

Abstract
Fulvestrant is a dose dependent selective estrogen receptor (ER) down‐regulator (SERD) used in ER‐positive metastatic breast cancer (MBC). Nearly all patients develop resistance. We performed molecular analysis of circulating tumor cells (CTC) to gain insight into fulvestrant resistance.
Preclinical studies were performed with cultured breast cancer cells spiked into human blood and analyzed on the CellSearch® system. Clinical data are limited to a subset of patients with ER‐positive MBC from a previously reported pilot trial whose disease was progressing on fulvestrant (N = 7) or aromatase inhibitors (AIs) (N = 10). CTCs were enumerated and phenotyped for ER and B‐cell lymphoma (BCL2) using the CellSearch® CXC kit.
In preclinical modeling, tamoxifen and AIs resulted in stabilized ER expression, whereas fulvestrant eliminated it. Five of seven patients progressing on fulvestrant had ≥5CTC/7.5 ml WB. Two of these five, treated with 500 mg/month fulvestrant, had no detectable CTC‐expression of ER and BCL2 (an ER regulated gene). Three patients had heterogeneous CTC‐ER and BCL2 expression indicating incomplete degradation of the ER target by fulvestrant. Two of these patients received 250 mg/month whereas the third patient received 500 mg/month fulvestrant. Her cancer harbored a mutation (Y537S) in the estrogen receptor alpha gene (ESR1). All seven ER positive patients progressing on AIs had heterogeneous CTC‐ER expression.
These results suggest heterogeneous mechanisms of resistance to fulvestrant, including insufficient dosage, ESR1 mutation, or conversion to dependence on non‐ER pathways. CTC enumeration, phenotyping, and genotyping might identify patients who would benefit from fulvestrant dose escalation versus switching to alternative therapies.
Keywords: Circulating tumor cells (CTC), Breast cancer, Hormone receptor positive, Fulvestrant, Aromatase inhibitors
Highlights
Heterogeneous CTC‐ER & BCL2 expression suggests diverse mechanisms of ET resistance.
CTC‐ER expression may be a pharmacodynamic monitoring tool for SERD therapy.
Liquid biopsy may help to understand the bases of resistance to ET.
Serial liquid biopsies may provide further insight into drug resistance.
Abbreviations
- AI
aromatase inhibitor
- BCL2
B-cell lymphoma 2
- CCS
charcoal stripped calf serum
- CTC
circulating tumor cells
- EpCAM
epithelial cell adhesion molecule
- ER
estrogen receptor
- ESR1
estrogen receptor alpha gene
- ETs
endocrine therapies
- E2
17β-estradiol
- HER2
human epidermal growth factor receptor 2
- HR
hormone receptor
- IM
intramuscular
- LBD
ligand binding domain
- MBC
metastatic breast cancer
- oSERD
oral selective estrogen receptor down-regulator (SERD)
- OS
overall survival
- pt-DNA
circulating plasma tumor DNA
- SERD
selective estrogen receptor down-regulator
- SERMs
selective estrogen receptor modulators
- WB
whole blood
1. Introduction
Multiple endocrine therapies (ETs) are effective in hormone receptor (HR)‐positive metastatic breast cancer (MBC). These drugs include selective estrogen receptor (ER) modulators (SERMs: tamoxifen, toremifene, or raloxifene), aromatase inhibitors (AIs: anastrozole, letrozole, exemestane), and selective ER down‐regulators (SERDs), such as fulvestrant.
Several mechanisms of resistance to ET have been proposed (Osborne and Schiff, 2011), including absence of ER expression by deletion or suppression, alteration of ER signaling pathway genes, or upregulation of multiple growth factor receptor pathways. Another possible mechanism of resistance includes mutations in the ligand binding domain (LBD) of estrogen receptor alpha gene (ESR1) (Jeselsohn et al., 2015; Robinson et al., 2013; Toy et al., 2013), which confers ligand‐independent ER signaling and therefore a relative and context‐specific resistance to ET.
Importantly, simple pharmacokinetic considerations are also important mechanisms of resistance. Clinical trials have shown little, if any, evidence of dose response to SERMs or AIs (Hayes et al., 1995, 1996, 1983, 1976). In contrast, the activity of fulvestrant is clearly dose dependent. The initially recommended dose of fulvestrant was 250 mg intramuscular (IM) once monthly, after a brief loading period, for all patients (Osborne et al., 2002). In the CONFIRM trial, Di Leo et al. demonstrated that 500 mg IM is superior to 250 mg IM, and the former has become the standard dose (Di Leo et al., 2010). Recently, fulvestrant 500 mg has been shown to improve overall survival (OS) when compared to AI in patients with HR positive MBC (Ellis et al., 2015). Nonetheless, even at the higher dose (500 mg/month), most HR‐positive MBC develop resistance and progress. Currently, there is no way to predict which patients, if any might benefit from even higher doses of fulvestrant, or how to monitor if dose adjustments have been effective. Further, there is also no means to predict if a patient on a SERD might be better treated with an alternative form of ET, addition of complementary treatments to ET, such as everolimus or palbociclib, or even proceed to chemotherapy for palliation.
