

Abstract
The DNA damage checkpoints provide an anti‐cancer barrier in diverse tumour types, however this concept has remained unexplored in prostate cancer (CaP). Furthermore, targeting DNA repair defects by PARP1 inhibitors (PARPi) as a cancer treatment strategy is emerging yet requires suitable predictive biomarkers. To address these issues, we performed immunohistochemical analysis of multiple markers of DNA damage signalling, oxidative stress, DNA repair and cell cycle control pathways during progression of human prostate disease from benign hyperplasia, through intraepithelial neoplasia to CaP, complemented by genetic analyses of TMPRSS2‐ERG rearrangement and NQO1, an anti‐oxidant factor and p53 protector. The DNA damage checkpoint barrier (γH2AX, pATM, p53) mechanism was activated during CaP tumorigenesis, albeit less and with delayed culmination compared to other cancers, possibly reflecting lower replication stress (slow proliferation despite cases of Rb loss and cyclin D1 overexpression) and progressive loss of ATM activator NKX3.1. Oxidative stress (8‐oxoguanine lesions) and NQO1 increased during disease progression. NQO1 genotypes of 390 men did not indicate predisposition to CaP, yet loss of NQO1 in CaP suggested potential progression‐opposing tumour suppressor role. TMPRSS2‐ERG rearrangement and PTEN loss, events sensitizing to PARPi, occurred frequently along with heterogeneous loss of DNA repair factors 53BP1, JMJD1C and Rev7 (all studied here for the first time in CaP) whose defects may cause resistance to PARPi. Overall, our results reveal an unorthodox DNA damage checkpoint barrier scenario in CaP tumorigenesis, and provide novel insights into oxidative stress and DNA repair, with implications for biomarker guidance of future targeted therapy of CaP.
Keywords: Prostate tumorigenesis, DNA damage response barrier, p53 and NKX3.1 tumour suppressors, NQO1 and oxidative stress, TMPRSS2‐ERG, PARP inhibitor biomarkers
Highlights
Activated ATM, γH2AX and p53 ‘checkpoint’ increases during prostate tumorigenesis.
Prostate DNA damage checkpoint barrier is delayed and less pronounced than in other cancers.
Oxidative stress and NQO1 levels increase with human prostate lesion progression.
NKX3.1 or DNA repair factors 53BP1, JMJD1C, Rev7 show focal loss in prostate cancer.
TMPRSS2‐ERG gain, loss of PTEN and DNA repair factors may help to predict response to PARP inhibitors.
1. Introduction
Prostate cancer (CaP) is the most frequently diagnosed malignancy in men and one of the major causes of cancer‐related death in developed countries (Siegel et al., 2014). There is currently some uncertainty about the value of systematic screening for prostate‐specific antigen (PSA), reflecting an ongoing debate on potential CaP overdiagnosis and overtreatment with no major benefits in terms of overall patient survival (Andriole et al., 2012; Schroder et al., 2014). Prostate cancer develops as androgen‐dependent and initially responds to androgen deprivation therapies, however, ultimately the disease progresses into a hormone‐independent and largely incurable stage with metastases to the bones, lung, brain or liver. Therefore, there is a pressing need for better understanding of molecular mechanisms underlying CaP progression to inspire discoveries and validation of new treatment strategies. Among the key factors implicated in CaP pathogenesis are genomic alterations such as the TMPRSS2‐ERG and related fusion oncogenes, loss of tumour suppressors such as PTEN, p53 or NKX3.1, inflammation, enhanced DNA damage and chromosomal instability (Felgueiras et al., 2014).
Cellular DNA damage response (DDR) represents a dynamic network of proteins capable of detecting DNA lesions and signalling their presence to numerous effector pathways including cell‐cycle checkpoints, regulation of transcription, chromatin remodelling, DNA repair and cell death mechanisms (Jackson and Bartek, 2009). Proper DDR is essential for genome maintenance, organismal development and tissue homeostasis, while DDR malfunction may lead to grave pathological consequences such as neurodegeneration, immunodeficiency, premature aging or cancer (Kastan and Bartek, 2004; Jackson and Bartek, 2009). During the early stages of cancer development, the DDR machinery becomes activated in response to oncogene‐triggered replication stress and ensuing DNA breaks, thereby providing an inducible biological barrier against malignant transformation and tumour progression (Bartkova et al., 2005; Gorgoulis et al., 2005; Halazonetis et al., 2008). Such DDR activation can be documented by immunohistochemical analyses of tissue sections from human clinical specimens, with antibodies against activated (phosphorylated) forms of checkpoint kinases ATM, Chk1 and Chk2, phosphorylated histone H2AX (γH2AX) and p53, as well as foci formation characteristic of response to DNA double strand breaks (DSBs), such as those of the 53BP1 protein (Bartkova et al., 2005; Evangelou et al., 2013). The observed DDR activation is at its maximum in the early, pre‐invasive stages of human tumours yet it also persists, at least in subsets of lesions at their fully developed malignant stages (Bartkova et al., 2010, 2005, 2006, 2006, 2005). The observed partial decrease of the DDR activation in advanced tumours likely reflects the selective pressure to bypass or inactivate p53, ATM, Chk2 or other components of the DNA damage checkpoints in the incipient cancer cells, as the fully operational DDR would otherwise induce cellular senescence or death and thus block further progression of the tumour (Bartkova et al., 2005, 2006, 2005, 2008). While the DDR barrier concept has been well documented for diverse types of major solid tumours derived from somatic cells, such as carcinomas of the colon, breast, lung, urinary bladder or brain gliomas (Bartkova et al., 2010, 2005, 2013, 2005), it does not seem to apply for germ‐cell tumours (Bartkova et al., 2014, 2007). Most relevant for our present study, the question to what extent is the DDR barrier model applicable to prostate cancer pathogenesis has remained largely unexplored.
Apart from oncogene‐induced replication stress, oxidative stress (reactive oxygen species) and the ensuing DNA damage is another source of genomic instability in cancer, a notion particularly relevant for prostate tumours that are commonly associated with chronic inflammation (Sfanos and De Marzo, 2012). One of the major factors involved in cellular protection against oxidative stress is NQO1 (Siegel et al, 1996, 2010, 1997, 2004), an enzyme that has also been implicated in carcinogenesis, including stabilization of the p53 (Anwar et al., 2003; Asher et al., 2001) tumour suppressor. NQO1‐deficient mice show reduced p53 induction and apoptosis, increased susceptibility to chemically induced tumours (Iskander et al., 2005; Long et al., 2000), and impaired NF‐kB function (Ahn et al., 2006). NQO1 C609T is a missense variant that is homozygous in 4–20% of human population (Kelsey et al., 1997). Cells with the homozygous NQO1 C609T genotype have no measurable NQO1 activity, reflecting the very low levels of the NQO1 P187S protein, which is inherently very unstable due to its rapid turnover via the ubiquitin proteasome pathway (Siegel et al., 2001). We previously reported this variant as a strong prognostic and predictive factor in breast cancer (Fagerhorlm et al., 2008). Although NQO1 status has not been directly linked to prostate carcinogenesis (Steiner et al., 1999) here we wished to analyse the patterns of NQO1 during the course of prostate tumorigenesis, along with assessment of a spectrum of other proteins involved in the maintenance of genome integrity (checkpoint signalling and DNA repair), and 8‐oxoguanine, an indicator of oxidative DNA lesions detectable directly in archival clinical specimens.
Another major motivation to carry out our present analyses was the emerging notion that defects in the DDR machinery, particularly those in some DNA repair pathways, represent vulnerabilities that can be effectively targeted by innovative cancer treatments. Possibly the best example of such treatment strategy is the application of small molecule inhibitors of PARP1 (Poly (ADP‐Ribose) Polymerase‐1), an enzyme that participates in several functions within the DDR network, and whose inhibition is highly toxic to cells that are defective in homologous recombination (HR) repair, such as cancer cells with mutant BRCA1 or BRCA2 repair genes (Lord and Ashworth, 2012). This concept, known as ‘synthetic lethality’, reflects selective killing of cells with a defective pathway (here HR) by inhibition of a complementary/redundant function (here PARP1 activity). Very relevant to prostate cancer, recent studies suggested that also some other features of cancer cells may sensitize to PARP inhibitors (PARPi), including aberrant transcription due to the androgen‐driven occurrence of fusion oncogenes such as TMPRSS2‐ERG (Brenner et al., 2011), or loss of the PTEN tumour suppressor (Mendes‐Pereira et al., 2009). Despite their promise, and recent approval of the first PARPi for clinical cancer treatment, both in the USA and Europe, there are also emerging aberrations that can cause enhanced resistance to PARPi in cancers that are otherwise inherently sensitive, such as those with mutant BRCA1. The latter scenario is often referred to as ‘synthetic viability’, enhancing fitness of cancer cells by a secondary mutation in a certain context, here the lack of BRCA1 function. Among examples of synthetic viability in BRCA1‐defective tumours, several of which have been identified in our laboratory, are loss of factors involved in regulation of DSB repair by HR versus non‐homologous end joining (NHEJ), including 53BP1 (Bouwman et al., 2010), Rif1 (Zimmermann et al., 2013), JMJD1C (Watanabe et al., 2013) or Rev7 (Xu et al., 2015). We have also documented that several of these factors are aberrantly lost or their abundance decreased in subsets of human breast carcinomas, especially those of the so‐called triple‐negative type (Bouwman et al., 2010; Watanabe et al., 2013; Xu et al., 2015) that have been regarded as promising for personalized treatment by PARPi (Anders et al., 2010). On the other hand, prevalence of potential aberrations of these DDR proteins remains unknown in prostate cancer, a gap in our knowledge that we wished to fill by our present study. Such analyses are even more desirable at present, as the most recent Phase II clinical trial showed high rate of positive responses to PARPi among prostate cancer patients whose metastatic tumours were no longer responding to standard treatments (Mateo et al., 2015). Overall, with this work, we hoped to provide insights into both patho‐biology of CaP progression (status of DDR activation, oxidative stress, tumour suppressors), as well as information about prevalence of candidate biomarkers of response to PARPi treatment, both in terms of predicted enhanced sensitivity (occurrence of TMPRSS2‐ERG fusions, defective PTEN) or resistance (decrease or loss of 53BP1, JMJD1C or Rev7).
2. Material and methods
2.1. Patients
The immunohistochemistry cohort study included 103 randomly selected patients with prostate cancer diagnosis, who underwent radical prostatectomy at the University Hospital in Olomouc between years 2003 and 2011. One of the two subsets of our cohort from this pathology database consisting of 68 patients from the years 2003 and 2010, was in the form of the archived material of prostate tissue stored as paraffin blocks, and fixed by standard procedures. Samples from 35 patients obtained between 2010 and 2011 include prostate tissue that was fixed in small pieces (see below and Table 1). NQO1 genotype was assessed in blood samples from 390 men (see below and Table 3). This study was approved by the ethical committee of the Faculty of Medicine and Dentistry, Palacky University.
