

Abstract
The kinase receptor encoded by the Met oncogene is a sensible target for cancer therapy. The chimeric monovalent Fab fragment of the DN30 monoclonal antibody (MvDN30) has an odd mechanism of action, based on cell surface removal of Met via activation of specific plasma membrane proteases. However, the short half‐life of the Fab, due to its low molecular weight, is a severe limitation for the deployment in therapy. This issue was addressed by increasing the Fab molecular weight above the glomerular filtration threshold through the duplication of the constant domains, in tandem (DCD‐1) or reciprocally swapped (DCD‐2). The two newly engineered molecules showed biochemical properties comparable to the original MvDN30 in vitro, acting as full Met antagonists, impairing Met phosphorylation and activation of downstream signaling pathways. As a consequence, Met‐mediated biological responses were inhibited, including anchorage‐dependent and ‐independent cell growth. In vivo DCD‐1 and DCD‐2 showed a pharmacokinetic profile significantly improved over the original MvDN30, doubling the circulating half‐life and reducing the clearance. In pre‐clinical models of cancer, generated by injection of tumor cells or implant of patient‐derived samples, systemic administration of the engineered molecules inhibited the growth of Met‐addicted tumors.
Keywords: Cancer targeted therapy, Met, Antibody, Fab, Half‐life, Protein engineering
Highlights
A new strategy to improve Fab pharmacokinetic has been conceived and developed.
The strategy is based on duplication of the constant domains within the Fab.
The strategy is potentially applicable to other Fab fragments.
The strategy increases the therapeutic potency of the Fab of origin.
The newly generated molecules are effective in preclinical models of cancer.
Abbreviations
- HGF
hepatocyte growth factor
- HGFR
hepatocyte growth factor receptor
- EGFR
epidermal growth factor receptor
- mAb
monoclonal antibody
- MvDN30
monovalent chimerized DN30 Fab
- NSCLC
non-small cell lung cancer
- CRC
colo rectal cancer
- PEG
poly ethylene glycol
- SEM
standard error of the mean
1. Introduction
The Hepatocyte Growth Factor Receptor encoded by the Met oncogene (HGFR/Met) is emerging as a cancer actionable target. During embryogenesis and tissue regeneration, the HGF‐Met axis controls the ‘invasive growth’, a complex network of cellular responses including proliferation, motility, invasion and protection from apoptosis (Trusolino and Comoglio, 2002; Birchmeier et al., 2003). The same program is harnessed by transformed cells to override barriers limiting tumor expansion and spreading (Trusolino et al., 2010; Skead and Govender, 2015). In most instances, Met activation, by overexpression and/or ligand activation, contributes to tumor progression, fostering an adaptive response to unfavorable micro environmental conditions (oncogene ‘expedience’) (Comoglio et al., 2008). In a limited number of cases (2–3%), a Met genetic lesion (mutation or amplification) directly drives transformation and the cancer cell relies on the oncogene for the long term maintenance of the malignant phenotype (oncogenic addiction) (Smolen et al., 2006; Lutterbach et al., 2007). Met oncogene overexpression is observed in many types of carcinomas and is strongly associated with poor prognosis (Blumenschein et al., 2012; Gao et al., 2015). An autocrine loop is more typical of non‐epithelial tumors such as osteosarcomas (Ferracini et al., 1995), rhabdomyosarcomas (Ferracini et al., 1996), glioblastomas (Moriyama et al., 1999), multiple myelomas (Börset et al., 1996), acute myeloid leukemias (Kentsis et al., 2012) and mesotheliomas (Mukohara et al., 2005). Point mutations were observed initially in sporadic and hereditary papillary renal carcinomas (Schmidt et al., 1997). High frequency of Met mutations has been scored in metastatic diseases (Lorenzato et al., 2002; Stella et al., 2011). Met gene amplification has been observed in cases of primary glioblastoma (Chi et al., 2012), gastric‐esophageal (Catenacci et al., 2011; Lennerz et al., 2011) and lung carcinomas (Ou et al., 2011). Moreover, Met amplification represents a mechanism responsible for resistance to Epithelial Growth Factor Receptor (EGFR) targeted therapies in Non‐Small Cell Lung Cancer (NSCLC) (Bean et al., 2007; Engelman et al., 2007) and Colo‐Rectal Cancer (CRC) patients (Bardelli et al., 2013).
A number of inhibitors blocking the HGF‐Met axis through a variety of mechanisms ‐ small molecule Met kinase inhibitors, monoclonal antibodies against HGF or Met, ligand competitors, receptor decoys and anticalins – are currently available (Skead and Govender, 2015). Among them, the monoclonal antibody DN30 displays an odd mechanism of action, enhancing the physiological rate of Met shedding (Petrelli et al., 2006). This leads concomitantly to reduction of the Met molecules exposed at the cell surface and to release, in the extracellular environment, of the entire N‐terminal Met domain (Lefebvre et al., 2012), which competes with the full size receptor dimerization (Michieli et al., 2004). As a consequence, Met‐mediated biological responses are strongly impaired. In its native bivalent form, the DN30 antibody displays a paradoxical Met agonist activity, resulting in a week activation of the invasive growth phenotype (Prat et al., 1998). Conversion to a monovalent form (Fab fragment – MvDN30) translates the antibody into a full antagonist, with potent inhibitory features, counteracting both the HGF‐dependent and ‐independent Met activation (Pacchiana et al., 2010; Vigna et al., 2015).
MvDN30‐Fab is thus an attractive drug for clinical applications, but the short plasma half‐life – typical of Fab fragments and mostly due to renal clearance – severely limits the clinical application. A rational approach to address this issue is to increase the molecular weight of the therapeutic protein above the threshold of the glomerular filtration. To this end we duplicated the constant domains of light and heavy chains. This new class of engineered proteins, named Dual Constant Domain‐Fab (DCD), was challenged against Met‐addicted cancer cells.
2. Material and methods
2.1. Construction, expression and purification of DCD molecules
Molecules have been designed by duplication of the human constant regions on the cDNA sequence of the MvDN30. cDNA synthesis, protein expression in mammalian HEK293E cells and subsequent Strep‐Tactin purification of MvDN30, DCD‐1, DCD‐2 and chimeric DN30 mAb were performed by U‐Protein Express BV (Utrecht, The Netherlands). Analysis of the purified molecules was performed by SDS‐PAGE of 200 ng of DCD‐1, DCD‐2 and MvDN30 as control under reducing and non‐reducing conditions, followed by GelCode Blue Stain reagent (Pierce, Waltham, MA).
2.2. ELISA assays
For affinity determinations, pure recombinant human Met‐Fc chimera (R&D Systems, Minneapolis, MN) (100 ng/well) was in solid phase and pure MvDN30, DCD‐1 or DCD‐2 (range of dilutions: 0–36 nM) were in liquid phase; for quantification of the molecules in the mice serum, collected samples (range of dilutions: 0–160000) were in liquid phase. Revealing was done with HRP‐conjugated anti‐strepTAG II antibody (IBA, Goettingen, Germany). Colorimetric assay was quantified by the multi‐label reader VICTOR X4 (Perkin Elmer Instrument INC., Whaltman, MA).