ER is clearly the target of ET, and ER is highly predictive of ET response or not (Davies et al., 2011). While not completely controlled by ER, BCL2 expression is strongly correlated with ER, and it is presumed to be, at least in part, an estrogen‐responsive gene (Teixeira et al., 1995). Thus, monitoring ER and downstream genes such as BCL‐2 might provide baseline and pharmacodynamic monitoring tools to predict response to any ET and to optimize the dose of fulvestrant, or other, newly developed SERDs. However, serial biopsies of metastatic disease to demonstrate loss of ER expression or changing expression of other markers is invasive, expensive, and logistically difficult. In this regard, circulating tumor cells (CTC) are currently being investigated as a type of “liquid biopsy” that might substitute for cancer tissue biopsy (Alix‐Panabieres and Pantel, 2013). We have recently reported the analytical validity of measuring CTC expression of markers of endocrine sensitivity (ER, BCL2) or resistance (HER2, Ki‐67) using the CellSearch® system (Janssen Diagnostics, LLC) (Paoletti et al., 2015). In that study, a subset of patients with ER‐positive MBC were progressing on fulvestrant or an AI, giving us the opportunity to examine expression of these biomarkers when the cancer had developed resistance to these agents. We report variable expression of CTC‐ER and CTC‐BCL2, suggesting multiple inter‐patient mechanisms of resistance to SERD therapy.
2. Materials and methods
2.1. In vitro preclinical studies of CTC‐biomarker expression
MCF‐7 and SKBR3 cells were originally obtained from the Tissue Culture Shared Resource (TCSR) at the Lombardi Comprehensive Cancer Center (LCCC; Georgetown University, Washington, DC) and routinely maintained as previously described (Rae et al., 2005). For assays in defined hormone‐free conditions, cells were repeatedly washed and grown in steroid depleted media (phenol red‐free IMEM supplemented with 10% charcoal‐stripped calf serum—CCS), and on the fifth day the cells were treated ex vivo for 24 h with the following ET drugs: tamoxifen (5 × 10−8 M); 17β‐estradiol (E2) (10−10 M); tamoxifen (5 × 10−8 M) + E2 (10−10 M); fulvestrant (5 × 10−8 M); fulvestrant (5 × 10−8 M) + E2 (10−10 M). MCF‐7 and SKBR3 cells cultured in steroid hormone‐free conditions were used as positive and negative controls for cellular ER expression, respectively. Approximately 150 of these treated cells were then spiked into 7.5 ml of normal human blood and processed using the CellSearch® System. The identity of the cells was confirmed by standard Short Tandem Repeat profiling (February 2011) and cultures were subjected to routine testing for mycoplasma contamination.
2.2. Patient accrual, blood collection, and processing, and CTC analysis
Fifty patients with progressive MBC scheduled to start a new therapeutic regimen of any type (ET or chemotherapy or other) were enrolled into a prospective single‐institution pilot study to establish the analytical validity of performing expression of selected biomarkers on CTC as previously reported (Paoletti et al., 2015). All patients provided a signed informed consent approved by the University of Michigan Institutional Review Board.
Blood specimens were drawn upon enrollment into 10 ml CellSave tubes, stored at room temperature, and processed for CTC enumeration and semi‐quantitative analysis of expression of ER, BCL2, HER2, and Ki‐67 using the using the CXC CellSearch® Kit and CellSearch® system within 96 h as previously reported (Paoletti et al., 2015). The pre‐analytical details follow the BRISQ criteria (Moore et al., 2011). The following antigen‐specific fluorescent‐labeled antibodies were used to characterize ER, HER2, BCL‐2, and Ki67 expression on CTC: ER‐α monoclonal murine ER‐119.3 antibody (Ab) (Janssen Diagnostics, LLC), HER2 monoclonal murine Her81 Ab (Janssen Diagnostics, LLC), BCL‐2, monoclonal murine Ab BCL‐2/(100) (BD Pharmingen), Ki67 monoclonal murine B56 Ab (BD Pharmingen). Only ER and BCL‐2 expression are included in this report. CTC‐staining for ER and BCL‐2 was determined visually by two readers. CTC‐staining was expressed on an arbitrary scale of 0–3+, as described previously (Paoletti et al., 2015). For each marker, 0–1+ was considered negative and 2–3+ was considered positive (Paoletti et al., 2015).
