0.333), relapse (P = 0.546), relapse within 18 months (P = 0.163), pN = 0 vs ≥ 1 (P = 0.558), or pWHO grade ≤2 vs > 2 (P = 0.104).
Figure 6.
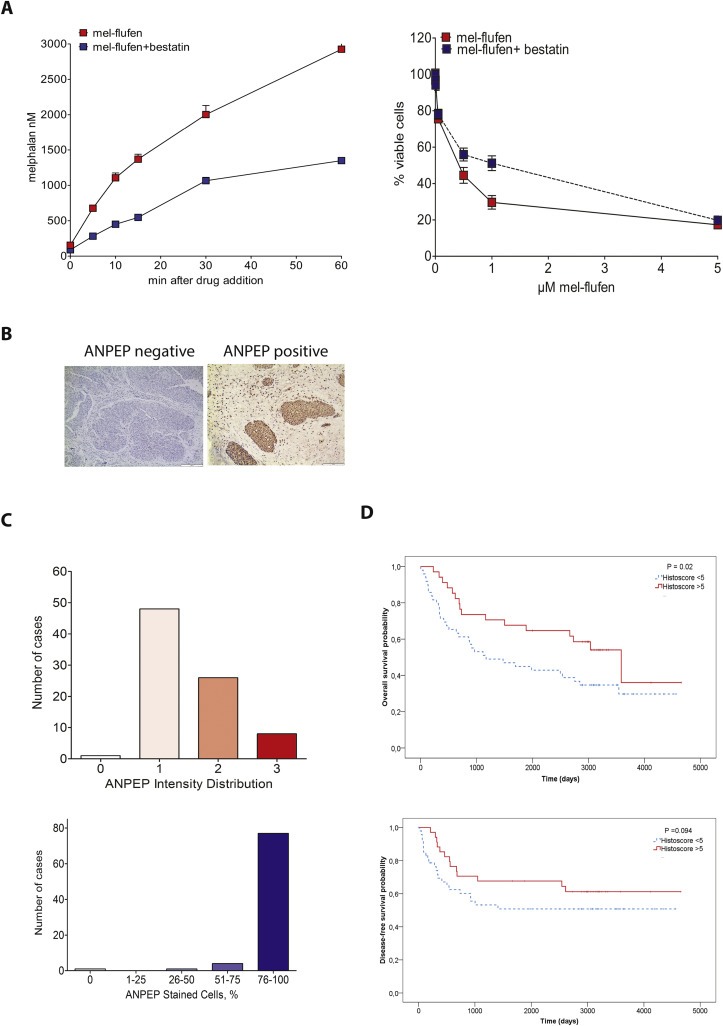
Mel‐flufen intracellular loading and cell death is controlled by aminopeptidases and Aminopeptidase N display increased expression in clinical UC specimens. (A) Left: J82 cells were treated with 1 μM mel‐flufen alone or in combination with 5 μM bestatin. The concentrations of melphalan were measured using HPLC‐MS/MS. Data shown is the mean of three biological replicates. Right: Mel‐flufen‐induced cytotoxicity ± bestatin (5 μM) was analysed in J82 cells at 72 h after 1 h pulse treatment with indicated doses of mel‐flufen using MTT‐assay. Data shown are the mean ± SD of three biological replicates. (B) Immunohistochemical staining of ANPEP in primary urothelial carcinoma. Micrographs of UC specimens determined as negative or positive for ANPEP staining. Scale bar = 200 and 100 μm, respectively. (C) Distribution of immunohistochemistry ANPEP staining intensity (upper) and semi‐quantitative scoring of number of positive cells (lower) in UC specimen from 83 patients. (D) ANPEP expression in relation to overall survival (top) and disease‐free survival (bottom) by Kaplan–Meier method. Days from cystectomy is stated. ANPEP expression was stratified as high (Histoscore > 5, red solid line) or low (Histoscore < 5, blue dotted line). High tumour expression was significantly associated with better OS (P = 0.02, upper panel), but not significant for DFS (P = 0.094, lower panel).
