

Abstract
Development of resistance to platinum compounds significantly hinders successful ovarian cancer (OVCA) treatment. In tumor cells, dysregulated pH gradient across cell membranes is a key physiological mechanism of metastasis/chemo‐resistance. These pH alterations are mediated by aberrant activation of key multi‐subunit proton pumps, Vacuolar‐ATPases (V‐ATPases). In tumor cells, its ‘a2’ isoform (V‐ATPase‐V0a2) is a component of functional plasma–membrane complex and promotes tumor invasion through tumor‐acidification and immuno‐modulation. Its involvement in chemo‐resistance has not been studied. Here, we show that V‐ATPase‐V0a2 is over‐expressed in acquired‐cisplatin resistant OVCA cells (cis‐A2780/cis‐TOV112D). Of all the ‘a’ subunit isoforms, V‐ATPase‐V0a2 exhibited an elevated expression on plasma membrane of cisplatin‐resistant cells compared to sensitive counterparts. Immuno‐histochemistry revealed V‐ATPase‐V0a2 expression in both low grade (highly drug‐resistant) and high grade (highly recurrent) human OVCA tissues indicating its role in a centralized mechanism of tumor resistance. In cisplatin resistant cells, shRNA mediated inhibition of V‐ATPase‐V0a2 enhanced sensitivity towards both cisplatin and carboplatin. This improved cytotoxicity was mediated by enhanced cisplatin‐DNA‐adduct formation and suppressed DNA‐repair pathway, leading to enhanced apoptosis. Suppression of V0a2 activity strongly reduced cytosolic pH in resistant tumor cells, which is known to enhance platinum‐associated DNA‐damage. As an indicator of reduced metastasis and chemo‐resistance, in contrast to plasma membrane localization, a diffused cytoplasmic localization of acidic vacuoles was observed in V0a2‐knockdown resistant cells. Interestingly, pre‐treatment with monoclonal V0a2‐inhibitory antibody enhanced cisplatin cytotoxicity in resistant cells. Taken together, our findings suggest that the isoform specific inhibition of V‐ATPase‐V0a2 could serve as a therapeutic strategy for chemo‐resistant ovarian carcinoma and improve efficacy of platinum drugs.
Keywords: Vacuolar ATPase, a2 isoform, Ovarian cancer, Cisplatin resistance
Highlights
V‐ATPase ‘a’ subunit is over expressed in platinum resistant ovarian cancer cells.
V‐ATPase‐a2 isoform inhibition sensitized cancer cells to platinum therapy.
Cytosolic acidification due to a2‐inhibition caused DNA damage‐induced apoptosis.
Combinatorial treatment of anti‐a2 with cisplatin improved treatment response.
V‐ATPase‐a2 is potential therapeutic target for cisplatin resistant ovarian cancer.
1. Introduction
Ovarian carcinoma (OVCA) is the most lethal gynecological malignancy and is the fifth leading cause of cancer related deaths in women (American Cancer Society (www.cancer.org); Guppy et al., 2005; Eltabbakh and Awtrey, 2001). Treatment failure in OVCA patients is mainly attributable to the emergence of chemo‐resistance towards the mainstay platinum drugs, cisplatin and carboplatin (Tummala, 2005; Kartalou and Essigmann, 2001). Moreover, these platinum resistant tumors exhibit cross‐resistance to mechanistically unrelated second line drugs, that further poses a challenge in effective treatment and survival of these patients. Studies have revealed the role of various molecular alterations acquired by the resistant tumor cells in order to escape the platinum mediated cell death (Kartalou and Essigmann, 2001). Some of these mechanisms include (i) increased drug efflux (ii) enhanced drug inactivation (iii) enhanced DNA damage repair, (iv) increase in anti‐apoptotic molecules and (v) deregulated signaling of platinum mediated DNA damage to the apoptotic machinery (Moreno‐Smith et al., 2013; Perego P., 1996; Altomare, 2004; Fais et al., 2014). However, the above mentioned mechanisms vary in different platinum resistant tumor phenotypes. In order to improve the treatment efficacy, it is crucial to investigate the precise molecular mechanism and to identify tumor specific targets that influence a more centralized mechanism of chemo‐resistance in OVCA.
Due to failure of many anti‐cancer approaches, recent focus has shifted towards targeting the physiological hallmark changes in tumor cells such as altered pH gradient as an alternate approach to overcome the chemotherapy resistance (Barar and Omidi, 2013). In tumor cells, protons produced by glycolysis are extruded extra‐cellularly which prevents the cell from acidosis‐induced apoptosis. As a consequence, the tumor cells maintain an acidic extracellular pH and an alkaline intracellular pH, both of which favor tumor cell proliferation, invasion and drug resistance (Federici et al., 2014; Nishi and Forgac, 2002). Recent reports suggest that platinum resistance in tumor cells is likely to be as a result of the changes in pH gradient between the extracellular environment and cytoplasm. V‐ATPases are the key proton pumps in the regulation of this pH gradient (Recchi and Chavrier, 2006). In normal cells, V‐ATPases extrude protons from the cytoplasm to the lumen of the acidic organelles and also regulate cytosolic pH. In tumor cells, plasma membrane V‐ATPases extrude protons and therefore acidify the extracellular matrix (Barar and Omidi, 2013; Recchi and Chavrier, 2006; Beyenbach and Wieczorek, 2006). Previous studies mainly focused on the role of V‐ATPase in cancer metastasis and invasion aspects. Plasma membrane associated V‐ATPases are shown to critically influence the malignant behavior of cancer (Nishisho et al., 2011; Capecci and Forgac, 2013; Sennoune et al., 2004; Hendrix et al., 2013; Lu et al., 2005). However, only limited reports are available on the relevance of V‐ATPase expression and drug resistance in tumor cells. Recently, the utility of proton pump inhibitors has been shown in sensitization of tumor cells to cisplatin therapy as well as in reducing metastasis (Spugnini et al., 2010; De Milito and Fais, 2005). Nevertheless, it is critical to identify molecules that selectively target tumor associated V‐ATPases and do not influence the normal cells.
Mammalian V‐ATPase complex contains fourteen different subunits organized into an ATP‐hydrolytic domain (V1) and a membrane bound proton‐translocation domain (V0) that work together as a rotary machine (Recchi and Chavrier, 2006). Several V‐ATPase subunit genes are upregulated in cisplatin resistant cells (Murakami et al., 2001; Sasazawa et al., 2009). V‐ATPase ‘V0a’ subunit is the major pH sensing unit and additionally plays a crucial role in subcellular localization of this proton pump and exists in four different isoforms (a1/a2/a3/a4) (Forgac M, 2007). Our previous studies have highlighted the profound role of ‘V0a2’ isoform in tumor progression, metastasis and immuno‐modulation (Kwong et al., 2014, 2015, 2015, 2011, 2007, 2007). In context of ovarian cancer, our recent study demonstrated a distinct surface expression of V0a2, the inhibition of which reduced tumor cell migration (Kulshrestha et al., 2015). However, its role in chemo‐resistance in tumors is still not explored.
In this study, the role of V0a2 in conferring chemotherapy resistance was further evaluated to understand its relevance as a targeted therapy to control platinum resistance. Of all the over‐expressed V‐ATPase‐V0 ‘a’ isoforms, V0a2 exhibited an elevated expression on the plasma membrane of cisplatin resistant cells. Additionally, enhanced V0a2 expression was observed in both low and high grade human serous carcinoma tissues. In vitro, a selective inhibition of V0a2 decreased the cytosolic pH that sensitized the resistant tumor cells towards cisplatin treatment through increased platinum mediated DNA damage and a downregulated DNA repair pathway that induced apoptotic cell death. Additionally, combination studies involving pre‐treatment of resistant cells with monoclonal V0a2 inhibitory antibody also demonstrated the sensitization to cisplatin treatment. Our findings suggest that selective inhibition of V‐ATPase‐V0a2 may provide a therapeutic strategy to overcome cisplatin resistance and has a potential as a combination therapy along with platinum drugs for treatment of chemo‐resistant ovarian carcinoma.
2. Material and methods
2.1. Cell lines and cell culture
Two cisplatin sensitive human ovarian carcinoma cell lines, A2780 and TOV112D were used in this study. A2780 cell line was procured from Sigma–Aldrich, St Louis, MO, USA and cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA). TOV‐112D cell line was obtained from American type Culture Collection [ATCC] Manassas, VA and cultured in CTOV medium [1:1 mixture of MCDB 105 medium and Medium 199 with sodium bicarbonate]. All media were supplemented with 10% (v/v) heat‐inactivated fetal bovine serum (Biowest LLC, MO, USA), 100 U/ml penicillin and 100 U/ml streptomycin (Sigma Aldrich). Acquired cisplatin resistant cell lines were generated by adapting A2780 and TOV112‐D cells to increasing cisplatin pressure (1, 2.5, up to 2.5 μM) up to 28 weeks in the laboratory as described in previous studies (Lundholm et al., 2013). The cisplatin resistant counter‐parts were designated as cis‐A2780 and cis‐TOV‐112D. The stability of cisplatin resistance was confirmed by passaging the resistant cells without drug and then exposing them to adapted cisplatin concentration to observe any killing. For routine culture, cells were grown until reaching approximately 80% confluency and then subcultured or plated for experiments.
2.2. Anti‐cancer drugs
Cisplatin was procured from Sigma–Aldrich, and dissolved in normal saline at 1 mM stock concentration (stored as aliquots at −20 °C up to 3 months) before further dilution into complete media. Carboplatin was obtained from Sigma and dissolved in PBS (10 mM stock concentration) and stored as aliquots at −20 °C up to 3 months.
2.3. Immunohistochemical staining of ovarian cancer tissue
The paraffin embedded human ovarian cancer tissue blocks from low and high grade serous ovarian cancer patients were obtained from Advocate Lutheran General Hospital (ALGH), Chicago, USA. The study was approved by the Ethics Committee of ALGH. Three samples from both the low and high tumor grade were selected. The specimens were prepared as 5‐μm serially sectioned slides and were used for HE and immuno‐histochemical staining. For controls, the normal ovarian tissue sections were obtained from Biochain Institute, Inc (Newark, CA, USA). The tissue sections were stained using a method based on horseradish peroxidase‐labeled polymer (EnVision+Dual Link System‐HRP; DAKO, USA) according to manufacturer's protocol, preceded by an antigen retrieval procedure by boiling the sections in sodium citrate buffer (pH = 6.0). Experiments were performed at least in duplicate. For detection of V0a2 expression, sections were incubated with 5 μg/ml of IgG antibodies in 1% BSA‐PBS overnight at 4 °C. Simultaneously, for negative controls, tissue sections were stained with mouse isotype‐control antibodies (R&D systems, USA) used at the same concentration as the primary antibodies. The sections were counterstained with Mayer's hematoxylin and mounted in Faramount aqueous mounting medium (Dako). The immunostaining was evaluated by light photomicroscopy (Carl Zeiss, Weesp, The Netherlands) using a high‐resolution camera.
For scoring of immuno‐histochemical data, the semi‐quantitative integration method was applied. For this, five random fields of view were observed for each specimen at high magnification (×400). The results were scored based on the following criteria: First, staining area score [SAS] (≤1%: 0; 2–25%: 1; 26–50%: 2; 51–75%: 3 and > 75%: 4). Second, staining intensity [SI] (light brown: 1; moderate brown: 2 and tan: 3). The IHC score was calculated using the formula: ICS score = SAS × SI.
