

Abstract
Cancer cell migration and invasion underlie metastatic dissemination, one of the major problems in cancer. Tumour cells exhibit a striking variety of invasion strategies. Importantly, cancer cells can switch between invasion modes in order to cope with challenging environments. This ability to switch migratory modes or plasticity highlights the challenges behind antimetastasis therapy design. In this Review, we present current knowledge on different tumour invasion strategies, the determinants controlling plasticity and arising therapeutic opportunities. We propose that targeting master regulators controlling plasticity is needed to hinder tumour dissemination and metastasis.
Keywords: actomyosin contractility, cancer metastasis, invasion, plasticity, Rho GTPases
Abbreviations
- Cdc42
cell division cycle 42
- ECM
extracellular matrix
- EMT
Epithelial‐to‐mesenchymal transition
- ERM
ezrin/radixin/moesin
- ERULS
ezrin‐rich uropod‐like structure
- ESCRT
endosomal sorting complexes required for transport
- GAP
GTPase‐activating proteins
- GEF
guanine exchange factor
- LIMK
LIM kinase
- MAT
mesenchymal‐to‐amoeboid transition
- MLCK
myosin light chain kinase
- MMP
matrix metalloproteinase
- MRCK
myotonic dystrophy kinase‐related Cdc42‐binding kinase
- MYPT1
myosin phosphatase target subunit‐1
- NMII
nonmuscle myosin II
- PAK
p21‐associated kinases
- PIG3
p53‐induced gene 3 protein
- Rac
Ras‐related C3 botulinum toxin substrate
- Rho
Ras homolog family member
- RLC
regulatory light chain
- ROCK
Rho‐associated coiled‐coil‐containing protein kinase
- ROS
reactive oxygen species
- SDF‐1
stromal cell‐derived factor 1
- uPA
urokinase plasminogen activator
- uPAR
uPA receptor
- ZIPK
zipper‐interacting protein kinase
1. Cancer cell invasion and dissemination
Abnormal tumour cell migration and invasion underlies metastatic dissemination, a major clinical problem in cancer (Sanz‐Moreno and Marshall, 2010). Metastasis is a multistage process involving cell migration and invasion, transit in the blood or lymph, extravasation and colonization in the secondary site. Acquisition of invasive behaviour involves activation of signalling pathways controlling cytoskeletal dynamics, as well as turnover of cell–matrix and cell–cell adhesions (Fig. 1; Friedl and Alexander, 2011). Cancer invasion is a heterogeneous and adaptive process involving changes in cell morphology and generation of cell polarity, resulting in translocation of the cell body. Cancer cells display exceptional ability to adapt to different environmental conditions engaging in different migration strategies, as reviewed in Clark and Vignjevic (2015); Friedl and Alexander (2011); Sahai (2005). Cancer cells can migrate either individually in the absence of cell–cell junctions, or collectively upon retention of cell–cell adhesions (Friedl and Alexander, 2011; Fig. 1). In turn, cancer cells can use a number of strategies when migrating individually (elongated‐mesenchymal, rounded‐amoeboid, spike‐mediated) or collectively (multicellular streaming, tumour budding, collective invasion; Fig. 1). Studies using histopathological human samples and intravital imaging of xenografted tumours in mice have shown that these strategies can be observed in vivo, as reviewed in Clark and Vignjevic (2015); Friedl and Gilmour (2009); Friedl et al. (2012). While collective cell migration allows entry into the lymphatic system, individual cell migration is essential for entry into the bloodstream and dissemination to distant sites (Giampieri et al., 2009).
Figure 1.
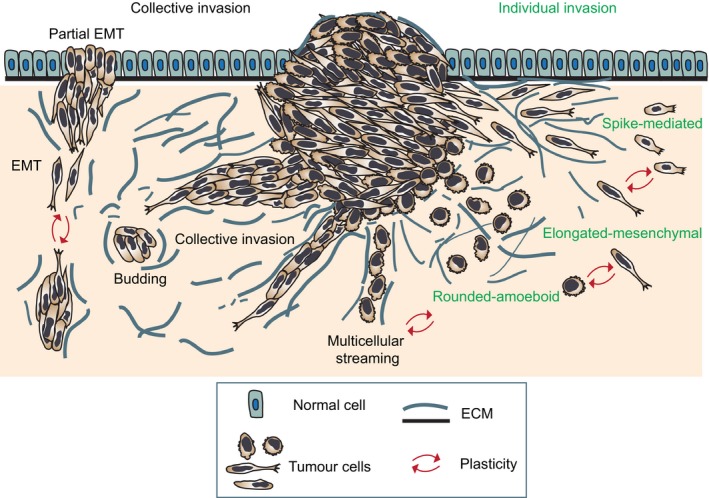
Modes of invasion during tumour dissemination. Diagram showing the main individual and collective modes of tumour invasion and plasticity that allows interconversion between modes. Cells invading individually can use protrusion‐based elongated‐mesenchymal, bleb‐ and contractility‐driven rounded‐amoeboid and filopodial spike‐mediated strategies. When cell–cell junctions are maintained, cells can move collectively as multicellular streams, budding or larger clusters (collective invasion). Migratory plasticity drives interconversion between the different modes.
In this Review, we describe the different individual and collective modes of invasion, the plasticity that cancer cells display, enabling them to switch between different migratory modes and the determinants of this plasticity. We also discuss the therapeutic challenges arising from migratory plasticity that could explain failure of some therapies, and the potential targets that could lead to a complete blockade in cancer cell migration and invasion. We propose that targeting master regulators controlling plasticity is needed to hinder tumour dissemination and metastasis. While this Review tries to cover the different modes of migration and key aspects of migratory plasticity during invasion and metastasis, it is beyond the scope of this work to provide detailed insight into each section. Hence, throughout the Review, readers are directed to other excellent reviews that cover the relevant topic in depth.
2. Cell migration mechanisms
The molecular interactions between F‐actin and nonmuscle myosin II (NMII) govern the generation of mechanical forces across diverse length scales, and these are important not only for migration (Murrell et al., 2015; Vicente‐Manzanares et al., 2009) but also for modulating cytokinesis (Green et al., 2012) and tissue morphogenesis (Murrell et al., 2015; Salbreux et al., 2012).
During cell migration (Fig. 1), directional polarity is achieved by cells generating a leading edge at the front and a lagging edge at the back (reviewed in Ridley, 2015). Protrusion and adhesion of the leading edge and retraction of the rear edge drive movement in the direction of locomotion (Richardson and Lehmann, 2010). The dynamics of cytoskeletal coupling with cell surface receptors that engage with surrounding tissue structures is the key process underlying all forms of migration (Friedl and Alexander, 2011).
Cell migration is a cyclic process (Friedl and Wolf, 2009; Lauffenburger and Horwitz, 1996) that begins with actin polymerization on one side of the cell resulting in actin‐rich protrusion at the leading edge. Migration is facilitated by the forward movement of the cell, which is achieved by the engagement of cell surface receptors with the extracellular matrix (ECM); the formation of leading edge adhesions associated with proteolytic degradation of the ECM; and actomyosin contractility‐mediated retraction of the rear edge of the cell.
Actin polymerization and organization into different cytoskeletal structures is regulated by the Rho family of proteins that play a central role in cell migration and has been extensively reviewed in Ridley (2015). Rho GTPases are molecular switches that cycle between active states when bound to GTP and inactive states when bound to GDP. This is regulated by activators or guanine exchange factors (GEFs) and inactivators or GTPase‐activating proteins (GAPs; Ridley, 2015). By interacting with specific downstream effectors, active GTPases induce diverse actin rearrangements (Heasman and Ridley, 2008).
Three prototypical members of the family, Ras‐related C3 botulinum toxin substrate (Rac), Ras homolog family member (Rho) and cell division cycle 42 (Cdc42), have been extensively linked to cell migration regulation (Ridley, 2015). Rho induces unbranched actin polymerization via formin mDia1, while Rho‐associated coiled‐coil‐containing protein kinase (ROCK) promotes bundling of actomyosin filaments resulting in either stress fibres or an actomyosin cortex (Kimura et al., 1996; Otomo et al., 2005). Activation of ROCK downstream of Rho results in activating phosphorylation of myosin II (Amano et al., 1996) and inactivation of myosin phosphatase target subunit‐1 (MYPT1; Kimura et al., 1996). Phosphorylated myosin II drives contraction of actin fibres in an ATP‐dependent manner (Scholey et al., 1980; Wang et al., 2003). In addition to myosin II, ROCK can also phosphorylate ezrin/radixin/moesin (ERM), LIM kinases (LIMK1, LIMK2), α‐adducin and several other proteins important for migration (Kimura et al., 1996; Matsui et al., 1998; Ohashi et al., 2000).
Rac and Cdc42 also regulate actin polymerization (Ridley et al., 2003; Wojciak‐Stothard and Leiper, 2008). Binding of Cdc42 to myotonic dystrophy kinase‐related Cdc42‐binding kinase (MRCK) results in phosphorylation of myosin II, MYPT1, LIMK1, LIMK2 and moesin (Leung et al., 1998; Nakamura et al., 2000; Scott and Olson, 2007; Tan et al., 2001b). The activation of LIM kinases by phosphorylation allows for the inactivating phosphorylation of actin‐severing protein cofilin, which inhibits actin depolymerization (Maekawa et al., 1999; Sumi et al., 1999). Rac proteins interact with lamellipodin and the WAVE complex that, in turn, promote actin nucleation by the Arp2/3 complex (Law et al., 2013; Ridley, 2015).
Another set of downstream effectors of Rac and Cdc42 include the p21‐associated kinases (PAKs). PAK1 promotes motility by inducing rapid turnover of focal contacts at leading edge of cells via phosphorylation of paxillin (Brown et al., 2002; Nayal et al., 2006; Premont et al., 2004). PAK‐mediated actin remodelling also involves LIMK1 (Edwards et al., 1999; Yang et al., 1998).
Actin polymerization by Rho GTPases directs the forces generated by actomyosin contractility needed for migration to take place. F‐actin polymers serve as the scaffold for myosin II motors and accessory proteins (Murrell et al., 2015; Vicente‐Manzanares et al., 2009) that can walk along, propel the sliding of or produce tension on actin filaments via ATPase activity (Vicente‐Manzanares et al., 2009). Depending on the location of myosin with respect to the middle filaments, this can result in the contraction or extension of two bound actin filaments. The contractile activity of NMII can be regulated via reversible phosphorylation of Ser19 on the regulatory light chain (RLC; Hirata et al., 2009) by ROCK, myosin light chain kinase (MLCK) and other kinases such as MRCK, citron kinase, LIMK, zipper‐interacting protein kinase (ZIP kinase) and Ca2+/calmodulin‐dependent protein (Endo et al., 2004; Kimura et al., 1996; Madaule et al., 1998; Poperechnaya et al., 2000; Tan et al., 2001a). Subsequent phosphorylation at Thr18 of the RLC further increases the contractile activity of myosin II (Hirata et al., 2009; Umemoto et al., 1989). For detailed function of myosin II, readers are referred to the review (Vicente‐Manzanares et al., 2009).
3. Collective modes of cancer invasion
While the models above tend to focus on cells migrating as separate entities, cancer cell invasion is not restricted to cells moving individually. Histopathological samples show invasion of normal tissue by compact groups or clusters of cells and strands or cords of connected tumour cells (Clark and Vignjevic, 2015; Friedl and Gilmour, 2009; Friedl et al., 2012; Leighton et al., 1960; Wang et al., 2016; Willis, 1952). Likewise, intravital microscopy and in vitro studies have shown that cancer cells can move as loosely/nonadherent ‘streams’ of cells or collective migration of cell strands and sheets (Alexander et al., 2008; Clark and Vignjevic, 2015; Friedl et al., 2012). At the invasive front (tumour border) of certain cancer types, such as some carcinomas, invasive cells are observed to migrate as collective groups (Christiansen and Rajasekaran, 2006; Friedl et al., 1995, 2004). Furthermore, collective cancer invasion can be seen as a dysregulated recapitulation of key steps that occur in many physiological processes such as embryonic morphogenesis or regeneration and tissue repair after wounding (Friedl and Gilmour, 2009).
Transition from collective to single‐cell invasion may enhance metastatic efficiency and has been reviewed in Friedl et al. (2012). However, intravasation into lymphatic vessels can be efficiently performed by cell groups or clusters (Byers et al., 1995; Giampieri et al., 2009; Hashizume et al., 1996; Madhavan et al., 2001). This is also supported by the existence of circulating tumour clusters from patient peripheral blood samples (Aceto et al., 2014; Brandt et al., 1996; Hart, 2009; Hou et al., 2011; Kats‐Ugurlu et al., 2009; Khoja et al., 2014).
Similar to single‐cell migration, collective cell movement results from the coordinated actions of the actin cytoskeleton, actomyosin contraction, cell polarity and cell surface receptors that engage with surrounding tissue structures (Friedl and Alexander, 2011; Ridley et al., 2003). While collective cell migration also follows the cyclical process described above for single‐cell migration (Friedl and Wolf, 2009; Lauffenburger and Horwitz, 1996), in collective movement cells remain grouped by cell–cell junctions (Friedl et al., 2004, 2012; Rorth, 2007). Protrusion extension and retraction are coordinated in a ‘supracellular manner’, in which cytoskeletal protrusion and contractility are mechanically mediated through cell–cell junctions and involve several cells (Friedl et al., 1995; Hegerfeldt et al., 2002; Hidalgo‐Carcedo et al., 2011; Tambe et al., 2011). Therefore, collective cell migration involves coordinating cell movement with ‘supracellular’ polarity, cytoskeletal organization and cell–cell junction stability (Friedl and Gilmour, 2009; Friedl et al., 2012).
