

Abstract
Background
In the pediatric cancer neuroblastoma (NB), patients are stratified into low, intermediate or high‐risk subsets based in part on MYCN amplification status. While MYCN amplification in general predicts unfavorable outcome, no clinical or genomic factors have been identified that predict outcome within these cohorts of high‐risk patients. In particular, it is currently not possible at diagnosis to determine which high‐risk neuroblastoma patients will ultimately fail upfront therapy.
Experimental design
We analyzed the prognostic potential of most published gene expression signatures for NB and developed a new prognostic signature to predict outcome for patients with MYCN amplification. Network and pathway analyses identified candidate therapeutic targets for this MYCN‐amplified patient subset with poor outcome.
Results
Most signatures have a high capacity to predict outcome of unselected NB patients. However, the majority of published signatures, as well as most randomly generated signatures, are highly confounded by MYCN amplification, and fail to predict outcome in subpopulations of high‐risk patients with MYCN‐amplified NB. We identify a MYCN module signature that predicts patient outcome for those with MYCN‐amplified tumors, that also predicts potential tractable therapeutic signaling pathways and targets including the DNA repair enzyme Poly [ADP‐ribose] polymerase 1 (PARP1).
Conclusion
Many prognostic signatures for NB are confounded by MYCN amplification and fail to predict outcome for the subset of high‐risk patients with MYCN amplification. We report a MYCN module signature that is associated with distinct patient outcomes, and predicts candidate therapeutic targets in DNA repair pathways, including PARP1 in MYCN‐amplified NB.
Keywords: Neuroblastoma, Gene signatures, Networks, Biomarkers, Confounding variables, MYCN
Highlights
Long‐term survival for patients with high‐risk neuroblastoma is <40%.
Almost ½ of high risk patients have tumors that harbor MYCN amplification (MYCNA).
Published neuroblastoma gene signatures are not prognostic for MYCNA subsets.
Novel signature identified to predict outcome for MYCNA patients.
MYCNA signature module identifies PARP‐1 as a therapeutic target.
1. Introduction
Neuroblastoma (NB), the most common pediatric extra‐cranial solid tumor and most frequent neoplasm of infancy (Irwin and Park, 2015; Maris, 2010), has a diverse pattern of clinical presentation that reflects its biological heterogeneity. The outcome of NB patients is highly variable, and clinical and biological risk factors, most commonly age, stage, histology, ploidy, segmental chromosome aberrations, and MYCN amplification (MNA), are used to stratify patients and tailor therapy (Cohn et al., 2009; Pinto et al., 2015; Brodeur et al., 1993). While survival rates for low and intermediate‐risk NB are >90%, more than half of patients present with high‐risk disease. Despite intensive multi‐modality therapies, survival for high‐risk NB is <50%, making it the most frequent cause of pediatric cancer‐related death. The majority of high‐risk NB include patients >18 months of age with metastatic disease, or patients of any age with loco‐regional or metastatic tumors that harbor MNA (Irwin and Park, 2015).
MNA, detected in approximately 20% of all NB tumors and 40% of those with high‐risk disease, is one of the strongest independent adverse prognostic factors (Brodeur et al., 1984). However, recent studies demonstrate divergent outcomes among populations of patients with MNA (Kushner et al., 2014). MNA patients either have an excellent response to induction chemotherapy that is often associated with long‐term survival, or develop early progressive disease with an unfavorable outcome. These observations suggest that molecular or genetic factors at diagnosis may discriminate patients with MNA who rapidly progress, from those who will be long‐term survivors. Many other molecular and pathologic factors have been identified that are associated with unfavorable outcome in NB, including segmental chromosomal aberrations such as 17q gain (Bown et al., 1999; Meddeb et al., 1996), 1p loss (Attiyeh et al., 2005; Caron et al., 1996), and 11q loss (Schleiermacher et al., 1996, 1999, 2010, 2012), diploid/tetraploid status (Ladenstein et al., 2001), and specific genetic alterations of ALK (Bresler et al., 2014) and TERT (Peifer et al., 2015). However, these genomic features and other well‐established clinical prognostic variables are highly correlated with MNA status (Thompson et al., 2016) and thus cannot be used to effectively predict outcome and potentially tailor therapy within this unfavorable prognosis subset of MNA patients. Therefore, alternative strategies are required to identify genes and pathways that are altered in subsets of MNA tumors to potentially uncover novel prognostic factors and therapeutic targets.
Recently, high‐throughput profiling techniques have enabled the identification of transcriptomic‐based predictors, commonly known as gene signatures. Signatures with reported predictive power in NB have been identified using a variety of techniques including manually curated gene sets that represent specific biological processes (Asgharzadeh et al., 2012), experimental perturbations to modulate gene expression (e.g., MYCN knockdown) (Barbieri et al., 2012, 2010, 2009, 2014, 2009, 2012, 2013), subtype analysis (Abel et al., 2011), or comparison of gene expression profiles derived from patients previously classified as high and low‐risk NB patients (Vermeulen et al., 2006, 2010, 2006, 2012, 2006, 2005, 2009, 2005, 2007, 2009, 2013, 2004). Reported signatures vary from 3 to 158 genes, comprise relatively few overlapping genes, and are associated with a diverse array of biological processes including differentiation, hypoxia, therapy resistance, migration/invasion, inflammation, and DNA repair.
The major objective of developing multi‐gene signatures is to build clinical tools to accurately predict outcome for NB patients and more precisely tailor treatment strategies. However, few signatures have been implemented clinically even though several signatures have undergone validation using clinically amenable technology platforms, such as RT‐PCR (Garcia et al., 2012) and the nanoString based technology nCounter® (Stricker et al., 2014). A current Children's Oncology Group (COG) pilot study is testing the feasibility of prospectively assessing RNA expression signatures using one of the most well‐validated signatures for the subset of high‐risk NB patients that lack MNA (Asgharzadeh et al., 2006, 2012). The majority of other NB signatures have been validated on large cohorts of NB patients that include both high‐risk and non‐high‐risk NB patients. To increase our understanding of the predictive potential and clinical utility of current NB gene signatures, and in particular for the high‐risk subset of patients with MNA, we analyzed the prognostic capacity of all reported prognostic signatures for NB. We find that although current signatures have a high capacity to predict outcome of unselected NB patients, randomly generated signatures are also highly associated with outcome in these patients. Both reported and randomly generated signatures are highly confounded by MNA, and no signature predicts outcome within the subset of patients with MNA tumors. To address this limitation, we identify a network‐based signature that specifically predicts outcome of MNA NB. Furthermore, the pathways identified using this signature in the unfavorable outcome MNA patients revealed upregulation of DNA damage‐related targets such as poly‐ADP ribose polymerase (PARP1), which plays a critical role in base excision repair. We further find that PARP1 inhibitors, which have shown efficacy in early phase clinical trials in tumors with BRCA1/2 mutations and other DNA‐repair defects (Kaufman et al., 2015; Mateo et al., 2015), more potently suppress the growth of MNA than non‐MYCN amplified (NA) NB cells, suggesting that PARP1 inhibition is a candidate therapy for this unfavorable subset of MNA tumors.
2. Methods
2.1. Patients and samples
All data was publically available and downloaded from either ArrayExpress (https://www.ebi.ac.uk/arrayexpress/, E‐TABM‐38, E‐MTAB‐179, E‐MTAB‐16), or the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/, GSE3446, GSE49710). For custom arrays, data was pre‐processed as described by the contributor, or raw data was pre‐processed using RMA (Irizarry et al., 2003). Gene level expression data was obtained by collapsing probe set IDs by Gene ID, based on maximum average expression across the entire dataset. Validation sets for the MYCN signature were combined through merging by Gene ID and removing batch effects using the ComBat algorithm (Johnson et al., 2007).
2.2. Signature evaluation
Signature genes were taken from relevant publications (ST1). To evaluate the capacity of a signature to stratify NB patients into low & high molecular risk groups we completed k‐means clustering (k = 2) based on signature gene expression and Cox‐regression to evaluate the differences in survival between patient subgroups, using the CoxPH package in R. p‐value scores were calculated as the −log(p‐value) based on the p‐value obtained using the Wald test.
2.3. Confounding analysis
We based our confounding analysis on a previous report that described the implementation of the Systematic MisPrediction (SMP) test to identify confounding factors in breast cancer (Tofigh et al., 2014). In short, this test identifies confounding variables by testing for a relationship between the proportion of times that a sample was correctly predicted to be high or low molecular risk across the NBsigDB set with clinical variables (age, MYCN amplification, stage, and gender). For binary variables (MYCN amplification, gender) we used the Wilcoxon rank sum test, otherwise we used Spearman's rank correlation.