4. Discussion
For UC patients with platinum‐refractory progressive disease, treatment options are limited, with vinflunine being the only approved drug in the EU at present and with taxanes and gemcitabine being used worldwide based on experience only (Bellmunt et al., 2009, 2013). In the present study we characterised mel‐flufen, a prodrug of melphalan, for its cytotoxic potential in UC. The superior efficacy of mel‐flufen as compared to melphalan has been demonstrated to involve increased melphalan delivery in several different tumour cell types (Chauhan et al., 2013; Wickstrom et al., 2007). A first‐in‐human phase I/IIa study has been conducted in solid tumour malignancies (Berglund et al., 2015), and a prospective phase I/II trial is ongoing in MM, which aims to define the maximum tolerated dose and response rate (NCT01897714).
We found that mel‐flufen reduced UC viability in vitro by approximately 70% in all the four cell lines examined. This effect required however different doses in the different UC cell lines with the RT4 cells being rather refractory yet more responsive to mel‐flufen than melphalan. Moreover, when comparing induction of apoptosis, mel‐flufen was found to be superior to melphalan in all four UC cell lines, which is in accordance with results from other tumour types (Chauhan et al., 2013; Wickstrom et al., 2007). Furthermore, we found that mel‐flufen in J82 UC cells induced a higher amount of DNA cross‐links, which subsequently and in a dose dependent relation, caused increased DNA DSB formation as reflected by the higher amount of the DNA DSB marker γH2AX as compared to melphalan. This finding is completely in line with very recently released data comparing mel‐flufen and melphalan in MM with respect to DNA damage and repair (Ray et al., 2014). Here we show, for the first time, that mel‐flufen treatment induced prominent activation of Bak and Bax, two Bcl‐2 proteins previously shown to be instrumental in controlling DNA‐damage‐induced apoptosis cisplatin (Mandic et al., 2002). Importantly, whereas melphalan and cisplatin to some extent caused Bak activation, we found that mel‐flufen at equimolar concentrations caused a more pronounced activation of Bak. Furthermore, mel‐flufen was the only agent that significantly triggered Bax activation in these UC cells. This activation of both Bak and Bax in response to mel‐flufen is well in line with the more pronounced PARP‐1 cleavage and induction of apoptotic cell nuclear morphology triggered by this agent in UC cells.
Cisplatin and gemcitabine are used in clinical routine for UC patients with metastatic disease. It was therefore relevant to analyse how these agents acted in combination with mel‐flufen. Indeed an augmented cytotoxic effect was found in UC cells when mel‐flufen was combined with either gemcitabine or cisplatin as compared to single drug treatment. For the gemcitabine and mel‐flufen combination this was also accompanied by increased PARP‐1 cleavage and hence increased apoptotic signalling. It is known that melphalan gives rise to DNA cross linking, which is repaired by unhooking of the DNA cross‐linking in a stepwise manner that involves several enzymatic steps of which polymerisation of the DNA strands is one (Hansson et al., 1991). Gemcitabine is a very efficient polymerase inhibitor, (Huang et al., 1991). In combination with mel‐flufen it may therefore inhibit completion of repair of damages triggered by melphalan, resulting in decreased cell viability and increased cell death. Cisplatin induces other type of damages as compared to melphalan which are repaired via a different repair system mainly operating in S‐phase (Galluzzi et al., 2012). Thus, one putative explanation for the observed decrease in cell viability is that mel‐flufen may overload UC cells with damages in S‐phase thereby tilting the cells towards cell death. In the context of chemosensitivity in UC, cisplatin has retained the key compound during the past half century. If the in vitro effects of mel‐flufen combined with cisplatin can be confirmed in vivo, this regimen may be explored in a prospective clinical trial.
