

Abstract
The p53 tumor suppressor gene encodes a sequence‐specific transcription factor. Mutations in the coding sequence of p53 occur frequently in human cancer and often result in single amino acid substitutions (missense mutations) in the DNA binding domain (DBD), blocking normal tumor suppressive functions. In addition to the loss of canonical functions, some missense mutations in p53 confer gain‐of‐function (GOF) activities to tumor cells. While many missense mutations in p53 cluster at six “hotspot” amino acids, the majority of mutations in human cancer occur elsewhere in the DBD and at a much lower frequency. We report here that mutations at K120, a non‐hotspot DNA contact residue, confer p53 with the previously unrecognized ability to bind and activate the transcription of the pro‐survival TNFAIP8 gene. Mutant K120 p53 binds the TNFAIP8 locus at a cryptic p53 response element that is not occupied by wild‐type p53. Furthermore, induction of TNFAIP8 is critical for the evasion of apoptosis by tumor cells expressing the K120R variant of p53. These findings identify induction of pro‐survival targets as a mechanism of gain‐of‐function activity for mutant p53 and will likely broaden our understanding of this phenomenon beyond the limited number of GOF activities currently reported for hotspot mutants.
Keywords: Mutant p53, Gain of function, Pro-survival, TNFα
Highlights
The tumor derived K120R mutation that drives a new p53 regulated transcriptome.
We discovered a novel mutant p53 activity, induction of pro‐survival target genes.
We demonstrate that non‐hotspot mutations in p53 have gain‐of‐function activities.
List of abbreviations
- Bax
BCL-2 associated X protein
- CDKN1A
cyclin-dependent kinase inhibitor 1A
- CPT
camptothecin
- NF-κB
nuclear factor κB
- qRT-PCR
quantitative real time polymerase chain reaction
- Tet
tetracycline
- TNFα
tumor necrosis factor alpha
- TNFAIP8
tumor necrosis factor alpha induced protein 8
1. Introduction
The p53 tumor suppressor is the most commonly lost or mutated gene in human cancer (Vogelstein et al., 2000). The majority (80%) of mutations that occur in p53 are missense, and typically result in the loss of tumor suppressor function (Weisz et al., 2007). These include six frequently occurring mutations that are referred to as “hotspot” mutants. Many p53 mutants not only lose activities possessed by WT p53, but also acquire distinct gain of function properties. In addition, it has become clear that there is a great deal of heterogeneity among the p53 mutants, displaying a wide array of oncogenic phenotypes. In vivo studies have demonstrated that mice expressing mutant variants of p53 have a more broad and aggressive tumor profile than p53 null mice, providing further evidence for the role of mutant p53 in malignant progression (Lang et al., 2004; Olive et al., 2004).
We and others have reported that the K120R mutation in p53, which occurs within the DNA‐binding domain, blocks acetylation at that residue (Sykes et al., 2006; Tang et al., 2006). Loss of acetylation potential at K120 results in the inability of p53 to induce apoptosis. We speculated, that while the K120R mutant does indeed lose selected WT activities, it might also acquire gain of function properties that contribute to the ability of a cell to evade apoptosis, thereby promoting tumorigenesis.
In addition to K120R, there are several other missense mutations at this residue in tumors, including K120M, K120E, K120N, and K120Q (Petitjean et al., 2007). What distinguishes K120R from these mutants is that it retains residual WT activity, whereas the others show a marked decrease or complete lack of transcriptional activity (Shiraishi et al., 2004). The lysine to arginine substitution at codon 120 is a conservative change, unlike the others, preserving the positive charge of the amino acid side chain. The K120R mutant can induce p53 cell cycle arrest targets, however it is defective for activating apoptosis genes, compromising its ability to initiate cell death (Sykes et al., 2006; Tang et al., 2006).
Lysine 120 is a residue that directly associates with DNA (Zupnick and Prives, 2006). The conservative lysine to arginine mutation preserves some DNA binding, evidenced by the fact that the K120R mutant retains the ability to transactivate some known p53 targets (Sykes et al., 2006). Given that it still retains some transcriptional ability, the K120R mutant may confer the acquisition of novel targets. Here we report that the K120R mutant induces a network of target genes that differ from those induced by WT p53. A pro‐survival gene, TNFAIP8 (tumor necrosis factor (TNF)‐α induced protein 8) was robustly induced by the K120R mutant, but not by WT p53, and has been well characterized as a negative regulator of apoptosis via its ability to inhibit Caspase‐8 activity (Kumar et al., 2000; You et al., 2001). Overexpression of TNFAIP8 has been observed in various cancers including ovary, non‐small‐cell lung (NSCL) and gastric adenocarcinoma (Liu et al., 2014; Wang et al., 2014; Yang et al., 2014). TNFAIP8 expression is also elevated in colon cancer specimens, where it is linked to poor prognosis and lymph node metastasis (Miao et al., 2012). Data presented here provide new insight regarding a non‐hotspot p53 mutation. First, they identify induction of pro‐survival targets as a mechanism of gain‐of‐function activity for mutant p53. Second, they illustrate that analysis of the gain‐of‐function activities conferred by non‐hotspot mutations in p53, which represent the majority of human cancer mutations, may broaden our understanding of this phenomenon beyond the limited number of GOF activities currently reported for hotspot mutants.
2. Materials and methods
2.1. Reagents
Tetracycline (Tet) and camptothecin (CPT) were purchased from Sigma–Aldrich (St. Louis, MO).
2.2. Cell culture
All cells were cultured in DMEM supplemented with 10% FBS in 5% C02. HCT116 cells and parental H1299 cells were obtained from ATCC. Tet‐ON p53 WT and K120 cells were generated via lenti‐viral infection and stable selection in H1299 cells. p53 expression was induced by treatment with 0.5 μg/mL tetracycline. DNA damage response was induced by camptothecin treatment at 2 μg/mL (5 μM) after 2 h (h) tetracycline.
2.3. Expression vectors and transient transfection
p53 expression vectors were generated in the pcDNA3.1 cloning vector (Addgene). Point mutations in p53 were introduced via site directed mutagenesis using the QuikChange II XL kit (Agilent). Transfections were performed using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions. Cells were harvested ∼48 h after transfection reaction.
2.4. Western blotting
Whole‐cell extracts were generated using E1A lysis buffer. 20–30 μg of protein was loaded per well, and transferred onto nitrocellulose membrane. Membranes were probed with the following antibodies for protein detection: p53 (DO‐1 Santa Cruz sc‐126), Actin (Santa Cruz sc‐8432), Caspase‐3 (Cell Signaling), TNFAIP8 (Prestige‐Sigma Aldrich). All quantifications shown were calculated using ImageJ software (Schneider et al., 2012).
2.5. Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation was performed as previously described (Zhang et al., 2005) using FL393R (Santa Cruz, sc‐6243 X) for p53 and H3K27Ac (Abcam, ab4729). ChIP products were quantified using the Step One Plus RT‐PCR system and Fast SYBR Green Master Mix (Applied Biosystems). Each immunoprecipitation was normalized to the amount of DNA detected in the input by RT‐PCR. The PCR reaction was carried out using the following primers TNFAIP8 K120R Binding site: F, 5′‐CGCACATACTGGCGAAAGC‐3′ and R, 5′‐CCAGCATCTCAGTCTGCAATCT‐3′.
