

Abstract
NADPH oxidase (NOX)-derived reactive oxygen species (ROS) have been demonstrated to mediate the activation of NOD-like receptor protein 3 (NLRP3) inflammasomes in podocytes in response to elevated levels of homocysteine (Hcys). However, it remains unknown how NLRP3 inflammasome activation is triggered by NOX. The present study tested whether the guanine nucleotide exchange factor Vav2 mediates Rac1-mediated NOX activation in response to elevated Hcys leading to NLRP3 inflammasome activation in podocytes and consequent glomerular injury. In a mouse model of hyperhomocysteinemia (hHcys), we found that mice with hHcys (on the FF diet) or oncoVav2 (a constitutively active form of Vav2) transfection in the kidney exhibited increased colocalization of NLRP3 with apoptosis-associated speck-like protein (ASC) or caspase-1 and elevated IL-1β levels in glomeruli, indicating the formation and activation of the NLRP3 inflammasome. This glomerular NLRP3 inflammasome activation was accompanied by podocyte dysfunction and glomerular injury, even sclerosis. Local transfection of Vav2 shRNA plasmids significantly attenuated hHcys-induced NLRP3 inflammasome activation, podocyte injury, and glomerular sclerosis. In cultured podocytes, Hcys treatment and oncoVav2 transfection were also found to increase NLRP3 inflammasome formation and activation, which were all inhibited by Vav2 shRNA. Furthermore, Vav2 shRNA prevented Hcys-induced podocyte damage as shown by restoring Hcys-impaired VEGF secretion and podocin production. This inhibitory action of Vav2 shRNA on Hcys-induced podocyte injury was associated with reduction of Rac1 activity and ROS production. These results suggest that elevated Hcys levels activate Vav2 and thereby increase NOX activity leading to ROS production, which triggers NLRP3 inflammasome activation, podocyte dysfunction and glomerular injury.
Keywords: Hyperhomocysteinemia, inflammatory machinery, podocytes, end-stage renal disease
INTRODUCTION
Hyperhomocysteinemia (hHcys) has been identified as one of the leading pathogenic factors in the progression of end-stage renal disease (ESRD) and is associated with an increased risk for cardiovascular complications. Amongst the growing prevalence of chronic kidney disease, hHcys is a prevalent comorbidity occurring in 85% of diagnosed ESRD patients1. The early mechanisms responsible for the pathogenic action of increased Hcys levels during ESRD or other kidney-associated diseases still remain unclear. Prior studies have demonstrated that a reduction in glomerular filtration rate (GFR) contributes to the increases in both serum creatinine and Hcys2–4. Elevated concentrations of Hcys leads to endothelial damage and dysfunction that progresses into a buildup of extracellular matrix proteins, ultimately inducing renal glomerular sclerosis and atherosclerosis5–7. The pathogenic role of elevated plasma Hcys has been attributed to its direct action on cells or organs and to effects of its metabolites. For example, Hcys has been reported to act on the the NMDA receptor to activate NADPH oxidase (NOX) producing local oxidative stress and thereby leading to tissue damage such as glomerular injury. Hcys can also be transported into the cells to induce endoplasmic reticulum stress, homocysteinylation, and hypomethylation leading to cell dysfunction8,9. Increases in tissue Hcys levels have also been shown to reduce the production of adenosine which has potential protetive effects in many tissues or organs upon different pathogenic challenges9. In addition, there is evidence that Hcys can be converted into homocysteine-thiolactone and homocystamides promoting free-radical production and oxidative damage to proteins10. With respect to the role of hHcys in chronic degenerative disease, Hcys-induced tissue oxidative stress through different pathways and consequent derangement of extracellular matrix metabolism are considered to be critical11, 12. In the kidney, hHcys has been reported to result in glomerular degenerative diseases mainly through podocyte dysfunction and injury similar to other metabolic disorders such as diabetes mellitus, where glomerular pathological modifications will develop even though the initiaing pathogenic factors have been removed or disappear. Understanding the mechanisms leading to hHcys-induced podocyte injury may facilitate new approaches to treatment of patients with hHcys-associated ESRD.
