

Abstract
Purpose:
To investigate mutations of visual system homeobox 1 (VSX1) and superoxide dismutase 1 (SOD1) in 20 patients with keratoconus in the south of Iran.
Methods:
Twenty patients with keratoconus who had a positive familial history were enrolled in this study and gave informed consent for DNA analysis. Genomic DNA was extracted from peripheral blood lymphocytes. Polymerase chain reaction (PCR) was carried out to amplify exon 2 of SOD1 and its exon-intron boundary for the detection of a seven-base deletion in intron 2 of SOD1, and also all five exons of VSX1 and their exon-intron boundaries. Amplified samples were then subjected to direct DNA sequencing.
Results:
Sequencing data were compared against reference sequences using NCBI basic local alignment search tool (BLAST), which revealed that our patients had no mutations in SOD1 and VSX1. Two single-nucleotide polymorphisms (SNPs), namely in VSX1(rs58752432 and rs59089167) were found in six patients.
Conclusion:
Mutations in VSX1 and SOD1 genes associated with keratoconus were not identified in our patients. Therefore, it will be necessary to investigate other chromosomal loci for potential causal mutations of keratoconus using next generation sequencing (NGS) methods in our population.
Keywords: Keratoconus, Superoxide Dismutase 1, Visual System Homeobox 1
INTRODUCTION
Keratoconus (KCN), which is a bilateral, noninflammatory corneal ectasia, is associated with a progressive increase in corneal curvature, apical thinning, and irregular corneal astigmatism. KCN is often asymmetric and an obvious cone-shaped protrusion of the corneal surface may develop in this condition.[1,2,3] Its worldwide prevalence has been estimated to be 5.4 cases per 10,000 individuals in the general population and the disease affects both genders and all ethnicities.[4,5]
The etiology of KCN is unknown and is believed to be multifactorial.[4] The hereditary form of KCN was estimated to account for between 6% and 23.5% of cases and its prevalence among first-degree relatives is up to 68 times higher compared to the general population.[4,6,7] It has been reported that KCN is associated with several systemic disorders, including atopy,[8,9] Down's syndrome,[10,11] floppy eyelid syndrome,[12] congenital hip dysplasia,[13] Rieger's syndrome,[14] focal dermal hypoplasia,[15] Crouzon's syndrome (craniofacial dysostosis)[16] and Marfan's syndrome.[4,17] Several studies have been conducted to identify the biochemical, histological, and genetic bases of KCN.[18,19]
Several genes with key roles in corneal development have been proposed as potential candidate disease-causing genes for KCN. Some studies have shown that mutations in a specific exon of visual system homeobox 1 (VSX1) are among the molecular bases of this disease.[20,21,22] However, other studies have not been able to find a disease-causing mutation in VSX1.[23] VSX1 is located on chromosome 20p11.21 and has important roles in craniofacial and ocular development. It encodes a protein consisting of a paired-like homeodomain that may regulate cone opsin expression during the early stages of ocular development.[24,25,26,27]
Another proposed candidate gene responsible for KCN is superoxide dismutase 1 (SOD1). This gene is located on chromosome 21q22.11 and provides instructions for making superoxide dismutase enzyme.[28,29] The enzyme, which binds zinc and copper ions, is responsible for destroying free superoxide radicals that can cause damage to cells.[30,31] Mutations in SOD1 are associated with amyotrophic lateral sclerosis.[29,32] Some studies identified a variant in SOD1, namely a seven-base deletion in intron 2 (IVS2+50del7bp), to be associated with KCN,[28] while other studies have not found any pathogenic mutations in SOD1 in patients with KCN.[33]
By the fact that some mutations in different exons of VSX1[21,22,34,35] and only in intron 2 of SOD1 were identified to be associated with KCN,[28] the purpose of this study was to investigate previously identified mutations of these two genes in 20 KCN patients in the south of Iran.
