

Abstract
Biglycan (BGN) is an important component of the extracellular matrix (ECM) that is implicated in a variety of human cancers. In our previous study, we reported that BGN was overexpressed in gastric cancer (GC) tissues and promoted cancer metastasis. Moreover, the tubular formation capacity in HUVECs was promoted by stimulation with culture media from BGN‐overexpressing GC cells, but the exact underlying mechanism is still unknown. The purpose of this study was to determine the role and molecular mechanism of BGN in VEGF expression in endothelial cells. We found that BGN stimulation of endothelial cells increased the interaction between NF‐kB and the HIF‐1α promoter, leading to enhanced promoter activity and increased HIF‐1α mRNA levels, as well as augmented HIF‐1 activity that resulted in VEGF expression. All of this was dependent on the interaction of BGN with its receptors, TLR2 and TLR4. Moreover, we found that BGN enhanced endothelial cell migration and proliferation, as well as tube formation, in a TLR signaling pathway‐dependent manner. In addition, endothelial cell‐derived VEGF in turn was found to act on GC cells and promotes their migration. The combined findings of our current and previous studies suggest that BGN secreted from GC cells into the tumor stroma promotes GC development, as well as its progression, potentially through the chronic activation of tumor angiogenesis.
Keywords: BGN, Endothelial cells, TLR2/4, VEGF, Angiogenesis, GC
Highlights
BGN is member of SLRPs, which is found in a variety of human cancers.
BGN stimulation of endothelial cells increased the expression of VEGF through NF‐kB and the HIF‐1α.
BGN enhanced endothelial cell migration, proliferation and tube formation in a TLR signaling pathway‐dependent manner.
Endothelial cell‐derived VEGF in turn acts on GC cells and promotes the migration of GC cells.
1. Introduction
Gastric cancer (GC) poses a great threat to the health of people worldwide and ranks third among all types of tumors in China (Piazuelo and Correa, 2013). Therefore, it is of great importance to explore the mechanisms of the occurrence and development of GC. Studies have proven that tumor inflammation is a key component of the tumor microenvironment, which contributes to tumor proliferation, survival and invasion (Mantovani et al., 2008). The most important factor causing inflammation in GC may be Helicobacter pylori (HP) infections (Horemans et al., 2016; Kazemi et al., 2016; Liang et al., 2014), especially with HP containing a contiguous cag pathogenicity island (cag‐PAI), which encodes a known virulence factor, CagA (Zhang et al., 2016). Biglycan (BGN) is a small leucine‐rich repeat proteoglycan (SLRP), which is found in a variety of human cancers, including GC (Hu et al., 2014), esophageal squamous cell carcinoma (Zhu et al., 2013), pancreatic cancer (Weber et al., 2001), colon tumors (Mikula et al., 2010) and tumors in blood vessels (Yamamoto et al., 2012). In our previous study, we found that BGN expression in GC tissues was significantly upregulated compared with its expression in adjacent normal gastric tissues and was correlated with axillary lymph node metastasis, the depth of tumor invasion and TNM stage. Moreover, BGN enhances the invasive capacity of GC by activating the FAK signaling pathway (Hu et al., 2014). In this study, we focused on its role as a mediator of inflammation in GC and its regulation of VEGF expression in endothelial cells.
Toll like receptor 2 (TLR2) and Toll like receptor 4 (TLR4), as members of TLR family, play essential roles in inflammation (Boehmer et al., 2005; Johnson et al., 2005). TLR2 and TLR4 are considered to be the receptors of BGN. The binding of BGN to TLR2/4 can boost inflammation and enhance the synthesis of TNF‐alpha and MIP‐2 (Schaefer et al., 2005). In inflammatory renal diseases, researchers have found that circulating BGN accumulates in the kidneys, where it causes the recruitment of leukocytes, which infiltrate the renal parenchyma concurrent with abnormal renal levels of the chemoattractants CXCL1, CXCL2, CCL2 and CCL5 (Hsieh et al., 2014; Moreth et al., 2014; Zeng‐Brouwers et al., 2014). Persistent NF‐kB activation and increased angiogenesis as a result of elevated vascular endothelial growth factor (VEGF) levels are considered to be hallmarks of inflammation in cancer (Riddell et al., 2012). Interestingly, NF‐kB is an essential component in the TLR signaling pathway (Xiao and Ghosh, 2005). Studies have shown that BGN can upregulate VEGF expression and promote angiogenesis in colon cancer (Xing et al., 2015), which is consistent with our previous findings that BGN could stimulate tubular formation activity in HUVECs. Nevertheless, the exact mechanism through which BGN regulates VEGF expression remains unclear.
In this study, we demonstrate that BGN induces VEGF mRNA levels and protein expression in endothelial cell through binding to its receptors TLR2 and TLR4 and that the tube‐forming, migratory and proliferative capacities of endothelial cell are enhanced in a TLR signaling pathway‐dependent manner. Endothelial cell‐derived VEGF secretion following stimulation by BGN acts on GC cells to promote GC cell migration.
2. Material and methods
2.1. Cell lines
The HUVEC and HAEC endothelial cell lines were kind gifts from the Shanghai Institute of Hypertension. The human gastric cancer cell line NCI‐N87 was purchased from the Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences. Cells were cultured at 37 °C in 5% CO2 at a saturating humidity in RPMI‐1640 medium containing 10% fetal bovine serum with 100 U/ml penicillin and 100 μg/ml streptomycin in a cell incubator. Exponentially growing cells were used for experiments.
2.2. Endothelial cell transfection
The shRNAs for HIF‐1α were purchased from Asia‐VectorBiotechnology (Shanghai) Co. Ltd. Cells in a logarithmic growth phase were trypsinized, counted, and seeded in 6‐well plates to ensure 50% cell confluence by the next day for transfection. The cells were transfected with shRNA using Lipofectamine 2000 transfection reagent according to manufacturer's protocol.
2.3. ELISA and quantitative realtime polymerase chain reaction
ELISA (Cat# ELH‐VEGF‐001) analyses of VEGF were performed at different times post‐stimulation with recombinant BGN (rBGN). The culture supernatants were collected, and secreted VEGF levels were quantified via ELISA according to the manufacturer's protocol. Total RNA was extracted from endothelial cells using TRIzol reagent (Invitrogen) according to the manufacturer's instructions. Reverse transcription was performed using a reverse transcription kit (Promega, Madison, WI, USA). Primers are listed in Table 1. PCR reactions were carried out, and GAPDH served as a constitutive control. PCRs for each sample were conducted in triplicate.