2.3. Cell culture
A549 human lung carcinoma cells, SNU‐5 human gastric carcinoma cells and HPAF‐II human pancreatic adenocarcinoma cells were obtained from ATCC/LGC Standards S.r.l. (Sesto San Giovanni, Italy). MKN‐45 human gastric carcinoma cells were obtained from the Japanese Collection of Research Bioresources (Osaka, Japan). GTL‐16 were derived from MKN‐45 cells (Giordano et al., 1988). Cells were maintained in RPMI medium (Sigma Life Science, St Louis, MO) as described (Pacchiana et al., 2010).
M162 colon cancer cells were derived from tumor material of a patient resistant to EGFR targeted therapy (Bardelli et al., 2013) propagated (one step) in mice. Tumor extracted from the animal was chopped, washed by PBS and centrifuged. Pellet was re‐suspended in Leibovitz's L‐15 medium (Gibco® Life technologies Italia, Monza, Italy) plus 2 mg/ml Collagenase I (Sigma Life Sciences) incubated for 1 h at 37 °C on shake. After a passage on 70 micron filter, tumor cells were subjected to Histopaque (Sigma Life Science) and collected from the mononucleated fraction. The obtained cells were seeded and cultured as monolayer in DMEM‐F12 medium with 20% FBS added of 1% N‐2 supplement (Thermo Fisher Scientific, Whaltman, MA) and 10 μM ROCK1 inhibitor Y‐27632‐2HCl (Selleckchem.com, Munich, Germany).
2.4. Met shedding analysis
Sub‐confluent A549 and GTL‐16 monolayers were incubated in serum‐free medium with increasing concentrations of MvDN30, DCD‐1 or DCD‐2 (range 62.5–500 nM). After 48 h (A549) or 18 h (GTL‐16), conditioned medium was collected and cells were lysed with Laemmli Buffer. Cell extracts and cell culture supernatants were resolved by SDS‐PAGE and analyzed by Western blotting.
2.5. Met activation analysis
For the analysis of the agonistic activity, sub‐confluent A549 monolayers were serum starved for 48 h. Then cells were stimulated for 15 min at 37 °C in serum‐free medium with two different concentrations of MvDN30, DCD‐1 and DCD‐2 (500 and 1000 nM). As positive control cells were also stimulated with 500 nM of DN30 mAb or 100 ng/ml (1.1 nM) of recombinant HGF (R&D Systems). Cells were lysed as described (Vigna et al., 2015). Total cell lysates were analyzed by Western blot.
For the analysis of the inhibitory activity, sub‐confluent A549 monolayers were incubated in serum‐free medium with a fixed concentration (1000 nM) of MvDN30, DCD‐1 or DCD‐2. After 24 h cells were stimulated with 100 ng/ml of HGF for 10 min at 37 °C and subsequently lysed and analyzed as above. For GTL‐16, sub‐confluent monolayers were incubated with the different tested molecules (500 nM) for 30 h, then lysed and analyzed as above.
2.6. Western blot analysis
Primary antibodies for Western blot detection were: anti‐human Met 3D4 mAb recognizing Met β chain intracellular domain (Invitrogen Corporation, Camarillo, CA), anti‐human Met DL21 mAb recognizing Met β chain extracellular domain (Vigna et al., 2008), anti‐pMet Tyr1234/1235 (D26), anti‐pAKT Ser473, anti‐AKT, anti‐pERK1/2 Thr202/Tyr204, anti‐ERK1/2 polyclonal Abs (all from Cell Signaling Technology, Beverly, MA); anti‐actin (I‐19) (Santa Cruz Biotechnology Inc., Heidelberg, Germany); anti‐Vinculin clone hVIN‐1 (Sigma Life Sciences). Secondary anti‐mouse IgG, anti‐rabbit IgG and anti‐goat IgG were from GE Healthcare (Freiburg, Germany).
2.7. In vitro biological assays
For scatter assay, HPAF‐II cells were seeded in a 96 well plate (8000 cells/well). After 24 h cells were stimulated with HGF (5 ng/ml) or treated with MvDN30, DCD‐1, DCD‐2, chimeric DN30 mAb (all molecules 200 nM) and stained with crystal violet (Sigma–Aldrich, St Louis, MO) 20 h later. Cell scattering was determined by microscopy.
For anchorage‐dependent cell growth, cells were seeded as described (Pacchiana et al., 2010). After 24 h, medium was replaced with a fresh one with 5% FBS plus the molecules to be tested (increasing concentrations – from 0.0032 to 10 μM – for Met‐addicted cell line analysis or a fixed dose −2 μM‐ for the M162 cells). Cell viability was evaluated after 72 h using the CellTiter‐Glo luminescent cell viability assay (Promega Corp., Madison, WI), according to the manufacturer's instructions. Chemiluminescence was detected with VICTOR X4.
For anchorage‐independent cell growth, cells (500 cells/well) were seeded in a 48 well plate in semisolid medium as described (Pacchiana et al., 2010). GTL‐16 were treated with two different concentrations of the tested molecules (1.5 and 0.75 μM), while A549 were treated with a single concentration of the tested molecules (1.5 μM), in absence or presence of HGF (50 ng/ml). Fresh medium was replaced every 3 days. Grown colonies were stained by tetrazolium salts (Sigma–Aldrich) after 11 days (GTL‐16) or 21 days (A549) of culture. Growth was evaluated analyzing the images from each sample with Metamorph software (Molecular Devices, Sunnyvale, CA).
2.8. Pharmacokinetic analysis
All animal procedures were performed according to protocols approved by Ethical Committee for animal experimentation of Fondazione Piemontese per la Ricerca sul Cancro and by Italian Ministry of Health. Adult immunodeficient NOD‐SCID mice (body weight between 18 and 22 gr, on average 20 gr) were injected intraperitoneally or intravenously (via tail vein) with 100 μg (5 mg/kg) of DCD‐1, DCD‐2, MvDN30 or chimeric DN30 mAb (the latter only via tail vein). Peripheral blood was collected at different time points. The amounts of the different molecules in the serum were evaluated by ELISA as described above. Concentrations of DN30‐derived molecules were obtained interpolating the absorbance values of the samples on the linear part of a standard curve obtained by serial dilutions of the different purified molecules. Each time point was the average value of at least 3 mice. Pharmacokinetic parameters were calculated analyzing the concentration values with the software Kinetica 4.1.1 (InnaPhase Corp, Philadelphia, PA).
2.9. Tumorigenesis assays
6 week‐old female NOD/SCID mice were intraperitoneally injected with luciferase‐expressing GTL‐16 cells (2 × 106 cells/mouse). Expression of the luciferase gene has been obtained by transduction with 100 ng/ml p24 of lentiviral vectors carrying the luciferase gene under the control of the CMV promoter as described (Amendola et al., 2005; Vigna et al., 2008). Mice were immediately randomized in four groups (n = 6) and treated twice a week from day 0 to the end of the experiment (day 24) with intravenous administrations of MvDN30, DCD‐1, DCD‐2 (8 nmol/mouse) or PBS (vehicle). At the end of the experiment, mice received XenoLight D‐Luciferin (PerkinElmer) (3 mg/mouse) by intraperitoneal injection. The luminescent signal was revealed and quantified in each single animal and in the omentum collected upon sacrifice.
M162 ‘xenopatient’ tumor samples were implanted in the right flank of 6 week‐old female NOD/SCID mice. Mice were immediately randomized in 3 groups (n = 7 Control and DCD‐2; n = 5 DCD‐1) and treated twice a week from day 0 to the end of the experiment (day 39) with intraperitoneal injections of DCD‐1 and DCD‐2 (7 nmol/mouse) or PBS (vehicle). Tumor size was evaluated periodically with a caliper; mice were considered tumor positive when masses reached a volume of 20 mm3.