METHODS
This study was designed as a mutation detection analysis in patients with KCN from the south of Iran. Twenty unrelated KCN patients with a familial history of the disease were recruited for this study. The study was approved by the Ethics Committee of Shiraz University of Medical Sciences, Shiraz, Iran. All patients gave informed consent before undergoing DNA tests for previously reportedmutations in SOD1 and VSX1. The patients were diagnosed by a group of ophthalmologists and the definitive diagnosis of KCN was made based on slit-lamp biomicroscopy, retinoscopy, and corneal topography. Additionally, other members of every affected patient were examined for KCN using imaging techniques such as corneal topography and Pentacam® (OCULUS, Wetzlar, Germany) scans. All 20 selected patients had at least one operation for KCN.
Three mL of whole blood samples from the patients were collected into ethylenediaminetetraacetic acid tubes (Vacutainer™ EDTA K3 Tubes) and genomic DNA was then extracted from peripheral blood lymphocytes using a CinnaPure® DNA extraction kit (SinaClon, Tehran, Iran) according to the manufacturer's instructions.
Polymerase chain reaction (PCR) oligonucleotide primers were designed to amplify exon 2 of SOD1 and its exon-intron boundary for the investigation of a genomic seven-base deletion in intron 2 of SOD1 (IVS2+50del7bp), and also all five exons of VSX1 and their exon-intron boundaries. All critical primer parameters were analyzed using online bioinformatics software programs (Primer 3, OligoAnalyzer, and OligoCalc) and compared against the NCBI database (BLASTn). All PCR primers used in the current study are listed in Table 1.
Table 1.
Polymerase chain reaction (PCR) primers used in this study

The PCR reactions were carried out in a total volume of 50 μl containing 1 μl of each primer (20 pmol/μl), 5 μl DNA template (50–200 ng), 5 μl PCR buffer (CinnaGen, Tehran, Iran), 0.5 μl dNTPs (10 mM), 1.5 μl MgCl2 (50 mM, CinnaGen), 0.2 μl Taq DNA Polymerase (CinnaGen), and 35.8 μl dH2O. The PCR reactions were performed using a Veriti™ Thermal Cycler (Applied Biosystems, Foster City, CA, USA) according to the Taq DNA polymerase protocol (CinnaGen) and the amplification products were then subjected to direct DNA sequencing.
RESULTS
Twenty unrelated patients (14 females and 6 males, mean ages: 22 years [females] and 45 years [males]) were evaluated using clinical and molecular approaches. DNA was extracted from peripheral blood lymphocytes, amplified, and subjected to direct DNA sequencing. The sequencing data were then compared against reference sequences (VSX1: NG_008101, SOD1: NG_008689) using NCBI BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). None of the patients showed mutations in these two genes. Only two single nucleotide polymorphisms (SNPs) were found in VSX1: g. 8326G>A (c.627+23G>A, rs58752432) and g. 7898 G>A (c.503+202G>A, rs59089167) [Table 2 and Figure 1].
Table 2.
Sequencing results for VSX1
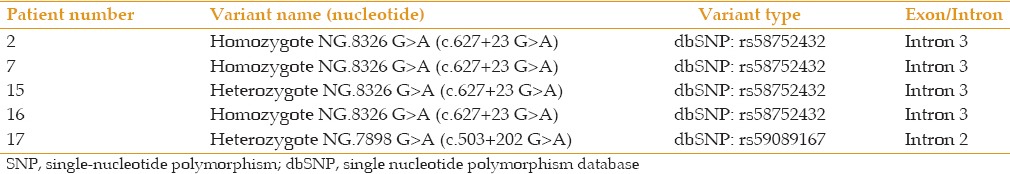
Figure 1.
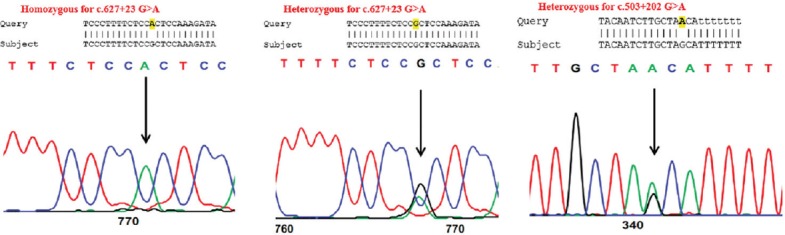
Sequencing results obtained in the current study.
