

Abstract
Breast cancer stem cells (CSCs) can be identified by increased Aldefluor fluorescence caused by increased expression of aldehyde dehydrogenase 1A3 (ALDH1A3), as well as ALDH1A1 and ALDH2. In addition to being a CSC marker, ALDH1A3 regulates gene expression via retinoic acid (RA) signaling and plays a key role in the progression and chemotherapy resistance of cancer. Therefore, ALDH1A3 represents a druggable anti‐cancer target of interest. Since to date, there are no characterized ALDH1A3 isoform inhibitors, drugs that were previously described as inhibiting the activity of other ALDH isoforms were tested for anti‐ALDH1A3 activity. Twelve drugs (3‐hydroxy‐dl‐kynurenine, benomyl, citral, chloral hydrate, cyanamide, daidzin, DEAB, disulfiram, gossypol, kynurenic acid, molinate, and pargyline) were compared for their efficacy in inducing apoptosis and reducing ALDH1A3, ALDH1A1 and ALDH2‐associated Aldefluor fluorescence in breast cancer cells. Citral was identified as the best inhibitor of ALDH1A3, reducing the Aldefluor fluorescence in breast cancer cell lines and in a patient‐derived tumor xenograft. Nanoparticle encapsulated citral specifically reduced the enhanced tumor growth of MDA‐MB‐231 cells overexpressing ALDH1A3. To determine the potential mechanisms of citral‐mediated tumor growth inhibition, we performed cell proliferation, clonogenic, and gene expression assays. Citral reduced ALDH1A3‐mediated colony formation and expression of ALDH1A3‐inducible genes. In conclusion, citral is an effective ALDH1A3 inhibitor and is able to block ALDH1A3‐mediated breast tumor growth, potentially via blocking its colony forming and gene expression regulation activity. The promise of ALDH1A3 inhibitors as adjuvant therapies for patients with tumors that have a large population of high‐ALDH1A3 CSCs is discussed.
Keywords: Breast cancer, ALDH1A3, Aldefluor, Cancer stem cell
Highlights
Citral is a specific inhibitor of cancer stem cell marker ALDH1A3.
Aldefluor fluorescence of breast tumor cells is blocked by citral.
Citral inhibits ALDH1A3‐driven breast tumor growth in vivo.
ALDH1A3‐mediated colony formation and gene expression is inhibited by citral.
Abbreviations
- ALDH
aldehyde dehydrogenase
- Citral‐NP
citral‐nanoparticle encapsulated
- CSC
cancer stem cell
- DEAB
4‐diethylaminobenzaldehyde
- FSC
forward scatter
- KD
knockdown
- NOD/SCID
non‐obese diabetic/severe combined immunodeficiency
- OE
overexpression
- PBS
phosphate buffered saline
- PDX
patient‐derived xenograft
- PEG‐b‐PCL
polyethylene glycol – block – polycaprolactone
- QPCR
quantitative polymerase chain reaction
- RA
retinoic acid
- SSC
side scatter
1. Introduction
First described in leukemia (Bonnet and Dick, 1997), and later in solid tumors, cancer stem cells (CSCs) are a highly tumorigenic subpopulation present within the heterogeneous tumors of many cancers including breast cancer (Al‐Hajj et al., 2003). These cells share certain characteristics with normal stem cells including the ability to self‐renew and to differentiate. CSCs also demonstrate a highly malignant phenotype, being able to initiate tumors, and promote epithelial‐to‐mesenchymal transition and metastasis (Al‐Hajj et al., 2003; Liu et al., 2014; Charafe‐Jauffret et al., 2009). Most concerning in terms of effective patient treatment and mitigating the risk of recurrence is the resistance of CSCs to common chemotherapies and radiotherapy (Shafee et al., 2008; Diehn et al., 2009). These characteristics suggest that CSCs must be eliminated during treatment to avoid risk of relapse, and that this subpopulation of cells is poised to avoid elimination. Thus, therapies that target CSC activities may improve cancer treatment efficacy and patient outcomes.
CSC‐associated enzymes and signaling pathways may provide novel avenues of therapeutic intervention, since these pathways (e.g. Notch, Wnt, and Hedgehog (Takebe et al., 2011)) and enzymes (e.g. aldehyde dehygrogenases; ALDHs) are also mediators of tumorigenicity, metastasis, and therapy resistance. A common biomarker for CSC identification is high Aldefluor fluorescence associated with increased ALDH activity (Ginestier et al., 2007). ALDHs are a superfamily of enzymes present in all three taxonomic domains with 19 isoforms expressed in humans (Vasiliou and Nebert, 2005). ALDHs convert aldehydes to carboxylic acids; metabolic processes generate toxic aldehydes and ALDHs are required to maintain cellular homeostasis. Furthermore, individual ALDH isoforms have varied substrate specificity and more specialized functions. Members of the ALDH1A family (ALDH1A1, ALDH1A2, ALDH1A3) oxidize the vitamin A metabolite, retinal, to retinoic acid (RA), a developmental cell signaling and gene expression induction molecule that also plays an important role in cancer (Coyle et al., 2013).
Both ALDH1A1 and ALDH1A3, as well as other isoforms (e.g. ALDH1A2, ALDH2 and ALDH7A1), have been implicated as contributors to CSC‐associated Aldefluor fluorescence, with specific isoforms playing a more predominant role in different cancers (Li et al., 2012; Marcato et al., 2011; Shao et al., 2014; Hartomo et al., 2015; Moreb et al., 2012; van den Hoogen et al., 2011). In particular, expression of the ALDH1A3 isoform is of primary importance for the Aldefluor fluorescence of breast cancer, lung cancer, melanoma, malignant pleural mesothelioma, and head and neck cancer (Marcato et al., 2015, 2015, 2012, 2011, 2015, 2014). In addition to being associated with CSCs, expression of ALDH1A1 and ALDH1A3 often correlates with poor prognosis in cancers such as breast, prostate and lung, kidney, esophageal, and head and neck (Charafe‐Jauffret et al., 2009; Ginestier et al., 2007; Li et al., 2012; Khoury et al., 2012; Wang et al., 2013; Yang et al., 2014; Qian et al., 2014). ALDH1A3 has also been directly implicated in tumor progression and therapy resistance. For breast cancer, ALDH1A3 been shown to promote tumor growth and metastasis though production of retinoic acid (RA) and expression of RA‐inducible genes (Marcato et al., 2015). ALDH1A3 also promotes the growth of lung tumors, glioblastoma, and melanoma (Shao et al., 2014; Mao et al., 2013; Luo et al., 2012). Furthermore, it is associated with the chemoresistant population of mesothelioma (Canino et al., 2015) and is a causative agent in the radioresistant population of head and neck cancer (Kurth et al., 2015). Together, these results suggest that targeting CSC‐associated ALDH1A enzymes, in particular ALDH1A3, may be an effective adjuvant cancer therapy (Luo et al., 2012; Mao et al., 2013).