In order to further understand which signalling pathways in UC cells that remained active and could drive refractoriness to mel‐flufen, profiling of the kinome was carried out. Albeit some kinases and growth factor receptors indeed showed decreased phosphorylation after mel‐flufen treatment, notably PDGFR, FGFR3, c‐Kit and InsR, the most striking finding of this analysis was that mel‐flufen (and also melphalan and cisplatin) failed to decrease Src phosphorylation in UC J82 cells. Src kinase expression and phosphorylation status has previously been described in both UC cells in vitro and in UC clinical specimen (Levitt et al., 2010). Furthermore, Src was found to control UC cell survival in vitro as addition of dasatinib which blocked Src phosphorylation also reduced cell viability when used in combination with cisplatin (Levitt et al., 2010). It has previously been shown that activation of PI3K/Akt pathway can be controlled by Src resulting in increased cell survival and proliferation capacity survival (Winograd‐Katz and Levitzki, 2006). Hence the observed the maintained phosphorylation of Src in J82 UC cells upon mel‐flufen treatment could render a cell death protective signal and maintain Akt and Erk proliferative signalling. Indeed, we showed that blocking Src activity with dasatinib sensitized for mel‐flufen treatment, hence further supporting Src as a driver of UC cell survival. Thus both the Levitt study and our data suggest Src to be an interesting target that could further be explored in chemotherapy regimens of UC as a way to combat chemotherapy refractoriness and improve chemotherapy efficacy.
Our analyses of intracellular accumulation of mel‐flufen, melphalan, and the de‐esterified form of mel‐flufen revealed an almost 20‐fold increase in melphalan loading by using mel‐flufen as compared to melphalan. Interestingly, in contrast to what is observed in other solid tumour cells, including neuroblastoma, breast‐, lung‐, and cervical carcinoma, both intact mel‐flufen and the de‐esterified form of mel‐flufen, were evident in UC cells. One may speculate that the intact mel‐flufen or the de‐esterified form of mel‐flufen could possibly function as a reservoir of mel‐flufen which gradually can be converted to the active melphalan and contribute to the observed increased cytotoxicity after mel‐flufen exposure in these cells. The conversion of mel‐flufen to melphalan has in other solid tumour cells been shown to rely on aminopeptidases, which are inhibited by bestatin (Chauhan et al., 2013; Wickstrom et al., 2010). Also in UC cells, we found evidence of aminopeptidases to be critically involved as bestatin blocked intracellular melphalan accumulation after mel‐flufen treatment and, accordingly, reduced mel‐flufen‐induced cell death.
One of the peptidases that bestatin inhibits is ANPEP and we next therefore analysed ANPEP expression in UC clinical specimen with the aim to reveal the expression pattern of this candidate biomarker for mel‐flufen and its prognostic significance in UC. In the majority of the analysed cases, a higher expression of ANPEP was found in the tumour tissue as compared to tumour associated stroma or normal non‐tumour tissue. Interestingly, we found that the normally membrane‐bound glycoprotein ANPEP was mainly confined to the cytoplasm of UC cells. The specific and, in most cases, abundant expression of ANPEP in UC cells which we here for the first time describe, noticeably illustrates activity of ANPEP in tumour and contrary not in the surrounding stromal cells as previous has been described (Goo et al., 2005; Liu et al., 2012). Taken together, the degree and proportion of ANPEP overexpression specifically in UC cells demonstrates activity of ANPEP and potentially a role in the development of this malignancy. Our findings considering ANPEP expression in UC tumour specimens and prognosis are consistent with previous data in gastric and prostate cancer, in which a longer OS was associated with higher ANPEP tumour expression as well (Kawamura et al., 2007; Sorensen et al., 2013). In addition, in the chi‐square analysis, high expression of ANPEP in UC was relatively more frequent in lower pT‐stages (≤1).
Interestingly, a connection between ANPEP and Src has previously been reported (Meyer et al., 2008; Subramani et al., 2013), suggesting role for Src alongside FAK and ERK in controlling ANPEP activity (Subramani et al., 2013). Thus it was found that Src‐mediated phosphorylation of ANPEP triggered ANPEP association with the scaffold protein IQGAP1 allowing it to link up towards cytoskeleton signalling thereby influencing migration and invasion. Since our data showed that both ANPEP and Src are active in UC, further analyses of their interconnection and their role in controlling UC cell signalling are warranted given the previously reported role of Src in contributing to a cisplatin refractory phenotype and given our findings of Src in controlling mel‐flufen sensitivity.