Both histopathological studies of cancer tissues and those using intravital microscopy have shown distinct modes of collective cancer migration (Fig. 1), as reviewed in Clark and Vignjevic (2015); Friedl and Gilmour (2009); Friedl et al. (2012). These sometimes overlapping strategies are determined by a combination of parameters such as degree of cell–cell adhesion, cellular morphology and supracellular coupling of cell–cell signalling (Friedl et al., 2012).
3.1. Multicellular streaming
During multicellular streaming, cells move one after the other in the same path within the tissue (Fig. 1; Friedl et al., 2012; Friedl and Wolf, 2003; Manning et al., 2015). In this migratory mode, cells are typically guided by chemokine or morphogen gradients or ECM structures (i.e. ‘microtracks’; Friedl et al., 1997; Haeger et al., 2015). Hence, coordinated migration takes place as directed movement of small strands of single cells, multicellular streams and as diffuse infiltration (‘chain‐ or swarm‐like’; Friedl and Alexander, 2011; Kedrin et al., 2008; Patsialou et al., 2013; Roussos et al., 2011; Seftor et al., 2002; Wyckoff et al., 2004). These chains (‘Indian files’) have been observed in infiltrating breast carcinoma (Page and Anderson, 1987; Pitts et al., 1991), ovarian cancer (Sood et al., 2001) and melanoma (Friedl and Wolf, 2008; Seftor et al., 2002). Importantly, in this mode of migration, each cells’ cytoskeleton acts independently to generate traction force on the matrix, while cell–cell adhesions are weak or short‐lived (Friedl et al., 2012), allowing velocities similar to those achieved by cells migrating individually (1–2 μm·min−1 or even faster; Clark and Vignjevic, 2015; Friedl et al., 2012). Streaming cells can display rounded‐amoeboid or elongated‐mesenchymal phenotypes (Clark and Vignjevic, 2015; Friedl and Alexander, 2011; Friedl et al., 2012). Intravital studies have shown that cells that display rounded‐amoeboid morphology in vitro, such as human and mouse melanoma cells, are more likely to migrate as single cells or as streams in vivo (Herraiz et al., 2016; Manning et al., 2015; Pinner and Sahai, 2008a,b; Sanz‐Moreno et al., 2008).
3.2. Tumour budding
Scattered clusters of approximately five cells (‘tumour buds’) located in close proximity ahead of the invasive front (Fig. 1) have also been observed in colorectal cancer (Brabletz et al., 2001; Bronsert et al., 2014; Carr et al., 1986; Prall et al., 2005) and carcinomas from the oesophagus, pancreas, lung and breast (reviewed in Grigore et al., 2016). Studies using 3D reconstructions from 2D serial sections of colorectal (Carr et al., 1986) and other cancer types (pancreatic, lung, breast; Bronsert et al., 2014) demonstrated that tumour budding is a dynamic process by which the tumour mass extends several finger‐like multicellular projections that, later, break away from the main tumour mass as small cell clusters (tumour buds; Bronsert et al., 2014; Carr et al., 1986). Importantly, tumour budding has been associated with poor cancer outcomes (Grigore et al., 2016).
3.3. Collective cell invasion
This mode involves compact and cohesive cell groups with two or more neighbouring cells (Fig. 1). Collective invasion is facilitated by long‐lived cell–cell junctions (Alexander et al., 2008; Friedl et al., 1995, 2012). Cells may adopt different morphologies depending on cell type and number and the structure of the tissue invaded (Friedl and Alexander, 2011).
These groups can be composed of small clusters, solid strands or files (1–2 cells in diameter) up to broad masses (Wolf et al., 2007) that can even form an inner lumen if epithelial polarity is maintained, as seen in some breast, prostate, pancreatic and colorectal tumours (Christiansen and Rajasekaran, 2006; Friedl and Gilmour, 2009; Friedl et al., 2012; Nabeshima et al., 1999). Protruding sheets and strands that remain in contact with the primary site and generate local invasion have been detected in invasive epithelial tumours such as oral squamous cell carcinoma and mammary carcinoma (Bell and Waizbard, 1986; Page and Anderson, 1987), colon carcinoma (Nabeshima et al., 1999), basal cell carcinoma and others (Friedl and Wolf, 2003). Cell clusters or ‘nests’ that detach from the primary tumour and extend into surrounding tissue have been described in epithelial cancers, melanoma and rhabdomyosarcoma (Ackerman and Ragaz, 1984; Bell and Waizbard, 1986; Nabeshima et al., 1999; Page and Anderson, 1987).
In the most cases, the leading edge of the multicellular group is composed of one or several leader cells with mesenchymal characteristics (Fig. 1). Leader cells extend actomyosin‐mediated actin‐rich protrusions that generate integrin‐mediated forward traction (Hegerfeldt et al., 2002) and pericellular proteolysis towards the tissue structure (Nabeshima et al., 2000; Wolf et al., 2007), which yields a re‐aligned ECM that guides the group (Fig. 2A; Gaggioli et al., 2007; Khalil and Friedl, 2010). Following cells are passively dragged behind along the established migration track by cell–cell adhesion (Fig. 2A; Friedl et al., 1995). Nevertheless, follower cells reinforce this ECM alignment and increase the diameter of the invading strand (Friedl and Wolf, 2008).
Figure 2.
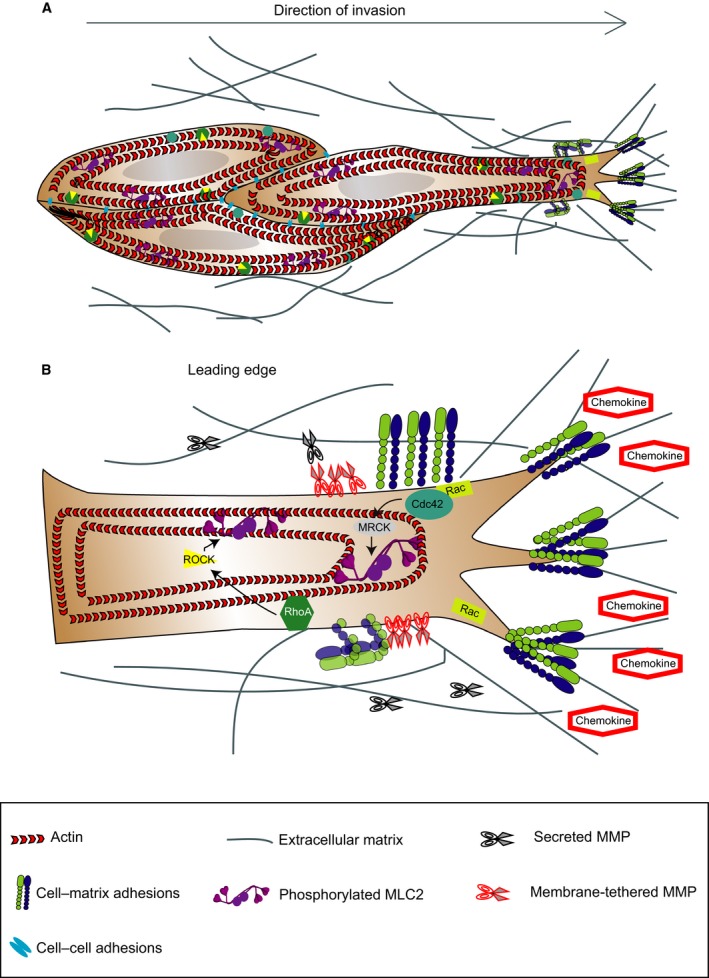
Signalling pathways controlling collective modes of invasion. (A) Diagram showing the key regulators of collective migration. The leading edge of the multicellular group comprises one (or several) leader cells with mesenchymal characteristics. Leader cells extend actomyosin‐mediated actin‐rich protrusions that generate integrin‐mediated forward traction and pericellular proteolysis yielding a re‐aligned ECM that guides the group. Following cells are passively dragged behind along the established migration track by cell–cell adhesion. (B) Diagram showing the intracellular pathways activated in response to external stimuli and proteolysis of ECM. Membrane receptors such as β1 integrins control migration of individual elongated‐mesenchymal cells. Rac activation at the leading edge allows for protrusion formation that is linked to a ‘supracellular’ cytoskeleton. Activation of myosin II‐based contractile forces by Rho‐ROCK and Cdc42‐MRCK signalling allows for contraction of cell body and retraction of the rear.
From studies using intravital microscopy, breast cancer cells or fibrosarcoma cells with predominantly individual phenotypes (Alexander et al., 2008; Giampieri et al., 2009; Roussos et al., 2011) are more prone to switching between single‐cell and collective migration modes (see ‘Plasticity during collective invasion’ section). Collective invasion is typically the slowest migratory mode (0.01–0.05 μm·min−1; Weigelin et al., 2012). Looking for advantages of this slower mode of migration, it has been suggested that the large cell mass could secrete high concentrations of promigratory factors and matrix proteases and protect inner cells from immune clearing. In addition, more migratory clones within the group could promote invasion of less motile cells, thereby increasing overall tumour invasion (Friedl and Wolf, 2003).
4. Plasticity during collective invasion
Extrinsic and intrinsic factors determine the adaptation of tumour cells to modify their migration mechanism (Friedl, 2004; Friedl and Alexander, 2011). This adaptive, dynamic behaviour is termed plasticity of tumour cell migration, and it is a combination of specific morphologic and mechanistic entities (Fig. 1). However, cells often display heterogeneity and can exhibit multiple modes of migration in 3D tissues (Fig. 1; Friedl and Wolf, 2010; Wolf et al., 2003). Furthermore, some cancer cells can spontaneously switch between different modes of migration (Sanz‐Moreno et al., 2008).
Extensive research has been performed in the last 15 years trying to understand the mechanisms supporting different types of migration and the signals and conditions that trigger tumour cell plasticity (Friedl and Alexander, 2011; Lauffenburger and Horwitz, 1996; Ridley et al., 2003; Sanz‐Moreno and Marshall, 2010). By understanding this complex array of extracellular and intracellular determinants, the general machinery governing most types of cancer migration could be identified holding promise to translation into therapeutic interventions.
4.1. Epithelial‐to‐mesenchymal transition (EMT) and partial EMT
In epithelial cancers, EMT is a molecular programme characterized by loss or weakening of cell–cell junctions, which disrupts apico‐basal polarity and cell anchoring to the basement membrane (Thiery et al., 2009). This leads, in turn, to individual cell migration with enhanced migratory and invasive capacity, increased resistance to apoptosis and augmented ECM production (Kalluri, 2009). EMT can be complete or partial depending on the degree of cell–cell adhesion (Fig. 1). Therefore, EMT‐like dissemination without the typical EMT‐associated gene expression patterns has been observed (Christiansen and Rajasekaran, 2006; Gavert et al., 2011; Wicki et al., 2006). Colorectal carcinomas often display cohesive cells at the leading edge, small groups of cells and individual cells scattered without connection to the main tumour, indicative of different degrees of EMT (Brabletz et al., 2001; Gavert et al., 2007).
4.2. Collective‐to‐individual transition
When cell–cell and cell–ECM interactions are simultaneously weakened, a transition from collective invasion to single‐cell migration takes place (Fig. 1; Friedl, 2004). In multicellular clusters invading away from melanoma explants, the inhibition of β1 integrin by blocking antibodies abolishes collective movement by inducing the detachment of individual cells (Hegerfeldt et al., 2002). This mechanism could involve an intermediate mesenchymal migration step that would later lead to rounded‐amoeboid single‐cell dissemination (Friedl, 2004). Collective invasion from fibrosarcoma and breast carcinoma spheroids can be abolished by proteolytic inhibition or by collagenase MT1‐MMP knock‐down, leading to nonproteolytic single‐cell dissemination (Wolf et al., 2007).
4.3. Determinants of plasticity
The ability to switch between various modes of migration is regulated by signalling pathways and sustained via transcriptional programmes. This, in turn, can facilitate efficient invasion and distant metastasis by conferring increased resistance to external stimuli and adaptability to different microenvironments. Plasticity requires integration of intracellular and extracellular physical and molecular cues (Friedl, 2004; Salbreux et al., 2012). In this section, we describe how cancer cells translate extracellular signals into intracellular responses that impact the mode of migration.
Factors determining plasticity during collective migration include physical cues and molecular cues (Fig. 2).
4.3.1. Physical cues
The molecular and physical characteristics of the ECM, such as composition, geometry, porosity, alignment and stiffness, strongly contribute to cell adhesion, migration and invasion (Wolf and Friedl, 2011). As such, pericellular proteolysis generated by tumour‐ and stromal cell‐derived proteases generates micro‐ and macrotracks (micro‐ and macropatterning, respectively; Friedl and Wolf, 2008) surrounded by collagen bundles that support collective invasion (Friedl et al., 1997; Gaggioli et al., 2007; Wolf et al., 2007). In addition, force‐mediated ECM remodelling favours collective breast carcinoma cell invasion (Provenzano et al., 2008; Fig. 2A). Mechanical cues affecting modes of cell migration include confinement and topology, among other factors (Kurniawan et al., 2016).
4.3.2. Molecular cues
4.3.2.1. Proteases
Tumour invasion and progression have been linked to upregulation of proteases (Egeblad and Werb, 2002; Wolf and Friedl, 2011) with highest levels of activated proteases expressed at the tumour–stromal interface (Sternlicht et al., 2000). These proteases include matrix metalloproteinases (MMPs), ADAMs, cathepsins, urokinase plasminogen activator (uPA) and its receptor uPAR (Mason and Joyce, 2011; Rizki et al., 2008). Proteases contribute towards ECM degradation and tissue remodelling to form ECM bundles as well as generation of active epitopes of ECM components (Gaggioli et al., 2007; Kenny et al., 2008). The localized cleavage of ECM fibres by proteases results in release of ECM‐imposed confinement, allowing the relaxation of the nucleus and enhancing migration speeds (Wolf et al., 2007, 2013). As a consequence, the degree of proteolytic cleavage of ECM determines the degree of deformation and the confinement experienced by the cell.