Table 1.
Patient cohort whose tumours were examined by immunohistochemistry (N = 103; distribution of the 35 samples processed by optimized ‘fast fixation' is indicated in parentheses).
| Characteristic range/Number of patients | |||
|---|---|---|---|
| Age | 51–60 | 61–70 | 71–81 |
| 32 (5) | 56 (24) | 15 (6) | |
| Serum PSA (ng/ml) | <4 | 4–10 | >10 |
| 15 (4) | 59 (23) | 29 (8) | |
| Gleason scores | <7 | 7 | >7 |
| 28 (8) | 51 (21) | 24 (6) | |
| Cancer stages | pT2a‐c | pT3a‐b | pT4 |
| 80 (29) | 20 (5) | 3 (1) | |
| Risk groups | low | intermediate | high |
| 24 (8) | 41 (16) | 38 (11) | |
| NQO1 Genotype | CC | CT | TT |
| 9 (6) | 13 (5) | 5 (4) | |
Cancer stages were evaluated as localized (pT2) and advanced (pT3 and pT4).
Risk categories were determined based on both clinical and pathological data: low (pT1–T2a, GS ≤ 6 and PSA ≤ 10 ng/ml), intermediate (at least one of the following: pT2b‐c, GS = 7, or 10 < PSA ≤ 20 ng/ml) and high (at least one of the following: pT3–T4, GS > 7, or PSA > 20 ng/ml).
See Table 3 for genotype details.
Table 3.
Frequencies of NQO1 genotypes and alleles.
| N | NQO1 genotype (rs1800566) | Allele frequency | ||||
|---|---|---|---|---|---|---|
| Pro187Pro | Pro187Ser | Ser187Ser | Wild type | Variant allele | ||
| 609 CC | 609 CT | 609 TT | C | T | ||
| Prostate cancer | 171 | 110 (64.3%) | 56 (32.8%) | 5 (2.9%) | 276 (80.7%) | 66 (19.3%) |
| Benign disease | 219 | 149 (68.1%) | 64 (29.2%) | 6 (2.7%) | 362 (82.6%) | 76 (17.4%) |
| Sum | 390 | 259 (66.4%) | 120 (30.8) | 11 (2.8%) | 638 (81.8%) | 144 (18.2%) |
2.2. Tissue processing and immunohistochemical staining and scoring
It is known that the method of fixation, that is, its duration and the fixation solution used, is very important in tissue processing and may influence results of examination methods such as PCR or immunohistochemistry (Howat and Wilson, 2014). In the standard procedure, the entire prostate, after its surgical removal, is immersed in 10% formaldehyde solution for 24‐h fixation. Then it is prepared for histological examination. It commonly happens, especially if the prostate is large, that its central parts are poorly fixed even after 24‐h fixation period, and such areas may undergo degenerative changes. To control for such unwanted fixation bias, a different approach to fixation was used in subset of samples (n = 35) to assess the effect of more optimal fixation on immunohistochemistry (IHC) staining results. In the latter protocol, the surgically removed prostate was measured and annotated; the apex and base were removed and subsequently divided into the right and left halves. The remaining tissue was divided into the right anterior and posterior quadrants and the left anterior and posterior quadrants. Then each prostate part was put in a separate labeled container with 10% formaldehyde and fixed until the following day. When immunohistochemistry results obtained with sections from the same prostate yet fixed in the two ways indicated above were compared, histoscores (see below for histoscore calculation) for Rb and Ki67 (both p < 0.01), but not percentage of cell positivity, were decreased in those tissues exposed to standard fixation as compared to the modified fixation protocol of small tissue pieces. On the other hand, both histoscore and percentage of cell positivity for NQO1, cyclin D1 and nuclear PTEN were increased in tissues with standard fixation (p values for histoscores were p = 0.029, p = 0.049 and p = 0.001, respectively). For consistency, data for these affected proteins that are shown in Figures and Tables, are calculated from the sub‐cohort subjected to the small‐piece faster fixation, although similar results could be seen across the entire cohort. In order to avoid any misinterpretation due to delayed fixation, several proteins were analysed only in the sub‐cohort (n = 35) the tissues from which were subject to the optimized fixation protocol in smaller tissue pieces (Supplementary Table 1).
After evaluating the slides stained with hematoxylin‐eosin, for each patient three blocks with predominance of benign hyperplasia (BPH), prostate intraepithelial neoplasia (PIN), and cancer (CaP), respectively, were selected for further study. The stage of disease progression was estimated according to morphological criteria recommended by the World Health Organization Classification of Tumours (Eble et al., 2004). Tissue sections of 5‐μm thickness were cut and subjected to optimized, sensitive immunoperoxidase staining protocols, performed either manually: using either the Vectastain Elite kit and nickel enhancement procedure without nuclear counterstaining (Bartkova et al., 2005) or by the Ventana Benchmark XT workstation, using a kit and DAB Detection View (see Supplementary Table 1 for antibody list and staining methods). Each stained tissue section was first examined at a low magnification, and slice margins, as well as potential areas of acute inflammation or necrosis were excluded from the scoring. Within the areas of BPH, PIN and CaP of each case, 3 microscope fields at the 200× magnification (for each of the BPH, PIN and CaP stages) were randomly chosen and carefully evaluated. In the slides stained with the antibodies against nuclear proteins, the percentage of positive nuclei was counted from the overall 300 nuclei from each of the structures of interest (hyperplasia, PIN and carcinoma, respectively). For slides stained with antibodies against largely cytosolic proteins, percentage of positive cells was assessed by examination of at least 200–300 cells (in 2 cases, only some 200 cells in the BPH structures could be found, while a minimum of 300 cells was examined in all other cases and in all cases for the PIN and CaP stage). The staining intensity for both nuclear and cytoplasmic antigens was scored as follows: 0 – negative, 1 – weak, 2 – moderate, 3 – strong signal. Histoscore was then calculated as percentage of cell/nuclear positivity multiplied by staining intensity, resulting in histoscore values ranging from 0 (minimum) to 300 (maximum). In the vast majority of cases (all but 2 specimens) 300 cells were examined for each of the stages. For 2 patients, the specimens contained enough PIN and CaP regions to evaluate 300 cells but only a small region of BPH that was sufficient for only 200 cells to be evaluated.
2.3. NQO1 genotyping
DNA samples were genotyped using a restriction fragment length polymorphism (RFLP) assay reported earlier (Fagerholm et al., 2008). Briefly, PCR amplicon (279 bp; forward primer 5′ – CCT GAG GCC TCC TTA TCA GA – 3′, reverse primer 5′ – AGG CTG CTT GGA GCA AAA TA – 3′) was designed to contain one HinfI restriction site specific to the C609T allele. After digestion according to the enzyme manufacturer's instructions (New England BioLabs), PCR products containing the C609T allele are cleaved into fragments of 152 and 127 base pairs, readily distinguishable on regular 2% agarose gels, whereas wild‐type amplicons remain intact. EtBr was used for staining the PCR products.
2.4. TMPRSS2‐ERG status
In a subset of patients (N = 12, Kolar et al., 2014), presence of the chromosomal rearrangement at 21q22 was evaluated by interphase fluorescence in situ hybridization using Poseidon TMPRSS2‐ERG (21q22) Del, Break, TC Probe (Kreatech Diagnostics). Additional patients were evaluated by quantitative PCR. With respect to the potential adverse effect of delayed fixation in large pieces of tissue (see section 2.2 above), the TMPRSS2‐ERG status as examined by PCR was evaluated only in the smaller cohort of patients (in 31 of the 35 samples) processed with the optimized faster fixation protocol of smaller tissue pieces. RNA was isolated from paraffin sections by RNeasy FFPE Mini Kit (Qiagen), quantified by Nanodrop, preamplified by Whole Transcriptome Amplification Kit (Sigma–Aldrich) and subjected to qPCR with specific primers and probes (TMPRSS2‐ERG: forward CTG GAG CGC GGC AGG AA, reverse GTC CAT AGT CGC TGG AGG AG, probe Cy5‐TGA GTG AGG ACC AGT CGT TG‐BHQ2; POLR2A: forward CAA GTT CAA CCA AGC CAT TG, reverse CCA GCA TAG TGG AAG GTA TTC A, UPL probe No. 87; LightCycler480, Roche). Samples with undetectable POLR2A were considered as non‐evaluable. In total, 10 patients were classified as TMPRSS2‐ERG positive and 18 as negative (8 positive and 11 negative within the fast fixation cohort).
2.5. Statistical analysis
Relationships among expression of the analysed proteins, NQO1 genotype and clinicopathological parameters were statistically evaluated using Statistica 12 software. Paired samples were assessed by Wilcoxon signed rank test while different subgroups were compared either by Mann–Whithey or Kruskal–Wallis test. Correlations were evaluated by Spearman correlation test.
3. Results
3.1. Markers and general trends of dynamic alterations during CaP progression
To assess disease progression‐related dynamics of potential alterations in several important pathways governing patho‐biology and genome (in)stability of CaP, we examined a total of 17 distinct antibody‐defined markers (Supplementary Table 1) in benign prostate hyperplasia (BPH), PIN and CaP lesions from a cohort of prostate cancer patients (Table 1). The markers studied here were selected to characterize aspects of the lesions including cell proliferation and cell cycle control (Ki67, cyclin A, and the key components of the RB pathway: cyclin D1, CDK inhibitor p16 and Rb tumour suppressor), genome integrity control (p53 tumour suppressor, ATM kinase and its regulator NKX3.1, general DNA damage signalling marker γH2AX, and DNA repair factors: 53BP1, Rev7 and JMJD1C), oxidative stress parameters (8‐oxoguanine lesions and the anti‐oxidant factor NQO1, along with p53), as well as the most CaP‐relevant oncogene (TMPRSS2‐ERG fusion) and tumour suppressors (PTEN and its related Smad4, along with p53, Rb and NKX3.1). While most of the parameters will be presented and evaluated in detail in the subsequent sections of the Results devoted to specific areas of CaP biology, here we mention some of the general trends related to disease progression, and comment on the Rb pathway and proliferation rate. In general, immunohistochemically assessed expression of NQO1, 8‐oxoguanine, activated ATM (phospho‐ATM), γH2AX, cyclin D1, p53, p16, SPP1, nuclear PTEN and nuclear Smad4 were mostly increasing during the disease progression, while abundance of Rb, Rev7, JMJD1C and 53BP1 were decreasing (Figure 1, Supplementary Figure 1A, Supplementary Table 2). Significant correlations were found between some pairs or among groups of the studied markers (Table 2) and these relationships as well as the pathogenetic relevance will be discussed in the following paragraphs. In the G1/S regulatory Rb pathway, levels of the oncogenic cyclin D1 increased from benign hyperplasia through PIN to the highest levels seen at the CaP stage, whereas Rb showed the opposite trend with CaP stage marked by the highest frequency of Rb loss (Figure 1). Consistent with previous studies, the CDK4/6 inhibitor p16 was barely detectable in benign hyperplasia, elevated in the PIN lesions, and reached maximum at the CaP stage (Figure 1). Aberrant decrease or loss of Rb correlated with increased abundance of p53, the pattern often reflecting protein stabilisation due to missense mutations of p53 (Iggo et al., 1990; Bartek et al., 1990). Furthermore, cyclin D1 expression correlated positively with Ki67 (general proliferation), with patients' age and serum PSA level (a commonly used marker of androgen receptor signalling), the latter also correlating with the age of the patients. Inspired by a 4‐marker signature of aggressive CaP (PTEN, Smad4, cyclin D1 and SPP1; Ding et al., 2011) we analysed also osteopontin (alias SPP1, secreted phosphoprotein 1), an important cytokine affecting cancer progression (Shevde and Samant, 2014). SPP1 was increased in PIN and CaP which is in line with its role in tumour microenvironment, however we did not observe any correlation with other markers in our study (data not shown).