Recent studies have reported that hHcys leads to inflammasome activation which is responsible for glomerular injury and ultimately leads to sclerosis13–15. The inflammasome is a multiprotein complex recognized as an intracellular machinery for activation of inflammatory responses and diseases16. One of the most important inflammasome isoforms is the NLRP3 inflammasome comprised of NOD-like receptor protein 3 (NLRP3), an adaptor protein called apoptosis-associated speck-like protein (ASC), and caspase-116. Detection of non-microbial reactive oxygen species (ROS), ATP, hypotonic stress, uric acid crystals, asbestos and UV radiation as well as invading pathogens, the formation and activation of the NLRP3 inflammasome are triggered, resulting in autocatalytic activation of caspase-1 and subsequent cleavage of pro-IL-1β and pro-IL-18 into their mature forms16. This NLRP3 inflammasome activation has been implicated in the pathogenesis of various metabolic diseases including diabetes, gout, silicosis, acute myocardial infarction and liver toxicity17–21. Our laboratory has recently demonstrated that increased Hcys around podocytes induces NLRP3 inflammasome activation, which contributes to glomerular injury and sclerosis during hHcys15. Moreover, we have shown that NADPH oxidase (NOX)-derived ROS mediates the activation of the NLRP3 inflammasome13,14, leading to glomerular injury and sclerosis11. Inhibition of NOX by different strategies resulted in protection from glomerular dysfunction and injury as seen by decreased urinary protein excretion, improved glomerular damage and increased matrix metalloproteinase-1 activity12. However, it remains unknown how NOX is activated by increased Hcys and consequently results in NLRP3 inflammasome formation in podocytes to produce glomerular injury and sclerosis.
There is evidence that NOX is comprised of membrane-bound subunits gp91phox and p22phox as well as cytosolic subunits p47phox, p40phox, and p67phox 22. The major regulatory step necessary for activation of NOX involves heterodimerization of gp91phox and p67phox mediated by the Rac protein. Conversion of GDP-bound Rac to activated GTP-bound Rac via a guanine nucleotide exchange factor (GNEF) initiates the translocation of the cytosolic subunit to the membrane23,24. It has been reported that the Vav subfamily represents distinct GNEFs with high specificity to Rac-mediated NOX activation25–27. It is possible that the activation of Vav2 through elevated Hcys accelerates the switch from GDP to GTP, thereby enhancing NOX activity and O2·− production, which may instigate the cascade to glomerular sclerosis by activation of the NLRP3 inflammasomes in podocytes. The present study was designed to test this hypothesis. During our experiments, we first used a hyperhomocysteinemic mouse model to test the effects of overexpression or inhibition of Vav2 on NLRP3 inflammasome formation and activation as well as on glomerular injury induced by hHcys. Then, we went on to explore the molecular mechanisms by which Hcys results in NLRP3 inflammasome activation and podocyte dysfunction or injury.
MATERIALS AND METHODS
Animals
Eight weeks old mice on a C57BL/6J background were purchased from Jackson Laboratories (Bar Harbor, ME). All protocols were approved by the Institutional Animal Care and Use Committee of Virginia Commonwealth University. To produce a rapid and advanced model of hHcys, mice were uninephrectomized and afterward allowed 1-week to recover. Short hairpin RNA (shRNA)-Vav2 or a dominant-active Vav2 variant (oncoVav2) plasmids with a luciferase expression vector were co-transfected into the kidneys via an intrarenal artery injection using the ultrasound-microbubble system. Plasmids containing scrambled small RNA (sRNA) were used as a control. After introduction of plasmids into the kidney, these uninephrectomized mice were fed either a normal diet (ND) or a folate-free (FF) diet to induce hHcys (Dyets Inc, Bethlehem, PA). One day before sacrifice, 24-hour urine samples were collected using mouse metabolic cages and afterward blood samples were collected. The mice were sacrificed and renal tissues were harvested for biochemical and molecular analysis as well as for morphological examinations.
Cell Culture
A conditionally immortalized mouse podocyte cell line gifted by Dr. Paul E. Klotman (Division of Nephrology, Department of Medicine, Mount Sinai School of Medicine, New York, NY, USA) was cultured undifferentiated with 10 U/mL recombinant mouse interferon-γ at 33°C on collagen I-coated flasks in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 mg/mL streptomycin. Prior to experiments, podocytes were thermoshifted at 37°C for 10–14 days to differentiate in the absence of interferon-γ and afterwards treated with 40 μM of L-Hcys for 24 hours.