2.4. RNA isolation and reverse transcription‐PCR
The cells were washed with HBSS (Gibco, USA) and harvested using accutase solution (Sigma–Aldrich, St Louis, MO, USA). The harvested cells were washed twice with HBSS by centrifugation at 200 ×g for 5 min. RNA isolation was performed using RNeasy® mini kit (Qiagen) according to the manufacturer's protocol. Samples were stored at −80 °C until further use. 2.5 μg of total RNA was reverse transcribed at 37 °C using random primers and M‐MLV Reverse transcriptase system using high capacity cDNA kit (Applied Biosystems, Foster City, CA) using conditions recommended by the manufacturer. At least three biological replicates were prepared for each of the samples. Duplex RT‐PCR was performed using the Step One Real‐Time PCR system (Applied Biosystems), with GAPDH as the internal reference. The pre‐validated TaqMan gene‐expression assays for V0a1 (Atp6v0a1; Hs00193110_m1); V0a2 (Atp6v0a2; Hs00429389_m1); V0a3 (Hs00990751_m1) V0a4 (Hs00220886_m1), and internal control Gapdh (4326317E) were purchased from Applied Biosystems (Foster City, CA). All Real time PCR reactions were performed in triplicate in 10 μl volumes using Universal fast PCR Master Mix reagent (Applied Biosystems, USA) according to the manufacturer's protocol. The results were analyzed using the ΔΔCt method. Similarly, for analysis of DNA damage repair pathway (RT2 profiler, SA Biosciences, Frederick, MD, USA), a SYBR‐Green (Applied Biosystems, USA) based Q‐PCR assay was performed and the results were analyzed using the ΔΔCt method.
2.5. Generation of stable V0a2 knockdown cells
The transfection assay for V0a2 knock down was performed as described previously (Katara et al., 2015). Briefly, the cisplatin resistant cells (cis‐A2780/cis‐TOV112D) were seeded onto twenty four‐well plates (TPP Techno Plastic Products, Switzerland) at 75,000 cells/well overnight. The medium was changed and cells were transfected with anti‐V0a2 construct (shRNA, Suresilencing Plasmid, Qiagen, Valencia, CA, USA) or scramble control using attractene transfection reagent (Qiagen) according to the manufacturer's instructions. After transfection for 24 h, cells were washed with respective media and new media with selection antibiotic (1 mg/ml G418) was added to the wells. Cells were replenished with new medium containing G418 every 72 h till the wells became confluent. The transfected cells were initially checked for V0a2 knockdown by Q‐RT PCR. The positive transfectants were cloned using limited dilution method. The knock down was confirmed at protein level by western blot analysis. The V0a2 knockdown cisplatin resistant cells were designated as sh‐V0a2‐cis and the scrambled control cells as sh‐scr‐cis. Additionally, cisplatin sensitive OVCA cells (A2780, TOV‐112D) were also knock down for V0a2 as described above to determine whether they exhibit an improved cisplatin efficacy.
With regard to other ‘a’ isoforms, we also performed knock down of V0a3 in cisplatin resistant OVCA cells (showing highest upregulation among all ‘a’ isoforms) as described above using anti‐V0a3 construct (shRNA, Suresilencing plasmid, Qiagen) or scramble control.
2.6. Cell survival assay
The exponentially growing ovarian tumor cells were seeded into 96‐well plate (1 × 104/well) overnight. The V0a2 shRNA transfected/un‐transfected cells were respectively exposed to two‐fold serial dilutions of 200 μM cisplatin (Range: 0.78–200 μM) or 750 μM carboplatin (Range: 1.46–750 μM) for 48 h at 37 °C in 5% CO2. After incubation, in vitro cell viability was measured using Alamar Blue reagent (Life Technologies, USA) or MTS assay (Promega, USA). Untreated cells were used as negative control. All experiments were performed in triplicate. The IC50 (inhibitory concentration of the drug that kills 50% of cell population) was calculated using Origin 6.0 software (Origin Lab) and semi‐log plots of dose response curves were generated using Prism 5.0 software (GraphPad Software, Inc.).
2.7. Western blot analysis
Cells were harvested, resuspended in NP‐40 lysis buffer with protease inhibitors (Pierce Protein Biology, USA). Cell lysates were incubated on ice for 30 min with intermittent shaking and then centrifuged for 30 min at 4 °C at 13 000 × rpm to remove cellular debris. Protein concentrations were determined using BCA assay (Pierce Protein Biology, USA). SDS sample buffer was added to the lysates and aliquots containing 40 μg of protein were separated by SDS‐PAGE on 4–20% gradient acrylamide gels. For detecting the presence of subunit ‘a2V’ isoform, antibody against V0a2 (2C1) was used as described previously (Kulshrestha et al., 2015) followed by IR secondary antibody (Licor) and visualized using Odyssey® infrared imaging system (LI‐COR Biotechnology Lincoln, NE, USA). To assess equivalent loading, blots were probed with a monoclonal β‐actin antibody (Calbiochem, La Jolla, CA).
2.8. Assessment of apoptotic cell death
The apoptosis rate was measured by flow cytometry by Annexin V‐FITC and PI staining kit (BD Biosciences, USA). Briefly, V0a2 shRNA transfected/untransfected OVCA cells were incubated with 20 μM cisplatin (near to IC25 for cisplatin resistant cells) for 24 h at 37 °C in 5% CO2. The cells were collected, washed twice with PBS. The cells were resuspended in Annexin V binding buffer at a concentration of 5 × 105/ml. FITC‐conjugated Annexin V (1 μg/ml) and PI (50 μg/ml) were added to the cells, and incubated for 15 min in the dark. Quantitative analysis of apoptotic level was performed using an LSR‐II flow cytometer (BD Biosciences). A minimum of 20,000 cells were analyzed in each sample, and all experiments were performed in triplicate.
2.9. Cisplatin‐DNA adduct quantification
To stain the cisplatin adducts, we followed the protocol as described by Lundholm et al. (2013), V0a2 knockdown or control cisplatin resistant OVCA cells were treated with cisplatin as described above for apoptosis experiment. Cells were fixed in 70% ethanol for 30 min at 4 °C and then washed thrice with PBS. For staining the DNA adducts, anti‐cisplatin‐modified DNA antibody (ab103261; Abcam, Cambridge, UK) was diluted in PBS containing 100 mg/ml digitonin and used to stain cells for overnight at 4 °C. Cells were then incubated with goat anti‐rat IgG H&L (DyLight 488) secondary antibody (ab96887; Abcam) for 1 h. Stained cells were washed with PBS and recorded using LSR‐II Flow cytometer and analyzed using the CellQuest Software (BD Bioscience, Franklin Lakes, NJ, USA) for cisplatin adduct formation. Further, the cisplatin adduct‐stained cells were analyzed microscopically by cyto‐spinning on glass slides and applying mounting media containing DAPI (Vectashield, Vector Labs Inc., Burlingame, CA, USA). Images were acquired at 20× magnification on a Nikon microscope and analyzed using metamorph software (Molecular Devices, CA, USA).
2.10. Cytosolic pH measurement
To evaluate the cytosolic pH of tumor cells, sh‐V0a2‐cis, sh‐scr‐cis and cisplatin sensitive (cis‐S) cells were grown in 75 cm2 tissue culture flasks (TPP, Trasadingen, Switzerland) in RPMI or CTOV medium supplemented with 10% FBS. When the flasks attained 80% confluency, 1 × 106 cells/ml PBS were loaded with 1 μm SNARF‐1 (5‐[and‐6] carboxy‐SNARF‐1‐AM), and cells were incubated for 30 min with the acetoxymethyl ester form of SNARF‐1 (Life technologies, USA). Depending on the cytosolic pH, cellular esterases cleave the acetoxymethyl groups leaving the charged form of SNARF‐1 in the cytosol. In situ calibration curves were generated. For this, the cells were treated with ionophore, nigericin (30 μg/ml) that equilibrates H+ and K+ across the membrane, rendering same intra‐ and extra‐cellular pH. The cells were subsequently perfused with high K+ calibration buffers of different pH range (pH 4.0–8.0; Life Technologies, USA). The excitation of SNARF‐1 loaded cells was performed at 534 nm, and the fluorescence emission signal was collected at 490 and 640 nm. The pH‐dependent spectral shifts exhibited by carboxy SNARF‐1 allow calibration of the pH response in terms of the ratio of fluorescence intensities measured at two different wave lengths. The ratios (R = 644/480) of SNARF‐1 were converted to pH using a modified Henderson–Hasselbalch equation.
2.11. Staining of acidic vesicles by LysoSensor probe
The LysoSensor Green DND‐189 probe (Molecular Probes) was used according to the manufacturer's instructions to measure the effects of cisplatin treatment on acidic vesicle. This dye accumulates in acidic vesicles and exhibits a pH dependent increase in fluorescence intensity on acidification. Briefly, 5 × 105 cells were collected after 24 h of cisplatin treatment [20 μM, 24 h] and washed twice in PBS. Cells were then incubated for 30 min at 37 °C in respective medium with 1 M LysoSensor probe and analyzed by flow cytometry for assessing the changes in endosomal acidification upon V0a2 knockdown. Untreated and unstained cells were used to set the background fluorescence. For analysis of the localization of LysoSensor positive vesicles, cells were incubated for 30 min at 37 °C with respective medium containing the LysoSensor probe and analyzed by live microscopy (Zeiss). The experiments were repeated twice in duplicate.
2.12. Statistical analysis
The means of two data sets were compared and significance was determined by two‐tailed Students t‐test. Differences were considered to be statistically significant where P < 0.05. Data is graphically represented as mean ± standard deviation of the mean (SD). Data was analyzed using GraphPad Prism (version 5) statistical software. Dose response curves were generated as semi‐log plots where the drug dose had a wide range. Origin 6.0 software was used for determination of the IC50 values using sigmoidal regression analysis. All experiments were repeated at least twice in duplicate.
3. Results
3.1. Generation and characterization of cisplatin‐resistant ovarian cancer cells
A2780 and TOV112D cells were exposed to cisplatin pressure by step wise increase in drug concentration up to of 2.5 μM cisplatin (1.25, 2 and 2.5 μM; over a period of 10 and 28 weeks respectively). The cisplatin resistant OVCA cells were denoted as cis‐A2780 and cis‐TOV‐112D. Cisplatin resistant cells exhibited a significantly higher IC50 (cis‐A2780 = 59.3 ± 5.1 μM, P < 0.001; cis‐TOV112D = 44.83 ± 6.30 μM, P < 0.0001) compared to their sensitive counterparts that were passaged simultaneously with resistant cells (IC50 = 6.85 ± 2.21 μM and 5.80 ± 3.21 μM; for A2780 and TOV112D respectively). Dose response curves were generated and the fold resistance after cisplatin treatment was calculated for cis‐A2780 as 8.5 fold (P < 0.01; Figure 1A) and for cis‐TOV‐112D as 7.5 fold (P < 0.01; Figure 1B). The stability of cisplatin resistance was confirmed by cultivation of these cells in drug‐free media for at least 12 passages (approximately 5 weeks). There was no significant difference in the growth profile of cisplatin sensitive and resistant OVCA cells [Supplementary Figure S1]. Next, we analyzed the status of Vacuolar ATPase ‘a’ subunit isoforms in cisplatin resistant cells. Comparative gene expression profiling by real time RT‐PCR revealed a significantly elevated expression of all three major V‐ATPase ‘a’ subunit isoforms in the two tested cisplatin resistant cells (V0a1 – 1.93 ± 0.05 fold, V0a2 – 2.6 ± 0.56 fold and V0a3 – 5.01 ± 1.4 fold) [Figure 1C]. This elevated V‐ATPase expression was further confirmed at protein level by immuno‐fluorescence analysis [Figure 1D]. Our previous studies have highlighted the relevance of surface V0a2 expression and its role in tumor invasion and metastasis (Kulshrestha et al., 2015). To validate the importance, V0a2 isoform was therefore selected to decipher its role in cisplatin resistance.
Figure 1.