2.10. Statistical analysis
To calculate Kd and Bmax, data from ELISA assays were analyzed and fitted according to nonlinear regression, one site binding (hyperbola) curve, using GraphPad Prism software (San Diego, CA). Data from growth assays were analyzed and fitted according to a nonlinear regression, sigmoidal dose response curve, using GraphPad Prism software. Averages, standard errors of the mean (SEM) and p values obtained by Student's t Test and by two‐way ANOVA were calculated using GraphPad Prism software.
3. Results
3.1. Design, synthesis and purification of the Dual Constant Domain Fab
To generate engineered molecules derived from the chimeric MvDN30, the constant domains in the light and heavy chains were duplicated (Dual Constant Domain‐MvDN30, DCD). The predicted molecular weight is 75 kDa, which is above the threshold of glomerular filtration. Two different molecules were engineered: (i) DCD‐1, built by duplication in tandem of the human constant domains, generating a VH‐CH1‐CH1 heavy chain and a VL‐CL‐CL light chain; (ii) DCD‐2, engineered by reciprocal swap of the terminal domains, generating a VH‐CH1‐CL heavy chain and a VL‐CL‐CH1 light chain (Figure 1A). The purified recombinant proteins, analyzed under reducing conditions, showed the expected molecular weight (i.e. two bands corresponding to the Fab light and heavy chains with the added sequences), while under non‐reducing conditions, DCD‐1 formed dimers and oligomers and DCD‐2 preferentially generated oligomers, as probably the swap between the terminal constant domains forced the joint between multiple chains (Figure 1B and C). Oligomerization results from inter‐molecule disulfide bonds between the cysteine residues of the heavy and light constant domains (data not shown).
Figure 1.
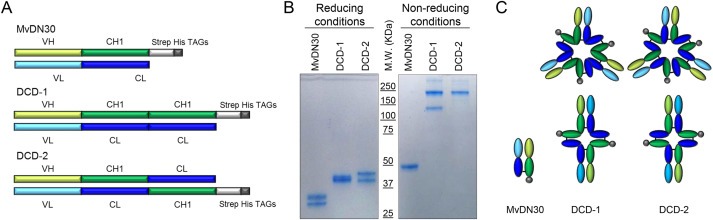
DCDs appear associated by disulfide bonds in dimers and oligomers. A. Schematic representation of MvDN30 and of the Dual Constant Domain molecules (DCD‐1 and DCD‐2). VH: variable domain derived from DN30 heavy chain. CH1: first constant domain derived from human IgG1 heavy chain. Strep His TAGs: sequences included for detection and purification of the proteins. VL: variable domain derived from DN30 light chain. CL: constant domain derived from human Igk light chain. B. SDS‐PAGE in polyacrylamide gel under reducing and non‐reducing conditions, followed by staining with GelCode Blue Stain reagent. C. Schematization of the hypothesized structures of the molecules.
3.2. DCD‐1 and DCD‐2 bind Met with high affinity, inducing Met shedding
Purified DCD‐1, DCD‐2 and MvDN30 as a control, analyzed by ELISA, bound Met with similar high affinity (Figure 2A). The maximal saturation values were higher for both DCDs versus the MvDN30, as expected by the conformation of the former, including more than one Strep‐TAG epitope per molecule (cfr Figure 1C). Upon binding to Met, both DCDs efficiently induced Met shedding in human cancer cells of different origin (A549 lung and GTL‐16 gastric carcinoma cells). As for the parental MvDN30, DCD binding to the surface resulted in decrease of Met levels in the cell and in release of soluble Met ectodomain in the extracellular space, accordingly to the antibody‐derivative given dose (Figure 2B).
Figure 2.
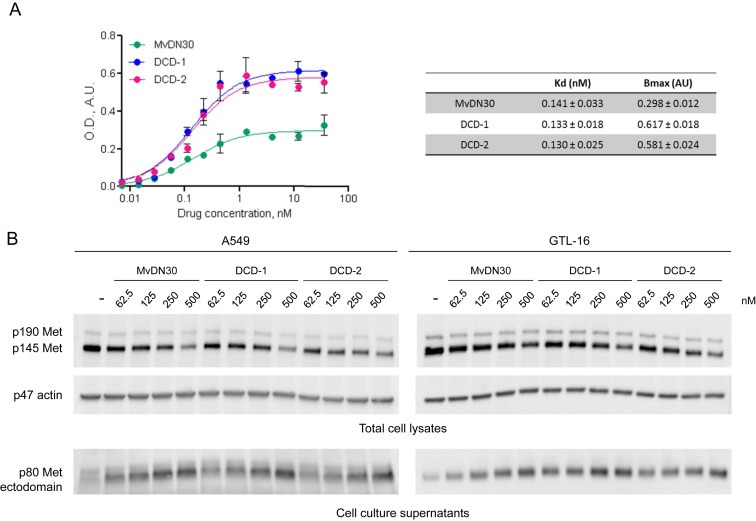
DCDs maintain high binding affinity to Met and efficient induction of receptor shedding. A. ELISA binding analysis of Met‐Fc chimera (solid phase) to the different DN30‐derived molecules (liquid phase). O.D.: Optical Density at 450 nm; A.U.: Arbitrary Unit. Each point is the mean of triplicate values. Bars represent SEM. Values of Affinity (Kd) and Maximal Binding (Bmax) are reported in the table. B. A549 (left panels) or GTL‐16 (right panels) cells were incubated with increasing concentrations of the indicated molecules for 48 h (A549) or 18 h (GTL‐16). Total Met levels in the cell lysates (upper panels) and in the cell culture supernatants (lower panels) were determined by Western blot using anti‐Met antibodies directed against epitopes located respectively at the c‐terminal tail or within the extracellular domain of the β chain. To normalize protein loading, the same filter was re‐probed with anti‐actin antibodies. p190 Met: unprocessed Met receptor; p145 Met: Met receptor β chain; p80 Met: Met extracellular domain. p47 actin: actin. Data reported in the figure are representative of at least three experiments done.
3.3. DCD‐1 and DCD‐2 act as pure Met antagonists
To assess if DCDs are endowed with residual agonist properties towards Met, A549 carcinoma cells, expressing Met receptors prone to activation by HGF or ligand‐mimetic molecules, were stimulated by increasing amounts of DCD‐1 or DCD‐2. HGF, DN30 bivalent mAb or monovalent MvDN30 were included as positive or negative controls. DCD‐1 and DCD‐2 retained a minimal residual agonist activity on Met phosphorylation, negligible compared to HGF or DN30 mAb. Activation of downstream signal transducers did not occur (Figure 3A). Accordingly, both molecules did not evoke biological responses, as assessed by analysis of the highly sensitive scatter assay (Figure 3B). On the other hand, a strong DCD inhibitory activity was measured in A549 stimulated with HGF and in GTL‐16 cells, featuring constitutively active Met. In both systems, DCD‐1 and DCD‐2 efficiently impaired the level of Met phosphorylation, resulting in inhibition of downstream activation of AKT and ERK (Figure 4A, B).
Figure 3.