DISCUSSION
Keratoconus has been described as a complex multifactorial disorder with involvement of both environmental and genetic elements. It has been shown that dizygotic twin discordance, familial inheritance, and association with other genetic diseases can provide insights into its genetic etiology. However, environmental conditions that are responsible for some clinical presentations of KCN have also been reported, including chronic eye rubbing, eye atopy, and wearing contact lenses. It is worth noting that genetic heterogeneity, reduced penetrance, and interactions between genes may also influence KCN.[4,36]
Until now, various loci have been proposed to be related to the molecular mechanism of KCN. Two main methods that have been used to investigate the disease-causing genes related to KCN are association studies and linkage analysis.[37,38] In the case of KCN, linkage analysis is usually difficult because this method of gene mapping is a model-based approach that requires inputs including the disease allele's frequency, penetration, and phenocopy. However, linkage analyses conducted for KCN have reported some loci related to the disease,[39] and one KCN locus, 5q21.2, was identified in two separate studies.[40]
The association study method has been used in several studies on KCN.[41,42] This method, which investigates the relationship between genotype and phenotype, is applied in two ways, namely direct association using SNPs as the causative factor or indirect association that reveals linkage disequilibrium with the causative SNP. In addition, a genome-wide association study was conducted to investigate candidate genes for KCN and a candidate gene, RAB3GAP1, was identified at locus 2q21.3.[42]
Different modes of inheritance have been identified in patients with KCN; for instance, 95% of cases with familial KCN have been reported to show autosomal dominant inheritance.[7] Other modes of inheritance such as autosomal recessive have also identified in the children of consanguineous parents.[43] Some studies have also reported that the relatives of KCN patients showed an increased prevalence of the disease.[44]
To date, a large number of loci have been identified in KCN, such as 16q22.3–q23.1 (autosomal dominant locus), 15q22.33–24.2, 17p13, 3p14–q13, 5q14.3–q21.1, 2p24, and 13q32.[45] However, until now, there have been no reports on the identification of mutations within genes at those loci.[45]
The only major genetic factor reported in the pathogenesis of KCN to date is VSX1. This gene encodes a protein that contains a paired-like homeodomain. The protein, which binds to the red/green visual pigment gene cluster region of genomic DNA, may play an important role in the regulation of cone opsin expression during the initial stages of development.[25,46] Posterior polymorphous and corneal dystrophies are two abnormalities that can result from mutations in VSX1.[47]
Several genetic variations (p.L17P, p.D144E, p.R166W, and others) in VSX1 have been identified to be deleterious in KCN patients [Figure 2].[22,48,49] Themutation frequency of VSX1 has been shown to be different in KCN patients, compared to general population, but the pathogenicity of these mutations has not been fully confirmed. It has been found that VSX1 plays a role in corneal wound healing by influencing the differentiation of corneal keratocytes into myofibroblasts.[50] This function of VSX1 may be connected with its involvement in the pathogenesis of KCN. In several studies, VSX1 has been shown to be responsible for causing KCN in some ethnicities and countries. For instance, VSX1 mutations were first identified in about 9% of 63 unrelated KCN patients in a study conducted by Heon et al in the United States.[20] In Iran, mutations of VSX1 were reported in two out of 26 unrelated patients with KCN.[49] In many studies performed to investigate VSX1 mutations, the variants responsible for KCN have not been found.[23,51]
Figure 2.

Variations reported in VSX1 protein.
To understand the pathogenic mechanism of KCN, it is essential to identify genes that may be associated with this disorder. It has been shown that various mutations in VSX1 have caused distinct disorders that may be due to disrupted interactions between this protein and its predicted partners in a complex protein network. The initial step in drug discovery research is to identify essential proteins or drug targets for a biological process. To identify interactions between this protein and other partners that may play important roles in the pathogeneses of KCN and other eye disorders, we used STRING software (Search tool for the Retrieval of Interacting Genes/Proteins: string.embl.de/) and we identified that VSX1 protein had interactions with several essential proteins expressed in eyes according to the National Eye Institute (https://neibank.nei.nih.gov/) such as ubiquitin-conjugating enzyme E2I (UBE2I) and NK2 homeobox 1 (NKX2-1) [Figure 3]. One of the predicted partners of VSX1 is UBE2I, which is crucial for nuclear architecture and chromosome segregation SUMOylates p53/TP53 at ‘Lys-386’. NKX2-1, which is another predicted partner of VSX1, is a transcription factor with a major role in the maintenance of the thyroid differentiation phenotype (data about predicted protein partners of VSX1 were extracted using the network analysis functionality of STRING software). Disruption of the interactions between VSX1 and these predicted partners may result in different clinical manifestations in patients with disease-causing mutations of VSX1 and should be investigated in future studies.