During collective migration, cells at the leading edge of collectively invading colorectal carcinomas show increased expression and activity of membrane‐tethered MT1‐MMP and secreted MMP2, leading to polarized ECM degradation (Nabeshima et al., 2000; Fig. 2B). MT1‐MMP is essential in collagen processing and multicellular strand formation during collective invasion of fibrosarcoma cells (Wolf et al., 2007).
4.3.2.2. Membrane receptors
Extracellular matrix‐binding molecules also determine the mode of invasion. Integrins couple the ECM to the actin cytoskeleton and develop small focal complexes (Friedl and Wolf, 2003; Hynes, 2002), which allow Rho GTPase‐mediated outside‐in signalling (Geiger and Peeper, 2009; Grashoff et al., 2010; Hodivala‐Dilke et al., 1999; Lee et al., 2009; Ridley et al., 2003; Fig. 2B).
β1 Integrins can control migration of multicellular melanoma (Hegerfeldt et al., 2002) and ovarian carcinoma (Casey et al., 2001).
CD44 binds to different ECM proteins (Zoller, 2011) and connects to the actin cytoskeleton through the ERM complex and ankyrin, signalling also through Rho GTPases (Zoller, 2011). CD44 serves also as a co‐receptor for other adhesion molecules such as integrins and podoplanin; the latter signals to enhance RhoA activity, increasing collective invasion of squamous cell carcinomas (Martin‐Villar et al., 2006).
DDR family of receptors interact with fibrillar collagen and signal through several intracellular pathways (STAT5, NF‐kB, p38 MAPK/ERK and Src‐family kinases; Neuhaus et al., 2011; Vogel et al., 2006). When co‐engaged with DDR1, E‐cadherin signalling limits excessive actomyosin contractility along cell–cell junctions; this stabilizes junctions and, in turn, maintains collective invasion (Hidalgo‐Carcedo et al., 2011).
In addition to cell–matrix adhesion, collective migration is also enabled by cell–cell adhesions through different adhesion systems, such as cadherins, tight junctions, gap junctions and others (Friedl et al., 2012; Hegerfeldt et al., 2002; Hidalgo‐Carcedo et al., 2011; Fig. 2A). Loss or downregulation of E‐cadherin expression that drives EMT seems to be tunable, therefore leading to complete or partial EMT. In the latter, different E‐cadherin levels that do not confound migration may be retained, or alternative proinvasive cadherins including N‐ or VE‐cadherin may be expressed (Yano et al., 2004). Collective invasion with E‐cadherin in cell–cell junctions can be facilitated upon upregulation of L1‐CAM (Gavert et al., 2011; Shtutman et al., 2006) and upregulation of podoplanin, which activates RhoA (Wicki et al., 2006).
4.3.2.3. Secreted factors
Extracellular chemokines, cytokines and growth factors secreted by tumour or stromal cells enable and promote migration in a paracrine and autocrine fashion (Friedl and Alexander, 2011; Haeger et al., 2015). In addition, ECM degradation allows the release of these factors that can also be processed by proteases resulting in their activation, inactivation or degradation (Dean et al., 2008; Mu et al., 2002; Shiao and Coussens, 2010; Sounni et al., 2010).
Invasion‐promoting chemokines, growth factors and their receptors engage intracellular signalling networks (JAK, PI3K, Src, ERK) and/or Rho GTPase activity (Friedl and Alexander, 2011; Fig. 2B). Collective invasion of oral squamous carcinoma cells is stimulated by stromal cell‐derived factor 1 (SDF‐1) and HGF secreted from stromal fibroblasts in response to tumour‐derived IL‐1α (Daly et al., 2008). Likewise, a paracrine loop between tumour‐associated macrophages secreting EGF and breast carcinoma cells secreting CSF‐1 drives cancer cell migration (Wyckoff et al., 2004).
4.3.2.4. Intracellular signalling pathways
Effective collective migration requires supracellular coordination of the cytoskeleton, which is controlled by Rho GTPase signalling (Friedl and Alexander, 2011). Leader cells generate actomyosin‐ and integrin‐mediated traction towards the ECM, controlling tensional regulation of ECM alignment (Hegerfeldt et al., 2002). High Cdc42/MRCK‐ and ROCK‐mediated actomyosin contractility levels are found at the edges of groups of invading cancer cells (Gaggioli et al., 2007; Fig. 2B). Actomyosin contractility generates pulling forces between the substrate and the follower cells, which, together with cortical actomyosin at lateral regions of the groups, maintain coupling between cells and collective forward movement in melanoma (Hegerfeldt et al., 2002) and squamous cell carcinoma (Gaggioli et al., 2007; Hidalgo‐Carcedo et al., 2011). Cell contractility mediated by Rho/ROCK/MLCK is also required for retraction of the tail in migrating groups and for lateral mechanocoupling via cadherin‐based adhesions (Vicente‐Manzanares et al., 2009).
5. Individual cancer cell invasion
Cancer cells can also invade individually in the absence of cell–cell junctions using a variety of strategies (Fig. 1).
5.1. Elongated‐mesenchymal mode of invasion
On stiff 2D matrices and 3D matrices such as collagen I, adherent cancer cells arising from connective tissues, such as sarcomas, gliomas and some epithelial cancers (Paulus et al., 1996; Polette et al., 1998; Wolf et al., 2003), can adopt actin‐rich protrusions for migration. During this mode of migration, cells have an elongated morphology (Fig. 3) that is characterized by focal adhesion formation, MMP activity and actomyosin contractility localized at the rear of the cells. The requirement of strong focal adhesion limits velocity for cells adopting elongated‐mesenchymal mode of migration resulting in relatively slow speed (0.1–2 μm·min−1 in vitro; Friedl, 2004).
Figure 3.
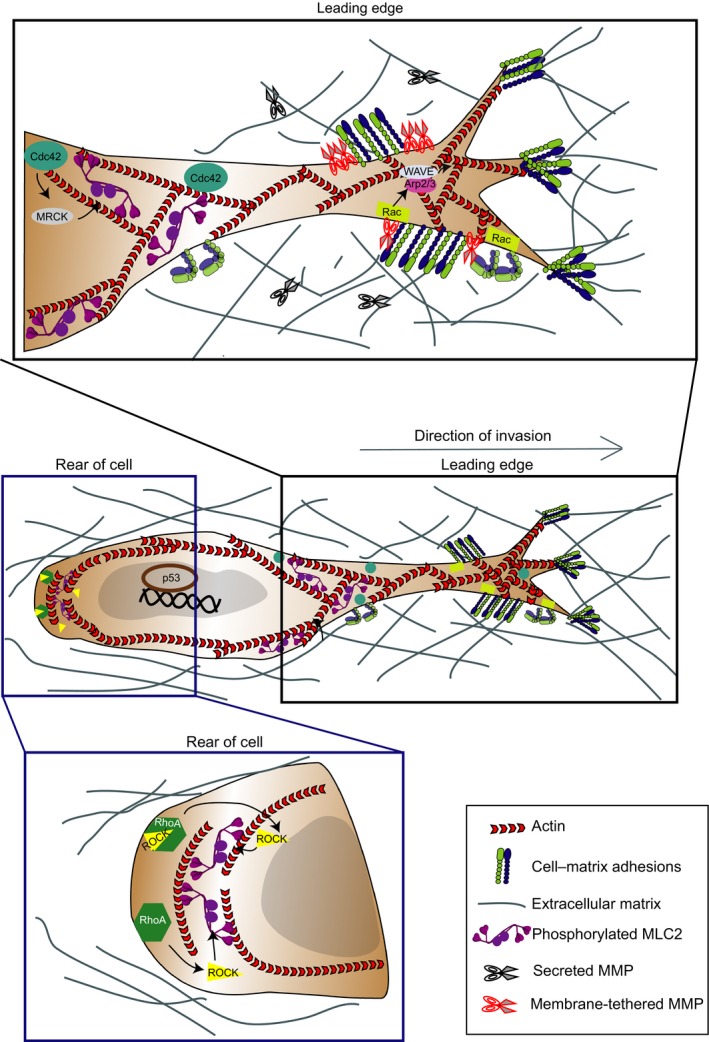
Signalling pathways controlling elongated‐mesenchymal mode of invasion. Diagram showing key regulators of elongated‐mesenchymal mode of migration in cells. During this mode, cells adopt an elongated morphology that is characterized by actin‐rich protrusions, focal adhesion formation, matrix metalloproteinase (MMP) activity and actomyosin contractility localized at the rear of the cells. Top inset: signalling activity at the leading edge of cells exhibiting elongated‐mesenchymal migration. Polarized signalling of GTPase Rac1 directs Arp2/3 via WAVE2 to drive actin polymerization in branched filaments against the plasma membrane. Bottom inset: signalling activity at the rear of cells exhibiting elongated‐mesenchymal migration. Rho‐ROCK signalling is required for the contractile activity of actomyosin scaffold to retract the cell rear. Transcription driven by p53 promotes elongated‐mesenchymal strategies.
Elongated‐mesenchymal migration is a protrusion‐dependent mode mediated by polarized signalling of GTPases Rac1 (Sanz‐Moreno et al., 2008; Yamazaki et al., 2009) and Cdc42 (Nalbant et al., 2004), which direct Arp2/3 to drive actin polymerization in branched filaments against the plasma membrane (Amann and Pollard, 2001; Giri et al., 2013; Machesky et al., 1999). Adhesion maturation is controlled by signalling activity of RhoA and effector proteins such as formin protein diaphanous homologs 1 and 2, while Rho‐ROCK signalling is required for the contractile activity of actomyosin scaffold to retract the cell rear (Friedl and Wolf, 2009; Ridley et al., 2003; Fig. 3).
5.2. Rounded‐amoeboid mode of invasion
Cancer cells migrating across pliable matrices can use rounded‐amoeboid strategies and squeeze through the matrix using small, unstable blebs present throughout the surface of the cell (Sahai and Marshall, 2003; Sanz‐Moreno and Marshall, 2010; Sanz‐Moreno et al., 2008) except at the rear, due to the presence of ezrin‐rich uropod‐like structures (ERULS; Lorentzen et al., 2011) that dictate cell polarity (Fig. 4). Blebs are a consequence of low membrane–cortex attachment, increased intracellular pressure, low degree of β1 integrin‐mediated adhesion, reduced focal adhesion size and force generation (Bergert et al., 2015; Charras and Paluch, 2008; Charras and Sahai, 2014; Petrie et al., 2012; Sahai and Marshall, 2003; Sanz‐Moreno et al., 2008; Wolf et al., 2003). Due to low reliance on focal adhesions and their deformability, the average speed during rounded‐amoeboid migration can be significantly faster (2–25 μm·min−1 in vitro, 1–15 μm·min−1 in vivo) than the mesenchymal type of cell migration (Pankova et al., 2010; Pinner and Sahai, 2008a; Sanz‐Moreno et al., 2008).
Figure 4.
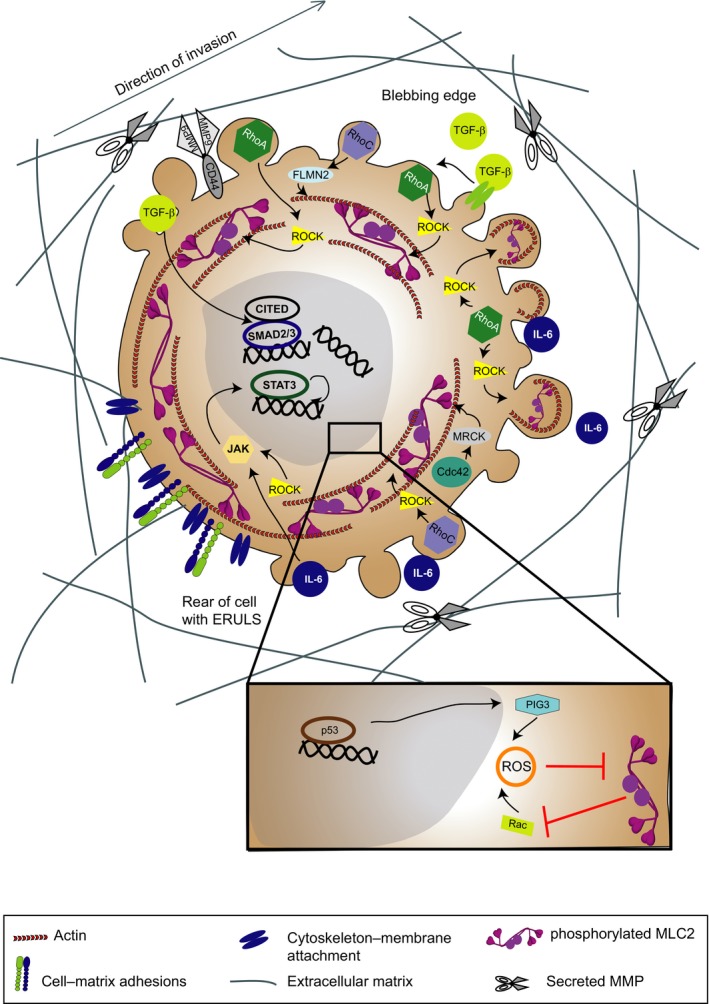
Signalling pathways controlling rounded‐amoeboid mode of invasion. Diagram showing key regulators of rounded‐amoeboid mode of migration. Rounded‐amoeboid cells squeeze through the matrix using small, unstable blebs present throughout the surface of the cells except at the rear, due to the presence of ezrin‐rich uropod‐like structures (ERULS) that determine polarity. Blebs are a consequence of low membrane–cortex attachment, increased intracellular pressure, high actomyosin contractility, low degree of β1 integrin‐mediated adhesion, reduced focal adhesion size and force generation. Rounded‐amoeboid motility is supported by high levels of actomyosin contractility downstream of Rho‐ROCK. While there is significant overlap in the RhoA‐ and RhoC‐mediated activation of actomyosin contractility, the assembly of cortical actin as a consequence of formin FLMN2 activation seems to be specific to RhoC. Maintenance of rounded‐amoeboid movement is driven by IL‐6 family of cytokines and the transcription factor STAT3. Conversely, ROCK can activate JAK/STAT3 signalling generating a positive feedback loop. TGF‐β promotes rounded‐amoeboid migration, which is perpetuated via SMAD2/CITED1‐mediated transcription. In addition, Rho/ROCK suppresses p53/PIG3‐mediated ROS production. On the other hand, Rac suppresses actomyosin contractility via ROS generation.