2.4. Network analysis
MYCN modules identification was implemented using the Cytoscape Reactome FI plug‐in (Wu and Stein, 2012). Briefly, outcome associated genes were mapped to nodes in Reactome (Wu et al., 2010). Weights were assigned to edges connecting interacting nodes based on the absolute value of the Pearson correlation co‐efficient of expression. Markov Clustering (MCL) network clustering was implemented to identify network modules, and we selected modules comprising at least 5 nodes with average Pearson correlation of at least 0.25.
2.5. Network index and signature score calculation
We calculated the module indices as the geometric mean (of Log2 expression) of the module genes, similar to previous reports (Hallett et al., 2012, 2012). This occurred as follows:
where x is the log2 expression, n is the number of probe sets, and P is the set of genes comprising the module.
2.6. In vitro cell culture, apoptosis assays, cell viability experiments
SK‐N‐AS, IMR‐32, SK‐N‐Be2(c), and SK‐N‐DZ NB cell lines were purchased from ATCC. SHEP, LAN5, and CHLA‐20 cell lines were kind gifts from Dr. Patrick Reynolds (Texas Tech University, Children's Oncology Group (COG) cell bank). BYK204165 was purchased from Tocris (Cat. 3734). For cell viability experiments, cells were treated with BYK204165 24‐h post‐seeding and cell growth was assessed by alamarBlue assay (Invitrogen) according to the manufacturer's instructions. For apoptosis experiments, cells treated with the indicated concentrations of BYK204165 and time‐points were harvested, lysed, and subjected to immunoblot analysis as previously described (Seong et al., 2015). Antibodies used included Cl‐PARP (#5625), Cl‐Caspase 3 (#9661) from Cell Signaling, MYCN (Millipore, CBL431), Vinculin (Upstate, 05‐386), and β‐actin (Sigma, A5316).
2.7. Statistical analysis
Cox‐regression, correlation, Wilcoxon rank sum and Fisher exact tests were completed in R. Kaplan–Meier analysis was completed using GraphPad Prism™ 5. p‐values less than 0.05 were taken to indicate significance.
3. Results
3.1. Outcome prediction is confounded in unstratified NB
We assembled all published gene signatures that were reported to have prognostic capacity in NB, that we collectively refer to as NB Signature database (NBsigDB) (n = 23, Supplementary Table 1 [ST1]). Together, the NBsigDB signatures comprise 627 unique genes, of which 70 appear in at least two independent signatures. Based on these data, we created two additional signatures, called MASTER and CORE, which comprise all of the unique genes present in NBsigDB and the 70 genes present in at least 2 NBsigDB signatures, respectively. Network and pathway analysis of the MASTER signature revealed that the genes comprising NBsigDB collectively interact in 9 modules which are enriched in a diverse array of biological processes (Supplementary Figure 1 [SF1, ST2]), many of which have previously been linked with NB outcome. Using a standardized technique based on k‐means clustering and Cox‐regression, each signature comprising NBsigDB was evaluated for its capacity to assign high and low molecular risk to patients that had either died from disease or survived during clinical follow‐up, from the E‐MTAB‐179 cohort (n = 477) (Oberthuer et al., 2006). Importantly, patients comprising the E‐MTAB‐179 test cohort have not been used to develop any of the signatures developed in NBsigDB, and provide an independent and relatively large group of NB patients in which to compare the relative accuracies of each signature. Based on p‐value score, represented as the negative log of the p‐value calculated from the Wald test, every signature was highly predictive of NB patient outcome (Figure 1A). Overall, the CORE signature provided the best capacity to predict patient outcome.
Figure 1.
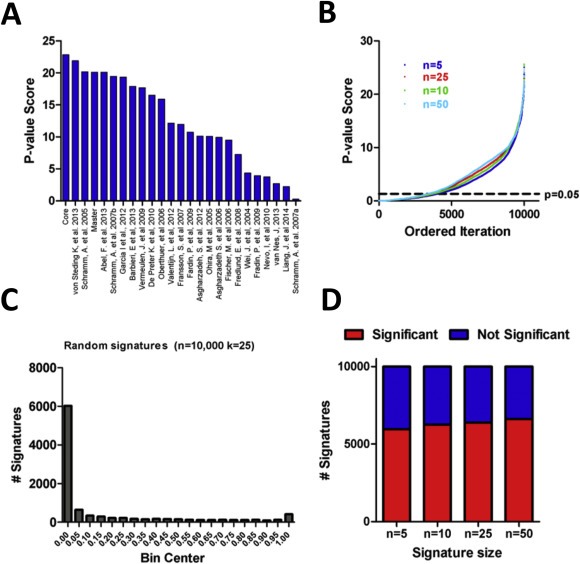
Assessment of NBsigDB and randomly generated signatures. A) P‐value scores of the signatures comprising NBsigDB, signatures are ranked in order of significance. B) p‐value scores for 10,000 randomly generated for k‐length signatures (k = 5, 10, 25, 50). C) Histogram of p‐values for 10,000 randomly generated 25 gene signatures. D) Proportion of significant and non‐significant signatures among 5‐, 10‐, 25‐, and 50‐gene signatures is similar among each tested signature size.
Previous work, mainly in breast cancer, has demonstrated that randomly selected genes/gene signatures can stratify patients on the basis of outcome (Tofigh et al., 2014; Venet et al., 2011). To investigate whether this also occurs in NB, we used a similar approach to assess the prognostic capacity of randomly generated gene signatures. Surprisingly, the majority of randomly generated signatures were significantly associated with outcome independent to the number of genes comprising the signature (Figure 1B, 10,000 iterations/n). For example, 6394 of 10,000 randomly generated 25‐gene signatures were significantly associated with patient outcome as assessed using Cox‐regression and a p‐value cut‐off of 0.05 (Figure 1C). Similar frequencies of randomly generated signatures significantly associated with outcome were observed for all signature sizes (Figure 1D). Interestingly, only 11 of 25 NBsigDB signatures were more accurate than 95% of the randomly generated signatures.
3.2. MYCN status confounds prognostic signatures in unselected pan‐NB populations
In breast cancer, the unexpected capacity of randomly selected signatures to predict outcome is attributed to the systematic confounding of prognostic signatures by either biological processes or clinico‐pathological variables that are also linked with outcome (Tofigh et al., 2014). To explore this possibility in NB, we examined the prediction accuracy of the 11 NBsigDB signatures that out‐performed 95% of the randomly selected signatures. Strikingly, these signatures displayed a remarkable degree of similarity in their predictions (Pearson r = 0.91, p < 0.0001), indicating high concordance between a signature's capacity to either correctly (Figure 2A) or incorrectly (Figure 2A, boxes i & ii), predict high or low molecular risk NB. Using the Systematic MisPrediction (SMP) test (Tofigh et al., 2014), we tested whether the difficulty of patient classification, calculated as the proportion of times a patient was correctly classified across the 11 NB signatures, was associated with standard clinical or biological variables that are known to be independently prognostic. Analysis using age, stage, gender, and MYCN status found that MNA consistently confounded the 11 NB signatures capacity to identify patients that either died from disease or experienced survival (ST3). The proportion of correct signature predictions among patients who survived was significantly higher in MYCN‐non‐amplified (NA) than MNA NB (Figure 3A). Conversely, among patients who died of disease, the proportion of correct predictions was significantly higher in MNA than NA tumors (Figure 3B). Overall, we found that the proportion of misclassified favorable outcome NB patients was highly enriched with MNA tumors, whereas among poor‐outcome patients, the misclassified NB patients were enriched in NA tumors (Figure 3C&D, p < 0.05, Fisher's exact test). We therefore concluded that the predictive capacity of the signatures is highly confounded by MNA.
Figure 2.
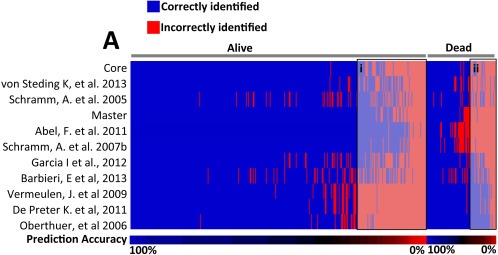
NBsigDB signatures identify a difficult to classify phenotype among NB patients. A) Blue and red indicate the accuracy of prediction. Patients are divided based on survival/death and ordered based on ease of prediction. Gray boxes highlight the patients that are generally miss‐classified by the NB signatures.