RNAi
Podocytes were transfected with scrambled sRNA, Vav2 shRNA or constitutively active form of Vav2 (oncoVav2) plasmids directly into the nucleus via 4D-Nucleofector Technology (Lonza, Basel, Switerland). Small pores in the cell membrane develop temporarily through electrical impulses and cell-specific solutions. Substrates are then delivered through the cytoplasm and into the nuclear membrane. 2 × 106 podocytes were resuspended in SF Cell Line nucleofector solution containing 2 μg plasmid DNA, subject to cell-type specific program CM-137, and transferred to cultured plates. An additional group of podocytes was transfected using the siLentFect Lipid Reagent (Bio Rad, Berkeley, CA) incubated in serum-free medium with siRNA for 25 minutes according to the manufacturer’s instructions.
Confocal Microscopy
Frozen tissue sections or podocytes seeded in 8-well chambers were used to detect the expression and colocalization of NLRP3 (Abcam, Cambridge, MA), ASC (Santa Cruz, Santa Cruz, CA), caspase-1 (Santa Cruz, Santa Cruz, CA), podocin (Santa Cruz, Santa Cruz, CA) and desmin (BD Biosciences, San Jose, CA) in glomeruli and podocytes, where Alexa-488-conjugated secondary antibody was used to label NLRP3 and Alex-555 was used to label ASC, caspase-1, podocin and desmin. In addition, gene transfection efficiency in mouse glomeruli was determined by measuring Vav2 (Santa Cruz, Santa Cruz, CA) expression levels in frozen kidney sections.
Immunohistochemical Staining
Kidneys were perfused in 4% paraformaldehyde, removed and fixed in 10% neutral buffered formalin. 5-μm tissue sections embedded in paraffin were cut and mounted onto microscope slides. Deparaffination was induced through heat, tissue cleaning agent, ethanol and water. To unmask antigen binding sites, slides were boiled in 0.01 M citrate buffer pH 6.0 for 20 minutes. Endogenous peroxidase activity was blocked by incubating slides in 3% H2O2 in 100% MeOH for 30 minutes. The sections were then incubated at room temperature for 30 minutes in 10% goat serum to block nonspecific binding and incubated overnight at 4°C in a humidified chamber with antibodies against IL-1β (Abcam, Cambridge, MA) diluted 1:50 in TBS-T containing 4% goat serum. Then, the slides were incubated for 30 minutes at room temperature in a humidified chamber with a biotinylated goat anti-rabbit IgG-B antibody diluted 1:200 in TBS-T. The slides were subsequently placed in streptavidin-horseradish peroxidase for 30 minutes at room temperature in a humidified chamber. Finally, the slides were incubated with 50 μL of diaminobenzadine (BioGenex, San Ramon, CA) as a substrate, counterstained with hematoxylin (Sigma-Aldrich, Saint Louis, MO), dehydrated, and fixed with Permount histological mounting medium (Fisher Scientific, Hampton, NH).
Caspase-1 Activity, urinary protein measurements, IL-1β and VEGF
Caspase-1 activity (Biovision, Mountain View, CA) was measured by a commercially available colorimetric assay kit. ELISA kits were used for quantitation of urinary albumin (Bethyl Laboratories Inc., Montgomery, TX), IL-1β production (R&D Systems, Minneapolis, MN) and VEGF (Bender Medsystems, San Diego, CA). All assays were performed according to the manufacturer’s instructions.
Morphological Examinations
Glomerular structure was examined using fixed paraffin-embedded kidneys, stained with a Periodic-Acid Schiff (PAS) stain. Glomeruli were scored on a scale of 0–4 depending on the extent of sclerotic changes. In general, 30–50 glomeruli were counted under the microscope, 0 represents no lesion, 1 represents sclerosis <25% of the glomerulus while 2, 3 and 4 represent sclerosis at 25% to 50%, >50% to 75%, and >75% of the glomerulus, respectively. A general glomerular damage index (GDI) was obtained by averaging scores from counted glomeruli. This observation was examined by at least 3 investigators and averaged under blind conditions.
