

Abstract
Mutations in TP53 gene play a pivotal role in tumorigenesis and cancer development. Here, we report that gain‐of‐function mutant p53 proteins inhibit the autophagic pathway favoring antiapoptotic effects as well as proliferation of pancreas and breast cancer cells. We found that mutant p53 significantly counteracts the formation of autophagic vesicles and their fusion with lysosomes throughout the repression of some key autophagy‐related proteins and enzymes as BECN1 (and P‐BECN1), DRAM1, ATG12, SESN1/2 and P‐AMPK with the concomitant stimulation of mTOR signaling. As a paradigm of this mechanism, we show that atg12 gene repression was mediated by the recruitment of the p50 NF‐κB/mutant p53 protein complex onto the atg12 promoter. Either mutant p53 or p50 NF‐κB depletion downregulates atg12 gene expression. We further correlated the low expression levels of autophagic genes (atg12, becn1, sesn1, and dram1) with a reduced relapse free survival (RFS) and distant metastasis free survival (DMFS) of breast cancer patients carrying TP53 gene mutations conferring a prognostic value to this mutant p53‐and autophagy‐related signature. Interestingly, the mutant p53‐driven mTOR stimulation sensitized cancer cells to the treatment with the mTOR inhibitor everolimus. All these results reveal a novel mechanism through which mutant p53 proteins promote cancer cell proliferation with the concomitant inhibition of autophagy.
Keywords: Autophagy, Mutant p53, Gain‐of‐function, Cancer, mTOR, AMPK
Highlights
GOF p53 mutant proteins inhibit the autophagic vesicle formation in cancer cells.
Mutant p53 proteins inhibit the expression of ATGs in cancer cells and patients.
Mutant p53/NF‐κB p50 complex inhibits atg12 gene expression.
Mutant p53 proteins stimulate mTOR and repress AMPK signaling.
The expression of mutant p53 proteins sensitizes cancer cells to mTOR inhibition.
1. Introduction
Mutations in the TP53 gene occur in over 50% of human cancers, where most of them are missense mutations resulting in the expression of mutant forms of p53 (Vousden and Lu, 2002; Waddell et al., 2015). In addition, p53 mutated proteins acquire new biological properties referred to as gain‐of‐function (GOF) that contribute to the induction and maintenance of cancer (Santoro et al., 2014). In many human tumors, p53 mutations are associated with high genomic instability, poor prognosis, poor response to chemotherapy and accelerated tumor recurrence (Ganci et al., 2013; Liu et al., 2012; Walerych et al., 2012). Different models have been proposed to explain the GOF activities of mutant p53, including binding and inactivation of the p53 family members p63 and p73, modulation of the activity of the transcription factors NF‐Y, E2F1, E2F4, p65 NF‐κB, and vitamin D receptor (VDR), or the inactivation of the DNA damage sensor ataxia telangiectasia mutated (ATM) (Di Agostino et al., 2006; Stambolsky et al., 2010; Strano et al., 2007; Valenti et al., 2015; Weisz et al., 2007). Recently, we have documented that DNA damage with gemcitabine stabilized mutant p53 protein in cell nuclei triggering chemoresistance and inducing the expression of cell cycle‐related genes, as Cdk1 and CCNB1 increased cell growth (Fiorini et al., 2015b). Autophagy is a self‐eating process by which eukaryotic cells degrade proteins and cytoplasmic organelles through lysosomal hydrolases, thus controlling quality of the cytoplasm and recycling macromolecules to extract energy and to overcome nutritional or microenvironmental stressful situations (Viry et al., 2014). Several studies indicate that wild type p53 triggers the autophagic machinery in human cancer cells involving many pathways as the stimulation of the nutrient energy sensor AMP‐activated protein kinase (AMPK), the inhibition of the mammalian target of rapamycin (mTOR) by up‐regulation of PTEN and TSC1, and the induction of the autophagy‐related gene DRAM1 (Comel et al., 2014; Crighton et al., 2007; Drakos et al., 2009; Gomes et al., 2015). Interestingly, autophagy regulation is strictly interconnected with the aberrant setting of cancer cell metabolism as revealed by the fact that mTOR and AMPK pathways are both the master regulators of autophagy and the most important sensors of the cellular energy status (Dodson et al., 2013). In particular, mTOR complex stimulates anabolic biosynthesis for cancer cell growth and inhibits autophagy, while AMPK signaling triggers degradation of macromolecules, including lysosomal autophagic catabolism (DeBerardinis and Thompson, 2012). Recently, Zhou et al. have shown that GOF mutant p53 inhibited AMPK signaling in head and neck cancer cells directly binding to the AMPKα subunit thus stimulating anabolic growth and gaining its oncogenic function (Zhou et al., 2014). Other studies demonstrated the involvement of mutant p53 in the regulation of cancer metabolism, throughout the transcriptional modulation of SREBP1, a downstream target of AMPK (Freed‐Pastor et al., 2012). However, despite a study describing a correlation between the subcellular localization of overexpressed p53 mutant proteins and the regulation of autophagy (Morselli et al., 2008), the functional involvement of endogenous mutant p53 in the regulation of autophagy in cancer cells and the identification of the associated molecular mechanisms remain largely unknown.
In the present work, we ascertain that GOF mutant p53 proteins are able to orchestrate a plethora of events addressed to counteract autophagy in the various phases of the process, thus contributing to inhibit apoptosis and to sustain oncogenic activity. Notably, we document that mutant p53 interacts with the p50 NF‐κB subunit onto the atg12 promoter inhibiting its expression and resulting in the repression of the autophagic machinery. Furthermore, we point out that cancer cells bearing endogenous mutant p53 are more sensitive to mTOR inhibition than cancer cells expressing wild type p53 protein, providing a conceivable personalized therapy for cancer patients carrying GOF mutations of TP53 gene.
2. Material and methods
2.1. Chemicals
Chloroquine diphosphate (CQ), 3‐methyladenine (3MA), and everolimus (EVE; RAD‐001) were obtained from Sigma (Milan, Italy). Compound C (CC) solution was obtained from Calbiochem (Merck; Darmstadt, Germany).
2.2. Cell culture
PaCa3 (WTp53), Panc1 (mutant p53‐R273H), PaCa44 (mutant p53‐C176S), MiaPaCa2 (mutant p53‐R248W), Suit‐2 (mutant p53‐R273H) and AsPC1 (p53‐null) human pancreatic adenocarcinoma cell lines were grown in RPMI medium (Life Technologies, Milan, Italy), while lung cancer H1299 (p53‐null), breast cancer MCF7 (WTp53), SKBr3 (mutant p53‐R175H), MDA‐MB‐468 (mutant p53‐R273H) and MDA‐MB‐231 (mutant p53‐R280K) cell lines were cultured in DMEM medium (Life Technologies, Milan, Italy). Both culture media were supplemented with 10% FBS, and 50 μg/ml gentamicin sulfate (BioWhittaker, Lonza, Bergamo, Italy). Cell lines were incubated at 37 °C with 5% CO2. The list of the cell lines used in this study and their p53 status are summarized in Table 1.
Table 1.
Tissue of origin and p53 status of cancer cell lines.
| Cancer cell lines | Tissue origin | p53 status | Mutation |
|---|---|---|---|
| PaCa3 | Pancreas | Wild‐type | None |
| MCF7 | Breast | Wild‐type | None |
| MiaPaca2 | Pancreas | Mutated | R248W |
| PaCa44 | Pancreas | Mutated | C176S |
| Panc1 | Pancreas | Mutated | R273H |
| Suit‐2 | Pancreas | Mutated | R273H |
| MDA‐MB‐468 | Breast | Mutated | R273H |
| SkBr3 | Breast | Mutated | R175H |
| MDA‐MB‐231 | Breast | Mutated | R280K |
| AsPC1 | Pancreas | Null | Gene deleted |
| H1299 | Lung | Null | Gene deleted |
2.3. Cell proliferation assay
Cells were seeded in 96‐well plates (5 × 103 cells/well) and the day after were incubated with various compounds at the indicated conditions or transfected with the indicated constructs (see figure legends). At the end of the treatments, cell growth was measured by Crystal Violet assay (Sigma, Milan, Italy) according to the manufacturer's protocol, and absorbance was measured by spectrophotometric analysis (A595nm).
2.4. Transient transfection assays
Exponentially growing cells were seeded at 5 × 103 cells/well in 96‐well plates or at 2.5 × 105 cells in 60 mm cell culture plates. The ectopic expression of mutant p53 in p53‐null cancer cells was carried out transfecting pcDNA3‐mutp53R273H or pcDNA3‐mutp53R175H expression vectors, or their relative negative control (pcDNA3). The ectopic expression of wild type or dominant negative (DN)‐AMPK subunit γ2 was obtained by transfection of the pcDNA5/FRT expression vector containing the human wild type AMPK γ2 subunit or the mutated R531G protein which were kindly provided by Dr. Hawley (University of Dundee, Scotland, UK). Wild‐type and mutant p53 protein expression was transiently knocked‐down by transfection with pRSUPER‐p53 or pLVTHM‐p53 vectors or their negative controls (pRSUPER or pLVTHM), kindly provided by Dr. Agami (The Netherlands Cancer Institute, Amsterdam) (Brummelkamp et al., 2002) and by Dr. Sergio Ruiz (CNIO, Madrid, Spain), respectively. Commercial siRNA smart pool of three oligonucleotides (si‐p53) transiently targeting p53 (Santa Cruz Biotech. sc‐29435) was used in the experiments of RT‐qPCR to exclude back‐side effects and to confirm the robustness of the data. A si‐GFP as non‐silencing control 5′‐GGCTACGTCCAGGAGCGCACC‐3′ was used as a negative control. The silencing transfections were carried out for 48 h using Lipofectamine 2000 (Life Technologies), according to the manufacturer's instructions. Knockdown of Atg5 expression was performed by shRNA‐Atg5 cloned in pMSCV‐Puro‐miR30 vector (or its negative empty vector) kindly provided by Dr. Hans‐Uwe Simon (University of Bern, Switzerland) (Maskey et al., 2013). Knock‐down of Beclin1 expression was obtained by transfecting cells with specific Beclin1 small interfering (si) RNA having the following sequences: 5′‐ACAGUGAAUUUAAACGACAGCAGCU‐3′ and 5′‐AGCUGCUGUCGUUUAAAUUCACUGU‐3′ and with a siRNA‐CTRL (negative control): 5′‐CAGUCGCGUUUGCGACUGG‐3′ purchased from Life Technologies. Knock‐down of p50 NF‐κB subunit expression was obtained by transfecting cells with a specific 21‐nt‐long interfering RNA duplex with two 3'‐end overhang dT nucleotides. The sequence of the antisense strand of the anti‐p50 siRNA was 5′‐AGUCCAGGAUUAUAGCCCCdTdT‐3' (MWG Biotec, Ebersberg, Germany; or Dharmacon, Lafayette, CO) as reported (Laderach et al., 2003). Cells were transfected by siRNAs at a final concentration of 50 nM using Lipofectamine 2000 (Life Technologies), according to the manufacturer's instructions.
2.5. RNA isolation and quantitative real‐time PCR analysis
Total RNA was extracted from cells using TRIZOL Reagent (Life Technologies) in accordance with the manufacturer's instructions. Five micrograms of total RNA were reverse‐transcribed at 37 °C for 60 min in the presence of random hexamers and Moloney murine leukemia virus reverse transcriptase (Life Technologies). Transcripts were measured by real‐time PCR using the SYBR Green assay (Applied Biosystems, Carlsbad, CA, USA) with a StepOne instrument (Applied Biosystems). PCR analysis was carried out using specific oligonucleotides for the genes listed in Supplementary Table 1. The primers were designed with Primer3 version 0.4.0 (http://frodo.wi.mit.edu/primer3/). All primer sets worked under identical quantitative PCR cycling conditions with similar efficiencies to obtain simultaneous amplification in the same run. The 2−ΔΔCT method for relative quantification of gene expression was used to determine mRNA expression levels. GAPDH gene expression was used as endogenous control to standardize mRNA expression. All reactions were performed in duplicate from three independent experiments.
2.6. ChIP experiments
1% formaldehyde cross‐linking and chromatin immunoprecipitations were performed as described in (Di Agostino et al., 2006; Valenti et al., 2011). The chromatin solution was immunoprecipitated with sheep anti‐p53 Ab7 (Millipore, Billerica, MA, USA), rabbit anti‐p50 (Santa Cruz Biotech. SC‐7178), rabbit anti‐p65 (Santa Cruz Biotech. SC‐372), or no antibody as negative control. The immunoprecipitations were performed using Pierce ChIP‐grade Protein A/G magnetic beads (Thermo Fisher Scientific, Rockford, IL, USA). Primers used for the amplification of the different regulatory regions are listed in Supplementary Table 2. NF‐κB p50 Region 2 consensus sequence on ATG12 promoter was selected from the literature and NF‐κB p50 Region 1, the consensus sequence was identified by MatInspector software (www.genomatix.de). The promoter occupancy was analyzed by RT‐qPCR using the Fast SYBR Green assay (Applied Biosystems, Carlsbad, CA, USA) and the StepOne™ Real‐Time PCR System (Applied Biosystems). Normalization was performed to the amount of input chromatin. The ChIP results were further normalized on the qPCR of a H1H2B2, the genic region resulted negative for the recruitment of mutant p53 (Supplementary Figure 1) (Biagioni et al., 2012).
2.7. Immunoblot analysis
Cells were harvested, washed in PBS, and re‐suspended in lysis buffer in the presence of phosphatase and protease inhibitors (50 mM Tris–HCl pH 8, 150 mM NaCl, 1% Igepal CA‐630, 0.5% Na‐Doc, 0.1% SDS, 1 mM Na3VO4, 1 mM NaF, 2.5 mM EDTA, 1 mM PMSF, and 1× protease inhibitor cocktail). After incubation on ice for 30 min, the lysates were centrifuged at 14,000 × g for 10 min at 4 °C and the supernatant fractions were used for Western blot analysis. Protein concentration was measured by Bradford reagent (Pierce, Milan, Italy) using bovine serum albumin as a standard. Protein extracts (50 μg/lane) were resolved on a 12% SDS‐polyacrylamide gel and electro‐blotted onto PVDF membranes (Millipore, Milan, Italy). Membranes were blocked in 5% low‐fat milk in TBST (50 mM Tris pH 7.5, 0.9% NaCl, 0.1% Tween 20) for 1 h at room temperature and probed overnight at 4 °C with a mouse polyclonal anti‐p53 (1:500) (Santa Cruz, #sc‐263), rabbit monoclonal anti‐LC3 (1:1000) (Cell Signaling, #2775), rabbit polyclonal anti‐phospho(Ser15)Beclin1 (1:1000) (Cell Signaling #13825) rabbit polyclonal anti‐Beclin1 (1:1000) (GeneTex, #GTX113039), rabbit monoclonal glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) (1:1000) (Cell Signaling, #5174S), rabbit monoclonal anti‐phospho (Thr172)AMPK (1:1000) (Cell Signaling #2535), rabbit monoclonal anti‐AMPK (1:1000) (Cell Signaling #2603), mouse monoclonal anti‐phospho (Thr389)p70S6K (1:1000) (Cell Signaling #9206), rabbit monoclonal anti‐p70S6K (1:1000) (Cell Signaling #2708), rabbit polyclonal anti‐SESN1 (1:1000) (GeneTex #GTX116926), rabbit polyclonal anti‐SESN2 (1:1000) (GeneTex #118141), rabbit polyclonal anti‐NF‐κB p50 (1:1000) (Santa Cruz #sc‐7178), rabbit polyclonal anti‐NF‐κB p65 (1:1000) (Santa Cruz #sc‐372), mouse monoclonal anti‐alpha‐tubulin (1:2500) (Oncogene #CP06), or rabbit monoclonal anti‐phospho (Tyr705)STAT3 (1:1000) (Cell Signaling #9131) antibodies.
Horseradish peroxidase conjugated anti‐mouse or anti‐rabbit IgGs (1:8000 in blocking solution) (Upstate Biotechnology, Milan, Italy) were used as secondary antibodies. Immunodetection was carried out using chemiluminescent substrates (Amersham Pharmacia Biotech, Milan, Italy) and recorded using a HyperfilmECL (Amersham Pharmacia Biotech). ECL results were scanned and the amount of each protein band quantified using NIH Image J software (http://rsb.info.nih.gov/nih‐image/).
2.8. Immunoprecipitation assay
Cell extracts were solubilized in lysis buffer with 150 mM Hepes pH 7.5, 300 mM NaCl, 1% Triton‐X100, phosphatase and protease inhibitors. Cells were harvested and lysed by sonication in lysis buffer and cleared by centrifugation. Protein concentrations were determined by colorimetric assay (Bio‐Rad, Hercules, CA, USA). For each immunoprecipitation, 1 μg of antibody and 1 μg of rabbit or sheep IgG (Santa Cruz Biotech) were used as controls. To immunoprecipitate, we used rabbit polyclonal anti‐p50 (sc‐7178) from Santa Cruz Biotech and sheep polyclonal anti‐p53 Ab7 (PC35‐1EA) from Millipore. 1 mg of pre‐cleared extracts were diluted in lysis buffer containing 0.05% BSA and incubated with protein A/G‐Agarose beads (Thermo Fisher Scientific, Rockford, IL, USA) and antibodies, under constant shaking at 4 °C for 3 h. Bead‐bound immunocomplexes were rinsed with lysis buffer and eluted in 50 μl of SDS sample buffer for Western blotting. Western blotting was performed using the following primary antibodies: mouse monoclonal p53 (DO1), GAPDH (Santa Cruz Biotech), rabbit polyclonal anti‐NF‐κB p65 (1:1000) (Santa Cruz #sc‐372), rabbit polyclonal anti‐p50 (sc‐7178) and actin (Sigma) antibodies. Western blot analysis was performed with the aid of the enhanced chemiluminescence system (Thermo Fisher Scientific, Rockford, IL, USA) and the acquisition of the images by Uvitec technology (Eppendorf).
2.9. Apoptosis assay
Cells were seeded in 96‐well plates (5× 103 cells/well). Twenty‐four hours later, cells were treated as indicated in the figure legends. At the end of the treatment, cells were fixed with 2% paraformaldehyde in PBS for 30 min at room temperature, washed twice with PBS and stained with annexinV/FITC (Bender MedSystem, Milan, Italy) in binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, and 2.5 mM CaCl2) for 10 min at room temperature in the dark. Cells were then washed with binding buffer and fluorescence was measured using a multimode plate reader (Ex485nm and Em535nm) (GENios Pro, Tecan, Milan, Italy). The values were normalized on cell proliferation by Crystal Violet assay.
2.10. Monodansylcadaverine staining and autophagosome formation assay
To quantify the induction of autophagy, cells were incubated with the fluorescent probe monodansylcadaverine (MDC; Sigma, Milan, Italy), accordingly with the guidelines for studying autophagy (Klionsky et al., 2016). MDC is a selective marker for acidic vesicular organelles, such as autophagic vacuoles. Briefly, cells were seeded in 96‐well plates (5 × 103 cells/well) and treated with various compounds as indicated in the figure legends. At the end of the treatments, cells were incubated in culture medium containing 50 μM MDC at 37 °C for 15 min. Cells were then washed with Hanks buffer (20 mM Hepes pH 7.2, 10 mM glucose, 118 mM NaCl, 4.6 mM KCl, and 1 mM CaCl2) and fluorescence was measured using a multimode plate reader (Ex340nm and Em535nm) (GENios Pro, Tecan, Milan, Italy). The values were normalized for cell proliferation by Crystal Violet assay.
2.11. Acridine orange staining and autolysosome detection
Cells were seeded in a 24‐well plate on glass cover‐slips at a density of 6 × 104/well. After 24 h, cells were transfected with pRSUPER‐p53 or with the empty pRSUPER vector by using Lipofectamine 2000 according to the manufacturer's instructions. 48 h after transfection cells were rinsed in phosphate buffer saline (PBS) and stained with 6 μg/ml acridine orange (AO). Cell images were captured using a fluorescence microscope Leica DM6000 (Leica Microsystem, Manheim, Germany) at 40× magnification and processed using Adobe Photoshop and NIH Image J software.