Figure 1.
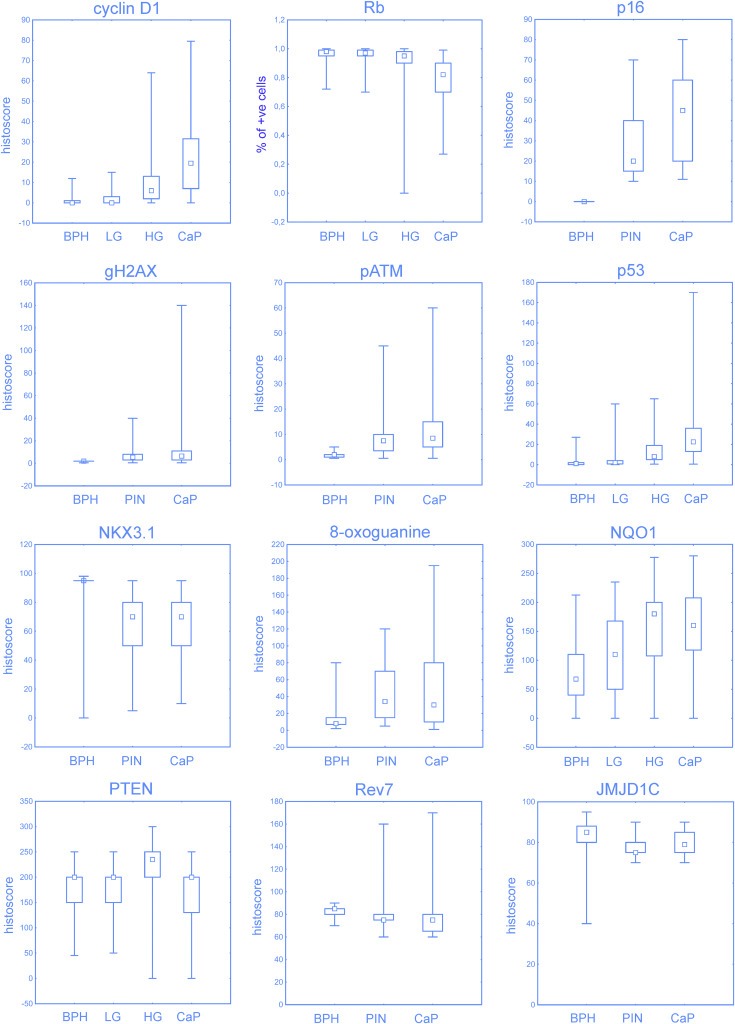
Expression of selected proteins in benign hyperplasia (BPH), prostate intraepithelial neoplasia (PIN; LG, low‐grade; HG, high‐grade) and prostate cancer (CaP). Low and high‐grade PINs were distinguished for indicated proteins. Percentage of positive cells is shown for Rb protein. Box‐plots represent median, 25%–75% percentiles and range of values. Expression of all proteins were significantly different between tumour and benign areas (p < 0.001; comparisons to PIN are available in Supplementary Table 2).
Table 2.
Spearman correlations within the cohort with fast fixation (N = 35, significant correlations with p value less than 0.05 are highlighted in bold).
| Age | NQO1 | 8‐oxo | p53 | Rb | Smad4 | D1 | SPP1 | PTEN | Ki67 | PSA | p16 | NKX3.1 | gH2AX | pATM | 53BPP1 | JMJD1C | Rev7 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Age | 1.000 | |||||||||||||||||
| NQO1 | −0.125 | 1.000 | ||||||||||||||||
| 8‐oxo | −0.280 | 0.527 | 1.000 | |||||||||||||||
| p53 | −0.094 | 0.301 | 0.189 | 1.000 | ||||||||||||||
| Rb | −0.052 | −0.180 | 0.096 | −0.350 | 1.000 | |||||||||||||
| Smad4 | 0.115 | −0.187 | 0.028 | 0.018 | −0.024 | 1.000 | ||||||||||||
| D1 | 0.352 | 0.089 | −0.150 | 0.275 | −0.091 | 0.228 | 1.000 | |||||||||||
| SPP1 | 0.130 | −0.330 | −0.151 | −0.145 | 0.223 | −0.171 | 0.108 | 1.000 | ||||||||||
| PTEN | −0.145 | −0.114 | −0.086 | 0.008 | 0.198 | 0.028 | −0.187 | 0.092 | 1.000 | |||||||||
| Ki67 | 0.126 | −0.118 | −0.177 | −0.023 | −0.176 | 0.330 | 0.506 | 0.004 | 0.023 | 1.000 | ||||||||
| PSA | 0.560 | −0.208 | −0.146 | 0.014 | 0.088 | −0.123 | 0.369 | 0.220 | −0.041 | 0.211 | 1.000 | |||||||
| p16 | 0.009 | −0.009 | 0.027 | 0.165 | 0.126 | −0.113 | 0.133 | 0.068 | −0.218 | 0.235 | 0.157 | 1.000 | ||||||
| NKX3.1 | 0.021 | −0.273 | −0.257 | 0.054 | −0.078 | 0.208 | −0.006 | −0.006 | −0.448 | 0.048 | −0.080 | 0.259 | 1.000 | |||||
| gH2AX | 0.115 | 0.125 | −0.027 | 0.303 | −0.022 | 0.146 | −0.060 | −0.036 | −0.260 | −0.150 | −0.117 | 0.018 | 0.076 | 1.000 | ||||
| pATM | 0.017 | 0.203 | 0.175 | 0.257 | −0.017 | 0.107 | −0.063 | 0.042 | −0.281 | −0.041 | −0.245 | 0.128 | 0.140 | 0.763 | 1.000 | |||
| 53BPP1 | −0.154 | −0.176 | −0.076 | −0.078 | 0.108 | 0.224 | −0.092 | 0.110 | −0.167 | −0.041 | −0.081 | 0.253 | 0.553 | −0.007 | 0.004 | 1.000 | ||
| JMJD1C | −0.121 | −0.256 | −0.104 | 0.111 | 0.201 | 0.223 | 0.314 | 0.208 | 0.092 | 0.263 | 0.140 | 0.087 | 0.424 | −0.209 | −0.210 | 0.410 | 1.000 | |
| Rev7 | 0.017 | −0.131 | −0.046 | −0.065 | 0.183 | −0.135 | 0.140 | 0.293 | −0.254 | −0.059 | 0.135 | 0.314 | 0.325 | −0.115 | −0.058 | 0.221 | 0.360 | 1.000 |
Finally not surprisingly for a CaP cohort, yet very relevant for the context with other markers, the overall proliferation rate deduced from the percentage of cells positive for the Ki67 marker was generally low (around 10% of cancer cells at the CaP stage). In addition, the fraction of S/G2 phase cells, as judged by cyclin A positivity, was even much lower than Ki67, within the range of 1–5% of cancer cells (data not shown), suggesting that most of the Ki67‐positive tumour cells are in the G1 phase.
Overall, we conclude that the proliferation rate of the lesions is generally very low and, more importantly, that the expression patterns of most of the examined markers show dynamic changes during progression of the disease from the benign lesions towards full malignancy.
3.2. DNA damage signalling barrier
One of the key issues, we wished to address in this study, was the extent and timing of potential DNA damage signalling activation during CaP progression, as a sign of the endogenous DNA damage response barrier against tumour progression analogous to other major solid tumours (Halazonetis et al., 2008). The level of DNA damage signalling, judged from the staining patterns of γH2AX as a general marker of active DDR kinases such as ATM or ATR, was detectable in a small fraction (around 2%) of cells at the BPH stage, usually increased several fold at the PIN stage of the same patient, and was highly variable, in a few cases even reaching 40–70% of positive tumour cells at the CaP stage. The graphical summary of the data is presented in Figure 1, and examples of immunohistochemical staining for γH2AX are shown in Figure 2A, along with aberrantly enhanced abundance of p53 protein, the major downstream effector of DDR signalling. Furthermore, we observed a very similar trend to that seen for γH2AX also for the activated, serine1981‐phosphorylated form of ATM (pATM), the major kinase responsible for histone H2AX phosphorylation, increasing from BPH through PIN towards the CaP stage (1, 2B, top images). Indeed, among all the biomarkers tested in our present study, γH2AX and pATM correlated most closely with each other (Table 2). Another aspect of DDR activation, especially response to DNA double strand breaks (DSBs) is the characteristic formation of so‐called nuclear foci that mark accumulation of DDR proteins such as 53BP1 in the DSB‐flanking chromatin (Lukas et al., 2011). Indeed, focal nuclear staining was apparent at closer inspection of slides stained for γH2AX and 53BP1, at least in some tumour cell nuclei (Figure 2B, lower images). Despite the detectable DDR activation, however, the overall extent of such signalling was clearly below that seen in other major solid tumour types, and the maximum was seen later during CaP progression. This intriguing difference can be exemplified by an overall graphical comparison of the extent of DDR signalling activation in CaP versus urinary bladder tumour progression (Bartkova et al., 2005), depicting subsets of cases with at least 10% of γH2AX‐positive tumour cells (Figure 2C).
Figure 2.