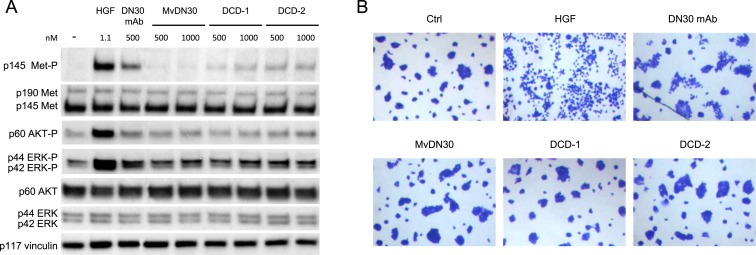
DCDs do not show Met agonistic properties A. A549 cells stimulated with increasing concentrations of MvDN30, DCD‐1 or DCD‐2 and analyzed by Western blot for Met, AKT and ERK activation. As positive control, cells were also stimulated with the indicated concentrations of pure HGF or chimeric DN30 mAb. To normalize protein loading, the same filter was re‐probed with anti‐vinculin antibodies. Data reported in the figure are representative of at least three experiments done. B. HPAF‐II cells treated with MvDN30, DCD‐1, DCD‐2 (200 nM), HGF (5 ng/ml) or chimeric DN30 mAb (200 nM) for 18 h. Pictures are representative of the cell phenotype: control cells grew in compact islands, while HGF‐treated cells appeared as single cells separated from each other (scattered). Some scattered islands were found in the chimeric DN30 mAb treated cells. Stimulation with MvDN30, DCD‐1 or DCD‐2 did not induce cell motility, appearing indistinguishable from controls. Pictures are representative of two experiments done.
Figure 4.

DCDs inhibit Met activation. A. A549 cells treated for 24 h with the indicated amount of MvDN30, DCD‐1 or DCD‐2 and then stimulated with HGF (100 ng/ml) for 15 min. B. GTL‐16 cells treated for 30 h with the indicated amount of the same above molecules. Activation of Met, AKT and ERK activation was analyzed for both cell lines. To normalize protein loading, the same filter was re‐probed with anti‐vinculin antibodies. Data reported in the figure are representative of at least three experiments done.
3.4. DCD‐1 and DCD‐2 inhibit Met‐mediated biological responses in vitro
Cell growth impairment was evaluated in a panel of Met‐addicted carcinoma cell lines featuring Met amplification (GTL‐16, MKN‐45, SNU‐5). DCD‐1 and DCD‐2 inhibited cell growth in a dose‐dependent manner, in most cases with a potency similar to MvDN30 (Figure 5A and Supplementary Figure 1A, B). As expected, cells that are not Met‐addicted were insensitive to MvDN30, DCD‐1 or DCD‐2 inhibition (Supplementary Figure 2A). Inhibition of anchorage‐independent cell growth was tested in A549 cells stimulated or not with HGF and in GTL‐16 cells carrying a constitutively phosphorylated Met. Against A549, DCD‐2 strongly reduced HGF‐induced colony formation in semisolid medium while DCD‐1 was less effective (Figure 5B). If HGF was not added, all the molecules did not display any inhibitor activity (Supplementary Figure 2B). Against GTL‐16, DCD‐1 and DCD‐2 were similarly potent, with a reduced performance compared to MvDN30 (Figure 5C).
Figure 5.
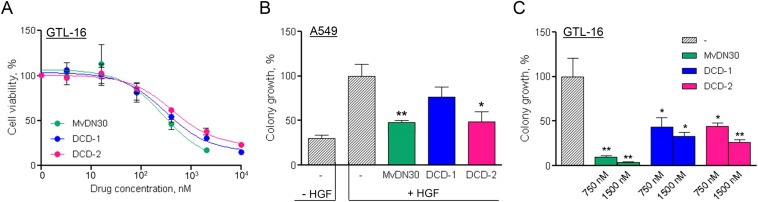
DCDs inhibit Met‐mediated anchorage‐dependent and ‐independent tumor cell growth in vitro. A. Growth of GTL‐16 Met‐addicted cells treated with increasing concentrations of MvDN30, DCD‐1 or DCD‐2 for 3 days. Graph represents percentage of average growth for each treatment with respect to untreated cells. Samples are in quadruplicates, bars represent SEM. B. Anchorage‐independent growth of HGF‐stimulated A549 cells treated with the different DN30‐derived molecules (1.5 μM). Graph represents the percentage of the average colony growth for each treatment with respect to HGF‐treated cells. C. Anchorage‐independent growth of GTL‐16 cells treated with the indicated concentrations of MvDN30, DCD‐1 or DCD‐2. Graph represents percentage of average colony growth for each treatment with respect to untreated cells. *, Student's t Test p < 0.05; **, Student's t Test p < 0.01. Each point is the mean of at least triplicate values, bars: SEM. Data reported in the figure are representative of at least three experiments done.
3.5. DCD‐1 and DCD‐2 show improved pharmacokinetics
To evaluate the pharmacokinetics of the two new molecules, a single dose of DCD‐1, DCD‐2 or MvDN30 – as control – was delivered, either intraperitoneally or intravenously, to immunocompromised mice and the serum concentrations were determined by ELISA assay. The kinetics of the anti‐Met molecules administered by intraperitoneal injection are reported in Figure 6A, as well as the corresponding pharmacokinetic parameters. Compared to MvDN30, the newly engineered molecules showed: (i) more than three folds higher maximal circulating concentration (Cmax); (ii) around two folds longer circulating half‐life (t1/2 elim); (iii) clearance and blood systemic exposure (tot AUC) ten or thirteen folds improved (DCD‐1 and DCD‐2 respectively) versus control. Intravenous administration confirmed a clear improvement of DCD‐1 and DCD‐2 pharmacokinetic profiles versus MvDN30. In the direct comparison with the chimeric DN30 full size antibody, included in the analysis as positive control, the new molecules showed a limited loss in distribution half‐life, clearance and blood systemic exposure, while elimination half‐life and terminal elimination rate (k elim) reached values similar to the full size antibody (Figure 6B).
Figure 6.
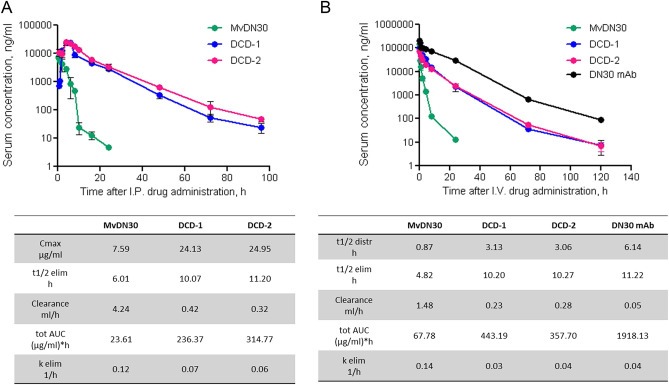
DCDs show improved pharmacokinetic profiles. A. Serum concentration kinetic of MvDN30, DCD‐1 and DCD‐2 after intraperitoneal delivery in immunocompromised mice. B. Serum concentration kinetic of MvDN30, DCD‐1, DCD‐2 and chimeric DN30 mAb after intravenous delivery in immunocompromised mice. Concentration values were determined by ELISA on mouse sera. Samples are in triplicates, bars represent standard deviation. I.P.: intraperitoneal injection; I.V.: intravenous injection. Obtained pharmacokinetic parameters in the tables (lower panels A and B). Cmax: maximal circulating concentration; t1/2 distr: distribution half‐life; t1/2 elim: elimination half‐life; tot AUC: total Area Under the Curve; k elim: elimination constant.