To quantify autolysosome formation, cells transfected with pRSUPER‐p53 vector or its negative control (pRSUPER) were stained with AO following the manufacturer's instructions. Quantification of autolysosomes was performed measuring the red/green fluorescence intensity ratio of AO staining (AO green fluorescence Ex485nm and Em535nm; AO red fluorescence Ex430nm and Em590nm) with a multimode plate reader (GENios Pro, Tecan, Milan, Italy). Values were normalized on cell proliferation by crystal violet assay.
2.12. LC3‐GFP and lysosome co‐localization analysis
Cells were seeded in a 24‐well plate on glass cover‐slips at a density of 6 × 104/well. After 24 h, cells were co‐transfected with pEGFP‐LC3B and pRSUPER‐p53 vector or with pEGFP‐LC3B and the pRSUPER empty vector by using Lipofectamine 2000, according to the manufacturer's instructions. 48 h after transfection, lysosomes were stained with 50 nM Lysotracker‐RED‐DN99 (Life Technologies) and nuclei were stained with 0.1 μg/ml Hoechst (Life Technologies). Cells were than rinsed in PBS and fixed in 4% (w/v) paraformaldehyde. Cover‐slips were mounted over slides in AF1 medium (Dako). Cell images were captured using a confocal laser‐scanning fluorescence microscope Leica SP5 (Leica Microsystem, Manheim, Germany) at 63× magnification and processed using Adobe Photoshop and NIH Image J software.
2.13. mRNA expression data
We used the publicly available data sets: GSE22358 (Gluck et al., 2012) and GSE31812 (Freed‐Pastor et al., 2012). The data sets were analyzed by the Oncomine platform www.oncomine.org (Rhodes et al., 2004). The mRNA expression data were measured using Agilent UNC Perou Lab Homo sapiens 1 × 44K Custom Array and Human Gene 1.0 ST arrays, respectively.
2.14. Kaplan‐Meier analysis
Gene expression dataset raw data and survival information of 108 BCL patients with TP53 mutated were downloaded from GEO (http://www.ncbi.nlm.nih.gov/geo/) and the Kaplan‐Meier analysis was performed by using the on‐line tool www.kmplot.com (Gyorffy et al., 2010) where eight public datasets were considered. Gene expression profiles were performed by using Affymetrix HG‐U133A (GPL96) and HG‐U133 Plus 2.0 (GPL570) microarrays were only considered. The latter are the most frequently used and have 22,277 probe sets in common. To analyze the prognostic value of genes, the cohorts were divided into two groups according to the median (or upper/lower quartile) expression of gene signature considered. The two groups were compared in terms of relapse free survival (RFS) and distant metastasis free survival (DMFS). A survival curve was displayed, and the hazard ratio with 95% confidence intervals and logrank P value are calculated. P values < 0.05 were considered statistically significant.
2.15. Statistical analysis
ANOVA (post hoc Bonferroni) analysis was performed by GraphPad Prism 5 software. Student's t‐test (two‐tailed) was also conducted. P value < 0.05 was indicated as statistically significant. Values are the means of three independent experiments (±SD).
3. Results
3.1. Gain of function p53 mutant proteins inhibit the autophagic vesicles formation in cancer cells
To study the functional role of GOF mutant p53 proteins in the autophagic mechanism, we first analyzed the endogenous level of autophagosome vesicle formation by staining diverse cancer cell lines with the fluorescent probe MDC. Cancer cells with different missense mutations of TP53 gene had the endogenous level of autophagosome vesicles about 5–6 folds lower than cells with wild type TP53 alleles (Figure 1A). When PaCa3 and MCF7 cell lines (expressing a wild type p53 protein) were knocked down for p53 expression, the autophagosome formation was inhibited, accordingly with the literature (Figure 1B and C) (Crighton et al., 2007). Conversely, the autophagosome formation was significantly increased after knock‐down of GOF mutant p53 in six cancer cell lines (Figure 1B and C). Consistent with this, overexpression of mutant p53 R175H or R273H proteins in AsPC1 cells (null for p53 expression) produced a drastic reduction of MDC probe incorporation revealing an inhibitory role of mutant p53 of the autophagic process (Figure 1B and C). To further support the idea that mutant p53 may have a role in the inhibition of the autophagic event, we analyzed the expression of the autophagic receptor/adaptor SQSTM1/p62 (sequestosome 1), whose massive accumulation is usually a consequence of autophagy impairment and accumulation of aggregated structures positive for ubiquitin (Katsuragi et al., 2015). Accordingly, we observed that mutant p53 knockdown triggered p62 expression decrease (Figure 1D). Furthermore, we evaluated the expression levels of the lipidated and truncated isoform II of light chain 3 protein (LC3‐II), which is functionally involved in the formation and maturation of autophagic vesicles. Our findings showed that LC3‐II protein expression levels considerably decreased in AsPC1 cells after ectopic expression of mutant R175H or R273H p53 proteins while increased in mutant p53‐knocked down cells (Figure 1D). We also observed that chloroquine (CQ) further increased the amount of LC‐II in silencing mutant p53 conditions (Figure 1D), accordingly with the hypothesis of the autophagic inhibitory flux inside the blockage of the lysosomal degradation. According to this, confocal microscopy analysis revealed that mutant p53 counteracts the fusion between autophagic vesicles (identified by LC3‐GFP overexpression) and lysosomes (identified by lysotracker red probe) since the merged image of Panc1‐shp53 cells showed a significant co‐localization of LC3‐II with lysosomes represented by yellow puncta and confirmed by the RGB profile (Figure 1E). In Supplementary Figure 2, we included a negative control about formation of autophagic vesicles after knock‐down of mutant p53 in the absence or presence of the autophagy inhibitor 3‐methyladenine. Furthermore, we have examined the formation of autolysosomes (autophagic vesicles joined with lysosomes) using the fluorescent probe acridine orange (AO), which changes its fluorescence emission from green to red upon accumulation into lysosomal acidic compartments. The typical pH acidification of lysosomes during autophagic stimulation has been examined by AO probe staining in knocked down mutant p53 conditions (Figure 1F and G). Figure 1F showed that mutant p53 cell lines (Panc1 and SKBr3) had a high red/green fluorescence ratio in the sh‐p53 condition, conversely wild type p53 cells (MCF7) showed a decrease of the autolysosome formation (Figure 1F). The microscopy experiments confirmed these results revealing the presence of red areas (acidic lysosomes containing accumulation of AO probe) into the cytoplasm of the cells interfered for mutant p53 expression (Figure 1G). Overall these data strongly support the hypothesis that GOF mutant p53 proteins may contribute to prevent both the formation of the autophagic vesicles and their fusion with lysosomes in cancer cells.
Figure 1.
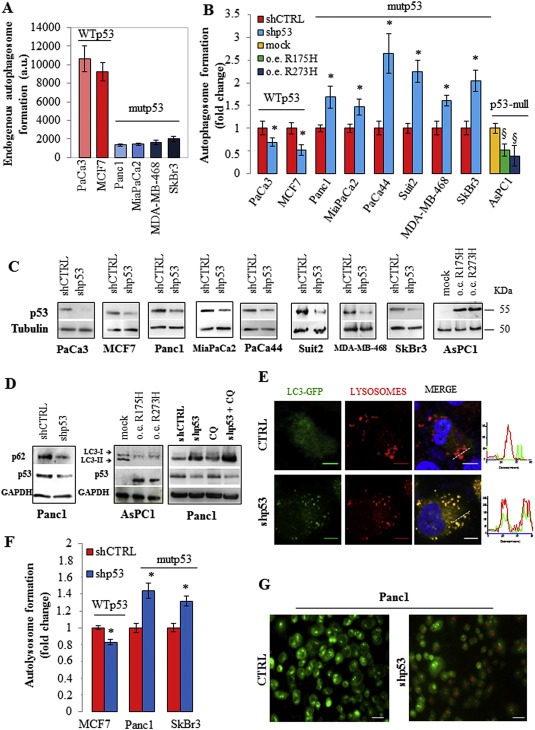
Mutant p53 counteracts autophagy. (A) The endogenous basal level of autophagosomes in cancer cells carrying WT or mutant p53 was analyzed using the incorporation of the fluorescent probe monodansylcadaverine (MDC). Cells were seeded in 96‐well plates, incubated overnight, and stained with MDC as described in Material and Methods. (B) Cells were transfected with the pRSuper‐p53 vector (shp53) to knockdown p53 expression (see Material and Methods) or with plasmids coding mutp53R175H, mutp53R273H proteins, or their relative negative controls (shCTRL or mock, respectively). Autophagosome formation assay was performed using the incorporation of MDC probe. (C) Western blot of p53 using the protein extract of the samples used for (B) to test the effective knock down of WT or mutant p53 and the overexpression of mutant p53 in the various cell lines indicated. (D) Panc1 cells were transfected with pRSuper‐p53 vector (shp53) or its negative control (shCTRL) and AsPC‐1 cells with plasmids for mutant p53 overexpression or its mock vector. Panc1 cells were pre‐treated with 5 μM chloroquine (CQ) for 1 h before cell transfection. Whole‐cell extracts were used for Western blot analysis of the autophagic proteins p62/SQSTM1 and LC3 (isoforms I and II), p53 and GAPDH (as control loading). (E) 48 h after co‐transfection with plasmids coding for LC3‐GFP and pRSuper‐p53 vector (shp53) or LC3‐GFP and the empty vector (negative control; CTRL), Panc1 cells were fixed. Lysosomes and nuclei were stained with Lysotracker (red) and Hoescht (blue), respectively. The RGB profile plotted along the dashed line drawn in the merge image is also shown. Merge and single channel images come from a single z‐plane. Scale bar 10 μm. (F) Autolysosome formation analyzed by red/green fluorescence intensity ratio quantification of acridine orange (AO) staining in the indicated cells transfected with pRSuper‐p53 vector (shp53) or with its negative control (shCTRL). (G) 48 h after the p53 depletion Panc1 cells were stained with an AO solution (1:1500 in PBS) and observed at 40× magnification. Scale bar 40 μm. All the experiments presented in this figure are representative of three biological replicates. P‐values were calculated with two‐tailed t‐test. Statistical analysis: *p < 0.05 shp53 vs shCTRL; §p < 0.05 R175H or R273H vs mock.
3.2. Autophagy inhibition by mutant p53 increases the proliferation of cancer cells
Mutant p53 proteins have been implicated in the induction of cell proliferation, chemoresistance, disruption of tissue architecture, promotion of migration, invasion and metastasis, and several other pro‐oncogenic properties (Adorno et al., 2009; Di Agostino et al., 2006; Freed‐Pastor and Prives, 2012; Girardini et al., 2011; Walerych et al., 2012). In this study, we considered cancer cell lines where the role of tumor suppressor wild type p53 and oncogenic mutant p53 in the regulation of cell proliferation was well assessed (Supplementary Figure 3) (Cadwell and Zambetti, 2001; Levine, 1997; Muller and Vousden, 2013). To unravel the possible biological role of autophagy inhibition in the aberrant proliferation induced by mutant p53, we knocked‐down the essential autophagic protein Atg5 in Panc1, MiaPaCa2, and MDA‐MB‐468 cells (Mizushima et al., 1998). The MDC assay established that Atg5 knockdown effectively repressed the autophagosome formation triggered by mutant p53 downregulation (Figure 2A; Supplementary Figure 4 ). Importantly, the depletion of Atg5 caused a reversion of cell growth inhibition (Figure 2B) and a decrease of the apoptosis (Figure 2C) stimulated by mutant p53 knockdown. These findings indicated that autophagy inhibition strongly contributed to the increased proliferation and to the concomitant down‐regulation of the apoptotic stimuli driven by mutant p53. Similar results are also observed using 3‐methyladenine (3MA) as autophagy inhibitor at non‐toxic concentrations (Supplementary Figure 2).
Figure 2.
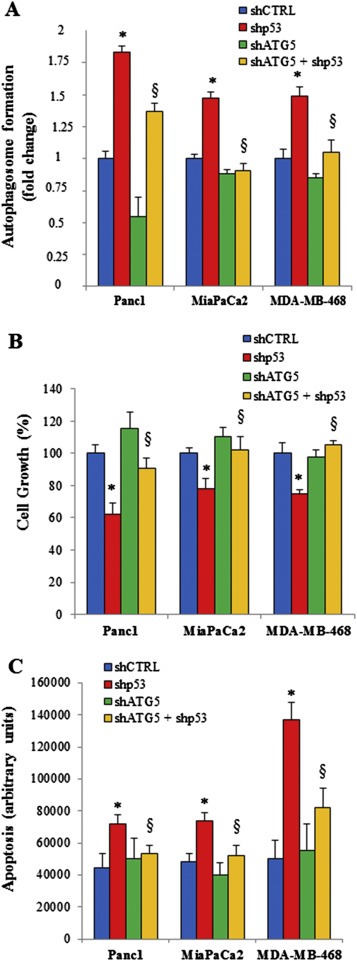
Impact of autophagy inhibition by mutant p53 on cell growth and apoptosis. Panc1, MiaPaCa2 and MDA‐MB‐468 cell lines were seeded in 96‐well plates and transfected with pRSuper‐p53 vector (shp53) and/or pMSCV‐Puro‐miR30‐Atg5 (shAtg5), or their relative negative controls (shCTRL) for 48 h. Autophagosome formation (A), cell growth (B), and apoptosis (C) were determined using MDC assay, crystal violet colorimetric assay and annexinV/FITC binding assay, respectively. All the experiments presented in this Figure are representative of three biological replicates. P‐values were calculated with two‐tailed t‐test. Statistical analysis: *p < 0.05 shp53 vs shCTRL; §p < 0.05 shp53+shATG5 vs shp53.
3.3. Mutant p53 protein inhibits the expression of autophagic genes in tumoral cell lines and cancer patients
Autophagic pathway is a complex sequence of biological events leading to formation, maturation and fusion of autophagosomes with lysosomes to allow the degradation and recycling of cellular components (Viry et al., 2014). These events are driven by a number of autophagy‐related genes (ATGs), which are mostly transcriptionally induced by autophagic stimuli such as nutritional deprivation, infections or metabolic and oncogenic stress (Galluzzi et al., 2015). In the present study, we have shown that tumor cells carrying mutant p53 proteins escaped autophagic activity (1, 2). In order to investigate the role of mutant p53 in autophagy inhibition, we studied whether mutant p53 proteins could control the expression of some crucial ATGs, as atg12, becn1, sesn1, sesn2 and dram1 genes. The depletion of mutant p53 by p53 siRNA smart pool oligos transfection (sip53), determined an increase of the mRNA expression in most of these ATGs in Panc1, MDA‐MB‐468 and SKBr3 cell lines (Figure 3A, Supplementary Figure 5). In these cell lines, we have checked the CCNB1 mRNA expression as a control of our cellular systems (Supplementary Figure 6). Cyclin B protein is related to the cell cycle proteins, important for the control of the cell cycle progression, and is a well‐established mutant p53 target gene (Di Agostino et al., 2015; Di Agostino et al., 2006). Conversely, the depletion of WTp53 expression in MCF7 breast cancer cell line led to a general decrease of the expression of ATGs (Figure 3A, Supplementary Figure 5). Accordingly, ectopic expression of mutp53R175H or mutp53R273H in p53‐null H1299 cells led to a significant reduction of ATGs transcripts (Figure 3A, Supplementary Figure 5, Supplementary Figure 6). To further investigate the inverse relationship between the expression of mutant p53 and two representative ATGs (atg12 and becn1 transcripts), we queried public gene expression data repositories (http://www.oncomine.org/) (Rhodes et al., 2004). In Figure 3B, we reported that atg12 and becn1 transcripts increased substantially in three separate biological clones of MDA‐MB‐468 breast cancer cells (carrying mutp53R273H) where the mutant p53 knock‐down was induced by doxycycline (DOX) [our elaboration of the data set from Freed‐Pastor and colleagues published in (Freed‐Pastor et al., 2012)]. This result was further corroborated by our additional analysis of the data set from Gluck et al. (Gluck et al., 2012). where atg12 and becn1 transcripts were significantly down‐regulated in breast cancer patients carrying mutant p53, compared to those with WTp53 (Figure 3C). Altogether these data strongly confirm the inhibitory role of mutant p53 in vitro and in vivo on the expression level of ATGs. Moreover, to test the hypothetical prognostic value of ATGs in cancer patients, we considered a gene expression signature (atg12, becn1, sesn1, and dram1) by selecting autophagy‐related genes that were up‐regulated by mutant p53 depletion in Figure 3A. About 70% of the breast cancer alterations in TP53 are missense mutations (TCGA) and where only missense mutations compared to the frame‐shift and non‐sense mutations have been demonstrated to have Gain‐Of‐Function activities (Bertheau et al., 2013). It is for this reason why we focused our attention only on a subgroup of breast cancer patients expressing mutant p53 proteins independent by intrinsic subtype (luminal, basal‐like, Her+) to assess the potential correlation between the experimental autophagic gene signature and the patient's clinical outcome (Figure 3D). We analyzed gene expression datasets and survival information downloaded from GEO and sorted for the presence of TP53 gene mutations (www.ncbi.nlm.nih.gov/geo/) (www.kmplot.com) (Gyorffy et al., 2010). To analyze the prognostic value of the signature, we split patients into two groups (high and low expression levels of autophagic gene signature) by the median of the expression values. The two groups were then compared in terms of relapse free survival (RFS) and distant metastasis free survival (DMFS). KM analysis in the Figure 3D showed that low atg12, dram1, sesn1 and becn1 signature expression was significantly associated with a poor prognosis in the mutant p53 breast cancer subgroup (RFS Statistically significant results with p‐value = 0.02, N = 188; DMFS Statistically significant results with p‐value = 0.0028, N = 83).
Figure 3.
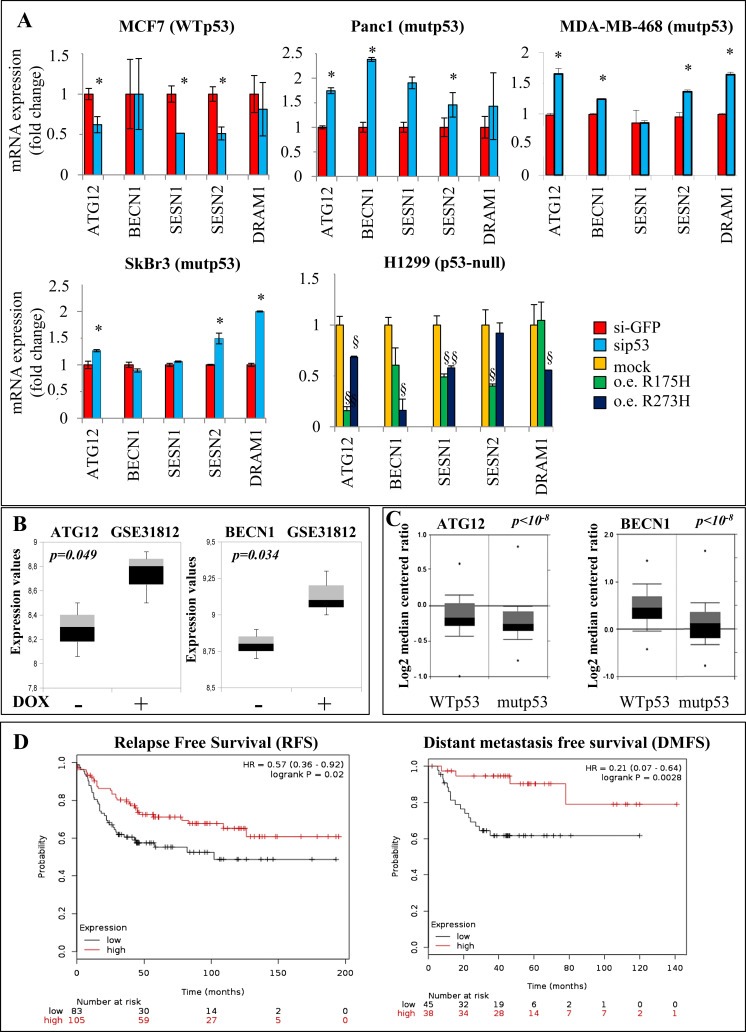
Autophagy‐related gene regulation by mutant p53. (A) The indicated cell lines were transfected with si‐p53 and si‐GFP (as control) oligonucleotides. H1299 cells were transfected with mutp53R175H, mutp53R273H or pcDNA3 (mock) vectors for 48 h. Gene expression analysis of the autophagy‐related genes ATG12, BECN1, SESN1, SESN2, and DRAM1 was performed by RT‐qPCR and was normalized to GAPDH mRNA. *p < 0.05 si‐p53 vs si‐GFP; §p < 0.05 R175H or R273H vs mock. All the experiments are representative of three biological replicates. P‐values were calculated with two‐tailed t‐test. (B) Box plot of ATG12 and BECN1 mRNAs expression obtained in MDA‐MB‐468 breast cancer cells by elaboration of data set from Freed‐Pastor and colleagues (Freed‐Pastor et al., 2012; Rhodes et al., 2004). (C) Box plots of ATG12 and BECN1 mRNAs expression in breast cancer patients (n = 72 for wt‐p53 and n = 72 for mut‐p53). Data from Gluck and colleagues were obtained from www.oncomine.org website (Gluck et al., 2012). (D) Kaplan‐Meier survival curves of relapse free survival (RFS) and distant metastasis free survival (DMFS) of breast cancer patients bearing mutant TP53 gene classified according to the expression of atg12, becn1, sesn1, and dram1 signature. RFS Statistically significant results with p‐value = 0.02, N = 188; DMFS Statistically significant results with p‐value = 0.0028, N = 83). The two compared groups are the patients with the highest expression (red) levels of the signature versus the patients with the lowest expression (black).