Figure 3.
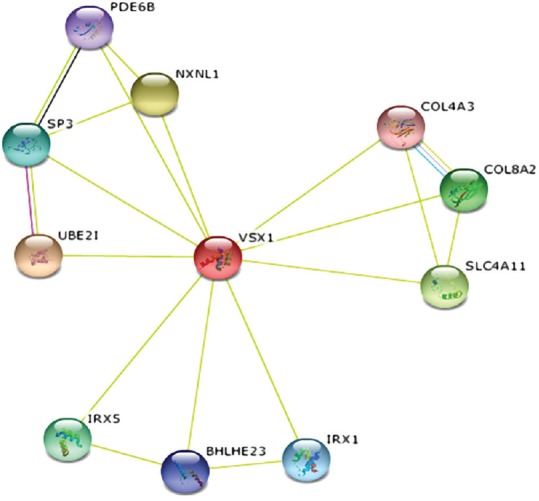
Functional and physical protein interactions of VSX1 identified using STRING9 software.
Another reported candidate disease-causing gene for KCN is SOD1.[28] The protein encoded by SOD1 is responsible for destroying superoxide free radicals in the body[52] and mutations of SOD1 result in postcholecystectomy syndrome, ocular colobomas, ichthyosis, brain malformations, and endocrine abnormalities.[53,54]
In a study conducted by Udar et al, mutation screening of SOD1 was carried out using DNA sequencing in 15 families with KCN and a seven-base deletion in intron 2 of the gene was identified in two families. Based on this observation, a pathogenic role was proposed for variants of SOD1 in KCN.[28] In Iran, a mutation analysis of SOD1 was performedin 26 unrelated families; however, no mutations were identified.[49]
To determine whether interactions between SOD1 protein and its partners may have important roles in the pathogeneses of KCN and other eye disorders, we conducted an analysis of protein-protein interactions using STRING software. This analysis revealed that SOD1 protein had essential interactions with several crucial partners [Figure 4] expressed in eyes according to the National Eye Institute. Identification of the main roles of these proteins may help to determine the mechanism by which a specific mutation in SOD1 can cause KCN, while other SOD1 variants do not have this consequence.
Figure 4.
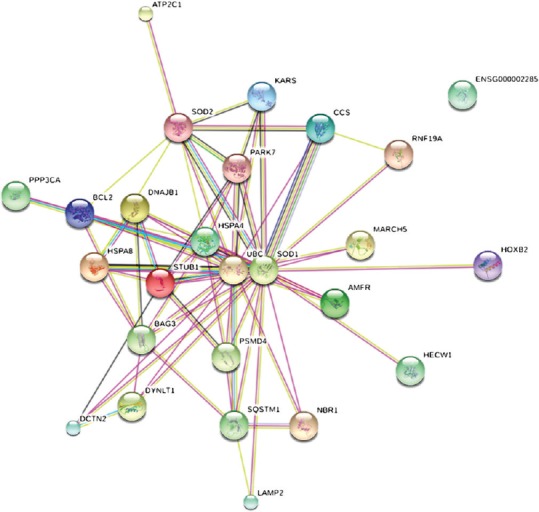
Functional and physical protein interactions of SOD1 identified using STRING9 software.
As described above, some studies have identified mutations of VSX1 and SOD1; however, other subsequent investigations have not been able to confirm the roles of these variants. These studies support the notion that several genes are involved in KCN pathophysiology, rather than a single gene. Therefore, more studies should be conducted to investigate the roles of other possible genes such as MIR184 which its mutations were reported in families affected by KCN.[55] However, in a study conducted By Farzadfard et al, they could not find any pathogenic mutations in their Iranian patients with KCN.[56] In conclusion, the present study showed that there were no mutations in VSX1 and the previously reported mutation of SOD1 was also not present in our patients. Therefore, we suggest that other genes may have essential roles in the pathogenesis of KCN in this population and VSX1 and SOD1 should be investigated in a large number of KCN patients in Iran.