3.6. DCD‐1 and DCD‐2 inhibit Met‐addicted tumor growth in vivo
Met‐addicted GTL‐16 cells, expressing luciferase by lentiviral vector transduction, were intraperitoneally injected in immunodeficient mice. The animals were randomized and treated by intravenous injection with MvDN30, DCD‐1, DCD‐2 or vehicle. The number and the volume of the tumor foci were evaluated by measuring the total luminescence with the IVIS imaging system. Treatments with the newly engineered molecules were effective: a significant reduction in number and size of tumor foci was scored, compared to both vehicle and MvDN30 administrations (Figure 7A, B).
Figure 7.
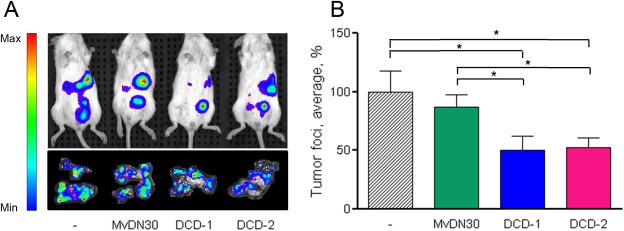
DCDs inhibit Met‐addicted tumor growth. Met‐addicted GTL16 tumor cells expressing luciferase were intraperitoneally injected in NOD/SCID mice. Animals were randomized and treated twice a week as follows: Vehicle, MvDN30, DCD‐1 or DCD‐2 (all molecules 8 nmol/mouse). A. IVIS Spectrum images of one representative mouse/group (upper panel) and of the tumor foci grown on the omentum collected after animal sacrifice (one/group, lower panel). B. Total bioluminescence derived from peritoneal tumor foci at the day of sacrifice. Graph represents the percentage of the average bioluminescent signal with respect to vehicle treated group. Bars represent SEM; *, Student's t Test p < 0.05. Data reported in the figure are representative of two experiments done.
3.7. DCD‐1 and DCD‐2 inhibit the growth of a Met‐amplified patient‐derived colon carcinoma in vivo
Taking advantage of the ‘xenopatients’ platform available in our institute (Bertotti et al., 2011), the engineered inhibitors were tested against M162, a patient‐derived colorectal cancer sample that has the peculiarity to be resistant to cetuximab treatment by Met amplification (Bardelli et al., 2013). First we analyzed the inhibitory properties in vitro, on cells derived from this tumor expanded in mice. In this setting DCD‐1 inhibited cell growth by 30% and DCD‐2 by 33% (Supplementary Figure 3). Then M162 tumor samples were subcutaneously implanted in the right flank of immunocompromised mice. Animals were randomized and treated by intraperitoneal injection with DCD‐1, DCD‐2 or vehicle. DCD‐1 and DCD‐2 delayed the appearance of palpable tumors – including one mouse/group not developing tumor at all – and reduced tumor growth (Figure 8A,B).
Figure 8.
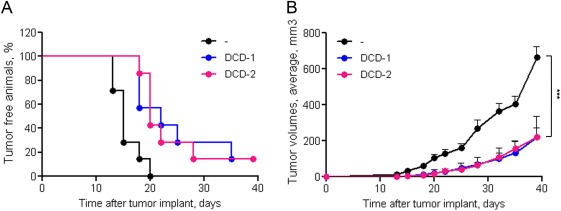
DCDs inhibit tumor implant and growth of a Met‐amplified patient‐derived colorectal carcinoma. Tumor specimens from the M162 ‘xenopatient’, were subcutaneously implanted into the flank of NOD/SCID mice; animals were randomized into three groups and treated twice a week as follows: Vehicle, DCD‐1 or DCD‐2 (both 7 nmol/mouse). A. Kinetic of tumor implant. Each symbol represents the percentage of tumor negative animals at the indicated time point B. Kinetic of tumor growth. Each symbol represents the average tumor volume at the indicated time point; bars: SEM. ***, two‐way ANOVA p < 0.0001.
4. Discussion
In recent years, targeted therapy became a major goal in cancer clinical management. The Met receptor – for its central role in regulation of the invasive growth program – is included in the panel of key oncogenes to be aimed (Smyth et al., 2014). As a consequence, several drugs against Met or HGF have been developed and a number entered in clinical trials (Cui, 2014; Parikh et al., 2014). Case reports of patients carrying Met‐amplified glioblastoma, gastric, esophageal or lung carcinomas showed a clear response to Met inhibitors (Catenacci et al., 2011; Lennerz et al., 2011; Ou et al., 2011; Chi et al., 2012). On the other hand, more than hundred ongoing Met‐targeted phase I/II/III trials are stuck on inconsistent results 1 . due to: (i) the use of wrong drugs, as in the case of molecules not targeting Met (Michieli and Di Nicolantonio, 2013); (ii) wrong patients, i.e. lack of selection in accrual, for inadequate/absent evaluation of the Met genetic status (Sheridan, 2014); (iii) wrong pharmacology, including unawareness of the ‘flare’ effect (Chaft et al., 2011). Given the fulfillment of the last two points, novel Met inhibitors are still an unmet medical need.
The antibody MvDN30, in principle, is a good candidate to enter clinical trials. Its mechanism of action, wiping out Met receptors from cell surface and releasing free Met extracellular domains (acting as a ‘decoys’ with inhibitory properties), proved to be effective against all forms of Met activation, both ligand‐dependent or ‐independent (Vigna and Comoglio, 2015). The limit of the molecule is the short half‐life due to the low molecular weight. To bypass this drawback, conventional technologies based on conjugation with Poly Ethylene Glycol (PEG) were exploited (Chapman, 2002). MvDN30 PEGylation not only reduced renal clearance but also increased water solubility, protected from enzymatic degradation and limited immunogenic and antigenic reactions (Vigna et al., 2015). Nevertheless, the process requires cumbersome synthetic efforts and it is associated with reduction of binding affinity. Alternatively, the size of a protein drug may be increased by molecular engineering. In this context, fusion proteins proved to be effective, as in the case of albuferones, i.e. interferons fused with human albumin, employed in the clinic for their anti‐viral activities (Chemmanur and Wu, 2006). Human albumin has been also genetically fused to coagulation factors (rVIIa‐FP and rIX‐FP) for the treatment of haemophilia patients (Metzner et al., 2013). A further example of a successful fusion protein is ‘Etanercept’, a biologic modulator of inflammation approved for the treatment of autoimmune diseases. This molecule is generated by a soluble form of tumor necrosis factor receptor linked to the Fc receptor for human IgG1 immunoglobulin (Scott, 2014). In our hands the attempt of increasing the size of MvDN30 by an extra‐tail of albumin proved to be awkward (Vigna, unpublished results). As an alternative, we modified the MvDN30 molecular weight by duplication of the Fab constant domains. In a different context (bispecific antibodies), fusion between variable regions has been demonstrated to be functional (Wu et al., 2007). MvDN30 derivatives with the predicted weight of 75 kDa were generated. As expected, in solution these molecules spontaneously dimerize and oligomerize. In fact, the second constant domain of both Fab chains gives rise to a second pair of free cysteine residues, available for inter‐molecule disulfide bonds. From one side, the multimeric molecules have the high molecular weight required for extending the half‐life in the circulation. From the other side, since the oligomers contain more than one binding site, they potentially re‐acquire agonistic properties. However, careful analysis of Met phosphorylation and activation of down‐stream pathways showed that DCD‐1 and DCD‐2, while retaining a minimal residual agonistic activity, were totally ineffective in activation of the intracellular signaling and the ensuing invasive growth biological response. It is thus concluded that multivalency of the Dual Constant Domain Fabs does not mean ‘per se’ acquisition of agonistic activity. This finding is in line with the notion that the ability to stably dimerize receptors (including Met) relies on the distance between the target molecules engaged by the antibody (Cochran et al., 2001; Kai et al., 2008). Moreover it is known that receptor dimerization – although required – is not necessarily sufficient for the activation of tyrosine kinase receptors (Schlessinger, 2014). A behavior similar to the DCDs has in fact also been reported for other inhibitory antibodies, e.g. matuzumab and cetuximab, inhibiting EGFR downstream signaling in the presence of subliminal receptor phosphorylation (Yoshida et al., 2008). We argue that, compared to a natural antibody, where the hinge region confers a certain grade of flexibility (Roux et al., 1997), DCD molecules are more rigid and therefore less prone to the adaptation in the space required for induction of functional dimerization.