We conducted this retrospective observation of two cohorts of patients who, when enrolled, were progressing on either fulvestrant (n = 7 out of 50 enrolled patients) or an AI (n = 10 out of 50 enrolled patients). Each CTC‐biomarker analysis requires a separate 7.5 ml aliquot of blood. Thus, to perform four different CTC‐biomarker analyses, four separate aliquots were processed for enumeration and phenotyping. In our prior analytical publication, we reported the number of CTC/7.5 ml WB as the mean of the enumeration of all 4 aliquots from a single blood draw, and we designated a cutoff of mean ≥5CTC/7.5 ml as elevated. Of the 17 patients in the current report, 13 had a mean of ≥5CTC/7.5 ml. Five patients, two in the fulvestrant and three in the AI group, had a mean <5CTC/7.5 ml whole blood and were considered to be uninformative for this analysis due to low/negative CTC. However, one patient (#21) had 4, 6, 4, and 5 CTC/7.5 ml WB in the ER, BCL2, HER2, and Ki‐67 aliquots, respectively, so that her mean CTC count was 4.8/7.5 ml WB, and she is included in the current report. Further, a second patient (#17) had ≥5CTC/7.5 ml WB in three aliquots (ER, HER2, and Ki‐67), but only 3 CTC/7.5 ml WB in the BCL2 aliquot. Her mean CTC/7.5 ml WB was 7, and she, too, is included in the current analysis.
3. Results
3.1. Pre‐clinical studies of effect of endocrine manipulation on CTC‐biomarker expression
There is an extensive body of literature studying ER expression under different endocrine therapy conditions (Wijayaratne and McDonnell, 2001; Wijayaratne et al., 1999). However, in order to establish analytical validity in the CellSearch® system of monitoring CTC‐biomarker expression in different hormonal milieus, ER‐positive human MCF‐7 breast cancer cells were cultured in steroid hormone‐free conditions (mimicking patients on AI therapy) or treated with tamoxifen or fulvestrant, were spiked into normal human blood, then cell number and ER was expression determined using the CellSearch® system as previously described (Paoletti et al., 2015). Examples of CTC‐ER staining in CellSearch® are shown in Supplementary Figure 1. Table 1, which is representative of three separate experiments, illustrates that CTC‐ER expression was similar for MCF‐7 cells grown in the absence of estrogen or treated with 5 × 10−8 M tamoxifen for 24 h (56% ± 0.3%). The CTC‐ER was slightly decreased if MCF‐7 cells were cultured in both E2 and tamoxifen (51%), mimicking the pre‐menopausal state. On the other hand, CTC‐ER was substantially suppressed when MCF‐7 cells were treated with E2 (10−10 M) alone, fulvestrant (5 × 10−8 M) alone, and the combination of E2 and fulvestrant (5.8%, 0.7%, and 2.7%, respectively). The ER‐negative SKBR3 cells, were used as a negative control with 0% of ER expression detected (Table 1). Together, these data suggest that monitoring ER expression in CTCs could serve as a useful pharmacodynamic tool in patients treated with different ETs when analyzed by CellSearch®.
Table 1.
In vitro preclinical studies of CTC‐ER expression.
| Cell line | Condition/treatment (dose) | % Of CTC‐ER positive (2+/3+) |
|---|---|---|
| MCF‐7a | CCSb | 56% |
| MCF‐7 | Tamoxifen (5 × 10−8 M) | 56% |
| MCF‐7 | E2c(10−10 M) + Tamoxifen (5 × 10−8 M) | 51.2% |
| MCF‐7 | E2c (10−10 M) | 5.8% |
| MCF‐7 | Fulvestrant (5 × 10−8M) | 0.7% |
| MCF‐7 | E2 (10−10 M) + Fulvestrant (5 × 10−8M) | 2.7% |
| SKBR3d | CCSb | 0% |
MCF‐7 = ER positive.
CCS = charcoal stripped calf serum.
E2 = estradiol.
SKBR3 = ER negative.
3.2. Patient results
3.2.1. CTC‐biomarker for patients progressing on fulvestrant
Of 50 enrolled and eligible patients, 19 were progressing on ET at the time of blood draw (Figure 1). Two of these were progressing on tamoxifen and are not included in this report. Seven were progressing on fulvestrant and 10 were progressing on AIs (N = 4 letrozole; N = 1 letrozole and PI3K inhibitor; N = 3 exemestane; N = 2 anastrozole). Of the seven patients progressing on fulvestrant, two patients had undetectable CTCs (#27 and 32), and therefore CTC ER, BCL2, HER2 and Ki‐67 expression could not be determined, whereas five patients with a mean of ≥5CTC/7.5 ml of WB were assessed for biomarker expression (Figure 2).
Figure 1.