The incentive for developing new therapies and enhancing the effects of available systemic chemotherapies in UC is significant. Accordingly, this is the first study to describe the efficacy of the alkylating agent mel‐flufen in UC cell lines and the mechanistic effects related to this melphalan prodrug, including its possible combination with existing chemotherapies of UC. Mouse toxicity and‐xenograft efficacy studies, are however needed to confirm the utility of mel‐flufen as a novel treatment regimen for UC either alone but more likely as part of a combination regimen with the already used agents i.e. cisplatin or gemcitabine. Our data showed ANPEP to be frequently expressed in UC and suggest it to be a candidate prognostic marker for outcome following cystectomy in this tumour type. As our in vitro data showed ANPEP to be involved in the biotransformation of mel‐flufen, ANPEP emerges as a potential predictive biomarker for mel‐flufen treatment in UC, which should be further explored in mouse xenograft studies as well as in tumour cells from UC patients treated in vitro with mel‐flufen. If successfully confirmed, such data could form the basis for a prospective clinical trial in UC which should include evaluation of ANPEP as a predictive marker for treatment outcome.
Contributions
KV, C‐HS, TJ, JS, RL, and AU designed the study and the experimental analyses. KV, TJ, PH, KZC, AS and JT performed the experiments on UC cell lines. LK, C‐HS and KV performed the immunohistochemical analyses. KV, C‐HS, PH, TJ, KZC, AS, LK, KH, JS, RL, and AU analysed and interpreted data. KV, C‐HS, TJ, and AU wrote the manuscript with the other authors revising the manuscript.
Conflicts of interest
TJ, KV and JS were partly employed by Oncopeptides AB during the initiation of the study. RL has shares in Oncopeptides AB. The other authors have no conflict of interest to declare.
Supporting information
The following are the supplementary data related to this article:
Figure S1. Relative mel‐flufen‐induced cytotoxicity in UC cells. J82, RT4, 5637 and TCC‐SUP UC cells were treated with mel‐flufen or melphalan for 72 h and cell viability was examined at 72 h of continuous treatments. The relative cytotoxic effect of mel‐flufen vs. melphalan at indicated concentrations were calculated from values presented in Figure 1B.
Supplementary Figure S2. Induction of apoptotic morphology by mel‐flufen or melphalan in RT4, 5637 and TCC‐SUP UC cells. The UC cell lines 5637, TCC‐SUP and RT4 were treated with mel‐flufen (red bars) or melphalan (grey bars) at indicated concentrations for 1 h and post incubated for 72 h. The nuclear morphology of the cells was assessed by DAPI‐staining and subsequent visualization in the FL‐1 channel of a Fluorescence microscope (A–C, left panels). The percentage of cells showing apoptotic nuclear morphology out of 200 cells examined is given. Results shown are the mean ± SD from three biological replicates. Statistical differences between mel‐flufen and melphalan were observed for 5637 at 1 μM and 5 μM (P = 0.02), for TCCsup at doses 0.5 μM and 1 μM (P = 0.02 and P = 0.03 respectively) and for RT4 at doses 0.5 μM, 1 μM and 5 μM (P = 0.002; P = 0.03; P = 0.007). PARP‐1 and caspase‐9 cleavage was also analysed by western blot in 5637 and TCC‐SUP cells treated as in above. β‐tubulin and GAPDH were used as loading controls, respectively.
Supplementary Figure S3. Mel‐flufen induces a more prominent s‐phase arrest than melphalan. Cell cycle profiling was carried out in J82 cells after 24 h post a 1 h pulse treatment with indicated doses of mel‐flufen or melphalan or after 24 h of continuous cisplatin treatment. Data shown are mean % distribution ±SD.
Acknowledgements
Financial support, including the source and number of grants, for each author: This study was supported by the Swedish Cancer Society: grant agreements 130277 (A.Ullén), 130550 (R.Lewensohn); Stockholm Cancer Society: grant agreements 141433, 144233 (A.Ullén), 131253 (K.Viktorsson), 131102 (P.Hååg), 111213 (R.Lewensohn); the Swedish National Board of Health and Welfare; the Stockholm County Council, and the Karolinska Institutet Research Fund.