3.4. Mutant p53 inhibits atg12 expression by interaction with NF‐κB p50 subunit
We and others have shown that mutant p53 binds to the promoters of its target genes through cooperating with other transcription factors, such as E2F1, NF‐Y, Sp1 and others (Di Agostino et al., 2006; Stambolsky et al., 2010; Strano et al., 2007; Valenti et al., 2015; Weisz et al., 2007). Recently, we have also shown that mutant p53 was able to repress the DNA repair gene expression by the association with the transcriptional repressor E2F4 (Valenti et al., 2015). Since ATG12 is an essential mediator of the initial phases of autophagosome vesicle formation, we studied this gene looking for promotorial consensus regions binding transcriptional repressors. Our in silico analysis by using MatInspector software (www.genomatix.de) revealed the presence of two NF‐κB p50 specific consensus sequences in the atg12 promoter: atg12 promoter Region 1 (−1055 bp) and atg12 promoter Region 2 (−513 bp) (Figure 4A). MatInspector software uses TRANSFAC matrix that was able to discriminate between the specificity of binding between NF‐κB p50 and NF‐κB p65 factors conferring different and specific position frequency to the nucleotides belonging to the promotorial consensus sequence (Supplementary Figure 7). As NF‐κB p50 usually acts as a transcriptional repressor (Elsharkawy et al., 2010; Tong et al., 2004), we studied its recruitment onto the atg12 promoter. Chromatin immunoprecipitation experiments (ChIP) in SKBr3 cells revealed that p50 was actually bound to both two binding regions of atg12 promoter and that this protein‐DNA interaction was removed by transfection of siRNAp50 oligos (Figure 4B; Supplementary Figure 1A). Notably, ChIP assay showed that also mutant p53 protein was recruited onto p50 consensus sites and its binding decreased after p50 depletion (Figure 4B). As expected by the in silico analysis, NF‐κB p65 did not bind the two atg12 promoter regions instead recruited NF‐κB p50 (Figure 4B). The same results were observed in the ChIP experiment in Panc1 cells (Supplementary Figure 1B). In order to support new evidence that mutant p53 and p50 bind onto the atg12 gene promoter, we aimed to investigate the existence of a novel floating mutp53/p50 protein complex. Coimmunoprecipitation experiments from whole protein extracts of Panc1 and SKBr3 cancer cell lines endogenously expressing p53 mutant proteins revealed the formation of a mutp53/p50 protein complex (Figure 4C). NF‐κB p65 was not detected in this protein complex in any coimmunoprecipitation conditions, confirming its absence on the atg12 promoter previously discussed (Figure 4C). The formation of endogenous mutant p53/p50 protein complex was further observed also in MDA‐MB‐468 and MDA‐MB‐231 cancer cells (Supplementary Figure 8). We supported this result by also showing the formation of the complex between the overexpressed mutp53R175H and endogenous p50 proteins in H1299 cells (Figure 4C). Strikingly, the depletion of p50 led to the resumption of atg12 mRNA transcription in mutant p53 Panc1 and MDA‐MB‐468 cells (Figure 4D). According to the above results, the p50 knock down had a functional effect on the increase of autophagosome formation only in SKBr3 and Panc1 cells (bearing mutant p53), thus supporting the inhibitory role of mutant p53/p50 complex in the autophagic process (Figure 4E).
Figure 4.
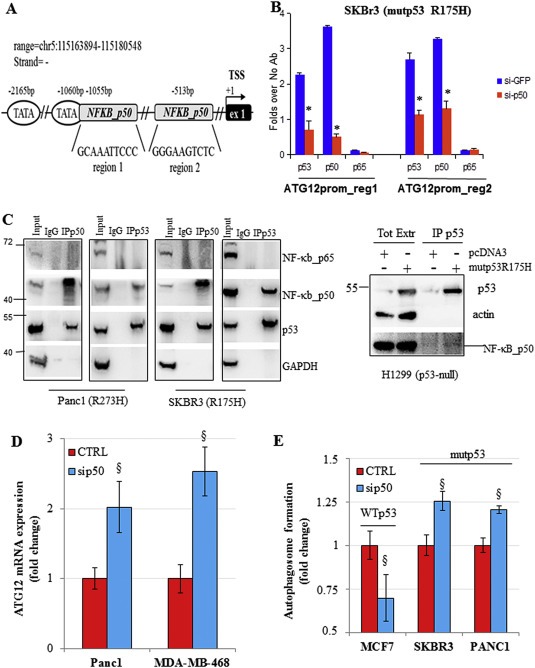
Mutant p53 inhibits atg12 expression by interaction with NF‐κB p50 subunit. (A) Schematic representation of ATG12 promoter gene regions containing NF‐κB p50 consensus box sequences analyzed in ChIP assays. ATG12 promoter: two regions at 1055 bp (region 1) and 513 bp (region 2) upstream the first exon of ATG12 gene. The TSS is indicated and it was predicted by Eponine software (Down and Hubbard, 2002). (B) Cross‐linked chromatin derived from sip50‐treated SKBr3 cells and from siGFP‐transfected control, was immunoprecipitated with the indicated antibodies or in the absence of antibody, and analyzed by RT‐qPCR with specific primers (see Material and Methods or Supplementary Table 2) for the indicated regions in the Figure 4A. Each point of the experiment was carried out in biological duplicate and each reaction of each sample in RT‐qPCR was carried out in technical replicate. The P‐values were calculated with two‐tailed t‐test. *p < 0.05. (C) (Left panel) Immunoprecipitations of NF‐κB p50 or NF‐κB p65 proteins and western blot analysis for p53 binding are performed from lysates of the indicated cancer cell lines expressing mutant p53 proteins (described in Material and Methods). NF‐κB p50 or NF‐κB p65 proteins were immunoprecipitated with their relative rabbit polyclonal antibody and the same amount of rabbit IgG was used as negative control of IP. (Right panel) Cell lysate from H1299 cells over‐expressing mutp53R175H protein was immunoprecipitated with p53 sheep polyclonal antibody. Cells transfected with the empty vector (pcDNA3) was used as negative control. (D) Cells were transfected with the siRNAp50 or its negative control for 48 h. Gene expression analysis of ATG12 mRNA was performed by RT‐qPCR and normalized to GAPDH mRNA. This experiment was a biological triplicate. The P‐values were calculated with two‐tailed t‐test. Statistically significant results were with p‐value<0.05. (E) Cells were transfected with siRNAp50 or its relative negative control. Autophagosome formation assay was performed using the incorporation of MDC probe. Statistical analysis: *p < 0.05 p53 or p50 vs No Ab; § sip50 vs CTRL
3.5. Mutant p53 proteins inhibit AMPK and stimulate mTOR signaling
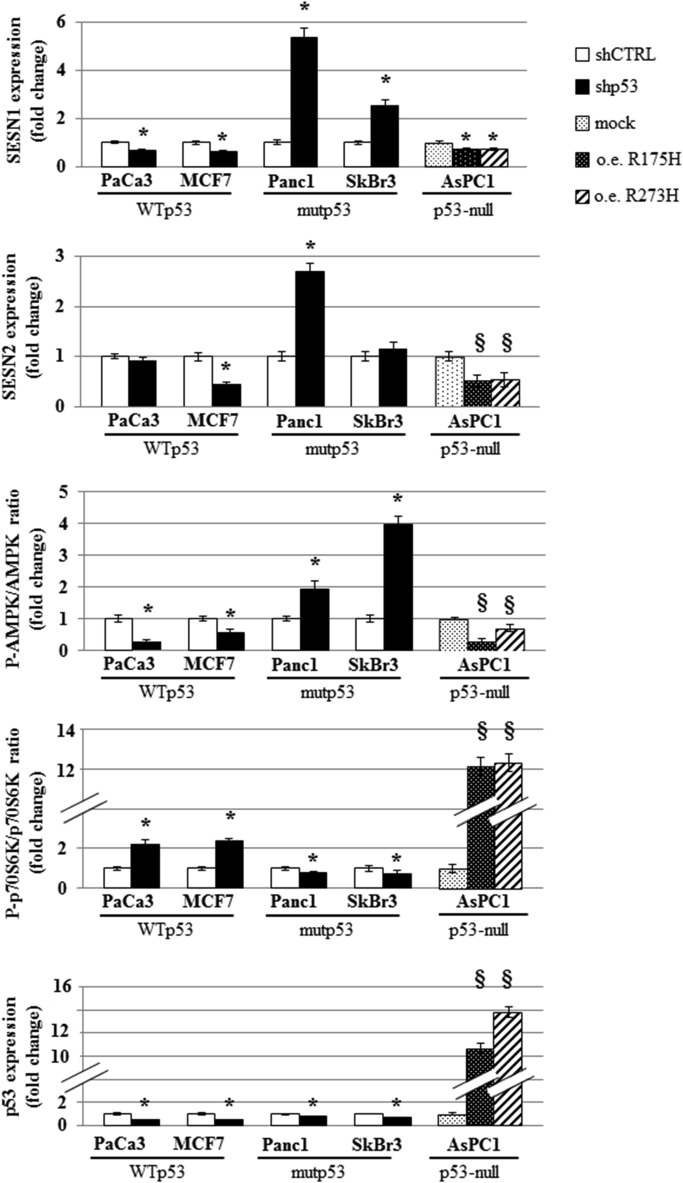
In order to examine whether mutant p53 impinged in the crucial regulatory kinase complexes of the autophagic process we analyzed the protein expression and the phosphorylation state in different cancer cell lines expressing wild type or p53 mutant proteins. We found that the knock down of mutant p53 in either Panc1 and SKBr3 cell lines led to an increase of Sestrin1 and Sestrin2 protein levels, while silencing of wild type p53 in PaCa3 and MCF7 cancer cells resulted in a general down‐regulation of Sestrin protein levels (Figure 5A). Sestrins are a class of proteins that can directly interact with AMPK subunits favoring their phosphorylation by upstream kinases and thereby resulting in AMPK signaling stimulation (Budanov and Karin, 2008; Morrison et al., 2015; Sanli et al., 2012). To determine whether mutant p53 could affect the phosphorylation of AMPK, we analyzed the level of phospho‐AMPKα (Thr172) after modulation of mutant or wild type p53 expression. We observed that depletion of mutant p53 was able to increase the phosphorylation level of AMPK [P(Thr172)‐AMPK/AMPK] and to decrease that of p70S6K [P(Thr)389‐p70S6K/p70S6K], a direct target of mTOR signaling (Figure 5A). Similar results were obtained with pLVTHM‐p53 vector confirming data observed with pRSUPER‐p53 vector (Supplementary Figure 9). Accordingly, the ectopic expression of mutp53R175H or mutp53R273H clearly determined the repression of AMPK phosphorylation and the increase of p70S6K phosphorylation (Figure 5A). To better represent our results, we quantified P‐AMPK/P‐p70S6K ratio (Figure 5B), highlighting that wild type and mutant p53 proteins produced inverse effects on these signaling pathways. Overall, quantification and statistical evaluation of the proteins representatively shown in Figure 5A are reported in Supplementary Figure 10. To address the role of AMPK phosphorylation mutant p53‐dependent, we inhibited AMPK activity treating the cancer cells with compound C (CC) or overexpressing a dominant negative (DN) AMPK isoform (Figure 5C and D). Strikingly, AMPK signaling inhibition by both CC and DN‐AMPK strongly rescued the autophagosome formation induced by mutant p53 knock down (Figure 5C). Consistently, in the same conditions of AMPK signaling inhibition the oncogenic proliferation impaired by mutant p53 knock down was restored in all three cell lines tested (Figure 5D). Overall, these results provide robust evidence that mutant p53 exerts its inhibitory activity on the autophagic signaling throughout the constitutive blockage of AMPK signaling.
Figure 5.
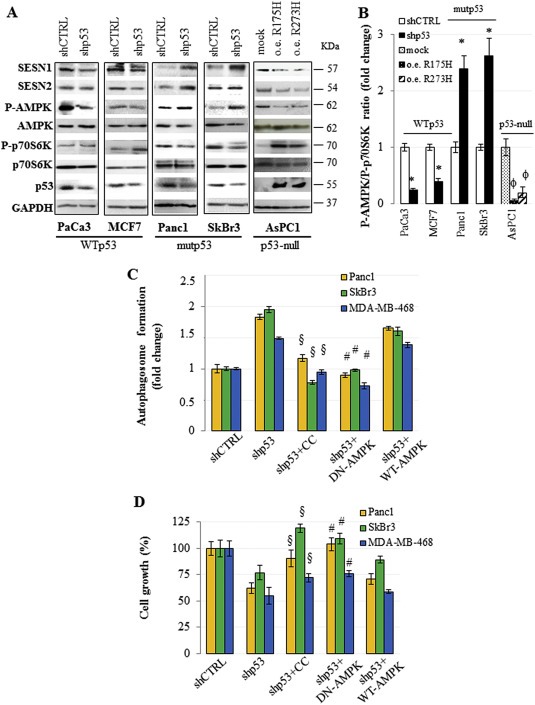
Mutant p53 inhibits AMPK and stimulates mTOR. (A) Cells were seeded in 100‐mm diameter culture dishes and transfected for 48 h with the pRSuper‐p53 vector (shp53) and with plasmids for the ectopic expression of mutant p53 (o.e. R175H; o.e. R273H), or their relative negative control (shCTRL or mock, respectively). Western blotting analysis from these cells was performed with 50 μg of whole cell extracts and probed with the indicated antibodies. This analysis was representative of three biological replicates. (B) Quantitative analysis of P‐AMPK/P‐p70S6K ratio. The bands shown in (A) were scanned as digital peaks and the areas of the peaks were reported as fold change, as described in Material and Methods. (C and D) The indicated cell lines were transfected for 48 h with the pRSuper‐p53 vector (shp53) or its negative control (shCTRL), in the absence or presence of 20 μM Compound C (CC) or together the expression vector coding for WT or dominant negative (DN; mutant R531G) AMPK γ2 subunit. Autophagosome formation (C) and cell growth (D) were analyzed by using MDC and crystal violet staining, respectively. Statistical analysis: *p < 0.05 shp53 vs CTRL; ϕ R175H or R273H vs mock; §p < 0.05 shp53+CC vs shp53; #p < 0.05 shp53+DN‐AMPK vs shp53+WT‐AMPK.
3.6. Mutant p53 inhibits Beclin1 phosphorylation by mTOR stimulation and its expression level
Given that mTOR pathway was controlled by mutant p53 (Figure 5), we speculated that our finding was in correlation with the fact that mTOR complex might de‐phosphorylate Beclin1 via ULK1 inhibition impairing the formation of Beclin1‐mediated autophagic protein complexes and autophagy maturation (Russell et al., 2013). To gain insight into the molecular mechanisms controlling the Beclin1 expression, we observed that the overexpression of mutp53R175H or mutp53R273H proteins in AsPC1 cells decreased both Beclin1 phosphorylation in Serine 15 and its total protein expression level (Figure 6A). Accordingly, the depletion of mutant p53 from Panc1 cells exerted the inverse results (Figure 6A). These data correlated with the Beclin1 mRNA expression shown in Figure 3.
Figure 6.
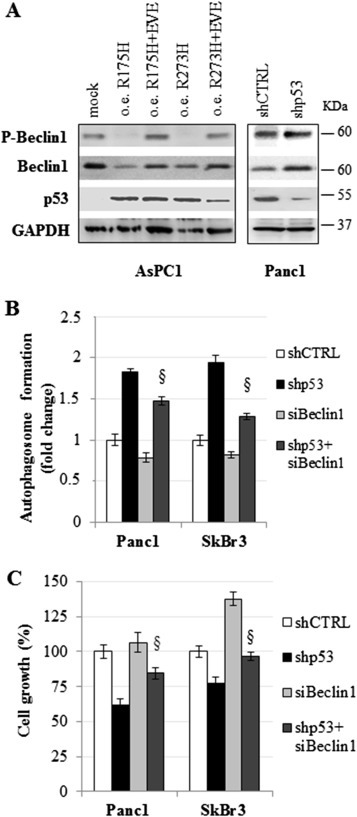
Mutant p53 inhibits Beclin1. (A) AsPC1 cells were seeded in 100‐mm diameter culture dishes and transfected for 48 h with the plasmids coding for mutant p53 proteins (R175H or R273H), or with pCDNA3 as negative control (mock), in the absence or presence of 5 μM everolimus (EVE). Panc1 cells were knocked‐down for mutant p53 expression by 48 h of pRSuper‐p53 vector transfection (shp53). Whole‐cell extracts were used for Western blot analysis using the indicated antibodies. (B and C) Panc1 and SkBr3 cells were seeded in 96‐well plates and transfected with the pRSuper‐p53 vector (shp53) and/or si‐Beclin1 oligos (siBeclin1). Autophagosome formation (B) and cell growth (C) were analyzed after 48 h by the transfection using MDC and crystal violet staining, respectively. These experiments were biological triplicates. The P‐values were calculated with two‐tailed t‐test. Statistical analysis: §p < 0.05 shp53+siBeclin1 vs shp53.
In line with the findings from Russel and colleagues, we also investigated whether the stimulation of mTOR signaling by mutant p53 (shown in Figure 5) may be responsible for the decrease in levels of Beclin1 phosphorylation. We observed that the addition of everolimus (RAD001), an inhibitor of mTOR currently used in the therapy of several human cancers, was able to completely restore Beclin1 phosphorylation repressed by overexpression of both R175H or R273H p53 mutant proteins, indicating that mTOR stimulation by mutant p53 is highly involved in Beclin1 de‐phosphorylation (Figure 6A). Quantification and statistical evaluation of Beclin1 and of its phosphorylated form are shown in Supplementary Figure 11. Since Beclin1 phosphorylation has been described to be crucial for ATG proteins interaction during autophagosome vesicles formation, we investigated the role of Beclin1 repression on overall autophagy inhibition by mutant p53. We observed that the knockdown of Beclin1 (Supplementary Figure 12) significantly reduced the autophagosome formation induced by the depletion of mutant p53 protein (Figure 6B), thus suggesting a role for Beclin1 repression on the autophagy pathway inhibition driven by mutant p53. According to this, the concomitant depletion of mutant p53 and Beclin1 led to a partial rescue of proliferation ability of cancer cells as compared to mutant p53 knockdown conditions (Figure 6C). Overall, our data strongly support the negative effect of mutant p53 on the Beclin1 functionality throughout the inhibition of its expression and phosphorylation via mTOR stimulation.