Rounded‐amoeboid motility is supported by high levels of actomyosin contractility downstream of Rho‐ROCK (Sahai and Marshall, 2003; Sanz‐Moreno et al., 2008; Wilkinson et al., 2005; Yamazaki et al., 2009). There is significant overlap in the RhoA‐ and RhoC‐mediated activation of actomyosin contractility. Nevertheless, the assembly of cortical actin as a consequence of formin FLMN2 activation seems to be specific to RhoC in rounded‐amoeboid cells (Kitzing et al., 2010). Furthermore, mDia2–Dip interaction induces the characteristic cell blebbing in rounded‐amoeboid movement (Eisenmann et al., 2007).
In rounded‐amoeboid migrating cells, a local decrease in attachment of the cell membrane to the actin cortex or local rupture of the actin cortex initiates a cycle of bleb expansion and retraction that allows cell movement (Charras and Paluch, 2008). Bleb expansion appears to be a direct mechanical consequence of intracellular pressure pushing the membrane outwards in the direction of motion. Bleb expansion is then slowed down and inhibited by recruitment of membrane–cortex linker proteins that facilitate actin recruitment underneath the membrane. The retraction phase begins with rapid assembly of actomyosin filaments beneath the bleb membrane (Charras and Paluch, 2008). Bleb‐based movement is generated by creating blebs at the leading edge and exerting force onto the substrate to translocate the cell body (Charras and Paluch, 2008). These forces could be achieved by weakly adhering to the ECM or to surrounding cells; by applying forces on the ECM perpendicular to the direction of movement; or through nonspecific substrate friction (Bergert et al., 2015; Charras and Paluch, 2008). Tumour xenograft intravital imaging studies have shown that melanoma and breast cancer cells in the invasive fronts predominantly move using rounded‐amoeboid strategies (Giampieri et al., 2009; Herraiz et al., 2016; Pinner and Sahai, 2008a,b; Sanz‐Moreno et al., 2008, 2011). Importantly, the invasive fronts of human melanoma primary tumours and metastases are enriched in rounded cells (Cantelli et al., 2015; Orgaz et al., 2014; Sanz‐Moreno et al., 2011).
5.3. Other modes of individual invasion
While elongated‐mesenchymal and rounded‐amoeboid modes of migration are extremes of the spectrum, intermediate modes of migration have been reported as cells transition between these modes (Yin et al., 2013). Glioblastoma‐initiating cells can efficiently invade exhibiting a round cell body aided by long or short protrusions (Ruiz‐Ontanon et al., 2013). Under confinement, breast cancer cells exhibit a mode of migration that is dependent on directed water permeation. This mode, termed the osmotic engine model, relies on aquaporin5 and Na+/H+ exchangers (Stroka et al., 2014).
Another mode of migration described in the recent years is the lobopodial mode of migration. This pressure‐based mode involves the use of the nucleus as a piston to generate intracellular pressure that drives forward a blunt cylindrical protrusion termed lobopodia (Petrie et al., 2012, 2014). This mode is characterized by nonpolarized distribution of active Rac1 at the plasma membrane and RhoA‐driven actomyosin contractility at the front of the nucleus. Actomyosin contraction pulls the nucleus towards the front, which poses a diffusion barrier and results in increased intracellular pressure that pushes the leading edge forwards (Petrie et al., 2012, 2014). However, this mode of migration has only been described in fibroblasts and its role in cancer cell invasion remains to be established.
Furthermore, filopodial spike‐based cancer cell invasion has also been recently described (Paul et al., 2015; Fig. 1). In this mode, α5β1 integrin recycling promotes RhoA‐ROCK‐FHOD3‐driven invasion independently of Arp2/3 activity.
6. Plasticity during individual cell invasion
6.1. Mesenchymal‐amoeboid plasticity
As noted earlier, ECM degradation and tissue remodelling by secreted proteases regulate invasion (Friedl and Alexander, 2011; Mantovani et al., 2008). Importantly, pioneer work in the cell migration field showed that upon inhibition of pericellular proteases, elongated‐mesenchymal cells still invaded as rounded‐amoeboid cells both in vitro and in vivo (Sahai and Marshall, 2003; Wolf et al., 2003; Wyckoff et al., 2006) while undergoing mesenchymal‐to‐amoeboid transition (MAT; Friedl, 2004; Wolf et al., 2003). This plasticity most likely contributes to the failure of therapies targeting proteases (see section ‘Therapeutic challenges posed by migratory plasticity’).
As both actin assembly and the actomyosin machinery can regulate cell morphology, modulation of actin organization can predict the type of protrusions formed by migrating cells (Bergert et al., 2012; Derivery et al., 2008; Langridge and Kay, 2006; Mierke, 2015). These changes in actin structures have been shown to be highly dependent on two key pathways that play compensatory roles and inhibit each other and that regulate the switch between rounded‐amoeboid and elongated‐mesenchymal migratory states (Fig. 1). The activation of Rac1‐WAVE2‐Arp2/3 drives elongated‐mesenchymal adhesive movement, while RhoA/C‐ROCK1/2 pathways drive rounded‐amoeboid migration (Sanz‐Moreno et al., 2008; Yamazaki et al., 2009) although some degree of Rho‐ROCK‐driven contractility is required also for elongated‐mesenchymal migration (Friedl and Wolf, 2009; Vicente‐Manzanares et al., 2009). Cdc42 is required for both elongated‐mesenchymal and rounded‐amoeboid movement depending on engagement of different effectors (Calvo et al., 2011; Gadea et al., 2008). Interestingly, loss of the Ras regulator RasGRF2 in melanoma cells induces MAT (Calvo et al., 2011).
6.2. Single‐to‐collective tumour invasion
In fibrosarcoma and breast carcinoma 3D‐spheroids, a spontaneous transition from individual mesenchymal invasion towards multicellular strands (Fig. 1) occurs in follower cells along the microtracks generated by leader cells (Wolf et al., 2007). These microtracks are occupied by following coupled cells and therefore, tracks increase in width, ultimately resulting in strand‐like collective invasion (Friedl and Wolf, 2008; Wolf et al., 2007).
The microenvironment, in particular ECM porosity, can regulate tumour plasticity and single‐to‐collective transition. Cell jamming is a collective mode of invasion of mesenchymal tumour cells that is imposed by tissue confinement. Dense matrix induces cell–cell interactions, leader–follower cell behaviour and collective migration as an obligate protease‐dependent process (Haeger et al., 2014). The conversion to collective invasion with increasing ECM confinement supports the concept of cell jamming as a guiding principle for melanoma and fibrosarcoma cells into dense tissue (Haeger et al., 2014; Sadati et al., 2013; Vedula et al., 2012). In addition, confinement modelled with micropillar arrays can also force collective migration of breast carcinoma cells (Wong et al., 2014).
Single‐to‐collective migration can also be induced by gradients or changes in adhesion molecules. For example, when individual cells become attracted by the same chemotactic source, they may first undergo multicellular streaming with short‐lived, dynamic cell–cell junctions. When cell–cell adhesion molecules are then upregulated, the cells may join each other and convert to a collective migration mode (Friedl and Alexander, 2011).
6.3. Determinants of plasticity
Determinants of plasticity in cells exhibiting individual mode of migration include physical and molecular cues (proteases, membrane receptors, secreted factors and intracellular signalling pathways), which are broadly highlighted in Figs 3 and 4.
6.3.1. Physical cues
Migration in discontinuous 3D substrates that allow cell–matrix adhesion results in a highly polarized spindle‐shaped morphology in elongated‐mesenchymal cells (Charras and Sahai, 2014; Starke et al., 2014; Fig. 3A). However, within discontinuous 3D matrices, if availability of small surface areas for attachment is low, such surfaces might not support adhesion formation and bleb‐based modes of migration are favoured (Petrie et al., 2012; Tozluoglu et al., 2013).
Another characteristic of ECM is porosity, which determines the confinement of migrating cells. Tissue confinement can also promote single‐to‐collective transitions such as cell jamming (Haeger et al., 2014; Sadati et al., 2013; Vedula et al., 2012). During individual migration, increasing confinement and decreasing adhesion result in increased deformability of the cell and MAT (Liu et al., 2015; Tozluoglu et al., 2013). The switch in these modes of migration is regulated by a delicate balance between adhesion and actomyosin contractility (Bergert et al., 2012).
While the cell cytoplasm is readily deformable in confined conditions, the nucleus is 2–10 times stiffer than the cytoplasm, thus generating a deformability barrier (Wolf et al., 2013). The deformability of the nucleus is dependent on the stiffness of nuclear lamina, which is regulated by lamin A/C levels (Lammerding et al., 2004, 2006). While low levels of lamins result in increased nuclear deformability, excessive softness of nuclear lamina decreases cell survival. In fact, cancer cells migrating in confined spaces experience nuclear envelope ruptures that result in DNA damage, which is solved using DNA repair machinery and endosomal sorting complexes required for transport (ESCRT; Denais et al., 2016; Raab et al., 2016). Cancer cells capable of resealing nuclear envelop rapidly could benefit from greater nuclear deformability, increased migration and survival. On the other hand, DNA damage responses induced by reactive oxygen species (ROS) dramatically reduce rounded‐amoeboid invasion in vitro and in vivo, by suppressing actomyosin contractility (Herraiz et al., 2016). In migrating cells, how different types of DNA damage are sensed and repaired will be an important question to solve.
In addition to ECM properties, mechanical perturbations such as interstitial flow can also affect cell migration. In fact, inflammation in cancer can dramatically increase fluid flow between the blood and lymphatic system (Dafni et al., 2002; Shieh and Swartz, 2011), causing an increase in migration speed of breast cancer cells (Haessler et al., 2012). Interestingly, for breast cancer cells able to migrate using both rounded‐amoeboid and elongated‐mesenchymal motility within 3D collagen type I matrix, interstitial flow favours a switch towards rounded‐amoeboid motility (Huang et al., 2015).
6.3.2. Molecular cues
6.3.2.1. Proteases
While pericellular proteolytic inhibition in elongated‐mesenchymal cells drives MAT and cells keep invading (Sahai and Marshall, 2003; Wolf et al., 2003), rounded‐amoeboid melanoma cells are able to degrade the matrix (Hooper et al., 2006), in some cases even more efficiently than elongated‐mesenchymal melanoma cells (Orgaz et al., 2014). This may be due to a higher secretion of certain MMPs such as MMP13 and MMP2. Furthermore, melanoma cells use MMP9 noncatalytic functions to sustain rounded‐amoeboid invasion (Orgaz et al., 2014) via regulation of actomyosin contractility.
6.3.2.2. Membrane receptors
Membrane receptors such as β1 integrins can also control migration of individual elongated‐mesenchymal cells (Ahn et al., 2012; Friedl, 2004; Wolf et al., 2007). Furthermore, CD44 has been shown to be required for individual rounded‐amoeboid invasion (Orgaz et al., 2014). CD44 forms a complex with MMP9, which results in the activation of actomyosin contractility in melanoma (Orgaz et al., 2014).
6.3.2.3. Secreted factors
Melanoma cells secrete high levels of IL‐6 family cytokines that promote individual rounded‐amoeboid invasion (Sanz‐Moreno et al., 2011). HGF receptor Met‐driven signalling has also been implicated in MAT via Rho‐ROCK pathway (Laser‐Azogui et al., 2014). Therefore, extracellular ligands govern how integration of signals is achieved in migrating cells travelling through different tumour microenvironments.
6.3.2.4. Intracellular signalling pathways
Actin dynamics determine the type of protrusions. Promotion of actin polymerization in carcinoma cells drives the formation of actin‐rich lamellipodia, whereas blebbing requires both actin polymerization and depolymerization (Bergert et al., 2012; Bovellan et al., 2014; Derivery et al., 2008; Langridge and Kay, 2006; Mierke, 2015).
The balance between antagonistic RhoA and Rac1 signalling determines the mode of migration and lies at the core of tumour cell plasticity in individual migration of several cancer cell types (Sanz‐Moreno et al., 2008; Yamazaki et al., 2009). Downstream of β3 integrin, adaptor NEDD9 activates Src signalling (involving also p130Cas, Crk) and the Rac GEF DOCK3 (Ahn et al., 2012; Carragher et al., 2006; Kiyokawa et al., 1998; Sanz‐Moreno et al., 2008). In turn, active Rac signals through WAVE‐2 promoting Arp2/3‐dependent actin assembly and protrusion formation, driving elongated‐mesenchymal migration (Sanz‐Moreno et al., 2008; Yamazaki et al., 2009; Fig. 3B). WAVE‐2 suppresses rounded‐amoeboid movement by inhibiting actomyosin contractility (Sanz‐Moreno et al., 2008; Yamazaki et al., 2009).