2.6. ChIP‐Seq sample generation and analysis
The samples used in the ChIP‐Seq dataset were generated as described previously (Lee et al., 2006) using FL393R (Santa Cruz) or total Pol II (Santa Cruz sc‐899). Illumina‐compatible sequencing libraries were prepared from immunoprecipitated DNA using NEBNext Ultra DNA Library (NEB e7370) with unique single indexes. Library insert size and molarity was quantified using the Agilent BioAnalyzer and KAPA Library Quantification methods. Multiplexed libraries were sequenced on the Illumina NextSeq 500 with V1 chemistry to generate 75 bp reads. Uniquely aligned reads were aligned to the hg19/NCBI37 reference genome using Bowtie2 (‐k1, ‐N1) and enriched regions were determined using MACS (v1.4, mfold 10,30). BedGraphs files for visualization on the UCSC Genome Browser were generated using HOMER (homer.salk.edu) with a visualization fragment length equal to the median estimated fragment length. ChIP‐Seq experiments were performed in biological duplicates.
2.7. shRNA mediated knockdown
To silence TNFAIP8 expression, short hairpin RNAs targeting the 3′UTR of the transcript were employed. TNFAIP8 shRNA (TRCN 0000116160) in the pLKO cloning vector was transfected into 293Ts with packaging plasmids to generate lentivirus. Viral media was transferred onto target cells, which were then subject to puromycin selection (5 μg/mL) for 3–4 days. shRNA constructs were provided by David Schultz (Wistar).
2.8. RNA extraction and RT‐PCR
Total RNA was isolated using TRIzol (Thermo Fisher) according to the manufacturer's instructions. The cDNA was generated using 200 ng of RNA using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher). For the RT‐PCR reaction, 2 μL of cDNA was used per reaction. Quantification of DNA was performed using the Step One Plus Real Time‐PCR system with Fast SYBR Green Master Mix (Applied Biosystems). Gene values were normalized to GAPDH. Primer sequences for each gene are indicated in Supplementary Table 2.
2.9. Microarray expression profiling and gene ontology
H1299 Tet‐ON p53 WT or K120R cells were treated with tetracycline for 8 h before harvest in parallel with untreated cells for both cell lines. Extracted RNA was utilized to generate end‐labeled biotinylated ssDNA. Labeled DNA was hybridized to the Exon Expression Chip HuGene 1.0 ST array oligonucleotide microarray (Affymetrix) according to manufacturer's instructions. The array chip was read by the GeneChip Scanner 3000 (Affymetrix). Processing of data was performed with GeneSpring (Agilent). The normalization algorithm utilized was Robust Multi‐array Average (RMA‐16) and log2 transformation. Genes that were up‐regulated ≥1.5 fold were considered for gene ontology analysis. Functional categories for each input group were generated using DAVID.
2.10. Flow cytometry
Cells were harvested and stained using either the Muse Caspase 3/7 or Annexin V staining kit, according to manufacturer's instructions (EMD Millipore). Stained cells were quantified using the Muse Cell Analyzer (EMD Millipore).
2.11. Luciferase reporter assay
The p53 response elements for CDKN1A (GAACATGTCCCAACATGTTG) or TNFAIP8 (GTACATGACTGCACATGTCG) were cloned into the pGL3 Luciferase Reporter Plasmid (Promega). Plasmids were transfected into H1299 TO‐p53 cells and harvested 48 h later. Cells were treated with tetracycline 24 h before harvest. Luminescence was measured using the Nano‐Glo Luciferase Assay System (Promega) on the FLUOstar Optima Microplate Reader (BMG Labtech).
3. Results
3.1. K120R is functionally unique among K120 tumor derived mutants
Loss of p53 tumor suppressor function is a crucial event in tumor formation. The high incidence of missense mutations in the DBD raises the possibility that mutations in this region of the peptide sequence are favorable for malignant progression via the acquisition of novel targets (Weisz et al., 2007). Within this domain are several mutations at K120 that have been documented in human cancer, including K120R, K120E, K120M, K120N and K120Q (Petitjean et al., 2007). To investigate the effect of these mutations on p53 function, we referred to the UMD TP53 mutation database. The transcriptional activity for p53 missense mutants was measured using a yeast assay, and reported as a percentage of WT activity on the promoters of several known p53 targets (Kato et al., 2003). We generated a heat map from these values, which includes all of the K120 mutants as well as the hotspot R175H mutant (Figure 1A). As shown in the heat map, the K120R mutant retains significant transcriptional activity, whereas the others have little or no activity. For example, K120R expression resulted in transcriptional activity of 120% and 105% of WT for the p53 targets Noxa and MDM2, respectively.
Figure 1.
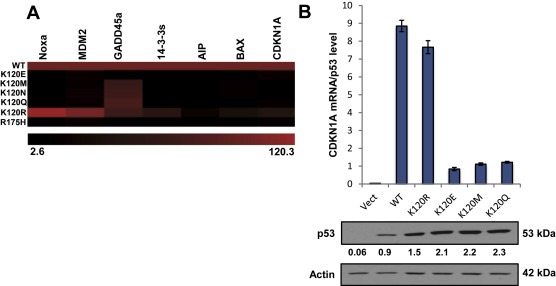
K120R is distinct from other K120 derived mutants. (A) Heat map representative of transcription activity of the p53 variant indicated on the left for various p53 targets, measured by fluorescence in a yeast screen assay (Kato et al., 2003) as referenced in the UMD TP53 Mutant Database. Values were taken as percentage of wild type (100%). (B) qRT‐PCR on RNA extracted from H1299 cells transfected with p53 for 48 h. CDKN1A‐mRNA levels were measured relative to GAPDH (bottom panel: Western blot of p53 expression for each transfection). CDKN1A‐mRNA levels shown were normalized to quantified p53 protein levels by Western blot. Error bars indicate ± S.D. Relative intensities, normalized to actin, for each p53 band are indicated.
To further explore the differences between the distinct missense mutants at K120, their ability to induce p53 target gene activation in human cells was tested. Expression plasmids encoding for one of the indicated p53 mutants were transiently transfected into H1299 human NSCLC cells (p53 null). Quantitative RT‐PCR analysis of RNA extracted from the cells was measured for CDKN1A expression, a bona fide p53 target gene with a high affinity p53 consensus binding site (el‐Deiry et al., 1992). CDKN1A transcript levels were normalized to the quantified p53 protein level for each transfection reaction. Both WT and K120R expression resulted in increased CDKN1A transcription while the other three mutants tested showed little, if any, increase in CDKN1A transcript over vector control (Figure 1B). Thus, the K120R mutant retains the ability to induce a subset of canonical p53 targets, distinguishing it from other tumor‐derived mutants at K120.
3.2. K120R mutant p53 induces a distinct group of target genes
Given the unique characteristic of the K120R p53 mutant, in that it retains certain p53 transcriptional abilities, we sought to investigate on a genome wide scale the differences between the transcriptional profiles induced by expression of WT versus K120R. To this end, transcriptional profiling in H1299 cells expressing WT or K120R p53 (referred to as TO‐WT or TO‐K120R) was performed, using a tetracycline (Tet) inducible system to control p53 expression (Figure 2A and 2B). Using a threshold of 1.5 fold induction, we found 377 genes up regulated in a p53‐dependent manner. Of these genes, 102 were induced by the WT exclusively, 259 by K120R exclusively, and 16 were induced by both (Figure 2C). As expected, based previous findings from our group and others (Sykes et al., 2006; Tang et al., 2006), gene ontology analysis of these subsets of target genes revealed that genes induced exclusively by WT p53 were clustered in functional categories involving induction of apoptosis, and the genes induced by both WT and the K120R mutant clustered in cell cycle arrest groups (Supplementary Table 1). Interestingly, functional groups associated with neurological processes were enriched in the K120R induced gene set. While lung cancer cell lines have been reported to have some features of neuronal lineage cells (Giaccone et al., 1992), it is unclear if this contributes to the pro‐survival attributes of the K120R mutant. Among the most strongly induced targets of K120R, TNFAIP8 was unique in that it has been characterized as a pro‐survival gene, potentially contributing to an oncogenic phenotype (Table 1).