3.7. Mutant p53 sensitizes cancer cells to mTOR inhibition
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR) routinely used in the targeted therapy of many cancers. We aimed to investigate whether cancer cells bearing mutant p53 might be more responsive to everolimus than wild type p53 cancer cells, accordingly with the inhibitory role of mutant p53 on autophagy via mTOR pathway induction (5, 6). To this end, we incubated everolimus Panc1, SkBr3 and MCF7 cell lines and observed that the cells expressing mutant p53 (Panc1 and SKBr3) were more sensitive than cells with wild type p53 (MCF7) to the treatment as reported by the IC25 and IC50 values (Figure 7A). To functionally demonstrate the involvement of mutant p53 on cancer cell sensitivity to everolimus, we evaluated the response of the cells after knockdown or overexpression of p53 mutant proteins. Our data reported in Figure 7B, showed that mutant p53 knockdown significantly decreased Panc1 and SkBr3 cell response to everolimus treatment if compared to controls. To corroborate this result, we assessed that the overexpression of R175H or R273H mutant p53 conferred to p53‐null AsPC1 cells a strong sensitization to everolimus incubation, as compared to its negative control (mock vector). Moreover, the dominant negative effect of p53 mutant proteins ectopically expressed in WTp53 cells (PaCa3), allowed a stronger impact resulting in a higher response to everolimus incubation in comparison to the PaCa3 cells under control conditions (Figure 7B). Overall, these results demonstrated that targeting the mTOR pathway may represent an effective therapeutic strategy for cancer patients bearing mutations in the TP53 gene.
Figure 7.
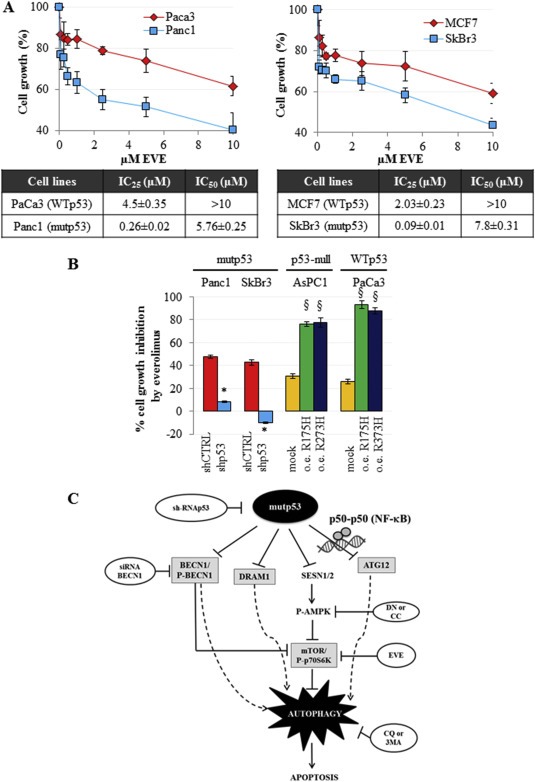
Mutant p53 sensitizes cancer cells to everolimus. (A) Cells were seeded in 96‐well plates and treated for 48 h with increasing concentrations of everolimus. Cell growth was measured using the crystal violet colorimetric assay and IC25 and IC50 values for each cell line tested have been reported. (B) Cells were seeded in 96‐well plates and transfected with pRSuper‐p53 vector (shp53) and with the plasmids coding for mutant p53 proteins (R175H or R273H), or with empty pRSuper (shCTRL) or pCDNA3 (mock), respectively, as negative controls. For each experimental condition, cells were untreated or treated with 5 μM everolimus for 48 h. Cell growth was determined using the crystal violet colorimetric assay. The rate of cell growth inhibition corresponds to the effect of everolimus, as compared to vehicle (untreated), in each indicated transfection condition. These experiments were at least biological triplicates. The P‐values were calculated with two‐tailed t‐test. Statistical analysis: *p < 0.05 shp53 vs shCTRL; §p < 0.05 R175H or R273H vs mock. (C) Model of the molecular mechanism unraveled in the present study.
4. Discussion
Autophagy is a tightly‐regulated catabolic process of cellular self‐digestion by which cellular components are targeted to lysosomes for their degradation. However, the role of autophagy in modulating cell death is highly dependent on the metabolic context and on the microenvironmental conditions in which the cells lie (Lorin et al., 2013). Key functions of autophagy are aimed to provide energy and metabolic precursors under conditions of starvation and to alleviate cellular stress removing damaged proteins and organelles, which are deleterious for cell survival. Therefore, autophagy appears to serve as a pro‐survival stress response in most settings (Fiorini et al., 2013; Matsuzawa et al., 2015). Increasing evidence show its pivotal role in cell death after treatment with anti‐cancer drugs or under particular stressful conditions (Fiorini et al., 2015a; Fulda and Kögel, 2015). Thus, despite the net effect of autophagy in cancer cells is highly contextual, substantial results describe that enforced over‐activation of autophagy can lead to excessive cellular self‐digestion via the autophagosomal‐lysosomal pathway determining autophagic cell death (also named cell death type II) (Galluzzi et al., 2012). The importance of autophagic cell death becomes evident by the consideration that it is able to bypass drug resistance, especially in apoptosis‐refractory tumors, offering new therapeutic options in overcoming cancer resistance mechanisms (Fulda and Kögel, 2015; Schonthal, 2009). Since Avantaggiati and colleagues have previously demonstrated that mutant p53 stability is controlled by lysosomal degradation and not by proteasomal involvement as occurs for wild type p53 (Choudhury et al., 2013), autophagy inhibition by mutant p53 may, at least partially, explain also the high expression levels of mutant p53 itself observed in cancer cells.
Recently Mathew and colleagues have clearly shown how defects in the autophagic pathway could diminish survival and enhance tumorigenesis with the concomitant accumulation in the DNA mutations (Mathew et al., 2007). The high mutation rate and the severe genomic instability are the most important features of the cancers expressing mutant p53 proteins (Negrini et al., 2010; Soussi and Wiman, 2007). Intriguingly, in this study we intended to deeply investigate the molecular mechanisms related to autophagy inhibition by GOF p53 mutants, which can be summarized with the following considerations schematically illustrated in Figure 7C: i) mutant p53 inhibits the expression of a panel of autophagy‐related genes, including becn1, dram1, sesn1/2, and atg12; ii) autophagy inhibition by mutant p53 is mediated by its interaction with p50 NF‐κB subunit on the atg12 gene promoter; iii) mutant p53 inhibits SESN1/AMPK axis and stimulates mTOR pathway; iv) mutant p53 inhibits P‐Beclin1 via mTOR sensitizing cancer cells to everolimus.
During the last several years, Morselli et al. stated that the cytoplasmic localization is the preponderant requirement for mutant p53 to inhibit autophagy indicating that those variants of mutant p53 that preferentially localize in the cytoplasm effectively repress autophagy, whereas p53 mutants that display a prominently nuclear distribution fail to inhibit it (Morselli et al., 2008). However, these previous observations exclusively reached by mutant p53 over‐expression assays are not confirmed by our investigation. Indeed, we reveal that all tested GOF p53 mutant variants, listed in Table 1, acquire the property to inhibit autophagy irrespectively of their tissue of origin, such as breast, pancreas or lung, or of their prevalent subcellular localization previously determined by Morselli et al. (Morselli et al., 2008). In addition, our results indicate that the repression of the autophagic machinery is functional to the hyper‐proliferating and anti‐apoptotic activities of these hot‐spot p53 mutants in cancer cells. Very recently, Gu et al. have identified an autophagy‐related prognostic signature for breast cancer (Gu et al., 2015) analyzing six independent public‐domain microarrays from GEO database. They reported that inactivation of autophagy was associated with shortened survival of breast cancer patients and that a set of eight genes (bcl2, birc5, eif4ebp1, ero1L, fos, gapdh, itpr1 and vefgA) were assigned as an autophagy‐related prognostic signature for breast cancer. However, despite that their statement is consistent with our data, the genes identified by Gu et al. have a high level of functional versatility thus lacking in autophagy specificity of the signature. In this study, we show that low expression of atg12, dram1, sesn1 and becn1 autophagy‐related signature was significantly associated with a poor prognosis in the mutant p53 breast cancer subgroup. This consideration strongly reinforces the statement of autophagy as an anti‐proliferative and pro‐apoptotic event that cancer cells try to escape in vivo. Furthermore, autophagy‐deficient cell lines increase DNA double‐strand breaks and other chromosome abnormalities suggesting a role for the autophagic pathway in suppressing genome damage and inhibition of pro‐tumorigenic process (Mathew et al., 2007). In addition, we demonstrated a protein‐protein interaction between p50 NF‐κB subunit and mutant p53 and their recruitment in two regions of atg12 promoter. Intriguingly, we observed that the depletion of p50 protein increased both atg12 mRNA expression and autophagosome formation in cancer cells bearing mutant p53. Therefore, it is tempting to speculate that the interaction between mutant p53 and the p50‐p50 homodimer could represent a novel molecular mechanism by which mutant p53 is able to repress the expression of its target genes (Guan et al., 2005; Tong et al., 2004). The existence of this functional interaction might have a great impact in cancer cell biology contributing to the discovery of new drugs potentially able to inhibit this oncogenic mechanism.
Concerning signal transduction pathways involved in autophagy inhibition, our data are in accordance with a recent study of Myers and colleagues which indicated that under conditions of energy stress, GOF p53 mutants, but not WTp53, can bind to the AMPKα subunit inhibiting AMPK signaling (Zhou et al., 2014). We further documented that mutant p53 suppressed the expression of Sestrins which have been described to act as AMPK activators through the direct interaction with AMPK complex favoring its phosphorylation by upstream kinases (Budanov and Karin, 2008; Morrison et al., 2015; Sanli et al., 2012). Overall, we showed that AMPK signaling inhibition was functionally implicated on autophagy counteraction by mutant p53. Moreover, we demonstrated that mTOR pathway stimulation by mutant p53 was able to repress Beclin‐1 phosphorylation, which is required for activating the ATG14L‐VPS34 complex during autophagosome formation (Russell et al., 2013), thus orchestrating autophagy machinery inhibition driven by mutant p53. Indeed, it is important to highlight that mutant p53 orchestrated a number of adjustments aimed at inhibiting the autophagic flow starting from ATG regulation to autophagic vesiscles formation and their fusion with lysosomes. We finally underline the translational relevance of our data concerning the potential use of the mTOR inhibitor everolimus for cancer patients carrying mutant TP53 gene.
5. Conclusions
Overall, we have observed that mutant p53: i) inhibited mRNA and protein expression levels of various key ATGs (3, 4, 5, 6); ii) inhibited the formation of autophagic vesicles and formation of the mature isoform of LC3 (Figure 1); iii) inhibited fusion of the autophagic vesicles with lysosomes, formation of autolysosomes and lysosomal acidification of the lumen (Figure 1); iv) inhibited AMPK signaling and stimulated mTOR pathway making cancer cells bearing mutant p53 susceptible to mTOR inhibition (5, 7). Altogether these observations also provided new prognostic tools and innovative insights into mutant p53 GOF activities in cancer cell biology, strongly supporting mTOR inhibition as a personalized therapeutic strategy for cancer patients carrying mutant p53.
Conflict of interest
The authors declare that they have no conflicts of interest.
Supporting information
The following are the supplementary data related to this article:
Suppl. Figure 1 (A) Cells were seeded in 100‐mm diameter culture dishes and transfected for 48 h with sip50 oligos or with negative control (siGFP). 40 μg of total protein extracts were analyzed by Western blotting using p50 NF‐κB and GAPDH or H3 (as normalizing factor) antibodies. (B) Cross‐linked chromatin derived from sip50‐treated Panc1 cells and from siGFP‐transfected control, was immunoprecipitated with the indicated antibodies or in the absence of antibody, and analyzed by RT‐qPCR with specific primers (see Material and Methods or Supplementary Table 2) for the regions of ATG12 promoter indicated in the Figure 4A or in H1H2B2 genic region resulted negative for the recruitment of mutant p53. *p < 0.05.
{kind=link}
Suppl. Figure 2 (A) Panc1 cells were pre‐treated with 1 mM 3MA for 1 h before cell transfection with plasmids coding for LC3‐GFP and pRSuper‐p53 vector (shp53) or empty vector (shCTRL) for 48 h. Cells were fixed and nuclei were stained with Hoescht (blue). Merge images come from a single z‐plane. Scale bar 10 μm. (B) Panc1 cells were pre‐treated with 1 mM 3MA for 1 h before cell transfection for 48 h. Whole‐cell extracts were used for Western blot analysis of the autophagic protein LC3 (isoforms I and II), p53 and GAPDH (as control loading). (C‐E) Panc1 and MDA‐MB‐468 cells were seeded in 96‐well plates and pre‐treated with 1 mM 3MA for 1 h before cell transfection for 48 h. Autophagosome formation (C), cell growth (D), and apoptosis (E) were determined using MDC assay, crystal violet colorimetric assay and annexinV/FITC binding assay, respectively. All the experiments presented in this figure are representative of three biological replicates. P‐values were calculated with two‐tailed t‐test. Statistical analysis: *p < 0.05 shp53 vs shCTRL; §p < 0.05 shp53+3MA vs shp53.
{kind=link}
Suppl. Figure 3 Cells were seeded in 96‐well plates and transfected with pRSuper‐p53 vector (shp53), with pCDNA‐p53R175H, pCDNA‐p53R273H plasmids or their negative controls (empty pRSuper and pCDNA3 mock vector, respectively). Cell growth was determined using the crystal violet colorimetric assay. Statistical analysis: *p < 0.05 shp53 vs CTRL; §p < 0.05 R175H or R273H vs mock.
{kind=link}
Suppl. Figure 4 Panc1 cells were transfected with pMSCV‐Puro‐miR30‐shATG5 vector (or its negative empty vector). Gene expression analysis of ATG5 was performed by RT‐qPCR and was normalized to GAPDH mRNA. *p < 0.05 shATG5 vs shCTRL.
{kind=link}
Suppl. Figure 5 Western blot of p53, and GAPDH as normalizing factor, performed in all cell lines used for RT‐qPCR shown in Figure 3A. To exclude back‐side effects of shp53 sequence (pRSuper‐p53 vector) and to confirm the robustness of the data, a commercial siRNA smart pool of three oligonucleotides (si‐p53) transiently targeting p53 (Santa Cruz Biotech. sc‐29435), and its si‐GFP negative control, were used in these experiments.
{kind=link}
Suppl. Figure 6 (A and B) Indicated cell lines were transfected with pRSuper‐p53 vector (shp53), with pCDNA‐p53R175H, pCDNA‐p53R273H plasmids or their negative controls (empty pRSuper and pCDNA3 mock vector, respectively). Gene expression analysis of CCNB1 was performed by RT‐qPCR and was normalized to GAPDH mRNA. *p < 0.05 sip53 vs siGFP; R175H or R273H vs vector.
{kind=link}
Suppl. Figure 7 TRANSFAC matrix of NF‐κB p50 and NF‐κB p65 consensus sequences used by MatInspector software.
{kind=link}
Suppl. Figure 8 Immunoprecipitations of NF‐κB p50 and western blot analysis for p53 binding are performed from lysates of the indicated cancer cell lines expressing mutant p53 proteins, as described in Material and Methods.
{kind=link}
Suppl. Figure 9 Panc1 cells were transfected with pLVTHM‐p53 vector (shp53) or its negative control pLVTHM (shCTRL) to confirm results obtained with pRSUPER‐p53 vector. (A) Autophagosome formation assay was performed using the incorporation of MDC probe. *p < 0.05 shp53 vs shCTRL (B) Western blot of P‐AMPK, AMPK, P‐p70S6K, p70S6Kp53 and GAPDH was performed as described in Material and Methods.
{kind=link}
Suppl. Figure 10 Quantitative analysis of SESN1/GAPDH, SESN2/GAPDH, P‐AMPK/AMPK, P‐p70S6K/p70S6K and p53/GAPDH ratios representatively shown in Figure 5A. The Western blot bands were scanned as digital peaks and the areas of the peaks were reported as fold change, as described in Material and Methods. *p < 0.05 shp53 vs shCTRL; §p < 0.05 R175H or R273H vs mock.
{kind=link}
Suppl. Figure 11 Quantitative analysis of P‐Beclin1, Becin1 and p53 normalized on GAPDH expression representatively shown in Figure 6A. The Western blot bands were scanned as digital peaks and the areas of the peaks were reported as fold change, as described in Material and Methods. §p < 0.05 R175H or R273H vs mock; *p < 0.05 R175H+EVE vs R175H or R273H+EVE vs R273H; # shp53 vs shCTRL.
{kind=link}
Suppl. Figure 12 Cells were seeded in 100‐mm diameter culture dishes and transfected for 48 h with siBeclin1 oligos or with negative control (siGFP). 40 μg of total protein extracts were analyzed by Western blot using Beclin1 and GAPDH (as normalizing factor) antibodies.
{kind=link}
Supplementary data
Supplementary data
Acknowledgments
We wish to thank Tania Merlino for manuscript editing. This work was supported by Italian Association for Cancer Research (AIRC)‐Fondazione CariPaRo, Padova, Italy (n. C98C13000210007); Joint Projects program 2015 from University of Verona to M. Donadelli (n. B12I15002320003); Italian Association for Cancer Research (AIRC) to S. Di Agostino (AIRC IG‐n.16984) and to G. Blandino (AIRC IG‐n.14455). M. Nadal‐Serrano was supported by a short‐term fellowship for the stay abroad (University of Verona, Italy) from European Molecular Biology Organization (EMBO). I. Dando is a fellow of AIRC (AIRC‐Fondazione CariPaRo, Padova, Italy) and NanoMedicine (Fondazione Cariverona, Project Verona Nanomedicine Initiative). E. Dalla Pozza is a fellow of ARC‐Net (Applied Research on Cancer Network), University of Verona, Italy. E. Butturini is a fellow of Joint Project (University of Verona, Italy and Aboca, Sansepolcro, Arezzo, Italy).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.04.001.
Cordani Marco, Oppici Elisa, Dando Ilaria, Butturini Elena, Dalla Pozza Elisa, Nadal-Serrano Mercedes, Oliver Jordi, Roca Pilar, Mariotto Sofia, Cellini Barbara, Blandino Giovanni, Palmieri Marta, Di Agostino Silvia, Donadelli Massimo, (2016), Mutant p53 proteins counteract autophagic mechanism sensitizing cancer cells to mTOR inhibition, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.04.001.
Contributor Information
Silvia Di Agostino, Email: diagostino@ifo.it.
Massimo Donadelli, Email: massimo.donadelli@univr.it.