Due to their promise as an anti‐CSC agent, several ALDH inhibitors have been explored for anti‐cancer activity. Pan‐ALDH inhibitor DEAB reduces growth of melanoma xenografts and the number of residual melanoma cells (Yue et al., 2015). However, DEAB has a very short duration of efficacy in vivo and probably requires modification or encapsulation to have therapeutic value (Mahmoud et al., 1993). Another ALDH inhibitor of considerable interest is disulfiram, which can inhibit TGF‐β induced “stem like” features of MDA‐MB‐231 breast cancer cells (Han et al., 2015), increase chemosensitivity (Raha et al., 2014), and also reduce mammosphere formation (Liu et al., 2014). However, the ability of disulfiram to directly inhibit ALDH in breast cancer cells was not confirmed, and though mammospheres had increased expression of ALDH1A3, disulfiram did not reduce mammosphere‐associated ALDH1A3 mRNA. Thus, the effect of specifically inhibiting ALDH1A3 has not been explored yet, nor is the specificity of disulfiram for ALDH1A3 known.
A panel of compounds known to inhibit at least one ALDH isoform and with unknown ALDH1A3 inhibitory activity were investigated for their potential as ALDH1A3 inhibitors in breast cancer. Citral was identified as a strong inhibitor of ALDH1A3 and reduced ALDH1A3‐dependent colony formation, gene expression and tumour growth. To our knowledge, this is the first study to characterize inhibitors of ALDH1A3 specifically, and is the first to show that inhibiting ALDH1A3 can slow breast tumor growth.
2. Materials and methods
2.1. ALDH inhibitors and cell lines
All ALDH inhibitors were acquired from Sigma and dissolved in the indicated vehicle (Supplementary Table 1). MDA‐MB‐231, MDA‐MB‐468, or SKBR3 cells were challenged with dissolved drug or vehicle alone at the indicated final concentration. The cells were obtained from American Type Culture Collection and the same ALDH isoform overexpression or knockdown clones generated and validated in our prior publications (ALDH1A1 shRNA1, ALDH1A3 shRNA3 and ALDH2 shRNA2; Supplementary Table 2; Supplementary Figures 2–4) were used (Marcato et al., 2011, 2015). All cells were cultured in Dulbecco's Modified Eagle Medium (Invitrogen) supplemented with 10% fetal bovine serum (Invitrogen), 1X antibiotic‐antimycotic (Invitrogen), and 0.25 mg/mL puromycin (Sigma Aldrich) in a 37°C humidified chamber with 5% CO2.
2.2. Aldefluor assay on patient‐derived xenograft and cell lines
A patient‐derived xenograft (PDX) previously established in female NOD/SCID mice (Marcato et al., 2011) was harvested to generate cell suspensions. Red blood cells were lysed and remaining cells were washed with PBS and Aldefluor assay performed as per the manufacturer's instructions (Stemcell Technologies), with or without the addition of one of the panel of drugs (Supplementary Table 1). To eliminate dead cells and non‐cancer cells of mouse origin, cells were stained with viability stain 7‐AAD (Biolegend) and anti‐H2Kd (mouse histocompatibility class I) conjugated to Alexafluor 647 nm (Biolegend), respectively. Cell populations were identified using a FACSCalibur flow cytometer (Becton Dickinson). Distinct Aldefluor‐positive and Aldefluor‐negative populations in the PDX were revealed after excluding debris, mouse, and dead cells. For cell line assays anti‐H2Kd stain was not used and Aldefluor levels were quantified via mean fluorescence intensity.
2.3. Quantifying live cells
Cells quantified for percentage of early and late apoptotic cells via Alexafluor 488 conjugated‐ Annexin V (Invitrogen) and 7‐AAD (Biolegend) staining following the manufacturer's protocol and analyzed with a FACSCalibur.
2.4. Generation of citral nanoparticles (Citral‐NP)
Nanoparticle encapsulated citral (citral‐NP) and empty nanoparticles (vehicle control) were generated based on Zeng et al., 2015 protocols with few modifications (Zeng et al., 2015). Briefly, 1 mL of a 0.5 mM polyethylene glycol‐block‐polycaprolactone (Polymer Source, Quebec, CAN; PEG‐b‐PCL; MW PEG: 10,000; MW PCL: 5000) solution was made in HPLC‐grade acetone (Thermofisher Scientific). Citral (16 mg/mL), followed by 1 mL of PBS was added while vortexing to form nanoparticles. Rotary evaporation and nitrogen gas flushing removed acetone. Samples were reconstituted and centrifuged at 8000 × g for 5 min for the separation of two distinct layers: an upper waxy layer containing polymer aggregates and excess unencapsulated citral, and a lower more fluid layer containing nanoparticles. The lower layer was collected and unencapsulated citral and nanoparticles >220 nm were removed by filtering (0.22 μm nylon syringe filter, Fisher Scientific).
2.5. HPLC determination of Citral‐NP concentration
High performance liquid chromatography (HPLC) was utilized to measure the concentration of nanoparticle encapsulated citral. A known dilution of citral‐NP was injected into a 18C 3.9 × 150 mm Symmetry column (Waters) and eluted at room temperature with isocratic 40% water and 60% acetonitrile (both, with 0.1% trifluoroacetic acid) for 10 min at a rate of 0.5 mL/min using a Waters 2695 separations module and a Waters 2487 dual λ absorbance detector set at 254 nm. No peaks were detected for PEG‐b‐PCL at this wavelength; whereas acetone eluted at ∼2.55 min (Supplementary Figure 5).
2.6. In vivo citral‐NP treatment
Eight week old NOD/SCID female mice were orthotopically injected with 2 × 106 MDA‐MB‐231 vector control or ALDH1A3 overexpression cells, admixed in 1:1 ratio with phenol red‐free high concentration matrigel (Fisher Scientific). Subsequently, the mice were randomly divided into empty‐NP (NT) or citral‐NP treatment groups (n = 6 per group) and were injected via tail vein every 3 or 4 days (alternating) with 100 μL of citral‐NP (0.4 mg/kg) or saline. Resulting tumor growth was quantified (length × width × height/2) and on day 38 the tumors were harvested and weighed from euthanized mice.
2.7. In vitro growth rate analysis
Seeded cells were treated with 100 μM citral or vehicle control and collected and counted 24 or 72 h later. Growth rate = number of cells at a time point/number of cells pre‐treatment, normalized to the vehicle control, no treatment (NT) growth rate.
2.8. Colony forming assay
Seeded cells were treated with 100 μM citral or vehicle for 24 h prior to re‐seeding at very low confluency for single cell generation of colonies (20 cells/cm2 for MDA‐MB‐231 cells, 50 cells/cm2 for MDA‐MB‐468 cells) and cultured for 13 days with media change every other day. Resulting colonies were visualized by methanol fixation and 0.5% crystal violet staining. Colonies >50 cells were counted, and colony forming efficiency = number of colonies/number of seeded cells.
2.9. Quantitative PCR
After treatment with the ALDH inhibitors for 24 h, RNA from the cells was extracted using TRIzol (Invitrogen) and the Purelink RNA kit (Invitrogen), and reverse transcribed with iScript™ cDNA Synthesis Kit (Bio‐Rad) as per manufacturer's instructions. Real‐time quantitative PCR (QPCR) used SsoAdvanced Universal SYBR supermix (Bio‐Rad) with gene‐specific primers (Supplementary Table 4) was performed as per manufacturer's instructions using a 96CFX Touch Real‐Time PCR Detection System (Bio‐Rad). Standard curves were generated to incorporate primer efficiencies and relative levels of mRNA were calculated utilizing internal reference gene B2M.