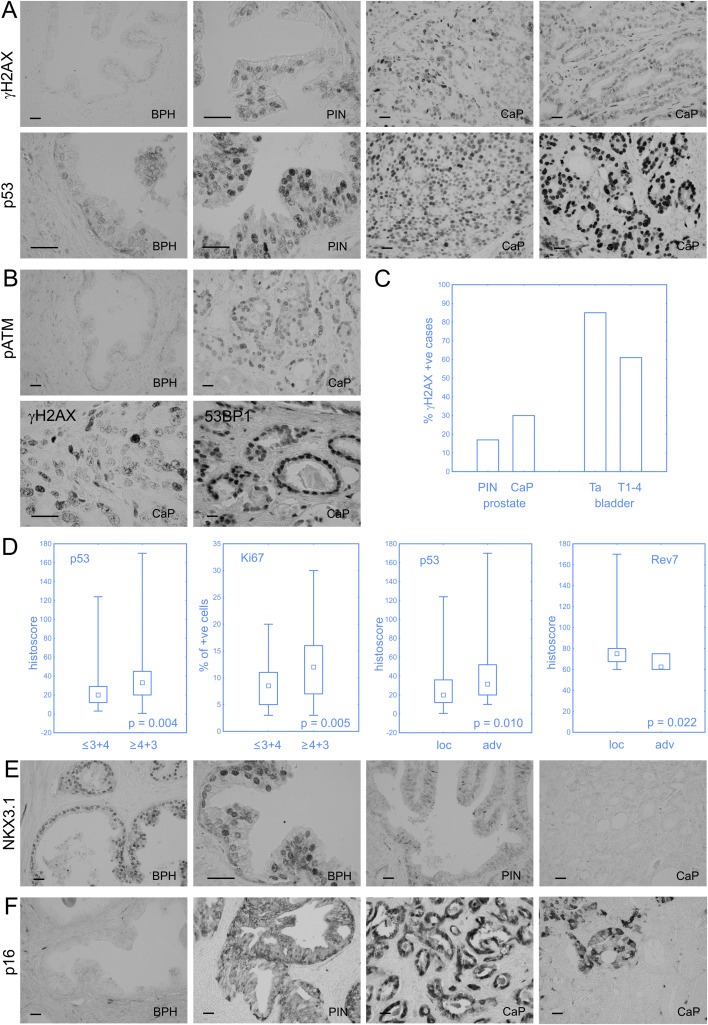
DNA damage response as an anticancer barrier in prostate cancer. Increasing genotoxic stress from BPH to PIN and CaP is illustrated by γH2AX, p53 and phospho‐ATM staining (A and B). Strong p53 positivity may indicate p53 mutation (A, far‐right image). Nuclear foci of H2AX and 53BP1 as markers of DNA damage response activation in CaP (B). The overall extent of DDR signalling is less pronounced in CaP than in other somatic tumour types (e.g. bladder cancer), and it culminates at the malignant, rather than pre‐malignant stage. (C). Higher Gleason score was associated with stronger staining of p53 (probably mutated) and faster proliferation (assessed by Ki67). Enhanced p53 and, more importantly, decreased Rev7 staining were observed in advanced CaP compared to localized ones (D). Complete (NKX3.1) and partial (p16) loss of tumour suppressors can be observed in invasive CaP (E and F). Scale bars represent 50 μm.
Regarding clinicopathological parameters, aberrantly enhanced expression of p53 and positivity for the proliferation marker Ki67 were significantly higher in tumours with Gleason score 4 + 3 or worse (Figure 2D). Furthermore, overabundant p53 (the highest levels of which likely reflect missense mutations of the p53 gene) was also higher in the advanced CaP tumours in comparison with the localized ones (Figure 2D).
Given that loss of the NKX3.1 is one of the characteristic features of CaP, and that one of the emerging functions of this tumour suppressor is a positive regulation of ATM kinase activation (Bowen et al., 2013), we regarded the status of NKX3.1 as a potential component of the DDR barrier mechanism. While the patterns of NKX3.1 were variable, there was a clear trend of maximum protein detection at the BPH stage, compared to generally aberrantly lower levels in both PIN and CaP lesions. The graphical summary of these results is presented in Figure 1 (bottom right graph) and representative examples of immunohistochemical staining patterns for NKX3.1 shown are shown in Figure 2E. Last but not least, since both the DDR checkpoint signalling and the p16 tumour suppressor can induce cellular senescence, we also considered p16 in the context of the potential anti‐tumour barriers. As briefly mentioned in section 3.1, abundance of p16 was increasing from BPH, through PIN to maximal levels in CaP lesions. Notably, this trend is overall reminiscent of the increasing DDR signalling, and examples of p16 staining patterns at various stages of CaP progression are shown in Figure 2F. What was also apparent at closer inspection, however, was that at both the PIN and the CaP stages, p16 expression was often heterogeneous and completely missing in some tumour areas (Figure 2F, far right), possibly reflecting focal loss of this tumour suppressor.
We conclude that the DNA damage checkpoint signalling is indeed activated in human prostate premalignant lesions and even more so in subsets of CaP lesions. On the other hand, the overall extent of DDR signalling is less pronounced than in other somatic tumour types such as carcinomas of the urinary bladder and colon (Bartkova et al., 2005), lung carcinomas (Gorgoulis et al., 2005), or gliomas (Bartkova et al., 2010), and it culminates at the malignant, rather than pre‐malignant stage, a pattern that is consistent also with the lower frequency of p53 overexpression (mutation) in CaP. The potential mechanisms and significance of these results including the apparent presence of DSB foci that we observe, will be considered in a broader context and more details in the Discussion section.
3.3. Oxidative stress and NQO1
Chronic inflammation and oxidative stress are among the hallmarks of CaP, and such conditions can also contribute to genomic instability. To gain a better insight into the kinetics and extent of oxidative stress in the course of human prostate tumorigenesis, we next examined 8‐oxoguanine (8‐oxoG, a key oxidative DNA lesion) and the status of NQO1, a cytoprotective enzyme of major importance in combating oxidative stress and a factor that stabilizes p53 (Dinkova‐Kostova and Talalay, 2010) by immunohistochemistry in our cohort of 103 prostatic lesions. In addition, we also established the genetic status of NQO1 with respect to its protein‐destabilizing polymorphism (C609T variant) as a candidate cancer‐predisposing or ‐promoting factor in the blood of a control cohort of 390 men (Table 3).
Our immunohistochemical analysis showed that abundance of 8‐oxoG as well as NQO1 was increasing from BPH, through PIN to CaP stages (see summary graphs in Figure 1). While both markers localized mainly in the cytoplasmic compartment of tumour cells (Figure 3A, B), nuclear positivity was also detectable and in a few cases, NQO1 was predominantly perinuclear (Figure 3B and data not shown). Consistent with their mutual functional link we also found a positive correlation between increasing NQO1 and 8‐oxoG (Table 2). As expected, all tumours from patients with the genetically confirmed NQO1 variant homozygote genotype (TT at position 609) lacked detectable NQO1 protein (see examples in Figure 3A, B). Notably, however, we also found several large NQO1 protein‐negative areas in tumours from patients with the heterozygous (CT) or wild‐type homozygous (CC) genotype (see Figure 3A, B for examples), a phenomenon that was particularly apparent in the immunoperoxidase‐stained sections without nuclear counterstaining, where the stromal blood vessels showed clear NQO1 positivity in contrast to surrounding negative cancer cells (Figure 3A, far‐right image). Whereas expression of the other protein markers examined in our present study remained apparently unaffected by the lack of NQO1 (see Supplementary Figure 1B for an example of parallel sections stained for NQO1 and 53BP1, respectively), in one case the area‐restricted lack of NQO1 correlated with the local loss of PTEN staining on parallel sections (Figure 3B, the two bottom right images). Notably, the area was positive for the other markers examined on parallel sections, thereby excluding false negative results for NQO1 and PTEN due to fixation problems or tissue necrosis. Furthermore, consistent with the role of NQO1 in protecting basal levels of p53 from proteolysis, tumours from patients with the homozygous TT variant of NQO1 featured significantly lower expression of p53 in comparison with both heterozygous and normal homozygous NQO1 genotypes (Figure 3C, p < 0.005). Patterns of other examined proteins were unaffected by the NQO1 genotype, further supporting the intimate functional relationship between NQO1 and p53. There was no association of NQO1 expression with clinicopathological parameters (serum PSA, Gleason score, or risk groups).
Figure 3.
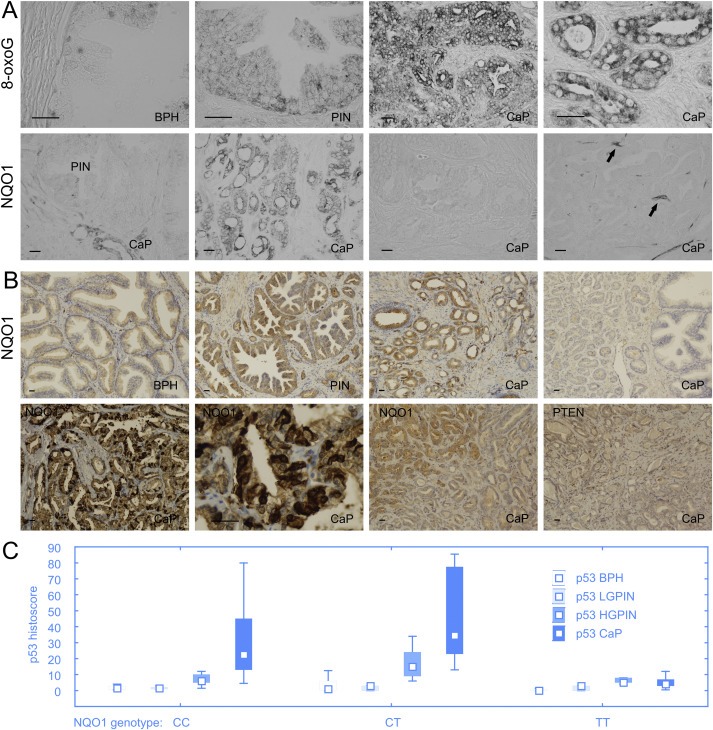
Oxidative stress and NQO1 in prostate cancer. Both 8‐oxoguanine and NQO1 increase from BPH to PIN and CaP (A; different staining method is shown in upper panel of B). All tumours from patients with the genetically confirmed NQO1 variant homozygote genotype (TT at position 609) lacked detectable NQO1 protein (A, the third image in the lower panel; B, far‐right image in the upper panel). Notably, we found several large NQO1 protein‐negative areas in tumours from patients with the heterozygous (CT) genotype, a phenomenon that was particularly apparent in the immunoperoxidase‐stained sections without nuclear counterstaining, where the stromal blood vessels showed clear NQO1 positivity (arrows) in contrast to surrounding negative cancer cells (A, far‐right image in the lower panel). Whereas expression of the other protein markers examined in our present study remained apparently unaffected by the lack of NQO1, in one case the area‐restricted lack of NQO1 correlated with the local loss of PTEN staining on parallel sections (B, the two bottom right images). Furthermore, strong perinuclear expression of NQO1 expression was observed in some patients (B, the two bottom left images). Scale bars represent 50 mm. Consistent with the role of NQO1 in protecting basal levels of p53 from proteolysis, tumours from patients with the homozygous TT variant of NQO1 featured significantly lower expression of p53 in comparison with both heterozygous and normal homozygous NQO1 genotypes (Figure 3C, p<0.005). Box‐plots represent median, 25%e75% percentiles and range of values.