References
- Adorno, M. , Cordenonsi, M. , Montagner, M. , Dupont, S. , Wong, C. , Hann, B. , Solari, A. , Bobisse, S. , Rondina, M.B. , Guzzardo, V. , Parenti, A.R. , Rosato, A. , Bicciato, S. , Balmain, A. , Piccolo, S. , 2009. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell. 137, 87–98. [DOI] [PubMed] [Google Scholar]
- Bertheau, P. , Lehmann-Che, J. , Varna, M. , Dumay, A. , Poirot, B. , Porcher, R. , Turpin, E. , Plassa, L.F. , de Roquancourt, A. , Bourstyn, E. , de Cremoux, P. , Janin, A. , Giacchetti, S. , Espie, M. , de The, H. , 2013. p53 in breast cancer subtypes and new insights into response to chemotherapy. Breast. 22, (Suppl 2) S27–S29. [DOI] [PubMed] [Google Scholar]
- Biagioni, F. , Bossel Ben-Moshe, N. , Fontemaggi, G. , Canu, V. , Mori, F. , Antoniani, B. , Di Benedetto, A. , Santoro, R. , Germoni, S. , De Angelis, F. , Cambria, A. , Avraham, R. , Grasso, G. , Strano, S. , Muti, P. , Mottolese, M. , Yarden, Y. , Domany, E. , Blandino, G. , 2012. miR-10b*, a master inhibitor of the cell cycle, is down-regulated in human breast tumours. EMBO Mol Med. 4, 1214–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummelkamp, T.R. , Bernards, R. , Agami, R. , 2002. A system for stable expression of short interfering RNAs in mammalian cells. Science. 296, 550–553. [DOI] [PubMed] [Google Scholar]
- Budanov, A.V. , Karin, M. , 2008. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell. 134, 451–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell, C. , Zambetti, G.P. , 2001. The effects of wild-type p53 tumor suppressor activity and mutant p53 gain-of-function on cell growth. Gene. 277, 15–30. [DOI] [PubMed] [Google Scholar]
- Choudhury, S. , Kolukula, V.K. , Preet, A. , Albanese, C. , Avantaggiati, M.L. , 2013. Dissecting the pathways that destabilize mutant p53: the proteasome or autophagy?. Cell Cycle. 12, 1022–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comel, A. , Sorrentino, G. , Capaci, V. , Del Sal, G. , 2014. The cytoplasmic side of p53's oncosuppressive activities. FEBS Lett. 588, 2600–2609. [DOI] [PubMed] [Google Scholar]
- Crighton, D. , Wilkinson, S. , Ryan, K.M. , 2007. DRAM links autophagy to p53 and programmed cell death. Autophagy. 3, 72–74. [DOI] [PubMed] [Google Scholar]
- DeBerardinis, R.J. , Thompson, C.B. , 2012. Cellular metabolism and disease: what do metabolic outliers teach us?. Cell. 148, 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Agostino, S. , Sorrentino, G. , Ingallina, E. , Valenti, F. , Ferraiuolo, M. , Bicciato, S. , Piazza, S. , Strano, S. , Del Sal, G. , Blandino, G. , 2015. YAP enhances the pro-proliferative transcriptional activity of mutant p53 proteins. EMBO Rep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Agostino, S. , Strano, S. , Emiliozzi, V. , Zerbini, V. , Mottolese, M. , Sacchi, A. , Blandino, G. , Piaggio, G. , 2006. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell. 10, 191–202. [DOI] [PubMed] [Google Scholar]
- Dodson, M. , Darley-Usmar, V. , Zhang, J. , 2013. Cellular metabolic and autophagic pathways: traffic control by redox signaling. Free Radic Biol Med. 63, 207–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Down, T.A. , Hubbard, T.J. , 2002. Computational detection and location of transcription start sites in mammalian genomic DNA. Genome Res. 12, 458–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drakos, E. , Atsaves, V. , Li, J. , Leventaki, V. , Andreeff, M. , Medeiros, L.J. , Rassidakis, G.Z. , 2009. Stabilization and activation of p53 downregulates mTOR signaling through AMPK in mantle cell lymphoma. Leukemia. 23, 784–790. [DOI] [PubMed] [Google Scholar]
- Elsharkawy, A.M. , Oakley, F. , Lin, F. , Packham, G. , Mann, D.A. , Mann, J. , 2010. The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J Hepatol. 53, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiorini, C. , Cordani, M. , Gotte, G. , Picone, D. , Donadelli, M. , 2015. Onconase induces autophagy sensitizing pancreatic cancer cells to gemcitabine and activates Akt/mTOR pathway in a ROS-dependent manner. Biochim Biophy Acta. 1854, 549–560. [DOI] [PubMed] [Google Scholar]
- Fiorini, C. , Cordani, M. , Padroni, C. , Blandino, G. , Di Agostino, S. , Donadelli, M. , 2015. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochim Biophy Acta. 1853, 89–100. [DOI] [PubMed] [Google Scholar]
- Fiorini, C. , Menegazzi, M. , Padroni, C. , Dando, I. , Dalla Pozza, E. , Gregorelli, A. , Costanzo, C. , Palmieri, M. , Donadelli, M. , 2013. Autophagy induced by p53-reactivating molecules protects pancreatic cancer cells from apoptosis. Apoptosis. 18, 337–346. [DOI] [PubMed] [Google Scholar]
- Freed-Pastor, W.A. , Mizuno, H. , Zhao, X. , Langerod, A. , Moon, S.H. , Rodriguez-Barrueco, R. , Barsotti, A. , Chicas, A. , Li, W. , Polotskaia, A. , Bissell, M.J. , Osborne, T.F. , Tian, B. , Lowe, S.W. , Silva, J.M. , Borresen-Dale, A.L. , Levine, A.J. , Bargonetti, J. , Prives, C. , 2012. Mutant p53 disrupts mammary tissue architecture via the mevalonate pathway. Cell. 148, 244–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed-Pastor, W.A. , Prives, C. , 2012. Mutant p53: one name, many proteins. Genes Develop. 26, 1268–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulda, S. , Kögel, D. , 2015 Oct 1. Cell death by autophagy: emerging molecular mechanisms and implications for cancer therapy. Oncogene. 34, (40) 5105–5113. 10.1038/onc.2014.458 [DOI] [PubMed] [Google Scholar]
- Galluzzi, L. , Pietrocola, F. , Bravo-San Pedro, J.M. , Amaravadi, R.K. , Baehrecke, E.H. , Cecconi, F. , Codogno, P. , Debnath, J. , Gewirtz, D.A. , Karantza, V. , Kimmelman, A. , Kumar, S. , Levine, B. , Maiuri, M.C. , Martin, S.J. , Penninger, J. , Piacentini, M. , Rubinsztein, D.C. , Simon, H.U. , Simonsen, A. , Thorburn, A.M. , Velasco, G. , Ryan, K.M. , Kroemer, G. , 2015. Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi, L. , Vitale, I. , Abrams, J.M. , Alnemri, E.S. , Baehrecke, E.H. , Blagosklonny, M.V. , Dawson, T.M. , Dawson, V.L. , El-Deiry, W.S. , Fulda, S. , Gottlieb, E. , Green, D.R. , Hengartner, M.O. , Kepp, O. , Knight, R.A. , Kumar, S. , Lipton, S.A. , Lu, X. , Madeo, F. , Malorni, W. , Mehlen, P. , Nunez, G. , Peter, M.E. , Piacentini, M. , Rubinsztein, D.C. , Shi, Y. , Simon, H.U. , Vandenabeele, P. , White, E. , Yuan, J. , Zhivotovsky, B. , Melino, G. , Kroemer, G. , 2012. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 19, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganci, F. , Sacconi, A. , Bossel Ben-Moshe, N. , Manciocco, V. , Sperduti, I. , Strigari, L. , Covello, R. , Benevolo, M. , Pescarmona, E. , Domany, E. , Muti, P. , Strano, S. , Spriano, G. , Fontemaggi, G. , Blandino, G. , 2013. Expression of TP53 mutation-associated microRNAs predicts clinical outcome in head and neck squamous cell carcinoma patients. Ann. Oncol. 24, 3082–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girardini, J.E. , Napoli, M. , Piazza, S. , Rustighi, A. , Marotta, C. , Radaelli, E. , Capaci, V. , Jordan, L. , Quinlan, P. , Thompson, A. , Mano, M. , Rosato, A. , Crook, T. , Scanziani, E. , Means, A.R. , Lozano, G. , Schneider, C. , Del Sal, G. , 2011. A Pin1/mutant p53 axis promotes aggressiveness in breast cancer. Cancer Cell. 20, 79–91. [DOI] [PubMed] [Google Scholar]
- Gluck, S. , Ross, J.S. , Royce, M. , McKenna, E.F. , Perou, C.M. , Avisar, E. , Wu, L. , 2012. TP53 genomics predict higher clinical and pathologic tumor response in operable early-stage breast cancer treated with docetaxel-capecitabine +/- trastuzumab. Breast Cancer Res. Treat. 132, 781–791. [DOI] [PubMed] [Google Scholar]
- Gomes, L.R. , Vessoni, A.T. , Menck, C.F. , 2015 Oct 6. Three-dimensional microenvironment confers enhanced sensitivity to doxorubicin by reducing p53-dependent induction of autophagy. Oncogene. 34, (42) 5329–5340. 10.1038/onc.2014.461 [DOI] [PubMed] [Google Scholar]
- Gu, Y. , Li, P. , Peng, F. , Zhang, M. , Zhang, Y. , Liang, H. , Zhao, W. , Qi, L. , Wang, H. , Wang, C. , Guo, Z. , 2015. Autophagy-related prognostic signature for breast cancer. Mol. Carcinog. 55, 292–299. [DOI] [PubMed] [Google Scholar]
- Guan, H. , Hou, S. , Ricciardi, R.P. , 2005. DNA binding of repressor nuclear factor-kappaB p50/p50 depends on phosphorylation of Ser337 by the protein kinase A catalytic subunit. J. Biol. Chem. 280, 9957–9962. [DOI] [PubMed] [Google Scholar]
- Gyorffy, B. , Lanczky, A. , Eklund, A.C. , Denkert, C. , Budczies, J. , Li, Q. , Szallasi, Z. , 2010. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res. Treat. 123, 725–731. [DOI] [PubMed] [Google Scholar]
- Katsuragi, Y. , Ichimura, Y. , Komatsu, M. , 2015. p62/SQSTM1 functions as a signaling hub and an autophagy adaptor. FEBS J. 282, 4672–4678. [DOI] [PubMed] [Google Scholar]
- Klionsky, D.J. , Abdelmohsen, K. , Abe, A. , Abedin, M.J. , Abeliovich, H. , Acevedo Arozena, A. , Adachi, H. , Adams, C.M. , Adams, P.D. , Adeli, K. , Adhihetty, P.J. , Adler, S.G. , Agam, G. , Agarwal, R. , Aghi, M.K. , Agnello, M. , Agostinis, P. , Aguilar, P.V. , Aguirre-Ghiso, J. , Airoldi, E.M. , Ait-Si-Ali, S. , Akematsu, T. , Akporiaye, E.T. , Al-Rubeai, M. , Albaiceta, G.M. , Albanese, C. , Albani, D. , Albert, M.L. , Aldudo, J. , Algul, H. , Alirezaei, M. , Alloza, I. , Almasan, A. , Almonte-Beceril, M. , Alnemri, E.S. , Alonso, C. , Altan-Bonnet, N. , Altieri, D.C. , Alvarez, S. , Alvarez-Erviti, L. , Alves, S. , Amadoro, G. , Amano, A. , Amantini, C. , Ambrosio, S. , Amelio, I. , Amer, A.O. , Amessou, M. , Amon, A. , An, Z. , Anania, F.A. , Andersen, S.U. , Andley, U.P. , Andreadi, C.K. , Andrieu-Abadie, N. , Anel, A. , Ann, D.K. , Anoopkumar-Dukie, S. , Antonioli, M. , Aoki, H. , Apostolova, N. , Aquila, S. , Aquilano, K. , Araki, K. , Arama, E. , Aranda, A. , Araya, J. , Arcaro, A. , Arias, E. , Arimoto, H. , Ariosa, A.R. , Armstrong, J.L. , Arnould, T. , Arsov, I. , Asanuma, K. , Askanas, V. , Asselin, E. , Atarashi, R. , Atherton, S.S. , Atkin, J.D. , Attardi, L.D. , Auberger, P. , Auburger, G. , Aurelian, L. , Autelli, R. , Avagliano, L. , Avantaggiati, M.L. , Avrahami, L. , Awale, S. , Azad, N. , Bachetti, T. , Backer, J.M. , Bae, D.H. , Bae, J.S. , Bae, O.N. , Bae, S.H. , Baehrecke, E.H. , Baek, S.H. , Baghdiguian, S. , Bagniewska-Zadworna, A. , Bai, H. , Bai, J. , Bai, X.Y. , Bailly, Y. , Balaji, K.N. , Balduini, W. , Ballabio, A. , Balzan, R. , Banerjee, R. , Banhegyi, G. , Bao, H. , Barbeau, B. , Barrachina, M.D. , Barreiro, E. , Bartel, B. , Bartolome, A. , Bassham, D.C. , Bassi, M.T. , Bast, R.C. , Basu, A. , Batista, M.T. , Batoko, H. , Battino, M. , Bauckman, K. , Baumgarner, B.L. , Bayer, K.U. , Beale, R. , Beaulieu, J.F. , Beck, G.R. , Becker, C. , Beckham, J.D. , Bedard, P.A. , Bednarski, P.J. , Begley, T.J. , Behl, C. , Behrends, C. , Behrens, G.M. , Behrns, K.E. , Bejarano, E. , Belaid, A. , Belleudi, F. , Benard, G. , Berchem, G. , Bergamaschi, D. , Bergami, M. , Berkhout, B. , Berliocchi, L. , Bernard, A. , Bernard, M. , Bernassola, F. , Bertolotti, A. , Bess, A.S. , Besteiro, S. , Bettuzzi, S. , Bhalla, S. , Bhattacharyya, S. , Bhutia, S.K. , Biagosch, C. , Bianchi, M.W. , Biard-Piechaczyk, M. , Billes, V. , Bincoletto, C. , Bingol, B. , Bird, S.W. , Bitoun, M. , Bjedov, I. , Blackstone, C. , Blanc, L. , Blanco, G.A. , Blomhoff, H.K. , Boada-Romero, E. , Bockler, S. , Boes, M. , Boesze-Battaglia, K. , Boise, L.H. , Bolino, A. , Boman, A. , Bonaldo, P. , Bordi, M. , Bosch, J. , Botana, L.M. , Botti, J. , Bou, G. , Bouche, M. , Bouchecareilh, M. , Boucher, M.J. , Boulton, M.E. , Bouret, S.G. , Boya, P. , Boyer-Guittaut, M. , Bozhkov, P.V. , Brady, N. , Braga, V.M. , Brancolini, C. , Braus, G.H. , Bravo-San Pedro, J.M. , Brennan, L.A. , Bresnick, E.H. , Brest, P. , Bridges, D. , Bringer, M.A. , Brini, M. , Brito, G.C. , Brodin, B. , Brookes, P.S. , Brown, E.J. , Brown, K. , Broxmeyer, H.E. , Bruhat, A. , Brum, P.C. , Brumell, J.H. , Brunetti-Pierri, N. , Bryson-Richardson, R.J. , Buch, S. , Buchan, A.M. , Budak, H. , Bulavin, D.V. , Bultman, S.J. , Bultynck, G. , Bumbasirevic, V. , Burelle, Y. , Burke, R.E. , Burmeister, M. , Butikofer, P. , Caberlotto, L. , Cadwell, K. , Cahova, M. , Cai, D. , Cai, J. , Cai, Q. , Calatayud, S. , Camougrand, N. , Campanella, M. , Campbell, G.R. , Campbell, M. , Campello, S. , Candau, R. , Caniggia, I. , Cantoni, L. , Cao, L. , Caplan, A.B. , Caraglia, M. , Cardinali, C. , Cardoso, S.M. , Carew, J.S. , Carleton, L.A. , Carlin, C.R. , Carloni, S. , Carlsson, S.R. , Carmona-Gutierrez, D. , Carneiro, L.A. , Carnevali, O. , Carra, S. , Carrier, A. , Carroll, B. , Casas, C. , Casas, J. , Cassinelli, G. , Castets, P. , Castro-Obregon, S. , Cavallini, G. , Ceccherini, I. , Cecconi, F. , Cederbaum, A.I. , Cena, V. , Cenci, S. , Cerella, C. , Cervia, D. , Cetrullo, S. , Chaachouay, H. , Chae, H.J. , Chagin, A.S. , Chai, C.Y. , Chakrabarti, G. , Chamilos, G. , Chan, E.Y. , Chan, M.T. , Chandra, D. , Chandra, P. , Chang, C.P. , Chang, R.C. , Chang, T.Y. , Chatham, J.C. , Chatterjee, S. , Chauhan, S. , Che, Y. , Cheetham, M.E. , Cheluvappa, R. , Chen, C.J. , Chen, G. , Chen, G.C. , Chen, G. , Chen, H. , Chen, J.W. , Chen, J.K. , Chen, M. , Chen, M. , Chen, P. , Chen, Q. , Chen, Q. , Chen, S.D. , Chen, S. , Chen, S.S. , Chen, W. , Chen, W.J. , Chen, W.Q. , Chen, W. , Chen, X. , Chen, Y.H. , Chen, Y.G. , Chen, Y. , Chen, Y. , Chen, Y. , Chen, Y.J. , Chen, Y.Q. , Chen, Y. , Chen, Z. , Chen, Z. , Cheng, A. , Cheng, C.H. , Cheng, H. , Cheong, H. , Cherry, S. , Chesney, J. , Cheung, C.H. , Chevet, E. , Chi, H.C. , Chi, S.G. , Chiacchiera, F. , Chiang, H.L. , Chiarelli, R. , Chiariello, M. , Chieppa, M. , Chin, L.S. , Chiong, M. , Chiu, G.N. , Cho, D.H. , Cho, S.G. , Cho, W.C. , Cho, Y.Y. , Cho, Y.S. , Choi, A.M. , Choi, E.J. , Choi, E.K. , Choi, J. , Choi, M.E. , Choi, S.I. , Chou, T.F. , Chouaib, S. , Choubey, D. , Choubey, V. , Chow, K.C. , Chowdhury, K. , Chu, C.T. , Chuang, T.H. , Chun, T. , Chung, H. , Chung, T. , Chung, Y.L. , Chwae, Y.J. , Cianfanelli, V. , Ciarcia, R. , Ciechomska, I.A. , Ciriolo, M.R. , Cirone, M. , Claerhout, S. , Clague, M.J. , Claria, J. , Clarke, P.G. , Clarke, R. , Clementi, E. , Cleyrat, C. , Cnop, M. , Coccia, E.M. , Cocco, T. , Codogno, P. , Coers, J. , Cohen, E.E. , Colecchia, D. , Coletto, L. , Coll, N.S. , Colucci-Guyon, E. , Comincini, S. , Condello, M. , Cook, K.L. , Coombs, G.H. , Cooper, C.D. , Cooper, J.M. , Coppens, I. , Corasaniti, M.T. , Corazzari, M. , Corbalan, R. , Corcelle-Termeau, E. , Cordero, M.D. , Corral-Ramos, C. , Corti, O. , Cossarizza, A. , Costelli, P. , Costes, S. , Cotman, S.L. , Coto-Montes, A. , Cottet, S. , Couve, E. , Covey, L.R. , Cowart, L.A. , Cox, J.S. , Coxon, F.P. , Coyne, C.B. , Cragg, M.S. , Craven, R.J. , Crepaldi, T. , Crespo, J.L. , Criollo, A. , Crippa, V. , Cruz, M.T. , Cuervo, A.M. , Cuezva, J.M. , Cui, T. , Cutillas, P.R. , Czaja, M.J. , Czyzyk-Krzeska, M.F. , Dagda, R.K. , Dahmen, U. , Dai, C. , Dai, W. , Dai, Y. , Dalby, K.N. , Dalla