2.10. Statistical analysis
GraphPad Prism Version 4 was used to perform one‐way ANOVAs and post‐hoc tests, or paired t‐tests as indicated in the figure legends. Significance is indicated as follows p < 0.05*, p < 0.01**, p < 0.001***, and unless specified otherwise, error bars represent SEM.
3. Results
3.1. Citral, DEAB, and benomyl eliminate the Aldefluor‐positive population of a breast cancer patient‐derived xenograft tumor
To identify a compound that would inhibit ALDH1A3 activity specifically and ALDH1A3‐mediated breast tumor growth, we compiled a group of previously described ALDH inhibitors, some of which had been tested for cytotoxic effects, tumor growth inhibition potential, and anti‐CSC activity (Supplementary Table 3) (Koppaka et al., 2012). As a first assessment of the ALDH inhibition capacity of the compounds, we tested their ability to reduce Aldefluor fluorescence associated with CSCs(Ginestier et al., 2007; Marcato et al., 2011). Using a PDX previously demonstrated to contain Aldefluor‐positive tumor initiating cells and high ALDH1A3 expression (Marcato et al., 2011), we quantified and compared the efficiency of the 12 compounds (Koppaka et al., 2012) to eliminate the Aldefluor‐positive population from the PDX tumor ex vivo. PDX cells from serial passages were incubated with the Aldefluor substrate with increasing concentrations of ALDH inhibitor. The mean percentage Aldefluor‐positive cells in this PDX was 4.56%. Of the 12 inhibitors tested, citral, DEAB and benomyl significantly reduced the Aldefluor‐positive population (Figure 1). Interestingly, disulfiram, which has previously been used as an ALDH inhibitor in cancer models (Raha et al., 2014), only partially eliminated the Aldefluor‐positive population (not significant). This suggests that at least for this PDX, citral, DEAB and benomyl have superior activity against the Aldefluor‐positive tumor population.
Figure 1.
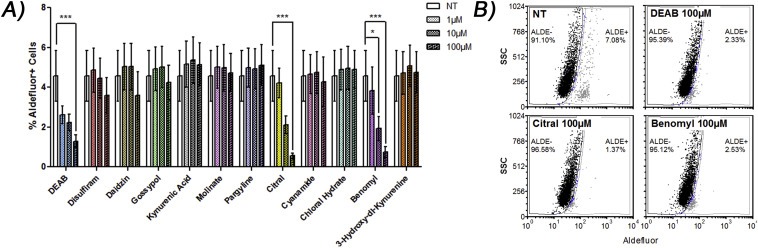
Citral, benomyl, and DEAB eliminate the Aldefluor + population in a patient‐derived xenograft (PDX) ex vivo. A) Indicated drugs were added to harvested tumor cells of a PDX and Aldefluor assay performed. Significance determined by one‐way ANOVA with Tukey post‐hoc test. B) Representative dot plots.
3.2. Citral is most effective at inhibiting Aldefluor fluorescence induced by ALDH1A3
Multiple ALDH isoforms can contribute to the Aldefluor fluorescence observed in breast cancer cells, with ALDH1A3, as well as ALDH1A1 and ALDH2 to a lesser degree, being the most important (Marcato et al., 2011). Therefore, cell lines with defined ALDH isoform expression were utilized to compare the specificity of the compounds for inhibiting the production of Aldefluor fluorescence by a specific isoform. For this purpose we used MDA‐MB‐231, MDA‐MB‐468 and SKBR3 breast cancer cells.
MDA‐MB‐231 cells have very low endogenous Aldefluor fluorescence (Marcato et al., 2011), and therefore served as a good model system to introduce isoform‐specific Aldefluor fluorescence by overexpressing individual ALDH isoforms. The low protein expression of ALDH1A1 and ALDH1A3 in MDA‐MB‐231 cells in comparison to MDA‐MB‐468 cells was previously reported via quantitative proteomics (Murphy et al., 2014) and is visualized here by western blotting in (Supplementary Figure 1). Overexpression of ALDH1A1 or ALDH1A3 to a greater degree, significantly increased mean Aldefluor fluorescence compared to that of cells with vector control (Figure 2A and B, respectively). Expression of the ALDH isoforms was previously shown (Marcato et al., 2015), and illustrated here in Supplementary Figure 2. ALDH1A3‐specific Aldefluor fluorescence can also be modeled with MDA‐MB‐468 breast cancer cells that have endogenously high Aldefluor fluorescence dependent upon ALDH1A3 expression (Marcato et al., 2011). Knockdown ALDH1A3 cells showed significantly reduced mean Aldefluor fluorescence compared to scramble control (Figure 2B). Similarly, ALDH2‐associated Aldefluor fluorescence can be modeled with SKBR3 breast cancer cells with high endogenous levels of ALDH2 activity (Marcato et al., 2011). Knockdown ALDH2 cells showed significantly lower mean Aldefluor fluorescence compared to cells with scramble control (Figure 2C). The reduction of ALDH1A3 and ALDH2 protein levels in the knockdown cell lines was shown previously (Marcato et al., 2011), and illustrated here in Supplementary Figures 3 and 4.
Figure 2.
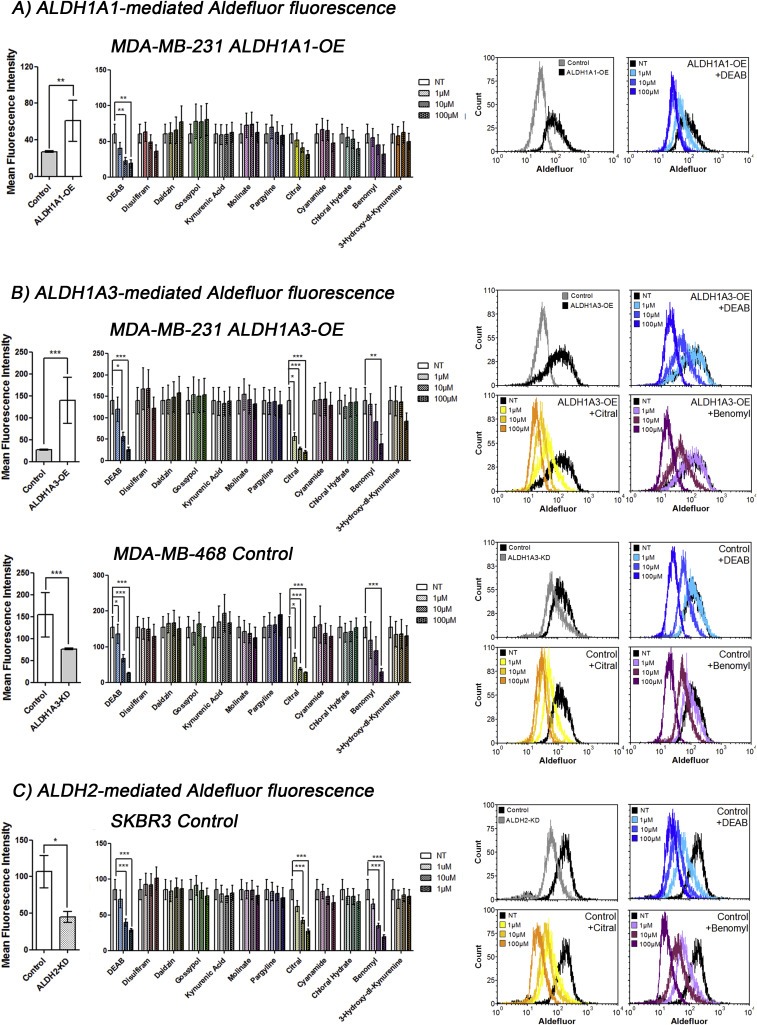
Citral is the best inhibitor of ALDH1A3‐mediated Aldefluor positivity quantified by mean fluorescence intensity. A, B, C) The effect of overexpression or knockdown of indicated ALDH isoform in Aldefluor positivity (relative mean fluorescence intensity, MFI) of the cell lines is show in the left panels. The effect of indicated drugs on Aldefluor positivity mediated by ALDH1A1 in MDA‐MB‐231 ALDH1A1 overexpression (OE) cells (A), or mediated by ALDH1A3 in MDA‐MB‐231 ALDH1A3‐OE cells and in MDA‐MB‐468 cells with intrinsic high ALDH1A3 (B), or mediated by ALDH2 in SKBR3 cells with intrinsic high ALDH2 (C). A, B, C) Significance determined by one‐way ANOVA with Tukey's post‐hoc test.