Assay of Rac-1 Activation
A GTPase linked immunosorbent assay was performed to determine Rac-1 activation in vitro using a colorimetric based assay kit (Cytoskeleton, Denver, CO). Podocytes were lysed in provided lysis buffer and clarified to remove cellular debris by centrifugation at 10,000 × g for 1 minute at 4°C. Equal amounts of protein (1 mg/mL) were loaded onto a Rac-1 GTP affinity plate for 30 minutes. Following incubation, the plate was washed with wash buffer, then incubated with a primary antibody against Rac-1 and then with a secondary antibody labeled with Horseradish Peroxidase (HRP). The activated Rac-1 was determined after exposure to HRP detection reagents by measuring absorbance at 490 nm using a microplate spectrophotometer.
Electromagnetic spin resonance (ESR) analysis of O2·− production
Cellular protein samples were prepared by using modified Kreb’s-Hepes buffer containing deferoximine (100 μM) and diethyldithio-carbamate (5 μM). NOX-dependent O2·− production was examined by addition of 1 mM NADPH as a substrate in 30 μg protein in the presence or absence of superoxide dismutase (SOD) (800 U/ml), and then supplied with 10 mM O2·− specific spin trapping compound, 1-hydroxy-3-methoxycarbonyl-2,2,5,5-tetramethylpyrro-lidine (CMH). The mixture was loaded in glass capillaries and immediately analyzed for O2·− production kinetically for 10 minutes in an ESR spectrometer. SOD sensitive components of ESR signals were used to calculate changes in O2·− production or its level, which were shown as the fold changes of control.
RESULTS
In vivo inhibition of Vav2 prevented glomerular NLRP3 inflammasome formation and activation
Using a hyperhomocysteinemic mouse model, we tested whether Vav2 facilitates hHcys-induced inflammasome formation and glomerular injury. By confocal microscopy, we found that hHcys and oncoVav2 transfected mice fed a normal or folate-free (FF) diet exhibited increased colocalization of NLRP3 with ASC and NLRP3 with caspase-1 (Figure 1). However, this colocalization was inhibited in mice transfected with Vav2 shRNA (Figure 1). The summarized data is shown in Figure 1C and 1D. Since activation of the NLRP3 inflammasome is associated with the maturation of the pro-inflammatory cytokine, IL-1β, we measured the IL-1β levels in mouse glomeruli using immunohistochemistry. We detected a significant increase in IL-1β levels in hHcys mice and mice receiving the oncoVav2 plasmids on both normal and FF diets (Figure 2A). However, blockade of Vav2 abolished further increases in hHcys-induced glomerular IL-1β (Figure 2A). This data indicates that Vav2 is involved in both assembling and activation of the NLRP3 inflammasome in glomeruli of mice with hHcys, which could be mimicked by overexpression of Vav2.
Figure 1. Inhibition of Vav2 abolished glomerular NLRP3 inflammasome formation in podocytes of the mouse kidney.
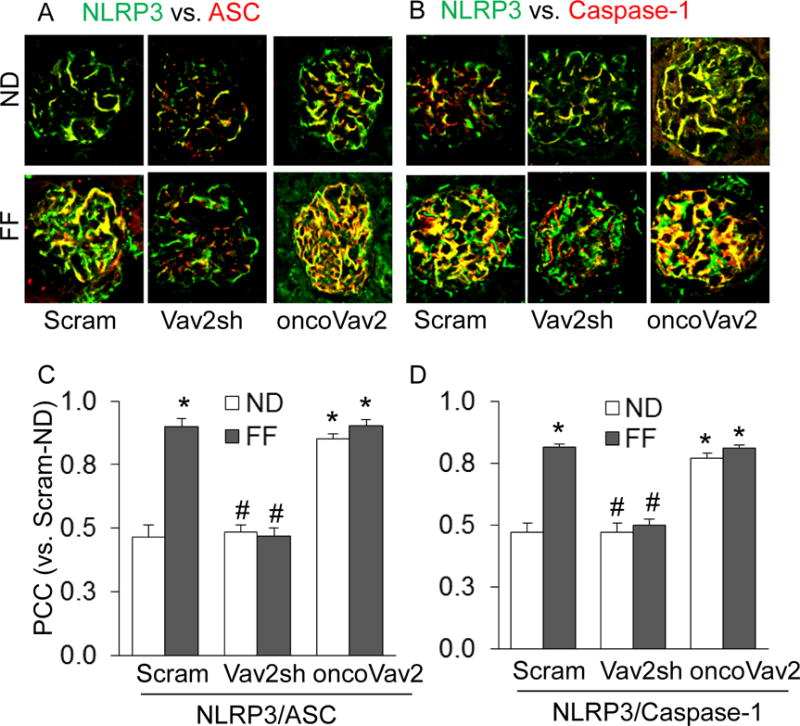
A. Colocalization of NLRP3 (green) with ASC (red) or caspase-1 (red) in mouse glomeruli fed a normal or FF diet. B. Summarized data showing the fold changes in PCC for the colocalization of NLRP3 with ASC or with Caspase-1 (n=6). Scram: Scramble, Vav2sh: Vav2 shRNA, ND: Normal Diet and FF: Folate-Free Diet. * p<0.05 vs. Scram on ND; # p<0.05 vs. Scram on FF Diet.