Valle, L. , Dalmasso, G. , D'Amelio, M. , Damme, M. , Darfeuille-Michaud, A. , Dargemont, C. , Darley-Usmar, V.M. , Dasarathy, S. , Dasgupta, B. , Dash, S. , Dass, C.R. , Davey, H.M. , Davids, L.M. , Davila, D. , Davis, R.J. , Dawson, T.M. , Dawson, V.L. , Daza, P. , de Belleroche, J. , de Figueiredo, P. , de Figueiredo, R.C. , de la Fuente, J. , De Martino, L. , De Matteis, A. , De Meyer, G.R. , De Milito, A. , De Santi, M. , de Souza, W. , De Tata, V. , De Zio, D. , Debnath, J. , Dechant, R. , Decuypere, J.P. , Deegan, S. , Dehay, B. , Del Bello, B. , Del Re, D.P. , Delage-Mourroux, R. , Delbridge, L.M. , Deldicque, L. , Delorme-Axford, E. , Deng, Y. , Dengjel, J. , Denizot, M. , Dent, P. , Der, C.J. , Deretic, V. , Derrien, B. , Deutsch, E. , Devarenne, T.P. , Devenish, R.J. , Di Bartolomeo, S. , Di Daniele, N. , Di Domenico, F. , Di Nardo, A. , Di Paola, S. , Di Pietro, A. , Di Renzo, L. , DiAntonio, A. , Diaz-Araya, G. , Diaz-Laviada, I. , Diaz-Meco, M.T. , Diaz-Nido, J. , Dickey, C.A. , Dickson, R.C. , Diederich, M. , Digard, P. , Dikic, I. , Dinesh-Kumar, S.P. , Ding, C. , Ding, W.X. , Ding, Z. , Dini, L. , Distler, J.H. , Diwan, A. , Djavaheri-Mergny, M. , Dmytruk, K. , Dobson, R.C. , Doetsch, V. , Dokladny, K. , Dokudovskaya, S. , Donadelli, M. , Dong, X.C. , Dong, X. , Dong, Z. , Donohue, T.M. , Doran, K.S. , D'Orazi, G. , Dorn, G.W. , Dosenko, V. , Dridi, S. , Drucker, L. , Du, J. , Du, L.L. , Du, L. , du Toit, A. , Dua, P. , Duan, L. , Duann, P. , Dubey, V.K. , Duchen, M.R. , Duchosal, M.A. , Duez, H. , Dugail, I. , Dumit, V.I. , Duncan, M.C. , Dunlop, E.A. , Dunn, W.A. , Dupont, N. , Dupuis, L. , Duran, R.V. , Durcan, T.M. , Duvezin-Caubet, S. , Duvvuri, U. , Eapen, V. , Ebrahimi-Fakhari, D. , Echard, A. , Eckhart, L. , Edelstein, C.L. , Edinger, A.L. , Eichinger, L. , Eisenberg, T. , Eisenberg-Lerner, A. , Eissa, N.T. , El-Deiry, W.S. , El-Khoury, V. , Elazar, Z. , Eldar-Finkelman, H. , Elliott, C.J. , Emanuele, E. , Emmenegger, U. , Engedal, N. , Engelbrecht, A.M. , Engelender, S. , Enserink, J.M. , Erdmann, R. , Erenpreisa, J. , Eri, R. , Eriksen, J.L. , Erman, A. , Escalante, R. , Eskelinen, E.L. , Espert, L. , Esteban-Martinez, L. , Evans, T.J. , Fabri, M. , Fabrias, G. , Fabrizi, C. , Facchiano, A. , Faergeman, N.J. , Faggioni, A. , Fairlie, W.D. , Fan, C. , Fan, D. , Fan, J. , Fang, S. , Fanto, M. , Fanzani, A. , Farkas, T. , Faure, M. , Favier, F.B. , Fearnhead, H. , Federici, M. , Fei, E. , Felizardo, T.C. , Feng, H. , Feng, Y. , Feng, Y. , Ferguson, T.A. , Fernandez, A.F. , Fernandez-Barrena, M.G. , Fernandez-Checa, J.C. , Fernandez-Lopez, A. , Fernandez-Zapico, M.E. , Feron, O. , Ferraro, E. , Ferreira-Halder, C.V. , Fesus, L. , Feuer, R. , Fiesel, F.C. , Filippi-Chiela, E.C. , Filomeni, G. , Fimia, G.M. , Fingert, J.H. , Finkbeiner, S. , Finkel, T. , Fiorito, F. , Fisher, P.B. , Flajolet, M. , Flamigni, F. , Florey, O. , Florio, S. , Floto, R.A. , Folini, M. , Follo, C. , Fon, E.A. , Fornai, F. , Fortunato, F. , Fraldi, A. , Franco, R. , Francois, A. , Francois, A. , Frankel, L.B. , Fraser, I.D. , Frey, N. , Freyssenet, D.G. , Frezza, C. , Friedman, S.L. , Frigo, D.E. , Fu, D. , Fuentes, J.M. , Fueyo, J. , Fujitani, Y. , Fujiwara, Y. , Fujiya, M. , Fukuda, M. , Fulda, S. , Fusco, C. , Gabryel, B. , Gaestel, M. , Gailly, P. , Gajewska, M. , Galadari, S. , Galili, G. , Galindo, I. , Galindo, M.F. , Galliciotti, G. , Galluzzi, L. , Galluzzi, L. , Galy, V. , Gammoh, N. , Gandy, S. , Ganesan, A.K. , Ganesan, S. , Ganley, I.G. , Gannage, M. , Gao, F.B. , Gao, F. , Gao, J.X. , Garcia Nannig, L. , Garcia Vescovi, E. , Garcia-Macia, M. , Garcia-Ruiz, C. , Garg, A.D. , Garg, P.K. , Gargini, R. , Gassen, N.C. , Gatica, D. , Gatti, E. , Gavard, J. , Gavathiotis, E. , Ge, L. , Ge, P. , Ge, S. , Gean, P.W. , Gelmetti, V. , Genazzani, A.A. , Geng, J. , Genschik, P. , Gerner, L. , Gestwicki, J.E. , Gewirtz, D.A. , Ghavami, S. , Ghigo, E. , Ghosh, D. , Giammarioli, A.M. , Giampieri, F. , Giampietri, C. , Giatromanolaki, A. , Gibbings, D.J. , Gibellini, L. , Gibson, S.B. , Ginet, V. , Giordano, A. , Giorgini, F. , Giovannetti, E. , Girardin, S.E. , Gispert, S. , Giuliano, S. , Gladson, C.L. , Glavic, A. , Gleave, M. , Godefroy, N. , Gogal, R.M. , Gokulan, K. , Goldman, G.H. , Goletti, D. , Goligorsky, M.S. , Gomes, A.V. , Gomes, L.C. , Gomez, H. , Gomez-Manzano, C. , Gomez-Sanchez, R. , Goncalves, D.A. , Goncu, E. , Gong, Q. , Gongora, C. , Gonzalez, C.B. , Gonzalez-Alegre, P. , Gonzalez-Cabo, P. , Gonzalez-Polo, R.A. , Goping, I.S. , Gorbea, C. , Gorbunov, N.V. , Goring, D.R. , Gorman, A.M. , Gorski, S.M. , Goruppi, S. , Goto-Yamada, S. , Gotor, C. , Gottlieb, R.A. , Gozes, I. , Gozuacik, D. , Graba, Y. , Graef, M. , Granato, G.E. , Grant, G.D. , Grant, S. , Gravina, G.L. , Green, D.R. , Greenhough, A. , Greenwood, M.T. , Grimaldi, B. , Gros, F. , Grose, C. , Groulx, J.F. , Gruber, F. , Grumati, P. , Grune, T. , Guan, J.L. , Guan, K.L. , Guerra, B. , Guillen, C. , Gulshan, K. , Gunst, J. , Guo, C. , Guo, L. , Guo, M. , Guo, W. , Guo, X.G. , Gust, A.A. , Gustafsson, A.B. , Gutierrez, E. , Gutierrez, M.G. , Gwak, H.S. , Haas, A. , Haber, J.E. , Hadano, S. , Hagedorn, M. , Hahn, D.R. , Halayko, A.J. , Hamacher-Brady, A. , Hamada, K. , Hamai, A. , Hamann, A. , Hamasaki, M. , Hamer, I. , Hamid, Q. , Hammond, E.M. , Han, F. , Han, W. , Handa, J.T. , Hanover, J.A. , Hansen, M. , Harada, M. , Harhaji-Trajkovic, L. , Harper, J.W. , Harrath, A.H. , Harris, A.L. , Harris, J. , Hasler, U. , Hasselblatt, P. , Hasui, K. , Hawley, R.G. , Hawley, T.S. , He, C. , He, C.Y. , He, F. , He, G. , He, R.R. , He, X.H. , He, Y.W. , He, Y.Y. , Heath, J.K. , Hebert, M.J. , Heinzen, R.A. , Helgason, G.V. , Hensel, M. , Henske, E.P. , Her, C. , Herman, P.K. , Hernandez, A. , Hernandez, C. , Hernandez-Tiedra, S. , Hetz, C. , Hiesinger, P.R. , Higaki, K. , Hilfiker, S. , Hill, B.G. , Hill, J.A. , Hill, W.D. , Hino, K. , Hofius, D. , Hofman, P. , Hoglinger, G.U. , Hohfeld, J. , Holz, M.K. , Hong, Y. , Hood, D.A. , Hoozemans, J.J. , Hoppe, T. , Hsu, C. , Hsu, C.Y. , Hsu, L.C. , Hu, D. , Hu, G. , Hu, H.M. , Hu, H. , Hu, M.C. , Hu, Y.C. , Hu, Z.W. , Hua, F. , Hua, Y. , Huang, C. , Huang, H.L. , Huang, K.H. , Huang, K.Y. , Huang, S. , Huang, S. , Huang, W.P. , Huang, Y.R. , Huang, Y. , Huang, Y. , Huber, T.B. , Huebbe, P. , Huh, W.K. , Hulmi, J.J. , Hur, G.M. , Hurley, J.H. , Husak, Z. , Hussain, S.N. , Hussain, S. , Hwang, J.J. , Hwang, S. , Hwang, T.I. , Ichihara, A. , Imai, Y. , Imbriano, C. , Inomata, M. , Into, T. , Iovane, V. , Iovanna, J.L. , Iozzo, R.V. , Ip, N.Y. , Irazoqui, J.E. , Iribarren, P. , Isaka, Y. , Isakovic, A.J. , Ischiropoulos, H. , Isenberg, J.S. , Ishaq, M. , Ishida, H. , Ishii, I. , Ishmael, J.E. , Isidoro, C. , Isobe, K.I. , Isono, E. , Issazadeh-Navikas, S. , Itahana, K. , Itakura, E. , Ivanov, A.I. , Iyer, A.K. , Izquierdo, J.M. , Izumi, Y. , Izzo, V. , Jaattela, M. , Jaber, N. , Jackson, D.J. , Jackson, W.T. , Jacob, T.G. , Jacques, T.S. , Jagannath, C. , Jain, A. , Jana, N.R. , Jang, B.K. , Jani, A. , Janji, B. , Jannig, P.R. , Jansson, P.J. , Jean, S. , Jendrach, M. , Jeon, J.H. , Jessen, N. , Jeung, E.B. , Jia, K. , Jia, L. , Jiang, H. , Jiang, H. , Jiang, L. , Jiang, T. , Jiang, X. , Jiang, X. , Jiang, X. , Jiang, Y. , Jiang, Y. , Jimenez, A. , Jin, C. , Jin, H. , Jin, L. , Jin, M. , Jin, S. , Jinwal, U.K. , Jo, E.K. , Johansen, T. , Johnson, D.E. , Johnson, G.V. , Johnson, J.D. , Jonasch, E. , Jones, C. , Joosten, L.A. , Jordan, J. , Joseph, A.M. , Joseph, B. , Joubert, A.M. , Ju, D. , Ju, J. , Juan, H.F. , Juenemann, K. , Juhasz, G. , Jung, H.S. , Jung, J.U. , Jung, Y.K. , Jungbluth, H. , Justice, M.J. , Jutten, B. , Kaakoush, N.O. , Kaarniranta, K. , Kaasik, A. , Kabuta, T. , Kaeffer, B. , Kagedal, K. , Kahana, A. , Kajimura, S. , Kakhlon, O. , Kalia, M. , Kalvakolanu, D.V. , Kamada, Y. , Kambas, K. , Kaminskyy, V.O. , Kampinga, H.H. , Kandouz, M. , Kang, C. , Kang, R. , Kang, T.C. , Kanki, T. , Kanneganti, T.D. , Kanno, H. , Kanthasamy, A.G. , Kantorow, M. , Kaparakis-Liaskos, M. , Kapuy, O. , Karantza, V. , Karim, M.R. , Karmakar, P. , Kaser, A. , Kaushik, S. , Kawula, T. , Kaynar, A.M. , Ke, P.Y. , Ke, Z.J. , Kehrl, J.H. , Keller, K.E. , Kemper, J.K. , Kenworthy, A.K. , Kepp, O. , Kern, A. , Kesari, S. , Kessel, D. , Ketteler, R. , Kettelhut, I.D. , Khambu, B. , Khan, M.M. , Khandelwal, V.K. , Khare, S. , Kiang, J.G. , Kiger, A.A. , Kihara, A. , Kim, A.L. , Kim, C.H. , Kim, D.R. , Kim, D.H. , Kim, E.K. , Kim, H.Y. , Kim, H.R. , Kim, J.S. , Kim, J.H. , Kim, J.C. , Kim, J.H. , Kim, K.W. , Kim, M.D. , Kim, M.M. , Kim, P.K. , Kim, S.W. , Kim, S.Y. , Kim, Y.S. , Kim, Y. , Kimchi, A. , Kimmelman, A.C. , Kimura, T. , King, J.S. , Kirkegaard, K. , Kirkin, V. , Kirshenbaum, L.A. , Kishi, S. , Kitajima, Y. , Kitamoto, K. , Kitaoka, Y. , Kitazato, K. , Kley, R.A. , Klimecki, W.T. , Klinkenberg, M. , Klucken, J. , Knaevelsrud, H. , Knecht, E. , Knuppertz, L. , Ko, J.L. , Kobayashi, S. , Koch, J.C. , Koechlin-Ramonatxo, C. , Koenig, U. , Koh, Y.H. , Kohler, K. , Kohlwein, S.D. , Koike, M. , Komatsu, M. , Kominami, E. , Kong, D. , Kong, H.J. , Konstantakou, E.G. , Kopp, B.T. , Korcsmaros, T. , Korhonen, L. , Korolchuk, V.I. , Koshkina, N.V. , Kou, Y. , Koukourakis, M.I. , Koumenis, C. , Kovacs, A.L. , Kovacs, T. , Kovacs, W.J. , Koya, D. , Kraft, C. , Krainc, D. , Kramer, H. , Kravic-Stevovic, T. , Krek, W. , Kretz-Remy, C. , Krick, R. , Krishnamurthy, M. , Kriston-Vizi, J. , Kroemer, G. , Kruer, M.C. , Kruger, R. , Ktistakis, N.T. , Kuchitsu, K. , Kuhn, C. , Kumar, A.P. , Kumar, A. , Kumar, A. , Kumar, D. , Kumar, D. , Kumar, R. , Kumar, S. , Kundu, M. , Kung, H.J. , Kuno, A. , Kuo, S.H. , Kuret, J. , Kurz, T. , Kwok, T. , Kwon, T.K. , Kwon, Y.T. , Kyrmizi, I. , La Spada, A.R. , Lafont, F. , Lahm, T. , Lakkaraju, A. , Lam, T. , Lamark, T. , Lancel, S. , Landowski, T.H. , Lane, D.J. , Lane, J.D. , Lanzi, C. , Lapaquette, P. , Lapierre, L.R. , Laporte, J. , Laukkarinen, J. , Laurie, G.W. , Lavandero, S. , Lavie, L. , LaVoie, M.J. , Law, B.Y. , Law, H.K. , Law, K.B. , Layfield, R. , Lazo, P.A. , Le Cam, L. , Le Roch, K.G. , Le Stunff, H. , Leardkamolkarn, V. , Lecuit, M. , Lee, B.H. , Lee, C.H. , Lee, E.F. , Lee, G.M. , Lee, H.J. , Lee, H. , Lee, J.K. , Lee, J. , Lee, J.H. , Lee, J.H. , Lee, M. , Lee, M.S. , Lee, P.J. , Lee, S.W. , Lee, S.J. , Lee, S.J. , Lee, S.Y. , Lee, S.H. , Lee, S.S. , Lee, S.J. , Lee, S. , Lee, Y.R. , Lee, Y.J. , Lee, Y.H. , Leeuwenburgh, C. , Lefort, S. , Legouis, R. , Lei, J. , Lei, Q.Y. , Leib, D.A. , Leibowitz, G. , Lekli, I. , Lemaire, S.D. , Lemasters, J.J. , Lemberg, M.K. , Lemoine, A. , Leng, S. , Lenz, G. , Lenzi, P. , Lerman, L.O. , Lettieri Barbato, D. , Leu, J.I. , Leung, H.Y. , Levine, B. , Lewis, P.A. , Lezoualc'h, F. , Li, C. , Li, F. , Li, F.J. , Li, J. , Li, K. , Li, L. , Li, M. , Li, M. , Li, Q. , Li, R. , Li, S. , Li, W. , Li, W. , Li, X. , Li, Y. , Lian, J. , Liang, C. , Liang, Q. , Liao, Y. , Liberal, J. , Liberski, P.P. , Lie, P. , Lieberman, A.P. , Lim, H.J. , Lim, K.L. , Lim, K. , Lima, R.T. , Lin, C.S. , Lin, C.F. , Lin, F. , Lin, F. , Lin, F.C. , Lin, K. , Lin, K.H. , Lin, P.H. , Lin, T. , Lin, W.W. , Lin, Y.S. , Lin, Y. , Linden, R. , Lindholm, D. , Lindqvist, L.M. , Lingor, P. , Linkermann, A. , Liotta, L.A. , Lipinski, M.M. , Lira, V.A. , Lisanti, M.P. , Liton, P.B. , Liu, B. , Liu, C. , Liu, C.F. , Liu, F. , Liu, H.J. , Liu, J. , Liu, J.J. , Liu, J.L. , Liu, K. , Liu, L. , Liu, L. , Liu, Q. , Liu, R.Y. , Liu, S. , Liu, S. , Liu, W. , Liu, X.D. , Liu, X. , Liu, X.H. , Liu, X. , Liu, X. , Liu, X. , Liu, Y. , Liu, Y. , Liu, Z. , Liu, Z. , Liuzzi, J.P. , Lizard, G. , Ljujic, M. , Lodhi, I.J. , Logue, S.E. , Lokeshwar, B.L. , Long, Y.C. , Lonial, S. , Loos, B. , Lopez-Otin, C. , Lopez-Vicario, C. , Lorente, M. , Lorenzi, P.L. , Lorincz, P. , Los, M. , Lotze, M.T. , Lovat, P.E. , Lu, B. , Lu, B. , Lu, J. , Lu, Q. , Lu, S.M. , Lu, S. , Lu, Y. , Luciano, F. , Luckhart, S. , Lucocq, J.M. , Ludovico, P. , Lugea, A. , Lukacs, N.W. , Lum, J.J. , Lund, A.H. , Luo, H. , Luo, J. , Luo, S. , Luparello, C. , Lyons, T. , Ma, J. , Ma, Y. , Ma, Y. , Ma, Z. , Machado, J. , Machado-Santelli, G.M. , Macian, F. , MacIntosh, G.C. , MacKeigan, J.P. , Macleod, K.F. , MacMicking, J.D. , MacMillan-Crow, L.A. , Madeo, F. , Madesh, M. , Madrigal-Matute, J. , Maeda, A. , Maeda, T. , Maegawa, G. , Maellaro, E. , Maes, H. , Magarinos, M. , Maiese, K. , Maiti, T.K. , Maiuri, L. , Maiuri, M.C. , Maki, C.G. , Malli, R. , Malorni, W. , Maloyan, A. , Mami-Chouaib, F. , Man, N. , Mancias, J.D. , Mandelkow, E.M. , Mandell, M.A. , Manfredi, A.A. , Manie, S.N. , Manzoni, C. , Mao, K. , Mao, Z. , Mao, Z.W. , Marambaud, P. , Marconi, A.M. , Marelja, Z. , Marfe, G. , Margeta, M. , Margittai, E. , Mari, M. , Mariani, F.V. , Marin, C. , Marinelli, S. , Marino, G. , Markovic, I. , Marquez, R. , Martelli, A.M. , Martens, S. , Martin, K.R. , Martin, S.J. , Martin, S. , Martin-Acebes, M.A. , Martin-Sanz, P. , Martinand-Mari, C. , Martinet, W. , Martinez, J. , Martinez-Lopez, N. , Martinez-Outschoorn, U. , Martinez-Velazquez, M. , Martinez-Vicente, M. , Martins, W.K. , Mashima, H. , Mastrianni, J.A. , Matarese, G. , Matarrese, P. , Mateo, R. , Matoba, S. , Matsumoto, N. , Matsushita, T. , Matsuura, A. , Matsuzawa, T. , Mattson, M.P. , Matus, S. , Maugeri, N. , Mauvezin, C. , Mayer, A. , Maysinger, D. , Mazzolini, G.D. , McBrayer, M.K. , McCall, K. , McCormick, C. , McInerney, G.M. , McIver, S.C. , McKenna, S. , McMahon, J.J. , McNeish, I.A. , Mechta-Grigoriou, F. , Medema, J.P. , Medina, D.L. , Megyeri, K. , Mehrpour, M. , Mehta, J.L. , Mei, Y. , Meier, U.C. , Meijer, A.J. , Melendez, A. , Melino, G. , Melino, S. , de