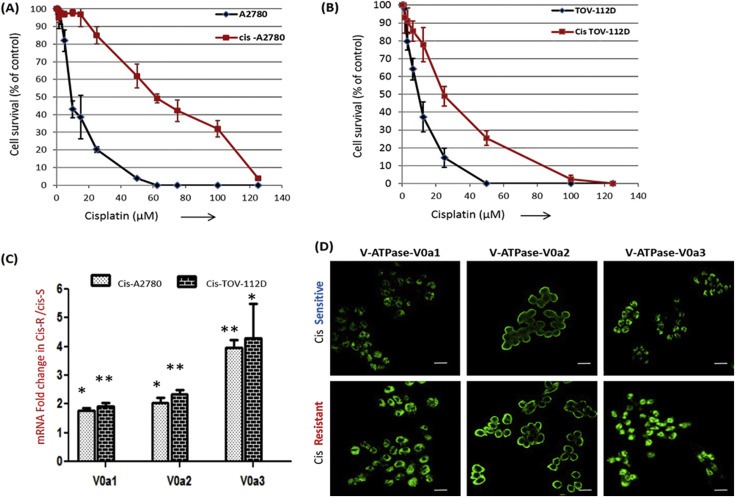
Vacuolar ATPase ‘a’ subunit isoforms are over‐expressed in acquired cisplatin resistant ovarian cancer cells. (A) Dose‐survival curves of cisplatin sensitive A2780 cell line and its resistant counterpart, cis‐A2780 following a 48 h incubation with different concentrations of cisplatin. (B) Similarly, dose–survival curves of cis‐TOV112D shows increase in cisplatin resistance compared to sensitive TOV‐112D. (C) Q‐RT‐PCR analysis showing mRNA levels of vacuolar ATPase‐V0 ‘a’ subunit isoforms (V‐ATPase‐V0a1, V0a2 and V0a3) in cisplatin resistant cell lines compared to respective sensitive counterparts. The Ct values were normalized against the Ct values for GAPDH from the same preparation. The data are provided as mRNA fold change in cisplatin resistant tumor cells over sensitive cells (**P < 0.001, *P < 0.05). (D) Immuno‐fluorescence analysis of cisplatin resistant cells using isoform specific antibodies against V0a1/V0a2/V0a3 compared to cisplatin sensitive ovarian tumor cells. Representative cis – A2780 (cis‐Resistant) and A2780 (cis‐Sensitive) cell lines are shown. Magnification (×200); scale bar – 10 μM. All experiments were performed at least in triplicate and data is provided as means with standard deviations.
3.2. Cisplatin resistant ovarian carcinoma cells exhibit elevated V‐ATPase–V0a2 expression on the tumor cell surface
To delineate the importance of V‐ATPase‐V0a2 in drug resistance, we examined the difference in protein expression of V0a2 between cisplatin‐resistant ovarian cancer cell lines cis‐A2780/cis‐TOV‐112D and sensitive cell lines A2780/TOV‐112D. Western blot analysis revealed a clear up regulation of V‐ATPase‐V0a2 protein expression in resistant cells [Figure 2A] compared with sensitive cells. Similarly, flow cytometry analysis of the non‐permeabilized cisplatin sensitive and resistant cells further confirmed elevated V0a2 expression on surface of cisplatin resistant cells [Figure 2B]. The immuno‐fluorescence analysis showed the over‐expression of V0a2 on the surface of both the cisplatin resistant cell lines [Figure 2C]. Further, confocal microscopy analysis confirmed the co‐localization of V‐ATPase‐V0a2 with the plasma membrane marker, pan‐cadherin, suggesting that the isoform is expressed on the plasma membrane of both cisplatin sensitive and resistant tumor cell, with elevated expression in cisplatin resistant cells [Figure 2D].
Figure 2.
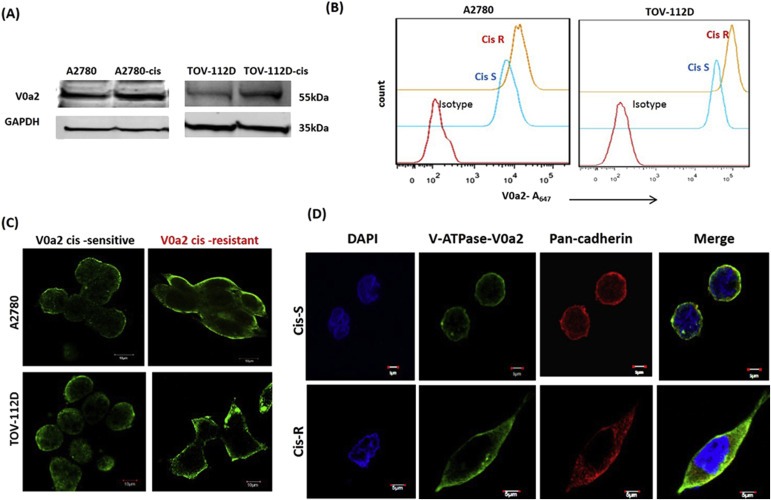
Cisplatin resistant ovarian carcinoma cells exhibit over‐expressed V‐ATPase‐V0a2 on the tumor cell surface. (A) Total protein from cisplatin resistant ovarian tumor cells and respective sensitive phenotypes were immuno‐blotted with anti‐V‐ATPase‐V0a2 antibody that showed V0a2 over expression in cisplatin resistant cells. (B) The surface expression of V‐ATPase‐V0a2 was also elevated in non‐permeabilized cis‐R (cis‐A2780/cis‐TOV112D; orange line) compared to cis‐S (A2780/TOV112D; blue line) as determined by fluorescence‐activated cell sorting (FACS). Red line indicates primary antibody isotype control. (C) Immuno‐fluorescence analysis showing elevated V‐ATPase‐V0a2 expression on the plasma membrane of cisplatin resistant OVCA cell lines as shown by green staining. Magnification – ×600; scale bars; 10 μm (D) Confocal microscopy analysis of V‐ATPase‐V0a2 (in green) and cells surface marker, pan cadherin (in red) in cisplatin sensitive ovarian cancer cells (Cis‐S) and cisplatin resistant ovarian cancer cells (Cis‐R). Merged images (yellow regions) reveal the co‐localization of V‐ATPase‐V0a2 with pan cadherin which is more pronounced in resistant cells. Original magnification – ×600; scale bars; 5 μm. Representative images from three independent experiments are shown.
3.3. V‐ATPase‐v0a2 is highly expressed in low and high grade ovarian carcinoma tissues: clinical relevance
To elucidate the clinical relevance of V‐ATPase‐V0a2 in ovarian cancer, we stained the serous cystadenocarcinoma tissues. The results of the immuno‐histochemical assay showed that the V‐ATPase‐V0a2 was expressed in both low and high grade serous ovarian carcinoma tissues [Figure 3A]. The expression of V‐ATPase‐V0a2 correlated with Ki67 antigen used to mark the tumor cells in the OVCA tissues [Supplementary Figure S2]. In contrast, the normal ovarian tissues showed weak staining for V0a2 [Figure 3B]. Among different pathological grades of serous ovarian cancer, the expression of V‐ATPase was significantly higher (P < 0.05) in high grade cancer (IHC score = 8.90 ± 0.1) than that of the low grade (IHC score = 5.6 ± 1.06) [Figure 3C]. This holds relevance as the high‐grade serous ovarian cancers account for most mortality due to frequent relapse and progression to chemotherapy resistance (Cooke and Brenton, 2011). However, given its prominent expression in low grade cancer also, V0a2 is a good target for treatment in both the cases. Additionally, as a prerequisite to form a functional V‐ATPase complex, V0 and V1 subunits should be in association with one another on the plasma membrane. V0 free of V1 is incompetent for proton translocation. To confirm that V0a2 is expressed as a part of plasma membrane associated V‐ATPase, immuno‐fluorescence assay was performed using ovarian cancer cell lines (A2780 and TOV112D). The V0a2 showed co‐localization with plasma membrane associated V1D subunit in both the cell lines [Figure 3D] indicating that it is a part of the plasma membrane V‐ATPase machinery. After confirming the clinical relevance of V0a2, we further investigated its possible association with cisplatin resistance in OVCA using cell lines.
Figure 3.
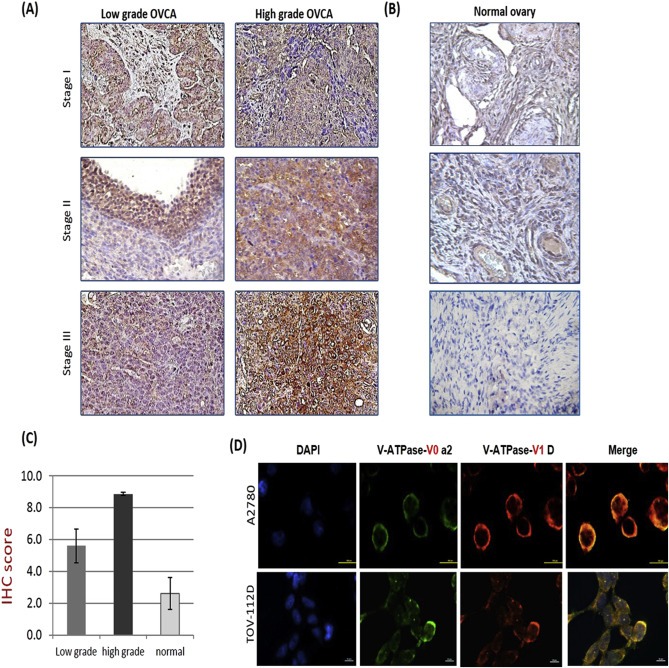
V‐ATPase‐V0a2 is over‐expressed in human ovarian carcinoma tissues. Positive staining (brown) observed for V‐ATPase‐V0a2 in (A): tissues from low and high grade serous ovarian cancer patients (B). Representative normal human ovarian cancer tissue showing very low V0a2 expression. Original magnification – ×400. (C) IHC score of V‐ATPase‐V0a2 expression revealed significantly higher expression in high grade carcinoma which are frequently associated with chemo‐resistance (n = 3 in respective group). (D) Immuno‐fluorescence analysis of V‐ATPase‐V0a2 (green) in the human ovarian cancer cell lines showing its co‐expression with plasma membrane associated V‐ATPase‐V1D subunit (red). Merged areas shown in yellow. Nuclear staining with DAPI. Original magnification: ×800; scale bars, 100 μm. Representative images from three independent experiments are shown. For normal tissues, the following tissue sections were used: human adult normal ovary; Biochain (Cat no: T2234183).
3.4. shRNA mediated V‐ATPase‐V0a2 knockdown in cisplatin resistant cells reveals reduced tumor cell proliferation
On the basis of an increased V0a2 expression in cisplatin resistant cell lines, we next examined the effect of V0a2 silencing on tumor cell proliferation in vitro. To suppress V0a2 expression in cisplatin resistant cells, we used a short hairpin RNA against V‐ATPase‐V0a2 (sha2) or a non targeting scrambled short hairpin RNA (sh‐scr). Representative data from cis‐A2780 cell line is shown here. The V0a2 shRNA transfected cells (sh‐V0a2‐cis) exhibited 3.8 fold inhibition at mRNA level compared with scrambled control (sh‐scr‐cis) [Figure 4A]. The V0a2 silencing was further confirmed at protein level and a 2.5 fold reduction in protein expression was observed by western blot analysis [Figure 4B]. The diminished V0a2 protein expression could also be observed by flow cytometry analysis [Figure 4C]. The expression of other isoforms (a1, a3 and a4) of ‘a’ subunit remained unaltered at mRNA level [Supplementary Figure S3]. After determining the stability of the V0a2 knockdown, we next examined if the inhibition of V0a2 expression has any effect on the tumor cell proliferative capacity. We therefore performed tumor cell proliferation assay for sh‐V0a2‐cis and sh‐scr‐cis. The results showed that the inhibition of V0a2 reduced the in vitro tumor cell proliferation in cisplatin resistant cells [Figure 4D] indicating that V0a2 plays a role in tumor cell growth.
Figure 4.
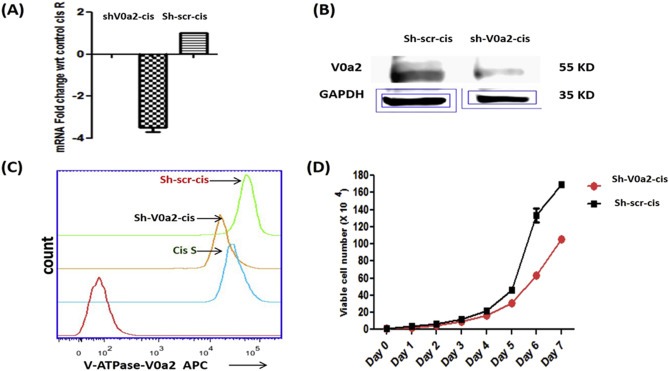
Stable knockdown of V‐ATPase‐V0a2 in cisplatin resistant ovarian cancer cells impairs cell growth. V‐ATPase‐V0a2 expression was knock down in cisplatin resistant cells (cis‐A2780) by transfection with SureSilencing shRNA plasmids (Qiagen, Valencia, CA, USA) Suppression of V0a2 expression was confirmed by (A) Quantitative real time PCR showing the mRNA levels of sh‐V0a2‐cis (V0a2 knock down cells) compared to scrambled control (sh‐scr‐cis). Data are expressed as fold change compared shV0a2‐cis‐control cells and as mean ± SD. *P < 0.05. (B) For determining protein levels, western blot was performed using isoform specific monoclonal anti‐V0a2 antibody. (C) FACS analysis was performed to validate the knock down of V0a2 in shV0a2‐cis cells. Sh‐scr‐cis (Control; green line), A2780 (Cis‐S; blue line), V0a2 knock down cells (sh‐V0a2‐cis; orange line). Red line indicates primary antibody isotype control. (D) In vitro tumor cell growth was monitored in shV0a2‐cis and shV0a2‐cis‐control cells. All experiments were performed at least three times in triplicate.