Conversely, the Rac‐specific GAPs ARHGAP22 and ARHGAP24 (also known as FilGAP), which are activated by high actomyosin contractility, maintain low levels of Rac activity in rounded‐amoeboid cells (Saito et al., 2012; Sanz‐Moreno et al., 2008). MAT can be induced through the inhibition of Rac activity (Sanz‐Moreno et al., 2008), or indirectly activating Rho by engaging EphA2 (Parri et al., 2009). Lowering the levels of RhoA‐negative regulator p27Kip1 (Besson et al., 2004) also promotes rounded‐amoeboid migration (Berton et al., 2009). The antagonistic interplay between Rho‐ROCK and Rnd3 (RhoE) at the cell membrane that regulates blebbing also drives cell plasticity. Absence of PDK1 allows for inhibitory binding of RhoE to ROCK leading to impaired actomyosin contractility and rounded‐amoeboid motility (Pinner and Sahai, 2008b). Importantly, Cdc42 has a dual role as it supports rounded‐amoeboid migration via DOCK10 and the Cdc42 effectors NWASP and PAK2 (Gadea et al., 2008). Supporting these data, blocking the Cdc42‐negative regulator and Ras GEF RasGRF2 ablates amoeboid invasion and metastatic colonization (Calvo et al., 2011). On the other hand, in elongated‐mesenchymal cells, Cdc42 promotes Rac activity by activating and recruiting ubiquitin ligase SMURF1 to the leading edge via a PAR6–aPKC polarity complex (Osmani et al., 2010).
Regulation of protein levels and protein localization drives plasticity. As such, downregulation of SMURF1, which targets RhoA for localized proteasomal degradation in Rac‐dependent protrusions, results in MAT (Sahai et al., 2007). Rab5‐dependent endocytosis regulates Rac localization to protrusions supporting therefore elongated‐mesenchymal movement (Palamidessi et al., 2008).
6.3.2.5. Transcriptional programmes
While individually invading cells can switch between blebs and protrusions in short timescales (Bergert et al., 2012), maintaining cell motility programmes requires a tight temporal coupling of actin dynamics and transcriptional activity (Olson and Nordheim, 2010). Hence, it is no surprise that several transcriptional factors have been implicated in different modes of migration and cellular plasticity. Loss of p53 function via mutant p53 overexpression results in MAT in melanoma cells (Gadea et al., 2007). Transcription driven by p53 further suppresses fast rounded‐amoeboid migration via induction of its transcriptional target p53‐induced gene 3 protein (PIG3). PIG3 is an oxidoreductase that produces ROS and further suppresses Rho activity via regulation of ARHGAP5 (Herraiz et al., 2016).
In contrast, maintenance of rounded‐amoeboid movement is driven by IL‐6 family of cytokines and the transcription factor STAT3. ROCK can activate JAK/STAT3 signalling generating a positive feedback loop (Sanz‐Moreno et al., 2011). As a result of high levels of STAT3 activity, rounded‐amoeboid melanoma cells secrete higher levels of most secreted MMPs (Orgaz et al., 2014).
MRTF‐ and SRF‐driven transcription can sustain high actomyosin contractility levels to promote metastasis in melanoma and breast carcinoma cells (Medjkane et al., 2009). In breast cancer models, TGF‐β/SMAD induces transcriptional changes that promote a cohesive‐to‐single invasion (Giampieri et al., 2009). Those transcriptional changes include genes that control actomyosin contractility (Giampieri et al., 2009). In melanoma, TGF‐β promotes rounded‐amoeboid migration, which is perpetuated via SMAD2/CITED1‐mediated transcription of LIF, JAK and the Rho GEF ARHGEF5 (Cantelli et al., 2015).
7. Therapeutic challenges posed by migratory plasticity
Plasticity or adaptability in terms of cell migration modes likely underlies the failure of some therapies aimed at blocking cancer invasion and metastasis. Several therapies targeting pericellular matrix‐degrading proteases were developed (Coussens et al., 2002; Overall and Kleifeld, 2006; Overall and Lopez‐Otin, 2002). However, extensive phase III clinical trials not only failed but even worsened metastatic processes (Coussens et al., 2002; Fingleton, 2003; Overall and Lopez‐Otin, 2002; Zucker et al., 2000). Such failure was attributed in part to the different roles of specific MMPs (Lopez‐Otin and Matrisian, 2007). However, the MAT that occurs upon pericellular proteolysis inhibition (Friedl, 2004; Sahai and Marshall, 2003; Wolf et al., 2003; Wyckoff et al., 2006) would add up to the reasons why therapies broadly targeting MMP functions were not successful. In addition, noncatalytic regulation of cell signalling (Orgaz et al., 2014) could be an additional reason for the failure of MMP inhibitor‐based therapies (Dufour and Overall, 2013; Overall and Kleifeld, 2006; Zucker et al., 2000). Therefore, targeting specific proteolytic and nonproteolytic functions of certain MMPs may provide better results in the clinic.
While targeting MMPs offers a singular therapeutic focal point, it is crucial to keep in mind that the tumour microenvironment presents a heterogeneous and discontinuous environment with varying matrix geometries and degree of stiffness. As a consequence, cells could exhibit MAT spontaneously in response to localized changes in stiffness and this plasticity can impact tumour dissemination in vivo. Thus, effective therapies should focus on blocking plasticity by inhibiting multiple intracellular and extracellular drivers of this mode of drug resistance.
8. Potential therapeutic targets to block migratory plasticity and tumour cell invasion
Adaptation of cancer cells to different environmental conditions is exemplified by the wide variety of invasion strategies they can adopt. More striking is their ability to switch from one strategy to another to keep on invading. This adaptability is complex, as tumour cell migration plasticity may not need fixed genetic drivers, but it may be aided by accumulated DNA damage in migrating cancer cells.
Such adaptability of cancer cells to change their mode of migration could be considered a type of drug resistance. Therefore, therapies should be aimed at targeting cytoskeletal regulators involved in multiple modes of migration, or combination of drugs aimed at different key targets (Figs 3 and 4). This goes in line with combinational therapies that are currently in clinical trials to stop primary tumour growth. Some key regulators could be β1 integrin, which controls single and collective invasion and the switch from one to another. Several therapeutic interventions are being clinically tested in patients with solid tumours, including peptide ATN‐161, which inhibits binding of α5β1 to fibronectin (Cianfrocca et al., 2006; Thundimadathil, 2012), and α5β1‐blocking antibody volociximab (Ricart et al., 2008). These therapies are also aimed to block tumour angiogenesis (Cianfrocca et al., 2006; Ricart et al., 2008).
ROCK lies at the core of cytoskeletal regulation in virtually all modes of migration, therefore appears as a good therapeutic target. Interestingly, a pan‐AGC kinase inhibitor that very effectively targets ROCK (Sadok et al., 2015) is being clinically evaluated in advanced solid tumours (ClinicalTrials.gov Identifier: NCT01585701).
Cdc42 or its effectors could be also suitable candidates given their involvement of both rounded‐amoeboid and elongated‐mesenchymal invasion strategies. A combination of drugs targeting cell adhesion and the actomyosin core machinery could also be considered.
Furthermore, careful attention should be given to targeting transcriptional programmes that self‐perpetuate invasion strategies (JAK/STAT3, TGF‐β/SMAD) and control processes such as tumour promoting inflammation and immunosuppression. Given the protumorigenic roles of the JAK/STAT3 pathway, inhibition of JAK/STAT3 in solid tumours is currently being evaluated (Buchert et al., 2016). Moreover, several inhibitors of the TGF‐β pathway are being developed and clinically tested for a number of cancers (Neuzillet et al., 2015). However, the dual role of TGF‐β as tumour suppressor or prometastatic (Massague, 2008) anticipates that targeting its transcriptional targets and/or regulators might be a better approach to block only its prometastatic effects.
9. Concluding remarks
Tumour cells usually encounter heterogeneous and discontinuous microenvironments. As a consequence, cancer cells need to adapt spontaneously in response to localized physical and chemical changes. The minimum machinery required to drive all different types of migration comprises the actomyosin cytoskeleton. Differential regulation of actomyosin machinery is what drives plasticity and different modes of migration, blockade of which is essential to prevent cancer invasion and metastasis. Thus, future therapies for preventing metastasis should focus on selective pharmacological inhibition of actomyosin machinery within cancer cells.
Acknowledgements
This work was supported by CRUK C33043/A12065 (VS‐M, JLO) and Royal Society RG110591 (VS‐M). PP is supported by King's Overseas Scholarship.
References
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H et al (2014) Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman AB and Ragaz A (1984) The Lives of Lesions. Masson Publishers, New York, NY. [Google Scholar]
- Ahn J, Sanz‐Moreno V and Marshall CJ (2012) The metastasis gene NEDD9 product acts through integrin beta3 and Src to promote mesenchymal motility and inhibit amoeboid motility. J Cell Sci 125, 1814–1826. [DOI] [PubMed] [Google Scholar]
- Alexander S, Koehl GE, Hirschberg M, Geissler EK and Friedl P (2008) Dynamic imaging of cancer growth and invasion: a modified skin‐fold chamber model. Histochem Cell Biol 130, 1147–1154. [DOI] [PubMed] [Google Scholar]
- Amann KJ and Pollard TD (2001) The Arp2/3 complex nucleates actin filament branches from the sides of pre‐existing filaments. Nat Cell Biol 3, 306–310. [DOI] [PubMed] [Google Scholar]
- Amano M, Ito M, Kimura K, Fukata Y, Chihara K, Nakano T, Matsuura Y and Kaibuchi K (1996) Phosphorylation and activation of myosin by Rho‐associated kinase (Rho‐kinase). J Biol Chem 271, 20246–20249. [DOI] [PubMed] [Google Scholar]
- Bell CD and Waizbard E (1986) Variability of cell size in primary and metastatic human breast carcinoma. Invasion Metastasis 6, 11–20. [PubMed] [Google Scholar]
- Bergert M, Chandradoss SD, Desai RA and Paluch E (2012) Cell mechanics control rapid transitions between blebs and lamellipodia during migration. Proc Natl Acad Sci USA 109, 14434–14439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergert M, Erzberger A, Desai RA, Aspalter IM, Oates AC, Charras G, Salbreux G and Paluch EK (2015) Force transmission during adhesion‐independent migration. Nat Cell Biol 17, 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berton S, Belletti B, Wolf K, Canzonieri V, Lovat F, Vecchione A, Colombatti A, Friedl P and Baldassarre G (2009) The tumor suppressor functions of p27(kip1) include control of the mesenchymal/amoeboid transition. Mol Cell Biol 29, 5031–5045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besson A, Gurian‐West M, Schmidt A, Hall A and Roberts JM (2004) p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev 18, 862–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovellan M, Romeo Y, Biro M, Boden A, Chugh P, Yonis A, Vaghela M, Fritzsche M, Moulding D, Thorogate R et al (2014) Cellular control of cortical actin nucleation. Curr Biol 24, 1628–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz‐Schughart LA, Knuechel R and Kirchner T (2001) Variable beta‐catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci USA 98, 10356–10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandt B, Junker R, Griwatz C, Heidl S, Brinkmann O, Semjonow A, Assmann G and Zanker KS (1996) Isolation of prostate‐derived single cells and cell clusters from human peripheral blood. Cancer Res 56, 4556–4561. [PubMed] [Google Scholar]
- Bronsert P, Enderle‐Ammour K, Bader M, Timme S, Kuehs M, Csanadi A, Kayser G, Kohler I, Bausch D, Hoeppner J et al (2014) Cancer cell invasion and EMT marker expression: a three‐dimensional study of the human cancer‐host interface. J Pathol 234, 410–422. [DOI] [PubMed] [Google Scholar]
- Brown MC, West KA and Turner CE (2002) Paxillin‐dependent paxillin kinase linker and p21‐activated kinase localization to focal adhesions involves a multistep activation pathway. Mol Biol Cell 13, 1550–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchert M, Burns CJ and Ernst M (2016) Targeting JAK kinase in solid tumors: emerging opportunities and challenges. Oncogene 35, 939–951. [DOI] [PubMed] [Google Scholar]
- Byers SW, Sommers CL, Hoxter B, Mercurio AM and Tozeren A (1995) Role of E‐cadherin in the response of tumor cell aggregates to lymphatic, venous and arterial flow: measurement of cell‐cell adhesion strength. J Cell Sci 108(Pt 5), 2053–2064. [DOI] [PubMed] [Google Scholar]
- Calvo F, Sanz‐Moreno V, Agudo‐Ibanez L, Wallberg F, Sahai E, Marshall CJ and Crespo P (2011) RasGRF suppresses Cdc42‐mediated tumour cell movement, cytoskeletal dynamics and transformation. Nat Cell Biol 13, 819–826. [DOI] [PubMed] [Google Scholar]