Melo, E.J. , Mena, M.A. , Meneghini, M.D. , Menendez, J.A. , Menezes, R. , Meng, L. , Meng, L.H. , Meng, S. , Menghini, R. , Menko, A.S. , Menna-Barreto, R.F. , Menon, M.B. , Meraz-Rios, M.A. , Merla, G. , Merlini, L. , Merlot, A.M. , Meryk, A. , Meschini, S. , Meyer, J.N. , Mi, M.T. , Miao, C.Y. , Micale, L. , Michaeli, S. , Michiels, C. , Migliaccio, A.R. , Mihailidou, A.S. , Mijaljica, D. , Mikoshiba, K. , Milan, E. , Miller-Fleming, L. , Mills, G.B. , Mills, I.G. , Minakaki, G. , Minassian, B.A. , Ming, X.F. , Minibayeva, F. , Minina, E.A. , Mintern, J.D. , Minucci, S. , Miranda-Vizuete, A. , Mitchell, C.H. , Miyamoto, S. , Miyazawa, K. , Mizushima, N. , Mnich, K. , Mograbi, B. , Mohseni, S. , Moita, L.F. , Molinari, M. , Molinari, M. , Moller, A.B. , Mollereau, B. , Mollinedo, F. , Mongillo, M. , Monick, M.M. , Montagnaro, S. , Montell, C. , Moore, D.J. , Moore, M.N. , Mora-Rodriguez, R. , Moreira, P.I. , Morel, E. , Morelli, M.B. , Moreno, S. , Morgan, M.J. , Moris, A. , Moriyasu, Y. , Morrison, J.L. , Morrison, L.A. , Morselli, E. , Moscat, J. , Moseley, P.L. , Mostowy, S. , Motori, E. , Mottet, D. , Mottram, J.C. , Moussa, C.E. , Mpakou, V.E. , Mukhtar, H. , Mulcahy Levy, J.M. , Muller, S. , Munoz-Moreno, R. , Munoz-Pinedo, C. , Munz, C. , Murphy, M.E. , Murray, J.T. , Murthy, A. , Mysorekar, I.U. , Nabi, I.R. , Nabissi, M. , Nader, G.A. , Nagahara, Y. , Nagai, Y. , Nagata, K. , Nagelkerke, A. , Nagy, P. , Naidu, S.R. , Nair, S. , Nakano, H. , Nakatogawa, H. , Nanjundan, M. , Napolitano, G. , Naqvi, N.I. , Nardacci, R. , Narendra, D.P. , Narita, M. , Nascimbeni, A.C. , Natarajan, R. , Navegantes, L.C. , Nawrocki, S.T. , Nazarko, T.Y. , Nazarko, V.Y. , Neill, T. , Neri, L.M. , Netea, M.G. , Netea-Maier, R.T. , Neves, B.M. , Ney, P.A. , Nezis, I.P. , Nguyen, H.T. , Nguyen, H.P. , Nicot, A.S. , Nilsen, H. , Nilsson, P. , Nishimura, M. , Nishino, I. , Niso-Santano, M. , Niu, H. , Nixon, R.A. , Njar, V.C. , Noda, T. , Noegel, A.A. , Nolte, E.M. , Norberg, E. , Norga, K.K. , Noureini, S.K. , Notomi, S. , Notterpek, L. , Nowikovsky, K. , Nukina, N. , Nurnberger, T. , O'Donnell, V.B. , O'Donovan, T. , O'Dwyer, P.J. , Oehme, I. , Oeste, C.L. , Ogawa, M. , Ogretmen, B. , Ogura, Y. , Oh, Y.J. , Ohmuraya, M. , Ohshima, T. , Ojha, R. , Okamoto, K. , Okazaki, T. , Oliver, F.J. , Ollinger, K. , Olsson, S. , Orban, D.P. , Ordonez, P. , Orhon, I. , Orosz, L. , O'Rourke, E.J. , Orozco, H. , Ortega, A.L. , Ortona, E. , Osellame, L.D. , Oshima, J. , Oshima, S. , Osiewacz, H.D. , Otomo, T. , Otsu, K. , Ou, J.J. , Outeiro, T.F. , Ouyang, D.Y. , Ouyang, H. , Overholtzer, M. , Ozbun, M.A. , Ozdinler, P.H. , Ozpolat, B. , Pacelli, C. , Paganetti, P. , Page, G. , Pages, G. , Pagnini, U. , Pajak, B. , Pak, S.C. , Pakos-Zebrucka, K. , Pakpour, N. , Palkova, Z. , Palladino, F. , Pallauf, K. , Pallet, N. , Palmieri, M. , Paludan, S.R. , Palumbo, C. , Palumbo, S. , Pampliega, O. , Pan, H. , Pan, W. , Panaretakis, T. , Pandey, A. , Pantazopoulou, A. , Papackova, Z. , Papademetrio, D.L. , Papassideri, I. , Papini, A. , Parajuli, N. , Pardo, J. , Parekh, V.V. , Parenti, G. , Park, J.I. , Park, J. , Park, O.K. , Parker, R. , Parlato, R. , Parys, J.B. , Parzych, K.R. , Pasquet, J.M. , Pasquier, B. , Pasumarthi, K.B. , Patschan, D. , Patterson, C. , Pattingre, S. , Pattison, S. , Pause, A. , Pavenstadt, H. , Pavone, F. , Pedrozo, Z. , Pena, F.J. , Penalva, M.A. , Pende, M. , Peng, J. , Penna, F. , Penninger, J.M. , Pensalfini, A. , Pepe, S. , Pereira, G.J. , Pereira, P.C. , Perez-de la Cruz, V. , Perez-Perez, M.E. , Perez-Rodriguez, D. , Perez-Sala, D. , Perier, C. , Perl, A. , Perlmutter, D.H. , Perrotta, I. , Pervaiz, S. , Pesonen, M. , Pessin, J.E. , Peters, G.J. , Petersen, M. , Petrache, I. , Petrof, B.J. , Petrovski, G. , Phang, J.M. , Piacentini, M. , Pierdominici, M. , Pierre, P. , Pierrefite-Carle, V. , Pietrocola, F. , Pimentel-Muinos, F.X. , Pinar, M. , Pineda, B. , Pinkas-Kramarski, R. , Pinti, M. , Pinton, P. , Piperdi, B. , Piret, J.M. , Platanias, L.C. , Platta, H.W. , Plowey, E.D. , Poggeler, S. , Poirot, M. , Polcic, P. , Poletti, A. , Poon, A.H. , Popelka, H. , Popova, B. , Poprawa, I. , Poulose, S.M. , Poulton, J. , Powers, S.K. , Powers, T. , Pozuelo-Rubio, M. , Prak, K. , Prange, R. , Prescott, M. , Priault, M. , Prince, S. , Proia, R.L. , Proikas-Cezanne, T. , Prokisch, H. , Promponas, V.J. , Przyklenk, K. , Puertollano, R. , Pugazhenthi, S. , Puglielli, L. , Pujol, A. , Puyal, J. , Pyeon, D. , Qi, X. , Qian, W.B. , Qin, Z.H. , Qiu, Y. , Qu, Z. , Quadrilatero, J. , Quinn, F. , Raben, N. , Rabinowich, H. , Radogna, F. , Ragusa, M.J. , Rahmani, M. , Raina, K. , Ramanadham, S. , Ramesh, R. , Rami, A. , Randall-Demllo, S. , Randow, F. , Rao, H. , Rao, V.A. , Rasmussen, B.B. , Rasse, T.M. , Ratovitski, E.A. , Rautou, P.E. , Ray, S.K. , Razani, B. , Reed, B.H. , Reggiori, F. , Rehm, M. , Reichert, A.S. , Rein, T. , Reiner, D.J. , Reits, E. , Ren, J. , Ren, X. , Renna, M. , Reusch, J.E. , Revuelta, J.L. , Reyes, L. , Rezaie, A.R. , Richards, R.I. , Richardson, D.R. , Richetta, C. , Riehle, M.A. , Rihn, B.H. , Rikihisa, Y. , Riley, B.E. , Rimbach, G. , Rippo, M.R. , Ritis, K. , Rizzi, F. , Rizzo, E. , Roach, P.J. , Robbins, J. , Roberge, M. , Roca, G. , Roccheri, M.C. , Rocha, S. , Rodrigues, C.M. , Rodriguez, C.I. , de Cordoba, S.R. , Rodriguez-Muela, N. , Roelofs, J. , Rogov, V.V. , Rohn, T.T. , Rohrer, B. , Romanelli, D. , Romani, L. , Romano, P.S. , Roncero, M.I. , Rosa, J.L. , Rosello, A. , Rosen, K.V. , Rosenstiel, P. , Rost-Roszkowska, M. , Roth, K.A. , Roue, G. , Rouis, M. , Rouschop, K.M. , Ruan, D.T. , Ruano, D. , Rubinsztein, D.C. , Rucker, E.B. , Rudich, A. , Rudolf, E. , Rudolf, R. , Ruegg, M.A. , Ruiz-Roldan, C. , Ruparelia, A.A. , Rusmini, P. , Russ, D.W. , Russo, G.L. , Russo, G. , Russo, R. , Rusten, T.E. , Ryabovol, V. , Ryan, K.M. , Ryter, S.W. , Sabatini, D.M. , Sacher, M. , Sachse, C. , Sack, M.N. , Sadoshima, J. , Saftig, P. , Sagi-Eisenberg, R. , Sahni, S. , Saikumar, P. , Saito, T. , Saitoh, T. , Sakakura, K. , Sakoh-Nakatogawa, M. , Sakuraba, Y. , Salazar-Roa, M. , Salomoni, P. , Saluja, A.K. , Salvaterra, P.M. , Salvioli, R. , Samali, A. , Sanchez, A.M. , Sanchez-Alcazar, J.A. , Sanchez-Prieto, R. , Sandri, M. , Sanjuan, M.A. , Santaguida, S. , Santambrogio, L. , Santoni, G. , Dos Santos, C.N. , Saran, S. , Sardiello, M. , Sargent, G. , Sarkar, P. , Sarkar, S. , Sarrias, M.R. , Sarwal, M.M. , Sasakawa, C. , Sasaki, M. , Sass, M. , Sato, K. , Sato, M. , Satriano, J. , Savaraj, N. , Saveljeva, S. , Schaefer, L. , Schaible, U.E. , Scharl, M. , Schatzl, H.M. , Schekman, R. , Scheper, W. , Schiavi, A. , Schipper, H.M. , Schmeisser, H. , Schmidt, J. , Schmitz, I. , Schneider, B.E. , Schneider, E.M. , Schneider, J.L. , Schon, E.A. , Schonenberger, M.J. , Schonthal, A.H. , Schorderet, D.F. , Schroder, B. , Schuck, S. , Schulze, R.J. , Schwarten, M. , Schwarz, T.L. , Sciarretta, S. , Scotto, K. , Scovassi, A.I. , Screaton, R.A. , Screen, M. , Seca, H. , Sedej, S. , Segatori, L. , Segev, N. , Seglen, P.O. , Segui-Simarro, J.M. , Segura-Aguilar, J. , Seki, E. , Seiliez, I. , Sell, C. , Semenkovich, C.F. , Semenza, G.L. , Sen, U. , Serra, A.L. , Serrano-Puebla, A. , Sesaki, H. , Setoguchi, T. , Settembre, C. , Shacka, J.J. , Shajahan-Haq, A.N. , Shapiro, I.M. , Sharma, S. , She, H. , Shen, C.J. , Shen, C.C. , Shen, H.M. , Shen, S. , Shen, W. , Sheng, R. , Sheng, X. , Sheng, Z.H. , Shepherd, T.G. , Shi, J. , Shi, Q. , Shi, Q. , Shi, Y. , Shibutani, S. , Shibuya, K. , Shidoji, Y. , Shieh, J.J. , Shih, C.M. , Shimada, Y. , Shimizu, S. , Shin, D.W. , Shinohara, M.L. , Shintani, M. , Shintani, T. , Shioi, T. , Shirabe, K. , Shiri-Sverdlov, R. , Shirihai, O. , Shore, G.C. , Shu, C.W. , Shukla, D. , Sibirny, A.A. , Sica, V. , Sigurdson, C.J. , Sigurdsson, E.M. , Sijwali, P.S. , Sikorska, B. , Silveira, W.A. , Silvente-Poirot, S. , Silverman, G.A. , Simak, J. , Simmet, T. , Simon, A.K. , Simon, H.U. , Simone, C. , Simons, M. , Simonsen, A. , Singh, R. , Singh, S.V. , Singh, S.K. , Sinha, D. , Sinha, S. , Sinicrope, F.A. , Sirko, A. , Sirohi, K. , Sishi, B.J. , Sittler, A. , Siu, P.M. , Sivridis, E. , Skwarska, A. , Slack, R. , Slaninova, I. , Slavov, N. , Smaili, S.S. , Smalley, K.S. , Smith, D.R. , Soenen, S.J. , Soleimanpour, S.A. , Solhaug, A. , Somasundaram, K. , Son, J.H. , Sonawane, A. , Song, C. , Song, F. , Song, H.K. , Song, J.X. , Song, W. , Soo, K.Y. , Sood, A.K. , Soong, T.W. , Soontornniyomkij, V. , Sorice, M. , Sotgia, F. , Soto-Pantoja, D.R. , Sotthibundhu, A. , Sousa, M.J. , Spaink, H.P. , Span, P.N. , Spang, A. , Sparks, J.D. , Speck, P.G. , Spector, S.A. , Spies, C.D. , Springer, W. , Clair, D.S. , Stacchiotti, A. , Staels, B. , Stang, M.T. , Starczynowski, D.T. , Starokadomskyy, P. , Steegborn, C. , Steele, J.W. , Stefanis, L. , Steffan, J. , Stellrecht, C.M. , Stenmark, H. , Stepkowski, T.M. , Stern, S.T. , Stevens, C. , Stockwell, B.R. , Stoka, V. , Storchova, Z. , Stork, B. , Stratoulias, V. , Stravopodis, D.J. , Strnad, P. , Strohecker, A.M. , Strom, A.L. , Stromhaug, P. , Stulik, J. , Su, Y.X. , Su, Z. , Subauste, C.S. , Subramaniam, S. , Sue, C.M. , Suh, S.W. , Sui, X. , Sukseree, S. , Sulzer, D. , Sun, F.L. , Sun, J. , Sun, J. , Sun, S.Y. , Sun, Y. , Sun, Y. , Sun, Y. , Sundaramoorthy, V. , Sung, J. , Suzuki, H. , Suzuki, K. , Suzuki, N. , Suzuki, T. , Suzuki, Y.J. , Swanson, M.S. , Swanton, C. , Sward, K. , Swarup, G. , Sweeney, S.T. , Sylvester, P.W. , Szatmari, Z. , Szegezdi, E. , Szlosarek, P.W. , Taegtmeyer, H. , Tafani, M. , Taillebourg, E. , Tait, S.W. , Takacs-Vellai, K. , Takahashi, Y. , Takats, S. , Takemura, G. , Takigawa, N. , Talbot, N.J. , Tamagno, E. , Tamburini, J. , Tan, C.P. , Tan, L. , Tan, M.L. , Tan, M. , Tan, Y.J. , Tanaka, K. , Tanaka, M. , Tang, D. , Tang, D. , Tang, G. , Tanida, I. , Tanji, K. , Tannous, B.A. , Tapia, J.A. , Tasset-Cuevas, I. , Tatar, M. , Tavassoly, I. , Tavernarakis, N. , Taylor, A. , Taylor, G.S. , Taylor, G.A. , Taylor, J.P. , Taylor, M.J. , Tchetina, E.V. , Tee, A.R. , Teixeira-Clerc, F. , Telang, S. , Tencomnao, T. , Teng, B.B. , Teng, R.J. , Terro, F. , Tettamanti, G. , Theiss, A.L. , Theron, A.E. , Thomas, K.J. , Thome, M.P. , Thomes, P.G. , Thorburn, A. , Thorner, J. , Thum, T. , Thumm, M. , Thurston, T.L. , Tian, L. , Till, A. , Ting, J.P. , Titorenko, V.I. , Toker, L. , Toldo, S. , Tooze, S.A. , Topisirovic, I. , Torgersen, M.L. , Torosantucci, L. , Torriglia, A. , Torrisi, M.R. , Tournier, C. , Towns, R. , Trajkovic, V. , Travassos, L.H. , Triola, G. , Tripathi, D.N. , Trisciuoglio, D. , Troncoso, R. , Trougakos, I.P. , Truttmann, A.C. , Tsai, K.J. , Tschan, M.P. , Tseng, Y.H. , Tsukuba, T. , Tsung, A. , Tsvetkov, A.S. , Tu, S. , Tuan, H.Y. , Tucci, M. , Tumbarello, D.A. , Turk, B. , Turk, V. , Turner, R.F. , Tveita, A.A. , Tyagi, S.C. , Ubukata, M. , Uchiyama, Y. , Udelnow, A. , Ueno, T. , Umekawa, M. , Umemiya-Shirafuji, R. , Underwood, B.R. , Ungermann, C. , Ureshino, R.P. , Ushioda, R. , Uversky, V.N. , Uzcategui, N.L. , Vaccari, T. , Vaccaro, M.I. , Vachova, L. , Vakifahmetoglu-Norberg, H. , Valdor, R. , Valente, E.M. , Vallette, F. , Valverde, A.M. , Van den Berghe, G. , Van Den Bosch, L. , van den Brink, G.R. , van der Goot, F.G. , van der Klei, I.J. , van der Laan, L.J. , van Doorn, W.G. , van Egmond, M. , van Golen, K.L. , Van Kaer, L. , van Lookeren Campagne, M. , Vandenabeele, P. , Vandenberghe, W. , Vanhorebeek, I. , Varela-Nieto, I. , Vasconcelos, M.H. , Vasko, R. , Vavvas, D.G. , Vega-Naredo, I. , Velasco, G. , Velentzas, A.D. , Velentzas, P.D. , Vellai, T. , Vellenga, E. , Vendelbo, M.H. , Venkatachalam, K. , Ventura, N. , Ventura, S. , Veras, P.S. , Verdier, M. , Vertessy, B.G. , Viale, A. , Vidal, M. , Vieira, H. , Vierstra, R.D. , Vigneswaran, N. , Vij, N. , Vila, M. , Villar, M. , Villar, V.H. , Villarroya, J. , Vindis, C. , Viola, G. , Viscomi, M.T. , Vitale, G. , Vogl, D.T. , Voitsekhovskaja, O.V. , von Haefen, C. , von Schwarzenberg, K. , Voth, D.E. , Vouret-Craviari, V. , Vuori, K. , Vyas, J.M. , Waeber, C. , Walker, C.L. , Walker, M.J. , Walter, J. , Wan, L. , Wan, X. , Wang, B. , Wang, C. , Wang, C.Y. , Wang, C. , Wang, C. , Wang, C. , Wang, D. , Wang, F. , Wang, F. , Wang, G. , Wang, H.J. , Wang, H. , Wang, H.G. , Wang, H. , Wang, H.D. , Wang, J. , Wang, J. , Wang, M. , Wang, M.Q. , Wang, P.Y. , Wang, P. , Wang, R.C. , Wang, S. , Wang, T.F. , Wang, X. , Wang, X.J. , Wang, X.W. , Wang, X. , Wang, X. , Wang, Y. , Wang, Y. , Wang, Y. , Wang, Y.J. , Wang, Y. , Wang, Y. , Wang, Y.T. , Wang, Y. , Wang, Z.N. , Wappner, P. , Ward, C. , Ward, D.M. , Warnes, G. , Watada, H. , Watanabe, Y. , Watase, K. , Weaver, T.E. , Weekes, C.D. , Wei, J. , Weide, T. , Weihl, C.C. , Weindl, G. , Weis, S.N. , Wen, L. , Wen, X. , Wen, Y. , Westermann, B. , Weyand, C.M. , White, A.R. , White, E. , Whitton, J.L. , Whitworth, A.J. , Wiels, J. , Wild, F. , Wildenberg, M.E. , Wileman, T. , Wilkinson, D.S. , Wilkinson, S. , Willbold, D. , Williams, C. , Williams, K. , Williamson, P.R. , Winklhofer, K.F. , Witkin, S.S. , Wohlgemuth, S.E. , Wollert, T. , Wolvetang, E.J. , Wong, E. , Wong, G.W. , Wong, R.W. , Wong, V.K. , Woodcock, E.A. , Wright, K.L. , Wu, C. , Wu, D. , Wu, G.S. , Wu, J. , Wu, J. , Wu, M. , Wu, M. , Wu, S. , Wu, W.K. , Wu, Y. , Wu, Z. , Xavier, C.P. , Xavier, R.J. , Xia, G.X. , Xia, T. , Xia, W. , Xia, Y. , Xiao, H. , Xiao, J. , Xiao, S. , Xiao, W. , Xie, C.M. , Xie, Z. , Xie, Z. , Xilouri, M. , Xiong, Y. , Xu, C. , Xu, C. , Xu, F. , Xu, H. , Xu, H. , Xu, J. , Xu, J. , Xu, J. , Xu, L. , Xu, X. , Xu, Y. , Xu, Y. , Xu, Z.X. , Xu, Z. , Xue, Y. , Yamada, T. , Yamamoto, A. , Yamanaka, K. , Yamashina, S. , Yamashiro, S. , Yan, B. , Yan, B. , Yan, X. , Yan, Z. , Yanagi, Y. , Yang, D.S. , Yang, J.M. , Yang, L. , Yang, M. , Yang, P.M. , Yang, P. , Yang, Q. , Yang, W. , Yang, W.Y. , Yang, X. , Yang, Y. , Yang, Y. , Yang, Z. , Yang, Z. , Yao, M.C. , Yao, P.J. , Yao, X. , Yao, Z. , Yao, Z. , Yasui, L.S. , Ye, M. , Yedvobnick, B. , Yeganeh, B. , Yeh, E.S. , Yeyati, P.L. , Yi, F. , Yi, L. , Yin, X.M. , Yip, C.K. , Yoo, Y.M. , Yoo, Y.H. , Yoon, S.Y. , Yoshida, K.I. , Yoshimori, T. , Young, K.H. , Yu, H. , Yu, J.J. , Yu, J.T. , Yu, J. , Yu, L. , Yu, W.H. , Yu, X.F. , Yu, Z. , Yuan, J. , Yuan, Z.M. , Yue, B.Y. , Yue, J. , Yue, Z. , Zacks, D.N. , Zacksenhaus, E. , Zaffaroni, N. , Zaglia, T. , Zakeri, Z. , Zecchini, V. , Zeng, J. , Zeng, M. , Zeng, Q. , Zervos, A.S. , Zhang, D.D. , Zhang, F. , Zhang, G. , Zhang, G.C. , Zhang, H. , Zhang, H. , Zhang, H. , Zhang, H. , Zhang, J. , Zhang, J. , Zhang, J. , Zhang, J. , Zhang, J.P. , Zhang, L. , Zhang, L. , Zhang, L. , Zhang, L. , Zhang, M.Y. , Zhang, X. , Zhang, X.D. , Zhang, Y. , Zhang, Y. , Zhang, Y. , Zhang, Y. , Zhang, Y. , Zhao, M. , Zhao, W.L. , Zhao, X. , Zhao, Y.G. , Zhao, Y. , Zhao, Y. , Zhao, Y.X. , Zhao, Z. , Zhao, Z.J. , Zheng, D. , Zheng, X.L. , Zheng, X. , Zhivotovsky, B. , Zhong, Q. , Zhou, G.Z. , Zhou, G. , Zhou, H. , Zhou, S.F. , Zhou, X.J. , Zhu, H. , Zhu, H. , Zhu, W.G. , Zhu, W. , Zhu, X.F. , Zhu, Y. , Zhuang, S.M. , Zhuang, X. , Ziparo, E. , Zois, C.E. , Zoladek, T. , Zong, W.X. , Zorzano, A. , Zughaier, S.M. , 2016. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laderach, D. , Compagno, D. , Danos, O. , Vainchenker, W. , Galy, A. , 2003. RNA interference shows critical requirement for NF-kappa B p50 in the production of IL-12 by human dendritic cells. Journal of Immunol. 171, 1750–1757. [DOI] [PubMed] [Google Scholar]