Both DCD molecules show improved pharmacokinetic profile when compared to the parental MvDN30. The half life was significantly extended after both intraperitoneal and intravenous administration. After intraperitoneal administration the DCDs maximal blood concentration was delayed but reached higher levels than MvDN30. This reflects the time lost in the transfer from the peritoneal cavity to the blood, counterbalanced by the extra‐time gained as a consequence of reduced renal clearance. In absolute terms, this strategy increases the half‐life of MvDN30 in the range obtained by PEGylation (Vigna et al., 2015). For the above considerations regarding the synthetic efforts to generate PEG derivatives and the reduction of their binding affinity, the described molecular engineering approach is preferable.
The improved pharmacokinetic profile of DCDs is followed by enhanced potency in vivo, in pre‐clinical settings of cancer of two different tissues of origin: stomach and colon. Notably, the ‘xenopatient’ model proved to be highly predictive of clinical outcomes (Hidalgo et al., 2014).
All together these data suggest that the strategy of duplicating the immunoglobulin constant domains by molecular engineering is potentially suitable to improve the therapeutic effectiveness of other Fab fragments.
Conflict of interest
PMC and EV are authors of the international patent WO2014108829 (“New antibody fragments, compositions and uses thereof”) owned by Metheresis Translational Research SA (Switzerland). The company did not interfere at all in the design of the study, collection/analysis of data and decision to publish. The other authors declare no potential conflict of interest.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
We thank Silvia Arpicco and Cristina Basilico for helpful scientific discussion, Livio Trusolino and Andrea Bertotti for providing M162 ‘xenopatient’.
This work was supported by AIRC grants (IG Project no 11852, IG Project no 15572 and 2010 Special Program Molecular Clinical Oncology 5xMille, Project no 9970) to PMC.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.03.004.
Cignetto Simona, Modica Chiara, Chiriaco Cristina, Fontani Lara, Milla Paola, Michieli Paolo, Comoglio Paolo M., Vigna Elisa, (2016), Dual Constant Domain‐Fab: A novel strategy to improve half‐life and potency of a Met therapeutic antibody, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.03.004.
Footnotes
Contributor Information
Paolo M. Comoglio, Email: pcomoglio@gmail.com
Elisa Vigna, Email: elisa.vigna@ircc.it.
References
- Amendola, M. , Venneri, M.A. , Biffi, A. , Vigna, E. , Naldini, L. , 2005. Coordinate dual-gene transgenesis by lentiviral vectors carrying synthetic bidirectional promoters. Nat. Biotechnol. 23, 108–116. [DOI] [PubMed] [Google Scholar]
- Bardelli, A. , Corso, S. , Bertotti, A. , Hobor, S. , Valtorta, E. , Siravegna, G. , Sartore-Bianchi, A. , Scala, E. , Cassingena, A. , Zecchin, D. , Apicella, M. , Migliardi, G. , Galimi, F. , Lauricella, C. , Zanon, C. , Perera, T. , Veronese, S. , Corti, G. , Amatu, A. , Gambacorta, M. , Diaz, L.A. , Sausen, M. , Velculescu, V.E. , Comoglio, P. , Trusolino, L. , Di Nicolantonio, F. , Giordano, S. , Siena, S. , 2013. Amplification of the MET receptor drives resistance to anti-EGFR therapies in colorectal cancer. Cancer Discov. 3, 658–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, J. , Brennan, C. , Shih, J.Y. , Riely, G. , Viale, A. , Wang, L. , Chitale, D. , Motoi, N. , Szoke, J. , Broderick, S. , Balak, M. , Chang, W.C. , Yu, C.J. , Gazdar, A. , Pass, H. , Rusch, V. , Gerald, W. , Huang, S.F. , Yang, P.C. , Miller, V. , Ladanyi, M. , Yang, C.H. , Pao, W. , 2007. MET amplification occurs with or without T790M mutations in EGFR mutant lung tumors with acquired resistance to gefitinib or erlotinib. Proc. Natl. Acad. Sci. U S A. 104, 20932–20937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertotti, A. , Migliardi, G. , Galimi, F. , Sassi, F. , Torti, D. , Isella, C. , Corà, D. , Di Nicolantonio, F. , Buscarino, M. , Petti, C. , Ribero, D. , Russolillo, N. , Muratore, A. , Massucco, P. , Pisacane, A. , Molinaro, L. , Valtorta, E. , Sartore-Bianchi, A. , Risio, M. , Capussotti, L. , Gambacorta, M. , Siena, S. , Medico, E. , Sapino, A. , Marsoni, S. , Comoglio, P.M. , Bardelli, A. , Trusolino, L. , 2011. A molecularly annotated platform of patient-derived xenografts (“xenopatients”) identifies HER2 as an effective therapeutic target in cetuximab-resistant colorectal cancer. Cancer Discov. 1, 508–523. [DOI] [PubMed] [Google Scholar]
- Birchmeier, C. , Birchmeier, W. , Gherardi, E. , Vande Woude, G.F. , 2003. Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 4, 915–925. [DOI] [PubMed] [Google Scholar]
- Blumenschein, G.R. , Mills, G.B. , Gonzalez-Angulo, A.M. , 2012. Targeting the hepatocyte growth factor-cMET axis in cancer therapy. J. Clin. Oncol. 30, 3287–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börset, M. , Hjorth-Hansen, H. , Seidel, C. , Sundan, A. , Waage, A. , 1996. Hepatocyte growth factor and its receptor c-met in multiple myeloma. Blood. 88, 3998–4004. [PubMed] [Google Scholar]
- Catenacci, D.V. , Henderson, L. , Xiao, S.Y. , Patel, P. , Yauch, R.L. , Hegde, P. , Zha, J. , Pandita, A. , Peterson, A. , Salgia, R. , 2011. Durable complete response of metastatic gastric cancer with anti-Met therapy followed by resistance at recurrence. Cancer Discov. 1, 573–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaft, J.E. , Oxnard, G.R. , Sima, C.S. , Kris, M.G. , Miller, V.A. , Riely, G.J. , 2011. Disease flare after tyrosine kinase inhibitor discontinuation in patients with EGFR-mutant lung cancer and acquired resistance to erlotinib or gefitinib: implications for clinical trial design. Clin. Cancer Res. 17, 6298–6303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman, A.P. , 2002. PEGylated antibodies and antibody fragments for improved therapy: a review. Adv. Drug Deliv. Rev. 54, 531–545. [DOI] [PubMed] [Google Scholar]