- Cantelli G, Orgaz JL, Rodriguez‐Hernandez I, Karagiannis P, Maiques O, Matias‐Guiu X, Nestle FO, Marti RM, Karagiannis SN and Sanz‐Moreno V (2015) TGF‐beta‐induced transcription sustains amoeboid melanoma migration and dissemination. Curr Biol 25, 2899–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr I, Levy M and Watson P (1986) The invasive edge: invasion in colorectal cancer. Clin Exp Metastasis 4, 129–139. [DOI] [PubMed] [Google Scholar]
- Carragher NO, Walker SM, Scott Carragher LA, Harris F, Sawyer TK, Brunton VG, Ozanne BW and Frame MC (2006) Calpain 2 and Src dependence distinguishes mesenchymal and amoeboid modes of tumour cell invasion: a link to integrin function. Oncogene 25, 5726–5740. [DOI] [PubMed] [Google Scholar]
- Casey RC, Burleson KM, Skubitz KM, Pambuccian SE, Oegema TR Jr, Ruff LE and Skubitz AP (2001) Beta 1‐integrins regulate the formation and adhesion of ovarian carcinoma multicellular spheroids. Am J Pathol 159, 2071–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charras G and Paluch E (2008) Blebs lead the way: how to migrate without lamellipodia. Nat Rev Mol Cell Biol 9, 730–736. [DOI] [PubMed] [Google Scholar]
- Charras G and Sahai E (2014) Physical influences of the extracellular environment on cell migration. Nat Rev Mol Cell Biol 15, 813–824. [DOI] [PubMed] [Google Scholar]
- Christiansen JJ and Rajasekaran AK (2006) Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res 66, 8319–8326. [DOI] [PubMed] [Google Scholar]
- Cianfrocca ME, Kimmel KA, Gallo J, Cardoso T, Brown MM, Hudes G, Lewis N, Weiner L, Lam GN, Brown SC et al (2006) Phase 1 trial of the antiangiogenic peptide ATN‐161 (Ac‐PHSCN‐NH(2)), a beta integrin antagonist, in patients with solid tumours. Br J Cancer 94, 1621–1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG and Vignjevic DM (2015) Modes of cancer cell invasion and the role of the microenvironment. Curr Opin Cell Biol 36, 13–22. [DOI] [PubMed] [Google Scholar]
- Coussens LM, Fingleton B and Matrisian LM (2002) Matrix metalloproteinase inhibitors and cancer: trials and tribulations. Science 295, 2387–2392. [DOI] [PubMed] [Google Scholar]
- Dafni H, Israely T, Bhujwalla ZM, Benjamin LE and Neeman M (2002) Overexpression of vascular endothelial growth factor 165 drives peritumor interstitial convection and induces lymphatic drain: magnetic resonance imaging, confocal microscopy, and histological tracking of triple‐labeled albumin. Cancer Res 62, 6731–6739. [PubMed] [Google Scholar]
- Daly AJ, McIlreavey L and Irwin CR (2008) Regulation of HGF and SDF‐1 expression by oral fibroblasts–implications for invasion of oral cancer. Oral Oncol 44, 646–651. [DOI] [PubMed] [Google Scholar]
- Dean RA, Cox JH, Bellac CL, Doucet A, Starr AE and Overall CM (2008) Macrophage‐specific metalloelastase (MMP‐12) truncates and inactivates ELR+ CXC chemokines and generates CCL2, ‐7, ‐8, and ‐13 antagonists: potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 112, 3455–3464. [DOI] [PubMed] [Google Scholar]
- Denais CM, Gilbert RM, Isermann P, McGregor AL, te Lindert M, Weigelin B, Davidson PM, Friedl P, Wolf K and Lammerding J (2016) Nuclear envelope rupture and repair during cancer cell migration. Science 352, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derivery E, Fink J, Martin D, Houdusse A, Piel M, Stradal TE, Louvard D and Gautreau A (2008) Free Brick1 is a trimeric precursor in the assembly of a functional wave complex. PLoS ONE 3, e2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufour A and Overall CM (2013) Missing the target: matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol Sci 34, 233–242. [DOI] [PubMed] [Google Scholar]
- Edwards DC, Sanders LC, Bokoch GM and Gill GN (1999) Activation of LIM‐kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol 1, 253–259. [DOI] [PubMed] [Google Scholar]
- Egeblad M and Werb Z (2002) New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer 2, 161–174. [DOI] [PubMed] [Google Scholar]
- Eisenmann KM, Harris ES, Kitchen SM, Holman HA, Higgs HN and Alberts AS (2007) Dia‐interacting protein modulates formin‐mediated actin assembly at the cell cortex. Curr Biol 17, 579–591. [DOI] [PubMed] [Google Scholar]
- Endo A, Surks HK, Mochizuki S, Mochizuki N and Mendelsohn ME (2004) Identification and characterization of zipper‐interacting protein kinase as the unique vascular smooth muscle myosin phosphatase‐associated kinase. J Biol Chem 279, 42055–42061. [DOI] [PubMed] [Google Scholar]
- Fingleton B (2003) Matrix metalloproteinase inhibitors for cancer therapy:the current situation and future prospects. Expert Opin Ther Targets 7, 385–397. [DOI] [PubMed] [Google Scholar]
- Friedl P (2004) Prespecification and plasticity: shifting mechanisms of cell migration. Curr Opin Cell Biol 16, 14–23. [DOI] [PubMed] [Google Scholar]
- Friedl P and Alexander S (2011) Cancer invasion and the microenvironment: plasticity and reciprocity. Cell 147, 992–1009. [DOI] [PubMed] [Google Scholar]
- Friedl P and Gilmour D (2009) Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10, 445–457. [DOI] [PubMed] [Google Scholar]
- Friedl P, Hegerfeldt Y and Tusch M (2004) Collective cell migration in morphogenesis and cancer. Int J Dev Biol 48, 441–449. [DOI] [PubMed] [Google Scholar]
- Friedl P, Locker J, Sahai E and Segall JE (2012) Classifying collective cancer cell invasion. Nat Cell Biol 14, 777–783. [DOI] [PubMed] [Google Scholar]
- Friedl P, Maaser K, Klein CE, Niggemann B, Krohne G and Zanker KS (1997) Migration of highly aggressive MV3 melanoma cells in 3‐dimensional collagen lattices results in local matrix reorganization and shedding of alpha2 and beta1 integrins and CD44. Cancer Res 57, 2061–2070. [PubMed] [Google Scholar]
- Friedl P, Noble PB, Walton PA, Laird DW, Chauvin PJ, Tabah RJ, Black M and Zanker KS (1995) Migration of coordinated cell clusters in mesenchymal and epithelial cancer explants in vitro. Cancer Res 55, 4557–4560. [PubMed] [Google Scholar]
- Friedl P and Wolf K (2003) Tumour‐cell invasion and migration: diversity and escape mechanisms. Nat Rev Cancer 3, 362–374. [DOI] [PubMed] [Google Scholar]
- Friedl P and Wolf K (2008) Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Res 68, 7247–7249. [DOI] [PubMed] [Google Scholar]
- Friedl P and Wolf K (2009) Proteolytic interstitial cell migration: a five‐step process. Cancer Metastasis Rev 28, 129–135. [DOI] [PubMed] [Google Scholar]
- Friedl P and Wolf K (2010) Plasticity of cell migration: a multiscale tuning model. J Cell Biol 188, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadea G, de Toledo M, Anguille C and Roux P (2007) Loss of p53 promotes RhoA‐ROCK‐dependent cell migration and invasion in 3D matrices. J Cell Biol 178, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadea G, Sanz‐Moreno V, Self A, Godi A and Marshall CJ (2008) DOCK10‐mediated Cdc42 activation is necessary for amoeboid invasion of melanoma cells. Curr Biol 18, 1456–1465. [DOI] [PubMed] [Google Scholar]
- Gaggioli C, Hooper S, Hidalgo‐Carcedo C, Grosse R, Marshall JF, Harrington K and Sahai E (2007) Fibroblast‐led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat Cell Biol 9, 1392–1400. [DOI] [PubMed] [Google Scholar]
- Gavert N, Sheffer M, Raveh S, Spaderna S, Shtutman M, Brabletz T, Barany F, Paty P, Notterman D, Domany E et al (2007) Expression of L1‐CAM and ADAM10 in human colon cancer cells induces metastasis. Cancer Res 67, 7703–7712. [DOI] [PubMed] [Google Scholar]
- Gavert N, Vivanti A, Hazin J, Brabletz T and Ben‐Ze'ev A (2011) L1‐mediated colon cancer cell metastasis does not require changes in EMT and cancer stem cell markers. Mol Cancer Res 9, 14–24. [DOI] [PubMed] [Google Scholar]
- Geiger TR and Peeper DS (2009) Metastasis mechanisms. Biochim Biophys Acta 1796, 293–308. [DOI] [PubMed] [Google Scholar]
- Giampieri S, Manning C, Hooper S, Jones L, Hill CS and Sahai E (2009) Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat Cell Biol 11, 1287–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri A, Bajpai S, Trenton N, Jayatilaka H, Longmore GD and Wirtz D (2013) The Arp2/3 complex mediates multigeneration dendritic protrusions for efficient 3‐dimensional cancer cell migration. FASEB J 27, 4089–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T et al (2010) Measuring mechanical tension across vinculin reveals regulation of focal adhesion dynamics. Nature 466, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RA, Paluch E and Oegema K (2012) Cytokinesis in animal cells. Annu Rev Cell Dev Biol 28, 29–58. [DOI] [PubMed] [Google Scholar]
- Grigore AD, Jolly MK, Jia D, Farach‐Carson MC and Levine H (2016) Tumor budding: the name is EMT. Partial EMT. J Clin Med 5, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeger A, Krause M, Wolf K and Friedl P (2014) Cell jamming: collective invasion of mesenchymal tumor cells imposed by tissue confinement. Biochim Biophys Acta 1840, 2386–2395. [DOI] [PubMed] [Google Scholar]
- Haeger A, Wolf K, Zegers MM and Friedl P (2015) Collective cell migration: guidance principles and hierarchies. Trends Cell Biol 25, 556–566. [DOI] [PubMed] [Google Scholar]
- Haessler U, Teo JC, Foretay D, Renaud P and Swartz MA (2012) Migration dynamics of breast cancer cells in a tunable 3D interstitial flow chamber. Integr Biol (Camb) 4, 401–409. [DOI] [PubMed] [Google Scholar]
- Hart IR (2009) New evidence for tumour embolism as a mode of metastasis. J Pathol 219, 275–276. [DOI] [PubMed] [Google Scholar]
- Hashizume R, Koizumi H, Ihara A, Ohta T and Uchikoshi T (1996) Expression of beta‐catenin in normal breast tissue and breast carcinoma: a comparative study with epithelial cadherin and alpha‐catenin. Histopathology 29, 139–146. [DOI] [PubMed] [Google Scholar]
- Heasman SJ and Ridley AJ (2008) Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol 9, 690–701. [DOI] [PubMed] [Google Scholar]
- Hegerfeldt Y, Tusch M, Brocker EB and Friedl P (2002) Collective cell movement in primary melanoma explants: plasticity of cell‐cell interaction, beta1‐integrin function, and migration strategies. Cancer Res 62, 2125–2130. [PubMed] [Google Scholar]
- Herraiz C, Calvo F, Pandya P, Cantelli G, Rodriguez‐Hernandez I, Orgaz JL, Kang N, Chu T, Sahai E and Sanz‐Moreno V (2016) Reactivation of p53 by a cytoskeletal sensor to control the balance between DNA damage and tumor dissemination. J Natl Cancer Inst 108, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidalgo‐Carcedo C, Hooper S, Chaudhry SI, Williamson P, Harrington K, Leitinger B and Sahai E (2011) Collective cell migration requires suppression of actomyosin at cell‐cell contacts mediated by DDR1 and the cell polarity regulators Par3 and Par6. Nat Cell Biol 13, 49–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata N, Takahashi M and Yazawa M (2009) Diphosphorylation of regulatory light chain of myosin IIA is responsible for proper cell spreading. Biochem Biophys Res Commun 381, 682–687. [DOI] [PubMed] [Google Scholar]
- Hodivala‐Dilke KM, McHugh KP, Tsakiris DA, Rayburn H, Crowley D, Ullman‐Cullere M, Ross FP, Coller BS, Teitelbaum S and Hynes RO (1999) Beta3‐integrin‐deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J Clin Invest 103, 229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper S, Marshall JF and Sahai E (2006) Tumor cell migration in three dimensions. Methods Enzymol 406, 625–643. [DOI] [PubMed] [Google Scholar]
- Hou JM, Krebs M, Ward T, Sloane R, Priest L, Hughes A, Clack G, Ranson M, Blackhall F and Dive C (2011) Circulating tumor cells as a window on metastasis biology in lung cancer. Am J Pathol 178, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YL, Tung CK, Zheng A, Kim BJ and Wu M (2015) Interstitial flows promote amoeboid over mesenchymal motility of breast cancer cells revealed by a three dimensional microfluidic model. Integr Biol (Camb) 7, 1402–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes RO (2002) Integrins: bidirectional, allosteric signaling machines. Cell 110, 673–687. [DOI] [PubMed] [Google Scholar]
- Kalluri R (2009) EMT: when epithelial cells decide to become mesenchymal‐like cells. J Clin Invest 119, 1417–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kats‐Ugurlu G, Roodink I, de Weijert M, Tiemessen D, Maass C, Verrijp K, van der Laak J, de Waal R, Mulders P, Oosterwijk E et al (2009) Circulating tumour tissue fragments in patients with pulmonary metastasis of clear cell renal cell carcinoma. J Pathol 219, 287–293. [DOI] [PubMed] [Google Scholar]
- Kedrin D, Gligorijevic B, Wyckoff J, Verkhusha VV, Condeelis J, Segall JE and van Rheenen J (2008) Intravital imaging of metastatic behavior through a mammary imaging window. Nat Methods 5, 1019–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny HA, Kaur S, Coussens LM and Lengyel E (2008) The initial steps of ovarian cancer cell metastasis are mediated by MMP‐2 cleavage of vitronectin and fibronectin. J Clin Invest 118, 1367–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil AA and Friedl P (2010) Determinants of leader cells in collective cell migration. Integr Biol (Camb) 2, 568–574. [DOI] [PubMed] [Google Scholar]