Figure 3.
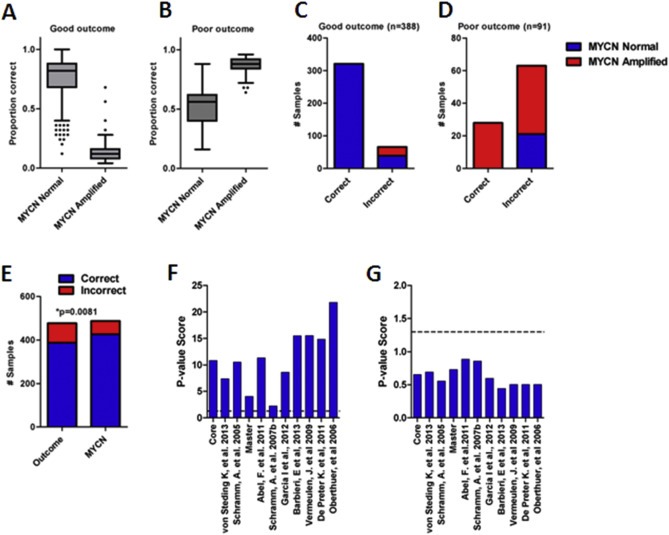
Identification of factors that confound NBsigDB signatures. A) Among good outcome patients MNA tumors are significantly more difficult to classify using the 11 NB signature panel (*p < 0.0001, Wilcoxon rank sum test). B) Among poor outcome patients, MNA tumors are significantly easier to classify using the 11 NB signature panel (*p < 0.0001, Wilcoxon rank sum test). C) Incorrectly classified good outcome tumors are enriched in MYCN amplified tumors (*p < 0.0001, Fisher's exact test). D) Correctly classified poor outcome tumors are enriched in MYCN amplified tumors (*p < 0.0001, Fisher's exact test). E) The CORE signatures is a better predictor of MYCN amplification than patient outcome (*p = 0.0081, Fisher's exact test). F) p‐value scores for NBsigDB signatures in NA NB. All signatures are significant (hashed line indicates p = 0.05). G) p‐value scores for NBsigDB signatures in MNA NB. None of the signatures are significant (hashed line indicates p = 0.05).
To examine why MNA confounds NBsigDB signatures, we further explored their relationship to MYCN status. Signatures that were most correlated with MYCN status generally also obtained the highest p‐value score based on Cox‐regression (Pearson r: 0.8, p < 0.0001), suggesting that much of the prognostic power of the signatures is derived from their capacity to identify MNA. Indeed, the capacity of CORE to predict MNA of NB tumors was superior to its capacity to predict patient outcome (Figure 3E, p = 0.0081, Fisher's exact test). Interestingly, this observation extended to a signature trained with data derived from NA patients (Asgharzadeh et al., 2006), as this signature was also highly correlated with MNA (p < 0.0001).
Finally, we examined the capacity of the 11 best NB signatures comprising NBsigDB to predict outcome in MNA and NA NB selected from the E‐MTAB‐179 cohort. Among NA samples, each of the 11 signatures was robustly associated with patient outcome (Figure 3F, p < 0.05). In contrast, not a single signature showed predictive capacity in the MNA patients (Figure 3G, lowest p‐value: 0.2). Taken with our previous data, these findings indicate that signatures comprising NBsigDB are generally only associated with NB patient outcome in the pan‐NB and NA NB cohorts. Furthermore, they highlight the need to identify prognostic signatures specifically for the MNA NB subset of patients, as at present, published signatures cannot readily distinguish between favorable and unfavorable prognosis NB within this subset.
3.3. Randomly generated signatures have a high probability of identifying MNA tumors
Given the surprising observation that most randomly generated signatures were predictive of NB patient outcome, we assessed whether they were also confounded by MNA. We randomly generated 10,000 k‐gene signatures (k = 5, 10, 25, 50), and examined the capacity of each signature to both predict outcome (Cox‐regression) or identify MNA tumors (Wilcoxon rank sum) (SF2). For each set of 10,000 n‐gene signatures, the predictive capacity of a signature was strongly linked to its capacity to identify MNA tumors (k = 5: r = 0.79, k = 10: r = 0.80, k = 25: r = 0.82, k = 50: r = 0.84). Hence, independent of signature size, the capacity of a random signature to predict NB outcome was strongly associated with its capacity to identify MNA tumors. We next tested what fraction of the NB transcriptome was associated with MNA. We found that transcript expression of some 60% of genes correlated with MYCN transcript expression in NB tumors (Pearson's correlation, p < 0.05), demonstrating that MNA expression has profound effects on the NB transcriptome. Based on these data, we concluded that the likelihood of randomly selecting a gene or gene set associated with MNA is high, and therefore also likely to be associated with patient outcome based on the robust link between MNA and poor outcome.
3.4. Identification of a prognostic signature for MNA NB
Given the limitations of previously reported signatures and our NBsigDB signatures in MNA NB, we sought to identify a prognostic gene signature specifically for this subgroup of NB. Briefly, we identified MNA tumors from GSE49710 (n = 93) with associated outcome data that identified patients with maximally divergent disease (experienced death within 1000 days of diagnosis or long‐term survival). To identify genes associated with outcome, we completed receiver‐operator characteristic (ROC) curve analyses across all genes. Using an AUC >0.7 as a cut‐off, we identified 474 outcome‐associated genes (Figure 4A). The 474 genes were mapped as nodes onto the human functional interaction network and clustered to identify candidate interaction modules associated with outcome (Wu et al., 2010; Wu and Stein, 2012). From this analysis, we identified 7 modules that each comprised at least 5 genes and displayed an average expression correlation of greater than 0.25 (based on expression), which were numbered 0–6 based on decreasing module size (Figure 4B, SF3, ST1). Based on the geometric mean of expression for the genes comprising each module, we calculated a module index and assessed the relationship to outcome. ROC analysis of the individual module indices demonstrated the capacity of each module to stratify NB patients into groups with maximally divergent outcome, and a combination index representing the mean of the individual modules indices was the nominally best predictor of patient outcome (Figure 4C).
Figure 4.
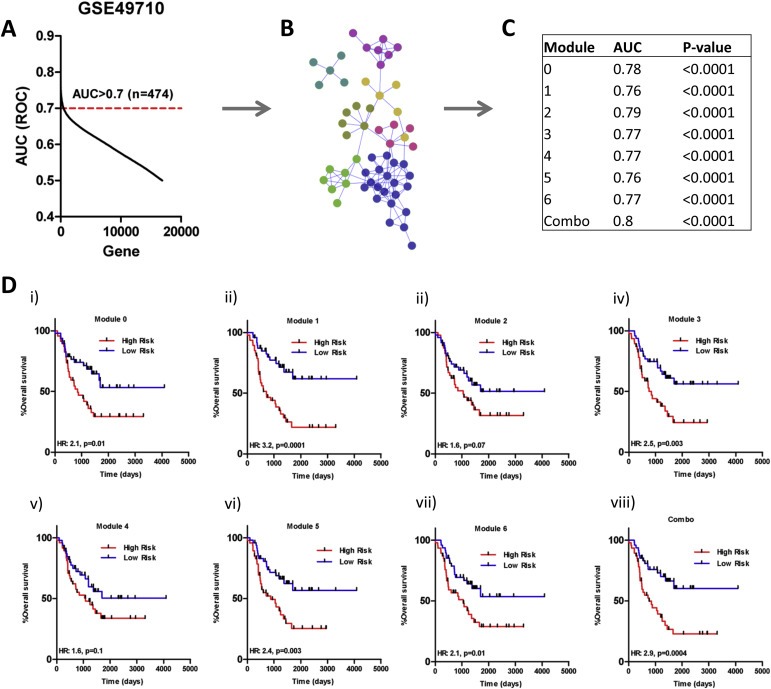
Identification of a module based signature that predicts outcome of MNA NB patients. A) AUC analysis to identify outcome associated genes in MNA NB (AUC > 0.7 used as cut‐off). B) Modules identified from network analysis of identified predictive genes. C) AUC values for each of the individual predictive modules, as well as a combination module. The combination module is the best predictor of outcome. D) Kaplan–Meier survival analysis of each of the MYCN modules in the validation cohort (i–viii: Modules 0–6, combination: *p = 0.01, p = 0.0001, p = 0.07, p = 0.003, p = 0.1, p = 0.003 p = 0.01, p = 0.0004, log‐rank test). High Risk: High molecular risk. Low Risk: Low molecular risk.
