

Abstract
Pancreatic ductal adenocarcinoma (PDAC) clinically has a very poor prognosis. No small molecule is available to reliably achieve cures. Meisoindigo is chemically related to the natural product indirubin and showed substantial efficiency in clinical chemotherapy for CML in China. However, its effect on PDAC is still unknown. Our results showed strong anti‐proliferation effect of meisoindigo on gemcitabine‐resistant PDACs. Using a recently established primary PDAC cell line, called Jopaca‐1 with a larger CSCs population as model, we observed a reduction of CD133+ and ESA+/CD44+/CD24+ populations upon treatment and concomitantly a decreased expression of CSC‐associated genes, and reduced cellular mobility and sphere formation. Investigating basic cellular metabolic responses, we detected lower oxygen consumption and glucose uptake, while intracellular ROS levels increased. This was effectively neutralized by the addition of antioxidants, indicating an essential role of the cellular redox balance. Further analysis on energy metabolism related signaling revealed that meisoindigo inhibited LKB1, but activated AMPK. Both of them were involved in cellular apoptosis. Additional in situ hybridization in tissue sections of PDAC patients reproducibly demonstrated co‐expression and ‐localization of LKB1 and CD133 in malignant areas. Finally, we detected that CD133+/CD44+ were more vulnerable to meisoindigo, which could be mimicked by LKB1 siRNAs. Our results provide the first evidence, to our knowledge, that LKB1 sustains the CSC population in PDACs and demonstrate a clear benefit of meisoindigo in treatment of gemcitabine‐resistant cells. This novel mechanism may provide a promising new treatment option for PDAC.
Keywords: Indirubin, Meisoindigo, Pancreatic cancer stem cell, CSC drug, LKB1 inactivation, AMPK activation, PDAC, CD133
List of abbreviations
- 2-DG
2-deoxyglucose
- ACC
acetyl-CoA carboxylase
- AMPK
5′ AMP-activated protein kinase
- CHK2
check point kinase 2
- DM
Dorsomorphin
- ESA
epithelial cell adhesion molecule
- GSH
glutathione
- HSP27
heat shock protein 2
- LKB1
liver kinase B1
- p38-MAPK
p38 mitogen-activated protein kinase
- Mei
meisoindigo/methylisoindigo
- NAC
N-acetyl-l-cysteine
- PDAC
pancreatic ductal adenocarcinoma
- PARP
poly ADP ribose polymerase
- RM
running medium
- ROS
reactive oxygen species
1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal diseases. More than 80% of patients suffering from PDAC are diagnosed at a clinically advanced state. PDAC exhibits high metastatic and aggressive phenotypes and presents with less than 5% in 5‐year survival (Hidalgo, 2010). Although intensive research efforts in the last decades provided a better biological understanding of PDAC and improved surgical and medical therapies (Wong and Lemoine, 2009), the high mortality among PDAC patients has not changed (Siegel et al., 2015). The molecular basis of such high resistance to almost all forms of therapies still needs to be elucidated further.
There is mounting evidence that subpopulations of PDAC and other cells display stem cell characteristics, consequently termed cancer stem cells (CSCs), which contribute to tumor initiation, progression, metastasis, invasion, resistance to therapies and also local recurrence after surgery and therefore should be an important target of these highly refractory malignant tumors (Clevers, 2011; Gattinoni et al., 2012; Magee et al., 2012). The concept that malignant cell populations are hierarchical was first pioneered by Dick and his colleagues in leukemia (Bonnet and Dick, 1997; Lapidot et al., 1994) and later by Al‐Hajj et al., using a combination of cell surface markers (Al‐Hajj et al., 2003). Weinberg and his coworkers further identified aberrant expression of CD44+/CD24− phenotype in epithelial cells with ongoing EMT, implicating this trans‐differentiation program confers epithelial cells CSC‐like traits (Mani et al., 2008). Very recently, they demonstrated that non‐CSCs could be converted to CSCs by aberrant expression of ZEB1, which highlights a potentially rapid transition and the importance of inhibiting stemness‐associated signaling in both non‐CSCs and CSCs (Chaffer et al., 2013). In PDAC, Li et al. discovered that PDACs also comprises heterogeneous populations and showed cells expressing CD44+/CD24+/ESA+ are self‐renewing and possess a 100‐fold higher tumorigenic potential (Li et al., 2007). Similarly, Moriyama et al. identified CD133 as a putative CSCs marker in PDACs and reported CD133+ PDAC cells to possess enhanced migration and invasion properties (Moriyama et al., 2010). Recently, Fredebohm et al. established a new primary PDAC cell line, Jopaca‐1, from a 46‐year old male patient and verified that in early passage (<19) it consisted of a high amount of CD133+ and CD44+/CD24+/ESA+ cells with a high mutant ratio, including TP53, showing poor response to gemcitabine and high clonogenic potential in vitro and in vivo (Fredebohm et al., 2012).
Peutz–Jeghers syndrome (PJS) is a hereditary disease caused by germ line mutations of liver kinase B1 (LKB1) and is closely associated with PDAC in patients (Alessi et al., 2006). Functional LKB1 is an upstream of AMPK family signaling and had been described as a master gene of cell metabolism. In malignancies, LKB1 often emerges as a loss‐of‐function mutation lacking regulation of cellular metabolism (Shackelford and Shaw, 2009). Recent studies revealed that LKB1 was also involved in quiescence of hematopoietic stem cells most likely in an AMPK‐independent manner (Gan et al., 2010; Gurumurthy et al., 2010; Nakada et al., 2010). Nevertheless, the role of LKB1 in cancer stem cells (CSCs) in PDAC is not well elucidated yet.
Indirubin, a 2′,3‐linked indigoid bisindole, is a major ingredient of a traditional Chinese herbal recipe, used for the treatment of chronic myeloid leukemia (CML) in China (Cheng et al., 2010, 2014, 2004, 2013, 2006, 2008). 1‐Methylisoindigo (N‐methylisoindigo), also known as meisoindigo, is a derivative of isoindigo, a 3,3′‐linked bisindole, that was first synthesized by condensation of oxindole and 1‐methylisatin (Wahl and Bagard, 1913), and later on, alternatively, by condensation of 1‐methyloxindole and isatin (Stolle et al., 1930). In comparison to indirubin, a well‐known ATP competitive protein kinase inhibitor (Davies et al., 2001; Hoessel et al., 1999; Meijer et al., 2003), meisoindigo did not show any significant activity against protein kinases (Bouchikhi et al., 2008; Wee et al., 2009). Several reports demonstrated that meisoindigo induced apoptosis, cell cycle arrest and differentiation in leukemic cell lines (Chen et al., 2010, 2010, 2002, 2006). However, the effect of meisoindigo on other tumors and its mechanism of action are still poorly understood.
In this work, we investigated the effect of meisoindigo on PDACs, and found that meisoindigo effectively inhibited growth of gemcitabine‐resistant PDAC cells, which included Panc1 and Jopaca‐1. Using Jopaca‐1 as a CSC model, we found that meisoindigo greatly ablated CSC populations, reduced CSC‐associated gene expression, suppressed self‐renewal, and reduced the tumorigenic potential. To gain insight into molecular mechanisms, we studied its impact on cell metabolism and observed activation of the AMPK cascade, while at the same time LKB1 was inhibited upon treatment. Further analysis unveiled that meisoindigo interrupted the cellular redox balance and consequently induced apoptosis, which could be compensated by antioxidants. Importantly, CD133+/CD44+ CSCs were more vulnerable to meisoindigo in comparison to other cell populations. Depletion of LKB1 by siRNAs mimicked this effect of meisoindigo treatment. Our results clearly demonstrate that meisoindigo preferentially killed CSCs in Jopaca‐1 by targeting LKB1 and AMPK signaling and interfering with the cellular metabolic balance. This may provide a new therapeutic option for treatment of CSCs in PDACs.
2. Materials and methods
2.1. Synthesis of meisoindigo
Under inert atmosphere, 1‐methyl‐isatin (1 mmol) and 2‐oxindole (1 mmol) were stirred in a mixture of glacial acetic acid (10 mL) and 1 mL of concentrated HCl (12 N) at room temperature for 24 h. After adding 100 mL of water, the precipitate was filtered and dried to achieve a reddish solid in a good yield (96%). Structures and purities were ascertained by 13C NMR, 1H NMR and HR‐MS spectra as previously reported (Wee et al., 2009). 1H NMR (300 MHz; DMSO‐d6): 3.20 (S, 3H), 6.83 (d, 3 J = 7.8 Hz), 6.95–7.02 (m, 3H), 7.33–7.41 (m, 2H), 9.06–9.09 (m, 2H), 10.87 (S, 1H). 13C NMR (DMSO‐d6): 26.0, 108.3, 109.5, 120.8, 121.1, 121.6, 121.7, 128.9, 132.2, 132.4, 132.7, 133.7, 144.2, 144.9, 167.1, 168.8. HRMS (ESI) m/z: 277.69 [M+H]+.
HPLC was performed and showed the purity of compound was at least higher than 96%.
2.2. Materials
2‐Deoxyglucose (2‐DG), N‐acetyl‐l‐cysteine (NAC), reduced glutathione (GSH) and gemcitabine (Gem) were purchased by Sigma–Aldrich (Germany). LKB1 (3047), pLKB1 (3482), AMPK1a (2603), pAMPK1a (2535), ACC (3676), pACC (3661), Caspase 3 (9662), PARP (9542), ZEB1 (3396), β‐Catenin (9582), N‐Cadherin (4061), c‐Myc, Bcl‐XL and surviving (2802), were obtained from cell signaling (NEB, Germany). Oct3/4 (sc‐5279) was from Santa Cruz (Germany). Nanog (ab62734) was from abcam (Germany). The study on patients' sample has been approved by ethics committee of the Medical Faculty Heidelberg of Heidelberg University (Ethikkommission I Heidelberg: Studienzeichen: S‐202/2012). In situ hybridization was performed as before (Ghafoory et al., 2012). The patients' information and primer sequences were listed in Supplementary information.
2.3. Cell culture
Jopaca‐1, Bxpc3 and NCCIT was cultured in RPMI1640 (Gibco, Germany) containing 10% FBS and 1% Pen/Strp (PS, Sigmal, Germany) under 5% CO2 at 37 °C in a humidified atmosphere. Panc1, Miapaca2, HeLa, MCF7 and HFF were cultured in DMEM (Gibco) containing 10% FBS and 1% PS. Transfections of siRNA oligonucleotides were reported previously (Stahmann et al., 2006) and synthesized by riboxx. Riboxx®FECT reagent was used to get maximal transfectional efficiency. The transfection was followed to the manufacturer's instructions (Cheng et al., 2012).
2.4. MTT assay
3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide (MTT) assay was performed to determine anti‐proliferative effect of compounds as previously reported (Cheng et al., 2014a). Briefly, cells were plated into 96‐well plates at a density of 10,000 cells/cm2 for 48 h and 72 h treatment and 50,000 cells/cm2 for 24 h MTT, and cultivated for 24 h. The cells were incubated with the respective compounds as indicated in completed medium in quadruplicate. Thereafter, the culture medium were replaced with a solution of MTT (25 mg/50 mL) in medium containing 1% FBS, incubated for 2 h and quantified photometrically at 595 nm. Cytotoxicity was determined as percent survival, determined by the number of treated over DMSO cells. The comparable results were obtained from three independent experiments.
2.5. ROS formation assay
Jopaca‐1 cells were incubated with compounds as described (Cheng et al., 2014a). Afterwards, cells were collected and re‐suspended in FACS (Fluorescence‐activated cell sorting) buffer (1% BSA in D‐PBS) containing 5 μM dihydroethidium (DHE, Sigma–Aldrich) at room temperature in the dark for 15 min, subsequently diluted with 500 μL FACS buffer, and immediately analyzed by FACS using a FACSCalibur (Becton Dickinson) and CellQuest Pro (BD) analysis software. Excitation and emission settings were 488 nm and 564–606 nm (FL2 filter), respectively. The comparable results were obtained from three‐independent experiments.