Our genetic analysis of NQO1 status in the cohort of 171 CaP patients and 219 healthy men did not reveal any increased prevalence of the variant NQO1 genotype with prostate cancer (Table 3). These results do not support a role of NQO1 ’insufficiency‘ in genetic predisposition to prostate cancer, at least not in the population of men in the Czech republic studied here. On the other hand, the NQO1 protein expression data that we obtained suggest a potential contribution to oxidative stress response and CaP progression (see Discussion for more details on the potential relevance and implications of these findings).
3.4. Treatment‐relevant aberrations in DNA repair
Treatment with PARP inhibitors takes advantage of certain cancer‐predisposing or –promoting abnormalities in the DDR machinery, particularly status of proteins that impact the choice of DNA repair pathway by deregulating the balance of DSB repair through homologous recombination (HR) versus non‐homologous end joining (NHEJ) mechanisms (Lord and Ashworth, 2012). Motivated by emerging promising results from clinical trials with PARPi in several types of malignancies including castration‐resistant CaP (Mateo et al., 2015), we examined 5 markers that are likely to impact response to PARPi in our CaP progression cohort. Two of these factors, occurrence of the TMPRSS2‐ERG fusion oncogene and loss of the PTEN tumour suppressor have been extensively studied and we chose these for comparison with the other markers and due to their relevance for genome instability. On the other hand, expression of the 3 chosen DNA repair proteins, 53BP1, JMJD1C and Rev7, whose aberrant loss in certain genetic backgrounds, as we recently showed for breast cancer (Bouwman et al., 2010; Watanabe et al., 2013; Xu et al., 2015), can cause resistance to PARPi, has not been examined in the context of CaP so far.
Based on our genetic data for TMPRSS2‐ERG, and immunohistochemistry analyses of PTEN and the 3 DNA repair proteins, we found that fusion of TMPRSS2‐ERG occurred in 42% of tumours (Figure 5c), while expression of PTEN, 53BP1, Rev7 and JMJD1C was decreasing during the progression from BPH, through PIN to CaP (see Figure 1 and Supplementary Figure 1A, for graphical overviews). The TMPRSS2‐ERG fusion was associated with lower expression of PTEN and with a trend towards higher expression of NQO1 and nuclear Smad4, a factor involved in PTEN‐related cellular signalling (Figure 4A). Furthermore, tumours positive for TMPRSS2‐ERG and low in PTEN expression (histoscore below 160) showed a trend towards higher proliferation compared to other combinations of the TMPRSS2‐ERG fusion status and PTEN expression (Figure 4B).
Figure 5.
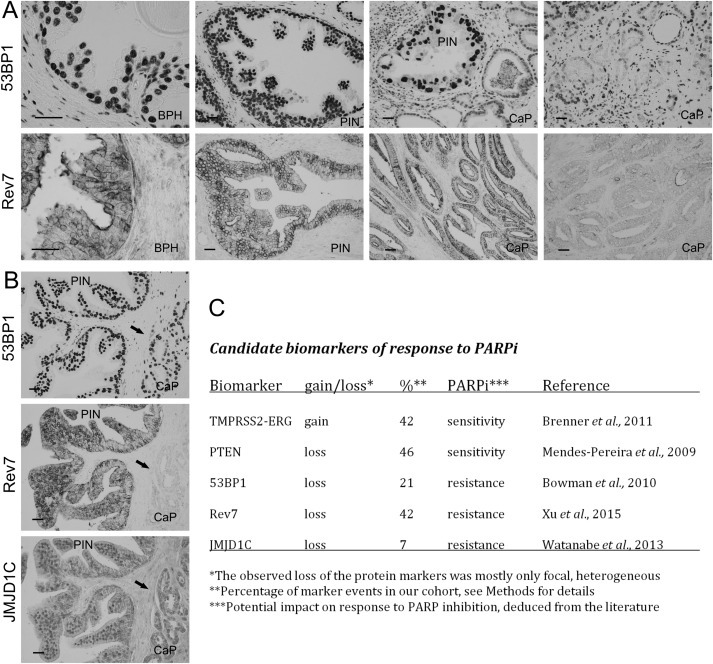
Potential predictive markers for prostate cancer therapy with PARP inhibitors. The aberrant decrease or loss of 53BP1 and Rev7 was heterogeneous, largely evident in only patches of cancer cells (A, upper panel), though for Rev7 such apparent loss could affect very large areas of the tumour (A, far‐right image in the lower panel). The observed lack of these repair factors was commonly restricted to only one of the proteins (B, parallel sections of a CaP stained for the indicated proteins with only Rev7 missing in the tumour area marked by the arrow) suggesting that such heterogeneous absence of these repair proteins was selective and did not simply reflect some tissue fixation problems or areas of necrosis. The summary of data for five potential predictive markers, showing prevalence of the aberrant cases within our cohort, along with their potential impact on response to PARPi treatment (C).
Figure 4.
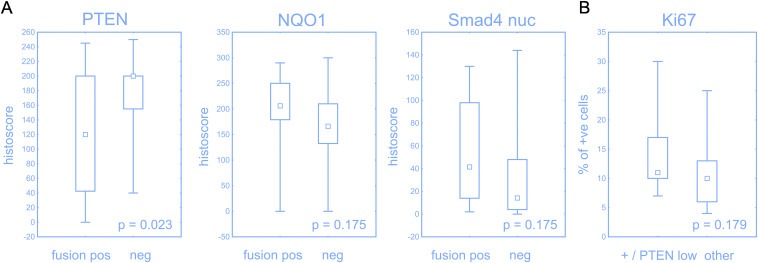
Effect of TMPRSS2‐ERG status. Expression of PTEN was significantly lower, while NQO1 and nuclear Smad4 had a trend towards higher expression in tumours with positive TMPRSS2‐ERG (A). Tumours with positive TMPRSS2‐ERG and low PTEN (histoscore less then 160) had a trend to a higher proliferation compared to other combination of fusion status and PTEN expression (B). Box‐plots represent median, 25%–75% percentiles and range of values.
As to the overall patterns of the 3 repair proteins, 53BP1 and JMJD1C correlated with each other, while JMJD1C correlated also with Rev7 (Table 2). Notably, the aberrant decrease or loss of 53BP1, Rev7 and JMJD1C was heterogeneous, largely evident in only patches of cancer cells, though for Rev7 such apparent loss could affect very large areas of the tumour (Figure 5A and B). The observed lack of these repair factors was commonly restricted to only one of the proteins (see Figure 5B for parallel sections of a CaP stained for the 3 factors, with only Rev7 missing in the tumour area marked by the arrow) suggesting that such heterogeneous absence of these repair proteins was selective and did not simply reflect some tissue fixation problems or areas of necrosis, for example.
The summary of the data for the 5 markers, showing prevalence of the aberrant cases within our cohort of CaP tumours, along with their potential impact on response to PARPi treatment of cases deemed suitable for this emerging therapy, is presented in Figure 5C. Functional ramifications and patho‐genetic as well as treatment‐related relevance of these results are discussed below.
4. Discussion
This study provides multiple insights into the dynamically changing landscape of key factors implicated in genome integrity control including DNA damage signalling and repair, oxidative stress and cell cycle regulation during the natural course of human prostate tumorigenesis. Apart from complementing previous studies (Aparicio et al., 2011) on CaP progression‐related alterations of the Rb pathway components (here cyclin D1, p16 and Rb), we wish to highlight the conceptual advances and potential clinical relevance of three separate yet related topics from the dataset we present: i) DNA damage response activation as a candidate biological barrier against CaP progression; ii) Oxidative stress and the role of NQO1; and iii) DNA repair‐related aberrations in CaP relevant for the emerging treatment strategy with PARP inhibitors.
First, based on the overall patterns of the DNA damage signalling markers γH2AX, pATM, focal pattern of 53BP1, and accumulation of p53, we conclude that in the course of human prostate tumour progression, the DNA damage checkpoint‘ barrier’ indeed becomes activated. While this phenomenon is conceptually broadly analogous to scenarios seen in progression of other somatic tumour types such as carcinomas of the lung, breast, colon or urinary bladder (Bartkova et al., 2010, 2005, 2013, 2005), we would like to emphasize some unorthodox features of this DDR barrier concept in prostate, distinct from other types of malignancies (see e.g. the comparison with urinary bladder tumours, Figure 2C). Such unorthodox aspects include the overall lesser extent, and later culmination of the DDR signalling during the progression from BPH and PIN, to culminate only at the CaP stage. We propose that the biological basis for this different DDR barrier scenario may reflect a combination of reasons, including the overall slower proliferation rate (documented here by the low Ki67 scores and very low fraction of S/G2 cells as revealed by cyclin A staining), possibly the low expression of histone H2AX per se, as reported for prostate luminal cells (Jaamaa et al., 2010), and also tumorigenic events characteristic for CaP, such as the very frequent loss of NKX3.1 tumour suppressor whose function is normally required for proper activation of the ATM kinase (Bowen et al., 2013). The observed slow proliferation rate of prostate tumours is furthermore likely to cause less replication stress compared to other major malignancies. Indeed, replication stress is commonly high in other types of tumours where it represents the major source of endogenous DNA damage, thereby inducing the DDR checkpoint barrier which consequently provides selective pressure to favour outgrowth of tumour clones with mutant p53 (Halazonetis et al., 2008). This line of thought appears consistent with the fact that the frequency of missense mutations of the p53 gene is generally much lower in CaP compared to most other carcinoma types. Indeed, based on a recent meta‐analysis of public cancer genetics databases the frequency of p53 mutations is only 17% in CaP (Williams et al., 2014), and this parallels very closely the subset of CaP cases (14%) which in our present study showed high percentage of p53‐positive cells, a feature that we showed in the past was characteristic for stabilized mutant p53 proteins (Iggo et al., 1990; Bartek et al., 1990). In this context, we propose that the DDR checkpoint barrier is generally less active in CaP (our current results), hence causing less pressure for checkpoint bypass, and thereby a lower ‘need’ to select for p53 mutations than in other tumours. This overall concept of the DDR barrier as a pressure selecting for p53 mutations is further supported by testicular germ cell tumours, which show hardly any evidence of the DDR barrier activation and, consistent with our arguments, show an exceptionally low p53 mutation rate (Bartkova et al., 2014, 2007). Also relevant to this topic is the positive correlation between aberrant loss of the Rb tumour suppressor and the high expression of p53 (the ‘mutation pattern’) in our cohort (Table 2). Since loss of Rb deregulates the G1/S controlling ‘restriction point’ (Bartek et al., 1996) and causes replication stress and DDR barrier activation (Tort et al., 2006), we would predict that such context should favour selection of p53‐mutant cancer cells in order to avoid DDR‐evoked senescence or apoptosis that would prevent tumour growth, a notion supported by the correlations between Rb loss and aberrant p53, and between aberrant p53 and advanced/aggressive CaP stage, observed in our present dataset.