Figure 2.
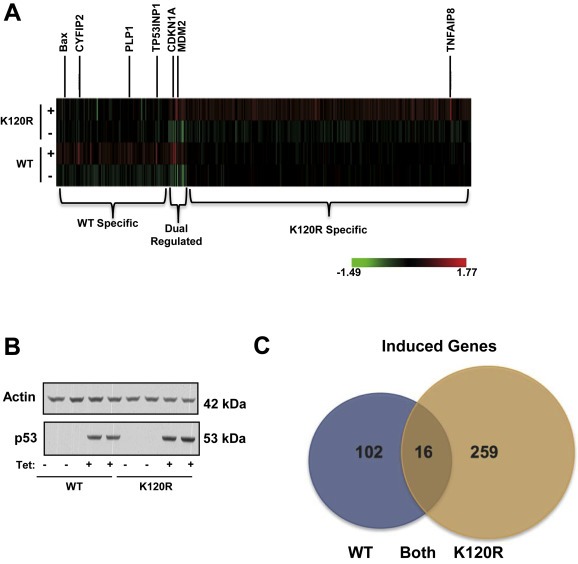
WT p53 and the K120R mutant induce distinct sets of target genes. (A) Expression profiling heat map generated with RNA extracted from H1299 cells with TO‐p53 (wild type (WT) or K120R). Cells were harvested 8 h after tetracycline treatment. Genes shown were up‐regulated ≥1.5 fold. (B) Western blot of whole cell lysates showing p53 induction in samples used to generate microarray profile. Profile was produced using biological replicates. (C) Quantification of heat map shown in (A). Number of genes induced ≥1.5 fold in each group are shown. Gene ontology analysis of the three groups is shown in Supplementary Table 1.
Table 1.
Genes induced most strongly by the K120R mutant, with induction measured as fold change from microarray, and gene function.
| Gene | Induction (Fold change) | Function |
|---|---|---|
| KCNJ8 | 2.48 | Potassium ion transport |
| TNFAIP8 | 2.40 | Apoptosis inhibitor |
| SPRN | 2.19 | Protein nuclear import |
| RYR1 | 2.18 | Calcium ion transport |
| NUPR1 | 2.15 | Inflammatory response |
| FHDC1 | 2.10 | Cytoskeleton |
| CPB1 | 2.03 | Proteolysis |
| CALCR | 1.95 | GPCR signaling |
| FCRL3 | 1.94 | Plasma membrane |
| DNHD1 | 1.93 | Microtubule activity |
3.3. Validation of TNFAIP8 as a target of mutant K120R
To confirm findings from the expression profiling, we measured TNFAIP8 transcript levels in the WT and K120R expressing H1299 cells after 24 h of tetracycline treatment. Quantitative RT‐PCR analysis showed TNFAIP8 mRNA levels increasing robustly after K120R induction, and no change in cells expressing WT p53 (Figure 3A). At the protein level, there is a slight increase in TNFAIP8 levels in cells after WT p53 induction, however these levels still do not reach that of the amount in K120R expressing cells. The increase in TNFAIP8 protein in WT expressing cells is not consistent (see Figure 6A), however the lack of TNFAIP8 transcript induction by WT p53 is consistent (Figures 3 and S2).
Figure 3.
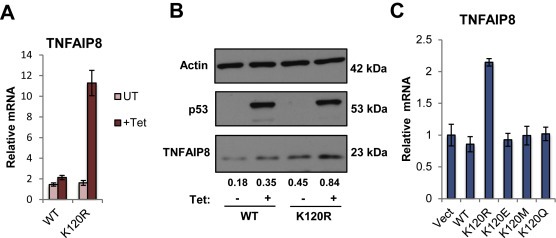
Validation of TNFAIP8 as a target of the mutant K120R. (A) qRT‐PCR on RNA extracted from H1299 TO‐p53 cells. Cells were harvested 24 h after tetracycline treatment to induce WT or K120R p53 expression. TNFAIP8‐mRNA levels were measured relative to GAPDH. Error bars indicate ± S.D. (B) Western blot of whole cell lysates from TO‐WT or K120R cells harvested 24 h after tetracycline treatment. Relative intensities, normalized to actin, for each TNFAIP8 band are indicated. (C) qRT‐PCR of TNFAIP8‐mRNA levels measured from p53 mutant expression experiment shown in Figure 1B. Values were normalized to GAPDH. Error bars indicate ± S.D.
Figure 6.
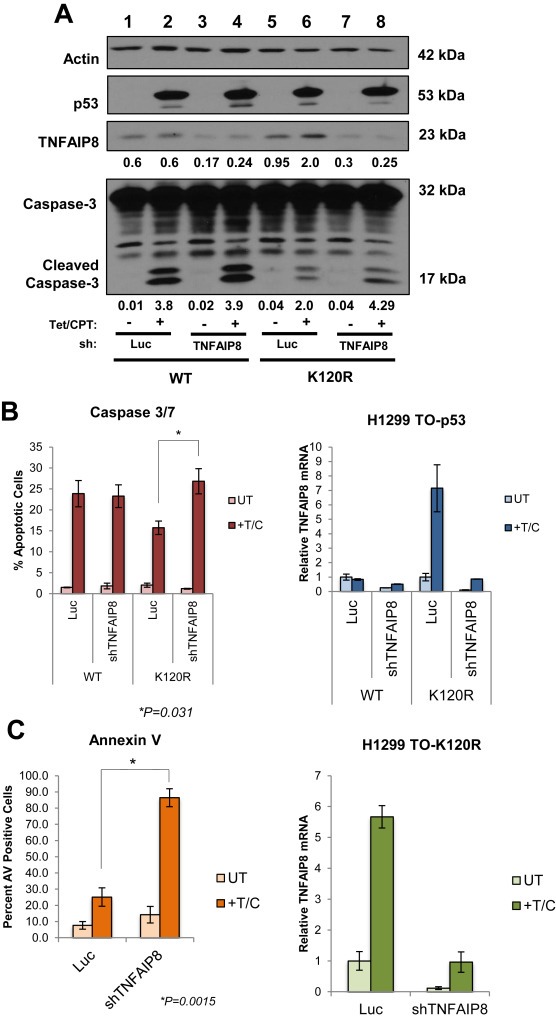
TNFAIP8 induction is important for evasion of apoptosis in K120R expressing cells. (A) Western blot of p53, TNFAIP8, Caspase‐3 and actin control from H1299 Tet‐inducible p53 cells (WT or K120R), 24 h after tetracycline and camptothecin treatment. Relative intensities, normalized to actin, for each TNFAIP8 and cleaved Caspase‐3 band are indicated. (B) Percentage of apoptotic cells treated as described in (A). Caspase 3/7 activity was measured by harvesting and staining cells then quantifying by flow cytometry. Percentage of apoptotic cells are represented as mean values, error bars indicate ± s.e. of three independent experiments, * indicates significance as calculated by Student's t‐test (p = 0.031). (C) Annexin V positivity was measured from stained cells treated as described in (A), then quantified as percentage of Annexin V positive cells by flow cytometry. Percentage of Annexin V positive cells are represented as mean values, error bars indicate ± s.e. of three independent experiments, * indicates significance as calculated by Student's t‐test (p = 0.0015). Right panel for (B) and (C): qRT‐PCR of TNFAIP8 mRNA levels measured from RNA extracted from cells used to asses apoptosis in (B) and (C). Values were normalized to GAPDH. Error bars indicate S.D. Data shown is representative of the triplicate experiments.