Financial Support and Sponsorship
Nil.
Conflicts of Interest
There are no conflicts of interest.
Acknowledgments
We acknowledge the Department of Ophthalmology at Shiraz University of Medical Sciences for funding this research. We also thank the patients for participating in this study. The study was conducted in the Comprehensive Medical Genetics Centre, Shiraz, Iran. The present article was extracted from a thesis written by Payam Naghash. This thesis was supervised by Dr. Mahmood Nejabat and Dr. Majid Fardaei.
REFERENCES
- 1.Zadnik K, Barr JT, Gordon MO, Edrington TB. Biomicroscopic signs and disease severity in keratoconus. Collaborative Longitudinal Evaluation of Keratoconus (CLEK) Study Group. Cornea. 1996;15:139–146. doi: 10.1097/00003226-199603000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Kennedy RH, Bourne WM, Dyer JA. A 48-year clinical and epidemiologic study of keratoconus. Am J Ophthalmol. 1986;101:267–273. doi: 10.1016/0002-9394(86)90817-2. [DOI] [PubMed] [Google Scholar]
- 3.Auffarth GU, Wang L, Volcker HE. Keratoconus evaluation using the Orbscan Topography System. J Cataract Refract Surg. 2000;26:222–228. doi: 10.1016/s0886-3350(99)00355-7. [DOI] [PubMed] [Google Scholar]
- 4.Rabinowitz YS. Keratoconus. Surv Ophthalmol. 1998;42:297–319. doi: 10.1016/s0039-6257(97)00119-7. [DOI] [PubMed] [Google Scholar]
- 5.Krachmer JH, Feder RS, Belin MW. Keratoconus and related noninflammatory corneal thinning disorders. Surv Ophthalmol. 1984;28:293–322. doi: 10.1016/0039-6257(84)90094-8. [DOI] [PubMed] [Google Scholar]
- 6.Wang Y, Rabinowitz YS, Rotter JI, Yang H. Genetic epidemiological study of keratoconus: Evidence for major gene determination. Am J Med Genet. 2000;93:403–409. [PubMed] [Google Scholar]
- 7.Edwards M, McGhee CN, Dean S. The genetics of keratoconus. Clin Exp Ophthalmol. 2001;29:345–351. doi: 10.1046/j.1442-9071.2001.d01-16.x. [DOI] [PubMed] [Google Scholar]
- 8.Gasset AR, Hinson WA, Frias JL. Keratoconus and atopic diseases. Ann Ophthalmol. 1978;10:991–994. [PubMed] [Google Scholar]
- 9.Wachtmeister L, Ingemansson SO, Moller E. Atopy and HLA antigens in patients with keratoconus. Acta Ophthalmol (Copenh) 1982;60:113–122. doi: 10.1111/j.1755-3768.1982.tb05787.x. [DOI] [PubMed] [Google Scholar]
- 10.Cullen JF, Butler HG. Mongolism (Down's Syndrome) and Keratoconus. Br J Ophthalmol. 1963;47:321–330. doi: 10.1136/bjo.47.6.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shapiro MB, France TD. The ocular features of Down's syndrome. Am J Ophthalmol. 1985;99:659–663. doi: 10.1016/s0002-9394(14)76031-3. [DOI] [PubMed] [Google Scholar]
- 12.Lee WJ, Kim JC, Shyn KH. Clinical evaluation of corneal diseases associated with floppy eyelid syndrome. Korean J Ophthalmol. 1996;10:116–121. doi: 10.3341/kjo.1996.10.2.116. [DOI] [PubMed] [Google Scholar]
- 13.Nucci P, Brancato R. Keratoconus and congenital hip dysplasia. Am J Ophthalmol. 1991;111:775–776. doi: 10.1016/s0002-9394(14)76792-3. [DOI] [PubMed] [Google Scholar]
- 14.Greenfield G, Stein R, Romano A, Goodman RM. Blue sclerae and keratoconus: Key features of a distinct heritable disorder of connective tissue. Clin Genet. 1973;4:8–16. doi: 10.1111/j.1399-0004.1973.tb01115.x. [DOI] [PubMed] [Google Scholar]