The technical support from Mrs Inger Bodin, Mrs Birgitta Mörk, and Mrs Lena Lennartsson is highly appreciated.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2015.12.013.
Viktorsson Kristina, Shah Carl-Henrik, Juntti Therese, Hååg Petra, Zielinska-Chomej Katarzyna, Sierakowiak Adam, Holmsten Karin, Tu Jessica, Spira Jack, Kanter Lena, Lewensohn Rolf, Ullén Anders, (2016), Melphalan‐flufenamide is cytotoxic and potentiates treatment with chemotherapy and the Src inhibitor dasatinib in urothelial carcinoma, Molecular Oncology, 10, doi: 10.1016/j.molonc.2015.12.013.
References
- Bambury, R.M. , Rosenberg, J.E. , 2013. Advanced urothelial carcinoma: overcoming treatment resistance through novel treatment approaches. Front Pharmacol. 4, 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellmunt, J. , Theodore, C. , Demkov, T. , Komyakov, B. , Sengelov, L. , Daugaard, G. , Caty, A. , Carles, J. , Jagiello-Gruszfeld, A. , Karyakin, O. , Delgado, F.M. , Hurteloup, P. , Winquist, E. , Morsli, N. , Salhi, Y. , Culine, S. , von der Maase, H. , 2009. Phase III trial of vinflunine plus best supportive care compared with best supportive care alone after a platinum-containing regimen in patients with advanced transitional cell carcinoma of the urothelial tract. J. Clin. Oncol. 27, 4454–4461. [DOI] [PubMed] [Google Scholar]
- Bellmunt, J. , Fougeray, R. , Rosenberg, J.E. , von der Maase, H. , Schutz, F.A. , Salhi, Y. , Culine, S. , Choueiri, T.K. , 2013. Long-term survival results of a randomized phase III trial of vinflunine plus best supportive care versus best supportive care alone in advanced urothelial carcinoma patients after failure of platinum-based chemotherapy. Ann. Oncol. 24, 1466–1472. [DOI] [PubMed] [Google Scholar]
- Berglund, Å. , Ullén, A. , Lisyanskaya, A. , Orlov, S. , Hagberg, H. , Tholander, B. , Lewensohn, R. , Nygren, P. , Spira, J. , Harmenberg, J. , Jerling, M. , Alvfors, C. , Ringbom, M. , Nordström, E. , Söderlind, K. , Gullbo, J. , 2015. First-in-human, phase I/IIa clinical study of the peptidase potentiated alkylator melflufen administered every three weeks to patients with advanced solid tumor malignancies. Invest. New Drugs. 33, 1232–1241. [DOI] [PubMed] [Google Scholar]
- Chauhan, D. , Ray, A. , Viktorsson, K. , Spira, J. , Paba-Prada, C. , Munshi, N. , Richardson, P. , Lewensohn, R. , Anderson, K.C. , 2013. In vitro and in vivo antitumor activity of a novel alkylating agent, melphalan-flufenamide, against multiple myeloma cells. Clin. Cancer Res. 19, 3019–3031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darzynkiewicz, Z. , Huang, X. , 2004. Analysis of cellular DNA content by flow cytometry. Curr. Protoc. Immunol. Chapter 5, section 7.1-5.7.18 [DOI] [PubMed] [Google Scholar]
- Ferlay, J. , Soerjomataram, I.I. , Dikshit, R. , Eser, S. , Mathers, C. , Rebelo, M. , Parkin, D.M. , Forman, D.D. , Bray, F. , 2014. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer. [DOI] [PubMed] [Google Scholar]
- Fogh, J. , Fogh, J.M. , Orfeo, T. , 1977. One hundred and twenty-seven cultured human tumor cell lines producing tumors in nude mice. J. Natl. Cancer Inst. 59, 221–226. [DOI] [PubMed] [Google Scholar]
- Galluzzi, L. , Senovilla, L. , Vitale, I. , Michels, J. , Martins, I. , Kepp, O. , Castedo, M. , Kroemer, G. , 2012. Molecular mechanisms of cisplatin resistance. Oncogene. 31, 1869–1883. [DOI] [PubMed] [Google Scholar]