- Khoja L, Shenjere P, Hodgson C, Hodgetts J, Clack G, Hughes A, Lorigan P and Dive C (2014) Prevalence and heterogeneity of circulating tumour cells in metastatic cutaneous melanoma. Melanoma Res 24, 40–46. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K et al (1996) Regulation of myosin phosphatase by Rho and Rho‐associated kinase (Rho‐kinase). Science 273, 245–248. [DOI] [PubMed] [Google Scholar]
- Kitzing TM, Wang Y, Pertz O, Copeland JW and Grosse R (2010) Formin‐like 2 drives amoeboid invasive cell motility downstream of RhoC. Oncogene 29, 2441–2448. [DOI] [PubMed] [Google Scholar]
- Kiyokawa E, Hashimoto Y, Kobayashi S, Sugimura H, Kurata T and Matsuda M (1998) Activation of Rac1 by a Crk SH3‐binding protein, DOCK180. Genes Dev 12, 3331–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurniawan NA, Chaudhuri PK and Lim CT (2016) Mechanobiology of cell migration in the context of dynamic two‐way cell‐matrix interactions. J Biomech 49, 1355–1368. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Fong LG, Ji JY, Reue K, Stewart CL, Young SG and Lee RT (2006) Lamins A and C but not lamin B1 regulate nuclear mechanics. J Biol Chem 281, 25768–25780. [DOI] [PubMed] [Google Scholar]
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, Stewart CL and Lee RT (2004) Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J Clin Invest 113, 370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langridge PD and Kay RR (2006) Blebbing of Dictyostelium cells in response to chemoattractant. Exp Cell Res 312, 2009–2017. [DOI] [PubMed] [Google Scholar]
- Laser‐Azogui A, Diamant‐Levi T, Israeli S, Roytman Y and Tsarfaty I (2014) Met‐induced membrane blebbing leads to amoeboid cell motility and invasion. Oncogene 33, 1788–1798. [DOI] [PubMed] [Google Scholar]
- Lauffenburger DA and Horwitz AF (1996) Cell migration: a physically integrated molecular process. Cell 84, 359–369. [DOI] [PubMed] [Google Scholar]
- Law AL, Vehlow A, Kotini M, Dodgson L, Soong D, Theveneau E, Bodo C, Taylor E, Navarro C, Perera U et al (2013) Lamellipodin and the Scar/WAVE complex cooperate to promote cell migration in vivo. J Cell Biol 203, 673–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HS, Lim CJ, Puzon‐McLaughlin W, Shattil SJ and Ginsberg MH (2009) RIAM activates integrins by linking talin to ras GTPase membrane‐targeting sequences. J Biol Chem 284, 5119–5127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leighton J, Kalla RL, Turner JM and Fennell RH Jr (1960) Pathogenesis of tumor invasion. II. Aggregate replication. Cancer Res 20, 575–586. [PubMed] [Google Scholar]
- Leung T, Chen XQ, Tan I, Manser E and Lim L (1998) Myotonic dystrophy kinase‐related Cdc42‐binding kinase acts as a Cdc42 effector in promoting cytoskeletal reorganization. Mol Cell Biol 18, 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YJ, Le Berre M, Lautenschlaeger F, Maiuri P, Callan‐Jones A, Heuze M, Takaki T, Voituriez R and Piel M (2015) Confinement and low adhesion induce fast amoeboid migration of slow mesenchymal cells. Cell 160, 659–672. [DOI] [PubMed] [Google Scholar]
- Lopez‐Otin C and Matrisian LM (2007) Emerging roles of proteases in tumour suppression. Nat Rev Cancer 7, 800–808. [DOI] [PubMed] [Google Scholar]
- Lorentzen A, Bamber J, Sadok A, Elson‐Schwab I and Marshall CJ (2011) An ezrin‐rich, rigid uropod‐like structure directs movement of amoeboid blebbing cells. J Cell Sci 124, 1256–1267. [DOI] [PubMed] [Google Scholar]
- Machesky LM, Mullins RD, Higgs HN, Kaiser DA, Blanchoin L, May RC, Hall ME and Pollard TD (1999) Scar, a WASp‐related protein, activates nucleation of actin filaments by the Arp2/3 complex. Proc Natl Acad Sci USA 96, 3739–3744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madaule P, Eda M, Watanabe N, Fujisawa K, Matsuoka T, Bito H, Ishizaki T and Narumiya S (1998) Role of citron kinase as a target of the small GTPase Rho in cytokinesis. Nature 394, 491–494. [DOI] [PubMed] [Google Scholar]
- Madhavan M, Srinivas P, Abraham E, Ahmed I, Mathew A, Vijayalekshmi NR and Balaram P (2001) Cadherins as predictive markers of nodal metastasis in breast cancer. Mod Pathol 14, 423–427. [DOI] [PubMed] [Google Scholar]
- Maekawa M, Ishizaki T, Boku S, Watanabe N, Fujita A, Iwamatsu A, Obinata T, Ohashi K, Mizuno K and Narumiya S (1999) Signaling from Rho to the actin cytoskeleton through protein kinases ROCK and LIM‐kinase. Science 285, 895–898. [DOI] [PubMed] [Google Scholar]
- Manning CS, Hooper S and Sahai EA (2015) Intravital imaging of SRF and Notch signalling identifies a key role for EZH2 in invasive melanoma cells. Oncogene 34, 4320–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A, Allavena P, Sica A and Balkwill F (2008) Cancer‐related inflammation. Nature 454, 436–444. [DOI] [PubMed] [Google Scholar]
- Martin‐Villar E, Megias D, Castel S, Yurrita MM, Vilaro S and Quintanilla M (2006) Podoplanin binds ERM proteins to activate RhoA and promote epithelial‐mesenchymal transition. J Cell Sci 119, 4541–4553. [DOI] [PubMed] [Google Scholar]
- Mason SD and Joyce JA (2011) Proteolytic networks in cancer. Trends Cell Biol 21, 228–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J (2008) TGFbeta in cancer. Cell 134, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui T, Maeda M, Doi Y, Yonemura S, Amano M, Kaibuchi K, Tsukita S and Tsukita S (1998) Rho‐kinase phosphorylates COOH‐terminal threonines of ezrin/radixin/moesin (ERM) proteins and regulates their head‐to‐tail association. J Cell Biol 140, 647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medjkane S, Perez‐Sanchez C, Gaggioli C, Sahai E and Treisman R (2009) Myocardin‐related transcription factors and SRF are required for cytoskeletal dynamics and experimental metastasis. Nat Cell Biol 11, 257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mierke CT (2015) Physical view on migration modes. Cell Adh Migr 9, 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC and Nishimura SL (2002) The integrin alpha(v)beta8 mediates epithelial homeostasis through MT1‐MMP‐dependent activation of TGF‐beta1. J Cell Biol 157, 493–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell M, Oakes PW, Lenz M and Gardel ML (2015) Forcing cells into shape: the mechanics of actomyosin contractility. Nat Rev Mol Cell Biol 16, 486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabeshima K, Inoue T, Shimao Y, Kataoka H and Koono M (1999) Cohort migration of carcinoma cells: differentiated colorectal carcinoma cells move as coherent cell clusters or sheets. Histol Histopathol 14, 1183–1197. [DOI] [PubMed] [Google Scholar]
- Nabeshima K, Inoue T, Shimao Y, Okada Y, Itoh Y, Seiki M and Koono M (2000) Front‐cell‐specific expression of membrane‐type 1 matrix metalloproteinase and gelatinase A during cohort migration of colon carcinoma cells induced by hepatocyte growth factor/scatter factor. Cancer Res 60, 3364–3369. [PubMed] [Google Scholar]
- Nakamura N, Oshiro N, Fukata Y, Amano M, Fukata M, Kuroda S, Matsuura Y, Leung T, Lim L and Kaibuchi K (2000) Phosphorylation of ERM proteins at filopodia induced by Cdc42. Genes Cells 5, 571–581. [DOI] [PubMed] [Google Scholar]
- Nalbant P, Hodgson L, Kraynov V, Toutchkine A and Hahn KM (2004) Activation of endogenous Cdc42 visualized in living cells. Science 305, 1615–1619. [DOI] [PubMed] [Google Scholar]
- Nayal A, Webb DJ, Brown CM, Schaefer EM, Vicente‐Manzanares M and Horwitz AR (2006) Paxillin phosphorylation at Ser273 localizes a GIT1‐PIX‐PAK complex and regulates adhesion and protrusion dynamics. J Cell Biol 173, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus B, Buhren S, Bock B, Alves F, Vogel WF and Kiefer F (2011) Migration inhibition of mammary epithelial cells by Syk is blocked in the presence of DDR1 receptors. Cell Mol Life Sci 68, 3757–3770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuzillet C, Tijeras‐Raballand A, Cohen R, Cros J, Faivre S, Raymond E and de Gramont A (2015) Targeting the TGFbeta pathway for cancer therapy. Pharmacol Ther 147, 22–31. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Nagata K, Maekawa M, Ishizaki T, Narumiya S and Mizuno K (2000) Rho‐associated kinase ROCK activates LIM‐kinase 1 by phosphorylation at threonine 508 within the activation loop. J Biol Chem 275, 3577–3582. [DOI] [PubMed] [Google Scholar]
- Olson EN and Nordheim A (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat Rev Mol Cell Biol 11, 353–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orgaz JL, Pandya P, Dalmeida R, Karagiannis P, Sanchez‐Laorden B, Viros A, Albrengues J, Nestle FO, Ridley AJ, Gaggioli C et al (2014) Diverse matrix metalloproteinase functions regulate cancer amoeboid migration. Nat Commun 5, 4255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmani N, Peglion F, Chavrier P and Etienne‐Manneville S (2010) Cdc42 localization and cell polarity depend on membrane traffic. J Cell Biol 191, 1261–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otomo T, Tomchick DR, Otomo C, Panchal SC, Machius M and Rosen MK (2005) Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature 433, 488–494. [DOI] [PubMed] [Google Scholar]
- Overall CM and Kleifeld O (2006) Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br J Cancer 94, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overall CM and Lopez‐Otin C (2002) Strategies for MMP inhibition in cancer: innovations for the post‐trial era. Nat Rev Cancer 2, 657–672. [DOI] [PubMed] [Google Scholar]
- Page DL and Anderson TJ (1987) Diagnostic Histopathology of the Breast. Churchill‐Livingstone, Edinburgh, UK. [Google Scholar]
- Palamidessi A, Frittoli E, Garre M, Faretta M, Mione M, Testa I, Diaspro A, Lanzetti L, Scita G and Di Fiore PP (2008) Endocytic trafficking of Rac is required for the spatial restriction of signaling in cell migration. Cell 134, 135–147. [DOI] [PubMed] [Google Scholar]
- Pankova K, Rosel D, Novotny M and Brabek J (2010) The molecular mechanisms of transition between mesenchymal and amoeboid invasiveness in tumor cells. Cell Mol Life Sci 67, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parri M, Taddei ML, Bianchini F, Calorini L and Chiarugi P (2009) EphA2 reexpression prompts invasion of melanoma cells shifting from mesenchymal to amoeboid‐like motility style. Cancer Res 69, 2072–2081. [DOI] [PubMed] [Google Scholar]
- Patsialou A, Bravo‐Cordero JJ, Wang Y, Entenberg D, Liu H, Clarke M and Condeelis JS (2013) Intravital multiphoton imaging reveals multicellular streaming as a crucial component of in vivo cell migration in human breast tumors. Intravital 2, e25294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul NR, Allen JL, Chapman A, Morlan‐Mairal M, Zindy E, Jacquemet G, Fernandez del Ama L, Ferizovic N, Green DM, Howe JD et al (2015) alpha5beta1 integrin recycling promotes Arp2/3‐independent cancer cell invasion via the formin FHOD3. J Cell Biol 210, 1013–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus W, Baur I, Beutler AS and Reeves SA (1996) Diffuse brain invasion of glioma cells requires beta 1 integrins. Lab Invest 75, 819–826. [PubMed] [Google Scholar]
- Petrie RJ, Gavara N, Chadwick RS and Yamada KM (2012) Nonpolarized signaling reveals two distinct modes of 3D cell migration. J Cell Biol 197, 439–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrie RJ, Koo H and Yamada KM (2014) Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 345, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinner S and Sahai E (2008a) Imaging amoeboid cancer cell motility in vivo. J Microsc 231, 441–445. [DOI] [PubMed] [Google Scholar]
- Pinner S and Sahai E (2008b) PDK1 regulates cancer cell motility by antagonising inhibition of ROCK1 by RhoE. Nat Cell Biol 10, 127–137. [DOI] [PubMed] [Google Scholar]
- Pitts WC, Rojas VA, Gaffey MJ, Rouse RV, Esteban J, Frierson HF, Kempson RL and Weiss LM (1991) Carcinomas with metaplasia and sarcomas of the breast. Am J Clin Pathol 95, 623–632. [DOI] [PubMed] [Google Scholar]
- Polette M, Gilles C, de Bentzmann S, Gruenert D, Tournier JM and Birembaut P (1998) Association of fibroblastoid features with the invasive phenotype in human bronchial cancer cell lines. Clin Exp Metastasis 16, 105–112. [DOI] [PubMed] [Google Scholar]
- Poperechnaya A, Varlamova O, Lin PJ, Stull JT and Bresnick AR (2000) Localization and activity of myosin light chain kinase isoforms during the cell cycle. J Cell Biol 151, 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prall F, Nizze H and Barten M (2005) Tumour budding as prognostic factor in stage I/II colorectal carcinoma. Histopathology 47, 17–24. [DOI] [PubMed] [Google Scholar]
- Premont RT, Perry SJ, Schmalzigaug R, Roseman JT, Xing Y and Claing A (2004) The GIT/PIX complex: an oligomeric assembly of GIT family ARF GTPase‐activating proteins and PIX family Rac1/Cdc42 guanine nucleotide exchange factors. Cell Signal 16, 1001–1011. [DOI] [PubMed] [Google Scholar]
- Provenzano PP, Inman DR, Eliceiri KW, Trier SM and Keely PJ (2008) Contact guidance mediated three‐dimensional cell migration is regulated by Rho/ROCK‐dependent matrix reorganization. Biophys J 95, 5374–5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab M, Gentili M, de Belly H, Thiam HR, Vargas P, Jimenez AJ, Lautenschlaeger F, Voituriez R, Lennon‐Dumenil AM, Manel N et al (2016) ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 352, 359–362. [DOI] [PubMed] [Google Scholar]