To test if induction of TNFAIP8 was unique to the K120R mutant, we measured changes in TNFAIP8 mRNA in response to expression of additional K120 mutants in H1299 cells (Figure 3C), using RNA from the panel of transfection reactions in Figure 1B. Only expression of K120R resulted in activation of TNFAIP8 transcription, confirming the specificity of this target gene to the K120R mutant. An identical transfection experiment was performed in the colon cancer cell line HCT116 (p53 −/−) cells, yielding the same result; i.e., that TNFAIP8 transcription increased exclusively with K120R expression (Supplemental Figure 1).
In human cancer, the majority of missense mutations that occur in p53 are located within the DBD, including six “hotspot” mutations (Olivier et al., 2010). Point mutations in the DBD of p53 can result in the acquisition of novel targets, presumably by altering the cognate DNA sequence recognized (Chin et al., 1992; Ludes‐Meyers et al., 1996; Pugacheva et al., 2002). Thus, it was possible that induction of TNFAIP8 by the K120R mutant could be a characteristic shared with hotspot p53 mutants. To investigate this possibility, various p53 proteins with hotspot mutations in the DNA binding domain were ectopically expressed in H1299 cells, and TNFAIP8 transcript levels measured. Similar to K120R, expression of the R282W mutant resulted in increased TNFAIP8 transcription, whereas expression of the other mutants had no effect on TNFAIP8 transcript levels (Supplemental Figure 2). At present, insufficient numbers of tumors from individual datasets carrying either the K120R or R282W mutations exist in The Cancer Genome Atlas (TCGA) samples to allow for assessing whether a correlation exists between the mutations and TNFAIP8 transcript levels.
3.4. Enhanced binding of K120R mutant at TNFAIP8 locus
p53 is a sequence specific transcription factor, and some mutant variants of p53 bind and activate transcriptional targets distinct from canonical p53 regulated genes (Chin et al., 1992; Cooks et al., 2013; Frazier et al., 1998; Pugacheva et al., 2002). To determine whether K120R binding patterns differed from those of WT p53, we generated parallel chromatin immunoprecipitation DNA sequencing (ChIP‐Seq) datasets utilizing the WT and K120R TO‐p53H1299s. Cells were treated with tetracycline for 24 h, harvested and cross‐linked, then subjected to immunoprecipitation with an antibody for p53 (Figure 4A). p53 was expressed at comparable levels in the WT and K120R expressing cells (Figure 4D). After induction, both K120R and WT p53 were bound at the cell cycle arrest target CDKN1A locus, as expected. The K120R mutant induced CDKN1A transcript levels to levels comparable to WT (Figure 4B). In contrast, the K120R mutant was defective for binding at the BAX locus, consistent with its defect in activating BAX transcription (Figure 4, middle panel) and previously published findings (Sykes et al., 2006; Tang et al., 2006). The peak pattern at the TNFAIP8 locus, ∼500 base pairs downstream of the transcriptional start site (TSS), revealed a robust signal for K120R but not for WT p53, consistent with the transcriptional patterns shown in which TNFAIP8 is induced by K120R but not WT p53. Binding at the TNFAIP8 locus by the K120R mutant was confirmed by empirical chromatin immunoprecipitation (Figure 4C). These data suggest that TNFAIP8 is a direct target of the K120R mutant, but not of WT p53.
Figure 4.

K120R binding is enhanced at TNFAIP8 locus. (A) TO‐p53 cells (WT or K120R) were treated with tetracycline or not treated, then harvested after 24 h. After crosslinking, cell lysates were subject to chromatin immunoprecipitation (ChIP) with antibody for p53, and purified DNA was sequenced and processed. Peaks for p53 at TNFAIP8, CDKN1A and Bax loci are shown, visualized using the UCSC Genome Browser. Peak scale is indicated to the left of the panel for each gene in normalized reads. (B) qRT‐PCR of RNA extracted from cells used in ChIP‐Seq experiment measuring transcript levels of CDKN1A, Bax and TNFAIP8. Values were normalized to GAPDH. Error bars indicate ± S.D. (C) p53 ChIP signal on DNA extracted from cells treated as described in (A) relative to input DNA at TNFAIP8 locus (+500 bp) for WT and K120R, −/+ Tet. Error bars indicate ± S.D. (D) Western blot for p53 and actin control from cells used to generate ChIP‐Seq data.
The UCSC Genome Browser, which displays transcript information using the UCSC Known Genes Dataset as a foundation (Hsu et al., 2006), identifies four different transcripts at the TNFAIP8 locus (Supplemental Figure 3). To determine if these transcripts were differentially regulated in response to p53 induction, we designed primers specific to each transcript, as well as to the 3′ exon common to all. Analysis by qRT‐PCR revealed that the K120R mutant induced all transcript variants, and WT p53 was uniformly defective for TNFAIP8 induction (Supplemental Figure 3).
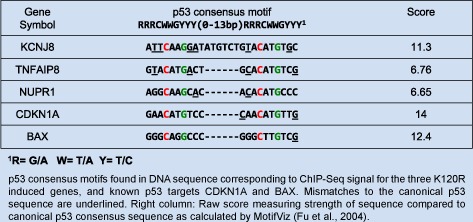
The DNA sequence contained within the K120R peak harbored a region matching the p53 consensus sequence as identified by the MotifViz motif discovery tool (Figure 5A and Table 2) (Fu et al., 2004). Interestingly, of the 10 genes induced exclusively and most robustly by K120R, only TNFAIP8 and two others were directly bound by K120R (KCNJ8 and NUPR1). The DNA sequence associated with these peaks contained p53 response element as well. At all three of these K120R binding sites, WT p53 was absent despite the presence of a p53 binding motif (Supplementary Figure 4 and Table 2).
Figure 5.
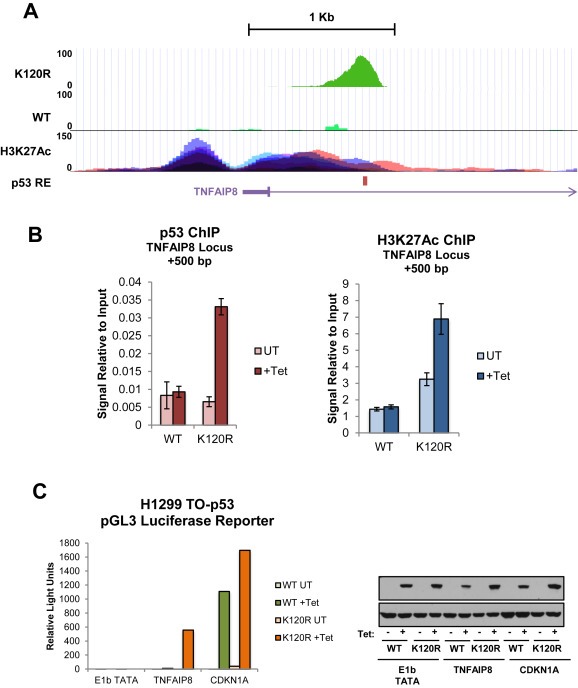
TNFAIP8 locus contains a functional, cryptic p53 response element (A) Higher resolution of TNFAIP8 locus shown in Figure 4A with ChIP‐Seq peaks for WT and K120R (Tetracycline treated condition only), location of p53 response element (RE) is indicated by red bar. H3K27Ac marks on seven cell lines (peaks are distinguished by color) from ENCODE are also shown (Encode Project Consortium, 2012). (B) ChIP signal at TNFAIP8 locus (+500 bp), for p53 or H3K27Ac from DNA extracted from cells as described in Figure 4. Signal is normalized to input DNA. (C) A luciferase reporter plasmid containing either the p53 response element associated with CDKN1A or TNFAIP8 was transfected into TO‐p53 WT or K120R cells. The plasmid containing the E1b TATA only was included as a negative control. After 24 h of tetracycline treatment, cells were harvested and luminescence was measured using the Nano‐Glo Luciferase Assay System. Right panel: Western blot showing p53 induction for cells used in reporter assay.