The second major topic addressed here was the character of progression‐related changes in oxidative DNA lesions and the role(s) of the major anti‐oxidant' enzyme NQO1 in predisposition to, or progression, of CaP. At the genetic level, we have not observed any association between NQO1 genotype and CaP incidence which is in line with other studies of Caucasian populations (Steiner et al., 1999; Ergen et al., 2007; Steinbrecher et al., 2010; Stoehr et al., 2012). At the biomarker level studied by immunohistochemistry, we found that both NQO1 expression and 8‐oxoG detection were increasing from BPH to PIN and CaP, indicating an increase of oxidative stress during carcinogenesis and a potential contribution of NQO1 to genome integrity protection. Increased 8‐oxoG with age and in CaP has been found by different methods in tissues, urine and serum (Malins et al., 2001; Miyake et al., 2004; Kosova et al., 2014). Also, a role of NQO1 in blocking CaP progression has been recently proposed for the mouse TRAMP CaP model (Thapa et al., 2014). Furthermore, NQO1 silencing in CaP cells enhanced the levels of nuclear IKKalpha and NF‐kB while decreasing the levels of p53, leading to interactions between NF‐kB and p300 that reinforce cellular pro‐survival signalling (Thapa et al., 2011, 2010, 2014). The role of NQO1 in protecting basal levels of p53 from proteasomal degradation is well established (Dinkova‐Kostova and Talalay, 2010) and consistent with our present findings of low p53 protein levels in patients with the homozygous C609T variant genotype of NQO1 that basically eliminates the function of NQO1 from such cells and tissues. What was intriguing and not reported before was the total absence of NQO1 protein in large tumour areas in which NQO1 was clearly expressed in blood vessel endothelium. We observed this scenario in several cases of CaP, and suggest that this cancer‐selective loss of NQO1 may reflect a pressure to mitigate the anti‐tumour functions of NQO1, including its ability to stabilize wild‐type p53 as a critical component of the cellular anti‐cancer mechanism. In other words, we speculate that such focal loss of NQO1 in advancing CaP lesions supports the candidacy of NQO1 for a tumour suppressor whose loss may facilitate tumour progression. This model is also consistent with the fact that NQO1 attenuation fueled proinflammatory signalling and promoted androgen‐independent prostate cancer cell survival (Thapa et al., 2014).
Last but not least, as prostate tumours feature extensive chromosomal instability, and patients with CaP tumours harbouring defective DSB repair have recently shown promising response to the emerging treatment with PARPi (Mateo et al., 2015), we wish to highlight our present results related to defects in DSB repair factors and aberrations relevant for cellular responses to PARPi, as candidate future predictive biomarkers. Highly relevant for CaP is also the fact that PARP1 enzymatic activity is required for AR‐driven gene expression and ensuing CaP cell proliferation in the context of both hormone therapy–sensitive and CRPC models of disease (Schiewer and Knudsen, 2014). PARP1 also regulates the activity of ETS transcription factors in models of prostate cancer, which is of clinical significance, given the high percentage of prostate tumours that harbor fusions that put ETS expression under the control of AR activity (as through the TMPRSS2–ERG fusion) (Brenner et al., 2011). As such, regulation of both AR and ETS transcription factors by PARP1 are now being exploited in a clinical trial combining PARPi and an AR‐directed drug (abiraterone acetate) for patients with metastatic CRPC (NCT01576172). Among the factors reported to sensitize prostate cancer cells to PARPi is the most frequent oncogenic TMPRSS2‐ERG rearrangement and loss of the PTEN tumour suppressor (Brenner et al., 2011; Mendes‐Pereira et al., 2009). Here, we have confirmed the occurrence of these pathogenic events in sizeable subsets of CaP cases in our cohort (Figure 5C), and showed that at least in terms of PTEN loss, such aberrations may occur in a heterogeneous manner within a given lesion, likely reflecting clonal evolution of the advancing tumours. Notably, nuclear PTEN is important for chromosome stability maintenance, regulation of DNA replication and replication fork recovery (Shen et al., 2007; He et al., 2015).
In addition, we have identified subsets of CaP cases with various degrees of aberrantly reduced or lost expression of DNA repair factors 53BP1, JMJD1C and Rev7, none of which has been examined in CaP to date. The reason for analysing these three repair proteins is that we previously found their defects in cohorts of human triple‐negative breast carcinomas (Bouwman et al., 2010; Watanabe et al., 2013; Xu et al., 2015) and showed that their aberrant loss impacts the balance between the HR and NHEJ repair pathways, thereby causing resistance to PARPi in HR‐defective tumours which are otherwise very sensitive to PARP inhibition. Our present work established that all 3 proteins show some focal defects in subsets of CaP cases, however a substantial loss of 53BP1 and JMJD1C was not observed, a feature different from more pronounced defects among triple negative breast cancers. This difference is in fact hopeful for responses to treatment with PARPi in prostate cancer patients, as the potential problem of treatment resistance caused by extensive loss of 53BP1 or JMJD1C is likely to be greater in breast cancer compared with CaP. On the other hand, we did find aberrant lack of Rev7 in large tumour areas in a small fraction of the tumours (Figure 5). Therefore, we suggest that aberrant reduction or loss of these factors, particularly of Rev7 might represent one of the hurdles for the new targeted therapy of CaP with PARP inhibitors. We furthermore note that tumours in our cohort were primary CaP lesions and by the time metastatic spread occurs, i.e. at the stage most relevant for PARPi treatment (Mateo et al., 2015) at least some of the secondary CaP clones may show more pronounced defects in the repair factors which show only patchy loss in the primary tumour. In addition, we suggest that also overexpression of cyclin D1 and loss of NKX3.1, which both occurred in our cohort and are frequent in CaP, should be considered as potential biomarkers of response to PARPi, as cyclin D1 promotes HR (Jirawatnotai et al., 2011; Bartek and Lukas, 2011) and NKX3.1 facilitates ATM signalling that is important for DNA repair (Bowen et al., 2013).
Taken together, our current study helps to elucidate the role of the genome integrity machinery and oxidative stress in the patho‐biology of CaP. In addition, given that the first PARPi called Lynparza (formerly olaparib) has recently been approved for clinical use (Kim et al., 2015), and also showed promise in treatment of a subset of castration‐resistant metastatic CaP (Mateo et al., 2015), our present findings can be of value in future biomarker studies, especially related to targeted treatment with the PARP inhibitors.
Disclosure statement
Nothing to declare.
Supporting information
The following are the supplementary data related to this article:
Supplementary Figure 1 (A) Expression of 53BP1 in benign hyperplasia (BPH), prostate intraepithelial neoplasia (PIN) and prostate cancer (CaP). Box‐plot represents median, 25%–75% percentiles and range of values. Expression of 53BP1 was significantly lower in tumour compared to benign and PIN areas (p < 0.001; Supplementary Table 2). (B) Expression of 53BP1 protein remained apparently unaffected by the lack of NQO1.
{kind=link}
Supplementary data
Acknowledgements
This work was supported by grants NT13573 from the Czech Ministry of Health, the Danish Cancer Society, the Danish Council for Independent Research (DFF‐1331‐00262B), the Danish National Research Foundation (Centre of Excellence: CARD), CancerFonden (grant 150733), the Swedish Research Council, NPU I LO1304 and RVO: 61989592 from the Czech Ministry of Education, Grant Agency of the Czech Republic13‐17555S, EU operation program CZ.1.07/2.3.00/30.0041, and the Kellner Family Foundation.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.02.005.
Kurfurstova Daniela, Bartkova Jirina, Vrtel Radek, Mickova Alena, Burdova Alena, Majera Dusana, Mistrik Martin, Kral Milan, Santer Frederic R., Bouchal Jan, Bartek Jiri, (2016), DNA damage signalling barrier, oxidative stress and treatment‐relevant DNA repair factor alterations during progression of human prostate cancer, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.02.005.
Contributor Information
Jirina Bartkova, Email: jib@cancer.dk.
Jan Bouchal, Email: jan.bouchal@upol.cz.
Jiri Bartek, Email: jb@cancer.dk.