3.5. Inhibition of V‐ATPase‐V0a2 and V0a3 sensitizes the cisplatin resistant cells to platinum drugs
Further, we determined the effect of V0a2 silencing on cisplatin sensitivity of resistant OVCA cells. After silencing of V‐ATPase‐V0a2, the cisplatin resistant cells exhibited a significantly improved sensitivity [Figure 5A ] towards cisplatin with the reduction in IC50 by 3.2 fold (sh‐V0a2‐cis, IC50 = 11.47 ± 3.02 μM, P < 0.05) compared to control resistant cells (sh‐scr‐cis, IC50 = 37.75 ± 6.62 μM) [Figure 5B] that was comparable to cis‐S cells (IC50 = 6.85 ± 2.21 μM). In addition, V0a2 silenced cis‐R cells also showed increased sensitivity towards carboplatin. The carboplatin IC50 of sh‐V0a2‐cis decreased to 290.90 ± 26.20 μM compared to 453.11 ± 24.46 uM in sh‐scr‐cis cells (P < 0.05) [Figure 5C]. The IC50 of carboplatin towards cis‐S was 244.17 ± 5.32 μM. Notably, knock down of V0a2 in A2780 and TOV‐112D sensitive cells also improved the sensitivity towards platinum drugs suggesting its utility in improving the platinum sensitivity [data not shown].
Figure 5.
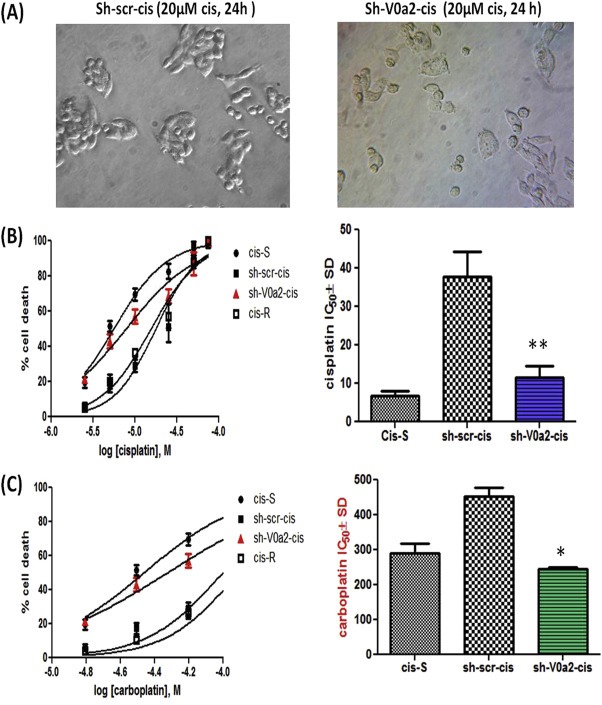
Inhibition of V‐ATPase‐V0a2 sensitizes the cisplatin resistant cells to platinum drugs. (A) Phase contrast images showing the cell morphology of control cis‐A2780 (sh‐scr‐cis) and V‐ATPase‐V0a2 knockdown cells (sh‐V0a2‐cis) upon treatment with cisplatin (20 μM, 24 h). (B) Dose–response curve of sh‐scr‐cis, sh‐V0a2‐cis, cisplatin resistant (cis‐R) and cisplatin sensitive cells (cis S) following 48 h treatment with cisplatin. The IC50 values are depicted in histograms in the right panel.(C) Dose response curves of cis‐S, sh‐scr‐cis, sh‐V0a2‐cis and cis‐R following 48 h treatment with carboplatin. The IC50 values are depicted in histograms in the right panel. Values are means (±S.D) of two independent experiments performed in triplicate. Compared with control **P < 0.01, *P < 0.05 (Mann–Whitney test).
Similarly, knock down of V0a3 in cis‐A2780 also sensitized the cells towards cisplatin [Supplementary Figure S4], indicating that the role of other isoforms and intracellular or endosomal V‐ATPases in cisplatin resistance cannot be ruled out.
3.6. V‐ATPase‐V‐0a2 inhibition improves cisplatin sensitivity by increasing platinum mediated DNA damage
We next asked as to why V0a2 inhibition is causing platinum sensitization in resistant cells. In cisplatin treated cells, DNA platination translates signal to damage recognition proteins, ultimately causing cell death. DNA platination is therefore the key factor in rendering cisplatin sensitivity in tumor cells (Mahoney et al., 2003). The cisplatin resistant cells exhibit decreased DNA adduct formation and therefore escape cell death. In the present study, we assessed the DNA adduct formation as a measure of cisplatin induced cell damage. For this, we performed FACS analysis by indirect staining using anti‐cisplatin‐modified DNA antibody, as described in methods section. Cisplatin adduct formation was significantly increased in sh‐V0a2‐cis cells compared to control sh‐scr‐cis cells (cisplatin 20 μM, 48 h treatment), hence indicating higher drug availability for DNA binding as the main factor for the increased cisplatin sensitivity [Figure 6A]. The mean fluorescence intensity was significantly higher in sh‐V0a2‐cis cells compared to control (P < 0.01) [Figure 6B]. The immuno‐fluorescence analysis further revealed enhanced green fluorescence from sh‐V0a2‐cis cells (as an indicator of high DNA adducts) compared to sh‐scr‐cis cells [Figure 6C].
Figure 6.
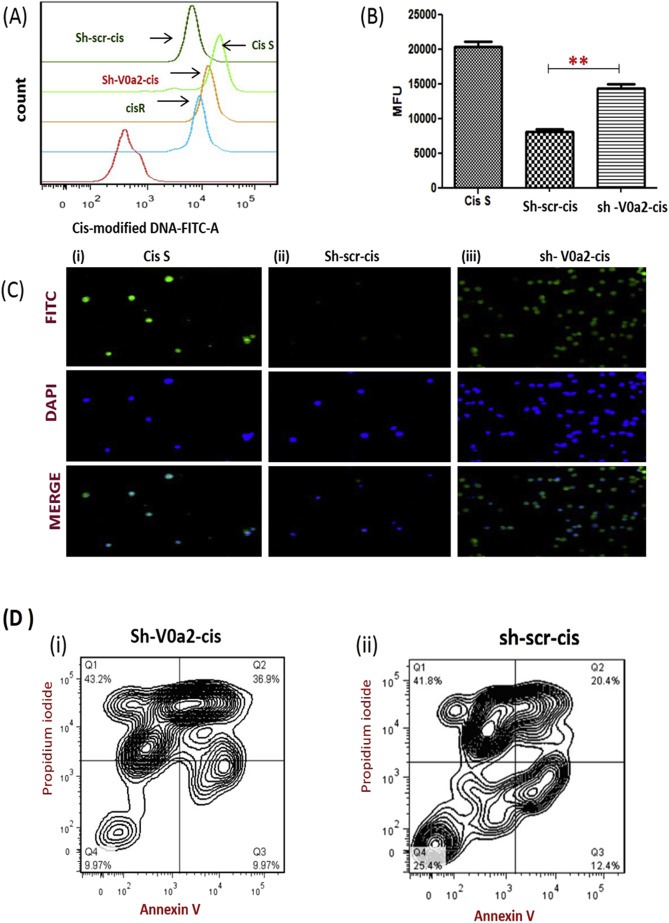
Inhibition of V‐ATPase‐V0a2 increases DNA damage and apoptosis in cisplatin resistant ovarian cancer cells. Cisplatin resistant ovarian cancer cells were stably knock down for V‐ATPase‐V0a2 and then treated with cisplatin for up to 48 h and fixed using ice‐cold 70% ethanol. Cells were stained overnight at 4 °C using a primary antibody that specifically recognizes cisplatin‐GpG DNA adducts. (A) Flow cytometry analysis was performed by indirect staining using secondary anti‐rat FITC®‐labeled antibody. Isotype control (Red line), Cisplatin sensitive cells (Cis‐S; light green line), Cisplatin resistant WT cells (Cis‐R; Blue line), knockdown scrambled control cisplatin resistant cells (sh‐scr‐cis control; dark green line), V‐ATPase‐V0a2 knock down cisplatin resistant cells (sh‐V0a2‐cis; Orange line) showing higher DNA damage in V0a2 knock down cells compared to cisplatin resistant control cells. (B) Histogram showing geometric mean fluorescence intensities of anti‐cisplatin DNA adduct antibody‐stained cells divided by isotype ± s.e.m (n = 3). (C) Same cells were subjected to Immunofluorescence staining; cisplatin adducts (green) and nuclear staining (blue) at 20× magnification. Cisplatin sensitive A2780 cells [panel (i)], Control cisplatin resistant cells [panel (ii)], and V0a2 knock down cisplatin resistant cells [panel (iii); showing higher DNA damage compared to control cells]. (D)The Annexin V/propidium iodide double staining assay following 20 μM cisplatin treatment for 48 h in (i) V0a2 knock down cisplatin resistant cells and (ii) cisplatin resistant control cells. The percentages in right quadrants represent percentage apoptosis over the blank and are an average of at least two independent trials. All experiments were repeated at least thrice in duplicate.
To further confirm that the cisplatin DNA adducts/lesions are not undergoing repair, we performed a PCR array for DNA damage repair pathway associated genes. We found that in a2 knock down cells (sh‐V0a2‐cis), upon cisplatin treatment, several nucleotide excision repair (NER) and base excision repair (BER) genes were significantly downregulated compared to the control cisplatin resistant cells (sh‐scr‐cis). This decreased expression of DNA repair genes may contribute to the observed increase in DNA damage and partly explain the cisplatin cytotoxicity in a2 inhibited cells (Supplementary Figure S5).
3.7. Inhibition of V‐ATPase‐V0a2 activity induces cell death pathway mediated by increased apoptosis upon cisplatin treatment
Once it was established that there is increased cisplatin‐DNA adduct formation in V‐ATPase‐V0a2 knockdown cells, we next wanted to decipher if this increased DNA adduct message is translated into cell death signal. To study the apoptosis mechanism, we treated sh‐V0a2‐cis and control sh‐scr‐cis with 20 μM cisplatin for 48 h and performed flow cytometric analysis to detect the cell apoptosis. We found that the late apoptotic rate in knock down sh‐V0a2‐cis cells (36.9 ± 2.12%) [Figure 6 (D i) ] was significantly higher than that of control sh‐scr‐cis cells (20.4 ± 1.56%) (P < 0.05) [Figure 6 (D ii)].
3.8. Inhibition of V‐ATPase‐V0a2 causes acidification of the cytoplasm of cisplatin resistant cells
Next, we wanted to understand the probable mechanism of V0a2 inhibition mediated cisplatin sensitivity in the resistant tumor cells. Cisplatin requires an acidic intracellular pH to convert to active protonated form that can bind to DNA (Kartalou and Essigmann, 2001). Since the key role of V‐ATPase‐V0a2 is in pH sensing and regulation, we measured the cytosolic pH in the V0a2 knock down cisplatin resistant cells using the fluorescence pH indicator SNARF‐1. sh‐V0a2‐cis and sh‐scr‐cis cells were loaded with SNARF‐1 and cytosolic pH was determined by measuring the ratio of fluorescence intensity at 640 and 480 nm upon excitation at 534 nm. A calibration curve was generated by incubating the cells in calibration buffers with known pH (pH 4.0, 5.0, 6.0, 7.0 and 8.0) that imparted known changes in pH in presence of ionophore, nigericin that equilibrates intra‐ and extra‐cellular pH [Figure 7A]. Control cisplatin resistant cells exhibited a more alkaline cytosolic pH (pH = 7.19 ± 0.03) compared to cisplatin sensitive cells (pH = 6.96 ± 0.01). Interestingly, knockdown of V‐ATPase‐V0a2 caused acidification of the cytosol in cisplatin resistant cells (pH = 6.87 ± 0.04) that was lower than cisplatin sensitive cells too [Figure 7B].