Table 1.
Primers used in qRT‐PCR.
| Primer name | Sequence |
|---|---|
| VEGF‐F | 5′‐AGGGCAGAATCATCACGAAGT‐3′ |
| VEGF‐R | 5′‐AGGGTCTCGATTGGATGGCA‐3′ |
| TLR2‐F | 5′‐ATCCTCCAATCAGGCTTCTCT‐3′ |
| TLR2‐R | 5′‐GGACAGGTCAAGGCTTTTTACA‐3′ |
| TLR4‐F | 5′‐AGTTGATCTACCAAGCCTTGAGT‐3′ |
| TLR4‐R | 5′‐GCTGGTTGTCCCAAAATCACTTT‐3′ |
| HIF1α‐F | 5′‐GAACGTCGAAAAGAAAAGTCTCG‐3′ |
| HIF1α‐R | 5′‐CCTTATCAAGATGCGAACTCACA‐3′ |
| GAPDH‐F | 5′‐GGACCTGACCTGCCGTCTAG‐3′ |
| GAPDH‐R | 5′‐GTAGCCCAGGATGCCCTTGA‐3′ |
2.4. Western blot analysis
Cells were harvested and lysed using RIPA buffer (Solarbio, Beijing, China) containing 1% PMSF protease inhibitors and phosphatase inhibitors. Equal protein amounts (50 μg) were electrophoresed on 10% SDS‐polyacrylamide gels. Proteins were electrotransferred to Immobilon‐P membranes. The membranes were blocked with 5% non‐fat milk in Tris‐buffered saline and then incubated with primary antibodies at 4 °C overnight. The primary antibodies used were anti‐NF‐kB (Cell Signaling Technology, USA), anti‐p‐NF‐kB (Cell Signaling Technology), anti‐HIF‐1α (Abcam, USA) and anti‐GAPDH (Cell Signaling Technology). Following incubation, membranes were then washed three times in TBST solution for 15 min each and then incubated with secondary antibodies. Band intensities were quantitated using a Tanon 2500 imaging system (TANON).
2.5. Luciferase reporter assay
The human VEGF or HIF‐1α gene promoters were amplified via PCR and cloned into the multi‐cloning site of a pGL3 reporter plasmid containing the WT firefly luciferase reporter gene. Mutations were introduced into the seed sequences of the VEGF or HIF‐1α promoter to generate VEGF or HIF‐1α mutation reporters. Cells seeded into 24‐well plates were co‐transfected with different plasmids (firefly reporter constructs containing the VEGF or HIF‐1α promoter, Renilla‐expressing plasmids, pRL‐TK, shRNA plasmids or control plasmids) or were treated with either TLR2/4 antibodies or PDTC. After a 24‐hour incubation, the transfectants were suspended in fresh medium and stimulated with rBGN (0 or 20 nM). The cells were lysed 6 h later, mixed with the dual luciferase assay reagent (Promega, USA). Relative luciferase activity was calculated by normalizing the firefly luminescence to the Renilla luminescence.
2.6. ChIP assay
To evaluate the interaction between HIF‐1α and the VEGF promoter region, ChIP assays were performed using a kit (Millipore) according to the manufacturer's instructions. After undergoing the different processing conditions, cells were fixed with formaldehyde for protein/DNA crosslinking and then lysed. The DNA was sheared via sonication (15 pulses; 35 s on, 35 s off) and then added to a well coated with an anti‐HIF‐1α antibody. Washes were performed to remove unbound material, and then HIF‐1α‐bound DNA was released via protein digestion with proteinase K. The DNA was purified by passing it through a column. PCRs were performed using primers designed to target the VEGF promoter region spanning the site of its interaction with HIF‐1α. The primers were as follows: VEGF‐F: 5′‐CTGTGTGTCCCTCTCCCC‐3′; VEGF‐R: 5′‐CCCACCAAGGTTCACAGC‐3′. Genomic DNA and IgG were used as controls.
2.7. Endothelial tube formation assay
Endothelial cells were transfected with different plasmids or treated with either TLR2/4 antibodies or the NF‐kB inhibitor PDTC (P8765, Sigma, USA) and plated in 96‐well plates coated with 50 μl Matrigel (BD Bioscience, CA, USA) at a concentration of 3 × 104 cells/well. Then, the cells were cultured in media with or without 20 nM rBGN. After a 12‐h incubation at 37 °C with 5% CO2, tubules were photographed via microscopy and evaluated using Image Pro Plus software.
2.8. Endothelial cell and GC cell migration assays
For the endothelial cell migration assay, endothelial cells were transfected with vectors engineered to express shRNA specific to HIF‐1α or treated with either TLR2/4 antibodies or PDTC. Then, 1 × 105 cells were added to serum‐free RPMI‐1640 with rBGN (0 or 20 nM) and plated in transwell chambers (8‐μm pore size, 24‐well plate; Corning Costar, NY, USA) according to the manufacturer's protocols. After a 12‐h incubation, cells were fixed with 10% formalin and stained with 0.5% crystal violet. Fort the GC cell migration assay, the method was the same as previously described (Hu et al., 2014). Finally, cells in the lower chamber were counted using an inverted microscope. Values are expressed as the mean cell number from 5 random fields of view (100×).
2.9. Endothelial cell proliferation assay
Cell proliferation was monitored using Cell Counting Kit‐8 (CCK‐8). Endothelial cells were transfected with vectors engineered to express shRNA specific to HIF‐1α or treated with either TLR2/4 antibodies or PDTC. Then, cells were plated in 96‐well plates at a concentration of 1500 cells/well. rBGN (0 or 20 nM) was used to stimulate the endothelial cells for 48 h. Cell proliferation was measured 48 h later after adding CCK‐8 for 2 h at an absorbance of 450 nm using an Epoch Microplate Spectrophotometer (Bio Tek).
2.10. Statistical analysis
The data are shown as the mean ± SD. Significant differences between the two groups were examined via Student's t‐test or one‐way ANOVA. P < 0.05 was considered significant, and P < 0.01 was considered highly significant. Statistical analyses were performed using IBM SPSS 19.0 software (SPSS Inc).