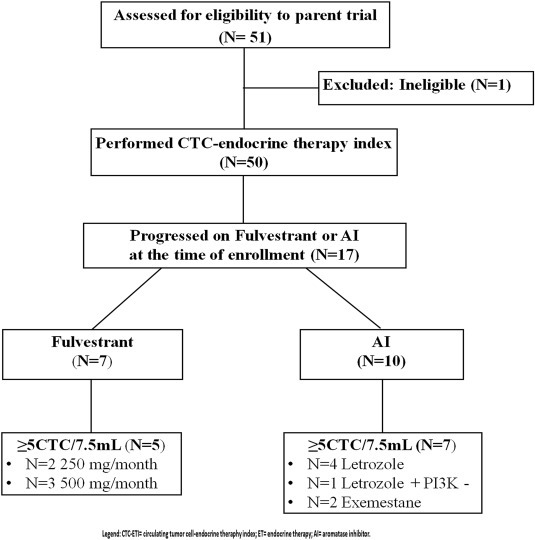
REMARK diagram for patient enrollment and distribution.
Figure 2.
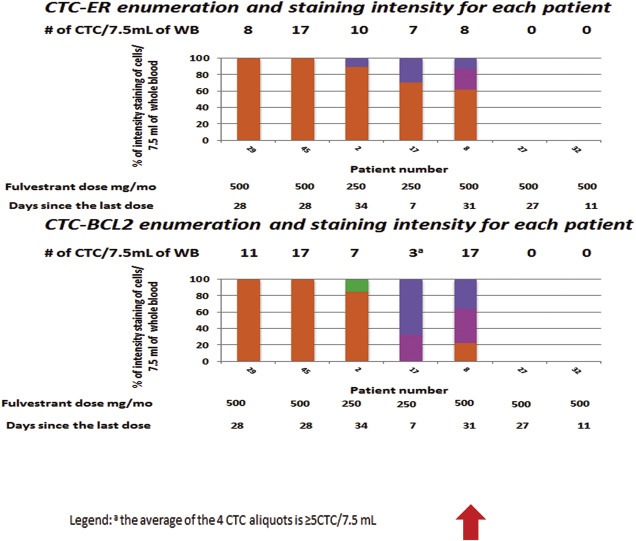
CTC‐biomarker enumeration and staining intensity for each patient progressing on fulvestrant. Each bar represents a different patient and the individual colors within each bar provide the percentage of CTC that stained 0 ( orange), 1+ (
orange), 1+ ( fuchsia), 2+ (
fuchsia), 2+ ( blue), or 3+ (
blue), or 3+ ( green) for ER and BCL2 expression within each patient. The number of CTC for each aliquot/marker is noted above each bar. The red arrow emphasizes patient #8. Although she was treated with fulvestrant at the dose 500 mg/month, she still yet had some CTC‐ER expression in CTC.
green) for ER and BCL2 expression within each patient. The number of CTC for each aliquot/marker is noted above each bar. The red arrow emphasizes patient #8. Although she was treated with fulvestrant at the dose 500 mg/month, she still yet had some CTC‐ER expression in CTC.
CTC in two of these five patients (#29, 45), who had 8 and 17 CTC/7.5 mL WB, were all negative for ER expression (0 or 1+) (Figure 2). Both of these patients were progressing on the now standard dose of fulvestrant (500 mg/month).
In contrast, CTC in three other patients treated with fulvestrant (#2, 17, and 8) were found to express ER, albeit heterogeneously within each patient. These patients had 10, 7, and 8 CTC/7.5 ml WB, with ER expression (2+ or 3+) of 10%, 28%, and 12%, respectively, with the remaining CTC being ER negative (0, 1+). In all three cases, BCL2 positive CTC were identified, suggesting intact ER signaling. Two of these patients (#2 and 17) were treated with the lower, less effective dose of fulvestrant (250 mg/month dose) (Di Leo et al., 2010). In contrast, patient #8 was treated with the recently‐established more effective higher dose of fulvestrant (500 mg/month dose). This patient received her treatment within a month of the time of blood draw, and yet she still had positive CTC‐ER and BCL2 expression in a portion, but not all, of her CTC.
3.2.2. CTC‐biomarker for patients progressing on AI
To contrast with progression on SERD, we examined CTC‐ER and other markers in 10 patients enrolled in the same pilot study, but who were progressing on AIs. As shown in Figure 3, three of them did not have CTC and thus the assay was uninformative. The other seven patients (#15, 21, 28, 34, 41, 16, and 49) had elevated CTC. As expected from our in vitro pre‐clinical data, CTC‐ER expression was present in every patient and, in general, the fraction of ER positive CTC was higher than in the three fulvestrant patients who had positive CTC‐ER (40%, 50%, 8%, 50%, 25%, 62%, and 8% of cells, respectively) (Figure 3). CTC‐BCL2 could not be performed on one patient due to technical failure (#15), but it was positive in all but one (#21) of the other patients, ranging from 14 to 100%.
Figure 3.