2.6. Apoptosis assay
The Jopaca‐1 cells were incubated with compounds as indicated. The cells were trypsinized, resuspended in 50 μL annexin V binding buffer and incubated with 5 μL FITC‐conjugated annexin V (BD Bioscience, Germany) for 15 min in the dark at room temperature. Afterwards, the suspension was diluted in 450 μL annexin V binding buffer containing 1.25 μL propidium iodide (PI, 1 mg/mL), incubated for 10 min in the dark at room temperature and analyzed on FACS as described (Cheng et al., 2014a). The comparable results were obtained from at least three independent experiments.
2.7. Cell cycle analysis
The Jopaca‐1 cells were treated with meisoindigo. After trypsinization, the pellets were fixed in 70% Ethanol for at least 24 h at −20 °C, washed two times with ice‐cold PBS and incubation with RNase A (50 μg/mL) for 1 h at 37 °C and PI (50 μg/mL) for 30 min. The mixture was washed with PBS and analyzed on FACS as described (Cheng et al., 2014a). The comparable results were obtained from two independent experiments. One experiment is depicted.
2.8. Detection of stem cell markers
PDAC cells were cultivated under standard condition and treated with compounds or siRNA as indicated. As described before (Fredebohm et al., 2012), cells were trypsinized, collected and blocked in FcR blocking buffer (Miltenyi, 130‐059‐901, Germany) at 4 °C for 10 min. PE‐Cy7‐CD44 (BD, C560533), PE‐CD133 (Miltenyi, 130‐098‐826), APC‐ESA (Miltenyi, 130‐091‐254.) and/or FITC‐CD24 (BD, C555427) antibodies were added according to manufacturer's recommendation and incubated 10 min at 4 °C.
2.9. Membrane potential measurement
The treated Jopaca‐1 cells were collected and stained with 1 μM JC‐1 (5,5,6,6‐tetrachloro‐1,1,3,3‐tetraethylbenzimidazolylcarbocyanineiodide, Sigma–Aldrich) in FACS buffer for 30 min at 37 °C and analyzed by FACS (Cheng et al., 2014a). Excitation and emission settings were 488 nm, 515–545 nm (FL1 channel) for JC monomers, and 564–606 nm (FL2 channel) for JC aggregates. The comparable results were obtained at least from two‐independent experiments. One experiment is depicted.
2.10. Immunoblot
Cell extracts were homogenized in urea‐lysis buffer (1 mM EDTA, 0.5% Triton X‐100, 5 mM NaF, 6 M Urea, 1 mM Na3VO4, 10 μg/mL Pepstatin, 100 μM PMSF and 3 μg/mL Aprotinin in PBS). The immunoblotting detected with ECL solution was performed as previous reported (Cheng et al., 2012). The fluorescent immunoblot was performed as following described. Briefly, the membrane was first blocked with fluorescent western blot blocking buffer (Rockland, Biomol, Germany). 40 μg of total protein was resolved on 10% SDS‐PAGE gels and immunoblotted with specific antibodies. Primary antibodies were incubated at a 1:1000 dilution in blocking buffer with gentle agitation overnight at 4 °C. The proper secondary antibodies (Dylight®680‐ or Dylight®800‐conjugated) were incubated in TBS (pH 7.5) at a 1:10,000 dilution for 1 h at room temperature. Licor®‐odeysee system was used.
2.11. qRT‐PCR
Quantitative real‐time reverse transcription‐PCR was performed according to manufacturing introduction (Light Cycler, Roche, Germany). Briefly, total RNA was isolated from cells using RNeasy kit from Qiagen. cDNA was generated by reverse‐transcription of equivalent quantities of RNA and qRT‐PCR was performed using SYBR Green PCR master mix on Light Cycler 480 (Roche, Germany). The following primer (Eurofins) pairs were used as previous reported (Cheng et al., 2015, 2015): Oct4 (5s: GAAGTTGGAGAAGGTGGAAC; 3as: GGTGATCCTCTTCTGCTTCAG), Sox2 (5s: CAAGACGCTCATGAAGAAGG; 3as: CATGTGCGCGTAACTGTCCATG), E‐Cadherin (5s: GAGAGACTGGGTTATTCCTC; 3as: GATGCTGTAGAAAACCTTGCC), N‐Cadherin (5s: GAAGATGGCATGGTGTATGC; 3as: CTCCTCAGTTAAGGTTGGCTTC), ZEB1 (5s: GCCATATGAATGCCCAAACTGC; 3as: CTTGAGTCCTGTTCTTGGTC); ASK1 (5s: AACTTCGGTGCTTCCTCTGTCA; 3as: CAGTGAAGTTCTGTGCAGAAG); HSP27 (5s: ACGAGCTGACGGTCAAGACCAA; 3as: AGCGTGTATTTCCGCGTGAAG); Axin2 (5s: AAGACGGTGCTTACCTGTTC; 3as: GGTATCCTTCAGGTTCATCTGC); Vimentin (5s: CAGACAGGATGTTGACAATGCG; 3as: CAGCTCCTGGATTTCCTCTTC). For the determination of Actin (5s: CTGACTACCTCATGAAGATCCTC; 3as: CATTGCCAATGGTGATGACCTG) was used as an endogenous control. The comparable results were obtained from three independent experiments.
2.12. Wound healing assay
Jopaca‐1 and BxPC3 cells were plated at a density of 300,000 cells/well in a 12‐well plate and grown to 85%–90% confluence. The scratch was made by a sterile P‐200 micropipette in the middle of each well. Cells were washed three times with PBS and treated with the compound. Photographs were taken at the beginning and the end of the experiments following the PBS wash.
2.13. Soft agar assay
Jopaca‐1 and BxPC3 cells were plated into 12‐well plate and treated with meisoindigo for 24 h. Cells were harvested and re‐plated into a 12‐well plate (1000 cells/well) at a bottom layer of 2% agar and upper layer 0.2% agar for 14 days. The medium were changed every 5 days. Afterwards cells were visualized by 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide for 2 h.
2.14. Tumor sphere formation assay
Jopaca‐1 and BxPC3 cells were treated with meisoindigo (20 μM) or DMSO 1% as control for 24 h in RPMI1640 (10% FBS, 1% PS). The sphere formation assay was performed as previously reported (Fredebohm et al., 2012). The cells were trypsinized and re‐plated in Ham's F12 medium supplemented with 20 ng/ml basic fibroblast growth factor (bFGF) (Biomol, Germany), 1× B‐27 supplements (Invitrogen, Germany), 2 mM l‐glutamine and 1% P/S on ultra‐low attachment plate (Corning, Germany) at a density of 2000 cells/well in a 6‐well plate for 21 days. The medium was changed every 5 days.
2.15. Clonogenic potential in vitro assay
Jopaca‐1 and BxPC3 cells were seeded in 96‐well plates in dilution series as indicated, starting at 125 cells per well (Fredebohm et al., 2012). After 14 days incubation without changing the medium, the cells were visualized with 3‐(4,5‐Dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide and spectrophotometrically qualified at 595 nm in the DMSO solution.
2.16. Glucose uptake assay and Bionas analysis in real‐time
Jopaca‐1 cells were treated with meisoindigo as designed. The cells were trypsinized, resuspended in 100 μL KRB buffer containing 10 μg 2‐NDBG (Cayman, Biomol, Germany) and incubated for 30 min. The uptake of 2‐NDBG was measured by FACS. Bionas analysis was performed as previously reported (Alborzinia et al., 2011).
2.17. ATP quantification assay
The cellular ATP concentration was quantitatively determined by using ATP determination kit (Invitrogen, Germany) according to the manufacturer's instructions. Briefly, Jopaca‐1 cells were treated with compound and collected in 1× reaction buffer at designed time points. 90 μL of standard reaction solution was added into 10 μL of the cell suspension in a new 96‐well white plate and the mixture was incubated for 15 min at room temperature. ATP was quantified using the Tecan infinite 2000 (Tecan, Germany).
2.18. Protein microarray analysis
Phosphorylated proteins were quantified using sandwich ELISA microarrays. The microarrays are based on the ArrayStrip™ platform (Alere Technologies GmbH, Jena, Germany). Detailed description of the assay protocol and information on reagents for this assay has been previously reported (Holenya et al., 2011). In brief, cellular samples were diluted 1:6 with dilution buffer (1 mM EDTA, 0.5% Triton X‐100, 5 mM sodium fluoride, 1 M urea in buffered saline, pH 7.2) and incubated with microarrays for 60 min. A detection cocktail of 15 biotin‐labeled phospho‐specific detection antibodies (R&D Systems) was used, with the concentration of each antibody at 18 ng/ml. Streptavidin–horseradish peroxidase conjugate (R&D Systems) was added and the enzymatic reaction was started with 3,3′,5,5′‐tetramethylbenzidine (TMB) (TrueBlue™, KPL Inc., Gaithersburg, USA). Colorimetric signals were detected by transmission measurements with the Arraymate™ reader (Alere Technology GmbH). TMB precipitation was monitored immediately over 60 min and proteins were quantified relative to total protein using the KOMA software package (Holenya et al., 2012, 2014).
2.19. Statistical analysis
The statistical significance of compared measurements was performed using the Student's one‐tailed or two‐tailed t‐test (Microsoft Excel).
3. Results
3.1. Meisoindigo kills gemcitabine‐resistant PDAC cells and reduces CSCs population
Meisoindigo (Figure 1A) is clinically used in China for CML treatment (Xiao et al., 2002, 2006), but its pharmaceutical activity against PDACs has not been elucidated. We evaluated its cytotoxicity by MTT assay after 72 h treatment in four pancreatic cancer cell lines, namely BxPC3, Panc1, Miapaca2 and Jopaca‐1. Gemcitabine (indicated as Gem in figures), a drug used for advanced PDAC (Mini et al., 2006), was used as a reference in parallel. Consistent with an earlier report (Wellner et al., 2009), gemcitabine showed a robust inhibition in BxPc3 and Miapaca2 with lower than 10 nM IC50 values. Jopaca‐1 was quite insensitive with a two orders of magnitude lower IC50 (4.32 ± 0.14 μM). Panc1 was the most resistant, even at a concentration of 50 μM (Figure 1B). By contrast, meisoindigo showed an advantage in Jopaca‐1 and Panc1 with IC50 values of 2.87 μM and 5.45 μM, respectively (Figure 1B). Shortening exposure time, less activity was detected in the case of gemcitabine due to its DNA synthesis interfering mechanism (Wellner et al., 2009), whereas the effect of meisoindigo (indicated as “Mei” in figures) was nearly unaffected (Figure 1B).
Figure 1.
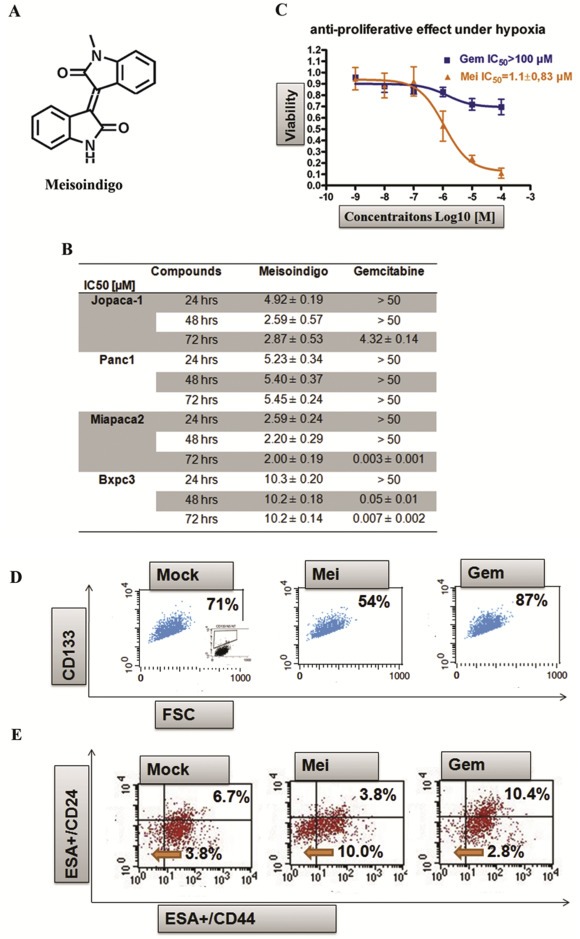
Meisoindigo effectively inhibits the growth of gemcitabine‐resistant cells and reduces CSC fractions. (A) Structure of meisoindigo. (B) Anti‐proliferative effect in PDACs. The cells were treated with increasing concentrations of meisoindigo. Cell viability was determined by the number of treated over control cells. The IC50 [μM] values were calculated from dose–response curves. (C) Anti‐proliferative effect in Jopaca‐1 under hypoxia (1% O2). (D) CD133+ population was declined after 24 h treatment with meisoindigo (Mei) (20 μM), while elevated by gemcitabine (Gem). The percentage of CD133+cell was highlighted. (E) Meisoindigo reduced ESA+/CD24+/CD44+ cell population. The percentage of ESA+/CD24+/CD44+ (upper right) and ESA+/CD24−/CD44− (lower left) were highlighted.