To assess the effect of the 12 compounds on Aldefluor fluorescence induced by ALDH1A1, MDA‐MB‐231 cells overexpressing ALDH1A1 (ALDH1A1‐OE) were incubated with Aldefluor substrate in the presence of increasing drug concentrations. Only DEAB significantly reduced the mean Aldefluor fluorescence of MDA‐MB‐231 ALDH1A1 cells (Figure 2A), suggesting the other drugs were ineffective ALDH1A1 inhibitors under the experimental conditions.
Similarly, to assess the effect of the inhibitors on Aldefluor fluorescence mediated by ALDH1A3, increasing concentrations of the compounds were added to MDA‐MB‐231 cells with or without overexpression of ALDH1A3 (ALDH1A3‐OE), and to MDA‐MB‐468 cells with high endogenous ALDH1A3‐dependent Aldefluor fluorescence. Of the 12 inhibitors tested, citral, DEAB and benomyl significantly reduced ALDH1A3‐mediated Aldefluor fluorescence production in the breast cancer cells (Figure 2B; Supplementary Figures 5 and 6); however, citral was the most effective, significantly inhibiting ALDH1A3 at the lowest concentration of 1 μM.
Finally, ALDH2‐dependent Aldefluor fluorescence production was challenged with increasing concentrations of the compounds applied to SKBR3 cells with predominately ALDH2‐dependent Aldefluor fluorescence. Citral, DEAB and benomyl significantly reduced Aldefluor fluorescence of SKBR3 cells to a similar extent, suggesting that all three compounds effectively inhibit ALDH2 (Figure 2C). Therefore, in terms of reducing Aldefluor fluorescence specifically associated with ALDH1A3 or ALDH2, citral was the most effective inhibitor, while DEAB was the most effective general ALDH inhibitor.
3.3. Disulfiram, gossypol, and citral induce apoptosis in MDA‐MB‐231 cells
To compare the effect of the inhibitors on apoptosis and also to test if ALDH1A3 expression has an effect on the sensitivity of cells to apoptosis, we next assessed the ability of the panel of inhibitors to induce apoptosis in MDA‐MB‐231, with or without overexpression of ALDH1A3. Of all the drugs, disulfiram most strongly induced apoptosis, which was magnified in the ALDH1A3‐OE cells (Figure 3). Gossypol also induced apoptosis in the cells, regardless of ALDH1A3 expression. At the highest concentration tested (100 μM), citral induced a low level of cell death in MDA‐MB‐231 ALDH1A3‐OE cells after 24 h, but did not appear to induce morphological changes (Figure 3; Supplementary Figure 7). Citral was shown to induce apoptosis and cell cycle arrest in other cancer cell lines (Supplementary Table 3), although this is the first indication that its effects on cell viability may be minimally related to ALDH activity.
Figure 3.
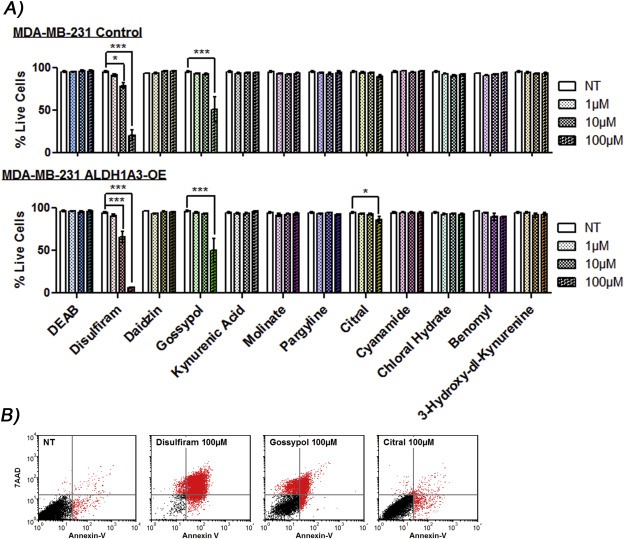
Disulfiram, gossypol, or citral induce apoptosis in MDA‐MB‐231 cells. A) MDA‐MB‐231 vector control and ALDH1A3‐OE cells were treated with indicated drugs for 24 h and assessed for live cells (percentage non‐apoptotic cells) by FACS analysis of annexin‐V‐Alexafluor 488 and 7‐AAD stained cells. Significance determined by one‐way ANOVA with Tukey's post‐hoc test. B) Representative dot plots.
Taking into consideration all four assays (i.e. elimination of Aldefluor‐positive population in a PDX, reduction of ALDH1A3‐specific Aldefluor fluorescence in breast cancer cells, and apoptosis induction which was associated with ALDH1A3), citral stood out among the ALDH inhibitor panel as a potentially effective ALDH1A3/Aldefluor fluorescence inhibitor. Therefore, we focused the remainder of our studies on the ability of citral to inhibit ALDH1A3‐mediated tumor growth.
3.4. Nanoparticle encapsulated citral reduces ALDH1A3‐mediated tumor growth of MDA‐MB‐231 cells
Citral is a common component of fragrances and flavor additives and is known to degrade at acidic pH and under oxidative stress (Ueno et al., 2004), suggesting it likely requires stabilization or encapsulation to enhance in vivo bioavailability. Zeng et al. reported a PEG‐b‐PCL micelle‐encapsulation method for citral (referred to as nanoparticle‐encapsulated NP) (Zeng et al., 2015). These authors also demonstrated that citral‐NP inhibited the tumor growth of murine mammary 4T1 tumor cells implanted in the BALB/C mice (Zeng et al., 2015). In the present study we found that intraperitoneal administration of free citral was ineffective at reducing tumor growth of MDA‐MB‐231 cells, regardless of ALDH1A3 expression (Supplementary Figure 8). Therefore, we adapted the encapsulation strategy devised by Zeng et al., to generate nanoparticle encapsulated citral (citral‐NP) and an empty nanoparticle control using PEG‐b‐PCL. HPLC confirmed that citral is composed of two E and Z isomers in a 2:1 ratio, as reported by Zeng et al. (Figure 4A). HPLC was used to quantify free citral and citral‐NP, and equivalent doses based on the HPLC quantification had similar effects on Aldefluor fluorescence, indicating citral‐NP was as bioactive as free citral (Figure 4B). The loading efficiency of this citral‐NP preparation was estimated to be 12.4% (Supplementary Materials and Methods). The mean citral concentration for an individual preparation of citral‐NP was 8.05 mg/mL. Subsequently, the nanoparticle preparations were administered to mice bearing MDA‐MB‐231 ALDH1A3‐OE or vector control tumors via tail vein injections. As we previously reported, overexpression of ALDH1A3 increased tumor growth of MDA‐MB‐231 cells (Marcato et al., 2015) (Figure 4C). Citral‐NP significantly reduced tumor volume and tumor weight of MDA‐MB‐231 ALDH1A3‐OE cells but not MDA‐MB‐231 vector control cells (Figure 4C and D). This suggests that the tumor growth inhibitory effect of citral is related to its anti‐ALDH activity, and in the case of ALDH1A3 expressing breast tumor cells, its specific inhibition of ALDH1A3.