- Goo, Y.A. , Goodlett, D.R. , Pascal, L.E. , Worthington, K.D. , Vessella, R.L. , True, L.D. , Liu, A.Y. , 2005. Stromal mesenchyme cell genes of the human prostate and bladder. BMC Urol. 5, 17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths, G.J. , Dubrez, L. , Morgan, C.P. , Jones, N.A. , Whitehouse, J. , Corfe, B.M. , Dive, C. , Hickman, J.A. , 1999. Cell damage-induced conformational changes of the pro-apoptotic protein Bak in vivo precede the onset of apoptosis. J. Cell Biol. 144, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gullbo, J. , Dhar, S. , Luthman, K. , Ehrsson, H. , Lewensohn, R. , Nygren, P. , Larsson, R. , 2003. Antitumor activity of the alkylating oligopeptides J1 (L-melphalanyl-p-L-fluorophenylalanine ethyl ester) and P2 (L-prolyl-m-L-sarcolysyl-p-L-fluorophenylalanine ethyl ester): comparison with melphalan. Anticancer Drugs. 14, 617–624. [DOI] [PubMed] [Google Scholar]
- Gullbo, J. , Wickstrom, M. , Tullberg, M. , Ehrsson, H. , Lewensohn, R. , Nygren, P. , Luthman, K. , Larsson, R. , 2003. Activity of hydrolytic enzymes in tumour cells is a determinant for anti-tumour efficacy of the melphalan containing prodrug J1. J. Drug Target. 11, 355–363. [DOI] [PubMed] [Google Scholar]
- Hansson, J. , Lewensohn, R. , Ringborg, U. , 1991. Cytotoxicity and DNA cross-linking induced by peptide conjugated m-L-sarcolysin in human melanoma cells. Anticancer Res. 11, 1725–1730. [PubMed] [Google Scholar]
- Huang, P. , Chubb, S. , Hertel, L.W. , Grindey, G.B. , Plunkett, W. , 1991. Action of 2',2'-difluorodeoxycytidine on DNA synthesis. Cancer Res. 51, 6110–6117. [PubMed] [Google Scholar]
- Kawamura, J. , Shimada, Y. , Kitaichi, H. , Komoto, I. , Hashimoto, Y. , Kaganoi, J. , Miyake, M. , Yamasaki, S. , Kondo, K. , Imamura, M. , 2007. Clinicopathological significance of aminopeptidase N/CD13 expression in human gastric carcinoma. Hepatogastroenterology. 54, 36–40. [PubMed] [Google Scholar]
- Lee, C.K. , Wang, S. , Huang, X. , Ryder, J. , Liu, B. , 2010. HDAC inhibition synergistically enhances alkylator-induced DNA damage responses and apoptosis in multiple myeloma cells. Cancer Lett. 296, 233–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitt, J.M. , Yamashita, H. , Jian, W. , Lerner, S.P. , Sonpavde, G. , 2010. Dasatinib is preclinically active against Src-overexpressing human transitional cell carcinoma of the urothelium with activated Src signaling. Mol. Cancer Ther. 9, 1128–1135. [DOI] [PubMed] [Google Scholar]
- Liu, A.Y. , Vencio, R.Z. , Page, L.S. , Ho, M.E. , Loprieno, M.A. , True, L.D. , 2012. Bladder expression of CD cell surface antigens and cell-type-specific transcriptomes. Cell Tissue Res. 348, 589–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandic, A. , Viktorsson, K. , Strandberg, L. , Heiden, T. , Hansson, J. , Linder, S. , Shoshan, M.C. , 2002. Calpain-mediated Bid cleavage and calpain-independent Bak modulation: two separate pathways in cisplatin-induced apoptosis. Mol. Cell Biol. 22, 3003–3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, R.D. , Sacks, D.B. , Rahimi, N. , 2008. IQGAP1-dependent signaling pathway regulates endothelial cell proliferation and angiogenesis. PLoS One. 3, e3848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak, S.K. , O'Toole, C. , Price, Z.H. , 1977. A cell line from an anaplastic transitional cell carcinoma of human urinary bladder. Br. J. Cancer. 