To confirm the predictive capacity of the modules, we identified a second cohort of 95 patients with MNA NB tumors by merging MNA NB patients from the E‐TABM‐179, E‐MTAB‐16, and E‐MTAB‐38 datasets, henceforth referred to as the validation set. Overlapping patients from these datasets identified on the basis of identical clinical annotations were removed. Univariate Cox‐regression analysis revealed a strong relationship between each module index and patient outcome. Within the validation set, each of the module indices was significantly associated with both OS and EFS (Table 1). As found within the training data, a combination index was nominally the best predictor of patient outcome (Table 1).
Table 1.
Prognostic capacity of the MYCN modules signature within the validation cohort.
| Module | Event‐free survival | Over‐free survival | ||
|---|---|---|---|---|
| HR | P | HR | P | |
| 0 | 1.7 | 0.0022 | 1.9 | 0.00025 |
| 1 | 1.7 | 0.00013 | 1.6 | 0.00022 |
| 2 | 1.8 | 2.40E‐06 | 1.6 | 1.02E‐05 |
| 3 | 1.9 | 0.00015 | 2.2 | 3.63E‐05 |
| 4 | 1.5 | 0.0019 | 1.4 | 0.0053 |
| 5 | 1.4 | 0.0024 | 1.4 | 0.0013 |
| 6 | 1.7 | 0.0014 | 1.7 | 0.00082 |
| Combo | 2 | 1.83E‐06 | 1.9 | 2.00E‐06 |
Survival curve analysis with validation patients, using the module indices to stratify MNA patients into molecular high and molecular low‐risk groups based on median module index value, revealed that with the exception of modules 2 and 4, each of the module indices stratified patients into subgroups with obviously disparate outcomes (Figure 4Di–viii). The combination index stratified patients into molecular high and low‐risk groups with the greatest difference in outcome (Figure 4Dviii, HR: 2.90, p = 0.0004). Whereas the Kaplan–Meier estimate for 2 year OS (730 days) among molecular high‐risk MNA patients was only 31%, it was 69% among molecular low‐risk MNA patients (Figure 4Dviii).
Finally, we also examined the predictive capacity of randomly generated signatures in MNA NB samples comprising the validation set. Initially, we attempted to identify modules using randomly selected sets of 474 genes, similar to how we identified the 7 MYCN modules using 474 outcome‐associated genes. However, this approach rarely identified more than a single module (<1%). As an alternative, we tested the capacity of 10,000 randomly generated signatures to predict MNA NB patient outcome, and found that none of the selected signatures outperformed the MYCN modules signatures (SF4). Accordingly, we concluded that the MYCN modules were significantly associated with the outcome of patients with MNA NB.
3.5. MNA NB modules are associated with distinct biological pathways and identify PARP1 as a therapeutic target
We performed pathway analyses on genes comprising each of the MYCN modules (ST4). Many of the identified pathways were not enriched in genes comprising the NBsigDB signature, suggesting that unique biological processes may regulate tumor progression and patient outcome for MNA NB. We observed a significant association between the MYCN modules and double stranded DNA repair pathways, including the Fanconi Anemia and BRCA1/2 pathways (Figure 5A). These findings are consistent with previous reports linking MYCN function with DNA repair in NB cell lines and tumors (Valentijn et al., 2012; Cole et al., 2011). Based on this data, we hypothesized that MNA NB cells might be susceptible to inhibitors of DNA repair. Recent studies have demonstrated that patients with ovarian or prostate tumors with genetic alterations in double strand break pathways are more sensitive to PARP1 inhibitors (Kaufman et al., 2015; Mateo et al., 2015). Thus, we asked whether these PARP1 inhibitors were selectively efficacious in MNA vs. NA NB cell lines. We assessed the capacity of the highly selective and potent PARP1 inhibitor BYK204165 to inhibit cell growth in a panel of cell lines with differing MYCN status (Figure 5B). We observed significantly lower IC50 values for the PARP1 inhibitor BYK204165 in NB cell lines with high MYCN expression as compared to those with low MYCN expression (Figure 5C), suggesting that MYCN amplification is associated with increased sensitivity to PARP1 inhibitors. Using LAN‐5 and SHEP cells as representative MNA and NA cell lines, respectively, we examined the expression of apoptotic markers in response to PARP1 inhibition. BYK204165 treatment was associated with dose‐dependent increase in the level of cleaved‐PARP and cleaved‐Caspase3, respectively (Figure 5D&E).
Figure 5.
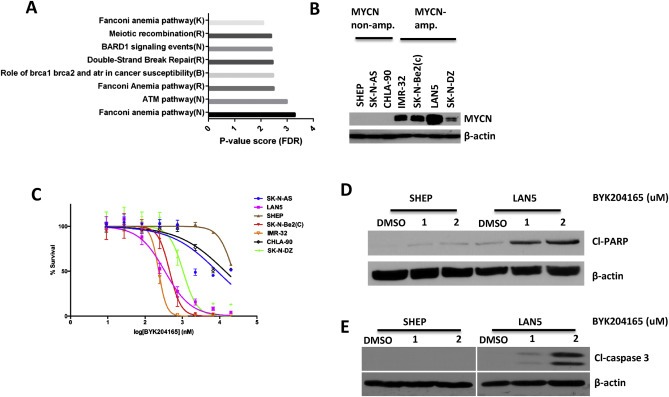
PARP1 inhibitors specifically target MNA NB cell lines. A) Pathway analysis of module 6 genes highlights enrichment in double stranded DNA repair pathways (R: Reactome, K: KEGG, N: NCI Pathways). B) Western blotting reveals high and low MNA NB cell lines. C) Dose response experiments reveal that the PARP1 inhibitor BYK204165 more potently suppresses the growth of MNA vs. NA NB cell lines. D&E) BYK204165 selectively induces the expression of the apoptotic markers cleaved‐PARP and cleaved‐Caspase3 (CASP3) in MNA NB cell lines.
In addition to hyperactivation of dsDNA repair pathways, we also observed enrichment of several signaling pathways for which potential therapeutic inhibitors are available. These included the TGFβ pathway enriched in module 3, and the RhoA, Insulin receptor (IGFR), and PDGFRβ signaling pathways enriched in module 4. Together, these observations suggest multiple potential therapeutic targets for MNA NB, and establish PARP1 inhibitors as candidate therapeutics for MNA NB.
4. Discussion
The outcome for NB patients is highly variable, and multiple clinical and biological risk factors are currently used to risk‐stratify patients and guide therapy. Whereas survival rates for low‐ and intermediate‐risk patients are excellent, half of patients present with high‐risk disease for which the survival rate is <50%. MNA is detected in approximately 20% of NB tumors and is among the most powerful adverse prognostic factor for NB. Recently, Kushner and colleagues used a large clinical database of patients treated since 2000 to identify a striking divergence in outcomes experienced by patients with MNA NB tumors (Kushner et al., 2014). Accordingly, even among high‐risk MNA NB, patients could benefit from stratification on the basis of additional molecular factors to identify those patients likely to rapidly progress from those who will be long‐term survivors. While many molecular and genetic factors have been reported that are associated with unfavorable outcome in NB, these factors and other well‐established clinical prognostic variables are highly correlated with MNA status (Thompson et al., 2016) and thus cannot be used to effectively predict outcome and potentially tailor therapy within this unfavorable prognosis subset of MNA patients. High‐throughput profiling techniques, such as microarrays and RNA‐seq, have also been used to identify molecular signatures that predict NB patient outcome. To date, more than 20 signatures have been reported, which collectively comprise relatively few overlapping genes, and that are associated with diverse biological processes and robustly associate with outcome in pan‐NB.