Figure 4.
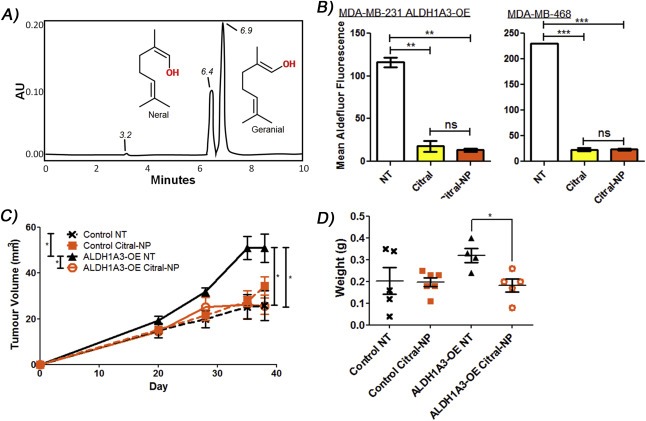
Nanoparticle encapsulated citral reduces ALDH1A3‐mediated MDA‐MB‐231 tumor growth. A) Nanoparticle encapsulated citral (citral‐NP) quantification via HPLC illustrated the two isomers present in citral and estimated the citral concentration. Three peaks present on the HPLC chromatogram represent the “non‐citral” products in the SIGMA citral (elute 3.2 min), the neral or E isomer (elute 6.4 min), and the geranial or Z isomer (elute 6.9 min). B) The effect of 100 μM citral‐NP and unencapsulated citral in the Aldefluor assay performed on MDA‐MB‐468 cells and MDA‐MB‐231 ALDH1A3‐OE cells. C) Tumor measurements in mice injected with MDA‐MB‐231 vector control or ALDH1A3‐OE cells, with or without citral‐NP treatment. D) The resulting tumor weights. B, C, D) Significance determined by one‐way ANOVA with Tukey's post‐hoc test.
3.5. Citral reduces ALDH1A3‐mediated colony formation
In order to further study the effect of citral on cancer cells, the potential mechanisms for the ALDH1A3‐specific tumor growth reduction caused by this compound were investigated. The minor apoptotic effects observed in Figure 3 upon treatment of cells with 100 μM of citral are probably insufficient to explain the observed tumor growth inhibition effects, since citral inhibits ALDH1A3 activity at much lower concentrations (i.e. 1 μM decreases Aldefluor fluorescence). Furthermore, the effect of citral on cancer cell proliferation over 72 h was not ALDH1A3‐dependent (Figure 5A). Therefore, we assessed the effects of citral on other cellular growth and signaling assays that may be specific to ALDH1A3 activity and related to the enhanced tumor growth mediated by the enzyme. Notably, the colony formation assay (clonogenic assay) quantifies the ability of a single cell to grow into a colony, and has been utilized to illustrate CSC phenotype since it measures the ability of the cells to undergo “unlimited” division (Fang et al., 2011). In both MDA‐MB‐231 and MDA‐MB‐468 cells, higher ALDH1A3 expression was associated with increased colony formation. Most importantly, citral reduced colony formation only in those cell lines with high ALDH1A3 expression (Figure 5B and C). The results from the colony formation assay mirror the tumor growth assay results (Figure 4C and D), and suggest citral may be inhibiting the enhanced ability of ALDH1A3 expressing MDA‐MB‐231 cells to form tumors.
Figure 5.
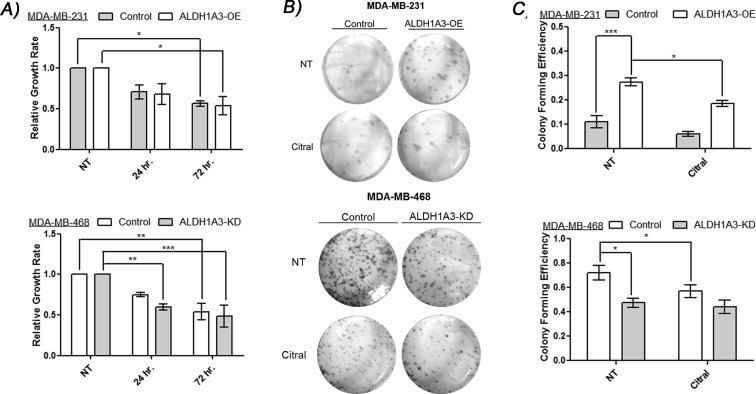
Citral reduces cell growth and ALDH1A3‐mediated colony formation. A) MDA‐MB‐231 cells with or without ALDH1A3‐OE and MDA‐MB‐468 cells with or without knockdown of ALDH1A3 were treated with citral for 72 h and their growth rate normalized to no treatment. B, C) MDA‐MB‐231 cells with or without ALDH1A3‐OE and MDA‐MB‐468 cells with or without knockdown of ALDH1A3 were pre‐treated with 100 μM citral or vehicle for 24 h prior to colony formation assay. B) Representative images of colonies. A, C) Significance determined by one‐way ANOVA with Tukey's post‐hoc test.
3.6. Citral inhibits ALDH1A3‐mediated gene expression
To investigate the mechanism by which citral exerts its anti‐tumorigenic and anti‐growth effects, we evaluated expression of genes inducible by retinoic acid, or pluripotency and markers associated with breast CSCs upon ALDH1A3 modulation and citral treatment. The effects of ALDH1A3 on MDA‐MB‐231 breast tumor growth and metastasis is dependent upon its induction of RA signaling via expression of RA‐inducible genes (Marcato et al., 2015). Furthermore, expression of RA‐inducible genes in breast cancer is specifically dependent upon expression ALDH1A3 (Marcato et al., 2015). Therefore, we evaluated the effect of citral on ALDH1A3‐induced expression of RARβ, RARRES1, and ELF3, all of which are RA‐inducible and contain retinoic acid response elements (RAREs) (Marcato et al., 2015). Overexpression of ALDH1A3 in MDA‐MB‐231 cells increased expression of RARβ, RARRES1, and ELF3 (Figure 6A), while knockdown of ALDH1A3 in MDA‐MB‐468 cells reduced expression of RARβ, RARRES1, and ELF3 (Figure 6B). Citral significantly reduced ALDH1A3‐dependent expression of RARβ, RARRES1, and ELF3 in MDA‐MB‐231 cells (Figure 6A), and reduced expression of RARβ and RARRES1 in MDA‐MB‐468 cells (Figure 6B).
Figure 6.