Hypoxia is considered a tumor associated condition promoting metastasis and mutation in cancer (Akakura et al., 2001). Indeed, continually incubating Jopaca‐1 cells under hypoxia (1% O2, a concentration common in many tissues) for 14 days resulted in an extraordinary chemotherapeutic resistance to gemcitabine (IC50hypoxia > 100 μM). In contrast an unexpected improvement in efficacy was observed in the presence of meisoindigo with an IC50 of 1.1 μM (Figure 1C).
Panc1 and Jopaca‐1 are quite heterogenic, possess aberrant stem cell‐associated gene expression and show migratory, invasive properties as well as resistance to gemcitabine (Fredebohm et al., 2012; Wellner et al., 2009). We therefore postulated that the inhibitory effect of meisoindigo on growth of gemcitabine‐resistant tumor cells may be ascribed to preferentially targeting CSCs. Labeling with the well‐established CSC surface marker CD133, a high amount of CD133+ cells (71%) could be detected in Jopaca‐1 cells, which increased to 87% after treatment with 20 μM gemcitabine for 24 h (Figure 1D) similar to previous data (Fredebohm et al., 2012), while was notably reduced to 54% in presence of 20 μM meisoindigo (Figure 1D). Comparably, the percentage of ESA+/CD24+/CD44+, another putative CSC population in PDACs, was increased from 6.7% to 10.4% upon addition of gemcitabine, but inversely decreased by more than 44% to only 3.8% in meisoindigo treatment (Figure 1E) with a concomitant enrichment of the CD24−/CD44− population (Figure 1E). In line with previous report (Fredebohm et al., 2012), a trace of CD133+ cells could be visualized (SI. 1; Supplemental Information) and further suppressed by meisoindigo in BxPC3 (from 1.04% to 0.22%), Panc1 (from 2.10% to 0.53%) and MiaPaCa‐2 (from 6.29% to 1.01%).
3.2. Meisoindigo represses activity of CSC‐associated genes and reduces stemness of Jopaca‐1
A body of experimental results indicates that genes involved in the regulation of pluripotency, such as Oct3/4, Nanog and Sox2 (Cheng et al., 2015, 2015, 2011), or abundantly expressed during EMT (Chaffer et al., 2013), such as N‐Cadherin and ZEB1, could play important roles in maintaining CSCs. Upon treatment with 20 μM meisoindigo for 24 h, we detected a significant reduction in the expression of CSC‐associated genes in Jopaca‐1 analyzed by qRT‐PCR, showing only 40% residual expression for ZEB1 and 10% for Oct3/4, Sox2 and N‐Cadherin (Figure 2A). By contrast, ASK1 and HSP27, which are closely related to apoptosis under oxidative stress (Cheng et al., 2014a), were elevated, while Axin2 and Vimentin were not affect (Figure 2A). Interestingly, the expression of epithelial marker, E‐Cadherin was also remarkably reduced to 50%. Since the recent results pointed out the involvement of E‐Cadherin in inducing chemotherapeutic resistance (Fischer et al., 2015; Zheng et al., 2015), E‐Cadherin could be a valuable target in cancer therapy. Immunoblotting results consistently showed that 48 h treatment at 0.625 μM and 1.25 μM also led to a notable reduction in protein level of Oct3/4 and N‐Cadherin (Figure 2B). Furthermore, time‐response study revealed that reduction of Nanog, ZEB1 and β‐Catenin occurred synchronously as early as 2 h when treated with 20 μM meisoindigo (Figure 2C). Taken together, our results clearly demonstrated that meisoindigo repressed activities of CSC‐associated molecules in Jopaca‐1.
Figure 2.
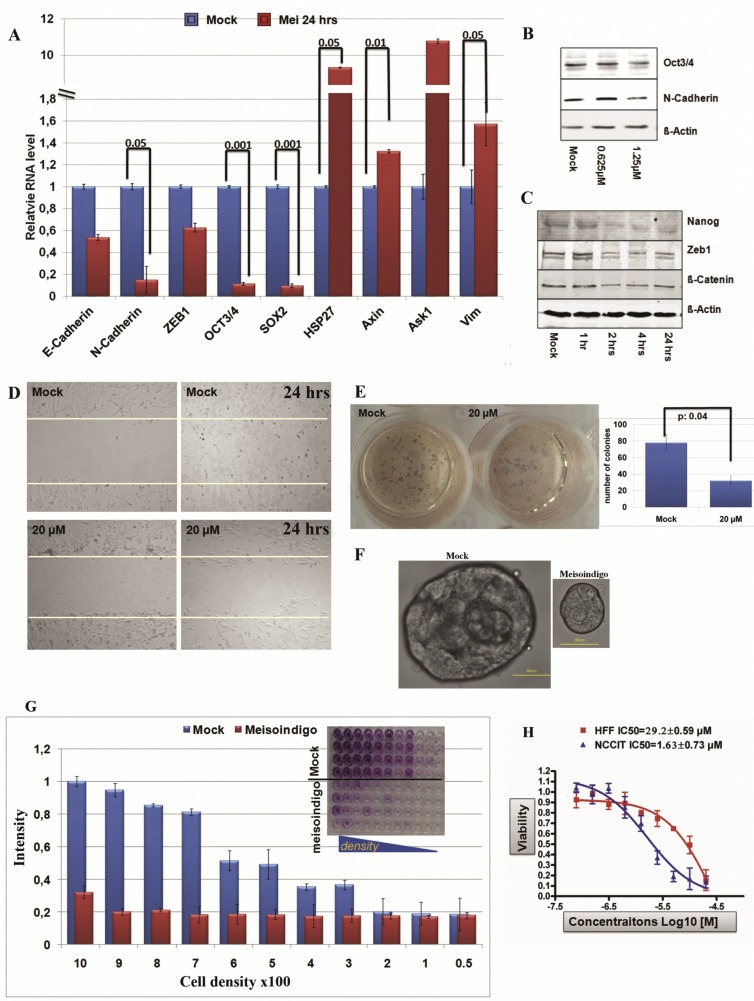
Meisoindigo repressed CSC phenotypes in Jopaca‐1. (A) Meisoindigo suppressed CSC‐associated gene expression analyzed by qRT‐PCR in Jopaca‐1 cells at 20 μM for 24 h. (B) Meisoindigo repressed CSC‐associated proteins. Jopaca‐1 cells were incubated with meisoindigo (0.625 μM and 1.25 μM) for 48 h. The whole cell lysate was subjected to immunoblot. Specific antibodies against Oct3/4 and N‐Cadherin were used. β‐Actin served as loading control and DMSO as mock. (C) Meisoindigo inhibited activities of CSC‐associated proteins in a time‐dependant manner. Jopaca‐1 cells were incubated with meisoindigo (20 μM) as indicated and analyzed by immunoblot. (D) Meisoindigo inhibited gap closure in wound healing assay of Jopaca‐1 cells treated at 20 μM concentration. Photos were taken at the beginning and after 24 h treatment. (E) Meisoindigo inhibited colony formation in soft agar assay. Jopaca‐1 cells were pre‐treated with meisoindigo (20 μM) or DMSO for 24 h. After removal of compound the cells were re‐plated in agar‐coated plates. The number of colonies (>50 cells) was scored using microscopy images. (F) Meisoindigo reduced cell self‐renewal in mammosphere assay (scale bar: 50 μm). (G) Meisoindigo reduced cellular clonogenic potential in the in vitro dilution assay. After 24 h treatment with 20 μM meisoindigo, cells were re‐plated in 96‐well plate at indicated cell density. Colonies were allowed to form within 14 days without changing the medium thereafter stained with MTT and counted by microscopy. The number of colonies was normalized to that obtained from plating 1000 cells. The dye was dissolved in DMSO and wells were photographed as depicted in the figure. (H) Cell viabilities of HFF and NCCIT were evaluated in a 72 h MTT assay.
Previous results showed a high tumorigenic ability of Jopaca‐1 cells in vitro and in vivo (Fredebohm et al., 2012). To examine if meisoindigo is able to suppress the aggressive growth phenotype of Jopaca‐1 cells, the following assays were carried out: (i) in a wound healing assay to analyze cellular migration into a gap generated with a 200 μL plastic pipette, we detected a rapid recovery in control experiments after 24 h (Figure 2D), while meisoindigo treatment inhibited recolonization; ii) in a soft agar assay pre‐treatment with 20 μM compound for 24 h resulted in a 3‐fold decrease in the number of colonies formed (Figure 2E); iii) we tested sphere‐forming ability of cells in suspension culture using serum replacement, bFGF2 and B27, which had been shown to rely on the number of CSCs (Gupta et al., 2009). Over a period of 21 days, the number of colonies derived from meisoindigo‐treated cells was much reduced compared to control; in addition, they were also 3‐fold smaller in size (Figure 2F); iv) In line with this, treated cells had a lower clonogenic potential clearly visible even in a dilution assay starting with 1000 cells analyzed after 14 days incubation without medium change. Similar results could be obtained starting with 300 mock treated cells (Figure 2G). In addition, we compared the anti‐proliferative effect of meisoindigo on human embryonic carcinoma NCCIT cells and human neonatal foreskin fibroblast HFF cells and found a biased anti‐proliferational effect (20‐fold) on stem cell‐like NCCIT cells (Figure 2H). For comparison, we examined the tumorigenic potential in meisoindigo‐treated BxPc3 cells, which expressed lower levels of CD133 (SI. 1) and were resistant to meisoindigo. We also did not find any effect of the compound in the in vitro dilution (SI. 2) and colony formation assays (SI. 3). However, the recovery in wound healing assay was remarkably inhibited (SI. 4).
These results demonstrated that meisoindigo may target CSC‐related signaling pathways, which leads to ablation of stem cell fractions, reduction of cellular mobility and repression of the tumorigenic potential in Jopaca‐1.
3.3. Meisoindigo targets mitochondrial activity
CSCs show different metabolic properties in comparison to normal tumor cells due to their particular demand for oxygen, glucose and mitochondrial activity (Ward and Thompson, 2012). To study the impact of meisoindigo on cellular metabolism, we used a micro‐fluidic biosensor system (Bionas 2500) for continuously on‐line recording metabolic parameters (Alborzinia et al., 2011) and monitored cellular respiration in real‐time with different concentrations of compounds in two pancreatic cancer cell lines, Panc1 and Miapaca2. Cellular respiration was immediately reduced after adding meisoindigo (Figure 3A), implicating a reduced mitochondrial capacity. In Jocapa‐1 cells, we examined the mitochondrial activity by studying membrane potential, measured with JC‐1, and ROS formation, visualized with ROS specific dye DHE (dihydroethidium). As shown in Figure 3B, the redhigh/greenlow population, reflecting high membrane potential, was reduced (Gate R2, 59% in mock versus 36% in the treatment), and the redlow/greenhigh population (blue arrow), indicating lower membrane potential in JC‐1 assay, was reduced as early as 30 min after treatment, and sustained over the test period of 4 h. Furthermore, the cellular ROS level was increased 3.5‐fold after 4 h (Figure 3C). The average mitochondrial mass labeled with Mito Tracker green and quantified by FACS increased from 100 in mock to 130 and 180 units after 4 h and 24 h treatments respectively (Figure 3D). This could reflect a rapid response to compensate and overcome the reduced, compound‐induced mitochondrial activity.
Figure 3.