- Levine, A.J. , 1997. p53, the cellular gatekeeper for growth and division. Cell. 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Liu, J. , Ma, Q. , Zhang, M. , Wang, X. , Zhang, D. , Li, W. , Wang, F. , Wu, E. , 2012. Alterations of TP53 are associated with a poor outcome for patients with hepatocellular carcinoma: evidence from a systematic review and meta-analysis. Eur. J. Cancer. 48, 2328–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorin, S. , Hamai, A. , Mehrpour, M. , Codogno, P. , 2013. Autophagy regulation and its role in cancer. Semin. Cancer Biol. 23, 361–379. [DOI] [PubMed] [Google Scholar]
- Maskey, D. , Yousefi, S. , Schmid, I. , Zlobec, I. , Perren, A. , Friis, R. , Simon, H.U. , 2013. ATG5 is induced by DNA-damaging agents and promotes mitotic catastrophe independent of autophagy. Nat. Commun. 4, 2130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathew, R. , Kongara, S. , Beaudoin, B. , Karp, C.M. , Bray, K. , Degenhardt, K. , Chen, G. , Jin, S. , White, E. , 2007. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes Development. 21, 1367–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa, Y. , Oshima, S. , Takahara, M. , Maeyashiki, C. , Nemoto, Y. , Kobayashi, M. , Nibe, Y. , Nozaki, K. , Nagaishi, T. , Okamoto, R. , Tsuchiya, K. , Nakamura, T. , Ma, A. , Watanabe, M. , 2015. TNFAIP3 promotes survival of CD4 T cells by restricting MTOR and promoting autophagy. Autophagy. 11, 1052–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima, N. , Noda, T. , Yoshimori, T. , Tanaka, Y. , Ishii, T. , George, M.D. , Klionsky, D.J. , Ohsumi, M. , Ohsumi, Y. , 1998. A protein conjugation system essential for autophagy. Nature. 395, 395–398. [DOI] [PubMed] [Google Scholar]
- Morrison, A. , Chen, L. , Wang, J. , Zhang, M. , Yang, H. , Ma, Y. , Budanov, A. , Lee, J.H. , Karin, M. , Li, J. , 2015. Sestrin2 promotes LKB1-mediated AMPK activation in the ischemic heart. FASEB J. 29, 408–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morselli, E. , Tasdemir, E. , Maiuri, M.C. , Galluzzi, L. , Kepp, O. , Criollo, A. , Vicencio, J.M. , Soussi, T. , Kroemer, G. , 2008. Mutant p53 protein localized in the cytoplasm inhibits autophagy. Cell Cycle. 7, 3056–3061. [DOI] [PubMed] [Google Scholar]
- Muller, P.A. , Vousden, K.H. , 2013. p53 mutations in cancer. Nat. Cell Biol. 15, 2–8. [DOI] [PubMed] [Google Scholar]
- Negrini, S. , Gorgoulis, V.G. , Halazonetis, T.D. , 2010. Genomic instability–an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228. [DOI] [PubMed] [Google Scholar]
- Rhodes, D.R. , Yu, J. , Shanker, K. , Deshpande, N. , Varambally, R. , Ghosh, D. , Barrette, T. , Pandey, A. , Chinnaiyan, A.M. , 2004. ONCOMINE: a cancer microarray database and integrated data-mining platform. Neoplasia. 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell, R.C. , Tian, Y. , Yuan, H. , Park, H.W. , Chang, Y.Y. , Kim, J. , Kim, H. , Neufeld, T.P. , Dillin, A. , Guan, K.L. , 2013. ULK1 induces autophagy by phosphorylating Beclin-1 and activating VPS34 lipid kinase. Nat. Cell Biol. 15, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanli, T. , Linher-Melville, K. , Tsakiridis, T. , Singh, G. , 2012. Sestrin2 modulates AMPK subunit expression and its response to ionizing radiation in breast cancer cells. PLoS One. 7, e32035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santoro, R. , Strano, S. , Blandino, G. , 2014. Transcriptional regulation by mutant p53 and oncogenesis. Sub-cellular Biochem. 85, 91–103. [DOI] [PubMed] [Google Scholar]
- Schonthal, A.H. , 2009. Endoplasmic reticulum stress and autophagy as targets for cancer therapy. Cancer Lett. 275, 163–169. [DOI] [PubMed] [Google Scholar]
- Soussi, T. , Wiman, K.G. , 2007. Shaping genetic alterations in human cancer: the p53 mutation paradigm. Cancer Cell. 12, 303–312. [DOI] [PubMed] [Google Scholar]
- Stambolsky, P. , Tabach, Y. , Fontemaggi, G. , Weisz, L. , Maor-Aloni, R. , Siegfried, Z. , Shiff, I. , Kogan, I. , Shay, M. , Kalo, E. , Blandino, G. , Simon, I. , Oren, M. , Rotter, V. , 2010. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell. 17, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strano, S. , Dell'Orso, S. , Di Agostino, S. , Fontemaggi, G. , Sacchi, A. , Blandino, G. , 2007. Mutant p53: an oncogenic transcription factor. Oncogene. 26, 2212–2219. [DOI] [PubMed] [Google Scholar]
- Tong, X. , Yin, L. , Washington, R. , Rosenberg, D.W. , Giardina, C. , 2004. The p50-p50 NF-kappaB complex as a stimulus-specific repressor of gene activation. Mol. Cell. Biochem. 265, 171–183. [DOI] [PubMed] [Google Scholar]
- Valenti, F. , Fausti, F. , Biagioni, F. , Shay, T. , Fontemaggi, G. , Domany, E. , Yaffe, M.B. , Strano, S. , Blandino, G. , Di Agostino, S. , 2011. Mutant p53 oncogenic functions are sustained by Plk2 kinase through an autoregulatory feedback loop. Cell Cycle. 10, 4330–4340. [DOI] [PubMed] [Google Scholar]
- Valenti, F. , Ganci, F. , Fontemaggi, G. , Sacconi, A. , Strano, S. , Blandino, G. , Di Agostino, S. , 2015. Gain of function mutant p53 proteins cooperate with E2F4 to transcriptionally downregulate RAD17 and BRCA1 gene expression. Oncotarget. 6, 5547–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viry, E. , Paggetti, J. , Baginska, J. , Mgrditchian, T. , Berchem, G. , Moussay, E. , Janji, B. , 2014. Autophagy: an adaptive metabolic response to stress shaping the antitumor immunity. Biochem. Pharmacol. 92, 31–42. [DOI] [PubMed] [Google Scholar]
- Vousden, K.H. , Lu, X. , 2002. Live or let die: the cell's response to p53. Nature reviews. Cancer. 2, 594–604. [DOI] [PubMed] [Google Scholar]
- Waddell, N. , Pajic, M. , Patch, A.M. , Chang, D.K. , Kassahn, K.S. , Bailey, P. , Johns, A.L. , Miller, D. , Nones, K. , Quek, K. , Quinn, M.C. , Robertson, A.J. , Fadlullah, M.Z. , Bruxner, T.J. , Christ, A.N. , Harliwong, I. , Idrisoglu, S. , Manning, S. , Nourse, C. , Nourbakhsh, E. , Wani, S. , Wilson, P.J. , Markham, E. , Cloonan, N. , Anderson, M.J. , Fink, J.L. , Holmes, O. , Kazakoff, S.H. , Leonard, C. , Newell, F. , Poudel, B. , Song, S. , Taylor, D. , Waddell, N. , Wood, S. , Xu, Q. , Wu, J. , Pinese, M. , Cowley, M.J. , Lee, H.C. , Jones, M.D. , Nagrial, A.M. , Humphris, J. , Chantrill, L.A. , Chin, V. , Steinmann, A.M. , Mawson, A. , Humphrey, E.S. , Colvin, E.K. , Chou, A. , Scarlett, C.J. , Pinho, A.V. , Giry-Laterriere, M. , Rooman, I. , Samra, J.S. , Kench, J.G. , Pettitt, J.A. , Merrett, N.D. , Toon, C. , Epari, K. , Nguyen, N.Q. , Barbour, A. , Zeps, N. , Jamieson, N.B. , Graham, J.S. , Niclou, S.P. , Bjerkvig, R. , Grutzmann, R. , Aust, D. , Hruban, R.H. , Maitra, A. , Iacobuzio-Donahue, C.A. , Wolfgang, C.L. , Morgan, R.A. , Lawlor, R.T. , Corbo, V. , Bassi, C. , Falconi, M. , Zamboni, G. , Tortora, G. , Tempero, M.A. , Australian Pancreatic Cancer Genome, I. , Gill, A.J. , Eshleman, J.R. , Pilarsky, C. , Scarpa, A. , Musgrove, E.A. , Pearson, J.V. , Biankin, A.V. , Grimmond, S.M. , 2015. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature. 518, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walerych, D. , Napoli, M. , Collavin, L. , Del Sal, G. , 2012. The rebel angel: mutant p53 as the driving oncogene in breast cancer. Carcinogenesis. 33, 2007–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz, L. , Damalas, A. , Liontos, M. , Karakaidos, P. , Fontemaggi, G. , Maor-Aloni, R. , Kalis, M. , Levrero, M. , Strano, S. , Gorgoulis, V.G. , Rotter, V. , Blandino, G. , Oren, M. , 2007. Mutant p53 enhances nuclear factor kappaB activation by tumor necrosis factor alpha in cancer cells. Cancer Res. 67, 2396–2401. [DOI] [PubMed] [Google Scholar]
- Zhou, G. , Wang, J. , Zhao, M. , Xie, T.X. , Tanaka, N. , Sano, D. , Patel, A.A. , Ward, A.M. , Sandulache, V.C. , Jasser, S.A. , Skinner, H.D. , Fitzgerald, A.L. , Osman, A.A. , Wei, Y. , Xia, X. , Songyang, Z. , Mills, G.B. , Hung, M.C. , Caulin, C. , Liang, J. , Myers, J.N. , 2014. Gain-of-function mutant p53 promotes cell growth and cancer cell metabolism via inhibition of AMPK activation. Mol. Cell. 54, 960–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Suppl. Figure 1 (A) Cells were seeded in 100‐mm diameter culture dishes and transfected for 48 h with sip50 oligos or with negative control (siGFP). 40 μg of total protein extracts were analyzed by Western blotting using p50 NF‐κB and GAPDH or H3 (as normalizing factor) antibodies. (B) Cross‐linked chromatin derived from sip50‐treated Panc1 cells and from siGFP‐transfected control, was immunoprecipitated with the indicated antibodies or in the absence of antibody, and analyzed by RT‐qPCR with specific primers (see Material and Methods or Supplementary Table 2) for the regions of ATG12 promoter indicated in the Figure 4A or in H1H2B2 genic region resulted negative for the recruitment of mutant p53. *p < 0.05.
Suppl. Figure 2 (A) Panc1 cells were pre‐treated with 1 mM 3MA for 1 h before cell transfection with plasmids coding for LC3‐GFP and pRSuper‐p53 vector (shp53) or empty vector (shCTRL) for 48 h. Cells were fixed and nuclei were stained with Hoescht (blue). Merge images come from a single z‐plane. Scale bar 10 μm. (B) Panc1 cells were pre‐treated with 1 mM 3MA for 1 h before cell transfection for 48 h. Whole‐cell extracts were used for Western blot analysis of the autophagic protein LC3 (isoforms I and II), p53 and GAPDH (as control loading). (C‐E) Panc1 and MDA‐MB‐468 cells were seeded in 96‐well plates and pre‐treated with 1 mM 3MA for 1 h before cell transfection for 48 h. Autophagosome formation (C), cell growth (D), and apoptosis (E) were determined using MDC assay, crystal violet colorimetric assay and annexinV/FITC binding assay, respectively. All the experiments presented in this figure are representative of three biological replicates. P‐values were calculated with two‐tailed t‐test. Statistical analysis: *p < 0.05 shp53 vs shCTRL; §p < 0.05 shp53+3MA vs shp53.
Suppl. Figure 3 Cells were seeded in 96‐well plates and transfected with pRSuper‐p53 vector (shp53), with pCDNA‐p53R175H, pCDNA‐p53R273H plasmids or their negative controls (empty pRSuper and pCDNA3 mock vector, respectively). Cell growth was determined using the crystal violet colorimetric assay. Statistical analysis: *p < 0.05 shp53 vs CTRL; §p < 0.05 R175H or R273H vs mock.
Suppl. Figure 4 Panc1 cells were transfected with pMSCV‐Puro‐miR30‐shATG5 vector (or its negative empty vector). Gene expression analysis of ATG5 was performed by RT‐qPCR and was normalized to GAPDH mRNA. *p < 0.05 shATG5 vs shCTRL.
Suppl. Figure 5 Western blot of p53, and GAPDH as normalizing factor, performed in all cell lines used for RT‐qPCR shown in Figure 3A. To exclude back‐side effects of shp53 sequence (pRSuper‐p53 vector) and to confirm the robustness of the data, a commercial siRNA smart pool of three oligonucleotides (si‐p53) transiently targeting p53 (Santa Cruz Biotech. sc‐29435), and its si‐GFP negative control, were used in these experiments.
Suppl. Figure 6 (A and B) Indicated cell lines were transfected with pRSuper‐p53 vector (shp53), with pCDNA‐p53R175H, pCDNA‐p53R273H plasmids or their negative controls (empty pRSuper and pCDNA3 mock vector, respectively). Gene expression analysis of CCNB1 was performed by RT‐qPCR and was normalized to GAPDH mRNA. *p < 0.05 sip53 vs siGFP; R175H or R273H vs vector.
Suppl. Figure 7 TRANSFAC matrix of NF‐κB p50 and NF‐κB p65 consensus sequences used by MatInspector software.
Suppl. Figure 8 Immunoprecipitations of NF‐κB p50 and western blot analysis for p53 binding are performed from lysates of the indicated cancer cell lines expressing mutant p53 proteins, as described in Material and Methods.
Suppl. Figure 9 Panc1 cells were transfected with pLVTHM‐p53 vector (shp53) or its negative control pLVTHM (shCTRL) to confirm results obtained with pRSUPER‐p53 vector. (A) Autophagosome formation assay was performed using the incorporation of MDC probe. *p < 0.05 shp53 vs shCTRL (B) Western blot of P‐AMPK, AMPK, P‐p70S6K, p70S6Kp53 and GAPDH was performed as described in Material and Methods.
Suppl. Figure 10 Quantitative analysis of SESN1/GAPDH, SESN2/GAPDH, P‐AMPK/AMPK, P‐p70S6K/p70S6K and p53/GAPDH ratios representatively shown in Figure 5A. The Western blot bands were scanned as digital peaks and the areas of the peaks were reported as fold change, as described in Material and Methods. *p < 0.05 shp53 vs shCTRL; §p < 0.05 R175H or R273H vs mock.
Suppl. Figure 11 Quantitative analysis of P‐Beclin1, Becin1 and p53 normalized on GAPDH expression representatively shown in Figure 6A. The Western blot bands were scanned as digital peaks and the areas of the peaks were reported as fold change, as described in Material and Methods. §p < 0.05 R175H or R273H vs mock; *p < 0.05 R175H+EVE vs R175H or R273H+EVE vs R273H; # shp53 vs shCTRL.
Suppl. Figure 12 Cells were seeded in 100‐mm diameter culture dishes and transfected for 48 h with siBeclin1 oligos or with negative control (siGFP). 40 μg of total protein extracts were analyzed by Western blot using Beclin1 and GAPDH (as normalizing factor) antibodies.
Supplementary data
Supplementary data