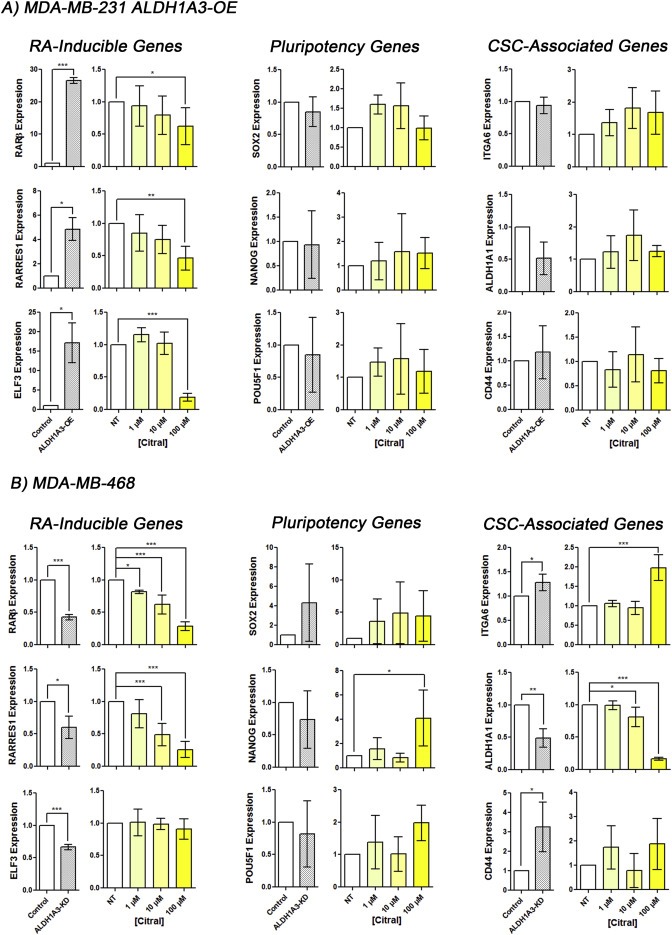
Citral reduces ALDH1A3‐mediated expression of retinoic acid‐inducible genes. Relative mRNA expression levels of retinoic acid‐inducible genes RARβ, RARRES1, and ELF3, pluripotency genes SOX2, NANOG, and POU5F1/Oct4, and CSC‐associated genes ITAG6, ALDH1A1, and CD44 was quantified by QPCR, normalized to control and B2M levels in MDA‐MB‐231 ALDH1A3‐OE cells (A) and MDA‐MB‐468 cells (B) with increasing citral treatment for 24 h compared to vehicle no treatment control. Significance of ALDH1A3‐OE compared to control MDA‐MB‐231 cells (A) and ALDH1A3 knockdown compared to control MDA‐MB‐468 cells (B) was determined by t‐test. A, B) Significance of citral treatments compared to vehicle no treatment determined by one‐way ANOVA with Dunnett's post‐hoc test.
In contrast, the expression of pluripotency and markers associated with breast CSCs was not consistently altered by ALDH1A3 expression or citral treatment MDA‐MB‐231 and MDA‐MB‐468 cells (Figure 6A and B) (Akrap et al., 2016). While ALDH1A3‐KD and citral treatment increased ITGA6 expression in MDA‐MB‐468 cells, no change in expression was observed in MDA‐MB‐231 cells. An opposite effect was seen for ALDH1A1 expression where ALDH1A3‐KD and citral treatment reduced ALDH1A1 expression in MDA‐MB‐468, though no change was observed in MDA‐MB‐231 cells. Gene expression of CSC‐associated/marker CD44 was also not consistently altered by ALDH1A3 expression or citral treatment. This result mirrors our cell surface expression analysis of breast CSC markers CD24 and CD44; neither ALDH1A3 expression nor citral treatment altered the percentage CD24−/CD44+ cells in MDA‐MB‐231 cells (Supplementary Figure 10). Together, the gene expression analysis strengthens the possibility that citral's inhibition of ALDH1A3‐mediated tumor growth is related to inhibition of ALDH1A3‐induced retinoic acid signaling.
4. Discussion
Breast CSCs are highly tumorigenic and resistant to conventional therapies (Al‐Hajj et al., 2003; Liu et al., 2014; Charafe‐Jauffret et al., 2009; Shafee et al., 2008; Diehn et al., 2009); therefore, the presence of a residual population of CSCs after treatment may increase a patient's risk for relapse and justifies a search for compounds that target CSC‐associated activity. High ALDH activity is used as a biomarker for many types of CSCs, and more recently it has been shown that ALDH1A3 has a functional role in breast cancer, lung cancer, melanoma, malignant pleural mesothelioma, and head and neck cancer (Marcato et al., 2015, 2015, 2012, 2011, 2015, 2014). To date, the inhibition of ALDH1A3 as a breast cancer therapy has not been explored.
We evaluated 12 compounds with known inhibitory activity for other ALDH isoforms (with unknown ALDH1A3 activity) (Koppaka et al., 2012), and found that citral effectively inhibited ALDH1A3. To our knowledge, this is the first time that these compounds have been comparatively investigated for ALDH inhibitory activity in live cells using the Aldefluor assay. Previous cell‐free enzymatic and/or in vivo assays implied that chloral hydrate, citral, cyanamide, daidzin, DEAB, disulfiram, gossypol, molinate, and pargyline inhibit ALDH1A1 or ALDH2 (reviewed in Koppaka et al., 2012) (Koppaka et al., 2012). Of the compounds we tested, only DEAB, citral, and benomyl significantly inhibited ALDH1A3, ALDH1A1, or ALDH2‐mediated Aldefluor fluorescence in breast cancer cells. The lack of positive results with the other compounds could be attributed to the fact that the previous studies reporting inhibition of ALDH isoforms were performed using cell‐free enzymatic assays or animal models. Several of these compounds potentially require in vivo processing, as in the case of cyanamide, which requires the action of catalase to inhibit ALDH2, or pargyline which is activated in liver microsomes. Furthermore, the efficiency by which these compounds can enter (and remain within) the cell is unknown and could contribute to their lack of effects in the present study.
Our 12 drug panel included disulfiram, a long‐used drug in the deterrence of alcohol abuse, which has also been extensively tested previously for its anti‐cancer properties (Supplementary Table 3). The most commonly reported anti‐cancer mechanism of disulfiram is the inhibition of the proteasome‐mediated degradation pathway (Chen et al., 2006). Specifically, when bound to copper, the disulfiram/copper complex is a potent proteasome inhibitor, thereby inducing apoptosis of cancer cells. In cancers with clearly elevated copper levels such as glioblastoma multiforme, disulfiram treatment induces apoptosis and re‐sensitizes the tumors to temozolomide therapy (Liu et al., 2012; Triscott et al., 2012). However, as illustrated by our findings here, it is possible that the apoptotic effects of disulfiram are not only attributable to its effects on proteasome‐inhibition, but may also be related to its ALDH enzyme inhibition activity.
Another drug tested here, gossypol, induced notable apoptosis in MDA‐MB‐231 cells. This is consistent with the previously described pro‐apoptotic activity of gossypol on breast cancer cells (Supplementary Table 3). Interest in gossypol as a breast cancer treatment stalled after a Phase I/II clinical trial determined the compound has negligible antitumor effects on refractory metastatic breast cancer. Furthermore, the lack of ALDH1A3‐specific apoptotic effects induced by gossypol decreased its relevance as a potential ALDH1A3‐specific inhibitor in this study.