3. Results
3.1. rBGN stimulates VEGF expression in endothelial cells of through TLR2 and TLR4
To investigate whether BGN is capable of stimulating endothelial expression of VEGF, the VEGF protein level in the culture media and the mRNA level in HUVECs treated with rBGN were tested via ELISA and qPCR. VEGF secretion increased within 3 h of stimulation and reached its peak within 9 h. After 24 h, the level of VEGF declined to baseline (Figure 1A). Consistent with the protein level, the mRNA level also increased after stimulation with rBGN and reached the highest value within 6 h (Figure 1B). BGN is considered to be the ligand of TLR2 and TLR4, so we investigated the expression of TLR2 and TLR4 in endothelial cells and whether the induction of VEGF in endothelial cells by rBGN was dependent upon TLR2 and TLR4. Endothelial cells were incubated with antibodies specific for TLR2 and/or TLR4, which abrogates the binding between the receptors and ligand. Both TLR2 and TLR4 were expressed in endothelial cells (Figure 1C and D). In addition, endothelial cells incubated with TLR2 and/or TLR4 antibodies prior to rBGN stimulation limited the increase in VEGF protein (Figure 1E) and mRNA levels (Figure 1F). In conclusion, these results suggest that BGN induction of VEGF is dependent upon its receptors, TLR2 and TLR4.
Figure 1.
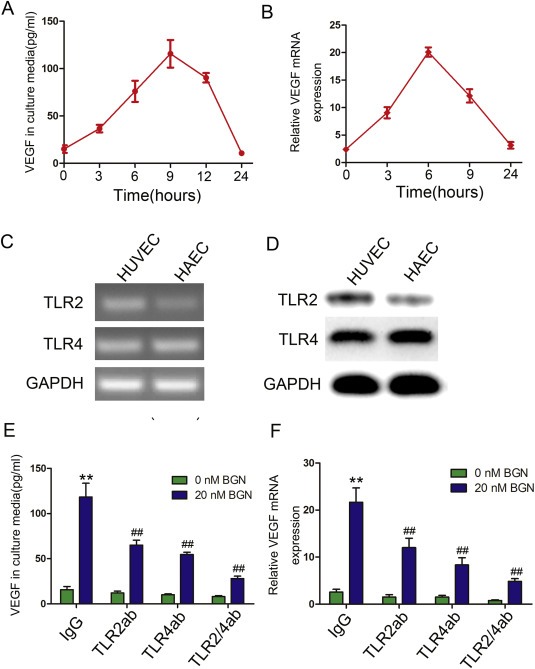
rBGN stimulates endothelial cell expression of VEGF. (A) VEGF protein expression in HUVEC culture media was determined via ELISA after cells were stimulated with 20 nM rBGN for various times. (B) VEGF mRNA expression in HUVECs was determined via qPCR after cells were stimulated with 20 nM rBGN for various times. (C–D) TLR2/4 mRNA and protein expression in endothelial cells were determined via PCR and western‐blot. (E–F) HUVECs were incubated with antibodies to neutralize TLR2 and/or TLR4 receptor(s). Then, VEGF protein and mRNA expression were determined via ELISA and qPCR after cells were stimulated with or without 20 nM BGN. Each value is the mean ± SD of three experiments. ** Represents P ≤ 0.01 when compared to untreated control levels; ## represents P ≤ 0.01 when compared to stimulated control values.
3.2. rBGN enhances VEGF expression dependent on HIF‐1 binding to its promoter
To investigate how BGN promotes VEGF expression in endothelial cells, luciferase assays were performed to determine if VEGF promoter activity was increased in HUVECs and HAECs when treated with rBGN (Figure 2A). The enhancement of VEGF promoter activity was limited during TLR2 and/or TLR4 antibody incubation prior to the rBGN stimulation of HUVECs (Figure 2B). As has been demonstrated, HIF‐1 is an important transcription factor that regulates VEGF. We analyzed the VEGF promoter region for potential HIF‐1α‐binding sites and found three putative sites (−981 to −965 nt, −516 to −500 nt and −317 to −300 nt). We found that luciferase activity was upregulated to a greater degree in HUVECs after rBGN stimulation of cells transfected with VEGF‐WT, VEGF‐MUT2 (−516 to −500 nt) or VEGF‐MUT3 (−981 to −965) than was observed in control cells without rBGN stimulation. However, the activity was significantly decreased when the cells were transfected with VEGF‐MUT1 (−317 to −300 nt), which indicates that VEGF‐MUT1 (−317 to −300 nt) might contain a HIF‐1α‐binding site (Figure 2C). We also transfected shRNA targeting HIF‐1α (shHIF‐1α) into cells prior to rBGN stimulation (Figure 2D and E). As expected, shHIF‐1α expression resulted in a reduction in the degree of rBGN‐induced VEGF promoter activity (Figure 2F) and protein expression in the culture media (Figure 2G). NF‐kB is an important downstream molecule in the TLR signaling pathway, so we constructed NF‐kB binding‐site mutants in the VEGF promoter region (MUT1: −652 to −638, MUT2: −1003 to −989). We found no difference in the luciferase activity between the VEGF‐WT and mutant groups (Figure 2H), which indicates that NF‐kB does not regulate VEGF expression by directly binding its promoter. ChIP analysis of the VEGF promoter using an antibody specific to HIF‐1α and qPCR primers specific to the VEGF promoter region containing the HIF‐1α binding site were performed. rBGN stimulation lead to increased HIF‐1 interaction with the VEGF promoter (Figure 2I). HIF‐1α protein expression in endothelial cells was significantly increased after stimulation with rBGN (Figure 2J). Thus, rBGN appears to regulate VEGF promoter activity through the HIF‐1α interaction with the HIF‐1α binding site, independent of the NF‐kB regulatory elements.
Figure 2.