In this study, we assembled essentially all previously reported prognostic gene signatures in NB and evaluated their capacity to identify high‐ and low‐molecular risk NB. We found that the signatures comprising NBsigDB were robust and concordant predictors of patient outcome in pan‐NB and NA NB, and generally poor predictors of outcome in patients with MNA NB. Hence, currently described signatures are likely best suited to assign molecular risk for patients with NA NB. Using the SMP test, we found that NBsigDB signatures were highly confounded by MNA. Previous findings suggest that large numbers of transcripts correlate with MNA (Huang and Weiss, 2013; Valentijn et al., 2012). Our data suggest that NBsigDB confounding by MNA is likely the result of both a large fraction of transcripts being highly correlated with MYCN expression and the robust predictive capacity of MNA itself. We believe that these factors also underpin the relationship between randomly selected gene signatures and patient outcome, which to our knowledge has not been previously reported. These findings, coupled with reports that MNA NB patients can experience highly divergent outcome (Kushner et al., 2014), prompted us to test whether a gene signature could be identified with prognostic capacity in MNA NB. Our experiments provide proof‐of‐principle for the identification of a molecular risk signature within MNA NB, which we validated in an independent patient cohort. Overall, our findings are consistent with previous studies in breast cancer demonstrating that prognostic gene signatures are easily confounded by clinical variables and that randomly generated signatures can robustly associate with outcome (Tofigh et al., 2014; Venet et al., 2011). However, we subsequently show that the identification of confounding factors enables the identification of robustly predictive signatures in previously difficult to predict tumor subsets, in this case MNA NB.
MYCN was originally identified as a homolog of MYC that was amplified in human NB (Schwab et al., 1983; Brodeur et al., 1984). Structurally, MYCN and MYC are highly similar, and both proteins heterodimerize with MAX to promote transcription (Huang and Weiss, 2013). Although MYC is over‐expressed in a limited number of NB tumors (Slavc et al., 1990) and NA NB patients with tumors with positive immunostaining for MYC protein have a poor prognosis (Wang et al., 2015), MYCN has historically been considered the dominant driver of the family in NB. MYCN is reported to play many roles during NB tumorigenesis, including the regulation of signaling pathways involved in immune surveillance, stem‐cell like properties, proliferation, survival, angiogenesis and metastasis among others (reviewed in (Huang and Weiss, 2013)). Despite intense investigation and the evidence that MYCN is a driver of NB in multiple animal models (Weiss et al., 1997; Zhu et al., 2012), MYCN itself has remained challenging to target therapeutically. However, recent publications have identified potential vulnerabilities of MNA neuroblastoma, including to bromodomain inhibitors and combinations of bcl2 and aurora kinase inhibitors (Ham et al., 2016; Puissant et al., 2013). Analysis of the genes comprising MYCN modules indicated enrichment for multiple DNA maintenance/repair pathways, including the BRCA1/BRCA2, ATM, BARD1 and Fanconi Anemia pathways. This finding is supported by previous reports that examined the therapeutic potential of targeting DNA repair in NB, including targeting PARP1, CHK1, and alternative non‐homologous end joining (NHEJ) elements (Newman et al., 2015; Cole et al., 2011; Norris et al., 2014). For example, Norris et al., showed that Olaparib, a PARP inhibitor, inhibited tumor growth of NB xenografts at clinically achievable concentrations (Norris et al., 2014) however, the authors did not report a mechanistic link between MNA status and PARP1 inhibitor sensitivity. Our data, from in vitro experiments with NB cell lines demonstrate that NB cell lines are sensitive to PARP1 inhibition, with more robust activity in MNA NB cells. In addition, it is known that somatic and/or germline alterations in many genes implicated in DNA repair are altered in rare subsets of patients with NB, including the BRCA binding partner PALB2 (FANCN), BARD1, and CHEK1 (Pugh et al., 2013; Capasso et al., 2009). Our data, combined with previous reports, provide a compelling rationale to investigate targeting genes involved in DNA repair as a potential treatment for MNA NB. In addition, since most PARP1 inhibitors are being assessed on tumors with DNA repair aberrations, our findings suggest that it may be informative to determine whether MYCN overexpression or amplification contributes to sensitivity to these inhibitors.
From a clinical standpoint, accurate predictors of NB patient outcome provide a means to precisely guide therapy based on patient risk. Overall, our data demonstrates that many previously reported signatures are confounded by MNA and these signatures are not able to predict outcome in patients with MNA NB. These findings have important implications for integrating gene signature‐based molecular risk predictors in the clinic, as they highlight the dependency between clinical context and predictor utility. Our findings indicate that current molecular risk signatures are best suited for predicting outcome in NA NB, including the signature currently being studied prospectively for NA NB patient (Park et al., 2013). Whereas the studies presented here provide proof‐of‐principle that predictive gene signatures can be identified for MNA NB, many additional developments would need to occur to enable implementation into the clinic. These include transfer of the signature to a clinically amenable technology, such as the nanoString nCounter® platform, which enables profiling of the expression of large numbers of genes on low amounts of suboptimal quality RNA, which is typical of clinical FFPE samples. Furthermore, methods to evaluate the signature in single samples as well as threshold development for patient stratification on the basis of risk would need to be established. Finally, prospective validation of this or similar MNA‐specific signatures will be a critical step to (1) identify a minimal number of predictive genes within the signature, and (2) determine the clinical utility of this approach for predicting outcome within the poor prognosis MNA subset of high‐risk NB patients using cohorts of patients treated with current era therapies.
Conflict of interest
None.
Supporting information
The following are the supplementary data related to this article:
Supplementary data
Supplementary data
Acknowledgements
This work was supported by grants from the Canadian Institute of Health Research, the James Birrell Fund and Lilah's Fund for Neuroblastoma Research to MSI and DRK, and the Canadian Stem Cell Network to DRK. MSI and DRK are Canada Research Chairs. We would like to thank Dr. Anna Goldenberg for valuable advice on the manuscript.
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.07.012.
Hallett Robin M., Seong Alex B.K., Kaplan David R., Irwin Meredith S., (2016), Transcript signatures that predict outcome and identify targetable pathways in MYCN-amplified neuroblastoma, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.07.012.
References