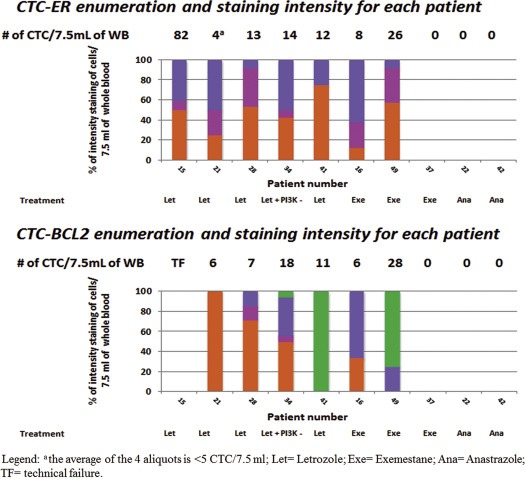
CTC‐Biomarker enumeration and staining intensity for each patient progressing on AI. Each bar represents a different patient and the individual colors within each bar provide the percentage of CTC that stained 0 ( orange), 1+ ( fuchsia), 2+ ( blue), or 3+ ( green) for ER and BCL2 expression within each patient. The number of CTC for each aliquot/marker is noted above each bar.
4. Discussion
In this paper, we investigated the potential use of CTC‐ER as a pharmacodynamic monitoring tool to better personalize management of patients with previously demonstrated ER positive MBC who are progressing on fulvestrant, or for that matter AIs. As expected, in our pre‐clinical experiment, we found that a SERM (tamoxifen) and E2 depletion (mimicking E2 depletion from AI therapy) resulted in stabilized expression of CTC‐ER (51.2%), as determined in a semi‐quantitative fashion using CellSearch®. In contrast, CTC‐ER was decreased in the presence of fulvestrant (0.7%), as measured in the Cellsearch® system. These data strongly suggest this assay can be used to monitor CTC‐biomarker results during ET.
We then retrospectively explored CTC‐ER within a subgroup of patients with HR positive MBC who were progressing on either fulvestrant or an AI at the time of blood draw. Since the CTC‐BCL2 data were available, we investigated whether there appeared to be a correlation, although not perfect, between CTC‐ER and BCL2. We assumed that since BCL2 is both an ER‐responsive gene and is overexpressed in approximately 85% of ER‐positive breast cancers (Johnston et al., 1994; Teixeira et al., 1995), its expression would represent intact ER signaling. Indeed BCL2 is expressed, although less commonly, in ER‐negative breast cancers (Dawson et al., 2010). Nonetheless, BCL2 was investigated in the parent trial because several studies have suggested that ER‐positive, BCL2‐positive breast cancers are more sensitive to endocrine therapies than ER‐positive, BCL2‐negative cancers, and therefore BCL2 was incorporated into the CTC‐endocrine therapy index (Paoletti et al., 2015).
Our exploratory data suggest diverse mechanisms of resistance to fulvestrant between and even within patients. Two of these patients (#2 and 17) were treated with the lower and now considered less effective dose of fulvestrant (250 mg/month). They still expressed CTC‐ER and BCL2. We hypothesize that the presence of BCL2 suggests ongoing signaling through ER. However, these patients also had ER‐negative and BCL2‐negative CTC. We speculate that the presence of ER‐positive CTC, with evidence of ER signaling, suggests that higher doses of fulvestrant might have been more effective. However, the presence of ER‐negative CTC in these patients raises concern that molecular heterogeneity may be one mechanism of resistance to targeted therapy. In other words, within each patient, there is evidence of both pharmacologic resistance, as exhibited by lack of ER downregulation, and molecular resistance, as exhibited by ER‐negative CTC.
The other 3 patients (#29, 45, and 8) were all treated with the higher dose of fulvestrant (500 mg/month). Two of them (#29 and 45) had elevated CTC but neither of these patients had CTC that expressed ER. Furthermore, CTC‐BCL2 was negative in those patients, which in this case we interpret to suggest that the ER signaling pathway was absent. We cannot determine from our data whether ER in these CTC had been down‐regulated by fulvestrant or if they were derived from ER negative subclones that were selected under ET pressure. Regardless, their presence, in patients progressing on fulvestrant, suggests that these cancers were driven by alternative pathways for growth and metastases.
Patient #8 was a particularly interesting “n of one” case. She was on the now widely accepted higher dose (500 mg/month), yet still had measurable ER and BCL2 in a portion, but not all, of the CTC in her blood, indicating ongoing signaling in at least some cells in her cancer. Interestingly, this patient was incidentally enrolled in a separate clinical study (Personalized Oncology Through High‐throughput Sequencing) conducted by the Michigan Oncology Sequencing Center (MI‐ONCOSEQ) at the University of Michigan (Robinson et al., 2013). Her cancer was found to harbor an ESR1 mutation (Y537S) in her metastatic tumor. In a separate study, she was also found to have both the Y537S and a separate ESR1 mutation (D538G) in circulating plasma tumor DNA (pt‐DNA) (Chu et al., 2015; Robinson et al., 2013). These results raise speculation that she might have benefitted from even higher doses of a SERD. This speculation is based on the observation that breast cancer cell lines that harbor certain ESR1 LBD mutations retain sensitivity, but may require higher doses, of SERDs to down‐regulate ESR1 transcriptional activity (Robinson et al., 2013).