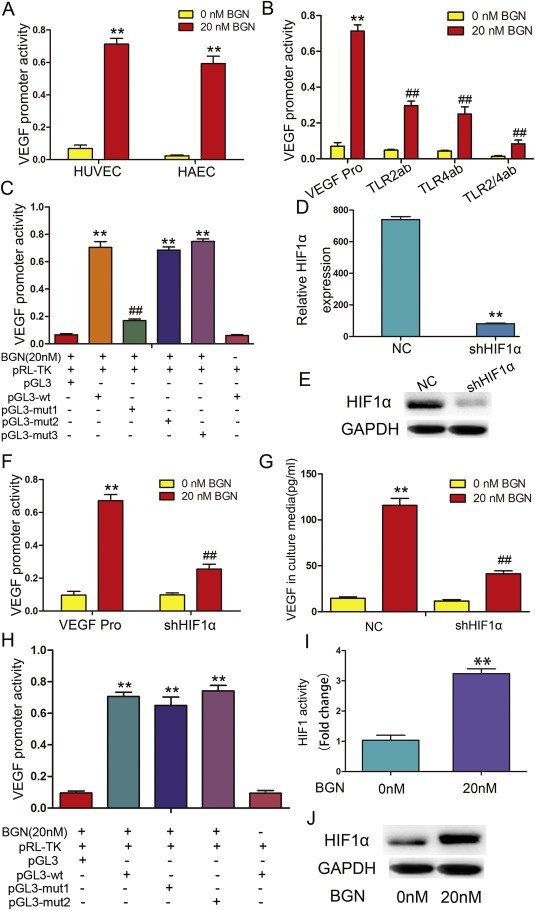
rBGN induction of VEGF is dependent upon HIF‐1α. (A) HUVECs and HAECs were transfected with a VEGF promoter reporter plasmid (pGL3‐VEGF). Cells were stimulated with 20 nM rBGN 24 h after transfection, and VEGF promoter activity was measured 6 h later. (B) HUVECs were transfected with a VEGF promoter reporter plasmid (pGL3‐VEGF) in the absence or presence of antibodies to neutralize TLR2 and/or TLR4 receptor(s). Cells were stimulated with 20 nM rBGN 24 h after transfection, and VEGF promoter activity was measured 6 h later. (C) A reporter plasmid for VEGF (pGL3‐VEGF) was generated by cloning the VEGF promoter region (wt) or its HIF‐1α binding site mutants (mut1, mut2 and mut3) into the pGL3‐basic vector. BGN significantly increased the luciferase activity of the VEGF promoter region (wt), while activity was significantly reduced when mut1 replaced the wt sequence. Mut1 may contain a HIF‐1α‐binding site. (D–E) The mRNA and protein expression of HIF1α after downregulation in endothelial cells were determined via qPCR and western‐blot. (F) HUVECs were transfected with a VEGF promoter reporter plasmid (pGL3‐VEGF) in the absence or presence of vectors engineered to express shHIF‐1α. Cells were stimulated with 20 nM rBGN 24 h after transfection, and VEGF promoter activity was measured 6 h later. (G) HUVECs were transfected with negative control vectors (NC) or with vector engineered to express shHIF‐1α. Cells were stimulated with 20 nM rBGN 24 h after transfection, and VEGF expression was measured 9 h later. (H) HUVECs were transfected with vectors containing the VEGF promoter region (wt) or its NF‐kB binding‐site mutants (mut1 and mut2). rBGN significantly increased the luciferase activity of the VEGF promoter region (wt), while there were no significant changes in the mut1‐ or mut2‐transfected cells. VEGF promoter region may not contain NF‐kB binding sites. (I) HUVECs were stimulated with rBGN. Then, ChIP assays were performed via immunoprecipitation with antibodies to HIF‐1α, along with qPCR amplification of the isolated DNA fragments using VEGF promoter‐specific primers. The results are reported as the fold change relative to untreated cells. (J) Western blot analysis of HIF‐1α after stimulation with or without rBGN. GAPDH was used as a loading control. Each value is the mean ± SD of three experiments. ** Represents P ≤ 0.01 when compared to untreated control levels; ## represents P ≤ 0.01 when compared to stimulated control values.
3.3. rBGN enhances HIF‐1α activity via NF‐kB
To determine whether NF‐kB is indirectly involved in the rBGN‐induced VEGF promoter activity, we treated HUVECs with PDTC, an inhibitor of NF‐kB. PDTC inhibited the rBGN induction of VEGF protein expression (Figure 3A) and promoter activity (Figure 3B), suggesting that the activation of NF‐kB by BGN indirectly regulates VEGF expression. Then, we analyzed NF‐kB binding sites in the HIF‐1α promoter region and constructed reporter vectors for this promoter, including the wild type (HIF‐1α‐WT) and the two mutants (MUT1: −79 to −65, MUT2: −500 to −486). To test the involvement of NF‐kB in HIF‐1α regulation, each HIF‐1α reporter was co‐transfected with pRL‐TK, and the promoter activities were measured after rBGN stimulation. HIF‐1α‐MUT1 reduced the rBGN activation of the HIF‐1α promoter when compared with the activation observed in response to HIF‐1α‐WT and HIF‐1α‐MUT2 (Figure 3C). NF‐kB, p‐NF‐kB and HIF‐1α protein levels were detected via western blotting. PDTC inhibited the activation of NF‐kB and consequently decreased the expression of HIF‐1α (Figure 3D). These results suggest that NF‐kB induces HIF‐1α expression upon rBGN stimulation.
Figure 3.
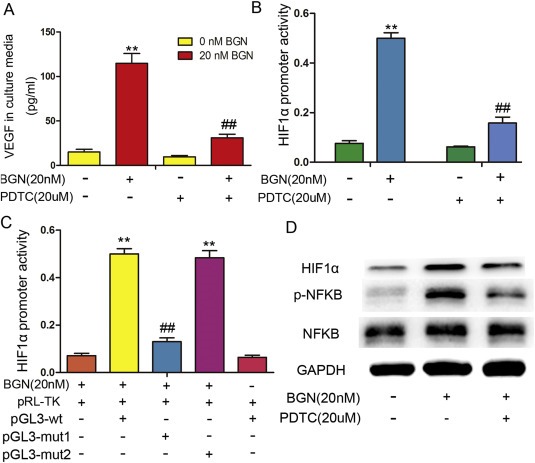
rBGN stimulates HIF‐1α activity and expression through NF‐kB. (A) VEGF levels in HUVEC culture media were determined via ELISA after cells were stimulated with or without PDTC (or rBGN). (B) HIF‐1α promoter activity was measured in HUVECs that were transfected with a HIF‐1α promoter reporter plasmid (pGL3‐HIF‐1α) in the absence or presence of PDTC (or rBGN). (C) A reporter plasmid for HIF‐1α (pGL3‐HIF‐1α) was generated by cloning the HIF‐1α promoter region (wt) or its NF‐kB binding‐site mutants (mut1and mut2) into the pGL3‐basic vector. rBGN significantly increased the luciferase activity of the HIF‐1α promoter region (wt), while activity was significantly reduced when using mut1 to replace the wt sequence. Mut1 may contain a HIF‐1α‐binding site. (D) Western blotting analysis of NF‐kB, phosphorylated NF‐kB and its downstream protein, HIF‐1α, in HUVECs after cells were stimulated with or without PDTC (or rBGN). GAPDH was used as a loading control. Each value is the mean ± SD of three experiments. ** Represents P ≤ 0.01 when compared to untreated control levels; ## represents P ≤ 0.01 when compared to stimulated control values.