- Abel, F. , Dalevi, D. , Nethander, M. , Jornsten, R. , De Preter, K. , Vermeulen, J. , Stallings, R. , Kogner, P. , Maris, J. , Nilsson, S. , 2011. A 6-gene signature identifies four molecular subgroups of neuroblastoma. Cancer Cell Int. 11, 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgharzadeh, S. , Pique-Regi, R. , Sposto, R. , Wang, H. , Yang, Y. , Shimada, H. , Matthay, K. , Buckley, J. , Ortega, A. , Seeger, R.C. , 2006. Prognostic significance of gene expression profiles of metastatic neuroblastomas lacking MYCN gene amplification. J. Natl. Cancer Inst. 98, 1193–1203. [DOI] [PubMed] [Google Scholar]
- Asgharzadeh, S. , Salo, J.A. , Ji, L. , Oberthuer, A. , Fischer, M. , Berthold, F. , Hadjidaniel, M. , Liu, C.W. , Metelitsa, L.S. , Pique-Regi, R. , Wakamatsu, P. , Villablanca, J.G. , Kreissman, S.G. , Matthay, K.K. , Shimada, H. , London, W.B. , Sposto, R. , Seeger, R.C. , 2012. Clinical significance of tumor-associated inflammatory cells in metastatic neuroblastoma. J. Clin. Oncol. 30, 3525–3532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attiyeh, E.F. , London, W.B. , Mosse, Y.P. , Wang, Q. , Winter, C. , Khazi, D. , McGrady, P.W. , Seeger, R.C. , Look, A.T. , Shimada, H. , Brodeur, G.M. , Cohn, S.L. , Matthay, K.K. , Maris, J.M. , 2005. Chromosome 1p and 11q deletions and outcome in neuroblastoma. N. Engl. J. Med. 353, 2243–2253. [DOI] [PubMed] [Google Scholar]
- Barbieri, E. , De Preter, K. , Capasso, M. , Johansson, P. , Man, T.K. , Chen, Z. , Stowers, P. , Tonini, G.P. , Speleman, F. , Shohet, J.M. , 2012. A p53 drug response signature identifies prognostic genes in high-risk neuroblastoma. PLoS One. 8, e79843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bown, N. , Cotterill, S. , Lastowska, M. , O'Neill, S. , Pearson, A.D. , Plantaz, D. , Meddeb, M. , Danglot, G. , Brinkschmidt, C. , Christiansen, H. , Laureys, G. , Speleman, F. , Nicholson, J. , Bernheim, A. , Betts, D.R. , Vandesompele, J. , Van Roy, N. , 1999. Gain of chromosome arm 17q and adverse outcome in patients with neuroblastoma. N. Engl. J. Med. 340, 1954–1961. [DOI] [PubMed] [Google Scholar]
- Bresler, S.C. , Weiser, D.A. , Huwe, P.J. , Park, J.H. , Krytska, K. , Ryles, H. , Laudenslager, M. , Rappaport, E.F. , Wood, A.C. , McGrady, P.W. , Hogarty, M.D. , London, W.B. , Radhakrishnan, R. , Lemmon, M.A. , Mosse, Y.P. , 2014. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 26, 682–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodeur, G.M. , Pritchard, J. , Berthold, F. , Carlsen, N.L. , Castel, V. , Castelberry, R.P. , De Bernardi, B. , Evans, A.E. , Favrot, M. , Hedborg, F. , 1993. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. J. Clin. Oncol. 11, 1466–1477. [DOI] [PubMed] [Google Scholar]
- Brodeur, G.M. , Seeger, R.C. , Schwab, M. , Varmus, H.E. , Bishop, J.M. , 1984. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 224, 1121–1124. [DOI] [PubMed] [Google Scholar]
- Capasso, M. , Devoto, M. , Hou, C. , Asgharzadeh, S. , Glessner, J.T. , Attiyeh, E.F. , Mosse, Y.P. , Kim, C. , Diskin, S.J. , Cole, K.A. , Bosse, K. , Diamond, M. , Laudenslager, M. , Winter, C. , Bradfield, J.P. , Scott, R.H. , Jagannathan, J. , Garris, M. , McConville, C. , London, W.B. , Seeger, R.C. , Grant, S.F. , Li, H. , Rahman, N. , Rappaport, E. , Hakonarson, H. , Maris, J.M. , 2009. Common variations in BARD1 influence susceptibility to high-risk neuroblastoma. Nat. Genet. 41, 718–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron, H. , van Sluis, P. , de Kraker, J. , Bokkerink, J. , Egeler, M. , Laureys, G. , Slater, R. , Westerveld, A. , Voute, P.A. , Versteeg, R. , 1996. Allelic loss of chromosome 1p as a predictor of unfavorable outcome in patients with neuroblastoma. N. Engl. J. Med. 334, 225–230. [DOI] [PubMed] [Google Scholar]
- Cohn, S.L. , Pearson, A.D. , London, W.B. , Monclair, T. , Ambros, P.F. , Brodeur, G.M. , Faldum, A. , Hero, B. , Iehara, T. , Machin, D. , Mosseri, V. , Simon, T. , Garaventa, A. , Castel, V. , Matthay, K.K. , 2009. The International Neuroblastoma Risk Group (INRG) classification system: an INRG Task Force report. J. Clin. Oncol. 27, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, K.A. , Huggins, J. , Laquaglia, M. , Hulderman, C.E. , Russell, M.R. , Bosse, K. , Diskin, S.J. , Attiyeh, E.F. , Sennett, R. , Norris, G. , Laudenslager, M. , Wood, A.C. , Mayes, P.A. , Jagannathan, J. , Winter, C. , Mosse, Y.P. , Maris, J.M. , 2011. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proc. Natl. Acad. Sci. U. S. A. 108, 3336–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Preter, K. , Vermeulen, J. , Brors, B. , Delattre, O. , Eggert, A. , Fischer, M. , Janoueix-Lerosey, I. , Lavarino, C. , Maris, J.M. , Mora, J. , Nakagawara, A. , Oberthuer, A. , Ohira, M. , Schleiermacher, G. , Schramm, A. , Schulte, J.H. , Wang, Q. , Westermann, F. , Speleman, F. , Vandesompele, J. , 2010. Accurate outcome prediction in neuroblastoma across independent data sets using a multigene signature. Clin. Cancer Res. 16, 1532–1541. [DOI] [PubMed] [Google Scholar]
- Fardin, P. , Barla, A. , Mosci, S. , Rosasco, L. , Verri, A. , Versteeg, R. , Caron, H.N. , Molenaar, J.J. , Ora, I. , Eva, A. , Puppo, M. , Varesio, L. , 2010. A biology-driven approach identifies the hypoxia gene signature as a predictor of the outcome of neuroblastoma patients. Mol. Cancer. 9, 185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fardin, P. , Cornero, A. , Barla, A. , Mosci, S. , Acquaviva, M. , Rosasco, L. , Gambini, C. , Verri, A. , Varesio, L. , 2009. Identification of multiple hypoxia signatures in neuroblastoma cell lines by l1-l2 regularization and data reduction. J. Biomed. Biotechnol. 2010, 878709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer, M. , Oberthuer, A. , Brors, B. , Kahlert, Y. , Skowron, M. , Voth, H. , Warnat, P. , Ernestus, K. , Hero, B. , Berthold, F. , 2006. Differential expression of neuronal genes defines subtypes of disseminated neuroblastoma with favorable and unfavorable outcome. Clin. Cancer Res. 12, 5118–5128. [DOI] [PubMed] [Google Scholar]
- Garcia, I. , Mayol, G. , Rios, J. , Domenech, G. , Cheung, N.K. , Oberthuer, A. , Fischer, M. , Maris, J.M. , Brodeur, G.M. , Hero, B. , Rodriguez, E. , Sunol, M. , Galvan, P. , de Torres, C. , Mora, J. , Lavarino, C. , 2012. A three-gene expression signature model for risk stratification of patients with neuroblastoma. Clin. Cancer Res. 18, 2012–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, C. , White, P.S. , Weiss, M.J. , Hogarty, M.D. , Thompson, P.M. , Stram, D.O. , Gerbing, R. , Matthay, K.K. , Seeger, R.C. , Brodeur, G.M. , Maris, J.M. , 1999. Allelic deletion at 11q23 is common in MYCN single copy neuroblastomas. Oncogene. 18, 4948–4957. [DOI] [PubMed] [Google Scholar]
- Hallett, R.M. , Dvorkin-Gheva, A. , Bane, A. , Hassell, J.A. , 2012. A gene signature for predicting outcome in patients with basal-like breast cancer. Sci. Rep. 2, 227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hallett, R.M. , Pond, G. , Hassell, J.A. , 2012. A target based approach identifies genomic predictors of breast cancer patient response to chemotherapy. BMC Med. Genomics. 5, 16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ham, J. , Costa, C. , Sano, R. , Lochmann, T.L. , Sennott, E.M. , Patel, N.U. , Dastur, A. , Gomez-Caraballo, M. , Krytska, K. , Hata, A.N. , Floros, K.V. , Hughes, M.T. , Jakubik, C.T. , Heisey, D.A. , Ferrell, J.T. , Bristol, M.L. , March, R.J. , Yates, C. , Hicks, M.A. , Nakajima, W. , Gowda, M. , Windle, B.E. , Dozmorov, M.G. , Garnett, M.J. , McDermott, U. , Harada, H. , Taylor, S.M. , Morgan, I.M. , Benes, C.H. , Engelman, J.A. , Mosse, Y.P. , Faber, A.C. , 2016. Exploitation of the apoptosis-primed state of MYCN-amplified neuroblastoma to develop a potent and specific targeted therapy combination. Cancer Cell. 29, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, M. , Weiss, W.A. , 2013. Neuroblastoma and MYCN. Cold Spring Harb. Perspect. Med. 3, a014415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry, R.A. , Hobbs, B. , Collin, F. , Beazer-Barclay, Y.D. , Antonellis, K.J. , Scherf, U. , Speed, T.P. , 2003. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 4, 249–264. [DOI] [PubMed] [Google Scholar]