Figure 7.
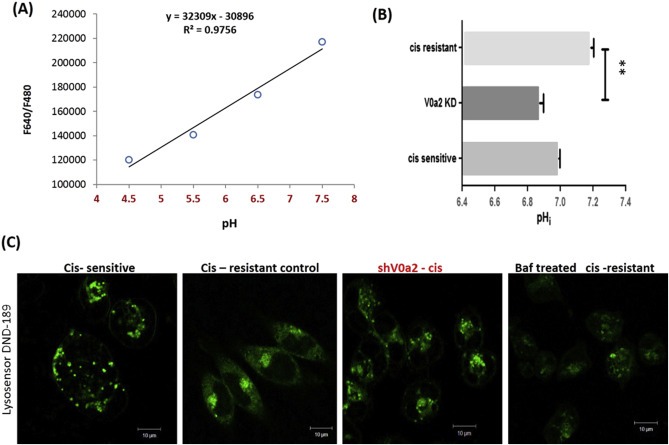
Cisplatin resistant ovarian cancer cells exhibit a decrease in cytosolic pH and alteration in localization of acidic vacuoles upon V‐ATPase‐V0a2 inhibition. The cytosolic pH was estimated using pH sensitive dye SNARF‐1 (Life Technologies). (A) cisplatin resistant A2780 cells were loaded with SNARF‐1 dye followed by imparting known changes in pH using calibration buffers (pH 4.5–8.0) and calibration curve was generated by recording ratio (480/640 nm) of SNARF‐1F (B) The fluorescence spectra of SNARF‐1 was obtained on cisplatin sensitive, cisplatin resistant and V‐ATPase V0a2 knock down cisplatin resistant cells. The corresponding intracellular pH was obtained from the pH calibration curve (Panel A). Values are means (±S.E.M) of two independent experiments performed in triplicate. **Compared with control P < 0.01 (Mann–Whitney test). (C) shV0a2‐cis knock down cells were treated with 1 μM Lysosensor Green DND‐189 (pH dependent acidic vesicle marker) for 30 min. To visualize the distribution pattern of acidic vesicles in V‐ATPase‐V0a2‐cis, immuno‐fluorescence analysis was performed by live cell imaging (Leica Microsystems). Magnification: 20×; scale bar‐10 μM. Cis‐sensitive‐ cisplatin sensitive A2780 ovarian cancer cells; cis‐resistant control – cisplatin resistant cells containing scrambled knockdown sequence, shV0a2‐cis – V0a2 knock down cells, Baf – Bafilomycin treated cells.
3.9. Suppression of V‐ATPase‐V0a2 activity alters intracellular localization of acidic vesicles in cisplatin resistant ovarian cancer cells
Vacuolar ATPases contribute to acidification of the endocytic compartment and is crucial for protein trafficking and processing (Barar and Omidi, 2013). To study the effect of V0a2 suppression on the lysosomal distribution and acidity, a vital dye, DND‐189 was employed, which selectively accumulates in the acidic compartments and emits green fluorescence as a function of acidic pH (lower the pH, more the intensity). As shown in [Figure 7C], suppression of V0a2 accelerated the redistribution of the lysosomes in cisplatin resistant cells as in sensitive counterpart. The extension of acidic vesicles to the cell periphery, indicative of high metastasis was not seen in V‐ATPase‐V0a2‐cis knock down cells compared to cisplatin resistant cells, where acidic vacuoles were present all through the cell periphery. Flow cytometry analysis did not show a significant change in the overall pH in knock down vs control cisplatin resistant cells [Supplementary Figure S6]. This indicates that targeting V0a2 in cisplatin resistance cells may play dual role of cisplatin sensitization and reduction in tumor cell migration and invasion.
3.10. Pre‐treatment with anti‐V‐ATPase‐V0a2 antibody sensitizes the resistant ovarian tumor cells to cisplatin treatment
To determine the functional implications of V‐ATPase and the V0a2 isoform inhibition on cisplatin sensitivity, we performed combination studies using anti‐V0a2 monoclonal antibody generated in our lab (Kwong et al., 2011; Kulshrestha et al., 2015). The V‐ATPase inhibitory activity of anti‐a2V antibody (also known as anti‐RTF antibody, from its previous name; for regeneration and tolerance factor) is well established in both in vitro and in vivo systems (Derks and Beaman., 2004; Boomer et al., 2000). Notably, pre‐treatment with anti‐V0a2 antibody (20 μg/ml, 6 h) augmented the cisplatin sensitivity in ovarian cancer cells. The effect on cisplatin sensitivity was significantly enhanced in A2780 cisplatin resistant cells upon pretreatment with anti‐V0a2 antibody (IC50 = 23.72 ± 2.66) compared to cisplatin treatment alone (IC50 = 52.74 ± 2.72) indicating a substantial reversal of cisplatin resistance [Figure 8A]. Anti‐V0a2 antibody treatment slightly enhanced cisplatin sensitivity in sensitive cells (IC50 = 5.66 ± 0.4) compared to cells subjected to monotherapy with cisplatin (IC50=7.50 ± 1.36) indicating that combination treatment using V‐ATpase‐V0a2 inhibitors could improve the overall efficacy of cisplatin treatment [Figure 8B]. Upon assessment of cytosolic pH in anti‐a2v treated cis‐R and cis‐S, we observed a decrease in pH upon treatment with anti‐a2v treatment further validating the role of intracellular pH in rendering cisplatin sensitivity [Supplementary Figure S7]. Interestingly, bafilomycin treated cisplatin sensitive and resistant cells did not show a significant change in cytosolic pH, indicating its mechanism of sensitization is different from anti‐a2v antibody or a2v inhibition and that the compensatory role of other proton pumps might be involved [Supplementary Figure S6]. This is in line with the previous observations that bafilomycin does not effect the cytosolic pH in tumor cells (Lucien et al., 2014).
Figure 8.
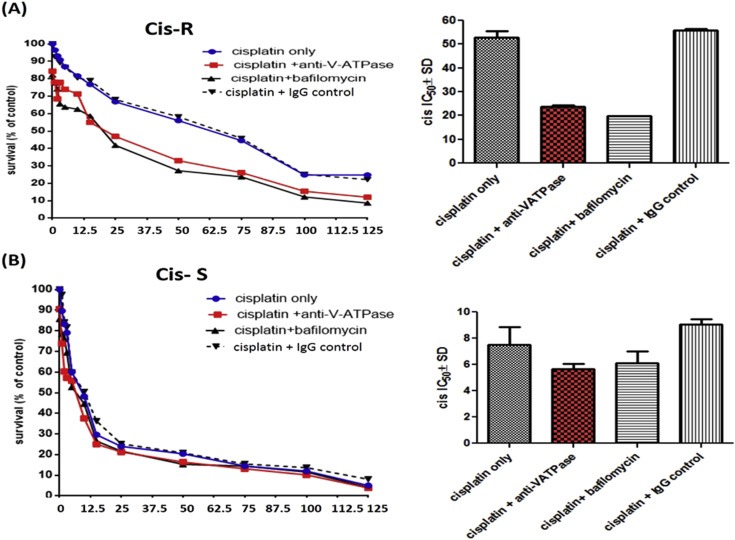
Combinatorial treatment with anti‐V‐ATPase‐V0a2 antibody and cisplatin is more effective in killing cisplatin resistant ovarian tumor cells. Ovarian cancer cells were pretreated with anti‐V‐ATPase‐V0a2 antibody following the treatment with cisplatin (A) Dose–response curves obtained by treating cisplatin sensitive cell line (A2780) or (B) cisplatin resistant cell line (cis‐A2780) with cisplatin alone (Cisplatin only) or pre‐treating them with anti‐V‐ATPase‐V0a2 antibody (cisplatin + anti‐V‐ATPase) or (IgG control antibody) for 6 h before cisplatin treatment. Bafilomycin, vacuolar ATPAse inhibitor, was used as the positive control. Cell death was assayed by using the fluoremetric alamar blue cell viability assay. The histograms represent mean ± SD of three different experiments.
4. Discussion
The therapeutic efficacy in ovarian carcinoma is commonly low due to the emergence of platinum resistance, leading to poor survival rates in the patients. The present study reports the involvement of Vacuolar‐ATPase ‘V0a2’ subunit isoform in rendering cisplatin resistance in ovarian cancer cells. Through a series of mechanistic analysis of acquired cisplatin resistant ovarian cancer cell lines, we observed a distinct over‐expression of Vacuolar‐ATPase ‘a2’ subunit isoform on the surface of cisplatin resistant cells. Selective V0a2 inhibition sensitized the resistant cells to platinum treatment through acidification of cytosol, thus inducing apoptotic cell death. Additionally, V0a2 inhibition further improved the cisplatin sensitivity in sensitive cells. In combinatorial studies, the pre‐treatment of resistant cells with V0a2 inhibitory antibody drastically improved the cisplatin sensitivity in these cells in vitro.
In tumor cells, the over‐expression of V‐ATPase proton pumps is a cell survival mechanism to prevent intra‐cellular acidosis, which otherwise is a trigger for apoptosis (Barar and Omidi, 2013). This in turn leads to an alkaline cytosolic pH and an acidic extracellular pH, both of which promote tumor growth, metastasis and chemo‐resistance (Sennoune et al., 2004; Hendrix et al., 2013). Additionally, V‐ATPases over‐expression alters the transmembrane electrochemical gradient that affects the accumulation, intracellular distribution and sensitivity of the anticancer drugs (Mahoney et al., 2003; Daniel et al., 2013). The model for the effect of V‐ATPase‐V0a2 inhibition on cisplatin resistant cells has been depicted in Figure 9.
Figure 9.
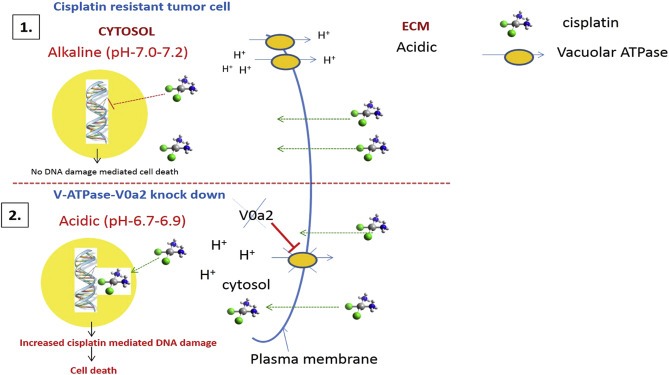
Inhibition of Vacuolar ATPase‐V0a2 in cisplatin resistant cells induces DNA damage mediated cell death through cytosolic acidification. Schematic representation of the mode of cisplatin sensitization in resistant cells upon V‐ATPase‐V0a2 inhibition. Aberrant activation of V‐ATPases in cisplatin resistant tumor cells causes de‐regulation of pH gradient across the cell membranes. (1) V0a2 isoform is expressed as part of the plasma membrane associated V‐ATPase machinery that leads to proton extrusion into extracellular environment (ECM), leading to an acidic extracellular and an alkaline cytosolic pH. The altered pH phenomenon interferes with cisplatin cytotoxicity. At an alkaline cytosolic pH, cisplatin is not effectively hydrolyzed to aqua ligand (active form) and therefore not able to impose DNA damage mediated cell death pathways. (2) Upon inhibition of V‐ATPase‐V0a2 isoform, the proton pump activity is inhibited, marked by the acidification of the cytosol. This low pH leads to effective conversion of cisplatin to aquated form, which causes increased cisplatin‐mediated DNA damage and activation of cell death pathways. Additionally, V0a2 inhibition leads to suppression of DNA damage repair pathways in cisplatin resistant cells, ultimately leading to enhanced cisplatin mediated toxicity.