Table 2.
p53 consensus motifs found in DNA sequence corresponding to ChIP‐Seq signal for the three K120R induced genes, and known p53 targets CDKN1A and BAX. Mismatches to the canonical p53 sequence are underlined. Right column: Raw score measuring strength of sequence compared to canonical p53 consensus sequence as calculated by MotifViz (Fu et al., 2004).
One mechanistic explanation for the different capacities of WT and K120R versions of p53 to induce TNFAIP8 transcription could be that the presence of p53 is required for recruitment of basal transcription factors. To this end, we assessed the differences in RNAPII at the TNFAIP8 locus in K120R expressing cells relative to those expressing WT p53 (Supplementary Figure 5, right panel). In all four conditions, RNAPII occupancy did not change, ruling out the possibility that the defect observed in TNFAIP8 induction by the WT p53 is due to a failure in recruitment of general transcriptional machinery.
An additional possibility for this phenomenon is the acquisition of a novel interaction between the K120R p53 variant and another transcription factor. To explore this possibility, motif search analysis for predicted transcription factor binding sites was performed on the peaks from the ChIP‐Seq experiment in the H1299 TO‐p53 cells (Table 3). First, all peaks that contained a p53 response element as identified by p53scan (Smeenk et al., 2008) were categorized into three groups: WT exclusive, K120R exclusive and common to both. Second, the DNA sequences associated with these peaks were analyzed using the HOMER motif discovery algorithm (Heinz et al., 2010). The results of the analysis revealed significant differences in transcription factor binding sites that were enriched in each group, however there does not appear to be a candidate that is over‐represented in the K120R peaks. It is therefore unlikely that the general mechanism for K120R DNA‐binding is via a novel interaction with another protein that WT does not exhibit.
Table 3.
Peaks containing a p53 response element, as identified by p53scan (Smeenk et al., 2008), from the ChIP‐Seq experiment in Figure 4 of the manuscript were divided into three groups (WT, K120R or common). Motif analysis using the HOMER motif discovery algorithm was performed to detect enrichment for other transcription factor binding sites (Heinz et al., 2010).
| WT | K120R | Common | ||||||
|---|---|---|---|---|---|---|---|---|
| A | Ba | C | A | Bb | C | A | Bc | C |
| Mef2c | 367 | 1 × 10−77 | NRF | 150 | 1 × 10−57 | Mef2a | 85 | 1 × 10−23 |
| GSC | 482 | 1 × 10−62 | BMAL1 | 498 | 1 × 10−46 | Mef2c | 87 | 1 × 10−23 |
| BMAL1 | 410 | 1 × 10−61 | HIF‐1b | 343 | 1 × 10−44 | Smad4 | 106 | 1 × 10−16 |
| Mef2a | 318 | 1 × 10−57 | CP2 | 61 | 1 × 10−42 | CP2 | 23 | 1 × 10−14 |
| NPAS2 | 308 | 1 × 10−57 | NPAS2 | 294 | 1 × 10−24 | GSC | 104 | 1 × 10−12 |
| TEAD4 | 190 | 1 × 10−39 | Arnt | 198 | 1 × 10−24 | Smad2 | 88 | 1 × 10−10 |
| CRX | 502 | 1 × 10−36 | Mef2a | 90 | 1 × 10−15 | CRX | 123 | 1 × 10−9 |
| Foxa2 | 133 | 1 × 10−30 | Pit1 | 160 | 1 × 10−15 | BMAL1 | 113 | 1 × 10−8 |
| TBX1 | 61 | 1 × 10−22 | Mef2c | 95 | 1 × 10−15 | Smad3 | 130 | 1 × 10−7 |
| CP2 | 46 | 1 × 10−21 | Egr1 | 137 | 1 × 10−14 | NPAS2 | 76 | 1 × 10−6 |
A: Motif Name; B: # of target sequences with motif; C: p‐value.
of 1464.
of 1406.
of 468.
To investigate the functionality of the p53 response element associated with the TNFAIP8 locus, we examined the levels of histone 3 lysine 27 acetylation (H3K27Ac) at this site in seven cell lines from datasets available through ENCODE (Encode Project Consortium, 2012) (Figure 5A, bottom panel). H3K27Ac, a mark associated with active enhancers (Creyghton et al., 2010), is enriched around the TNFAIP8 locus. To assess if there were differences in H3K27Ac at the TNFAIP8 site after WT or K120R induction, chromatin immunoprecipitation experiments were conducted in the p53‐Tet inducible cells. Indeed, occupancy of the K120R p53 mutant is correlated with an increase in H3K27Ac, whereas the presence of WT p53 is not.
In addition, the functionality of the p53 response element discovered within the TNFAIP8 gene was tested empirically using a luciferase reporter system. A reporter plasmid containing the p53 response element associated with CDKN1A or TNFAIP8 was transfected into WT or K120R TO‐p53 cells. After 24 h of tetracycline treatment, luminescence was measured and p53 induction was confirmed by Western blot (Figure 5C). Both WT and K120R expression activated the reporter plasmid containing the CDKN1A p53 response element, whereas only the K120R variant was able to activate the reporter with the TNFAIP8 p53 response element. This further validates the selectivity of K120R recognition of the TNFAIP8 locus, and demonstrates that it is a functional response element.
3.5. TNFAIP8 induction by the K120R mutant contributes to cell survival
Previously, we and others reported that cells expressing the K120R mutant are defective for inducing p53‐mediated apoptosis (Sykes et al., 2006; Tang et al., 2006). TNFAIP8 inhibits the catalytic activity of Caspase‐8 and attenuates apoptosis (You et al., 2001). We therefore investigated the role of TNFAIP8 activation in the ability of K120R expressing cells to evade p53‐mediated cell death. To test the contribution of TNFAIP8 induction to the survival of K120R expressing cells, shRNA‐mediated depletion of TNFAIP8 was performed in the H1299 TO‐p53 system (Figure 6). Depletion of TNFAIP8 resulted in enhanced Caspase‐3 cleavage in K120R cells, compared to luciferase control, as shown by western blot (Figure 6A, compare lanes 6 and 8) or by flow cytometry (Figure 6B). An increase in Annexin V positive cells was also observed upon depletion of TNFAIP8 in TO‐K120R cells (Figure 6C). These data suggest that TNFAIP8 activation is a gain of function property contributing to cell survival that is acquired by the tumor‐derived K120R mutant.
4. Discussion
Missense mutations in p53 often result in the absence of its normal functions and consequently, loss of tumor suppressor ability. Some p53 mutants also gain new functions, such as the ability to drive enhanced invasion or deregulate cell growth (Muller and Vousden, 2014). For example, the hotspot p53 mutant, R175H, augments tumor invasiveness via increased c‐Met tyrosine kinase activation and enhanced TGFβ‐dependent metastasis; and expression of the R273H mutant results in aberrant cell cycle regulation through up‐regulation of NF‐Kβ2 (Adorno et al., 2009; Grugan et al., 2013; Vaughan et al., 2012). Although there is a wealth of information regarding the oncogenic phenotypes exhibited by mutant p53, the mechanisms by which it confers cell survival are not well characterized. One recent study showed that the R273H mutant can promote cell survival and anoikis resistance by repressing BMF, an apoptotic activator that binds BCL2 proteins. However, repression of BMF by the R273H mutant was not direct, and was dependent on alterations in the PI3/AKT signaling pathway that were also regulated by the presence of the R273H mutation (Tan et al., 2015). Here we show that the K120R mutant, which is defective for inducing p53 mediated apoptosis, also confers oncogenic properties via direct regulation of an anti‐apoptotic gene, TNFAIP8 (Figure 7). Thus, our data implicate induction of an anti‐apoptotic gene product as a new gain of function mechanism by which mutant p53 contributes to tumorigenesis.