- 15.Zala L, Ettlin C, Krebs A. [Focal dermal hypoplasia with keratoconus, papillomatosis of esophagus and hidrocystomas (author's transl)] Dermatologica. 1975;150:176–185. [PubMed] [Google Scholar]
- 16.Perlman JM, Zaidman GW. Bilateral keratoconus in Crouzon's syndrome. Cornea. 1994;13:80–81. doi: 10.1097/00003226-199401000-00014. [DOI] [PubMed] [Google Scholar]
- 17.Austin MG, Schaefer RF. Marfan's syndrome, with unusual blood vessel manifestations. AMA Arch Pathol. 1957;64:205–209. [PubMed] [Google Scholar]
- 18.Ihalainen A. Clinical and epidemiological features of keratoconus genetic and external factors in the pathogenesis of the disease. Acta Ophthalmol Suppl. 1986;178:1–64. [PubMed] [Google Scholar]
- 19.Tang YG, Rabinowitz YS, Taylor KD, Li X, Hu M, Picornell Y, et al. Genomewide linkage scan in a multigeneration Caucasian pedigree identifies a novel locus for keratoconus on chromosome 5q14.3-q21.1. Genet Med. 2005;7:397–405. doi: 10.1097/01.gim.0000170772.41860.54. [DOI] [PubMed] [Google Scholar]
- 20.Heon E, Greenberg A, Kopp KK, Rootman D, Vincent AL, Billingsley G, et al. VSX1: A gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet. 2002;11:1029–1036. doi: 10.1093/hmg/11.9.1029. [DOI] [PubMed] [Google Scholar]
- 21.Bisceglia L, Ciaschetti M, De Bonis P, Campo PA, Pizzicoli C, Scala C, et al. VSX1 mutational analysis in a series of Italian patients affected by keratoconus: Detection of a novel mutation. Invest Ophthalmol Vis Sci. 2005;46:39–45. doi: 10.1167/iovs.04-0533. [DOI] [PubMed] [Google Scholar]
- 22.Mok JW, Baek SJ, Joo CK. VSX1 gene variants are associated with keratoconus in unrelated Korean patients. J Hum Genet. 2008;53:842–849. doi: 10.1007/s10038-008-0319-6. [DOI] [PubMed] [Google Scholar]
- 23.Abu-Amero KK, Kalantan H, Al-Muammar AM. Analysis of the VSX1 gene in keratoconus patients from Saudi Arabia. Mol Vis. 2011;17:667–672. [PMC free article] [PubMed] [Google Scholar]
- 24.Hayashi T, Huang J, Deeb SS. RINX(VSX1), a novel homeobox gene expressed in the inner nuclear layer of the adult retina. Genomics. 2000;67:128–139. doi: 10.1006/geno.2000.6248. [DOI] [PubMed] [Google Scholar]
- 25.Semina EV, Mintz-Hittner HA, Murray JC. Isolation and characterization of a novel human paired-like homeodomain-containing transcription factor gene, VSX1, expressed in ocular tissues. Genomics. 2000;63:289–293. doi: 10.1006/geno.1999.6093. [DOI] [PubMed] [Google Scholar]
- 26.Chow RL, Volgyi B, Szilard RK, Ng D, McKerlie C, Bloomfield SA, et al. Control of late off-center cone bipolar cell differentiation and visual signaling by the homeobox gene Vsx1. Proc Natl Acad Sci U S A. 2004;101:1754–1759. doi: 10.1073/pnas.0306520101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ohtoshi A, Wang SW, Maeda H, Saszik SM, Frishman LJ, Klein WH, et al. Regulation of retinal cone bipolar cell differentiation and photopic vision by the CVC homeobox gene Vsx1. Curr Biol. 2004;14:530–536. doi: 10.1016/j.cub.2004.02.027. [DOI] [PubMed] [Google Scholar]
- 28.Udar N, Atilano SR, Brown DJ, Holguin B, Small K, Nesburn AB, et al. SOD1: A candidate gene for keratoconus. Invest Ophthalmol Vis Sci. 2006;47:3345–3351. doi: 10.1167/iovs.05-1500. [DOI] [PubMed] [Google Scholar]
- 29.Noor R, Mittal S, Iqbal J. Superoxide dismutase-applications and relevance to human diseases. Med Sci Monit. 2002;8:RA210–215. [PubMed] [Google Scholar]