- Irwin, M.S. , Park, J.R. , 2015. Neuroblastoma: paradigm for precision medicine. Pediatr. Clin. North Am. 62, 225–256. [DOI] [PubMed] [Google Scholar]
- Johnson, W.E. , Li, C. , Rabinovic, A. , 2007. Adjusting batch effects in microarray expression data using empirical Bayes methods. Biostatistics. 8, 118–127. [DOI] [PubMed] [Google Scholar]
- Kaufman, B. , Shapira-Frommer, R. , Schmutzler, R.K. , Audeh, M.W. , Friedlander, M. , Balmana, J. , Mitchell, G. , Fried, G. , Stemmer, S.M. , Hubert, A. , Rosengarten, O. , Steiner, M. , Loman, N. , Bowen, K. , Fielding, A. , Domchek, S.M. , 2015. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 33, 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner, B.H. , Modak, S. , Kramer, K. , LaQuaglia, M.P. , Yataghene, K. , Basu, E.M. , Roberts, S.S. , Cheung, N.K. , 2014. Striking dichotomy in outcome of MYCN-amplified neuroblastoma in the contemporary era. Cancer. 120, 2050–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladenstein, R. , Ambros, I.M. , Potschger, U. , Amann, G. , Urban, C. , Fink, F.M. , Schmitt, K. , Jones, R. , Slociak, M. , Schilling, F. , Ritter, J. , Berthold, F. , Gadner, H. , Ambros, P.F. , 2001. Prognostic significance of DNA di-tetraploidy in neuroblastoma. Med. Pediatr. Oncol. 36, 83–92. [DOI] [PubMed] [Google Scholar]
- Liang, J. , Tong, P. , Zhao, W. , Li, Y. , Zhang, L. , Xia, Y. , Yu, Y. , 2014. The REST gene signature predicts drug sensitivity in neuroblastoma cell lines and is significantly associated with neuroblastoma tumor stage. Int. J. Mol. Sci. 15, 11220–11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maris, J.M. , 2010. Recent advances in neuroblastoma. N. Engl. J. Med. 362, 2202–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo, J. , Carreira, S. , Sandhu, S. , Miranda, S. , Mossop, H. , Perez-Lopez, R. , Nava Rodrigues, D. , Robinson, D. , Omlin, A. , Tunariu, N. , Boysen, G. , Porta, N. , Flohr, P. , Gillman, A. , Figueiredo, I. , Paulding, C. , Seed, G. , Jain, S. , Ralph, C. , Protheroe, A. , Hussain, S. , Jones, R. , Elliott, T. , McGovern, U. , Bianchini, D. , Goodall, J. , Zafeiriou, Z. , Williamson, C.T. , Ferraldeschi, R. , Riisnaes, R. , Ebbs, B. , Fowler, G. , Roda, D. , Yuan, W. , Wu, Y.M. , Cao, X. , Brough, R. , Pemberton, H. , A'Hern, R. , Swain, A. , Kunju, L.P. , Eeles, R. , Attard, G. , Lord, C.J. , Ashworth, A. , Rubin, M.A. , Knudsen, K.E. , Feng, F.Y. , Chinnaiyan, A.M. , Hall, E. , de Bono, J.S. , 2015. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 373, 1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meddeb, M. , Danglot, G. , Chudoba, I. , Venuat, A.M. , Benard, J. , Avet-Loiseau, H. , Vasseur, B. , Le Paslier, D. , Terrier-Lacombe, M.J. , Hartmann, O. , Bernheim, A. , 1996. Additional copies of a 25 Mb chromosomal region originating from 17q23.1-17qter are present in 90% of high-grade neuroblastomas. Genes Chromosomes Cancer. 17, 156–165. [DOI] [PubMed] [Google Scholar]
- Nevo, I. , Oberthuer, A. , Botzer, E. , Sagi-Assif, O. , Maman, S. , Pasmanik-Chor, M. , Kariv, N. , Fischer, M. , Yron, I. , Witz, I.P. , 2009. Gene-expression-based analysis of local and metastatic neuroblastoma variants reveals a set of genes associated with tumor progression in neuroblastoma patients. Int. J. Cancer. 126, 1570–1581. [DOI] [PubMed] [Google Scholar]
- Newman, E.A. , Lu, F. , Bashllari, D. , Wang, L. , Opipari, A.W. , Castle, V.P. , 2015. Alternative NHEJ pathway components are therapeutic targets in high-risk neuroblastoma. Mol. Cancer Res. 13, 470–482. [DOI] [PubMed] [Google Scholar]
- Norris, R.E. , Adamson, P.C. , Nguyen, V.T. , Fox, E. , 2014. Preclinical evaluation of the PARP inhibitor, olaparib, in combination with cytotoxic chemotherapy in pediatric solid tumors. Pediatr. Blood Cancer. 61, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberthuer, A. , Berthold, F. , Warnat, P. , Hero, B. , Kahlert, Y. , Spitz, R. , Ernestus, K. , Konig, R. , Haas, S. , Eils, R. , Schwab, M. , Brors, B. , Westermann, F. , Fischer, M. , 2006. Customized oligonucleotide microarray gene expression-based classification of neuroblastoma patients outperforms current clinical risk stratification. J. Clin. Oncol. 24, 5070–5078. [DOI] [PubMed] [Google Scholar]
- Ohira, M. , Oba, S. , Nakamura, Y. , Isogai, E. , Kaneko, S. , Nakagawa, A. , Hirata, T. , Kubo, H. , Goto, T. , Yamada, S. , Yoshida, Y. , Fuchioka, M. , Ishii, S. , Nakagawara, A. , 2005. Expression profiling using a tumor-specific cDNA microarray predicts the prognosis of intermediate risk neuroblastomas. Cancer Cell. 7, 337–350. [DOI] [PubMed] [Google Scholar]
- Park, J.R. , Bagatell, R. , London, W.B. , Maris, J.M. , Cohn, S.L. , Mattay, K.K. , Hogarty, M. , 2013. Children's Oncology Group's 2013 blueprint for research: neuroblastoma. Pediatr. Blood Cancer. 60, 985–993. [DOI] [PubMed] [Google Scholar]
- Peifer, M. , Hertwig, F. , Roels, F. , Dreidax, D. , Gartlgruber, M. , Menon, R. , Kramer, A. , Roncaioli, J.L. , Sand, F. , Heuckmann, J.M. , Ikram, F. , Schmidt, R. , Ackermann, S. , Engesser, A. , Kahlert, Y. , Vogel, W. , Altmuller, J. , Nurnberg, P. , Thierry-Mieg, J. , Thierry-Mieg, D. , Mariappan, A. , Heynck, S. , Mariotti, E. , Henrich, K.O. , Gloeckner, C. , Bosco, G. , Leuschner, I. , Schweiger, M.R. , Savelyeva, L. , Watkins, S.C. , Shao, C. , Bell, E. , Hofer, T. , Achter, V. , Lang, U. , Theissen, J. , Volland, R. , Saadati, M. , Eggert, A. , de Wilde, B. , Berthold, F. , Peng, Z. , Zhao, C. , Shi, L. , Ortmann, M. , Buttner, R. , Perner, S. , Hero, B. , Schramm, A. , Schulte, J.H. , Herrmann, C. , O'Sullivan, R.J. , Westermann, F. , Thomas, R.K. , Fischer, M. , 2015. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 526, 700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto, N.R. , Applebaum, M.A. , Volchenboum, S.L. , Matthay, K.K. , London, W.B. , Ambros, P.F. , Nakagawara, A. , Berthold, F. , Schleiermacher, G. , Park, J.R. , Valteau-Couanet, D. , Pearson, A.D. , Cohn, S.L. , 2015. Advances in risk classification and treatment strategies for neuroblastoma. J. Clin. Oncol. 33, 3008–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh, T.J. , Morozova, O. , Attiyeh, E.F. , Asgharzadeh, S. , Wei, J.S. , Auclair, D. , Carter, S.L. , Cibulskis, K. , Hanna, M. , Kiezun, A. , Kim, J. , Lawrence, M.S. , Lichenstein, L. , McKenna, A. , Pedamallu, C.S. , Ramos, A.H. , Shefler, E. , Sivachenko, A. , Sougnez, C. , Stewart, C. , Ally, A. , Birol, I. , Chiu, R. , Corbett, R.D. , Hirst, M. , Jackman, S.D. , Kamoh, B. , Khodabakshi, A.H. , Krzywinski, M. , Lo, A. , Moore, R.A. , Mungall, K.L. , Qian, J. , Tam, A. , Thiessen, N. , Zhao, Y. , Cole, K.A. , Diamond, M. , Diskin, S.J. , Mosse, Y.P. , Wood, A.C. , Ji, L. , Sposto, R. , Badgett, T. , London, W.B. , Moyer, Y. , Gastier-Foster, J.M. , Smith, M.A. , Guidry Auvil, J.M. , Gerhard, D.S. , Hogarty, M.D. , Jones, S.J. , Lander, E.S. , Gabriel, S.B. , Getz, G. , Seeger, R.C. , Khan, J. , Marra, M.A. , Meyerson, M. , Maris, J.M. , 2013. The genetic landscape of high-risk neuroblastoma. Nat. Genet. 45, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puissant, A. , Frumm, S.M. , Alexe, G. , Bassil, C.F. , Qi, J. , Chanthery, Y.H. , Nekritz, E.A. , Zeid, R. , Gustafson, W.C. , Greninger, P. , Garnett, M.J. , McDermott, U. , Benes, C.H. , Kung, A.L. , Weiss, W.A. , Bradner, J.E. , Stegmaier, K. , 2013. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 3, 308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiermacher, G. , Janoueix-Lerosey, I. , Ribeiro, A. , Klijanienko, J. , Couturier, J. , Pierron, G. , Mosseri, V. , Valent, A. , Auger, N. , Plantaz, D. , Rubie, H. , Valteau-Couanet, D. , Bourdeaut, F. , Combaret, V. , Bergeron, C. , Michon, J. , Delattre, O. , 2010. Accumulation of segmental alterations determines progression in neuroblastoma. J. Clin. Oncol. 28, 3122–3130. [DOI] [PubMed] [Google Scholar]