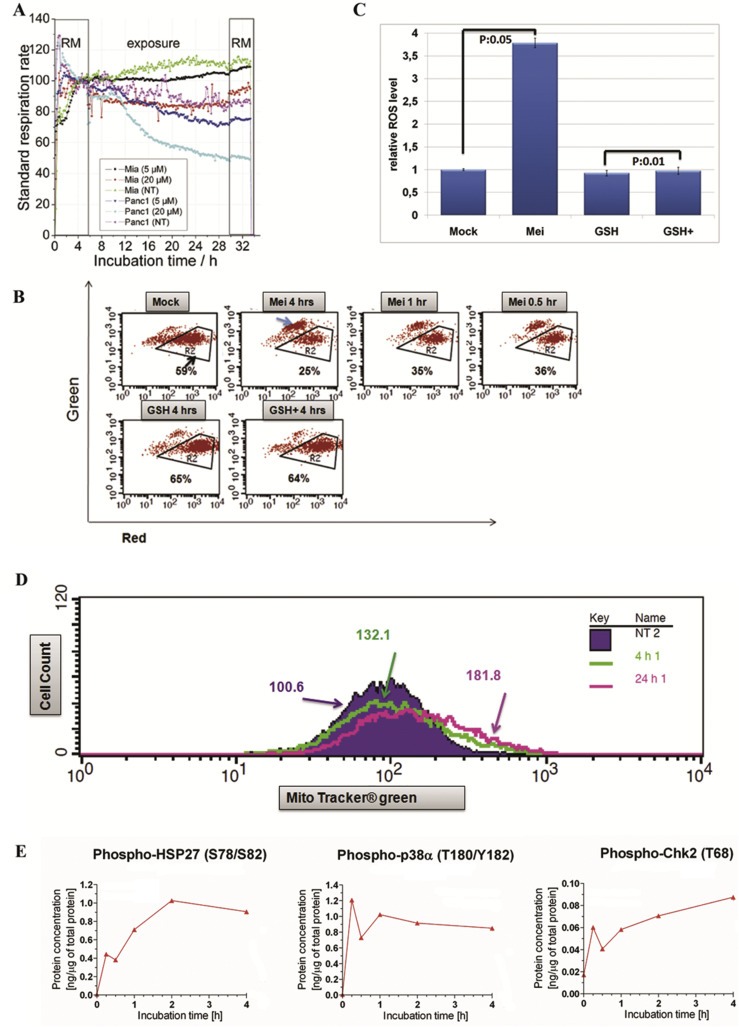
Meisoindigo regulated mitochondrial activity. (A) Meisoindigo reduced cell respiration in Miapaca2 and Panc1 cells monitored by Bionas 2500. The cells were incubated in Bionas running medium (RM) for 6 h and treated with compound at two different concentrations for 24 h. The data of cellular respiration was normalized to corresponding mock treatment. (B) Meisoindigo reduced mitochondrial membrane potential. Jopaca‐1 cells were incubated with meisoindigo (20 μM), antioxidant glutathione (GSH, 5 mM) or with a combination (all combination of agents with meisoindigo were abbreviated as agent+). The cells were collected at indicated time points, stained with JC‐1 and analyzed by FACS. Region R2 was gated to indicate redhigh/greenlow signal, while blue arrow pointed to redlow/greenhigh signal. (C) Meisoindigo enhanced cellular ROS formation. The level of ROS was determined by using ROS specific dye DHE in Jopaca‐1 treated with DMSO as mock or meisoindigo 20 μM for 4 h. (D) Total amount of mitochondrial mass was increased upon meisoindigo. Mito tracker green was used to stain mitochondria in living cells incubated with meisoindigo 20 μM for 4 h and 24 h and analyzed by FACS. The mean value of mitochondrial mass is highlighted. (E) Meisoindigo activated p38, HSP27 and CHK2 analyzed by protein microarray.
In a previous study we showed that glutathione (GSH) is able to compensate excessive ROS production (Cheng et al., 2014a). Here, adding this antioxidant normalized the membrane potential (Figure 3B) and completely neutralized meisoindigo‐mediated excessive ROS production (Figure 3C). To see how these changes are linked to cellular signaling pathways, we screened phosphorylation of several associated signaling proteins using our phospho‐protein microarray (Holenya et al., 2011) and found increased phosphorylation of ROS‐associated kinases, p38 and HSP27 (Cheng et al., 2014a) as well as the check point kinase 2 (Figure 3E and SI. 5). These results indicate that excessive ROS may be required to mediate meisoindigo‐induced cell damage.
3.4. Antioxidants rescue meisoindigo mediated cell apoptosis
Given the importance of phosphorylation of CHK2 at Tyr68 in cell cycle arrest (Niida and Nakanishi, 2006), we studied cell cycle distribution by staining with propidium iodide (PI) and detected DNA accumulation at S and G2/M phase after a 24 h treatment at both given concentrations (Figure 4A). Correspondingly, annexin v positive apoptotic cells emerged concentration‐dependently in the presence of meisoindigo for 24 h, which was inversely related to the number of trypan blue negative cells (Figure 4B). In good agreement with a previous report (Lee et al., 2010), meisoindigo‐triggered apoptosis involved caspase and PARP cleavage (Figure 4C and SI. 6), which could be inhibited by adding the pan‐caspase inhibitor Z‐VAD‐FMK (Figure 4B and D). Further analysis of several apoptosis‐related proteins (Yip and Reed, 2008) by immunoblot revealed a fast depletion of survivin, a caspase inhibitor protein, and a decrease in the level of the anti‐apoptotic protein Bcl‐xL with 15 min delay, as well as of the oncogene c‐Myc after 2 h treatment (SI. 7).
Figure 4.
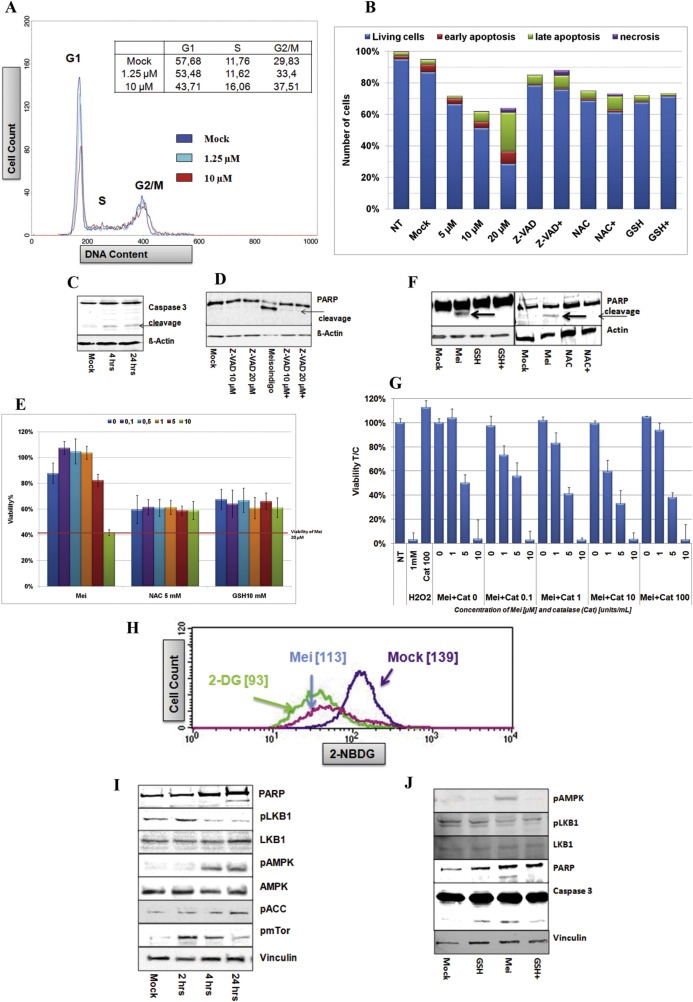
ROS scavengers neutralize meisoindigo‐associated cell apoptosis. (A) Meisoindigo induced cell cycle arrest. Jopaca‐1 cells were treated with meisoindigo (1.25 μM and 10 μM) for 24 h. The cells were fixed with methanol, stained with PI (propidium iodide) and analyzed by FACS. (B) N‐acetyl‐l‐cysteine (NAC), glutathione (GSH) and Z‐VAD‐FMK (Z‐VAD) rescued meisoindigo‐mediated apoptosis. Jopaca‐1 cells were incubated with meisoindigo (5 μM, 10 μM and 20 μM), Z‐VAD (10 μM), NAC (10 mM), GSH (5 mM) or in combination of 20 μM meisoindigo (agent+) with inhibitors for 24 h. The cells were stained with FITC‐annexin v antibody and PI, and analyzed by FACS. DMSO was used as mock and NT indicated non‐treatment. Double negative cells (blue), annexin v positive cells (red), double positive cells (green) and PI positive cells (violet) were defined as living cells, early apoptotic cells, late apoptotic cells and necrotic cells, respectively. The number of cells were counted using trypan blue assay. (C) Jopaca‐1 cells incubated with 20 μM meisoindigo for 4 h and 24 h. The cleavage of Caspase 3 was examined by immunoblot. β‐Actin was used as loading control. (D) The cells were incubated with Z‐VAD (10 μM and 20 μM) or meisoindigo (20 μM) or in combination. The cleavage of PARP was examined by immunoblot. (E) The cells were incubated with increasing concentrations of meisoindigo or in combination with antioxidants (NAC 10 mM or GSH 5 mM) for 48 h. The viability of cells was evaluated in MTT assay. (F) The cells were incubated with meisoindigo (20 μM), NAC (10 mM), GSH (5 mM) or in combination for 24 h. Immunoblot was performed to detect cleavage of PARP. (G) Cells were incubated with meisoindigo (1 μM, 5 μM and 10 μM) or in combination with catalase (0.1 U/mL, 1/mL, 10 U/mL and 100/mL). Cell viability was evaluated in MTT assay for 48 h incubation. H2O2 (1 mM) was used as positive control. (H) Glucose uptake was inhibited by meisoindigo in Jopaca‐1 cells. Cells were exposed to meisoindigo (20 μM) for 24 h and glucose internalization was measured using fluorescently labeled glucose, 2‐NBDG ([2‐(N‐(7‐nitrobenz‐2‐oxa‐1,3‐diazol‐4‐yl)amino)‐2‐deoxyglucose]). 2‐DG (2‐deoxyglucose) was used as positive control. The mean‐value of cellular glucose uptake is depicted. (I) Meisoindigo regulated LKB1 and AMPK signaling. Jopaca‐1 cells were treated with meisoindigo (20 μM), harvested at indicated time points and analyzed for activity of LKB1, AMPK1alpha and ACC using the corresponding specific antibodies. (J) Glutathione did not impact on meisoindigo‐induced inactivation of LKB1.
To further elucidate the role of drug‐induced excessive ROS in cellular damage, we added antioxidants, NAC and GSH, to balance ROS levels and found that these combinations efficiently declined the number of annexin v positive cells in a 24 h apoptotic assay and protected cells from damage in a 48 h MTT assay in comparison to the respective mock treatment (Figure 4B and E). In accordance, PARP cleavage was remarkably ablated (Figure 4F). Adding cell‐impermeable catalase (100 U/mL) antagonized toxicity of 1 mM H2O2, but not of meisoindigo (Figure 4G), confirming that intracellular ROS formation was essential in meisoindigo‐induced apoptosis.
3.5. Meisoindigo alters cellular metabolism by targeting LKB1 and AMPK signaling
Since mitochondria are the major energy house of cells, their dysregulation can directly influence metabolism‐related cellular events (Fulda et al., 2010). We studied the effect of meisoindigo on glucose uptake in Jopaca‐1 by exploiting 2‐NBDG, a fluorescent glucose derivative. After 4 h treatment with 20 μM meisoindigo, glucose uptake was reduced by 20% (mean value: 119 versus 133) in comparison to untreated control. We recruited 2‐deoxyglucose (2‐DG), a glucose uptake inhibitor, as positive control (Jeon et al., 2012) and observed 33% reduction (mean value: 93) at 5 mM, 250‐fold higher concentration to meisoindigo (Figure 4H).