Several V‐ATPase subunit genes have been shown to be upregulated in cisplatin resistant tumor cells (Torigoe et al., 2002). However, apart from the role in cisplatin resistant tumor cells, the reported V‐ATPase subunits are also involved in normal physiological functions and therefore targeting these subunits poses cytotoxicity issues. Given the major role of V‐ATPase ‘a’ subunit in pH sensing and intracellular targeting of the V‐ATPase complex (Capecci and Forgac, 2013), we focused our study on this subunit. Our results provide evidence that all Vacuolar‐ATPases V0 ‘a’ subunit isoforms (V0a1/V0a2/V0a3) are over‐expressed during cisplatin resistance in human ovarian cancer. Previous studies from our lab have established the role of V‐ATPase‐V0a2 in promoting tumor growth, migration and metastasis (Katara et al., 2015; Kulshrestha et al., 2015). V0a2 was shown to confer immune privilege to human malignant glioma (Roth et al., 2006). Further, our recent study suggested the presence of V0a2 exclusively on tumor cell surface and not on the normal ovarian epithelia. In this study, given the selective localization and over expression of V0a2 on the plasma membrane of the cisplatin resistant OVCA cells, we further explored the role of this isoform in mediating cisplatin resistance. Surface localization of V0a2 is particularly important as this suggests it to be a part of plasma membrane V‐ATPase machinery that regulates the tumor pH gradient.
Upon stable shRNA mediated V0a2 inhibition, cisplatin resistant cells exhibited increased sensitivity to platinum drugs, cisplatin and carboplatin, suggesting an enhanced platinum mediated cytotoxicity. In order to induce cytotoxicity, cisplatin is known to form double stranded DNA‐breaks by binding to the N7 atom of the guanine residues on the tumor cell DNA. This DNA platination causes recruitment of several damage recognition proteins, that culminate in an irreversible cell death programme (Chu G, 1994; Basu and Krishnamurthy, 2010). Reduced cisplatin‐DNA adduct formation and increased DNA damage repair is observed in the cisplatin resistant tumor cells (Johnson et al., 1994; Yang et al., 2000). Here, the V0a2 silenced cisplatin resistant cells exhibited a significantly enhanced DNA adduct formation that correlated with the cisplatin sensitivity. This enhanced DNA adduct mediated cytotoxicity in V0a2 silenced resistant cells suggests greater availability of active drug form. Further, a clear relationship between DNA repair and reduced cisplatin cytotoxicity is known and DNA repair has been proposed to represent a major mechanism underlying cisplatin resistance. Interestingly, we observed a suppressed DNA repair pathway in a2 inhibited cisplatin resistant cells. Since the intrastrand cross‐link is the major lesion caused by cisplatin‐induced DNA damage, it is primarily repaired via the nucleotide excision repair (NER) system in resistant cells (Welsh et al., 2004; Wu et al., 2003). Here, several NER genes such as ERCC2, ERCC5 and RAD23A were significantly downregulated, which further explains why the cisplatin mediated DNA damage is translated into cell death signal in the a2 inhibited resistant cells.
To understand the mechanism behind the increased DNA platination and subsequently enhanced cytotoxicity, we further investigated the effect of V0a2 knock down on cytosolic pH. The present study involving the inhibition of V0a2 shows to sensitize the cisplatin resistant cells by decreasing the cytosolic pH which is known to favor efficient cisplatin activation. In line with the previous studies (Huang and Huang, 2005; Lozupone et al., 2015), we found that the cytosolic pH of the cisplatin resistant OVCA cells is slightly alkaline (pH 7.1–7.3). The inhibition of V‐ATPase‐V0a2 acidified the cytosolic pH (∼pH 6.95) near to the pH in cisplatin sensitive cells. Chemical activity of cisplatin is greater at lower cytosolic pH, promoting DNA platination (Moreno et al., 1998). Plasma membrane associated V‐ATPases are the key regulators of cytoplasmic pH. Our results are in line with the previous studies that show a reduction in cytosolic pH when the function of the V‐ATPase is compromised (Moreno et al., 1998; Petrangolini et al., 2006). At lower pH, a greater proportion of hydrolyzed cisplatin has an aqua ligand [active form], rather than a hydroxo‐ligand [inactive drug] (Chau and Stewart, 1999). Further, at low cytosolic pH, DNA is more prone to damage by DNA‐damaging drugs that can be due to either an altered DNA conformation or a defective DNA damage repair mechanisms (Robinson et al., 1992; Liao et al., 2007; De Milito et al., 2010). However, previous study highlighted that the V‐ATPase mutants are more prone to DNA damage by enhanced activation of the DNA damage checkpoint and not due to defect in DNA repair (Liao et al., 2007). Recently, proton pump inhibitors (omeprazole, esomeprazole) have also been shown to normalize the pHi and pHe of human tumors, thus rendering the tumor cells sensitive to the action of several cytotoxic drugs to which they are normally refractory. When used in combination, the PPIs improve the efficacy of the anti‐cancer drugs (Luciani et al., 2004). This study further supports these previous observations. However, given the broad impact of PPIs on all cell types, our findings suggest that isoform specific inhibition of V0a2 is more selective towards tumor cells and is therefore more effective in overcoming cisplatin resistance while minimizing cytotoxicity to normal cells. It will be of interest to further elucidate whether out of cisplatin accumulation or DNA repair, which mechanism is under the direct effect of altered pH.
We further investigated the mechanism of cell death adopted by V0a2 knock down resistant cells. In V0a2 knockdown resistant cells, significantly high level of apoptosis was observed upon cisplatin treatment. This is particularly important as the cisplatin resistant cells exhibit an impaired apoptotic machinery as a mechanism of drug tolerance (Schneider et al., 2015). Additionally, since V‐ATPases regulate autophagy through endolysosomal acidification (Pamarthy et al., 2015), it will be interesting to decipher the involvement of autophagic cell death mechanism in sensitization of cisplatin treatment in resistant cells.
Upon monitoring the endosomal pH, in agreement with the previous studies (Luciani et al., 2004), we observed that cisplatin resistant cells exhibit an increased endosomal acidity compared to cisplatin sensitive counterpart. Additionally, the cisplatin resistant cells exhibited lysosomal localization toward the plasma membrane. This altered localization may help in secreting endosomal proteases that increases the metastatic potential (Liu et al., 2015) and exocytosis of the sequestered cisplatin, thus contributing to cisplatin resistance (Lozupone et al., 2015). Interestingly, though the depletion of V‐ATPase‐V0a2 in cisplatin resistant cells did not significantly change the pH of acidic vacuoles, it altered the localization of acidic vesicles from plasma membrane to the cytosol, as in case of the cisplatin sensitive cells. This altered lysosomal distribution may be one of the contributing factors towards cisplatin sensitization in these cells that may be responsible for reduced exocytosis of drug.
Our lab has generated an isoform specific monoclonal antibody (2C1) against the C‐ terminal end of plasma membrane associated V‐ATPase‐V0a2. The V‐ATPase inhibitory activity of anti‐a2V antibody has already been established. This antibody has been shown to inhibit the V‐ATPase function in macrophages (Derks and Beaman, 2004 as well as in activated T cells where neutralizing anti‐RTF antibodies induced apoptosis in activated T cells (Boomer et al., 2000). Here, we further expanded the knowledge of the inhibitory functions of anti‐a2v in relation to tumor cell chemo‐sensitivity. The advantage of isoform specific antibody for targeted V0a2 inhibition is that unlike other chemical reagents such as bafilomycin A or concanamycin A that also block the function of intracellular V‐ATPases, this antibody specifically recognizes the functional membrane bound form of V‐ATPases present on malignant cells (Kwong et al., 2011). In this study, we found that the pretreatment with 2C1 sensitized the OVCA cells to the cytotoxic effect of cisplatin in vitro. The cisplatin sensitization was more pronounced in the cisplatin resistant ovarian cancer cells than cisplatin sensitive cells indicating its potential utility in overcoming cisplatin resistance as well as increasing the treatment efficiency of the platinum based drugs and has been extensively shown to target lysosomal V‐ATPase, thereby impairing its acidification. Notably, V‐ATPase inhibitor bafilomycin that targets the ‘c’ subunit of the V‐ATPase V0 domain also showed a sensitizing effect towards cisplatin, suggesting that the role of other subunits and endosomal V‐ATPases cannot be excluded. However, we suggest that 2C1 antibody may be superior to chemical V‐ATPase inhibitors such as concanamycin A and bafilomycin A1 that block the function of intracellular V‐ATPases as this antibody specifically inhibits the functional cell membrane form of V‐ATPase present on malignant cells and therefore pose less cytotoxicity to normal cells. The treatment of OVCA cells with 2C1 is known to reduce the tumor cell migration by suppressing the MMP‐9 activity in tumor micro‐environment in vitro (Kulshrestha et al., 2015). 2C1 mediated V‐ATPase inhibition affects the surface ATPase activity in activated macrophages (Runkle et al., 2012) and has been shown to induce apoptosis in activated T cells and lymphocytes in a concentration dependent manner (Derks and Beaman, 2004). In glioma cells, the inhibition of V0a2 reduced the tumorigenicity in vivo (Chu, 1994). We therefore conclude that the cell surface form of the V‐ATPase a2 subunit is a potential target for overcoming cisplatin resistance in ovarian cancer. However, it will be of interest to explore the role of played by other up‐regulated isoforms (specifically V0a3) in chemo‐resistance.
In conclusion, we have demonstrated that the silencing of V‐ATPase‐V0a2 isoform sensitizes ovarian cancer cells to cisplatin. The sensitization is mediated by increased cytosolic acidification, culminating in enhanced cisplatin mediated DNA damage leading to apoptosis. Combinatorial treatment of V‐ATpase‐V0a2 antibody and cisplatin enhanced the cytotoxicity in cisplatin resistant cells. Our findings highlight the role of V‐ATPase‐V0a2 in mediating cisplatin resistance and a rationale for the use of V‐ATPase‐V0a2 as a promising therapeutic target for overcoming cisplatin resistance.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following are the supplementary data related to this article:
Figure S1 Characterization of the lab generated cisplatin resistant ovarian carcinoma cell lines. The comparative growth profile of lab generated acquired cisplatin resistant and cisplatin sensitive ovarian carcinoma cell lines.
{kind=link}
Figure S2 V‐ATPase‐V0a2 is expressed on tumor cells in serous ovarian cancer tissues. Immuno‐histochemical staining was performed for anti‐V‐ATPase‐V0a2 antibody and anti‐Ki67 antibody (proliferative/tumor cell marker) in serial sections of ovarian cancer tissues. Representative images from two different samples of tumor sections (A, B) are shown here. Magnification – ×400.
{kind=link}
Figure S3 Expression pattern of other V0a subunit isoforms upon V0a2 Knock down in cisplatin resistant ovarian cancer cells. The gene expression analysis revealed no significant alteration in the expression of other V‐ATPase‐V0a isoforms (V0a1/V0a3/V0a4) upon V0a2 knockdown. The fold change expression with respect to scrambled control cells was calculated using 2‐ddCt method.
{kind=link}
Figure S4 Stable knockdown of V‐ATPase‐V0a3 sensitizes the cisplatin resistant cells to platinum. V‐ATPase‐V0a3 expression was knock down in cisplatin resistant cells (A2780‐cis) by transfection with SureSilencing shRNA plasmids (Qiagen, Valencia, CA, USA). (A) Suppression of V0a3 expression was confirmed by Quantitative real time PCR showing the mRNA levels of sh‐V0a3‐cis (V0a3 knock down cells) compared to scrambled control (sh‐scr‐cis). Data are expressed as fold change compared shV0a3‐cis‐control cells. (B) Dose–response curves obtained by treating sh‐V0a3‐cis or cis‐A2780 with cisplatin. Cell death was assayed by using the MTS cell viability assay.