References
- Ahn, K.S. , Sethi, G. , Jain, A.K. , Jaiswal, A.K. , Aggarwal, B.B. , 2006. Genetic deletion of NAD(P)H:quinone oxidoreductase 1 abrogates activation of nuclear factor-kappaB, IkappaBalpha kinase, c-Jun N-terminal kinase, Akt, p38, and p44/42 mitogen-activated protein kinases and potentiates apoptosis. J. Biol. Chem. 281, 19798–19808. [DOI] [PubMed] [Google Scholar]
- Anders, C.K. , Winer, E.P. , Ford, J.M. , Dent, R. , Silver, D.P. , Sledge, G.W. , Carey, L.A. , 2010. Poly(ADP-Ribose) polymerase inhibition: “targeted” therapy for triple-negative breast cancer. Clin. Cancer Res. 16, 4702–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriole, G.L. , Crawford, E.D. , Grubb, R.L. , Buys, S.S. , Chia, D. , Church, T.R. , Fouad, M.N. , Isaacs, C. , Kvale, P.A. , Reding, D.J. , Weissfeld, J.L. , Yokochi, L.A. , O'Brien, B. , Ragard, L.R. , Clapp, J.D. , Rathmell, J.M. , Riley, T.L. , Hsing, A.W. , Izmirlian, G. , Pinsky, P.F. , Kramer, B.S. , Miller, A.B. , Gohagan, J.K. , Prorok, P.C. , 2012. Prostate cancer screening in the randomized prostate, lung, colorectal, and ovarian cancer screening trial: mortality results after 13 years of follow-up. J. Natl. Cancer Inst. 104, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar, A. , Dehn, D. , Siegel, D. , Kepa, J.K. , Tang, L.J. , Pietenpol, J.A. , Ross, D. , 2003. Interaction of human NAD(P)H:quinone oxidoreductase 1 (NQO1) with the tumor suppressor protein p53 in cells and cell-free systems. J. Biol. Chem. 278, 10368–10373. [DOI] [PubMed] [Google Scholar]
- Aparicio, A. , Den, R.B. , Knudsen, K.E. , 2011. Time to stratify? The retinoblastoma protein in castrate-resistant prostate cancer. Nat. Rev. Urol. 8, 562–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher, G. , Lotem, J. , Cohen, B. , Sachs, L. , Shaul, Y. , 2001. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proc. Natl. Acad. Sci. U. S. A. 98, 1188–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartek, J. , Bartkova, J. , Lukas, J. , 1996. The retinoblastoma protein pathway and the restriction point. Curr. Opin. Cell Biol. 8, 805–814. [DOI] [PubMed] [Google Scholar]
- Bartek, J. , Iggo, R. , Gannon, J. , Lane, D.P. , 1990. Genetic and immunochemical analysis of mutant p53 in human breast cancer cell lines. Oncogene. 5, 893–899. [PubMed] [Google Scholar]
- Bartek, J. , Lukas, J. , 2011. DNA repair: cyclin D1 multitasks. Nature. 474, 171–172. [DOI] [PubMed] [Google Scholar]
- Bartkova, J. , Hamerlik, P. , Stockhausen, M.T. , Ehrmann, J. , Hlobilkova, A. , Laursen, H. , Kalita, O. , Kolar, Z. , Poulsen, H.S. , Broholm, H. , Lukas, J. , Bartek, J. , 2010. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene. 29, 5095–5102. [DOI] [PubMed] [Google Scholar]
- Bartkova, J. , Hoei-Hansen, C.E. , Krizova, K. , Hamerlik, P. , Skakkebaek, N.E. , Rajpert-De, M.E. , Bartek, J. , 2014. Patterns of DNA damage response in intracranial germ cell tumors versus glioblastomas reflect cell of origin rather than brain environment: implications for the anti-tumor barrier concept and treatment. Mol. Oncol. 8, 1667–1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova, J. , Horejsi, Z. , Koed, K. , Kramer, A. , Tort, F. , Zieger, K. , Guldberg, P. , Sehested, M. , Nesland, J.M. , Lukas, C. , Orntoft, T. , Lukas, J. , Bartek, J. , 2005. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 434, 864–870. [DOI] [PubMed] [Google Scholar]
- Bartkova, J. , Horejsi, Z. , Sehested, M. , Nesland, J.M. , Rajpert-De, M.E. , Skakkebaek, N.E. , Stucki, M. , Jackson, S. , Lukas, J. , Bartek, J. , 2007. DNA damage response mediators MDC1 and 53BP1: constitutive activation and aberrant loss in breast and lung cancer, but not in testicular germ cell tumours. Oncogene. 26, 7414–7422. [DOI] [PubMed] [Google Scholar]
- Bartkova, J. , Rezaei, N. , Liontos, M. , Karakaidos, P. , Kletsas, D. , Issaeva, N. , Vassiliou, L.V. , Kolettas, E. , Niforou, K. , Zoumpourlis, V.C. , Takaoka, M. , Nakagawa, H. , Tort, F. , Fugger, K. , Johansson, F. , Sehested, M. , Andersen, C.L. , Dyrskjot, L. , Orntoft, T. , Lukas, J. , Kittas, C. , Helleday, T. , Halazonetis, T.D. , Bartek, J. , Gorgoulis, V.G. , 2006. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 444, 633–637. [DOI] [PubMed] [Google Scholar]
- Beyer, R.E. , Segura-Aguilar, J. , Di, B.S. , Cavazzoni, M. , Fato, R. , Fiorentini, D. , Galli, M.C. , Setti, M. , Landi, L. , Lenaz, G. , 1996. The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc. Natl. Acad. Sci. U. S. A. 93, 2528–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchal, J. , Santer, F.R. , Hoschele, P.P. , Tomastikova, E. , Neuwirt, H. , Culig, Z. , 2011. Transcriptional coactivators p300 and CBP stimulate estrogen receptor-beta signaling and regulate cellular events in prostate cancer. Prostate. 71, 431–437. [DOI] [PubMed] [Google Scholar]
- Bouwman, P. , Aly, A. , Escandell, J.M. , Pieterse, M. , Bartkova, J. , van der Gulden, H. , Hiddingh, S. , Thanasoula, M. , Kulkarni, A. , Yang, Q. , Haffty, B.G. , Tommiska, J. , Blomqvist, C. , Drapkin, R. , Adams, D.J. , Nevanlinna, H. , Bartek, J. , Tarsounas, M. , Ganesan, S. , Jonkers, J. , 2010. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 17, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen, C. , Ju, J.H. , Lee, J.H. , Paull, T.T. , Gelmann, E.P. , 2013. Functional activation of ATM by the prostate cancer suppressor NKX3.1. Cell Rep. 4, 516–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner, J.C. , Ateeq, B. , Li, Y. , Yocum, A.K. , Cao, Q. , Asangani, I.A. , Patel, S. , Wang, X. , Liang, H. , Yu, J. , Palanisamy, N. , Siddiqui, J. , Yan, W. , Cao, X. , Mehra, R. , Sabolch, A. , Basrur, V. , Lonigro, R.J. , Yang, J. , Tomlins, S.A. , Maher, C.A. , Elenitoba-Johnson, K.S. , Hussain, M. , Navone, N.M. , Pienta, K.J. , Varambally, S. , Feng, F.Y. , Chinnaiyan, A.M. , 2011. Mechanistic rationale for inhibition of poly(ADP-ribose) polymerase in ETS gene fusion-positive prostate cancer. Cancer Cell. 19, 664–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Micco, R. , Fumagalli, M. , Cicalese, A. , Piccinin, S. , Gasparini, P. , Luise, C. , Schurra, C. , Garre', M. , Nuciforo, P.G. , Bensimon, A. , Maestro, R. , Pelicci, P.G. , d'Adda di, F.F. , 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 444, 638–642. [DOI] [PubMed] [Google Scholar]
- Ding, Z. , Wu, C.-J. , Chu, G.C. , Xiao, Y. , Ho, D. , Zhang, J. , Perry, S.R. , Labrot, E.S. , Wu, X. , Lis, R. , Hoshida, Y. , Hiller, D. , Hu, B. , Jiang, S. , Zheng, H. , Stegh, A.H. , Scott, K.L. , Signoretti, S. , Bardeesy, N. , Wang, Y.A. , Hill, D.E. , Golub, T.R. , Stampfer, M.J. , Wong, W.H. , Loda, M. , Mucci, L. , Chin, L. , DePinho, R.A. , 2011. SMAD4-dependent barrier constrains prostate cancer growth and metastatic progression. Nature. 470, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinkova-Kostova, A.T. , Talalay, P. , 2010. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch. Biochem. Biophys. 501, 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eble, J.N. , Sauter, G. , Epstein, J.I. , Sesterhenn, I.A. , 2004. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Urinary System and Male Genital Organs IARCPress; Lyon: [Google Scholar]
- Ergen, H.A. , Gormus, U. , Narter, F. , Zeybek, U. , Bulgurcuoglu, S. , Isbir, T. , 2007. Investigation of NAD(P)H:quinone oxidoreductase 1 (NQO1) C609T polymorphism in prostate cancer. Anticancer Res. 27, 4107–4110. [PubMed] [Google Scholar]
- Evangelou, K. , Bartkova, J. , Kotsinas, A. , Pateras, I.S. , Liontos, M. , Velimezi, G. , Kosar, M. , Liloglou, T. , Trougakos, I.P. , Dyrskjot, L. , Andersen, C.L. , Papaioannou, M. , Drosos, Y. , Papafotiou, G. , Hodny, Z. , Sosa-Pineda, B. , Wu, X.R. , Klinakis, A. , Orntoft, T. , Lukas, J. , Bartek, J. , Gorgoulis, V.G. , 2013. The DNA damage checkpoint precedes activation of ARF in response to escalating oncogenic stress during tumorigenesis. Cell Death Differ. 20, 1485–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerholm, R. , Hofstetter, B. , Tommiska, J. , Aaltonen, K. , Vrtel, R. , Syrjakoski, K. , Kallioniemi, A. , Kilpivaara, O. , Mannermaa, A. , Kosma, V.M. , Uusitupa, M. , Eskelinen, M. , Kataja, V. , Aittomaki, K. , von, S.K. , Heikkila, P. , Lukas, J. , Holli, K. , Bartkova, J. , Blomqvist, C. , Bartek, J. , Nevanlinna, H. , 2008. NAD(P)H:quinone oxidoreductase 1 NQO1*2 genotype (P187S) is a strong prognostic and predictive factor in breast cancer. Nat. Genet. 40, 844–853. [DOI] [PubMed] [Google Scholar]
- Felgueiras, J. , Silva, J.V. , Fardilha, M. , 2014. Prostate cancer: the need for biomarkers and new therapeutic targets. J. Zhejiang Univ. Sci. B. 15, 16–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis, V.G. , Vassiliou, L.V. , Karakaidos, P. , Zacharatos, P. , Kotsinas, A. , Liloglou, T. , Venere, M. , Ditullio, R.A. , Kastrinakis, N.G. , Levy, B. , Kletsas, D. , Yoneta, A. , Herlyn, M. , Kittas, C. , Halazonetis, T.D. , 2005. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature. 434, 907–913. [DOI] [PubMed] [Google Scholar]
- Halazonetis, T.D. , Gorgoulis, V.G. , Bartek, J. , 2008. An oncogene-induced DNA damage model for cancer development. Science. 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- He, J. , Kang, X. , Yin, Y. , Chao, K.S.C. , Shen, W.H. , 2015. PTEN regulates DNA replication progression and stalled fork recovery. Nat. Commun. 6, 7620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howat, W.J. , Wilson, B.A. , 2014. Tissue fixation and the effect of molecular fixatives on downstream staining procedures. Methods. 70, 12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iggo, R. , Gatter, K. , Bartek, J. , Lane, D. , Harris, A.L. , 1990. Increased expression of mutant forms of p53 oncogene in primary lung cancer. Lancet. 335, 675–679. [DOI] [PubMed] [Google Scholar]