3.4. rBGN promotes tube formation, migration and proliferation through the TLR signaling pathway in endothelial cells
In our previous studies, we showed that BGN stimulated tubular formation activity in HUVECs (Hu et al., 2014). To test whether this function was dependent upon TLR2, TLR4 and its downstream signaling molecule, NF‐kB. HUVECs were incubated with antibodies to neutralize TLR2/4 or were treated with PDTC/shHIF‐1α prior to the addition of rBGN. The inhibition of TLR2/4 or NF‐kB/shHIF‐1α resulted in a decrease in tube formation in endothelia cells (Figure 4A–F). rBGN promoted the migration of HUVECs, but this effect was also limited by TLR2/4 antibodies and PDTC/shHIF‐1α (Figure 5A–D). To explore the potential role of BGN in endothelial cell proliferation, we stimulated HUVECs with rBGN. Then, we utilized a CCK8 analysis to evaluate the effect of BGN stimulation on cell proliferation. As indicated in Figure 5E and F, rBGN stimulation significantly promoted HUVECs growth. Additionally, the growth of endothelial cells incubated with TLR2/4 antibodies or treated with PDTC/shHIF‐1α was dramatically inhibited. These results suggest that BGN promotes endothelial cell tube formation, migration and proliferation through the TLR signaling pathway.
Figure 4.
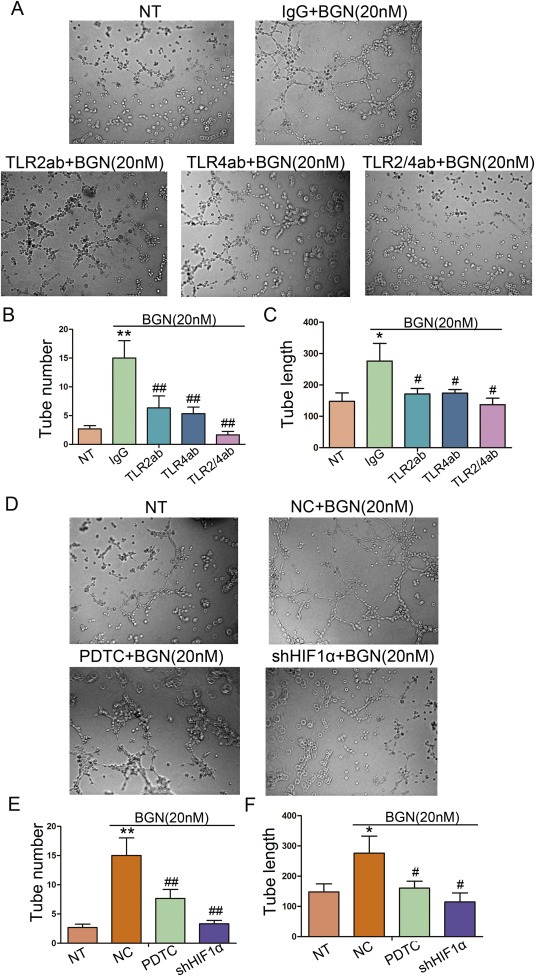
rBGN activates endothelial cell tube formation through the TLR signaling pathway. (A) Richly formed tubular structures were observed in the groups stimulated with rBGN when compared with the observations of the untreated group, whereas HUVECs formed less well‐formed tubular structures when incubated with antibodies to neutralize TLR2 and/or TLR4 receptor(s). (B–C) Bar charts show the number of tubules (B) and the mean tubular lengths (C) for the different groups, which were incubated with antibodies to neutralize TLR2 and/or TLR4 receptor(s). (D) Richly formed tubular structures were observed in the groups stimulated with rBGN when compared with the observations of the untreated group, whereas HUVECs formed less well‐formed tubular structures when treated with PDTC or vectors encoding for shHIF‐1α. (E–F) Bar charts show the number of tubules (E) and the mean tubular lengths (F) for the different groups, which were treated with PDTC or vectors encoding for shHIF‐1α. Each value is the mean ± SD of three experiments. * Represents P ≤ 0.05 when compared to untreated control groups; ** Represents P ≤ 0.01 when compared to untreated control groups. # represents P ≤ 0.05 when compared to stimulated control groups; ## represents P ≤ 0.01 when compared to stimulated control groups.
Figure 5.
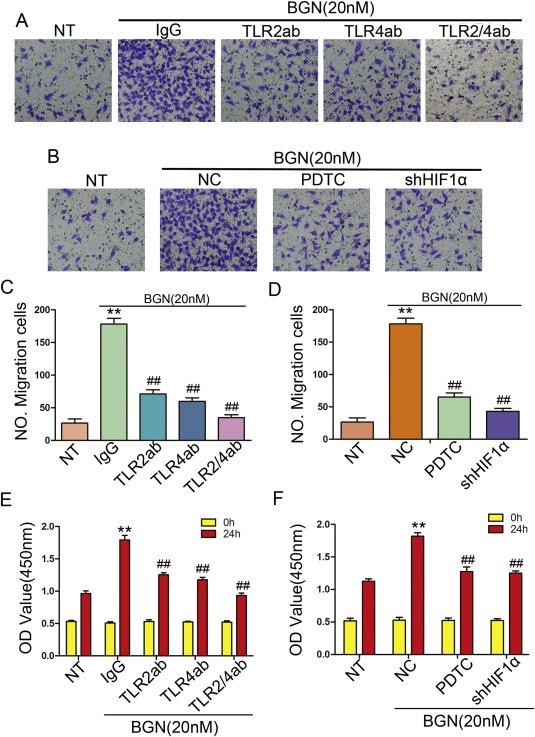
rBGN promotes endothelial cells migration and proliferation through the TLR signaling pathway. (A–B) Transwell assays. Photographs show HUVECs that traveled through the micropore‐membrane in the absence or presence of different types of stimulation. (C–D) Histograms show the numbers of migrating cells for the different groups. (E–F) Cell proliferation assay in HUVECs comparing the different groups. Each value is the mean ± SD of three experiments. ** Represents P ≤ 0.01 when compared to untreated control groups; ## represents P ≤ 0.01 when compared to stimulated control groups.