Importantly, in addition to inhibiting Aldefluor fluorescence, citral treatment resulted in direct inhibition of ALDH1A3‐mediated breast tumor growth. It is probable that citral inhibits general tumor growth by several mechanisms such as autophagy or apoptosis (Zeng et al., 2015; Chaouki et al., 2009), but this is the first report of ALDH1A3‐specific inhibition resulting in decreased tumor growth. Since ALDH1A3 can mediate breast cancer growth through upregulating RA signaling (Marcato et al., 2015), and citral can inhibit ALDH1A3‐mediated expression of RA‐inducible genes, this is a likely mechanism for citral‐mediated tumor growth inhibition. The ideal cancer therapy would reduce tumor mass as well as CSCs while sparing normal cells. Notably, citral is reported to be less cytotoxic towards normal mammary epithelial cells (MCF10A) than breast cancer cell lines MCF7 and MDA‐MB‐231, suggesting a cancer‐specific effect (Patel et al., 2015). A recent report demonstrated that citral reduced tumor growth in the 4T1 syngeneic tumor model; this growth inhibition was not associated with CSC targeting (Zeng et al., 2015). In the present study we report that citral reduced tumor growth driven by the CSC marker ALDH1A3 as well as ALDH1A3‐mediated colony formation. Finally, while free citral was ineffective in vivo, nanoparticle encapsulated citral reduced ALDH1A3‐mediated tumor growth, illustrating the beneficial effects of encapsulation in the in vivo delivery of drugs in the treatment of cancer. Nanoparticle encapsulation for the delivery of anti‐cancer drugs has several advantages: particles are too large for renal clearance or penetration of the endothelial junctions of normal blood vessels, yet the particles are small enough to extravasate the “leaky” vessels surrounding tumors (Greish, 2010). These characteristics define the enhanced‐permeability and retention effect, and are likely responsible for the improved efficacy of citral‐NP relative to free citral observed in this study.
In conclusion this study conceptualizes the use of ALDH1A3‐specific inhibitors in the treatment of cancer and illustrates the proof of principle that this enzyme may be targeted to reduce tumor growth associated with ALDH1A3. It would be worthwhile to investigate whether the anti‐cancer activity of citral that is observed in endometrial cancer, ovarian cancer, cervical cancer, B‐lymphoma, and glioblastoma (Supplementary Table 3), can be partially attributed to ALDH1A3 inhibition. Having illustrated the proof of principle with citral, small molecule library screening may identify ALDH1A3 inhibitors which are effective at nM concentrations, and therefore would likely be superior novel drug candidates. Future studies with citral and other ALDH1A3‐specific inhibitors will determine if the tumor‐initiating potential and therapy resistance of Aldefluor‐positive identified CSCs can be abrogated with such inhibitors. This could lead to the adjuvant application of ALDH1A3‐specific inhibition in the treatment of certain cancers where ALDH1A3 plays a functional role in tumor growth, metastasis and chemo‐ or radio‐resistance (Marcato et al., 2015, 2015, 2012, 2011, 2015).
Financial support
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) to PM (grant # MOP‐130304) and RD (grant #'s MOP‐13723 and MOP‐57881). MLT is supported by trainee awards from the Beatrice Hunter Cancer Research Institute (BHCRI), the Canadian Breast Cancer Foundation (CBCF), the Nova Scotia Health Research Foundation, CIHR, and the Nova Scotia government. KMC is supported by trainee awards from CIHR, BHCRI, and the Canadian Imperial Bank of Commerce (CIBC). MS is supported by a traineeship award from BHCRI, CIBC, and CBCF.
Conflict(s) of interest
None.
Supporting information
The following is the supplementary data related to this article:
Supplementary data
Supplementary data 1.
1.1.
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.molonc.2016.08.004.
Thomas Margaret Lois, de Antueno Roberto, Coyle Krysta Mila, Sultan Mohammad, Cruickshank Brianne Marie, Giacomantonio Michael Anthony, Giacomantonio Carman Anthony, Duncan Roy, Marcato Paola, (2016), Citral reduces breast tumor growth by inhibiting the cancer stem cell marker ALDH1A3, Molecular Oncology, 10, doi: 10.1016/j.molonc.2016.08.004.
References
- Akrap, N. , Andersson, D. , Bom, E. , Gregersson, P. , Ståhlberg, A. , Landberg, G. , 2016. Identification of distinct breast cancer stem cell populations based on single-cell analyses of functionally enriched stem and progenitor pools. Stem cell Rep. 6, (1) 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Hajj, M. , Wicha, M. , Benito-Hernandez, A. , Morrison, S. , Clarke, M. , 2003. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. U. S. A. 100, (7) 3983–3988. 10.1073/pnas.0530291100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet, D. , Dick, J.E. , 1997. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 3, (7) 730–737. [DOI] [PubMed] [Google Scholar]
- Canino, C. , Luo, Y. , Marcato, P. , Blandino, G. , Pass, H.I. , Cioce, M. , 2015. A STAT3-NFkB/DDIT3/CEBPbeta axis modulates ALDH1A3 expression in chemoresistant cell subpopulations. Oncotarget. 6, (14) 12637–12653. 10.18632/oncotarget.3703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaouki, W. , Leger, D.Y. , Liagre, B. , Beneytout, J. , Hmamouchi, M. , 2009. Citral inhibits cell proliferation and induces apoptosis and cell cycle arrest in MCF-7 cells. Fundam. Clin. Pharmacol. 23, (5) 549–556. [DOI] [PubMed] [Google Scholar]
- Charafe-Jauffret, E. , Ginestier, C. , Iovino, F. , 2009. Breast cancer cell lines contain functional cancer stem cells with metastatic capacity and a distinct molecular signature. Cancer Res. 69, (4) 1302–1313. 10.1158/0008-5472.CAN-08-2741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, D. , Cui, Q.C. , Yang, H. , Dou, Q.P. , 2006. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 66, (21) 10425–10433. [DOI] [PubMed] [Google Scholar]
- Coyle, K.M. , Sultan, M. , Thomas, M. , Marcato, A.V.P. , 2013. Retinoid signaling in cancer and its promise for therapy. J. Carcinog. Mutagen. 2013 [Google Scholar]
- Diehn, M. , Cho, R.W. , Lobo, N.A. , 2009. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 458, (7239) 780–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, X. , Cai, Y. , Liu, J. , 2011. Twist2 contributes to breast cancer progression by promoting an epithelial–mesenchymal transition and cancer stem-like cell self-renewal. Oncogene. 30, (47) 4707–4720. [DOI] [PubMed] [Google Scholar]
- Ginestier, C. , Hur, M.H. , Charafe-Jauffret, E. , 2007. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 1, (5) 555–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greish, K. , 2010. Enhanced permeability and retention (EPR) effect for anticancer nanomedicine drug targeting. Cancer Nanotechnology: Methods Protoc. 25–37. [DOI] [PubMed] [Google Scholar]