We observed that all the patients progressing on AI still maintained ER although, again, with enormous intra‐patient heterogeneity. Furthermore, BCL2 was also present in the CTC of all but one patient. These results suggest that signaling through ER is taking place despite absence of estrogen due to AI therapy; such ligand‐independent ER signaling is seen with ESR1 mutation and has been hypothesized through other mechanisms (Robinson et al., 2013). Furthermore, two patients (#41 and 49, Figure 3) had more CTC‐BCL2 positive than CTC‐ER positive cells, which is consistent with BCL‐2 being expressed occasionally even in the absence of ER (Dawson et al., 2010). Nonetheless, our data suggest a correlation between ER and BCL2 expression in CTCs, and further, support the hypothesis that the patients whose CTC lost ER also lost ER signaling.
Our study is hampered by the small sample size, and by its retrospective nature. Ideally, measuring both ER and BCL2 expression in the same cells would have been preferred but this is unfortunately not possible due to the current configuration of the CellSearch® platform, which does not permit such an analysis.
We believe these provocative, albeit preliminary data, provide insights into the mechanism of resistance to fulvestrant in ER positive MBC. A larger prospective study, including serial CTC‐expression monitoring, is underway (COMETI P2, ClinicalTrials.gov: NCT01701050).
These exploratory data suggest widely different mechanisms of resistance to fulvestrant in patients with ER positive MBC: pharmacologic, genetic, and biologic. However, higher doses of fulvestrant are impractical due to limitations of large volume required for intramuscular injection. In this regard, development of oral SERDs (oSERDs) is ongoing in several pharmaceutical companies. In the future, CTC‐markers, particularly ER and BCL2, may serve as pharmacodynamic monitoring tools for dose escalation of fulvestrant or oSERDs, or combination therapies. Indeed, we are currently studying the effects of increasing higher doses of an oSERD (AZD9496) on CTC‐ER and Ki‐67 and pt‐DNA ESR1 mutation status within a Phase I trial.
5. Conclusions
In this study we have shown exploratory data suggesting heterogeneous mechanisms of resistance to fulvestrant, including insufficient dosage, ESR1 mutation, or conversion to dependence on non‐ER pathways. These data suggest that more extensive, and serial, genomic and phenotypic evaluation, optimally using the “liquid biopsy” approach with CTC and circulating ptDNA, is needed in order to have a better understanding of the bases of resistance to fulvestrant and other endocrine therapies.
Conflict of interest
D.F.H. received support from Janssen Diagnostics, LLC, the commercial vendor of CellSearch®, to support clinical and laboratory research. M.C.C., and D.A.C. are paid as employees of Janssen Diagnostics, LLC. D.F.H. is the inventor named on a patent held by the University of Michigan and licensed to Janssen Diagnostics, LLC regarding CTC‐Endocrine Therapy Index (CTC‐ETI). The remaining authors have no conflict of interest to disclose. D.F.H. receives research funding from AstraZeneca, the manufacturer of fulvestrant.
Funding
This work was supported by Veridex/Janssen, LLC, Fashion Footwear Charitable Foundation of New York/QVC Presents Shoes on Sale™ (DFH), the Breast Cancer Research Foundation (BCRF) (N003173 JMR and DFH), National Institute of General Medical Sciences (1RO1GM099143 to JMR), and by a studentship from the Italian Foundation for Cancer Research (FIRC) – Milan Italy and the Associazione “Sandro Pitigliani”‐Prato Italy (CP).
Presentation/publications
Data from this work have been partially reported in Paoletti et al. Clin Cancer Res. 2015 Jun 1;21(11):2487–98. doi: 10.1158/1078‐0432.CCR‐14‐1913. Epub 2014 Nov 7; Robinson D et al., Nat Genet. 2013 Dec;45(12):1446–51. doi: 10.1038/ng.2823. Epub 2013 Nov 3; Chu D. et al., Clin Cancer Res. 2016 Feb 15;22(4):993–999. http://dx.doi.org/10.1158/1078‐0432.CCR‐15‐0943 [Epub 2015 Aug 10]; as a poster discussion at SABCS 2013 meeting (# PD6‐4).
Supporting information
The following is the supplementary data related to this article:
Figure S1 Examples of CTC‐ER staining in CellSearch® from in vitro experiments using MCF‐7 cell lines treated with different ETs.
Acknowledgements
We are grateful to all the patients who generously volunteered to participate in the study. We thank the research nurses, and study coordinators for their efforts on the behalf of the patients. We would like to thank the MI‐ONCOSEQ team (Michigan Oncology Sequencing Center) in particular Drs. Daniel Robinson and Arul Chinnaiyan as well as our collaborators Drs. Ben H. Park and David Chu from the Department of Oncology at Johns Hopkins University.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.04.006.