3.5. rBGN stimulates endothelial cell‐derived VEGF, promoting gastric cancer cell migration
To define the effect of the VEGF from endothelial cells induced by rBGN on the migration ability of GC cells, we established an in vitro transwell co‐culture system (Figure 6A). As shown in Figure 6B and C, HUVECs co‐cultured with the GC cell line NCI‐N87 promoted GC cell migration, but its function was abrogated by adding neutralized VEGF and/or a BGN antibody into the co‐culture system.
Figure 6.
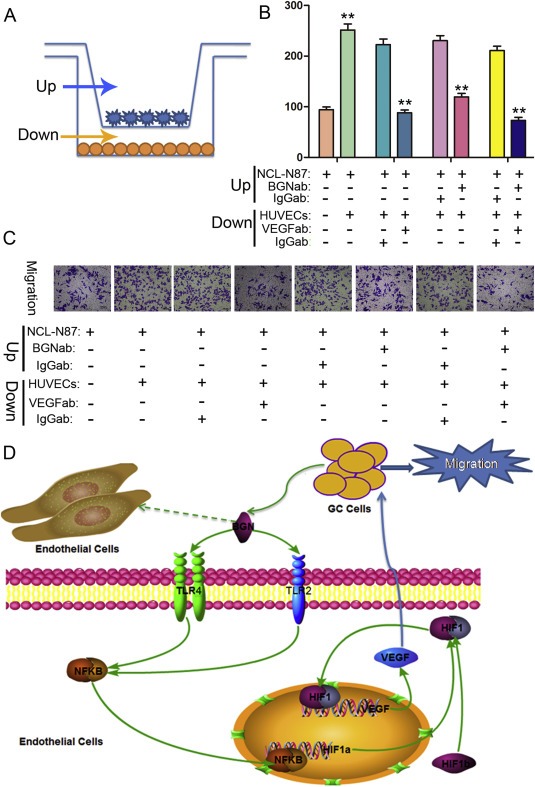
rBGN activates endothelial cells to promote GC cell migration through the secretion of VEGF. (A) In vitro transwell co‐culture system of endothelial cells and GC cells. (B) Histograms show the numbers of migrating GC cells in the presence of neutralizing antibody or the IgG isotype control antibody. (C) Representative photographs of migratory GC cells on the membrane are shown. (D) A schematic model is shown in which BGN secreted from GC cells interacts with TLR2 and TLR4 receptors expressed on the surface of endothelial cells. The interaction of BGN with TLR2/4 results in the activation of NF‐kB and the stimulation of HIF‐1α gene expression. HIF‐1α interacts with HIF‐1b to form HIF‐1. Then, HIF‐1 migrates into the nucleus and stimulates the expression of VEGF, which in turn stimulates the migration of GC cells. Each value is the mean ± SD of three experiments. ** Represents P ≤ 0.01 when compared to control groups.
Taking the results of our previous study together with the present results, BGN secreted by GC cells interacts with TLR2 and TLR4, which results in the activation of NF‐kB and the inducement of HIF‐1α gene expression. Then, HIF‐1α interacts with HIF‐1b to form HIF‐1, migrating into the nucleus to stimulate the expression of VEGF, which finally stimulates tube formation, migration and proliferation in endothelial cells. In addition, endothelial cell‐derived VEGF in turn acts on GC cells and promotes the migration of GC cells (Figure 6D).
4. Discussion
Tumor tissue consists of tumor parenchyma and stroma. Tumor parenchyma cells are the main component of a tumor and determine its biological characteristics and tumor specificity. The stroma section contains lymphocytes, myofibroblasts, and endothelial cells. Endothelial cells that make up blood vessels that provide nutrients to the tumor. Tumor stromal cells interactions with other tumor cells together determine the biological behavior of a tumor (growth, invasion and metastasis) (Tlsty and Coussens, 2006; Zigrino et al., 2005), and stromal cells also actively express extracellular matrix (ECM) components. The extracellular matrix (ECM) is a complex of proteins that plays an integral role in tissue repair and calcification, embryo development, cell migration and proliferation, and cancer development (Martinez and Araujo, 2004; Sommer et al., 1996; Zhao et al., 1999). BGN is a component of the ECM and is considered to be a new mesenchymal marker for the epithelial–mesenchymal transition (EMT) (Ellenrieder et al., 2001), which is the process by which malignant cancer cells obtained migratory and invasive abilities (Biddle and Mackenzie, 2012). In our previous study, we found that BGN expression in GC tissues was significantly upregulated compared with its expression in adjacent normal gastric tissues, which may represent an increase in the EMT in GC cells.