- Iskander, K. , Gaikwad, A. , Paquet, M. , Long, D.J. , Brayton, C. , Barrios, R. , Jaiswal, A.K. , 2005. Lower induction of p53 and decreased apoptosis in NQO1-null mice lead to increased sensitivity to chemical-induced skin carcinogenesis. Cancer Res. 65, 2054–2058. [DOI] [PubMed] [Google Scholar]
- Jaamaa, S. , Af Hallstrom, T.M. , Sankila, A. , Rantanen, V. , Koistinen, H. , Stenman, U.H. , Zhang, Z. , Yang, Z. , De Marzo, A.M. , Taari, K. , Ruutu, M. , Andersson, L.C. , Laiho, M. , 2010. DNA damage recognition via activated ATM and p53 pathway in nonproliferating human prostate tissue. Cancer Res. 70, 8630–8641. [DOI] [PubMed] [Google Scholar]
- Jackson, S.P. , Bartek, J. , 2009. The DNA damage response in human biology and disease. Nature. 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jirawatnotai, S. , Hu, Y. , Michowski, W. , Elias, J.E. , Becks, L. , Bienvenu, F. , Zagozdzon, A. , Goswami, T. , Wang, Y.E. , Clark, A.B. , Kunkel, T.A. , van, H.T. , Xia, B. , Correll, M. , Quackenbush, J. , Livingston, D.M. , Gygi, S.P. , Sicinski, P. , 2011. A function for cyclin D1 in DNA repair uncovered by protein interactome analyses in human cancers. Nature. 474, 230–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastan, M.B. , Bartek, J. , 2004. Cell-cycle checkpoints and cancer. Nature. 432, 316–323. [DOI] [PubMed] [Google Scholar]
- Kelsey, K.T. , Ross, D. , Traver, R.D. , Christiani, D.C. , Zuo, Z.F. , Spitz, M.R. , Wang, M. , Xu, X. , Lee, B.K. , Schwartz, B.S. , Wiencke, J.K. , 1997. Ethnic variation in the prevalence of a common NAD(P)H quinone oxidoreductase polymorphism and its implications for anti-cancer chemotherapy. Br. J. Cancer. 76, 852–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, G. , Ison, G. , McKee, A.E. , Zhang, H. , Tang, S. , Gwise, T. , Sridhara, R. , Lee, E. , Tzou, A. , Philip, R. , Chiu, H.J. , Ricks, T.K. , Palmby, T. , Russell, A.M. , Ladouceur, G. , Pfuma, E. , Li, H. , Zhao, L. , Liu, Q. , Venugopal, R. , Ibrahim, A. , Pazdur, R. , 2015. FDA approval summary: olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 21, 4257–4261. [DOI] [PubMed] [Google Scholar]
- Kolar, Z. , Burdova, A. , Jamaspishvili, T. , Bouchal, J. , Kucerova, R. , Bienova, M. , Kral, M. , Student, V. , 2014. Relation of ETS transcription factor family member ERG, androgen receptor and topoisomerase 2beta expression to TMPRSS2-ERG fusion status in prostate cancer. Neoplasma. 61, 9–16. [PubMed] [Google Scholar]
- Kosova, F. , Temeltas, G. , Ari, Z. , Lekili, M. , 2014. Possible relations between oxidative damage and apoptosis in benign prostate hyperplasia and prostate cancer patients. Tumour. Biol. 35, 4295–4299. [DOI] [PubMed] [Google Scholar]
- Long, D.J. , Waikel, R.L. , Wang, X.J. , Perlaky, L. , Roop, D.R. , Jaiswal, A.K. , 2000. NAD(P)H:quinone oxidoreductase 1 deficiency increases susceptibility to benzo(a)pyrene-induced mouse skin carcinogenesis. Cancer Res. 60, 5913–5915. [PubMed] [Google Scholar]
- Lord, C.J. , Ashworth, A. , 2012. The DNA damage response and cancer therapy. Nature. 481, 287–294. [DOI] [PubMed] [Google Scholar]
- Lukas, J. , Lukas, C. , Bartek, J. , 2011. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 13, 1161–1169. [DOI] [PubMed] [Google Scholar]
- Malins, D.C. , Johnson, P.M. , Wheeler, T.M. , Barker, E.A. , Polissar, N.L. , Vinson, M.A. , 2001. Age-related radical-induced DNA damage is linked to prostate cancer. Cancer Res. 61, 6025–6028. [PubMed] [Google Scholar]
- Mateo, J. , Carreira, S. , Sandhu, S. , Miranda, S. , Mossop, H. , Perez-Lopez, R. , Nava, R.D. , Robinson, D. , Omlin, A. , Tunariu, N. , Boysen, G. , Porta, N. , Flohr, P. , Gillman, A. , Figueiredo, I. , Paulding, C. , Seed, G. , Jain, S. , Ralph, C. , Protheroe, A. , Hussain, S. , Jones, R. , Elliott, T. , McGovern, U. , Bianchini, D. , Goodall, J. , Zafeiriou, Z. , Williamson, C.T. , Ferraldeschi, R. , Riisnaes, R. , Ebbs, B. , Fowler, G. , Roda, D. , Yuan, W. , Wu, Y.M. , Cao, X. , Brough, R. , Pemberton, H. , A'Hern, R. , Swain, A. , Kunju, L.P. , Eeles, R. , Attard, G. , Lord, C.J. , Ashworth, A. , Rubin, M.A. , Knudsen, K.E. , Feng, F.Y. , Chinnaiyan, A.M. , Hall, E. , de Bono, J.S. , 2015. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 373, 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes-Pereira, A.M. , Martin, S.A. , Brough, R. , McCarthy, A. , Taylor, J.R. , Kim, J.S. , Waldman, T. , Lord, C.J. , Ashworth, A. , 2009. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 1, 315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake, H. , Hara, I. , Kamidono, S. , Eto, H. , 2004. Oxidative DNA damage in patients with prostate cancer and its response to treatment. J. Urol. 171, 1533–1536. [DOI] [PubMed] [Google Scholar]
- Schiewer, M.J. , Knudsen, K.E. , 2014. Transcriptional roles of PARP1 in cancer. Mol. Cancer Res. 12, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder, F.H. , Hugosson, J. , Roobol, M.J. , Tammela, T.L. , Zappa, M. , Nelen, V. , Kwiatkowski, M. , Lujan, M. , Maattanen, L. , Lilja, H. , Denis, L.J. , Recker, F. , Paez, A. , Bangma, C.H. , Carlsson, S. , Puliti, D. , Villers, A. , Rebillard, X. , Hakama, M. , Stenman, U.H. , Kujala, P. , Taari, K. , Aus, G. , Huber, A. , van der Kwast, T.H. , van Schaik, R.H. , de Koning, H.J. , Moss, S.M. , Auvinen, A. , 2014. Screening and prostate cancer mortality: results of the European Randomised Study of Screening for Prostate Cancer (ERSPC) at 13 years of follow-up. Lancet. 384, 2027–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfanos, K.S. , De Marzo, A.M. , 2012. Prostate cancer and inflammation: the evidence. Histopathology. 60, 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, W.H. , Balajee, A.S. , Wang, J. , Wu, H. , Eng, C. , Pandolfi, P.P. , Yin, Y. , 2007. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. 128, 157–170. [DOI] [PubMed] [Google Scholar]
- Shevde, L.A. , Samant, R.S. , 2014. Role of osteopontin in the pathophysiology of cancer. Matrix Biol. 37, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel, D. , Anwar, A. , Winski, S.L. , Kepa, J.K. , Zolman, K.L. , Ross, D. , 2001. Rapid polyubiquitination and proteasomal degradation of a mutant form of NAD(P)H:quinone oxidoreductase 1. Mol. Pharmacol. 59, 263–268. [DOI] [PubMed] [Google Scholar]
- Siegel, D. , Bolton, E.M. , Burr, J.A. , Liebler, D.C. , Ross, D. , 1997. The reduction of alpha-tocopherolquinone by human NAD(P)H: quinone oxidoreductase: the role of alpha-tocopherolhydroquinone as a cellular antioxidant. Mol. Pharmacol. 52, 300–305. [DOI] [PubMed] [Google Scholar]
- Siegel, D. , Gustafson, D.L. , Dehn, D.L. , Han, J.Y. , Boonchoong, P. , Berliner, L.J. , Ross, D. , 2004. NAD(P)H:quinone oxidoreductase 1: role as a superoxide scavenger. Mol. Pharmacol. 65, 1238–1247. [DOI] [PubMed] [Google Scholar]
- Siegel, R. , Ma, J. , Zou, Z. , Jemal, A. , 2014. Cancer statistics, 2014. CA Cancer J. Clin. 64, 9–29. [DOI] [PubMed] [Google Scholar]
- Steinbrecher, A. , Rohrmann, S. , Timofeeva, M. , Risch, A. , Jansen, E. , Linseisen, J. , 2010. Dietary glucosinolate intake, polymorphisms in selected biotransformation enzymes, and risk of prostate cancer. Cancer Epidemiol. Biomarkers Prev. 19, 135–143. [DOI] [PubMed] [Google Scholar]
- Steiner, M. , Hillenbrand, M. , Borkowsi, M. , Seiter, H. , Schuff-Werner, P. , 1999. 609 C--> T polymorphism in NAD(P)H:quinone oxidoreductase gene in patients with prostatic adenocarcinoma or benign prostatic hyperplasia. Cancer Lett. 135, 67–71. [DOI] [PubMed] [Google Scholar]
- Stoehr, C.G. , Nolte, E. , Wach, S. , Wieland, W.F. , Hofstaedter, F. , Hartmann, A. , Stoehr, R. , 2012. NAD(P)H:quinone oxidoreductase 1 (NQO1) P187S polymorphism and prostate cancer risk in Caucasians. Int. J. Mol. Sci. 13, 10959–10969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa, D. , Meng, P. , Bedolla, R.G. , Reddick, R.L. , Kumar, A.P. , Ghosh, R. , 2014. NQO1 suppresses NF-kappaB-p300 interaction to regulate inflammatory mediators associated with prostate tumorigenesis. Cancer Res. 74, 5644–5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tort, F. , Bartkova, J. , Sehested, M. , Orntoft, T. , Lukas, J. , Bartek, J. , 2006. Retinoblastoma pathway defects show differential ability to activate the constitutive DNA damage response in human tumorigenesis. Cancer Res. 66, 10258–10263. [DOI] [PubMed] [Google Scholar]
- Watanabe, S. , Watanabe, K. , Akimov, V. , Bartkova, J. , Blagoev, B. , Lukas, J. , Bartek, J. , 2013. JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1-mediated chromatin response to DNA breaks. Nat. Struct. Mol. Biol. 20, 1425–1433. [DOI] [PubMed] [Google Scholar]
- Williams, J.L. , Greer, P.A. , Squire, J.A. , 2014. Recurrent copy number alterations in prostate cancer: an in silico meta-analysis of publicly available genomic data. Cancer Genet. 207, 474–488. [DOI] [PubMed] [Google Scholar]
- Xu, G. , Chapman, J.R. , Brandsma, I. , Yuan, J. , Mistrik, M. , Bouwman, P. , Bartkova, J. , Gogola, E. , Warmerdam, D. , Barazas, M. , Jaspers, J.E. , Watanabe, K. , Pieterse, M. , Kersbergen, A. , Sol, W. , Celie, P.H.N. , Schouten, P.C. , van den Broek, B. , Salman, A. , Nieuwland, M. , de Rink, I. , de Ronde, J. , Jalink, K. , Boulton, S.J. , Chen, J. , van Gent, D.C. , Bartek, J. , Jonkers, J. , Borst, P. , Rottenberg, S. , 2015. REV7 counteracts DNA double-strand break resection and affects PARP inhibition. Nature. 521, 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann, M. , Lottersberger, F. , Buonomo, S.B. , Sfeir, A. , de Lange, T. , 2013. 53BP1 regulates DSB repair using Rif1 to control 5′ end resection. Science. 339, 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary Figure 1 (A) Expression of 53BP1 in benign hyperplasia (BPH), prostate intraepithelial neoplasia (PIN) and prostate cancer (CaP). Box‐plot represents median, 25%–75% percentiles and range of values. Expression of 53BP1 was significantly lower in tumour compared to benign and PIN areas (p < 0.001; Supplementary Table 2). (B) Expression of 53BP1 protein remained apparently unaffected by the lack of NQO1.
Supplementary data