Paoletti Costanza, Larios Jose M., Mñiz Maria C., Aung Kimberly, Cannell Emily M., Darga Elizabeth P., Kidwell Kelley M., Thomas Dafydd G., Tokudome Nahomi, Brown Martha E., Connelly Mark C., Chianese David A., Schott Anne F., Henry N. Lynn, Rae James M., Hayes Daniel F., (2016), Heterogeneous estrogen receptor expression in circulating tumor cells suggests diverse mechanisms of fulvestrant resistance, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.04.006.
References
- Alix-Panabieres, C. , Pantel, K. , 2013. Circulating tumor cells: liquid biopsy of cancer. Clin. Chem. 59, 110–118. [DOI] [PubMed] [Google Scholar]
- Chu, D. , Paoletti, C. , Gersch, C. , VanDenBerg, D. , Zabransky, D. , Cochran, R. , Wong, H.Y. , Valda Toro, P. , Cidado, J. , Croessmann, S. , Erlanger, B. , Cravero, K. , Kyker-Snowman, K. , Button, B. , Parsons, H. , Dalton, W.B. , Gillani, R. , Medford, A. , Aung, K. , Tokudome, N. , Chinnaiyan, A. , Schott, A.F. , Robinson, D.R. , Jacks, K. , Lauring, J. , Hurley, P.J. , Hayes, D.F. , Rae, J.M. , Park, B.H. , 2016 Feb 15. ESR1 mutations in circulating plasma tumor DNA from metastatic breast cancer patients. Clin. Cancer Res. 22, (4) 993–999. 10.1158/1078-0432.CCR-15-0943 [Epub 2015 Aug 10] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies, C. , Godwin, J. , Gray, R. , Clarke, M. , Cutter, D. , Darby, S. , McGale, P. , Pan, H.C. , Taylor, C. , Wang, Y.C. , Dowsett, M. , Ingle, J. , Peto, R. , 2011. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 378, 771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, S.J. , Makretsov, N. , Blows, F.M. , Driver, K.E. , Provenzano, E. , Le Quesne, J. , Baglietto, L. , Severi, G. , Giles, G.G. , McLean, C.A. , Callagy, G. , Green, A.R. , Ellis, I. , Gelmon, K. , Turashvili, G. , Leung, S. , Aparicio, S. , Huntsman, D. , Caldas, C. , Pharoah, P. , 2010. BCL2 in breast cancer: a favourable prognostic marker across molecular subtypes and independent of adjuvant therapy received. Br. J. Cancer. 103, 668–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Leo, A. , Jerusalem, G. , Petruzelka, L. , Torres, R. , Bondarenko, I.N. , Khasanov, R. , Verhoeven, D. , Pedrini, J.L. , Smirnova, I. , Lichinitser, M.R. , Pendergrass, K. , Garnett, S. , Lindemann, J.P. , Sapunar, F. , Martin, M. , 2010. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor-positive advanced breast cancer. J. Clin. Oncol. 28, 4594–4600. [DOI] [PubMed] [Google Scholar]
- Ellis, M.J. , Llombart-Cussac, A. , Feltl, D. , Dewar, J.A. , Jasiowka, M. , Hewson, N. , Rukazenkov, Y. , Robertson, J.F. , 2015 Nov 10. Fulvestrant 500 mg versus anastrozole 1 mg for the first-line treatment of advanced breast cancer: overall survival analysis from the phase II first study. J. Clin. Oncol. 33, (32) 3781–3787. 10.1200/JCO.2015.61.5831 [Epub 2015 Sep 14] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes, D.F. , Van Zyl, J.A. , Hacking, A. , Goedhals, L. , Bezwoda, W.R. , Mailliard, J.A. , Jones, S.E. , Vogel, C.L. , Berris, R.F. , Shemano, I. , 1995. Randomized comparison of tamoxifen and two separate doses of toremifene in postmenopausal patients with metastatic breast cancer. J. Clin. Oncol. 13, 2556–2566. [DOI] [PubMed] [Google Scholar]
- Jeselsohn, R. , Buchwalter, G. , De Angelis, C. , Brown, M. , Schiff, R. , 2015 Oct. ESR1 mutations-a mechanism for acquired endocrine resistance in breast cancer. Nat. Rev. Clin. Oncol. 12, (10) 573–583. 10.1038/nrclinonc.2015.117 [Epub 2015 Jun 30]. Review [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston, S.R. , MacLennan, K.A. , Sacks, N.P. , Salter, J. , Smith, I.E. , Dowsett, M. , 1994. Modulation of Bcl-2 and Ki-67 expression in oestrogen receptor-positive human breast cancer by tamoxifen. Eur. J. Cancer. 30A, 1663–1669. [DOI] [PubMed] [Google Scholar]