Several reports implicated that the reduction of cellular glucose concentration can positively regulate AMPK signaling, a master cellular metabolism regulator (Jeon et al., 2012; Madiraju et al., 2014). A time‐dependent study revealed an ascending phosphorylation of AMPK, which occurred as early as 4 h after addition of meisoindigo (Figure 4I). Acetyl‐CoA carboxylase (ACC) is a putative AMPK substrate and its inhibitory phosphorylation at Ser79 is sensitively correlated to AMPK activity (Jeon et al., 2012; Stahmann et al., 2006). Thus, phosphorylation of ACC was commonly used to indicate activity of AMPK (Jeon et al., 2012; Stahmann et al., 2006), As shown in Figure 5A, increased levels of phospho‐ACC were clearly detected after 24 h of treatment, whereas in AMPK siRNA‐transfected cells they remained non‐phosphorylated, as showed in SI. 8 and Figure 5B (Stahmann et al., 2006). Since activity of AMPK is associated with cellular ATP level (Hardie et al., 2012), we decided to measure alternations of ATP and observed its reduction initiated as early as 1 h (up to 30%) upon treatment (SI. 9). This trend could be sustained at least for 4 h and the ATP level was remained lower level even after 24 h (60%) in comparison to mock.
Figure 5.
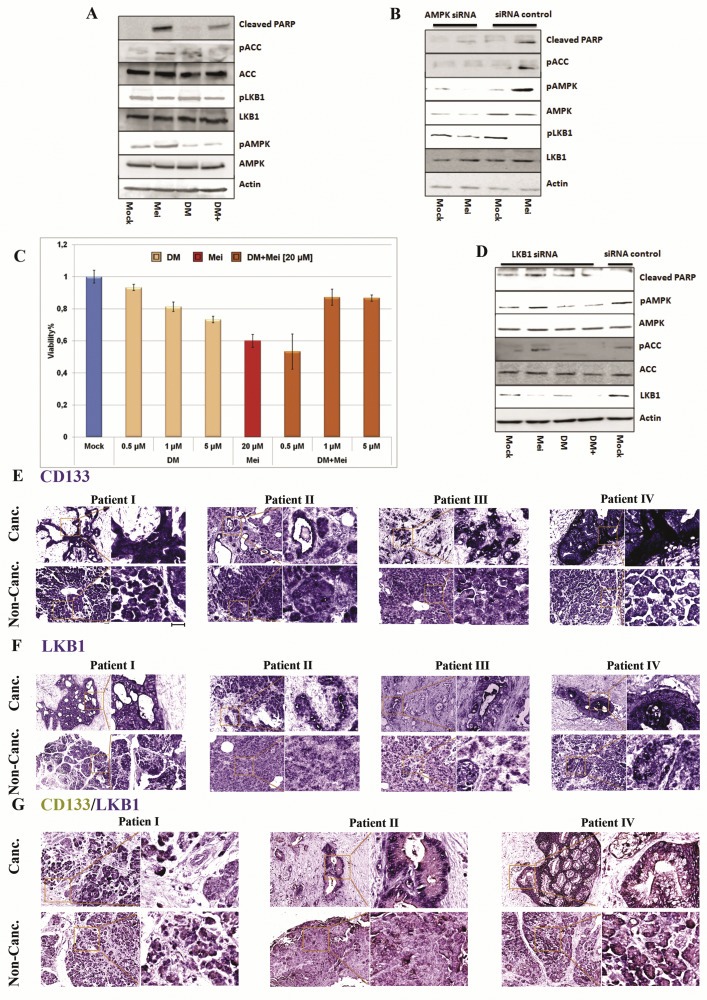
The role of LKB1 and AMPK in meisoindigo‐mediated apoptosis. (A) AMPK inhibitor counteracted the anti‐proliferative effect of meisoindigo. Cells were treated with meisoindigo (20 μM) alone or in combination with Dorsomorphin (DM), an AMPK inhibitor. DM prevented meisoindigo‐mediated AMPK activation and partially reduced cleavage of PARP. (B) AMPK associated signaling pathway was blocked by using AMPK siRNA. (C) Effects of DM and meisoindigo on cell viability counteracted each other. (D) Meisoindigo‐induced AMPK activation was independent on LKB1. (E) The expression of CD133 in cancerous (canc.) and non cancerous (non‐canc.) areas of PDAC patient tissue analyzed by in situ hybridization. (F) The expression of LKB1 analyzed by in situ hybridization. (G) Co‐staining for LKB1 (blue) and CD133 (yellow) in cancerous and non‐cancerous areas (scale bar: 40 μm).
LKB1 is one of the major AMPK activators and plays an important role in starvation‐induced AMPK activation (Shackelford and Shaw, 2009). We tested the phosphorylation of LKB1 at Ser428, an essential site for activating AMPK as well as inhibiting cell growth (Shackelford and Shaw, 2009). To our surprise, the level of phosphorylation was unexpectedly reduced, while pan LKB1 remained unaffected (4, 5B). We analyzed metabolic response in LKB1‐deficient Hela cells (Jeon et al., 2012) and reduced glucose uptake (SI. 10) and enhanced AMPK activity (SI. 11). Further analysis using BIONAS sensor chips showed stable reduction of glycolysis measured as acidification rate (SI. 12) and respiration (SI. 13) over a period of 24 h. Thus, meisoindigo‐mediated inhibition of glucose uptake and AMPK activation was independent of LKB1. Interestingly, glutathione (GSH) was able to compensate meisoindigo‐mediated cellular apoptosis (Figure 4F and J), alternation in cellular metabolism (SI. 12 and SI. 13), and activation of AMPK (Figure 4J), but failed to rescue phosphorylation of LKB1 (Figure 4J), suggesting that the imbalance in cellular redox homeostasis might be a consequence of altering cell metabolism by meisoindigo.
Very recent results reported by Vincent et al. indicated that the anti‐proliferative effect of a number of AMPK agonists was not associated with AMPK signaling (Vincent et al., 2015). Hence, we recruited Dorsomorphin (DM), a well‐known AMPK inhibitor, also known as compound C, and studied the influence of DM on meisoindigo‐treated Jopaca‐1 cells. The results demonstrated that 1 μM DM was sufficient to partially neutralize meisoindigo‐mediated anti‐proliferative effect in a 24 h MTT assay (Figure 5C). Analysis on the protein level by immunoblotting confirmed that DM is an effective AMPK antagonist (Figure 5A) and inhibited meisoindigo‐mediated AMPK activation, but the reduction of phosphorylated LKB1 was not effected (Figure 5A). Expectedly, DM partially rescued the cleavage of PARP at the given concentration (Figure 5A). Since DM is a protein kinase inhibitor and inhibits a wide range of protein kinases (Cheng et al., 2012), we used siRNA to genetically knockdown the level of cellular AMPK. A comparable result was obtained, including inactivation of AMPK cascade and rescue of PARP cleavage, both without reduction of LKB1 phosphorylation (Figure 5B). Finally, we attempted to knockdown LKB1 by using a mixture of two LKB1 siRNAs. Phosphorylation of AMPK and ACC was remarkably repressed in LKB1‐deficient Jopaca‐1, which is consistent with previous results that LKB1 is upstream of AMPK signaling (Figure 5D). Nevertheless, activation of AMPK signaling in the presence of meisoindigo was not influenced, confirming that meisoindigo‐mediated AMPK activation is independent of LKB1 (Figure 5D).
3.6. Meisoindigo preferentially targets CSCs through interfering LKB1
Experimental evidence documented a pivotal role of LKB1 in maintaining hematopoietic stem cells in an AMPK‐independent manner (Gan et al., 2010; Gurumurthy et al., 2010; Nakada et al., 2010). We assumed that LKB1 in PDAC might support CSCs maintenance. Using our previously reported in situ hybridization approach we compared the mRNA expression of CD133 (Figure 5E) and LKB1 (Figure 5F) in cancer and non‐cancer areas visualized in complementary pancreatic sections from PDAC patients (Ghafoory et al., 2015). As shown in Figure 5E and F, the expressions of CD133 and LKB1 were quite homogenous in non‐cancer area, while heterogeneous in cancer areas with very similar patterns in all four PDAC patients: the lack of expression in fibrosis‐like areas and ectopic expression in residual places, where cancer cells were assumed. Co‐staining demonstrated that aberrant expressions of LKB1 and CD133 were co‐localized in non‐fibrotic areas of cancer sections (Figure 5G). To study the relationship between LKB1 and CD133, we exploited two LKB1 siRNAs (siRNA a and b), which were reported previously (Stahmann et al., 2006), to knockdown LKB1 in Jopaca‐1 cells and performed immunoblotting to monitor the knockdown efficiency (Figure 6A) with individual or mixture of both (siRNA m). As a result, the CD133 positive population was reduced to 40%, 30% and 20% respectively in the presence of siRNA a, siRNA b and siRNA m, comparable to that in meisoindigo‐treated cells (Figure 6B). Interestingly, recovery of CD133 positive populations (from 20% to 80%) could be clearly observed by replacing treatment conditions with fresh medium for 48 h in our transient knockdown experiments. The recovery following removal of meisoindigo reached merely upto 50% even after 120 h in fresh medium (Figure 6B).
Figure 6.
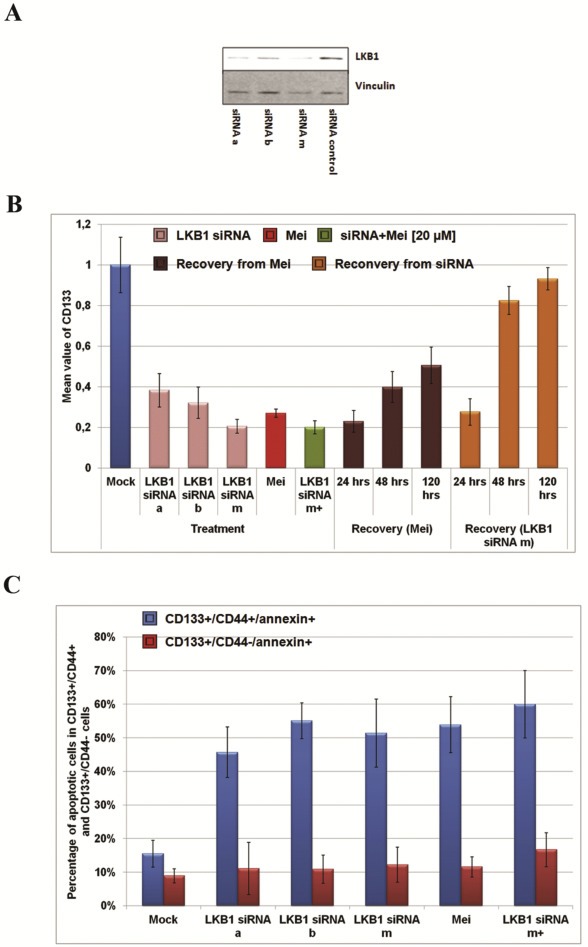
Meisoindigo selectively inhibited growth of CSCs. (A) The efficiency of LKB1 siRNA in Jopaca‐1 cells analyzed by immunoblotting. Jopaca‐1 cells were transfected with LKB1 siRNA a, b or both for 48 h. (B) The percentage of CD133 positive cells was quantified by FACS and the mean value obtained from individual treatment was normalized to that in mock. (C) The population of CD133+/annexin v+/CD44− and CD133+/annexin v+/CD44+ cells was analyzed by FACS.
Moreover, we established an in vitro assay with antibodies for CD133, CD44 and annexin v to examine apoptotic cells in CSC populations (SI. 14). Analysis of the entire CD133+ population, revealed two annexin v+ fractions in respect to CD44 expression, highlighted as annexin v+/CD44− and annexin v+/CD44+ (SI. 14). Treatments with meisoindigo or LKB1 siRNAs (a, b and m) significantly elevated the percentage of annexin v+/CD44+ cells about 3‐fold (from 15.5% to 50%), which was even further increased to nearly 4‐fold (60%) in the combination (Figure 6C). Nevertheless, the negligible alternations in annexin v+/CD44− population would be detected in the single treatments (less than 5%). These finding demonstrated that meisoindigo preferentially killed cells expressing CD133 and CD44 by interfering with LKB1 signaling in Jopaca‐1 cells.