- Ricart AD, Tolcher AW, Liu G, Holen K, Schwartz G, Albertini M, Weiss G, Yazji S, Ng C and Wilding G (2008) Volociximab, a chimeric monoclonal antibody that specifically binds alpha5beta1 integrin: a phase I, pharmacokinetic, and biological correlative study. Clin Cancer Res 14, 7924–7929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson BE and Lehmann R (2010) Mechanisms guiding primordial germ cell migration: strategies from different organisms. Nat Rev Mol Cell Biol 11, 37–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ (2015) Rho GTPase signalling in cell migration. Curr Opin Cell Biol 36, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley AJ, Schwartz MA, Burridge K, Firtel RA, Ginsberg MH, Borisy G, Parsons JT and Horwitz AR (2003) Cell migration: integrating signals from front to back. Science 302, 1704–1709. [DOI] [PubMed] [Google Scholar]
- Rizki A, Weaver VM, Lee SY, Rozenberg GI, Chin K, Myers CA, Bascom JL, Mott JD, Semeiks JR, Grate LR et al (2008) A human breast cell model of preinvasive to invasive transition. Cancer Res 68, 1378–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorth P (2007) Collective guidance of collective cell migration. Trends Cell Biol 17, 575–579. [DOI] [PubMed] [Google Scholar]
- Roussos ET, Balsamo M, Alford SK, Wyckoff JB, Gligorijevic B, Wang Y, Pozzuto M, Stobezki R, Goswami S, Segall JE et al (2011) Mena invasive (MenaINV) promotes multicellular streaming motility and transendothelial migration in a mouse model of breast cancer. J Cell Sci 124, 2120–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐Ontanon P, Orgaz JL, Aldaz B, Elosegui‐Artola A, Martino J, Berciano MT, Montero JA, Grande L, Nogueira L, Diaz‐Moralli S et al (2013) Cellular plasticity confers migratory and invasive advantages to a population of glioblastoma‐initiating cells that infiltrate peritumoral tissue. Stem Cells 31, 1075–1085. [DOI] [PubMed] [Google Scholar]
- Sadati M, Taheri Qazvini N, Krishnan R, Park CY and Fredberg JJ (2013) Collective migration and cell jamming. Differentiation 86, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadok A, McCarthy A, Caldwell J, Collins I, Garrett MD, Yeo M, Hooper S, Sahai E, Kuemper S, Mardakheh FK et al (2015) Rho kinase inhibitors block melanoma cell migration and inhibit metastasis. Cancer Res 75, 2272–2284. [DOI] [PubMed] [Google Scholar]
- Sahai E (2005) Mechanisms of cancer cell invasion. Curr Opin Genet Dev 15, 87–96. [DOI] [PubMed] [Google Scholar]
- Sahai E, Garcia‐Medina R, Pouyssegur J and Vial E (2007) Smurf1 regulates tumor cell plasticity and motility through degradation of RhoA leading to localized inhibition of contractility. J Cell Biol 176, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahai E and Marshall CJ (2003) Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat Cell Biol 5, 711–719. [DOI] [PubMed] [Google Scholar]
- Saito K, Ozawa Y, Hibino K and Ohta Y (2012) FilGAP, a Rho/Rho‐associated protein kinase‐regulated GTPase‐activating protein for Rac, controls tumor cell migration. Mol Biol Cell 23, 4739–4750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salbreux G, Charras G and Paluch E (2012) Actin cortex mechanics and cellular morphogenesis. Trends Cell Biol 22, 536–545. [DOI] [PubMed] [Google Scholar]
- Sanz‐Moreno V, Gadea G, Ahn J, Paterson H, Marra P, Pinner S, Sahai E and Marshall CJ (2008) Rac activation and inactivation control plasticity of tumor cell movement. Cell 135, 510–523. [DOI] [PubMed] [Google Scholar]
- Sanz‐Moreno V, Gaggioli C, Yeo M, Albrengues J, Wallberg F, Viros A, Hooper S, Mitter R, Feral CC, Cook M et al (2011) ROCK and JAK1 signaling cooperate to control actomyosin contractility in tumor cells and stroma. Cancer Cell 20, 229–245. [DOI] [PubMed] [Google Scholar]
- Sanz‐Moreno V and Marshall CJ (2010) The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr Opin Cell Biol 22, 690–696. [DOI] [PubMed] [Google Scholar]
- Scholey JM, Taylor KA and Kendrick‐Jones J (1980) Regulation of non‐muscle myosin assembly by calmodulin‐dependent light chain kinase. Nature 287, 233–235. [DOI] [PubMed] [Google Scholar]
- Scott RW and Olson MF (2007) LIM kinases: function, regulation and association with human disease. J Mol Med (Berl) 85, 555–568. [DOI] [PubMed] [Google Scholar]
- Seftor EA, Meltzer PS, Kirschmann DA, Pe'er J, Maniotis AJ, Trent JM, Folberg R and Hendrix MJ (2002) Molecular determinants of human uveal melanoma invasion and metastasis. Clin Exp Metastasis 19, 233–246. [DOI] [PubMed] [Google Scholar]
- Shiao SL and Coussens LM (2010) The tumor‐immune microenvironment and response to radiation therapy. J Mammary Gland Biol Neoplasia 15, 411–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh AC and Swartz MA (2011) Regulation of tumor invasion by interstitial fluid flow. Phys Biol 8, 015012. [DOI] [PubMed] [Google Scholar]
- Shtutman M, Levina E, Ohouo P, Baig M and Roninson IB (2006) Cell adhesion molecule L1 disrupts E‐cadherin‐containing adherens junctions and increases scattering and motility of MCF7 breast carcinoma cells. Cancer Res 66, 11370–11380. [DOI] [PubMed] [Google Scholar]
- Sood AK, Seftor EA, Fletcher MS, Gardner LM, Heidger PM, Buller RE, Seftor RE and Hendrix MJ (2001) Molecular determinants of ovarian cancer plasticity. Am J Pathol 158, 1279–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sounni NE, Dehne K, van Kempen L, Egeblad M, Affara NI, Cuevas I, Wiesen J, Junankar S, Korets L, Lee J et al (2010) Stromal regulation of vessel stability by MMP14 and TGFbeta. Dis Model Mech 3, 317–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starke J, Wehrle‐Haller B and Friedl P (2014) Plasticity of the actin cytoskeleton in response to extracellular matrix nanostructure and dimensionality. Biochem Soc Trans 42, 1356–1366. [DOI] [PubMed] [Google Scholar]
- Sternlicht MD, Bissell MJ and Werb Z (2000) The matrix metalloproteinase stromelysin‐1 acts as a natural mammary tumor promoter. Oncogene 19, 1102–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroka KM, Jiang H, Chen SH, Tong Z, Wirtz D, Sun SX and Konstantopoulos K (2014) Water permeation drives tumor cell migration in confined microenvironments. Cell 157, 611–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumi T, Matsumoto K, Takai Y and Nakamura T (1999) Cofilin phosphorylation and actin cytoskeletal dynamics regulated by rho‐ and Cdc42‐activated LIM‐kinase 2. J Cell Biol 147, 1519–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tambe DT, Hardin CC, Angelini TE, Rajendran K, Park CY, Serra‐Picamal X, Zhou EH, Zaman MH, Butler JP, Weitz DA et al (2011) Collective cell guidance by cooperative intercellular forces. Nat Mater 10, 469–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan I, Ng CH, Lim L and Leung T (2001a) Phosphorylation of a novel myosin binding subunit of protein phosphatase 1 reveals a conserved mechanism in the regulation of actin cytoskeleton. J Biol Chem 276, 21209–21216. [DOI] [PubMed] [Google Scholar]
- Tan I, Seow KT, Lim L and Leung T (2001b) Intermolecular and intramolecular interactions regulate catalytic activity of myotonic dystrophy kinase‐related Cdc42‐binding kinase alpha. Mol Cell Biol 21, 2767–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY and Nieto MA (2009) Epithelial‐mesenchymal transitions in development and disease. Cell 139, 871–890. [DOI] [PubMed] [Google Scholar]
- Thundimadathil J (2012) Cancer treatment using peptides: current therapies and future prospects. J Amino Acids 2012, 967347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tozluoglu M, Tournier AL, Jenkins RP, Hooper S, Bates PA and Sahai E (2013) Matrix geometry determines optimal cancer cell migration strategy and modulates response to interventions. Nat Cell Biol 15, 751–762. [DOI] [PubMed] [Google Scholar]
- Umemoto S, Bengur AR and Sellers JR (1989) Effect of multiple phosphorylations of smooth muscle and cytoplasmic myosins on movement in an in vitro motility assay. J Biol Chem 264, 1431–1436. [PubMed] [Google Scholar]
- Vedula SR, Leong MC, Lai TL, Hersen P, Kabla AJ, Lim CT and Ladoux B (2012) Emerging modes of collective cell migration induced by geometrical constraints. Proc Natl Acad Sci USA 109, 12974–12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente‐Manzanares M, Ma X, Adelstein RS and Horwitz AR (2009) Non‐muscle myosin II takes centre stage in cell adhesion and migration. Nat Rev Mol Cell Biol 10, 778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel WF, Abdulhussein R and Ford CE (2006) Sensing extracellular matrix: an update on discoidin domain receptor function. Cell Signal 18, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Wang X, Enomoto A, Asai N, Kato T and Takahashi M (2016) Collective invasion of cancer: perspectives from pathology and development. Pathol Int 66, 183–192. [DOI] [PubMed] [Google Scholar]
- Wang F, Kovacs M, Hu A, Limouze J, Harvey EV and Sellers JR (2003) Kinetic mechanism of non‐muscle myosin IIB: functional adaptations for tension generation and maintenance. J Biol Chem 278, 27439–27448. [DOI] [PubMed] [Google Scholar]
- Weigelin B, Bakker G and Friedl P (2012) Intravital third harmonic generation microscopy of collective melanoma cell invasion. Intravital 1, 32–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wicki A, Lehembre F, Wick N, Hantusch B, Kerjaschki D and Christofori G (2006) Tumor invasion in the absence of epithelial‐mesenchymal transition: podoplanin‐mediated remodeling of the actin cytoskeleton. Cancer Cell 9, 261–272. [DOI] [PubMed] [Google Scholar]
- Wilkinson S, Paterson HF and Marshall CJ (2005) Cdc42‐MRCK and Rho‐ROCK signalling cooperate in myosin phosphorylation and cell invasion. Nat Cell Biol 7, 255–261. [DOI] [PubMed] [Google Scholar]
- Willis R (1952) The Spread of Tumors in the Human Body. Butterworth, London. [Google Scholar]
- Wojciak‐Stothard B and Leiper J (2008) Rho GTPases and hypoxia in pulmonary vascular endothelial cells. Methods Enzymol 439, 267–283. [DOI] [PubMed] [Google Scholar]
- Wolf K and Friedl P (2011) Extracellular matrix determinants of proteolytic and non‐proteolytic cell migration. Trends Cell Biol 21, 736–744. [DOI] [PubMed] [Google Scholar]
- Wolf K, Mazo I, Leung H, Engelke K, von Andrian UH, Deryugina EI, Strongin AY, Brocker EB and Friedl P (2003) Compensation mechanism in tumor cell migration: mesenchymal‐amoeboid transition after blocking of pericellular proteolysis. J Cell Biol 160, 267–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Te Lindert M, Krause M, Alexander S, Te Riet J, Willis AL, Hoffman RM, Figdor CG, Weiss SJ and Friedl P (2013) Physical limits of cell migration: control by ECM space and nuclear deformation and tuning by proteolysis and traction force. J Cell Biol 201, 1069–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, Stack MS and Friedl P (2007) Multi‐step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol 9, 893–904. [DOI] [PubMed] [Google Scholar]
- Wong HS, Jaumouille V, Heit B, Doodnauth SA, Patel S, Huang YW, Grinstein S and Robinson LA (2014) Cytoskeletal confinement of CX3CL1 limits its susceptibility to proteolytic cleavage by ADAM10. Mol Biol Cell 25, 3884–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyckoff JB, Pinner SE, Gschmeissner S, Condeelis JS and Sahai E (2006) ROCK‐ and myosin‐dependent matrix deformation enables protease‐independent tumor‐cell invasion in vivo. Curr Biol 16, 1515–1523. [DOI] [PubMed] [Google Scholar]
- Wyckoff J, Wang W, Lin EY, Wang Y, Pixley F, Stanley ER, Graf T, Pollard JW, Segall J and Condeelis J (2004) A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res 64, 7022–7029. [DOI] [PubMed] [Google Scholar]
- Yamazaki D, Kurisu S and Takenawa T (2009) Involvement of Rac and Rho signaling in cancer cell motility in 3D substrates. Oncogene 28, 1570–1583. [DOI] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E and Mizuno K (1998) Cofilin phosphorylation by LIM‐kinase 1 and its role in Rac‐mediated actin reorganization. Nature 393, 809–812. [DOI] [PubMed] [Google Scholar]
- Yano H, Mazaki Y, Kurokawa K, Hanks SK, Matsuda M and Sabe H (2004) Roles played by a subset of integrin signaling molecules in cadherin‐based cell‐cell adhesion. J Cell Biol 166, 283–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Z, Sadok A, Sailem H, McCarthy A, Xia X, Li F, Garcia MA, Evans L, Barr AR, Perrimon N et al (2013) A screen for morphological complexity identifies regulators of switch‐like transitions between discrete cell shapes. Nat Cell Biol 15, 860–871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller M (2011) CD44: can a cancer‐initiating cell profit from an abundantly expressed molecule? Nat Rev Cancer 11, 254–267. [DOI] [PubMed] [Google Scholar]
- Zucker S, Cao J and Chen WT (2000) Critical appraisal of the use of matrix metalloproteinase inhibitors in cancer treatment. Oncogene 19, 6642–6650. [DOI] [PubMed] [Google Scholar]