35, 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Toole, C. , Price, Z.H. , Ohnuki, Y. , Unsgaard, B. , 1978. Ultrastructure, karyology and immunology of a cell line originated from a human transitional-cell carcinoma. Br. J. Cancer. 38, 64–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray, A. , Ravillah, D. , Sharma Das, D. , Song, Y. , Nordström, E. , Lindberg, J. , Richardson, P.G. , Chauhan, D. , Anderson, K.C. , 2014. A Novel Alkylating Agent Melphalan Flufenamide Ethyl Ester Induces an Irreversible DNA Damage in Multiple Myeloma Cells 56th ASH Annual Meeting and Exposition Program: Oral and Poster Abstracts Session: 652. Myeloma: Pathophysiology and Pre-clinical Studies, Excluding Therapy: Poster I. 2086. [Google Scholar]
- Rigby, C.C. , Franks, L.M. , 1970. A human tissue culture cell line from a transitional cell tumour of the urinary bladder: growth, chromosome pattern and ultrastructure. Br. J. Cancer. 24, 746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah, C.H. , Viktorsson, K. , Kanter, L. , Sherif, A. , Asmundsson, J. , Rosenblatt, R. , Lewensohn, R. , Ullen, A. , 2014. Vascular endothelial growth factor receptor 2, but not S100A4 or S100A6, correlates with prolonged survival in advanced urothelial carcinoma. Urol. Oncol. 32, 1215–1224. [DOI] [PubMed] [Google Scholar]
- Sorensen, K.D. , Abildgaard, M.O. , Haldrup, C. , Ulhoi, B.P. , Kristensen, H. , Strand, S. , Parker, C. , Hoyer, S. , Borre, M. , Orntoft, T.F. , 2013. Prognostic significance of aberrantly silenced ANPEP expression in prostate cancer. Br. J. Cancer. 108, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sternberg, C.N. , Yagoda, A. , Scher, H.I. , Watson, R.C. , Geller, N. , Herr, H.W. , Morse, M.J. , Sogani, P.C. , Vaughan, E.D. , Bander, N. , 1989. Methotrexate, vinblastine, doxorubicin, and cisplatin for advanced transitional cell carcinoma of the urothelium. Efficacy and patterns of response and relapse. Cancer. 64, 2448–2458. [DOI] [PubMed] [Google Scholar]
- Subramani, J. , Ghosh, M. , Rahman, M.M. , Caromile, L.A. , Gerber, C. , Rezaul, K. , Han, D.K. , Shapiro, L.H. , 2013. Tyrosine phosphorylation of CD13 regulates inflammatory cell-cell adhesion and monocyte trafficking. J. Immunol. 191, 3905–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surowiak, P. , Drag, M. , Materna, V. , Suchocki, S. , Grzywa, R. , Spaczynski, M. , Dietel, M. , Oleksyszyn, J. , Zabel, M. , Lage, H. , 2006. Expression of aminopeptidase N/CD13 in human ovarian cancers. Int. J. Gynecol. Cancer. 16, 1783–1788. [DOI] [PubMed] [Google Scholar]
- Tokuhara, T. , Adachi, M. , Hashida, H. , Ishida, H. , Taki, T. , Higashiyama, M. , Kodama, K. , Tachibana, S. , Sasaki, S. , Miyake, M. , 2001. Neutral endopeptidase/CD10 and aminopeptidase N/CD13 gene expression as a prognostic factor in non-small cell lung cancer. Jpn. J. Thorac. Cardiovasc. Surg. 49, 489–496. [DOI] [PubMed] [Google Scholar]
- Tokuhara, T. , Hattori, N. , Ishida, H. , Hirai, T. , Higashiyama, M. , Kodama, K. , Miyake, M. , 2006. Clinical significance of aminopeptidase N in non-small cell lung cancer. Clin. Cancer Res. 12, 3971–3978. [DOI] [PubMed] [Google Scholar]