4. Discussion
PDAC is one of the most difficult curable malignancies and the effective therapy with significant improved survival outcome is not available, yet (Siegel et al., 2015). Meisoindigo is the next‐generation of indirubin, an ATP‐competitive inhibitor (Eisenbrand et al., 2004; Hoessel et al., 1999), which is currently used for the treatment of CML in China and has been associated with an advanced therapeutic outcome due to its improved bioavailability (Xiao et al., 2002). However, very distinct binding position between the two indol moieties via a double‐bond in comparison to indirubin causes an exhaustive structural alternation, which results in much less inhibitory activity in in vitro kinase assays (Wee et al., 2009). Therefore, its biological mechanism is still a mystery. In this article, we studied the feasibility of using meisoindigo in treatment of PDACs, and showed a selective inhibition of gemcitabine‐resistant Panc1 and Jopaca‐1 cells possibly by targeting metabolic signaling of CSCs.
A body of experimental evidence suggests that CSCs are a subpopulation of cancer cells, which possess stem cell traits and majorly contribute to tumor generation, metastasis, and resistance to chemotherapy and radiation, as well as recurrence (Gattinoni et al., 2012; Magee et al., 2012; Wong and Lemoine, 2009). Previous research reported the enrichment of CSC populations in gemcitabine‐treated PDACs, which has been related to largely unsatisfying clinical results (Fredebohm et al., 2012; Hermann et al., 2007). In our experiments, we confirmed that CSC populations, defined as CD133+ and ESA+/CD24+/CD44+, increased in Jopaca‐1 cells in response to gemcitabine, while these populations were considerably reduced upon meisoindigo.
Pluripotency associated genes, including Oct3/4, Nanog and Sox2, have been identified to be aberrantly expressed and play important roles in CSC generation and maintenance (Lonardo et al., 2011). As anticipated, high levels of Oct3/4, Nanog and Sox2 were found in this heterogeneous primary cell line, which were significantly repressed by meisoindigo. In a seminal finding, Mani et al. revealed that cells with ongoing EMT in response to TGFβ or other cellular events exhibited stem cell‐like properties and high tumor incidence (Mani et al., 2008). We observed expression of EMT markers, N‐Cadherin and ZEB1 (Lonardo et al., 2011; Mani et al., 2008), in Jopaca‐1 cells. Both of them were drastically inhibited. As a result, the metastatic ability of Jopaca‐1 was clearly suppressed in various assays. To get insight in the molecular mechanism of cell death, we developed an apoptotic assay by using a set of antibodies that targets CD133, CD44 and annexin v to cover two CSCs populations. An up to 3‐fold increase of apoptotic cells could be detected in the CD133+/CD44+ fraction. Thus, our data demonstrate that meisoindigo preferentially kills CSCs and suppresses activity of CSC‐associated genes, in association with reduced cellular mobility, proliferation, self‐renewal and tumorigenesis. Strikingly, the expression of E‐Cadherin, a hallmark of epithelial cells, was also dramatically repressed upon treatment. Considering two very recent reports that EMT cells were more chemotherapeutic resistant, while non‐EMT cells rather contributed to tumor proliferation, progression and metastasis (Fischer et al., 2015; Zheng et al., 2015), our results indicate a potential chemotherapeutic benefit of meisoindigo by synchronously targeting EMT and non‐EMT cells.
Enhanced lactate production from glucose even under oxygen rich conditions, the so‐called Warburg effect, is a hallmark of tumor cells (Ward and Thompson, 2012). In this aspect, the function of mitochondria is reprogrammed to adapt to this biological alternation. Several small molecules, like metformin, which hypothetically target the function of mitochondria and cellular metabolism, haven been discovered as highly selective agents for inhibiting CSCs (Hirsch et al., 2009; Skrtic et al., 2011). Accordingly, we investigated the effect of meisoindigo on cell metabolism and found a constant reduction in cellular respiration, mitochondrial activity and mitochondrial membrane potential in Jopaca‐1 with a concomitant increase in ROS levels. In line with this, antioxidants completely prevented caspase 3 and PARP cleavages and protected compound‐induced cell damage, illustrating the crucial role of alternating the cellular redox balance in meisoindigo‐mediated cell apoptosis.
Since ATP is mostly synthesized in mitochondria, inhibition of mitochondrial respiration and nutrient‐starvation induced by meisoindigo may reduce ATP production resulting in an increase in AMP/ATP ratio and subsequent activation of the AMPK cascade (Hardie et al., 2012; Shackelford and Shaw, 2009). We found that meisoindigo clearly interfered with cellular glucose uptake in Jopaca‐1 and reduced cellular ATP levels, which might explain the activated AMPK cascade (Hirsch et al., 2009). Interestingly, repression of AMPK either by chemical inhibitor, DM, or siRNA, partially compensated the toxic effect of meisoindigo.
LKB1 is one of the major upstream kinases for activating AMPK signaling in the regulation of cellular energy metabolism and acts as a tumor‐suppressor (Alessi et al., 2006; Shackelford and Shaw, 2009). Loss of its metabolic regulatory mutations has been found in a number of carcinomas, including lung, liver, breast, colon and pancreas (Dancey et al., 2012). Albeit meisoindigo activated AMPK cascade, a reduction in the level of phosphorylated LKB1 was observed. Remarkably, AMPK was even activated in LKB1‐deficient cells, including HeLa and Jopaca‐1 treated with LKB1 siRNA, confirming that meisoindigo‐induced AMPK activation is LKB1‐independent, most likely due to its impact on cellular metabolism, like inhibiting glucose uptake. This is in line with a recent finding that 2‐DG, a glucose inhibitor, enhanced the activity of AMPK in HeLa cells (Jeon et al., 2012). Very recently, the anti‐proliferative effect of six commercially available chemical AMPK agonists, including metformin, phenformin, AICAR, 2‐DG, salicylate and A‐769662, was compared in‐depth. In this study AMPK activation associated toxicity was only observed in presence of the synthetic chemical, A‐76922 (Vincent et al., 2015). In our experiments, the toxicity of meisoindigo was significantly reduced in AMPK inactive Jopaca‐1 cells treated either with chemical inhibitor or siRNA, which clearly confirmed that AMPK was at least partially involved in meisoindigo‐induced apoptosis.
LKB1 is one of the most frequently mutated genes in tumors (Carretero et al., 2004) and acts as tumor suppressor due to its essential role in energy homeostasis (Dancey et al., 2012; Faubert et al., 2014). However, little is known why LKB1‐deficient cells are resistant to malignant transformation (Bardeesy et al., 2002; Shaw et al., 2004). The recent observations that LKB1‐associated cellular homeostasis is of importance in HSC maintenance in an AMPK‐independent manner (Gan et al., 2010; Gurumurthy et al., 2010; Nakada et al., 2010) and LKB1‐deficiency induces malignant transdifferentiation resulting in chemotherapeutic resistance (Li et al., 2015) opened new insights in the correlation between LKB1 and (cancer) stem cells. In this aspect, we investigated the expression of LKB1 and CD133 in tumor sections and detected reproducible patterns in samples from different PDAC patients that LKB1 and CD133 were overexpressed and co‐localized in malignant places. Thus, the robust elimination of CD133+ populations upon treatment might be due to inhibition of LKB1 signaling. In good agreement, siRNA mediated LKB1‐knockdown facilitated cell apoptosis specifically in CD133+/CD44+ population. It is well‐known that fibrosis often occurs in PDACs through activation of stellate cells and protects the tumor from chemotherapeutic agents, but also prevents the occurrence of EMT (Ozdemir et al., 2014; Rhim et al., 2014; Yang et al., 2015). The lack of LKB1 in fibrotic parts is also not surprising, since they behave very distinct to PDACs in metabolic profiling (Ozdemir et al., 2014; Rhim et al., 2014). Thus, conclusions in regard to the role of LKB1 deduced from using larger patient tumor sections or heterogeneous tissues or cell populations should be carefully re‐investigated.
In the current study, Panc1 is one of the most resistant cell lines to gemcitabine (IC50 > 50 μM), in which CD133+ cells comprised less than 3% of the cells. Nevertheless, meisoindigo showed a potent anti‐proliferative effect (Figure 1B). Although the number of CD133+ cells was considerably reduced upon meisoindigo (SI 1), the major cause of gemcitabine resistance in Panc1 should involve other CSC populations or CSC‐independent resistant mechanism. For the resistance of Panc1 to gemcitabine a pivotal role of ZEB1 had been demonstrated by Brabletz and co‐workers (Wellner et al., 2009). Interestingly, we observed a reduction of ZEB1 in Jopaca‐1 cells upon meisoindigo (Figure 2A). Thus, a possible explanation of the apparent discrepancy between the lack of CD133+ cells and strong meisoindigo response in Panc1 may be inhibition of cancer stem cells or “stem cell like cells” requiring ZEB1 expression (Moustakas and Heldin, 2012), which however needs to be investigated further.
Taken together, our data demonstrated that AMPK was involved in meisoindigo‐induced cell apoptosis in a LKB1‐independent manner, while LKB1 played a crucial role in sustaining CSCs. With these results, we can clearly show that a small molecule can preferentially target CSCs by interfering with LKB1 signaling, which could provide a potential advantage in clinical cancer therapy.
Authors' contributions
J.Y.K., S.G., T.D., and R.R., J.T. performed experiments. A.M., M.H. and A.S. prepared patients' samples. A.H. performed Bionase experiment. P.H. performed microarray experiment. J.F. isolated and characterized Jopaca‐1 cells. K.‐H. Merz and G.E. helped synthesized compound and write the manuscript. J.D.H. isolated Jopaca‐1 and helped write the manuscript. S.W. designed experiments, analyzed data and wrote the manuscript. X.C. designed experiments, synthesized and characterized meisoindigo, performed experiments, analyzed data and wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
We thank Elke Lederer, Marie‐Sofie Karipidis and Holger Treiber for technical assistance. This work was supported by DFG FOR630, the BMBF grant programs SysToxChip 031A202E and 0315398B (Drugs‐iPS), and PaCaNet as well as BfR.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.01.008.
Cheng Xinlai, Kim Jee Young, Ghafoory Shahrouz, Duvaci Tijen, Rafiee Roya, Theobald Jannick, Alborzinia Hamed, Holenya Pavlo, Fredebohm Johannes, Merz Karl-Heinz, Mehrabi Arianeb, Hafezi Mohammadreza, Saffari Arash, Eisenbrand Gerhard, Hoheisel Jörg D., Wölfl Stefan, (2016), Methylisoindigo preferentially kills cancer stem cells by interfering cell metabolism via inhibition of LKB1 and activation of AMPK in PDACs, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.01.008.
Contributor Information
Xinlai Cheng, Email: x.cheng@uni-heidelberg.de.
Jee Young Kim, Email: J.Kim@uni-heidelberg.de.
Shahrouz Ghafoory, Email: shahrouzghafoory@yahoo.com.
Tijen Duvaci, Email: tijenduvaci@gmail.com.
Roya Rafiee, Email: roya.rafie@gmail.com.
Jannick Theobald, Email: jannick.theobald@gmail.com.
Hamed Alborzinia, Email: Hamed.alborzinia@uni-heidelberg.de.
Pavlo Holenya, Email: Holenya@stud.uni-heidelberg.de.
Johannes Fredebohm, Email: J.Fredelbohm@dkfz.de.
Karl-Heinz Merz, Email: khmerz@rhrk.uni-kl.de.
Arianeb Mehrabi, Email: arianeb.mehrabi@med.uni-heidelberg.de.
Mohammadreza Hafezi, Email: mohammadreza.hafezi@med.uni-heidelberg.de.
Arash Saffari, Email: arash.saffari@med.uni-heidelberg.de.
Gerhard Eisenbrand, Email: eisenbra@rhrk.uni-kl.de.
Jörg D. Hoheisel, Email: J.Hoheisel@dkfz-heidelberg.de
Stefan Wölfl, Email: wolfl@uni-hd.de.