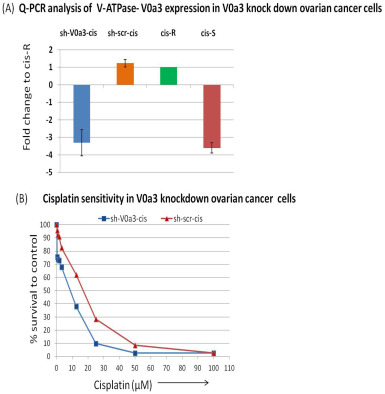{kind=link}
Figure S5 Inhibition of V‐ATPase‐V0a2 down regulates DNA repair pathway in cisplatin resistant ovarian cancer cells. mRNA levels of the DNA repair genes related to Base excision repair (BER) and nucleotide excision repair (NER) are reduced in cisplatin resistant ovarian cancer cells (A2780‐cis) following V‐ATPase‐V0a2 silencing using shRNA. GAPDH and ACT were used as endogenous control genes. Data expressed as mean ± SD of the two experiments.
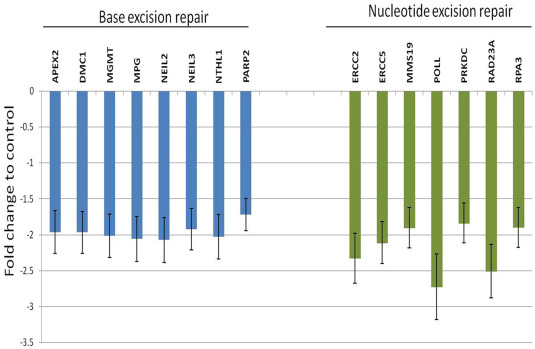{kind=link}
Figure S6 Analysis of lysosomal acidification upon V‐ATPase‐V0a2 knockdown in ovarian cancer cells. To measure any difference in acidification of the vesicles, flow cytometry analysis was performed to quantitate the green fluorescent intensity of DND‐189 stained ovarian cancer cells. The cisplatin resistant cells exhibited higher lysosomal acidification compared to sensitive counterpart. The knock down of V‐ATPase‐V0a2 however did not alter the lysosomal acidification. In contrast, chemical V‐ATPase inhibitor, bafilomycin treated cells exhibited a reduced lysosomal acidification.
{kind=link}
Figure S7 SNARF assay to determine the changes in cytosolic pH upon anti‐V‐ATPase a2v antibody (A) cisplatin resistant A2780 (cis‐A2780) cells were loaded with SNARF‐1 dye and the fluorescence spectra of SNARF‐1 was obtained on anti‐a2v antibody treated cisplatin sensitive and cisplatin resistant cells (20 ug/ml; 6 h, 37 °C, 5% CO2). Bafilomycin was used as the positive control. The corresponding intracellular pH was obtained from the pH calibration curve based on known changes imparted by buffers of different pH (4.5, 5.5, 6.5 and 7.5) in presence of nigericin. Values are means (±S.E.M) of two independent experiments performed in duplicate.
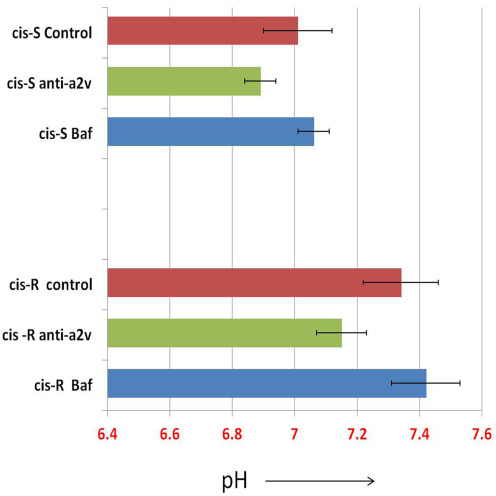{kind=link}
Acknowledgments
This work was partially supported by grants from the ALGH‐RFUMS collaborative research [Grant number: 27‐00‐612223‐111103] involving Department of Gynecologic Oncology, Advocate Lutheran General Hospital, Chicago and the grant from Clinical Immunology Laboratory, Rosalind Franklin University of Medicine and Science, North Chicago. We thank Dr. Patricia Loomis and Robert Dickinson for technical assistance and the Rosalind Franklin University of Medicine and Science Confocal microscopy, Live cell imaging and Flow cytometry core facility.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.01.003.
Kulshrestha Arpita, Katara Gajendra K., Ginter Jordyn, Pamarthy Sahithi, Ibrahim Safaa A., Jaiswal Mukesh K., Sandulescu Corina, Periakaruppan Ramayee, Dolan James, Gilman-Sachs Alice, Beaman Kenneth D., (2016), Selective inhibition of tumor cell associated Vacuolar‐ATPase ‘a2’ isoform overcomes cisplatin resistance in ovarian cancer cells, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.01.003.
References
- Altomare, D.A. , 2004. AKT and mTOR phosphorylation is frequently detected in ovarian cancer and can be targeted to disrupt ovarian tumor cell growth. Oncogene. 23, 5853–5857. [DOI] [PubMed] [Google Scholar]
- American Cancer Society Statistics for 2014. [www.cancer.org].
- Barar, J. , Omidi, Y. , 2013. Dysregulated pH in tumor microenvironment checkmates cancer therapy. Bioimpacts. 3, 149–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu, A. , Krishnamurthy, S. , 2010. Cellular responses to cisplatin-induced DNA damage. J. Nucleic Acids. 2010, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyenbach, K.W. , Wieczorek, H. , 2006. The V-type H+ ATPase: molecular structure and function, physiological roles and regulation. J. Exp. Biol. 209, 577–589. [DOI] [PubMed] [Google Scholar]
- Boomer, J.S. , Lee, G.W. , Givens, T.S. , Gilman-Sachs, A. , Beaman, K.D. , 2000. Regeneration and tolerance factor's potential role in T-cell activation and apoptosis. Hum. Immunol. 61, 959–971. [DOI] [PubMed] [Google Scholar]
- Capecci, J. , Forgac, M. , 2013. The function of vacuolar ATPase (V-ATPase) a subunit isoforms in invasiveness of MCF10a and MCF10CA1a human breast cancer cells. J. Biol. Chem. 288, 32731–32741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau, Q. , Stewart, D.J. , 1999. Cisplatin efflux, binding and intracellular pH in the HTB56 human lung adenocarcinoma cell line and the E-8/0.7 cisplatin-resistant variant. Cancer Chemother. Pharmacol. 44, 193–202. [DOI] [PubMed] [Google Scholar]
- Chu, G. , 1994. Cellular responses to cisplatin. The roles of DNA-binding proteins and DNA repair. J. Biol. Chem. 269, 787–790. [PubMed] [Google Scholar]
- Cooke, S.L. , Brenton, J.D. , 2011. Evolution of platinum resistance in high-grade serous ovarian cancer. Lancet Oncol. 12, 1169–1174. [DOI] [PubMed] [Google Scholar]
- Daniel, C. , Bell, C. , Burton, C. , Harguindey, S. , Reshkin, S.J. , Rauch, C. , 2013. The role of proton dynamics in the development and maintenance of multidrug resistance in cancer. Biochim. Biophys. Acta. 1832, 606–617. [DOI] [PubMed] [Google Scholar]
- De Milito, A. , Fais, S. , 2005. Tumor acidity, chemoresistance and proton pump inhibitors. Future Oncol. 1, 779–786. [DOI] [PubMed] [Google Scholar]
- De Milito, A. , Canese, R. , Marino, M.L. , Borghi, M. , Iero, M. , Villa, A. , 2010. pH-dependent antitumor activity of proton pump inhibitors against human melanoma is mediated by inhibition of tumor acidity. Int. J. Cancer. 127, 207–219. [DOI] [PubMed] [Google Scholar]
- Derks, R. , Beaman, K. , 2004. Regeneration and tolerance factor modulates the effect of adenosine triphosphate-induced interleukin 1 beta secretion in human macrophages. Hum. Immunol. 65, 676–682. [DOI] [PubMed] [Google Scholar]
- Eltabbakh, G.H. , Awtrey, C.S. , 2001. Current treatment for ovarian cancer. Expert Opin. Pharmacother. 2, 109–124. [DOI] [PubMed] [Google Scholar]
- Fais, S. , Venturi, G. , Gatenby, B. , 2014. Microenvironmental acidosis in carcinogenesis and metastases: new strategies in prevention and therapy. Cancer Metastasis Rev. 33, 1095–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federici, C. , Petrucci, F. , Caimi, S. , Cesolini, A. , Logozzi, M. , Borghi, M. , 2014. Exosome release and low pH belong to a framework of resistance of human melanoma cells to cisplatin. PLoS One. 9, e88193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgac, M. , 2007. Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8, 9917–9929. [DOI] [PubMed] [Google Scholar]
- Guppy, A.E. , Nathan, P.D. , Rustin, G.J.S. , 2005. Epithelial ovarian cancer: a review of current management. Clin. Oncol. 17, 399–411. [DOI] [PubMed] [Google Scholar]
- Hendrix, A. , Sormunen, R. , Westbroek, W. , Lambein, K. , Denys, H. , Sys, G. , 2013. V-ATPase expression and activity is required for Rab27B-dependent invasive growth and metastasis of breast cancer. Int. J. Cancer. 133, 843–845. [DOI] [PubMed] [Google Scholar]
- Huang, Z. , Huang, Y. , 2005. The change of intracellular pH is involved in the cisplatin-resistance of human lung adenocarcinoma A549/DDP cells. Cancer Invest. 23, 26–32. [PubMed] [Google Scholar]
- Johnson, S.W. , Swiggard, P.A. , Handel, L.M. , Brennan, J.M. , Godwin, A.K. , Ozols, R.F. , 1994. Relationship between platinum-DNA adduct formation and removal and cisplatin cytotoxicity in cisplatin-sensitive and -resistant human ovarian cancer cells. Cancer Res. 54, 5911–5916. [PubMed] [Google Scholar]
- Kartalou, M. , Essigmann, J.M. , 2001. Mechanisms of resistance to cisplatin. Mutat. Res. 478, 23–43. [DOI] [PubMed] [Google Scholar]
- Katara, G.K. , Jaiswal, M.K. , Kulshrestha, A. , Kolli, B. , Gilman-Sachs, A. , Beaman, K.D. , 2014. Tumor-associated vacuolar ATPase subunit promotes tumorigenic characteristics in macrophages. Oncogene. 33, 5649–5654. [DOI] [PubMed] [Google Scholar]
- Katara, G.K. , Kulshrestha, A. , Jaiswal, M.K. , Pamarthy, S. , Gilman-Sachs, A. , Beaman, K.D. , 2015. Inhibition of vacuolar ATPase subunit in tumor cells delays tumor growth by decreasing the essential macrophage population in the tumor microenvironment. Oncogene. 10.1038/onc.2015.159 (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- Kulshresth, A. , Katara, G.K. , Ibrahim, S. , Pamarthy, S. , Jaiswal, M.K. , Gilman Sachs, A. , Beaman, K.D. , 2015. Vacuolar ATPase ‘a2’ isoform exhibits distinct cell surface accumulation and modulates matrix metalloproteinase activity in ovarian cancer. Oncotarget. 6, 3797–3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong, C. , Gilman-Sachs, A. , Beaman, K. , 2011. Tumor-associated a2 vacuolar ATPase acts as a key mediator of cancer-related inflammation by inducing pro-tumorigenic properties in monocytes. J. Immunol. 186, 1781–1789. [DOI] [PubMed] [Google Scholar]
- Liao, C. , Hu, B. , Arno, M.J. , Panaretou, B. , 2007. Genomic screening in vivo reveals the role played by vacuolar H+ ATPase and cytosolic acidification in sensitivity to DNA-damaging agents such as cisplatin. Mol. Pharmacol. 71, 416–425. [DOI] [PubMed] [Google Scholar]
- Liu, Z. , Liu, J. , Li, L. , Nie, D. , Tao, Q. , Wu, J. , 2015. Inhibition of autophagy potentiated the antitumor effect of nedaplatin in cisplatin-resistant nasopharyngeal carcinoma cells. PLoS One. 10, e0135236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone, F. , Borghi, M. , Marzoli, F. , Azzarito, T. , Matarrese, P. , Iessi, E. , 2015. TM9SF4 is a novel V-ATPase-interacting protein that modulates tumor pH alterations associated with drug resistance and invasiveness of colon cancer cells. Oncogene. 34, 5163–5174. [DOI] [PubMed] [Google Scholar]
- Lu, X. , Qin, W. , Li, J. , Tan, N. , Pan, D. , Zhang, H. , 2005. The growth and metastasis of human hepatocellular carcinoma xenografts are inhibited by small interfering RNA targeting to the subunit ATP6L of proton pump. Cancer Res. 65, 6843–6849. [DOI] [PubMed] [Google Scholar]