Figure 2. Vav2 overexpression induced NLRP3 inflammasome activation in podocytes of the mouse kidney.
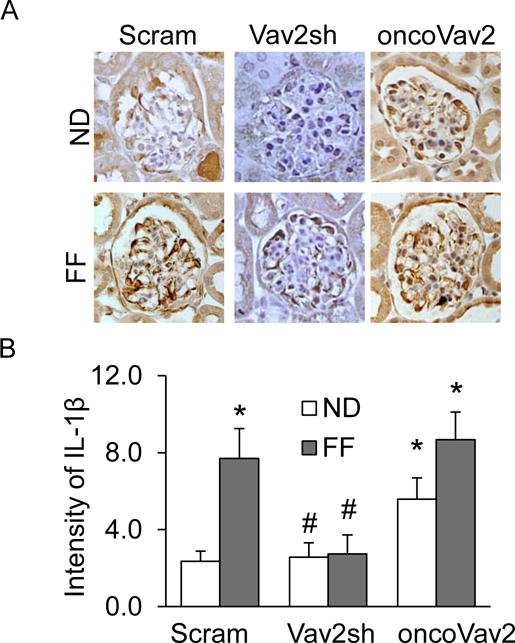
A. Representative images of immunohistochemical staining of IL-1β in glomerular podocytes in mice on the normal or FF diet. B. Summarized data depicting relative intensity of IL-1β brown staining (n=5). Scram: Scramble, Vav2sh: Vav2 shRNA, ND: Normal Diet and FF: Folate-Free Diet. * p<0.05 vs. Scram on ND; # p<0.05 vs. Scram on FF Diet.
Glomerular protection by Vav2 inhibition
It is well recognized that protein in the urine is a hallmark of many glomerular diseases. After a 24-hour urine collection, protein and albumin levels were evaluated in collected mouse urine to serve as an indicator of glomerular injury. As illustrated in Figures 3A and 3B, the FF diet produced both proteinuria and albuminuria in scramble sRNA-transfected mice, and the increased urinary excretion of protein and albumin was also observed in oncoVav2 transfected mice on a normal diet (ND), but albumin excretion in mice with oncoVav2 transfection and FF diet consumption was similar to mice on the ND. However, this increased albuminuria induced by the FF diet or oncoVav2 alone was attenuated in mice receiving intrarenal Vav2 shRNA transfection (Figures 3A and 3B). Furthermore, this glomerular dysfunction was correlated to modifications in the glomerular architecture as shown by morphological examinations. It was found that scramble sRNA and oncoVav2 plasmid-transfected mice fed a FF diet developed increased sclerotic damage to their glomeruli, as shown by mesangial expansion, fibrosis and hypercellularity with an increased glomerular damage index (GDI) (Figures 3C and 3D). Inhibition of Vav2 by shRNA protected mice from this glomerular sclerotic pathology. Immunofluorescence analysis demonstrated that the expression of podocin decreased in oncoVav2 transfected mice on either the ND or the FF diet, but this decrease in podocin levels was not seen in Vav2 shRNA transfected mice on the same diet (Figure 4A). In contrast, the levels of desmin, a marker of podocyte injury, increased in oncoVav2 transfected mice, but not in Vav2 shRNA transfected mice (Figure 4B). Furthermore, as shown in Figure 4C, Vav2 protein levels were significantly lower in scramble sRNA and Vav2 shRNA transfected mice, as depicted by green staining. Figure 4D showed the relative intensity of Vav2 staining in oncoVav2 transfected mice, which exhibited a two-fold increase in comparison to that seen in scramble sRNA transfected mice.