We have shown that culture media from BGN stably transfected GC cells can stimulate tubular formation in HUVECs (Hu et al., 2014), but the direct influence of BGN secreted from GC cells on endothelial cells is not clear. In this study, we demonstrate that BGN binds to its receptors TLR2/4 and induces VEGF expression by endothelial cells in normoxic settings via the activation of NF‐kB and HIF‐1α. The tubule formation, migratory and proliferative capacities of cells were promoted by direct rBGN stimulation. VEGF secreted from endothelial cells can promote GC cell migration. The role of VEGF cannot be ignored in tumor growth and metastasis, and the abnormal expression of VEGF has been found in many types of tumors, such as colon cancer (Tokunaga et al., 1998), breast cancer (Skobe et al., 2001), lung cancer (Salven et al., 1998) and hepatic carcinomas (Moon et al., 2003). It has clearly been shown that VEGF expression was also upregulated in GC, and high expression levels of VEGF are associated with GC metastasis and growth (Kakeji et al., 2002; Yonemura et al., 1999; Zheng et al., 2006). VEGF expression is mostly elevated under hypoxic conditions as a result of increased transcriptional activity mediated by the transcription factor HIF‐1 (hypoxia inducible factor‐1) (Pugh and Ratcliffe, 2003). Studies have shown that HIF‐1 can also be activated under normoxic conditions, stimulating VEGF expression (Riddell et al., 2012; Shi et al., 2005). HIF‐1 comprises of two subunits, a constitutively expressed β (HIF‐1β) subunit and an inducible labile α (HIF‐1α) subunit. It has been demonstrated that pro‐inflammatory cytokines and TLR signaling can induce HIF‐1α expression and increase HIF‐1α stability under normoxic conditions (Imtiyaz and Simon, 2010). As an important inflammatory factor, NF‐kB has also been found to be abnormally expressed in many types of cancer and plays a key role in cancer development and progression (Dolcet et al., 2005; Karin, 2006; Wu et al., 2005). NF‐kB is also a transcription factor that can promote the expression of specific genes, such as HIF‐1α. NF‐kB is one of the core components in TLR signaling, which is involved in the inflammatory and immune responses of our bodies. The results from this study show that rBGN stimulates VEGF expression in endothelial cells via the TLR2/4‐dependent activation of HIF‐1α. BGN is highly expressed in GC cells and is secreted into the stroma. Subsequently, BGN acts on endothelial cells, causing the expression of VEGF. Finally, VEGF secreted by endothelial cells can in turn act on GC cells, affecting their development and progression. This proves once again the interactions between the whole tumor and the stroma can influence tumor development and progression.
In conclusion, we found that BGN, an endogenous ligand for TLR2/4 that is secreted by GC cells, induces VEGF expression in endothelial cells via the NF‐kB‐dependent activation of HIF‐1α. BGN promotes tube formation, migration and proliferation in endothelial cells through interactions with TLR2/4 and the activation of a downstream signaling pathway. These results suggest a mechanism by which BGN induces angiogenesis in GC and suggests a scenario of continuous interactions between the tumor parenchyma and stroma in GC.
Conflict of interests
We have no potential conflicts of interest to declare.
Acknowledgments
This study was supported by grants from National Natural Science foundation of China (No. 91529302, No. 81572798, and No. 81272749), Key Projects in the National Science & Technology Pillar Program of China (No. 2014BAI09B03).
Hu Lei, Zang Ming-de, Wang He-xiao, Li Jian-fang, Su Li-ping, Yan Min, Li Chen, Yang Qiu-meng, Liu Bing-ya, Zhu Zheng-gang, (2016), Biglycan stimulates VEGF expression in endothelial cells by activating the TLR signaling pathway, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.08.002.
Contributor Information
Bing-ya Liu, Email: liubingya@sjtu.edu.cn.
Zheng-gang Zhu, Email: zzg1954@hotmail.com.
References
- Biddle, A. , Mackenzie, I.C. , 2012. Cancer stem cells and EMT in carcinoma. Cancer Metastasis Rev. 31, 285–293. [DOI] [PubMed] [Google Scholar]
- Boehmer, E.D. , Meehan, M.J. , Cutro, B.T. , 2005. Aging negatively skews macrophage TLR2- and TLR4-mediated pro-inflammatory responses without affecting the IL-2-stimulated pathway. Mech. Ageing Dev. 126, 1305–1313. [DOI] [PubMed] [Google Scholar]
- Dolcet, X. , Llobet, D. , Pallares, J. , 2005. NF-kB in development and progression of human cancer. Virchows Arch. 446, 475–482. [DOI] [PubMed] [Google Scholar]
- Ellenrieder, V. , Hendler, S.F. , Boeck, W. , 2001. Transforming growth factor beta1 treatment leads to an epithelial-mesenchymal transdifferentiation of pancreatic cancer cells requiring extracellular signal-regulated kinase 2 activation. Cancer Res. 61, 4222–4228. [PubMed] [Google Scholar]
- Horemans, T. , Boulet, G. , van Kerckhoven, M. , 2016. In-vivo evaluation of apocynin for prevention of Helicobacter pylori-induced gastric carcinogenesis. Eur. J. Cancer Prev. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Hsieh, L.T. , Nastase, M.V. , Zeng-Brouwers, J. , 2014. Soluble biglycan as a biomarker of inflammatory renal diseases. Int. J. Biochem. Cell Biol. 54, 223–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, L. , Duan, Y.T. , Li, J.F. , 2014. Biglycan enhances gastric cancer invasion by activating FAK signaling pathway. Oncotarget. 5, 1885–1896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imtiyaz, H.Z. , Simon, M.C. , 2010. Hypoxia-inducible factors as essential regulators of inflammation. Curr. Top. Microbiol. Immunol. 345, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, A.C. , Heinzel, F.P. , Diaconu, E. , 2005. Activation of toll-like receptor (TLR)2, TLR4, and TLR9 in the mammalian cornea induces MyD88-dependent corneal inflammation. Invest. Ophthalmol. Vis. Sci. 46, 589–595. [DOI] [PubMed] [Google Scholar]
- Kakeji, Y. , Koga, T. , Sumiyoshi, Y. , 2002. Clinical significance of vascular endothelial growth factor expression in gastric cancer. J. Exp. Clin. Cancer Res. 21, 125–129. [PubMed] [Google Scholar]