References
- Akakura, N. , Kobayashi, M. , Horiuchi, I. , Suzuki, A. , Wang, J. , Chen, J. , Niizeki, H. , Kawamura, K. , Hosokawa, M. , Asaka, M. , 2001. Constitutive expression of hypoxia-inducible factor-1alpha renders pancreatic cancer cells resistant to apoptosis induced by hypoxia and nutrient deprivation. Cancer Res. 61, (17) 6548–6554. [PubMed] [Google Scholar]
- Al-Hajj, M. , Wicha, M.S. , Benito-Hernandez, A. , Morrison, S.J. , Clarke, M.F. , 2003. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 100, (7) 3983–3988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alborzinia, H. , Can, S. , Holenya, P. , Scholl, C. , Lederer, E. , Kitanovic, I. , Wolfl, S. , 2011. Real-time monitoring of cisplatin-induced cell death. PLoS One. 6, (5) e19714 10.1371/journal.pone.0019714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alessi, D.R. , Sakamoto, K. , Bayascas, J.R. , 2006. LKB1-dependent signaling pathways. Annu. Rev. Biochem. 75, 137–163. [DOI] [PubMed] [Google Scholar]
- Bardeesy, N. , Sinha, M. , Hezel, A.F. , Signoretti, S. , Hathaway, N.A. , Sharpless, N.E. , Loda, M. , Carrasco, D.R. , DePinho, R.A. , 2002. Loss of the Lkb1 tumour suppressor provokes intestinal polyposis but resistance to transformation. Nature. 419, (6903) 162–167. [DOI] [PubMed] [Google Scholar]
- Bonnet, D. , Dick, J.E. , 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, (7) 730–737. [DOI] [PubMed] [Google Scholar]
- Bouchikhi, F. , Anizon, F. , Moreau, P. , 2008. Synthesis and antiproliferative activities of isoindigo and azaisoindigo derivatives. Eur. J. Med. Chem. 43, (4) 755–762. [DOI] [PubMed] [Google Scholar]
- Carretero, J. , Medina, P.P. , Pio, R. , Montuenga, L.M. , Sanchez-Cespedes, M. , 2004. Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene. 23, (22) 4037–4040. [DOI] [PubMed] [Google Scholar]
- Chaffer, C.L. , Marjanovic, N.D. , Lee, T. , Bell, G. , Kleer, C.G. , Reinhardt, F. , D'Alessio, A.C. , Young, R.A. , Weinberg, R.A. , 2013. Poised chromatin at the ZEB1 promoter enables breast cancer cell plasticity and enhances tumorigenicity. Cell. 154, (1) 61–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, F. , Li, L. , Ma, D. , Yan, S. , Sun, J. , Zhang, M. , Ji, C. , Hou, M. , 2010. Imatinib achieved complete cytogenetic response in a CML patient received 32-year indirubin and its derivative treatment. Leuk. Res. 34, (2) e75–77. [DOI] [PubMed] [Google Scholar]
- Cheng, X. , Rasque, P. , Vatter, S. , Merz, K.H. , Eisenbrand, G. , 2010. Synthesis and cytotoxicity of novel indirubin-5-carboxamides. Bioorg. Med. Chem. 18, (12) 4509–4515. 10.1016/j.bmc.2010.04.066 [DOI] [PubMed] [Google Scholar]
- Cheng, X. , Alborzinia, H. , Merz, K.H. , Steinbeisser, H. , Mrowka, R. , Scholl, C. , Kitanovic, I. , Eisenbrand, G. , Wolfl, S. , 2012. Indirubin derivatives modulate TGFbeta/BMP signaling at different levels and trigger ubiquitin-mediated depletion of nonactivated R-Smads. Chem. Biol. 19, (11) 1423–1436. [DOI] [PubMed] [Google Scholar]
- Cheng, X. , Holenya, P. , Can, S. , Alborzinia, H. , Rubbiani, R. , Ott, I. , Wolfl, S. , 2014. A TrxR inhibiting gold(I) NHC complex induces apoptosis through ASK1-p38-MAPK signaling in pancreatic cancer cells. Mol. Cancer. 13, 221 10.1186/1476-4598-13-221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, X. , Merz, K.H. , Vatter, S. , Christ, J. , Wolfl, S. , Eisenbrand, G. , 2014. 7,7'-Diazaindirubin – a small molecule inhibitor of casein kinase 2 in vitro and in cells. Bioorg. Med. Chem. 22, (1) 247–255. 10.1016/j.bmc.2013.11.031 [DOI] [PubMed] [Google Scholar]
- Cheng, X. , Dimou, E. , Alborzinia, H. , Wenke, F. , Gohring, A. , Reuter, S. , Mah, N. , Fuchs, H. , Andrade-Navarro, M.A. , Adjaye, J. , Gul, S. , Harms, C. , Utikal, J. , Klipp, E. , Mrowka, R. , Wolfl, S. , 2015. Identification of 2-[4-[(4-Methoxyphenyl)methoxy]-phenyl]acetonitrile and derivatives as potent Oct3/4 inducers. J. Med. Chem. 58, (12) 4976–4983. 10.1021/acs.jmedchem.5b00144 [DOI] [PubMed] [Google Scholar]
- Cheng, X. , Yoshida, H. , Raoofi, D. , Saleh, S. , Alborzinia, H. , Wenke, F. , Gohring, A. , Reuter, S. , Mah, N. , Fuchs, H. , Andrade-Navarro, M.A. , Adjaye, J. , Gul, S. , Utikal, J. , Mrowka, R. , Wolfl, S. , 2015. Ethyl 2-((4-Chlorophenyl)amino)thiazole-4-carboxylate and derivatives are potent inducers of Oct3/4. J. Med. Chem. 58, (15) 5742–5750. 10.1021/acs.jmedchem.5b00226 [DOI] [PubMed] [Google Scholar]
- Clevers, H. , 2011. The cancer stem cell: premises, promises and challenges. Nat. Med. 17, 313–319. [DOI] [PubMed] [Google Scholar]
- Dancey, J.E. , Bedard, P.L. , Onetto, N. , Hudson, T.J. , 2012. The genetic basis for cancer treatment decisions. Cell. 148, (3) 409–420. [DOI] [PubMed] [Google Scholar]
- Davies, T.G. , Tunnah, P. , Meijer, L. , Marko, D. , Eisenbrand, G. , Endicott, J.A. , Noble, M.E. , 2001. Inhibitor binding to active and inactive CDK2: the crystal structure of CDK2-cyclin A/indirubin-5-sulphonate. Structure. 9, (5) 389–397. [DOI] [PubMed] [Google Scholar]
- Eisenbrand, G. , Hippe, F. , Jakobs, S. , Muehlbeyer, S. , 2004. Molecular mechanisms of indirubin and its derivatives: novel anticancer molecules with their origin in traditional Chinese phytomedicine. J. Cancer Res. Clin. Oncol. 130, (11) 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert, B. , Vincent, E.E. , Griss, T. , Samborska, B. , Izreig, S. , Svensson, R.U. , Mamer, O.A. , Avizonis, D. , Shackelford, D.B. , Shaw, R.J. , Jones, R.G. , 2014. Loss of the tumor suppressor LKB1 promotes metabolic reprogramming of cancer cells via HIF-1alpha. Proc. Natl. Acad. Sci. U. S. A. 111, 2554–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, K.R. , Durrans, A. , Lee, S. , Sheng, J. , Li, F. , Wong, S.T.C. , Choi, H. , El Rayes, T. , Ryu, S. , Troeger, J. , Schwabe, R.F. , Vahdat, L.T. , Altorki, N.K. , Mittal, V. , Gao, D. , 2015. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 527, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredebohm, J. , Boettcher, M. , Eisen, C. , Gaida, M.M. , Heller, A. , Keleg, S. , Tost, J. , Greulich-Bode, K.M. , Hotz-Wagenblatt, A. , Lathrop, M. , Giese, N.A. , Hoheisel, J.D. , 2012. Establishment and characterization of a highly tumourigenic and cancer stem cell enriched pancreatic cancer cell line as a well defined model system. PLoS One. 7, (11) e48503 10.1371/journal.pone.0048503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda, S. , Galluzzi, L. , Kroemer, G. , 2010. Targeting mitochondria for cancer therapy. Nat. Rev. Drug Discov. 9, (6) 447–464. [DOI] [PubMed] [Google Scholar]
- Gan, B.Y. , Hu, J.A. , Jiang, S. , Liu, Y.C. , Sahin, E. , Zhuang, L. , Fletcher-Sananikone, E. , Colla, S. , Wang, Y.A. , Chin, L. , DePinho, R.A. , 2010. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature. 468, (7324) 701–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni, L. , Klebanoff, C.A. , Restifo, N.P. , 2012. Paths to stemness: building the ultimate antitumour T cell. Nat. Rev. Cancer. 12, (10) 671–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghafoory, S. , Breitkopf-Heinlein, K. , Li, Q. , Dzieran, J. , Scholl, C. , Dooley, S. , Wolfl, S. , 2012. A fast and efficient polymerase chain reaction-based method for the preparation of in situ hybridization probes. Histopathology. 61, (2) 306–313. [DOI] [PubMed] [Google Scholar]
- Ghafoory, S. , Mehrabi, A. , Hafezi, M. , Cheng, X. , Breitkopf-Heinlein, K. , Hick, M. , Huichalaf, M. , Herbel, V. , Saffari, A. , Wolfl, S. , 2015. Nuclear accumulation of CDH1 mRNA in hepatocellular carcinoma cells. Oncogenesis. 4, e152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta, P.B. , Onder, T.T. , Jiang, G. , Tao, K. , Kuperwasser, C. , Weinberg, R.A. , Lander, E.S. , 2009. Identification of selective inhibitors of cancer stem cells by high-throughput screening. Cell. 138, (4) 645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurumurthy, S. , Xie, S.Z. , Alagesan, B. , Kim, J. , Yusuf, R.Z. , Saez, B. , Tzatsos, A. , Ozsolak, F. , Milos, P. , Ferrari, F. , Park, P.J. , Shirihai, O.S. , Scadden, D.T. , Bardeesy, N. , 2010. The Lkb1 metabolic sensor maintains haematopoietic stem cell survival. Nature. 468, (7324) 659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie, D.G. , Ross, F.A. , Hawley, S.A. , 2012. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 13, (4) 251–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann, P.C. , Huber, S.L. , Herrler, T. , Aicher, A. , Ellwart, J.W. , Guba, M. , Bruns, C.J. , Heeschen, C. , 2007. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 1, (3) 313–323. [DOI] [PubMed] [Google Scholar]
- Heshmati, N. , Cheng, X. , Eisenbrand, G. , Fricker, G. , 2013. Enhancement of oral bioavailability of E804 by self-nanoemulsifying drug delivery system (SNEDDS) in rats. J. Pharm. Sci. 102, (10) 3792–3799. [DOI] [PubMed] [Google Scholar]
- Hidalgo, M. , 2010. Pancreatic cancer. N. Engl. J. Med. 362, (17) 1605–1617. [DOI] [PubMed] [Google Scholar]
- Hirsch, H.A. , Iliopoulos, D. , Tsichlis, P.N. , Struhl, K. , 2009. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 69, (19) 7507–7511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoessel, R. , Leclerc, S. , Endicott, J.A. , Nobel, M.E. , Lawrie, A. , Tunnah, P. , Leost, M. , Damiens, E. , Marie, D. , Marko, D. , Niederberger, E. , Tang, W. , Eisenbrand, G. , Meijer, L. , 1999. Indirubin, the active constituent of a Chinese antileukaemia medicine, inhibits cyclin-dependent kinases. Nat. Cell Biol. 1, (1) 60–67. [DOI] [PubMed] [Google Scholar]
- Holenya, P. , Kitanovic, I. , Heigwer, F. , Wolfl, S. , 2011. Microarray-based kinetic colorimetric detection for quantitative multiplex protein phosphorylation analysis. Proteomics. 11, 2129–2133. [DOI] [PubMed] [Google Scholar]
- Holenya, P. , Heigwer, F. , Wolfl, S. , 2012. KOMA: ELISA-microarray calibration and data analysis based on kinetic signal amplification. J. Immunol. Methods. 380, 10–15. [DOI] [PubMed] [Google Scholar]