- 30.Behndig A, Svensson B, Marklund SL, Karlsson K. Superoxide dismutase isoenzymes in the human eye. Invest Ophthalmol Vis Sci. 1998;39:471–475. [PubMed] [Google Scholar]
- 31.Zelko IN, Mariani TJ, Folz RJ. Superoxide dismutase multigene family: A comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic Biol Med. 2002;33:337–349. doi: 10.1016/s0891-5849(02)00905-x. [DOI] [PubMed] [Google Scholar]
- 32.Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62. doi: 10.1038/362059a0. [DOI] [PubMed] [Google Scholar]
- 33.Stabuc-Silih M, Strazisar M, Hawlina M, Glavac D. Absence of pathogenic mutations in VSX1 and SOD1 genes in patients with keratoconus. Cornea. 2010;29:172–176. doi: 10.1097/ICO.0b013e3181aebf7a. [DOI] [PubMed] [Google Scholar]
- 34.Mintz-Hittner HA, Semina EV, Frishman LJ, Prager TC, Murray JC. VSX1 (RINX) mutation with craniofacial anomalies, empty sella, corneal endothelial changes, and abnormal retinal and auditory bipolar cells. Ophthalmology. 2004;111:828–836. doi: 10.1016/j.ophtha.2003.07.006. [DOI] [PubMed] [Google Scholar]
- 35.Eran P, Almogit A, David Z, Wolf HR, Hana G, Yaniv B, et al. The D144E substitution in the VSX1 gene: A non-pathogenic variant or a disease causing mutation>? Ophthalmic Genet. 2008;29:53–59. doi: 10.1080/13816810802008242. [DOI] [PubMed] [Google Scholar]
- 36.Barr JT, Wilson BS, Gordon MO, Rah MJ, Riley C, Kollbaum PS, et al. Estimation of the incidence and factors predictive of corneal scarring in the Collaborative Longitudinal Evaluation of Keratoconus (CLEK) Study. Cornea. 2006;25:16–25. doi: 10.1097/01.ico.0000164831.87593.08. [DOI] [PubMed] [Google Scholar]
- 37.Brancati F, Valente EM, Sarkozy A, Feher J, Castori M, Del Duca P, et al. A locus for autosomal dominant keratoconus maps to human chromosome 3p14-q13. J Med Genet. 2004;41:188–192. doi: 10.1136/jmg.2003.012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gajecka M, Radhakrishna U, Winters D, Nath SK, Rydzanicz M, Ratnamala U, et al. Localization of a gene for keratoconus to a 5.6-Mb interval on 13q32. Invest Ophthalmol Vis Sci. 2009;50:1531–1539. doi: 10.1167/iovs.08-2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X, Rabinowitz YS, Tang YG, Picornell Y, Taylor KD, Hu M, et al. Two-stage genome-wide linkage scan in keratoconus sib pair families. Invest Ophthalmol Vis Sci. 2006;47:3791–3795. doi: 10.1167/iovs.06-0214. [DOI] [PubMed] [Google Scholar]
- 40.Bisceglia L, De Bonis P, Pizzicoli C, Fischetti L, Laborante A, Di Perna M, et al. Linkage analysis in keratoconus: Replication of locus 5q21.2 and identification of other suggestive Loci. Invest Ophthalmol Vis Sci. 2009;50:1081–1086. doi: 10.1167/iovs.08-2382. [DOI] [PubMed] [Google Scholar]
- 41.Li X, Bykhovskaya Y, Tang YG, Picornell Y, Haritunians T, Aldave AJ, et al. An association between the calpastatin (CAST) gene and keratoconus. Cornea. 2013;32:696–701. doi: 10.1097/ICO.0b013e3182821c1c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li X, Bykhovskaya Y, Haritunians T, Siscovick D, Aldave A, Szczotka-Flynn L, et al. A genome-wide association study identifies a potential novel gene locus for keratoconus, one of the commonest causes for corneal transplantation in developed countries. Hum Mol Genet. 2012;21:421–429. doi: 10.1093/hmg/ddr460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jacobs DS, Dohlman CH. Is keratoconus genetic? Int Ophthalmol Clin. 1993;33:249–260. doi: 10.1097/00004397-199303320-00023. [DOI] [PubMed] [Google Scholar]