Figure 3. In vivo inhibition of Vav2 attenuated hHcys-induced glomerular damage.
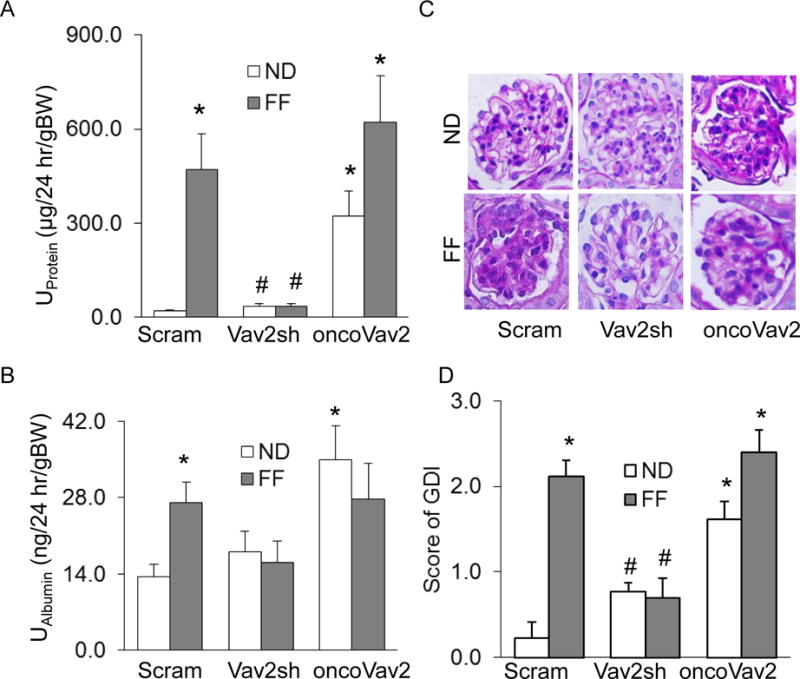
Urinary protein (A) and albumin (B) measurements in 24 hr urine samples of hHcys mice receiving shRNA- and onco-Vav2 transfection (n=5). C. Microscopic microphotographs of glomerular structure in PAS stained kidney sections. D. Semiquantitative assessment of glomerular damage index (GDI) from PAS images (n=4). Scram: Scramble, Vav2sh: Vav2 shRNA, ND: Normal Diet, FF: Folate-Free Diet, U: urinary, and GDI: Glomerular Damage Index. * p<0.05 vs. Scram on ND; # p<0.05 vs. Scram on the FF Diet.
Figure 4. Glomerular dysfunction associated with overexpressed Vav2 in mouse glomeruli.