Figure 7.
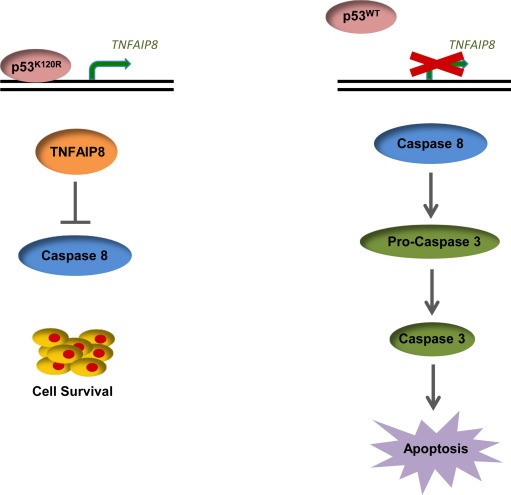
K120R induces TNFAIP8, resulting in cell survival. Brief model illustrating differential effects of WT and K120R p53 on TNFAIP8 induction and the biological outcomes.
Several mutations at K120, which makes direct contact with the p53 response element in the major groove of the DNA helix, have been documented in human cancer, including K120R, K120M, K120E, K120N and K120Q. Of these mutants, K120R retains the most residual WT p53 activity (Figure 1A). Since the K120R mutant exhibits a specific deficiency in p53 apoptotic gene induction (Figure 2A and Table S1) (Sykes et al., 2006; Tang et al., 2006), we explored the differences in the transcriptional networks regulated by WT and K120R p53. Of the unique K120R induced transcriptional network, one of the most highly induced genes, TNFAIP8, had previously been linked to inhibition of apoptosis (You et al., 2001). Since other p53 mutants regulate genes that promote oncogenesis (Muller and Vousden, 2014), we postulated that induction of TNFAIP8 by the K120R mutant contributes to the apoptotic deficiency in cells expressing this mutation. Results of TNFAIP8 RNAi studies revealed that TNFAIP8 induction contributes to cell survival in K120R expressing cells, as depletion of TNFAIP8 transcript resulted in increased apoptosis. As mentioned, K120R cells are defective for activating pro‐apoptotic genes that are normally induced by WT p53 (Sykes et al., 2006; Tang et al., 2006). Combined, these effects may contribute to the ability of a tumor expressing the K120R variant of p53 to survive under stress conditions.
While the K120R mutation itself occurs only rarely in human tumors, there are several hotspot mutations that occur elsewhere in the DNA binding domain of p53. We found that one of these p53 hotspot mutants, R282W, also acquires the ability to induce TNFAIP8 transcription. Further, p53 structural studies show R282 is in proximity to the L1 loop (which contains K120) when p53 is bound to DNA (Calhoun and Daggett, 2011). Analysis of the R282W mutation revealed that the L1 loop becomes distorted in the presence of this mutation, disrupting its typical interaction with DNA (Calhoun and Daggett, 2011; Joerger et al., 2006). It is therefore possible that the structural effects of the R282W and K120R mutations are similar in the context of p53's ability to bind DNA, with both causing misalignment of the L1 loop. Interestingly, R282 is one of the two mutational hotspots that has been linked to shorter survival in patients harboring p53 mutations (Xu et al., 2014). The R282W mutant however, has not previously been described to confer cell survival, and these data suggest that induction of TNFAIP8 and the resulting evasion of cell death could be a mechanism that contributes to how the R282W mutant promotes tumorigenesis.
In addition to discovering a transcriptional network independently regulated by the human cancer derived K120R mutant, we also found in our ChIP‐Seq data that K120R binds the genome at sites (TNFAIP8, KCNJ8 and NUPR1) where WT p53 does not. Motif analysis of the DNA sequences contained within the peaks associated with these genes revealed that each peak harbored a p53 response element, although there were several mismatches (Table 2). The p53 consensus sequence however, is highly degenerate and allows for some variance (Hoh et al., 2002). Regardless, de novo binding acquired by K120R and lack of WT p53 binding at these loci is surprising. One possible explanation for this observation could be a novel protein–protein interaction mediated by mutant p53 that is not exhibited by WT p53. Recruitment of other mutant forms of p53 to gene promoters via protein–protein interactions is a molecular mechanism for some gain of function activities (Do et al., 2012; Liu et al., 2011; Stambolsky et al., 2010). For example, various p53 mutants form a complex with the sequence specific transcription factor NF‐Y and subsequently bind to NF‐Y target gene promoters. Mutant p53 then recruits the histone acetyltransferase p300 to NF‐Y target genes in response to DNA damage, resulting in transcriptional activation and aberrant cell cycle regulation (Di Agostino et al., 2006). As discussed previously, the K120R mutant retains some transcriptional competency, and therefore its ability to bind a p53 response element is not unexpected. Perhaps the explanation underlying transcriptional activation of TNFAIP8 by K120R but not WT p53, could be the result of a novel protein interaction modulated by the K120R mutation combined with residual DNA binding ability. Previously, mutant p53 has been demonstrated to augment NF‐κB activity and promote chronic inflammation in colitis‐associated colorectal cancer, and mutant R175H p53 was detected at NF‐κB binding sites by ChIP‐on‐chip studies in SKBr3 breast cancer cells (Cooks et al., 2013; Dell'Orso et al., 2011). The potential for crosstalk between mutant K120R p53 and another transcription factor seems plausible, and further exploration of this model may reveal a mechanism for recruitment of K120R to the TNFAIP8 locus.
Our study is the first to link p53 with TNFAIP8 (also known as SCC‐2 and NDED), a death effector domain containing protein (Kumar et al., 2000). Typically, TNFAIP8 is a target of nuclear factor‐κB (NF‐κB), a transcription factor that is potently activated by the cytokine tumor necrosis factor (TNF) (You et al., 2001). NF‐κB plays a primary role in attenuating TNF‐mediated cell death via induction of anti‐apoptotic genes, including TNFAIP8. TNFAIP8 antagonizes the extrinsic pathway of apoptosis by inhibiting the catalytic activity of Caspase‐8, which is the effector of the executioner Caspase, Caspase‐3. Consequently, TNF‐mediated apoptosis is inhibited by overexpression of TNFAIP8 (You et al., 2001). Recently, the role of TNFAIP8 expression in malignant progression has been examined. Elevated levels of TNFAIP8 are associated with poor prognosis, drug resistance and metastasis in various tumor types (Liu et al., 2014; Miao et al., 2012; Wang et al., 2014; Yang et al., 2014). This raises the possibility that for K120R, and perhaps R282W, induction of TNFAIP8 contributes to oncogenesis in patients expressing this mutant variant of p53.
The heterogeneity among tumor‐associated p53 mutants is emphasized by the wide array of acquired functions displayed by the distinct p53 mutants (Muller and Vousden, 2014). There is significant knowledge pertaining to the gain of function properties among the hotspot p53 mutants, however the relevance of the less frequently occurring mutations in tumor biology is not as well characterized. Exploring the impact of a broader range of p53 mutations may allow the ability to use p53 mutational status for patient stratification in cancer therapy. In this study, we found that the K120R mutant induces a large set of target genes distinct from those induced by the wild type allele. Of the genes most robustly activated, a pro‐survival gene TNFAIP8 was identified as a direct target of mutant K120R. Its induction partially blocks apoptosis, providing cells expressing this mutant of p53 the ability to survive after exposure to DNA damage. The findings presented here contribute to the growing understanding of mutant p53, and identify pro‐survival gene induction as a gain of function activity acquired by specific mutants.