- Luciani, F. , Spada, M. , De Milito, A. , Molinari, A. , Rivoltini, L. , Montinaro, A. , 2004. Effect of proton pump inhibitor pretreatment on resistance of solid tumors to cytotoxic drugs. J. Natl. Cancer Inst. 96, 1702–1713. [DOI] [PubMed] [Google Scholar]
- Lucien, F. , Harper, K. , Pelletier, P.P. , Volkov, L. , Dubois, C.M. , 2014. Simultaneous pH measurement in endocytic and cytosolic compartments in living cells using confocal microscopy. J. Vis. Exp. e51395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundholm, L. , Haag, P. , Zong, D. , Juntti, T. , Mörk, B. , Lewensohn, R. , 2013. Resistance to DNA-damaging treatment in non-small cell lung cancer tumor-initiating cells involves reduced DNA-PK/ATM activation and diminished cell cycle arrest. Cell Death Dis. 4, e478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahoney, B.P. , Raghunand, N. , Baggett, B. , Gillies, R.J. , 2003. Tumor acidity, ion trapping and chemotherapeutics. I. Acid pH affects the distribution of chemotherapeutic agents in vitro. Biochem. Pharmacol. 66, 1207–1218. [DOI] [PubMed] [Google Scholar]
- Moreno, S.N. , Zhong, L. , Lu, H.G. , Souza, W.D. , Benchimol, M. , 1998. Vacuolar-type H+-ATPase regulates cytoplasmic pH in Toxoplasma gondii tachyzoites. Biochem. J. 330, 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Smith, M. , Halder, J.B. , Meltzer, P.S. , Gonda, T.A. , Mangala, L.S. , Rupaimoole, R. , 2013. ATP 11B mediates platinum resistance in ovarain cancer. J. Clin. Invest. 123, 2119–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami, T. , Shibuya, I. , Ise, T. , Chen, Z.S. , Akiyama, S. , Nakagawa, M. , 2001. Elevated expression of vacuolar proton pump genes and cellular pH in cisplatin resistance. Int. J. Cancer. 93, 869–874. [DOI] [PubMed] [Google Scholar]
- Nishi, T. , Forgac, M. , 2002. The vacuolar (H+)-ATPases- nature's most versatile proton pumps. Nat. Rev. Mol. Cell Biol. 3, 94–103. [DOI] [PubMed] [Google Scholar]
- Nishisho, T. , Hata, K. , Nakanishi, M. , Morita, Y. , Sun-Wada, G.H. , Wada, Y. , 2011. The a3 isoform vacuolar type H+-ATPase promotes distant metastasis in the mouse B16 melanoma cells. Mol. Cancer Res. 9, 845–855. [DOI] [PubMed] [Google Scholar]
- Ntrivalas, E. , Gilman-Sachs, A. , Kwak-Kim, J. , Beaman, K. , 2007. The N-terminus domain of the a2 isoform of vacuolar ATPase can regulate interleukin-1beta production from mononuclear cells in co-culture with JEG-3 choriocarcinoma cells. Am. J. Reprod. Immunol. 57, 201–209. [DOI] [PubMed] [Google Scholar]
- Ntrivalas, E. , Derks, R. , Gilman-Sachs, A. , Kwak-Kim, J. , Levine, R. , Beaman, K. , 2007. Novel role for the N-terminus domain of the a2 isoform of vacuolar ATPase in interleukin-1beta production. Hum. Immunol. 68, 469–477. [DOI] [PubMed] [Google Scholar]
- Pamarthy, S. , Jaiswal, M.K. , Kulshreshtha, A. , Katara, G.K. , Gilman-Sachs, A. , Beaman, K.D. , 2015. The vacuolar ATPase a2-subunit regulates notch signaling in triple-negative breast cancer cells. Oncotarget. 6, 34206–34220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perego, P. , 1996. Association between cisplatin resistance and P53 gene. Cancer Res. 56, 556–562. [PubMed] [Google Scholar]
- Petrangolini, G. , Supino, R. , Pratesi, G. , Dal Bo, L. , Tortoreto, M. , Croce, A.C. , Misiano, P. , Belfiore, P. , Farina, C. , Zunino, F. , 2006. Effect of novel vacular-H+-ATPase inhibitor on cell and tumor response to camptothecins. J. Pharmacol. Exp. Ther. 318, 939–946. [DOI] [PubMed] [Google Scholar]
- Recchi, C. , Chavrier, P. , 2006. V-ATPase: a potential pH sensor. Nat. Cell Biol. 8, 107–109. [DOI] [PubMed] [Google Scholar]
- Robinson, H. , van der Marel, G.A. , van Boom, J.H. , Wang, A.H. , 1992. Unusual DNA conformation at low pH revealed by NMR: parallel-stranded DNA duplex with homo base pairs. Biochemistry. 31, 10510–10517. [DOI] [PubMed] [Google Scholar]
- Roth, P. , Aulwurm, S. , Gekel, I. , Beier, D. , Sperry, R.G. , Mittelbronn, M. , 2006. Regeneration and tolerance factor: a novel mediator of glioblastoma-associated immunosuppression. Cancer Res. 66, 3852–3858. [DOI] [PubMed] [Google Scholar]
- Runkle, K.B. , Meyerkord, C.L. , Desai, N.V. , Takahashi, Y. , Wang, H.G. , 2012. Bif-1 suppresses breast cancer cell migration by promoting EGFR endocytic degradation. Cancer Biol. Ther. 13, 956–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasazawa, Y. , Futamura, Y. , Tashiro, E. , Imoto, M. , 2009. Vacuolar H+-ATPase inhibitors overcome Bcl-xL-mediated chemoresistance through restoration of a caspase-independent apoptotic pathway. Cancer Sci. 100, 1460–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider, L.S. , von Schwarzenberg, K. , Lehr, T. , Ulrich, M. , Kubisch-Dohmen, R. , Liebl, J. , Trauner, D. , 2015. Vacuolar-ATPase inhibition blocks iron metabolism to mediate therapeutic effects in breast Cancer. Cancer Res. 75, 2863–2874. [DOI] [PubMed] [Google Scholar]
- Sennoune, S.R. , Bakunts, K. , Martínez, G.M. , Chua-Tuan, J.L. , Kebir, Y. , Attaya, M.N. , 2004. Vacuolar H+-ATPase in human breast cancer cells with distinct metastatic potential: distribution and functional activity. Am. J. Physiol. Cell Physiol. 286, C1443–C1452. [DOI] [PubMed] [Google Scholar]
- Spugnini, E.P. , Citro, G. , Fais, S. , 2010. Proton pump inhibitors as anti vacuolar-ATPases drugs: a novel anticancer strategy. J. Exp. Clin. Cancer Res. 29, 44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torigoe, T. , Izumi, H. , Ishiguchi, H. , Uramoto, H. , Murakami, T. , Ise, T. , 2002. Enhanced expression of the human vacuolar H+ −ATPase c subunit gene (ATP6L) in response to anticancer agents. J. Biol. Chem. 277, 36534–36543. [DOI] [PubMed] [Google Scholar]
- Tummala, M.K. , 2005. Recurrent ovarian cancer. Clin. Adv. Hematol. Oncol. 3, 723–736. [PubMed] [Google Scholar]
- Welsh, C. , Day, R. , McGurk, C. , Masters, J.R.W. , Wood, R.D. , Köberle, B. , 2004. Reduced levels of XPA, ERCC1 and XPF DNA repair proteins in testis tumor cell lines. Int. J. Cancer. 110, 352–361. [DOI] [PubMed] [Google Scholar]
- Wu, X. , Fan, W. , Xu, S. , Zhou, Y. , 2003. Sensitization to the cytotoxicity of cisplatin by transfection with nucleotide excision repair gene xeroderma pigmentosun group A antisense RNA in human lung adenocarcinoma cells. Clin. Cancer Res. 9, 5874–5879. [PubMed] [Google Scholar]
- Yang, Z. , Faustino, P.J. , Andrews, P.A. , Monastra, R. , Rasmussen, A.A. , Ellison, C.D. , 2000. Decreased cisplatin/DNA adduct formation is associated with cisplatin resistance in human head and neck cancer cell lines. Cancer Chemother. Pharmacol. 46, 255–262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Figure S1 Characterization of the lab generated cisplatin resistant ovarian carcinoma cell lines. The comparative growth profile of lab generated acquired cisplatin resistant and cisplatin sensitive ovarian carcinoma cell lines.
Figure S2 V‐ATPase‐V0a2 is expressed on tumor cells in serous ovarian cancer tissues. Immuno‐histochemical staining was performed for anti‐V‐ATPase‐V0a2 antibody and anti‐Ki67 antibody (proliferative/tumor cell marker) in serial sections of ovarian cancer tissues. Representative images from two different samples of tumor sections (A, B) are shown here. Magnification – ×400.
Figure S3 Expression pattern of other V0a subunit isoforms upon V0a2 Knock down in cisplatin resistant ovarian cancer cells. The gene expression analysis revealed no significant alteration in the expression of other V‐ATPase‐V0a isoforms (V0a1/V0a3/V0a4) upon V0a2 knockdown. The fold change expression with respect to scrambled control cells was calculated using 2‐ddCt method.
Figure S4 Stable knockdown of V‐ATPase‐V0a3 sensitizes the cisplatin resistant cells to platinum. V‐ATPase‐V0a3 expression was knock down in cisplatin resistant cells (A2780‐cis) by transfection with SureSilencing shRNA plasmids (Qiagen, Valencia, CA, USA). (A) Suppression of V0a3 expression was confirmed by Quantitative real time PCR showing the mRNA levels of sh‐V0a3‐cis (V0a3 knock down cells) compared to scrambled control (sh‐scr‐cis). Data are expressed as fold change compared shV0a3‐cis‐control cells. (B) Dose–response curves obtained by treating sh‐V0a3‐cis or cis‐A2780 with cisplatin. Cell death was assayed by using the MTS cell viability assay.
Figure S5 Inhibition of V‐ATPase‐V0a2 down regulates DNA repair pathway in cisplatin resistant ovarian cancer cells. mRNA levels of the DNA repair genes related to Base excision repair (BER) and nucleotide excision repair (NER) are reduced in cisplatin resistant ovarian cancer cells (A2780‐cis) following V‐ATPase‐V0a2 silencing using shRNA. GAPDH and ACT were used as endogenous control genes. Data expressed as mean ± SD of the two experiments.
Figure S6 Analysis of lysosomal acidification upon V‐ATPase‐V0a2 knockdown in ovarian cancer cells. To measure any difference in acidification of the vesicles, flow cytometry analysis was performed to quantitate the green fluorescent intensity of DND‐189 stained ovarian cancer cells. The cisplatin resistant cells exhibited higher lysosomal acidification compared to sensitive counterpart. The knock down of V‐ATPase‐V0a2 however did not alter the lysosomal acidification. In contrast, chemical V‐ATPase inhibitor, bafilomycin treated cells exhibited a reduced lysosomal acidification.
Figure S7 SNARF assay to determine the changes in cytosolic pH upon anti‐V‐ATPase a2v antibody (A) cisplatin resistant A2780 (cis‐A2780) cells were loaded with SNARF‐1 dye and the fluorescence spectra of SNARF‐1 was obtained on anti‐a2v antibody treated cisplatin sensitive and cisplatin resistant cells (20 ug/ml; 6 h, 37 °C, 5% CO2). Bafilomycin was used as the positive control. The corresponding intracellular pH was obtained from the pH calibration curve based on known changes imparted by buffers of different pH (4.5, 5.5, 6.5 and 7.5) in presence of nigericin. Values are means (±S.E.M) of two independent experiments performed in duplicate.