- Vishnu, P. , Mathew, J. , Tan, W.W. , 2011. Current therapeutic strategies for invasive and metastatic bladder cancer. Onco Targets Ther. 4, 97–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von der Maase, H. , Sengelov, L. , Roberts, J.T. , Ricci, S. , Dogliotti, L. , Oliver, T. , Moore, M.J. , Zimmermann, A. , Arning, M. , 2005. Long-term survival results of a randomized trial comparing gemcitabine plus cisplatin, with methotrexate, vinblastine, doxorubicin, plus cisplatin in patients with bladder cancer. J. Clin. Oncol. 23, 4602–4608. [DOI] [PubMed] [Google Scholar]
- Wickstrom, M. , Johnsen, J.I. , Ponthan, F. , Segerstrom, L. , Sveinbjornsson, B. , Lindskog, M. , Lovborg, H. , Viktorsson, K. , Lewensohn, R. , Kogner, P. , Larsson, R. , Gullbo, J. , 2007. The novel melphalan prodrug J1 inhibits neuroblastoma growth in vitro and in vivo. Mol. Cancer Ther. 6, 2409–2417. [DOI] [PubMed] [Google Scholar]
- Wickstrom, M. , Viktorsson, K. , Lundholm, L. , Aesoy, R. , Nygren, H. , Sooman, L. , Fryknas, M. , Vogel, L.K. , Lewensohn, R. , Larsson, R. , Gullbo, J. , 2010. The alkylating prodrug J1 can be activated by aminopeptidase N, leading to a possible target directed release of melphalan. Biochem. Pharmacol. 79, 1281–1290. [DOI] [PubMed] [Google Scholar]
- Wickstrom, M. , Larsson, R. , Nygren, P. , Gullbo, J. , 2011. Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 102, 501–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winograd-Katz, S.E. , Levitzki, A. , 2006. Cisplatin induces PKB/Akt activation and p38(MAPK) phosphorylation of the EGF receptor. Oncogene. 25, 7381–7390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Figure S1. Relative mel‐flufen‐induced cytotoxicity in UC cells. J82, RT4, 5637 and TCC‐SUP UC cells were treated with mel‐flufen or melphalan for 72 h and cell viability was examined at 72 h of continuous treatments. The relative cytotoxic effect of mel‐flufen vs. melphalan at indicated concentrations were calculated from values presented in Figure 1B.
Supplementary Figure S2. Induction of apoptotic morphology by mel‐flufen or melphalan in RT4, 5637 and TCC‐SUP UC cells. The UC cell lines 5637, TCC‐SUP and RT4 were treated with mel‐flufen (red bars) or melphalan (grey bars) at indicated concentrations for 1 h and post incubated for 72 h. The nuclear morphology of the cells was assessed by DAPI‐staining and subsequent visualization in the FL‐1 channel of a Fluorescence microscope (A–C, left panels). The percentage of cells showing apoptotic nuclear morphology out of 200 cells examined is given. Results shown are the mean ± SD from three biological replicates. Statistical differences between mel‐flufen and melphalan were observed for 5637 at 1 μM and 5 μM (P = 0.02), for TCCsup at doses 0.5 μM and 1 μM (P = 0.02 and P = 0.03 respectively) and for RT4 at doses 0.5 μM, 1 μM and 5 μM (P = 0.002; P = 0.03; P = 0.007). PARP‐1 and caspase‐9 cleavage was also analysed by western blot in 5637 and TCC‐SUP cells treated as in above. β‐tubulin and GAPDH were used as loading controls, respectively.
Supplementary Figure S3. Mel‐flufen induces a more prominent s‐phase arrest than melphalan. Cell cycle profiling was carried out in J82 cells after 24 h post a 1 h pulse treatment with indicated doses of mel‐flufen or melphalan or after 24 h of continuous cisplatin treatment. Data shown are mean % distribution ±SD.