Financial support
JM is supported by the Ruth L. Kirschstein National Service Award, National Institutes of Health (NIH) (F31 CA174199‐0), JM and SM are supported by the NIH (R21 CA152786). MS and SB are supported by the NIH (R01 CA078831). J Manfredi is supported by the NIH (R01 CA200256).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Acknowledgments
The authors wish to thank Dr. Michael Root for providing technical assistance and equipment. We thank Dr. Adam Ertel and the Sidney Kimmel Cancer Center Cancer Genomics Shared Resource for data analysis. The Flag‐TNFAIP8 expression plasmid was a gift from Dr. Paul R Albert (University of Ottawa).
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.05.007.
Monteith Jessica A., Mellert Hestia, Sammons Morgan A., Kuswanto Laudita A., Sykes Stephen M., Resnick-Silverman Lois, Manfredi James J., Berger Shelley L., McMahon Steven B., (2016), A rare DNA contact mutation in cancer confers p53 gain-of-function and tumor cell survival via TNFAIP8 induction, Molecular Oncology 10, doi: 10.1016/j.molonc.2016.05.007.
Contributor Information
Jessica A. Monteith, Email: monteith.jessica@gmail.com
Hestia Mellert, Email: hestiamellert@gmail.com.
Morgan A. Sammons, Email: msammons@mail.med.upenn.edu
Laudita A. Kuswanto, Email: laudita.kuswanto@utsouthwestern.edu
Stephen M. Sykes, Email: Stephen.sykes@fccc.edu
James J. Manfredi, Email: james.manfredi@mssm.edu
Shelley L. Berger, Email: berger@upenn.edu
Steven B. McMahon, Email: Steven.mcmahon@jefferson.edu
References
- Adorno, M. , Cordenonsi, M. , Montagner, M. , Dupont, S. , Wong, C. , Hann, B. , Solari, A. , Bobisse, S. , Rondina, M.B. , Guzzardo, V. , Parenti, A.R. , Rosato, A. , Bicciato, S. , Balmain, A. , Piccolo, S. , 2009. A Mutant-p53/Smad complex opposes p63 to empower TGFβ-induced metastasis. Cell 137, 87–98. [DOI] [PubMed] [Google Scholar]
- Calhoun, S. , Daggett, V. , 2011. Structural effects of the L145Q, V157F, and R282W cancer-associated mutations in the p53 DNA-binding core domain. Biochemistry 50, 5345–5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin, K.V. , Ueda, K. , Pastan, I. , Gottesman, M.M. , 1992. Modulation of activity of the promoter of the human MDR1 gene by Ras and p53. Science (New York, N.Y.) 255, 459–462. [DOI] [PubMed] [Google Scholar]
- Cooks, T. , Pateras, I.S. , Tarcic, O. , Solomon, H. , Schetter, A.J. , Wilder, S. , Lozano, G. , Pikarsky, E. , Forshew, T. , Rosenfeld, N. , Harpaz, N. , Itzkowitz, S. , Harris, C.C. , Rotter, V. , Gorgoulis, V.G. , Oren, M. , 2013. Mutant p53 prolongs NF-kappaB activation and promotes chronic inflammation and inflammation-associated colorectal cancer. Cancer Cell 23, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton, M.P. , Cheng, A.W. , Welstead, G.G. , Kooistra, T. , Carey, B.W. , Steine, E.J. , Hanna, J. , Lodato, M.A. , Frampton, G.M. , Sharp, P.A. , Boyer, L.A. , Young, R.A. , Jaenisch, R. , 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U. S. A. 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Orso, S. , Fontemaggi, G. , Stambolsky, P. , Goeman, F. , Voellenkle, C. , Levrero, M. , Strano, S. , Rotter, V. , Oren, M. , Blandino, G. , 2011. ChIP-on-chip analysis of in vivo mutant p53 binding to selected gene promoters. OMICS 15, 305–312. [DOI] [PubMed] [Google Scholar]
- Di Agostino, S. , Strano, S. , Emiliozzi, V. , Zerbini, V. , Mottolese, M. , Sacchi, A. , Blandino, G. , Piaggio, G. , 2006. Gain of function of mutant p53: the mutant p53/NF-Y protein complex reveals an aberrant transcriptional mechanism of cell cycle regulation. Cancer Cell 10, 191–202. [DOI] [PubMed] [Google Scholar]
- Do, P.M. , Varanasi, L. , Fan, S. , Li, C. , Kubacka, I. , Newman, V. , Chauhan, K. , Daniels, S.R. , Boccetta, M. , Garrett, M.R. , Li, R. , Martinez, L.A. , 2012. Mutant p53 cooperates with ETS2 to promote etoposide resistance. Genes Dev. 26, 830–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encode Project Consortium, 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- el-Deiry, W.S. , Kern, S.E. , Pietenpol, J.A. , Kinzler, K.W. , Vogelstein, B. , 1992. Definition of a consensus binding site for p53. Nat. Genet. 1, 45–49. [DOI] [PubMed] [Google Scholar]
- Frazier, M.W. , He, X. , Wang, J. , Gu, Z. , Cleveland, J.L. , Zambetti, G.P. , 1998. Activation of c-myc gene expression by tumor-derived p53 mutants requires a discrete C-terminal domain. Mol. Cell. Biol. 18, 3735–3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y. , Frith, M.C. , Haverty, P.M. , Weng, Z. , 2004. MotifViz: an analysis and visualization tool for motif discovery. Nucleic Acids Res. 32, W420–W423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccone, G. , Battey, J. , Gazdar, A.F. , Oie, H. , Draoui, M. , Moody, T.W. , 1992. Neuromedin B is present in lung cancer cell lines. Cancer Res. 52, 2732s–2736s. [PubMed] [Google Scholar]
- Grugan, K.D. , Vega, M.E. , Wong, G.S. , Diehl, J.A. , Bass, A.J. , Wong, K.K. , Nakagawa, H. , Rustgi, A.K. , 2013. A common p53 mutation (R175H) activates c-Met receptor tyrosine kinase to enhance tumor cell invasion. Cancer Biol. Ther. 14, 853–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz, S. , Benner, C. , Spann, N. , Bertolino, E. , Lin, Y.C. , Laslo, P. , Cheng, J.X. , Murre, C. , Singh, H. , Glass, C.K. , 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoh, J. , Jin, S. , Parrado, T. , Edington, J. , Levine, A.J. , Ott, J. , 2002. The p53MH algorithm and its application in detecting p53-responsive genes. Proc. Natl. Acad. Sci. U. S. A. 99, 8467–8472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu, F. , Kent, W.J. , Clawson, H. , Kuhn, R.M. , Diekhans, M. , Haussler, D. , 2006. The UCSC known genes. Bioinformatics (Oxford, England) 22, 1036–1046. [DOI] [PubMed] [Google Scholar]
- Joerger, A.C. , Ang, H.C. , Fersht, A.R. , 2006. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc. Natl. Acad. Sci. U. S. A. 103, 15056–15061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato, S. , Han, S.Y. , Liu, W. , Otsuka, K. , Shibata, H. , Kanamaru, R. , Ishioka, C. , 2003. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc. Natl. Acad. Sci. U. S. A. 100, 8424–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, D. , Whiteside, T.L. , Kasid, U. , 2000. Identification of a novel tumor necrosis factor-α-inducible gene, SCC-S2, containing the consensus sequence of a death effector domain of Fas-associated death domain-like interleukin-1β-converting enzyme-inhibitory protein. J. Biol. Chem. 275, 2973–2978. [DOI] [PubMed] [Google Scholar]