- Jonat, W. , Howell, A. , Blomqvist, C. , Eiermann, W. , Winblad, G. , Tyrrell, C. , Mauriac, L. , Roche, H. , Lundgren, S. , Hellmund, R. , Azab, M. , 1996. A randomised trial comparing two doses of the new selective aromatase inhibitor anastrozole (Arimidex) with megestrol acetate in postmenopausal patients with advanced breast cancer. Eur. J. Cancer. 32A, 404–412. [DOI] [PubMed] [Google Scholar]
- Moore, H.M. , Kelly, A.B. , Jewell, S.D. , McShane, L.M. , Clark, D.P. , Greenspan, R. , Hayes, D.F. , Hainaut, P. , Kim, P. , Mansfield, E. , Potapova, O. , Riegman, P. , Rubinstein, Y. , Seijo, E. , Somiari, S. , Watson, P. , Weier, H.U. , Zhu, C. , Vaught, J. , 2011. Biospecimen reporting for improved study quality (BRISQ). J. Proteome Res. 10, 3429–3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborne, C.K. , Pippen, J. , Jones, S.E. , Parker, L.M. , Ellis, M. , Come, S. , Gertler, S.Z. , May, J.T. , Burton, G. , Dimery, I. , Webster, A. , Morris, C. , Elledge, R. , Buzdar, A. , 2002. Double-blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: results of a North American trial. J. Clin. Oncol. 20, 3386–3395. [DOI] [PubMed] [Google Scholar]
- Osborne, C.K. , Schiff, R. , 2011. Mechanisms of endocrine resistance in breast cancer. Annu. Rev. Med. 62, 233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti, C. , Muniz, M.C. , Thomas, D.G. , Griffith, K.A. , Kidwell, K.M. , Tokudome, N. , Brown, M.E. , Aung, K. , Miller, M.C. , Blossom, D.L. , Schott, A.F. , Henry, N.L. , Rae, J.M. , Connelly, M.C. , Chianese, D.A. , Hayes, D.F. , 2015. Development of circulating tumor cell-endocrine therapy index in patients with hormone receptor-positive breast cancer. Clin. Cancer Res. 21, 2487–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rae, J.M. , Johnson, M.D. , Scheys, J.O. , Cordero, K.E. , Larios, J.M. , Lippman, M.E. , 2005. GREB 1 is a critical regulator of hormone dependent breast cancer growth. Breast Cancer Res. Treat. 92, 141–149. [DOI] [PubMed] [Google Scholar]
- Robinson, D.R. , Wu, Y.M. , Vats, P. , Su, F. , Lonigro, R.J. , Cao, X. , Kalyana-Sundaram, S. , Wang, R. , Ning, Y. , Hodges, L. , Gursky, A. , Siddiqui, J. , Tomlins, S.A. , Roychowdhury, S. , Pienta, K.J. , Kim, S.Y. , Roberts, J.S. , Rae, J.M. , Van Poznak, C.H. , Hayes, D.F. , Chugh, R. , Kunju, L.P. , Talpaz, M. , Schott, A.F. , Chinnaiyan, A.M. , 2013. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat. Genet. 45, 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira, C. , Reed, J.C. , Pratt, M.A. , 1995. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving Bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res. 55, 3902–3907. [PubMed] [Google Scholar]
- Tormey, D.C. , Lippman, M.E. , Edwards, B.K. , Cassidy, J.G. , 1983. Evaluation of tamoxifen doses with and without fluoxymesterone in advanced breast cancer. Ann. Intern. Med. 98, 139–144. [DOI] [PubMed] [Google Scholar]
- Tormey, D.C. , Simon, R.M. , Lippman, M.E. , Bull, J.M. , Myers, C.E. , 1976. Evaluation of tamoxifen dose in advanced breast cancer: a progress report. Cancer Treat Rep. 60, 1451–1459. [PubMed] [Google Scholar]
- Toy, W. , Shen, Y. , Won, H. , Green, B. , Sakr, R.A. , Will, M. , Li, Z. , Gala, K. , Fanning, S. , King, T.A. , Hudis, C. , Chen, D. , Taran, T. , Hortobagyi, G. , Greene, G. , Berger, M. , Baselga, J. , Chandarlapaty, S. , 2013. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat. Genet. 45, 1439–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijayaratne, A.L. , McDonnell, D.P. , 2001. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J. Biol. Chem. 276, 35684–35692. [DOI] [PubMed] [Google Scholar]
- Wijayaratne, A.L. , Nagel, S.C. , Paige, L.A. , Christensen, D.J. , Norris, J.D. , Fowlkes, D.M. , McDonnell, D.P. , 1999. Comparative analyses of mechanistic differences among antiestrogens. Endocrinology. 140, 5828–5840. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Figure S1 Examples of CTC‐ER staining in CellSearch® from in vitro experiments using MCF‐7 cell lines treated with different ETs.