- Karin, M. , 2006. Nuclear factor-kappaB in cancer development and progression. Nature. 441, 431–436. [DOI] [PubMed] [Google Scholar]
- Kazemi, E. , Kahrizi, D. , Moradi, M.T. , 2016. Gastric cancer and helicobacter pylori: impact of hopQII gene. Cell. Mol. Biol. (Noisy-le-grand). 62, 107–110. [PubMed] [Google Scholar]
- Liang, X. , Zeng, J. , Wang, L. , 2014. Histone demethylase RBP2 induced by helicobactor pylori CagA participates in the malignant transformation of gastric epithelial cells. Oncotarget. 5, 5798–5807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani, A. , Allavena, P. , Sica, A. , 2008. Cancer-related inflammation. Nature. 454, 436–444. [DOI] [PubMed] [Google Scholar]
- Martinez, E.F. , Araujo, V.C. , 2004. In vitro immunoexpression of extracellular matrix proteins in dental pulpal and gingival human fibroblasts. Int. Endod. J. 37, 749–755. [DOI] [PubMed] [Google Scholar]
- Mikula, M. , Rubel, T. , Karczmarski, J. , 2010. Integrating proteomic and transcriptomic high-throughput surveys for search of new biomarkers of colon tumors. Funct. Integr. Genomics. 11, 215–224. [DOI] [PubMed] [Google Scholar]
- Moon, W.S. , Rhyu, K.H. , Kang, M.J. , 2003. Overexpression of VEGF and angiopoietin 2: a key to high vascularity of hepatocellular carcinoma?. Mod. Pathol. 16, 552–557. [DOI] [PubMed] [Google Scholar]
- Moreth, K. , Frey, H. , Hubo, M. , 2014. Biglycan-triggered TLR-2- and TLR-4-signaling exacerbates the pathophysiology of ischemic acute kidney injury. Matrix Biol. 35, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazuelo, M.B. , Correa, P. , 2013. Gastric cancer: overview. Colomb. Med. (Cali). 44, 192–201. [PMC free article] [PubMed] [Google Scholar]
- Pugh, C.W. , Ratcliffe, P.J. , 2003. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat. Med. 9, 677–684. [DOI] [PubMed] [Google Scholar]
- Riddell, J.R. , Maier, P. , Sass, S.N. , 2012. Peroxiredoxin 1 stimulates endothelial cell expression of VEGF via TLR4 dependent activation of HIF-1alpha. PLoS One. 7, e50394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salven, P. , Ruotsalainen, T. , Mattson, K. , 1998. High pre-treatment serum level of vascular endothelial growth factor (VEGF) is associated with poor outcome in small-cell lung cancer. Int. J. Cancer. 79, 144–146. [DOI] [PubMed] [Google Scholar]
- Schaefer, L. , Babelova, A. , Kiss, E. , 2005. The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages. J. Clin. Invest. 115, 2223–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, Y.H. , Wang, Y.X. , Bingle, L. , 2005. In vitro study of HIF-1 activation and VEGF release by bFGF in the T47D breast cancer cell line under normoxic conditions: involvement of PI-3K/Akt and MEK1/ERK pathways. J. Pathol. 205, 530–536. [DOI] [PubMed] [Google Scholar]
- Skobe, M. , Hawighorst, T. , Jackson, D.G. , 2001. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat. Med. 7, 192–198. [DOI] [PubMed] [Google Scholar]
- Sommer, B. , Bickel, M. , Hofstetter, W. , 1996. Expression of matrix proteins during the development of mineralized tissues. Bone. 19, 371–380. [DOI] [PubMed] [Google Scholar]
- Tlsty, T.D. , Coussens, L.M. , 2006. Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 1, 119–150. [DOI] [PubMed] [Google Scholar]
- Tokunaga, T. , Oshika, Y. , Abe, Y. , 1998. Vascular endothelial growth factor (VEGF) mRNA isoform expression pattern is correlated with liver metastasis and poor prognosis in colon cancer. Br. J. Cancer. 77, 998–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber, C.K. , Sommer, G. , Michl, P. , 2001. Biglycan is overexpressed in pancreatic cancer and induces G1-arrest in pancreatic cancer cell lines. Gastroenterology. 121, 657–667. [DOI] [PubMed] [Google Scholar]
- Wu, C.Y. , Wang, C.J. , Tseng, C.C. , 2005. Helicobacter pylori promote gastric cancer cells invasion through a NF-kappaB and COX-2-mediated pathway. World J. Gastroenterol. 11, 3197–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, C. , Ghosh, S. , 2005. NF-kappaB, an evolutionarily conserved mediator of immune and inflammatory responses. Adv. Exp. Med. Biol. 560, 41–45. [DOI] [PubMed] [Google Scholar]
- Xing, X. , Gu, X. , Ma, T. , 2015. Biglycan up-regulated vascular endothelial growth factor (VEGF) expression and promoted angiogenesis in colon cancer. Tumour Biol. 36, 1773–1780. [DOI] [PubMed] [Google Scholar]
- Yamamoto, K. , Ohga, N. , Hida, Y. , 2012. Biglycan is a specific marker and an autocrine angiogenic factor of tumour endothelial cells. Br. J. Cancer. 106, 1214–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yonemura, Y. , Endo, Y. , Fujita, H. , 1999. Role of vascular endothelial growth factor C expression in the development of lymph node metastasis in gastric cancer. Clin. Cancer Res. 5, 1823–1829. [PubMed] [Google Scholar]
- Zeng-Brouwers, J. , Beckmann, J. , Nastase, M.V. , 2014. De novo expression of circulating biglycan evokes an innate inflammatory tissue response via MyD88/TRIF pathways. Matrix Biol. 35, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B.G. , Hu, L. , Zang, M.D. , 2016. Helicobacter pylori CagA induces tumor suppressor gene hypermethylation by upregulating DNMT1 via AKT-NFkappaB pathway in gastric cancer development. Oncotarget. 7, 9788–9800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, M. , Lu, Y. , Takata, T. , 1999. Immunohistochemical and histochemical characterization of the mucosubstances of odontogenic myxoma: histogenesis and differential diagnosis. Pathol. Res. Pract. 195, 391–397. [DOI] [PubMed] [Google Scholar]
- Zheng, H. , Takahashi, H. , Murai, Y. , 2006. Expressions of MMP-2, MMP-9 and VEGF are closely linked to growth, invasion, metastasis and angiogenesis of gastric carcinoma. Anticancer Res. 26, 3579–3583. [PubMed] [Google Scholar]
- Zhu, Y.H. , Yang, F. , Zhang, S.S. , 2013. High expression of biglycan is associated with poor prognosis in patients with esophageal squamous cell carcinoma. Int. J. Clin. Exp. Pathol. 6, 2497–2505. [PMC free article] [PubMed] [Google Scholar]
- Zigrino, P. , Loffek, S. , Mauch, C. , 2005. Tumor-stroma interactions: their role in the control of tumor cell invasion. Biochimie. 87, 321–328. [DOI] [PubMed] [Google Scholar]