- Lang, G.A. , Iwakuma, T. , Suh, Y.A. , Liu, G. , Rao, V.A. , Parant, J.M. , Valentin-Vega, Y.A. , Terzian, T. , Caldwell, L.C. , Strong, L.C. , El-Naggar, A.K. , Lozano, G. , 2004. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 119, 861–872. [DOI] [PubMed] [Google Scholar]
- Lee, T.I. , Johnstone, S.E. , Young, R.A. , 2006. Chromatin immunoprecipitation and microarray-based analysis of protein location. Nat. Protoc. 1, 729–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, K. , Ling, S. , Lin, W.C. , 2011. TopBP1 mediates mutant p53 gain of function through NF-Y and p63/p73. Mol. Cell. Biol. 31, 4464–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, T. , Xia, B. , Lu, Y. , Xu, Y. , Lou, G. , 2014. TNFAIP8 overexpression is associated with platinum resistance in epithelial ovarian cancers with optimal cytoreduction. Hum. Pathol. 45, 1251–1257. [DOI] [PubMed] [Google Scholar]
- Ludes-Meyers, J.H. , Subler, M.A. , Shivakumar, C.V. , Munoz, R.M. , Jiang, P. , Bigger, J.E. , Brown, D.R. , Deb, S.P. , Deb, S. , 1996. Transcriptional activation of the human epidermal growth factor receptor promoter by human p53. Mol. Cell. Biol. 16, 6009–6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, Z. , Zhao, T. , Wang, Z. , Xu, Y. , Song, Y. , Wu, J. , Xu, H. , 2012. SCC-S2 is overexpressed in colon cancers and regulates cell proliferation. Tumour Biol. 33, 2099–2106. [DOI] [PubMed] [Google Scholar]
- Muller, P.A. , Vousden, K.H. , 2014. Mutant p53 in cancer: new functions and therapeutic opportunities. Cancer Cell 25, 304–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olive, K.P. , Tuveson, D.A. , Ruhe, Z.C. , Yin, B. , Willis, N.A. , Bronson, R.T. , Crowley, D. , Jacks, T. , 2004. Mutant p53 gain of function in two mouse models of Li-Fraumeni syndrome. Cell 119, 847–860. [DOI] [PubMed] [Google Scholar]
- Olivier, M. , Hollstein, M. , Hainaut, P. , 2010. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor Perspect. Biol. 2, a001008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean, A. , Mathe, E. , Kato, S. , Ishioka, C. , Tavtigian, S.V. , Hainaut, P. , Olivier, M. , 2007. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 28, 622–629. [DOI] [PubMed] [Google Scholar]
- Pugacheva, E.N. , Ivanov, A.V. , Kravchenko, J.E. , Kopnin, B.P. , Levine, A.J. , Chumakov, P.M. , 2002. Novel gain of function activity of p53 mutants: activation of the dUTPase gene expression leading to resistance to 5-fluorouracil. Oncogene 21, 4595–4600. [DOI] [PubMed] [Google Scholar]
- Schneider, C.A. , Rasband, W.S. , Eliceiri, K.W. , 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi, K. , Kato, S. , Han, S.Y. , Liu, W. , Otsuka, K. , Sakayori, M. , Ishida, T. , Takeda, M. , Kanamaru, R. , Ohuchi, N. , Ishioka, C. , 2004. Isolation of temperature-sensitive p53 mutations from a comprehensive missense mutation library. J. Biol. Chem. 279, 348–355. [DOI] [PubMed] [Google Scholar]
- Smeenk, L. , van Heeringen, S.J. , Koeppel, M. , van Driel, M.A. , Bartels, S.J. , Akkers, R.C. , Denissov, S. , Stunnenberg, H.G. , Lohrum, M. , 2008. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res. 36, 3639–3654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolsky, P. , Tabach, Y. , Fontemaggi, G. , Weisz, L. , Maor-Aloni, R. , Siegfried, Z. , Shiff, I. , Kogan, I. , Shay, M. , Kalo, E. , Blandino, G. , Simon, I. , Oren, M. , Rotter, V. , 2010. Modulation of the vitamin D3 response by cancer-associated mutant p53. Cancer Cell 17, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sykes, S.M. , Mellert, H.S. , Holbert, M.A. , Li, K. , Marmorstein, R. , Lane, W.S. , McMahon, S.B. , 2006. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol. Cell 24, 841–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan, B.S. , Tiong, K.H. , Choo, H.L. , Fei-Lei Chung, F. , Hii, L.W. , Tan, S.H. , Yap, I.K. , Pani, S. , Khor, N.T. , Wong, S.F. , Rosli, R. , Cheong, S.K. , Leong, C.O. , 2015. Mutant p53-R273H mediates cancer cell survival and anoikis resistance through AKT-dependent suppression of BCL2-modifying factor (BMF). Cell Death Dis. 6, e1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Y. , Luo, J. , Zhang, W. , Gu, W. , 2006. Tip60-dependent acetylation of p53 modulates the decision between cell-cycle arrest and apoptosis. Mol. Cell 24, 827–839. [DOI] [PubMed] [Google Scholar]
- Vaughan, C.A. , Singh, S. , Windle, B. , Sankala, H.M. , Graves, P.R. , Andrew Yeudall, W. , Deb, S.P. , Deb, S. , 2012. p53 mutants induce transcription of NF-κB2 in H1299 cells through CBP and STAT binding on the NF-κB2 promoter and gain of function activity. Arch. Biochem. Biophys. 518, 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein, B. , Lane, D. , Levine, A.J. , 2000. Surfing the p53 network. Nature 408, 307–310. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Song, Y. , Men, X. , 2014. Variance of TNFAIP8 expression between tumor tissues and tumor-infiltrating CD4+ and CD8+ T cells in non-small cell lung cancer. Tumour Biol. 35, 2319–2325. [DOI] [PubMed] [Google Scholar]
- Weisz, L. , Oren, M. , Rotter, V. , 2007. Transcription regulation by mutant p53. Oncogene 26, 2202–2211. [DOI] [PubMed] [Google Scholar]
- Xu, J. , Wang, J. , Hu, Y. , Qian, J. , Xu, B. , Chen, H. , Zou, W. , Fang, J.Y. , 2014. Unequal prognostic potentials of p53 gain-of-function mutations in human cancers associate with drug-metabolizing activity. Cell Death Dis. 5, e1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, M. , Zhao, Q. , Wang, X. , Liu, T. , Yao, G. , Lou, C. , Zhang, Y. , 2014. TNFAIP8 overexpression is associated with lymph node metastasis and poor prognosis in intestinal-type gastric adenocarcinoma. Histopathology 65, 517–526. [DOI] [PubMed] [Google Scholar]
- You, Z. , Ouyang, H. , Lopatin, D. , Polver, P.J. , Wang, C.Y. , 2001. Nuclear factor-κB-inducible death effector domain-containing protein suppresses tumor necrosis factor-mediated apoptosis by inhibiting caspase-8 activity. J. Biol. Chem. 276, 26398–26404. [DOI] [PubMed] [Google Scholar]
- Zhang, X.Y. , DeSalle, L.M. , Patel, J.H. , Capobianco, A.J. , Yu, D. , Thomas-Tikhonenko, A. , McMahon, S.B. , 2005. Metastasis-associated protein 1 (MTA1) is an essential downstream effector of the c-MYC oncoprotein. Proc. Natl. Acad. Sci. U. S. A. 102, 13968–13973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupnick, A. , Prives, C. , 2006. Mutational analysis of the p53 core domain L1 loop. J. Biol. Chem. 281, 20464–20473. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data