- Chemmanur, A.T. , Wu, G.Y. , 2006. Drug evaluation: Albuferon-alpha–an antiviral interferon-alpha/albumin fusion protein. Curr. Opin. Investig. Drugs. 7, 750–758. [PubMed] [Google Scholar]
- Chi, A.S. , Batchelor, T.T. , Kwak, E.L. , Clark, J.W. , Wang, D.L. , Wilner, K.D. , Louis, D.N. , Iafrate, A.J. , 2012. Rapid radiographic and clinical improvement after treatment of a MET-amplified recurrent glioblastoma with a mesenchymal-epithelial transition inhibitor. J. Clin. Oncol. 30, e30–33. [DOI] [PubMed] [Google Scholar]
- Cochran, J.R. , Cameron, T.O. , Stone, J.D. , Lubetsky, J.B. , Stern, L.J. , 2001. Receptor proximity, not intermolecular orientation, is critical for triggering T-cell activation. J. Biol. Chem. 276, 28068–28074. [DOI] [PubMed] [Google Scholar]
- Comoglio, P.M. , Giordano, S. , Trusolino, L. , 2008. Drug development of MET inhibitors: targeting oncogene addiction and expedience. Nat. Rev. Drug Discov. 7, 504–516. [DOI] [PubMed] [Google Scholar]
- Cui, J.J. , 2014. Targeting receptor tyrosine kinase MET in cancer: small molecule inhibitors and clinical progress. J. Med. Chem. 57, 4427–4453. [DOI] [PubMed] [Google Scholar]
- Engelman, J.A. , Zejnullahu, K. , Mitsudomi, T. , Song, Y. , Hyland, C. , Park, J.O. , Lindeman, N. , Gale, C.M. , Zhao, X. , Christensen, J. , Kosaka, T. , Holmes, A.J. , Rogers, A.M. , Cappuzzo, F. , Mok, T. , Lee, C. , Johnson, B.E. , Cantley, L.C. , Jänne, P.A. , 2007. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 316, 1039–1043. [DOI] [PubMed] [Google Scholar]
- Ferracini, R. , Di Renzo, M.F. , Scotlandi, K. , Baldini, N. , Olivero, M. , Lollini, P. , Cremona, O. , Campanacci, M. , Comoglio, P.M. , 1995. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene. 10, 739–749. [PubMed] [Google Scholar]
- Ferracini, R. , Olivero, M. , Di Renzo, M.F. , Martano, M. , De Giovanni, C. , Nanni, P. , Basso, G. , Scotlandi, K. , Lollini, P.L. , Comoglio, P.M. , 1996. Retrogenic expression of the MET proto-oncogene correlates with the invasive phenotype of human rhabdomyosarcomas. Oncogene. 12, 1697–1705. [PubMed] [Google Scholar]
- Gao, H. , Guan, M. , Sun, Z. , Bai, C. , 2015. High c-Met expression is a negative prognostic marker for colorectal cancer: a meta-analysis. Tumour Biol. 36, 515–520. [DOI] [PubMed] [Google Scholar]
- Giordano, S. , Di Renzo, M.F. , Ferracini, R. , Chiadò-Piat, L. , Comoglio, P.M. , 1988. p145, a protein with associated tyrosine kinase activity in a human gastric carcinoma cell line. Mol. Cell. Biol. 8, 3510–3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo, M. , Amant, F. , Biankin, A.V. , Budinská, E. , Byrne, A.T. , Caldas, C. , Clarke, R.B. , de Jong, S. , Jonkers, J. , Mælandsmo, G.M. , Roman-Roman, S. , Seoane, J. , Trusolino, L. , Villanueva, A. , 2014. Patient-derived xenograft models: an emerging platform for translational cancer research. Cancer Discov. 4, 998–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kai, M. , Motoki, K. , Yoshida, H. , Emuta, C. , Chisaka, Y. , Tsuruhata, K. , Endo, C. , Muto, M. , Shimabe, M. , Nishiyama, U. , Hagiwara, T. , Matsumoto, A. , Miyazaki, H. , Kataoka, S. , 2008. Switching constant domains enhances agonist activities of antibodies to a thrombopoietin receptor. Nat. Biotechnol. 26, 209–211. [DOI] [PubMed] [Google Scholar]
- Kentsis, A. , Reed, C. , Rice, K.L. , Sanda, T. , Rodig, S.J. , Tholouli, E. , Christie, A. , Valk, P.J. , Delwel, R. , Ngo, V. , Kutok, J.L. , Dahlberg, S.E. , Moreau, L.A. , Byers, R.J. , Christensen, J.G. , Vande Woude, G. , Licht, J.D. , Kung, A.L. , Staudt, L.M. , Look, A.T. , 2012. Autocrine activation of the MET receptor tyrosine kinase in acute myeloid leukemia. Nat. Med. 18, 1118–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre, J. , Ancot, F. , Leroy, C. , Muharram, G. , Lemière, A. , Tulasne, D. , 2012. Met degradation: more than one stone to shoot a receptor down. FASEB J. 26, 1387–1399. [DOI] [PubMed] [Google Scholar]
- Lennerz, J.K. , Kwak, E.L. , Ackerman, A. , Michael, M. , Fox, S.B. , Bergethon, K. , Lauwers, G.Y. , Christensen, J.G. , Wilner, K.D. , Haber, D.A. , Salgia, R. , Bang, Y.J. , Clark, J.W. , Solomon, B.J. , Iafrate, A.J. , 2011. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 29, 4803–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzato, A. , Olivero, M. , Patanè, S. , Rosso, E. , Oliaro, A. , Comoglio, P.M. , Di Renzo, M.F. , 2002. Novel somatic mutations of the MET oncogene in human carcinoma metastases activating cell motility and invasion. Cancer Res. 62, 7025–7030. [PubMed] [Google Scholar]
- Lutterbach, B. , Zeng, Q. , Davis, L.J. , Hatch, H. , Hang, G. , Kohl, N.E. , Gibbs, J.B. , Pan, B.S. , 2007. Lung cancer cell lines harboring MET gene amplification are dependent on Met for growth and survival. Cancer Res. 67, 2081–2088. [DOI] [PubMed] [Google Scholar]
- Metzner, H.J. , Pipe, S.W. , Weimer, T. , Schulte, S. , 2013. Extending the pharmacokinetic half-life of coagulation factors by fusion to recombinant albumin. Thromb. Haemost. 110, 931–939. [DOI] [PubMed] [Google Scholar]
- Michieli, P. , Di Nicolantonio, F. , 2013. Targeted therapies: Tivantinib–a cytotoxic drug in MET inhibitor's clothes?. Nat. Rev. Clin. Oncol. 10, 372–374. [DOI] [PubMed] [Google Scholar]
- Michieli, P. , Mazzone, M. , Basilico, C. , Cavassa, S. , Sottile, A. , Naldini, L. , Comoglio, P.M. , 2004. Targeting the tumor and its microenvironment by a dual-function decoy Met receptor. Cancer Cell. 6, 61–73. [DOI] [PubMed] [Google Scholar]
- Moriyama, T. , Kataoka, H. , Koono, M. , Wakisaka, S. , 1999. Expression of hepatocyte growth factor/scatter factor and its receptor c-Met in brain tumors: evidence for a role in progression of astrocytic tumors (Review). Int. J. Mol. Med. 3, 531–536. [DOI] [PubMed] [Google Scholar]