- 44.Karimian F, Aramesh S, Rabei HM, Javadi MA, Rafati N. Topographic evaluation of relatives of patients with keratoconus. Cornea. 2008;27:874–878. doi: 10.1097/ICO.0b013e31816f5edc. [DOI] [PubMed] [Google Scholar]
- 45.Nowak DM, Gajecka M. The genetics of keratoconus. Middle East Afr J Ophthalmol. 2011;18:2–6. doi: 10.4103/0974-9233.75876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dorval KM, Bobechko BP, Ahmad KF, Bremner R. Transcriptional activity of the paired-like homeodomain proteins CHX10 and VSX1. J Biol Chem. 2005;280:10100–10108. doi: 10.1074/jbc.M412676200. [DOI] [PubMed] [Google Scholar]
- 47.Clausen I, Weidle E, Duncker G, Grunauer-Kloevekorn C. [Mutational analysis of VSX-1 in one patient with posterior polymorphous corneal dystrophy and in three families with hereditary Fuchs endothelial dystrophy] Klin Monbl Augenheilkd. 2009;226:466–469. doi: 10.1055/s-0028-1109427. [DOI] [PubMed] [Google Scholar]
- 48.De Bonis P, Laborante A, Pizzicoli C, Stallone R, Barbano R, Longo C, et al. Mutational screening of VSX1, SPARC, SOD1, LOX, and TIMP3 in keratoconus. Mol Vis. 2011;17:2482–2494. [PMC free article] [PubMed] [Google Scholar]
- 49.Saee-Rad S, Hashemi H, Miraftab M, Noori-Daloii MR, Chaleshtori MH, Raoofian R, et al. Mutation analysis of VSX1 and SOD1 in Iranian patients with keratoconus. Mol Vis. 2011;17:3128–3136. [PMC free article] [PubMed] [Google Scholar]
- 50.Barbaro V, Di Iorio E, Ferrari S, Bisceglia L, Ruzza A, De Luca M, et al. Expression of VSX1 in human corneal keratocytes during differentiation into myofibroblasts in response to wound healing. Invest Ophthalmol Vis Sci. 2006;47:5243–5250. doi: 10.1167/iovs.06-0185. [DOI] [PubMed] [Google Scholar]
- 51.Tanwar M, Kumar M, Nayak B, Pathak D, Sharma N, Titiyal JS, et al. VSX1 gene analysis in keratoconus. Mol Vis. 2010;16:2395–2401. [PMC free article] [PubMed] [Google Scholar]
- 52.Hennig J, Andresen C, Museth AK, Lundstrom P, Tibell LA, Jonsson BH. Local destabilization of the metal-binding region in human copper-zinc superoxide dismutase by remote mutations is a possible determinant for progression of ALS. Biochemistry. 2015;54:323–333. doi: 10.1021/bi500606j. [DOI] [PubMed] [Google Scholar]
- 53.Kunst CB, Mezey E, Brownstein MJ, Patterson D. Mutations in SOD1 associated with amyotrophic lateral sclerosis cause novel protein interactions. Nat Genet. 1997;15:91–94. doi: 10.1038/ng0197-91. [DOI] [PubMed] [Google Scholar]
- 54.Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem. 2005;74:563–593. doi: 10.1146/annurev.biochem.72.121801.161647. [DOI] [PubMed] [Google Scholar]
- 55.Lechner J, Bae HA, Guduric-Fuchs J, Rice A, Govindarajan G, Siddiqui S, et al. Mutational analysis of MIR184 in sporadic keratoconus and myopia. Invest Ophthalmol Vis Sci. 2013;54:5266–5272. doi: 10.1167/iovs.13-12035. [DOI] [PubMed] [Google Scholar]
- 56.Farzadfard A, Nassiri N, Moghadam TN, Paylakhi SH, Elahi E. Screening for MIR184 Mutations in Iranian Patients with Keratoconus. J Ophthalmic Vis Res. 2016;11:3–7. doi: 10.4103/2008-322X.180715. [DOI] [PMC free article] [PubMed] [Google Scholar]