- Han, D. , Wu, G. , Chang, C. , 2015. Disulfiram inhibits TGF-beta-induced epithelial-mesenchymal transition and stem-like features in breast cancer via ERK/NF-kappaB/snail pathway. Oncotarget. 6, (38) 40907–40919. 10.18632/oncotarget.5723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartomo, T.B. , Van Huyen Pham, T. , Yamamoto, N. , 2015. Involvement of aldehyde dehydrogenase 1A2 in the regulation of cancer stem cell properties in neuroblastoma. Int. J. Oncol. 46, (3) 1089–1098. [DOI] [PubMed] [Google Scholar]
- Khoury, T. , Ademuyiwa, F.O. , Chandraseekhar, R. , 2012. Aldehyde dehydrogenase 1A1 expression in breast cancer is associated with stage, triple negativity, and outcome to neoadjuvant chemotherapy. Mod. Pathol. 25, (3) 388–397. 10.1038/modpathol.2011.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppaka, V. , Thompson, D.C. , Chen, Y. , 2012. Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 64, (3) 520–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurth, I. , Hein, L. , Mabert, K. , 2015. Cancer stem cell related markers of radioresistance in head and neck squamous cell carcinoma. Oncotarget. 6, (33) 34494–34509. 10.18632/oncotarget.5417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, X. , Wan, L. , Geng, J. , Wu, C. , Bai, X. , 2012. Aldehyde dehydrogenase 1A1 possesses stem-like properties and predicts lung cancer patient outcome. J. Thorac. Oncol. 7, (8) 1235–1245. 10.1097/JTO.0b013e318257cc6d [DOI] [PubMed] [Google Scholar]
- Liu, P. , Brown, S. , Goktug, T. , 2012. Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br. J. Cancer. 107, (9) 1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Cong, Y. , Wang, D. , 2014. Breast cancer stem cells transition between epithelial and mesenchymal states reflective of their normal counterparts. Stem Cell Rep. 2, (1) 78–91. 10.1016/j.stemcr.2013.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P. , Wang, Z. , Brown, S. , 2014. Liposome encapsulated disulfiram inhibits NFκB pathway and targets breast cancer stem cells in vitro and in vivo. Oncotarget. 5, (17) 7471–7485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, Y. , Dallaglio, K. , Chen, Y. , 2012. ALDH1A isozymes are markers of human melanoma stem cells and potential therapeutic targets. Stem Cells. 30, (10) 2100–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoud, M.I. , Potter, J.J. , Colvin, O.M. , Hilton, J. , Mezey, E. , 1993. Effect of 4-(diethylamino) benzaldehyde on ethanol metabolism in mice. Alcohol. Clin. Exp. Res. 17, (6) 1223–1227. [DOI] [PubMed] [Google Scholar]
- Mao, P. , Joshi, K. , Li, J. , 2013. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc. Natl. Acad. Sci. U. S. A. 110, (21) 8644–8649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcato, P. , Dean, C.A. , Pan, D. , 2011. Aldehyde dehydrogenase activity of breast cancer stem cells is primarily due to isoform ALDH1A3 and its expression is predictive of metastasis. Stem Cells. 29, (1) 32–45. [DOI] [PubMed] [Google Scholar]
- Marcato, P. , Dean, C.A. , Liu, R. , 2015. Aldehyde dehydrogenase 1A3 influences breast cancer progression via differential retinoic acid signaling. Mol. Oncol. 9, (1) 17–31. 10.1016/j.molonc.2014.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreb, J.S. , Ucar, D. , Han, S. , 2012. The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance. Chem. Biol. Interact. 195, (1) 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy, J.P. , Everley, R.A. , Coloff, J.L. , Gygi, S.P. , 2014. Combining amine metabolomics and quantitative proteomics of cancer cells using derivatization with isobaric tags. Anal. Chem. 86, (7) 3585–3593. 10.1021/ac500153a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel, P.B. , Thakkar, V.R. , Patel, J.S. , 2015. Cellular effect of curcumin and citral combination on breast cancer cells: induction of apoptosis and cell cycle arrest. J. Breast Cancer. 18, (3) 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian, X. , Wagner, S. , Ma, C. , 2014. Prognostic significance of ALDH1A1-positive cancer stem cells in patients with locally advanced, metastasized head and neck squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 140, (7) 1151–1158. 10.1007/s00432-014-1685-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raha, D. , Wilson, T.R. , Peng, J. , 2014. The cancer stem cell marker aldehyde dehydrogenase is required to maintain a drug-tolerant tumor cell subpopulation. Cancer Res. 74, (13) 3579–3590. 10.1158/0008-5472.CAN-13-3456 [DOI] [PubMed] [Google Scholar]
- Shafee, N. , Smith, C.R. , Wei, S. , 2008. Cancer stem cells contribute to cisplatin resistance in Brca1/p53–Mediated mouse mammary tumors. Cancer Res. 68, (9) 3243–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao, C. , Sullivan, J.P. , Girard, L. , 2014. Essential role of aldehyde dehydrogenase 1A3 for the maintenance of non-small cell lung cancer stem cells is associated with the STAT3 pathway. Clin. Cancer Res. 20, (15) 4154–4166. 10.1158/1078-0432.CCR-13-3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takebe, N. , Harris, P.J. , Warren, R.Q. , Ivy, S.P. , 2011. Targeting cancer stem cells by inhibiting wnt, notch, and hedgehog pathways. Nat. Rev. Clin. Oncol. 8, (2) 97–106. [DOI] [PubMed] [Google Scholar]
- Triscott, J. , Lee, C. , Hu, K. , 2012. Disulfiram, a drug widely used to control alcoholism, suppresses the self-renewal of glioblastoma and over-rides resistance to temozolomide. Oncotarget. 3, (10) 1112–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno, T. , Masuda, H. , Ho, C. , 2004. Formation mechanism of p-methylacetophenone from citral via a tert-alkoxy radical intermediate. J. Agric. Food Chem. 52, (18) 5677–5684. [DOI] [PubMed] [Google Scholar]
- van den Hoogen, C. , van der Horst, G. , Cheung, H. , Buijs, J.T. , Pelger, R.C. , van der Pluijm, G. , 2011. The aldehyde dehydrogenase enzyme 7A1 is functionally involved in prostate cancer bone metastasis. Clin. Exp. Metastasis. 28, (7) 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasiliou, V. , Nebert, D.W. , 2005. Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum. Genomics. 2, (2) 138–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Chen, X. , Zhan, Y. , 2013. Increased expression of ALDH1A1 protein is associated with poor prognosis in clear cell renal cell carcinoma. Med. Oncol. 30, (2) UNSP 574 10.1007/s12032-013-0574-z [DOI] [PubMed] [Google Scholar]
- Yang, L. , Ren, Y. , Yu, X. , 2014. ALDH1A1 defines invasive cancer stem-like cells and predicts poor prognosis in patients with esophageal squamous cell carcinoma. Mod. Pathol. 27, (5) 775–783. 10.1038/modpathol.2013.189 [DOI] [PubMed] [Google Scholar]
- Yue, L. , Huang, Z.M. , Fong, S. , 2015. Targeting ALDH1 to decrease tumorigenicity, growth and metastasis of human melanoma. Melanoma Res. 25, (2) 138–148. 10.1097/CMR.0000000000000144 [DOI] [PubMed] [Google Scholar]
- Zeng, S. , Kapur, A. , Patankar, M.S. , Xiong, M.P. , 2015. Formulation, characterization, and antitumor properties of trans- and cis-citral in the 4T1 breast cancer xenograft mouse model. Pharm. Res. 32, (8) 2548–2558. 10.1007/s11095-015-1643-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The following is the supplementary data related to this article:
Supplementary data