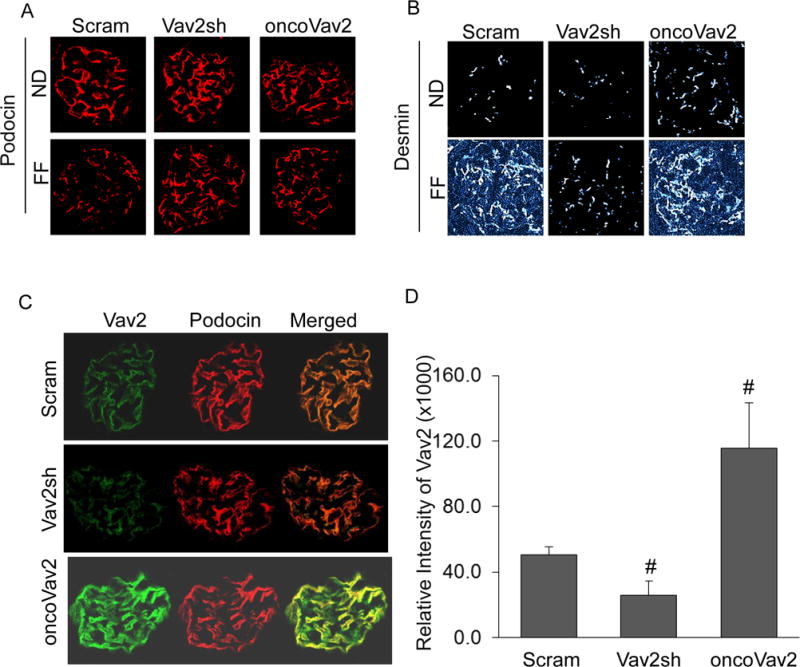
A. Fluorescent staining of podocyte markers, podocin (A) and desmin (B) in mouse glomeruli (n = 5). C. Confirmation of Vav2 (green) expression in glomerular podocytes. D. Quantification of relative intensity of Vav2 in glomeruli (n=4). Scram: Scramble, Vav2sh: Vav2 shRNA, ND: Normal Diet and FF: Folate-Free Diet. * p<0.05 vs. Scram on ND; # p<0.05 vs. Scram on the FF Diet.
Role of Vav2 in Hcys-induced NLRP3 inflammasome formation in podocytes
The glomerulus is comprised of specialized cells that operate in a partnership to preserve the integrity of the kidney. Evidence strongly links podocyte injury to the pathogenesis of many glomerular diseases28,29. To better understand the impact of glomerular dysfunction during hHcys, we chose to further investigate the molecular mechanisms initiating injury in podocytes by employing cultured podocytes. Our initial studies determined if Vav2 is involved in Hcys-induced inflammasome formation in these cells. By confocal microscopic analysis, we detected increased colocalization of NLRP3 with ASC or caspase-1 in Hcys-treated podocytes when compared with untreated cells (Figure 5). However, this Hcys-induced increase in colocalization was inhibited in podocytes transfected with Vav2 shRNA (Figure 5). Additionally, oncoVav2 overexpression increased colocalization of inflammasome molecules in both control and Hcys-treated groups of podocytes. The quantitative colocalization of NLRP3 with ASC or caspase-1 is summarized in Figure 5C and 5D.
Figure 5. Hcys treatment and oncoVav2 transfection increased NLRP3 inflammasome formation in podocytes.
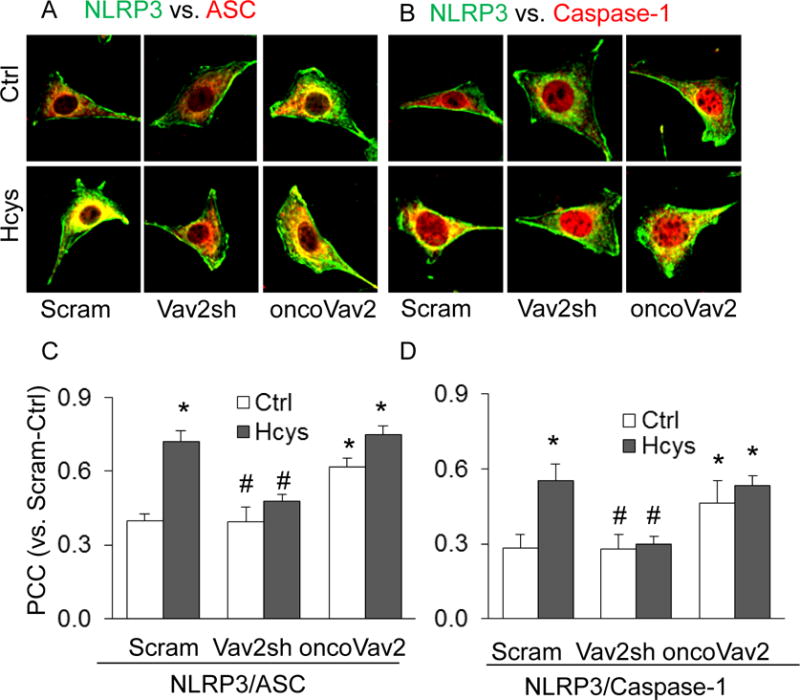
A. Confocal images signifying the colocalization of NLRP3 (green) with ASC (red) and NLRP3 (green) with caspase-1 (red) in cultured podocytes transfected with Vav2sh or oncoVav2. B. Summarized data showing the fold change in PCC colocalization (n=6). Scram: Scramble, Vav2sh: Vav2 shRNA and Ctrl: Control. * p<0.05 vs. Ctrl; # p<0.05 vs. Hcys.