- Schleiermacher, G. , Mosseri, V. , London, W.B. , Maris, J.M. , Brodeur, G.M. , Attiyeh, E. , Haber, M. , Khan, J. , Nakagawara, A. , Speleman, F. , Noguera, R. , Tonini, G.P. , Fischer, M. , Ambros, I. , Monclair, T. , Matthay, K.K. , Ambros, P. , Cohn, S.L. , Pearson, A.D. , 2012. Segmental chromosomal alterations have prognostic impact in neuroblastoma: a report from the INRG project. Br. J. Cancer. 107, 1418–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm, A. , Mierswa, I. , Kaderali, L. , Morik, K. , Eggert, A. , Schulte, J.H. , 2009. Reanalysis of neuroblastoma expression profiling data using improved methodology and extended follow-up increases validity of outcome prediction. Cancer Lett. 282, 55–62. [DOI] [PubMed] [Google Scholar]
- Schramm, A. , Schulte, J.H. , Klein-Hitpass, L. , Havers, W. , Sieverts, H. , Berwanger, B. , Christiansen, H. , Warnat, P. , Brors, B. , Eils, J. , Eils, R. , Eggert, A. , 2005. Prediction of clinical outcome and biological characterization of neuroblastoma by expression profiling. Oncogene. 24, 7902–7912. [DOI] [PubMed] [Google Scholar]
- Schramm, A. , Vandesompele, J. , Schulte, J.H. , Dreesmann, S. , Kaderali, L. , Brors, B. , Eils, R. , Speleman, F. , Eggert, A. , 2007. Translating expression profiling into a clinically feasible test to predict neuroblastoma outcome. Clin. Cancer Res. 13, 1459–1465. [DOI] [PubMed] [Google Scholar]
- Schwab, M. , Alitalo, K. , Klempnauer, K.H. , Varmus, H.E. , Bishop, J.M. , Gilbert, F. , Brodeur, G. , Goldstein, M. , Trent, J. , 1983. Amplified DNA with limited homology to myc cellular oncogene is shared by human neuroblastoma cell lines and a neuroblastoma tumour. Nature. 305, 245–248. [DOI] [PubMed] [Google Scholar]
- Seong, B.K. , Lau, J. , Adderley, T. , Kee, L. , Chaukos, D. , Pienkowska, M. , Malkin, D. , Thorner, P. , Irwin, M.S. , 2015. SATB2 enhances migration and invasion in osteosarcoma by regulating genes involved in cytoskeletal organization. Oncogene. 34, 3582–3592. [DOI] [PubMed] [Google Scholar]
- Slavc, I. , Ellenbogen, R. , Jung, W.H. , Vawter, G.F. , Kretschmar, C. , Grier, H. , Korf, B.R. , 1990. myc gene amplification and expression in primary human neuroblastoma. Cancer Res. 50, 1459–1463. [PubMed] [Google Scholar]
- Stricker, T.P. , Morales La Madrid, A. , Chlenski, A. , Guerrero, L. , Salwen, H.R. , Gosiengfiao, Y. , Perlman, E.J. , Furman, W. , Bahrami, A. , Shohet, J.M. , Zage, P.E. , Hicks, M.J. , Shimada, H. , Suganuma, R. , Park, J.R. , So, S. , London, W.B. , Pytel, P. , Maclean, K.H. , Cohn, S.L. , 2014. Validation of a prognostic multi-gene signature in high-risk neuroblastoma using the high throughput digital NanoString nCounter system. Mol. Oncol. 8, 669–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, D. , Vo, K.T. , London, W.B. , Fischer, M. , Ambros, P.F. , Nakagawara, A. , Brodeur, G.M. , Matthay, K.K. , DuBois, S.G. , 2016 Mar 15. Identification of patient subgroups with markedly disparate rates of MYCN amplification in neuroblastoma: a report from the International Neuroblastoma Risk Group project. Cancer. 122, (6) 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofigh, A. , Suderman, M. , Paquet, E.R. , Livingstone, J. , Bertos, N. , Saleh, S.M. , Zhao, H. , Souleimanova, M. , Cory, S. , Lesurf, R. , Shahalizadeh, S. , Garcia Lopez, N. , Riazalhosseini, Y. , Omeroglu, A. , Ursini-Siegel, J. , Park, M. , Dumeaux, V. , Hallett, M. , 2014. The prognostic ease and difficulty of invasive breast carcinoma. Cell Rep. 9, 129–142. [DOI] [PubMed] [Google Scholar]
- Valentijn, L.J. , Koster, J. , Haneveld, F. , Aissa, R.A. , van Sluis, P. , Broekmans, M.E. , Molenaar, J.J. , van Nes, J. , Versteeg, R. , 2012. Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc. Natl. Acad. Sci. U. S. A. 109, 19190–19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Nes, J. , Chan, A. , van Groningen, T. , van Sluis, P. , Koster, J. , Versteeg, R. , 2013. A NOTCH3 transcriptional module induces cell motility in neuroblastoma. Clin. Cancer Res. 19, 3485–3494. [DOI] [PubMed] [Google Scholar]
- Venet, D. , Dumont, J.E. , Detours, V. , 2011. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Comput. Biol. 7, e1002240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen, J. , De Preter, K. , Laureys, G. , Speleman, F. , Vandesompele, J. , 2009. 59-gene prognostic signature sub-stratifies high-risk neuroblastoma patients. Lancet Oncol. 10, 1030 [DOI] [PubMed] [Google Scholar]
- von Stedingk, K. , Koster, J. , Piqueras, M. , Noguera, R. , Navarro, S. , Pahlman, S. , Versteeg, R. , Ora, I. , Gisselsson, D. , Lindgren, D. , Axelson, H. , 2013. snoRNPs regulate telomerase activity in neuroblastoma and are associated with poor prognosis. Transl. Oncol. 6, 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L.L. , Teshiba, R. , Ikegaki, N. , Tang, X.X. , Naranjo, A. , London, W.B. , Hogarty, M.D. , Gastier-Foster, J.M. , Look, A.T. , Park, J.R. , Maris, J.M. , Cohn, S.L. , Seeger, R.C. , Asgharzadeh, S. , Shimada, H. , 2015. Augmented expression of MYC and/or MYCN protein defines highly aggressive MYC-driven neuroblastoma: a Children's Oncology Group study. Br. J. Cancer. 113, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei, J.S. , Greer, B.T. , Westermann, F. , Steinberg, S.M. , Son, C.G. , Chen, Q.R. , Whiteford, C.C. , Bilke, S. , Krasnoselsky, A.L. , Cenacchi, N. , Catchpoole, D. , Berthold, F. , Schwab, M. , Khan, J. , 2004. Prediction of clinical outcome using gene expression profiling and artificial neural networks for patients with neuroblastoma. Cancer Res. 64, 6883–6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss, W.A. , Aldape, K. , Mohapatra, G. , Feuerstein, B.G. , Bishop, J.M. , 1997. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 16, 2985–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. , Feng, X. , Stein, L. , 2010. A human functional protein interaction network and its application to cancer data analysis. Genome Biol. 11, R53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, G. , Stein, L. , 2012. A network module-based method for identifying cancer prognostic signatures. Genome Biol. 13, R112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, S. , Lee, J.S. , Guo, F. , Shin, J. , Perez-Atayde, A.R. , Kutok, J.L. , Rodig, S.J. , Neuberg, D.S. , Helman, D. , Feng, H. , Stewart, R.A. , Wang, W. , George, R.E. , Kanki, J.P. , Look, A.T. , 2012. Activated ALK collaborates with MYCN in neuroblastoma pathogenesis. Cancer Cell. 21, 362–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following are the supplementary data related to this article:
Supplementary data
Supplementary data