- Holenya, P. , Can, S. , Rubbiani, R. , Alborzinia, H. , Junger, A. , Cheng, X. , Ott, I. , Wolfl, S. , 2014. Detailed analysis of pro-apoptotic signaling and metabolic adaptation triggered by a N-heterocyclic carbene-gold(I) complex. Metallomics. 6, (10) 1591–1601. [DOI] [PubMed] [Google Scholar]
- Jeon, S.M. , Chandel, N.S. , Hay, N. , 2012. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 485, (7400) 661–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidot, T. , Sirard, C. , Vormoor, J. , Murdoch, B. , Hoang, T. , Cacerescortes, J. , Minden, M. , Paterson, B. , Caligiuri, M.A. , Dick, J.E. , 1994. A cell initiating human acute myeloid-leukemia after transplantation into SCID mice. Nature. 367, (6464) 645–648. [DOI] [PubMed] [Google Scholar]
- Lee, C.C. , Lin, C.P. , Lee, Y.L. , Wang, G.C. , Cheng, Y.C. , Liu, H.E. , 2010. Meisoindigo is a promising agent with in vitro and in vivo activity against human acute myeloid leukemia. Leuk. Lymphoma. 51, 897–905. [DOI] [PubMed] [Google Scholar]
- Li, C. , Heidt, D.G. , Dalerba, P. , Burant, C.F. , Zhang, L. , Adsay, V. , Wicha, M. , Clarke, M.F. , Simeone, D.M. , 2007. Identification of pancreatic cancer stem cells. Cancer Res. 67, (3) 1030–1037. [DOI] [PubMed] [Google Scholar]
- Li, F. , Han, X. , Li, F. , Wang, R. , Wang, H. , Gao, Y. , Wang, X. , Fang, Z. , Zhang, W. , Yao, S. , Tong, X. , Wang, Y. , Feng, Y. , Sun, Y. , Li, Y. , Wong, K.K. , Zhai, Q. , Chen, H. , Ji, H. , 2015. LKB1 inactivation elicits a redox imbalance to modulate non-small cell lung cancer plasticity and therapeutic response. Cancer Cell. 27, (5) 698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonardo, E. , Hermann, P.C. , Mueller, M.T. , Huber, S. , Balic, A. , Miranda-Lorenzo, I. , Zagorac, S. , Alcala, S. , Rodriguez-Arabaolaza, I. , Ramirez, J.C. , Torres-Ruiz, R. , Garcia, E. , Hidalgo, M. , Cebrian, D.A. , Heuchel, R. , Lohr, M. , Berger, F. , Bartenstein, P. , Aicher, A. , Heeschen, C. , 2011. Nodal/Activin signaling drives self-renewal and tumorigenicity of pancreatic cancer stem cells and provides a target for combined drug therapy. Cell Stem Cell. 9, (5) 433–446. [DOI] [PubMed] [Google Scholar]
- Madiraju, A.K. , Erion, D.M. , Rahimi, Y. , Zhang, X.M. , Braddock, D.T. , Albright, R.A. , Prigaro, B.J. , Wood, J.L. , Bhanot, S. , MacDonald, M.J. , Jurczak, M.J. , Camporez, J.P. , Lee, H.Y. , Cline, G.W. , Samuel, V.T. , Kibbey, R.G. , Shulman, G.I. , 2014. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 510, (7506) 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee, J.A. , Piskounova, E. , Morrison, S.J. , 2012. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 21, (3) 283–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani, S.A. , Guo, W. , Liao, M.J. , Eaton, E.N. , Ayyanan, A. , Zhou, A.Y. , Brooks, M. , Reinhard, F. , Zhang, C.C. , Shipitsin, M. , Campbell, L.L. , Polyak, K. , Brisken, C. , Yang, J. , Weinberg, R.A. , 2008. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 133, (4) 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer, L. , Skaltsounis, A.L. , Magiatis, P. , Polychronopoulos, P. , Knockaert, M. , Leost, M. , Ryan, X.P. , Vonica, C.A. , Brivanlou, A. , Dajani, R. , Crovace, C. , Tarricone, C. , Musacchio, A. , Roe, S.M. , Pearl, L. , Greengard, P. , 2003. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem. Biol. 10, (12) 1255–1266. [DOI] [PubMed] [Google Scholar]
- Mini, E. , Nobili, S. , Caciagli, B. , Landini, I. , Mazzei, T. , 2006. Cellular pharmacology of gemcitabine. Ann. Oncol. 17, (Suppl. 5) v7–v12. [DOI] [PubMed] [Google Scholar]
- Moriyama, T. , Ohuchida, K. , Mizumoto, K. , Cui, L. , Ikenaga, N. , Sato, N. , Tanaka, M. , 2010. Enhanced cell migration and invasion of CD133+ pancreatic cancer cells cocultured with pancreatic stromal cells. Cancer. 116, (14) 3357–3368. [DOI] [PubMed] [Google Scholar]
- Moustakas, A. , Heldin, C.H. , 2012. Induction of epithelial-mesenchymal transition by transforming growth factor beta. Semin. Cancer Biol. 22, 446–454. [DOI] [PubMed] [Google Scholar]
- Nakada, D. , Saunders, T.L. , Morrison, S.J. , 2010. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature. 468, (7324) 653–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida, H. , Nakanishi, M. , 2006. DNA damage checkpoints in mammals. Mutagenesis. 21, (1) 3–9. [DOI] [PubMed] [Google Scholar]
- Ozdemir, B.C. , Pentcheva-Hoang, T. , Carstens, J.L. , Zheng, X. , Wu, C.C. , Simpson, T.R. , Laklai, H. , Sugimoto, H. , Kahlert, C. , Novitskiy, S.V. , De Jesus-Acosta, A. , Sharma, P. , Heidari, P. , Mahmood, U. , Chin, L. , Moses, H.L. , Weaver, V.M. , Maitra, A. , Allison, J.P. , LeBleu, V.S. , Kalluri, R. , 2014. Depletion of carcinoma-associated fibroblasts and fibrosis induces immunosuppression and accelerates pancreas cancer with reduced survival. Cancer Cell. 25, (6) 719–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhim, A.D. , Oberstein, P.E. , Thomas, D.H. , Mirek, E.T. , Palermo, C.F. , Sastra, S.A. , Dekleva, E.N. , Saunders, T. , Becerra, C.P. , Tattersall, I.W. , Westphalen, C.B. , Kitajewski, J. , Fernandez-Barrena, M.G. , Fernandez-Zapico, M.E. , Iacobuzio-Donahue, C. , Olive, K.P. , Stanger, B.Z. , 2014. Stromal elements act to restrain, rather than support, pancreatic ductal adenocarcinoma. Cancer Cell. 25, (6) 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas, J. , Bettayeb, K. , Ferandin, Y. , Knockaert, M. , Garrofe-Ochoa, X. , Totzke, F. , Schachtele, C. , Mester, J. , Polychronopoulos, P. , Magiatis, P. , Skaltsounis, A.L. , Boix, J. , Meijer, L. , 2006. 7-Bromoindirubin-3′-oxime induces caspase-independent cell death. Oncogene. 25, (47) 6304–6318. [DOI] [PubMed] [Google Scholar]
- Shackelford, D.B. , Shaw, R.J. , 2009. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat. Rev. Cancer. 9, (8) 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, R.J. , Kosmatka, M. , Bardeesy, N. , Hurley, R.L. , Witters, L.A. , DePinho, R.A. , Cantley, L.C. , 2004. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc. Natl. Acad. Sci. U. S. A. 101, (10) 3329–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, R.L. , Miller, K.D. , Jemal, A. , 2015. Cancer statistics, 2015. CA – Cancer J. Clin. 65, (1) 5–29. [DOI] [PubMed] [Google Scholar]
- Skrtic, M. , Sriskanthadevan, S. , Jhas, B. , Gebbia, M. , Wang, X. , Wang, Z. , Hurren, R. , Jitkova, Y. , Gronda, M. , Maclean, N. , Lai, C.K. , Eberhard, Y. , Bartoszko, J. , Spagnuolo, P. , Rutledge, A.C. , Datti, A. , Ketela, T. , Moffat, J. , Robinson, B.H. , Cameron, J.H. , Wrana, J. , Eaves, C.J. , Minden, M.D. , Wang, J.C. , Dick, J.E. , Humphries, K. , Nislow, C. , Giaever, G. , Schimmer, A.D. , 2011. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 20, (5) 674–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahmann, N. , Woods, A. , Carling, D. , Heller, R. , 2006. Thrombin activates AMP-activated protein kinase in endothelial cells via a pathway involving Ca2+/calmodulin-dependent protein kinase kinase beta. Mol. Cell. Biol. 26, 5933–5945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolle, R. , Bergdoll, R. , Luther, M. , Auerhahn, A. , Wacker, W. , 1930. On N-substituted oxindol and isatine. J. Prakt. Chem. 128, 1–43. 10.1002/prac.19301280101 (in German) [Google Scholar]
- Vincent, E.E. , Coelho, P.P. , Blagih, J. , Griss, T. , Viollet, B. , Jones, R.G. , 2015. Differential effects of AMPK agonists on cell growth and metabolism. Oncogene. 34, (28) 3627–3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vougogiannopoulou, K. , Ferandin, Y. , Bettayeb, K. , Myrianthopoulos, V. , Lozach, O. , Fan, Y. , Johnson, C.H. , Magiatis, P. , Skaltsounis, A.L. , Mikros, E. , Meijer, L. , 2008. Soluble 3′,6-substituted indirubins with enhanced selectivity toward glycogen synthase kinase-3 alter circadian period. J. Med. Chem. 51, (20) 6421–6431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl, A. , Bagard, P. , 1913. A synthesis in the group of indigoids. C. R. Hebd. Acad. Sci. 156, 898–901. (in French) [Google Scholar]
- Ward, P.S. , Thompson, C.B. , 2012. Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell. 21, (3) 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee, X.K. , Yeo, W.K. , Zhang, B. , Tan, V.B. , Lim, K.M. , Tay, T.E. , Go, M.L. , 2009. Synthesis and evaluation of functionalized isoindigos as antiproliferative agents. Bioorg. Med. Chem. 17, (21) 7562–7571. [DOI] [PubMed] [Google Scholar]
- Wellner, U. , Schubert, J. , Burk, U.C. , Schmalhofer, O. , Zhu, F. , Sonntag, A. , Waldvogel, B. , Vannier, C. , Darling, D. , zur Hausen, A. , Brunton, V.G. , Morton, J. , Sansom, O. , Schuler, J. , Stemmler, M.P. , Herzberger, C. , Hopt, U. , Keck, T. , Brabletz, S. , Brabletz, T. , 2009. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 11, (12) 1487–1495. [DOI] [PubMed] [Google Scholar]
- Wong, H.H. , Lemoine, N.R. , 2009. Pancreatic cancer: molecular pathogenesis and new therapeutic targets. Nat. Rev. Gastroenterol. Hepatol. 6, (7) 412–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, Z. , Hao, Y. , Liu, B. , Qian, L. , 2002. Indirubin and meisoindigo in the treatment of chronic myelogenous leukemia in China. Leuk. Res. 30, (1) 54–59. [DOI] [PubMed] [Google Scholar]
- Xiao, Z. , Wang, Y. , Lu, L. , Li, Z. , Peng, Z. , Han, Z. , Hao, Y. , 2006. Anti-angiogenesis effects of meisoindigo on chronic myelogenous leukemia in vitro. Leuk. Res. 30, 54–59. [DOI] [PubMed] [Google Scholar]
- Yang, J.Y. , Jiang, S.H. , Liu, D.J. , Yang, X.M. , Huo, Y.M. , Li, J. , Hua, R. , Zhang, Z.G. , Sun, Y.W. , 2015. Decreased LKB1 predicts poor prognosis in pancreatic ductal adenocarcinoma. Sci. Rep. 5, 10575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip, K.W. , Reed, J.C. , 2008. Bcl-2 family proteins and cancer. Oncogene. 27, (50) 6398–6406. [DOI] [PubMed] [Google Scholar]
- Zheng, X. , Carstens, J.L. , Kim, J. , Scheible, M. , Kaye, J. , Sugimoto, H. , Wu, C.-C. , LeBleu, V.S. , Kalluri, R. , 2015. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 527, 525 10.1038/nature16064 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data