- Mukohara, T. , Civiello, G. , Davis, I.J. , Taffaro, M.L. , Christensen, J. , Fisher, D.E. , Johnson, B.E. , Jänne, P.A. , 2005. Inhibition of the met receptor in mesothelioma. Clin. Cancer Res. 11, 8122–8130. [DOI] [PubMed] [Google Scholar]
- Ou, S.H. , Kwak, E.L. , Siwak-Tapp, C. , Dy, J. , Bergethon, K. , Clark, J.W. , Camidge, D.R. , Solomon, B.J. , Maki, R.G. , Bang, Y.J. , Kim, D.W. , Christensen, J. , Tan, W. , Wilner, K.D. , Salgia, R. , Iafrate, A.J. , 2011. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J. Thorac. Oncol. 6, 942–946. [DOI] [PubMed] [Google Scholar]
- Pacchiana, G. , Chiriaco, C. , Stella, M.C. , Petronzelli, F. , De Santis, R. , Galluzzo, M. , Carminati, P. , Comoglio, P.M. , Michieli, P. , Vigna, E. , 2010. Monovalency unleashes the full therapeutic potential of the DN-30 anti-Met antibody. J. Biol. Chem. 285, 36149–36157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parikh, R.A. , Wang, P. , Beumer, J.H. , Chu, E. , Appleman, L.J. , 2014. The potential roles of hepatocyte growth factor (HGF)-MET pathway inhibitors in cancer treatment. Onco Targets Ther. 7, 969–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli, A. , Circosta, P. , Granziero, L. , Mazzone, M. , Pisacane, A. , Fenoglio, S. , Comoglio, P.M. , Giordano, S. , 2006. Ab-induced ectodomain shedding mediates hepatocyte growth factor receptor down-regulation and hampers biological activity. Proc. Natl. Acad. Sci. U S A. 103, 5090–5095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prat, M. , Crepaldi, T. , Pennacchietti, S. , Bussolino, F. , Comoglio, P.M. , 1998. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. J. Cell Sci. 111, (Pt 2) 237–247. [DOI] [PubMed] [Google Scholar]
- Roux, K.H. , Strelets, L. , Michaelsen, T.E. , 1997. Flexibility of human IgG subclasses. J. Immunol. 159, 3372–3382. [PubMed] [Google Scholar]
- Schlessinger, J. , 2014. Receptor tyrosine kinases: legacy of the first two decades. Cold Spring Harb. Perspect. Biol. 6, [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, L. , Duh, F.M. , Chen, F. , Kishida, T. , Glenn, G. , Choyke, P. , Scherer, S.W. , Zhuang, Z. , Lubensky, I. , Dean, M. , Allikmets, R. , Chidambaram, A. , Bergerheim, U.R. , Feltis, J.T. , Casadevall, C. , Zamarron, A. , Bernues, M. , Richard, S. , Lips, C.J. , Walther, M.M. , Tsui, L.C. , Geil, L. , Orcutt, M.L. , Stackhouse, T. , Lipan, J. , Slife, L. , Brauch, H. , Decker, J. , Niehans, G. , Hughson, M.D. , Moch, H. , Storkel, S. , Lerman, M.I. , Linehan, W.M. , Zbar, B. , 1997. Germline and somatic mutations in the tyrosine kinase domain of the MET proto-oncogene in papillary renal carcinomas. Nat. Genet. 16, 68–73. [DOI] [PubMed] [Google Scholar]
- Scott, L.J. , 2014. Etanercept: a review of its use in autoimmune inflammatory diseases. Drugs. 74, 1379–1410. [DOI] [PubMed] [Google Scholar]
- Sheridan, C. , 2014. Genentech to salvage anti-MET antibody with subgroup analysis. Nat. Biotechnol. 32, 399–400. [DOI] [PubMed] [Google Scholar]
- Skead, G. , Govender, D. , 2015. Gene of the month: MET. J. Clin. Pathol. 68, 405–409. [DOI] [PubMed] [Google Scholar]
- Smolen, G.A. , Sordella, R. , Muir, B. , Mohapatra, G. , Barmettler, A. , Archibald, H. , Kim, W.J. , Okimoto, R.A. , Bell, D.W. , Sgroi, D.C. , Christensen, J.G. , Settleman, J. , Haber, D.A. , 2006. Amplification of MET may identify a subset of cancers with extreme sensitivity to the selective tyrosine kinase inhibitor PHA-665752. Proc. Natl. Acad. Sci. U S A. 103, 2316–2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth, E.C. , Sclafani, F. , Cunningham, D. , 2014. Emerging molecular targets in oncology: clinical potential of MET/hepatocyte growth-factor inhibitors. Onco Targets Ther. 7, 1001–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stella, G.M. , Benvenuti, S. , Gramaglia, D. , Scarpa, A. , Tomezzoli, A. , Cassoni, P. , Senetta, R. , Venesio, T. , Pozzi, E. , Bardelli, A. , Comoglio, P.M. , 2011. MET mutations in cancers of unknown primary origin (CUPs). Hum. Mutat. 32, 44–50. [DOI] [PubMed] [Google Scholar]
- Trusolino, L. , Bertotti, A. , Comoglio, P.M. , 2010. MET signalling: principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 11, 834–848. [DOI] [PubMed] [Google Scholar]
- Trusolino, L. , Comoglio, P.M. , 2002. Scatter-factor and semaphorin receptors: cell signalling for invasive growth. Nat. Rev. Cancer. 2, 289–300. [DOI] [PubMed] [Google Scholar]
- Vigna, E. , Chiriaco, C. , Cignetto, S. , Fontani, L. , Basilico, C. , Petronzelli, F. , Comoglio, P.M. , 2015. Inhibition of ligand-independent constitutive activation of the Met oncogenic receptor by the engineered chemically-modified antibody DN30. Mol. Oncol. 9, 1760–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigna, E. , Comoglio, P.M. , 2015. Targeting the oncogenic Met receptor by antibodies and gene therapy. Oncogene. 34, 1883–1889. [DOI] [PubMed] [Google Scholar]
- Vigna, E. , Pacchiana, G. , Mazzone, M. , Chiriaco, C. , Fontani, L. , Basilico, C. , Pennacchietti, S. , Comoglio, P.M. , 2008. “Active” cancer immunotherapy by anti-Met antibody gene transfer. Cancer Res. 68, 9176–9183. [DOI] [PubMed] [Google Scholar]
- Wu, C. , Ying, H. , Grinnell, C. , Bryant, S. , Miller, R. , Clabbers, A. , Bose, S. , McCarthy, D. , Zhu, R.R. , Santora, L. , Davis-Taber, R. , Kunes, Y. , Fung, E. , Schwartz, A. , Sakorafas, P. , Gu, J. , Tarcsa, E. , Murtaza, A. , Ghayur, T. , 2007. Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin. Nat. Biotechnol. 25, 1290–1297. [DOI] [PubMed] [Google Scholar]
- Yoshida, T. , Okamoto, I. , Okabe, T. , Iwasa, T. , Satoh, T. , Nishio, K. , Fukuoka, M. , Nakagawa, K. , 2008. Matuzumab and cetuximab activate the epidermal growth factor receptor but fail to trigger downstream signaling by Akt or Erk. Int. J. Cancer. 122, 1530–1538. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data