Activation of the NLRP3 inflammasome in vitro
We confirmed the role of Vav2 in Hcys-induced inflammasome activation by measuring the levels of active or cleaved caspase-1 and the level of secreted IL-1β. As shown in Figure 6, Hcys treatment increased caspase-1 activity and IL-1β production in comparison to control podocytes. Hcys-induced increase in caspase-1 activity and elevation in IL-1β levels were significantly attenuated in podocytes transfected with Vav2 shRNA (Figure 6). Transfection with oncoVav2 mimicked the effects of Hcys which increased caspase-1 activity and elevated IL-1β levels in cultured podocytes (Figure 6).
Figure 6. In vitro induction of NLRP3 inflammasome activation in podocytes, independent of Hcys treatment.
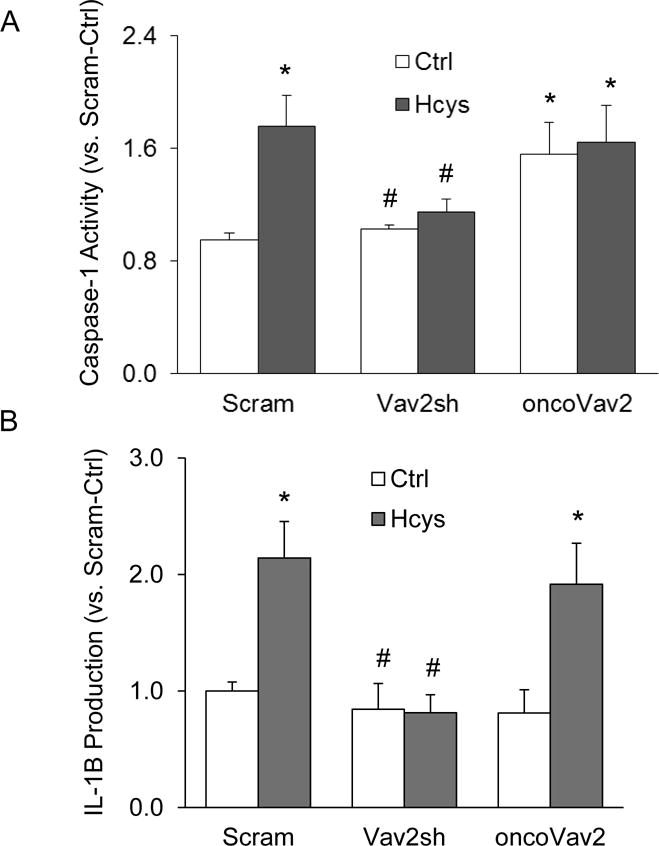
A. Caspase-1 activity, shown as fold vs. Ctrl, measured in transfected podocytes treated with Hcys (n=5). B. IL-1β production measured in the supernatant of transfected podocytes treated with Hcys (n=6). Scram: Scramble; Vav2sh: Vav2 shRNA. * p<0.05 vs. Ctrl; # p<0.05 vs. Hcys.
Inhibition of Vav2 improved podocytes damage
We further assessed the participation of Vav2 in podocyte dysfunction induced by Hcys. It was found that upon stimulation with Hcys, there was decreased staining of the podocyte marker podocin in Hcys-treated and oncoVav2-transfected podocytes, whereas desmin levels increased in these groups, suggesting damage to podocytes (Figure 7A). However, Vav2 shRNA reversed Hcys-induced podocyte damage (Figure 7A). Additionally, we observed that Hcys treatment and oncoVav2 transfection resulted in significant impairment of podocytes to secrete vascular endothelial growth factor (VEGF), a major product of healthy and mature podocytes (Figure 7B). Inhibition of Vav2 protected podocytes from such injury or dysfunction, which was seen by restored levels of VEGF secretion (Figure 7B).
Figure 7. NOX activation by Vav2 overexpression promoted podocyte dysfunction.
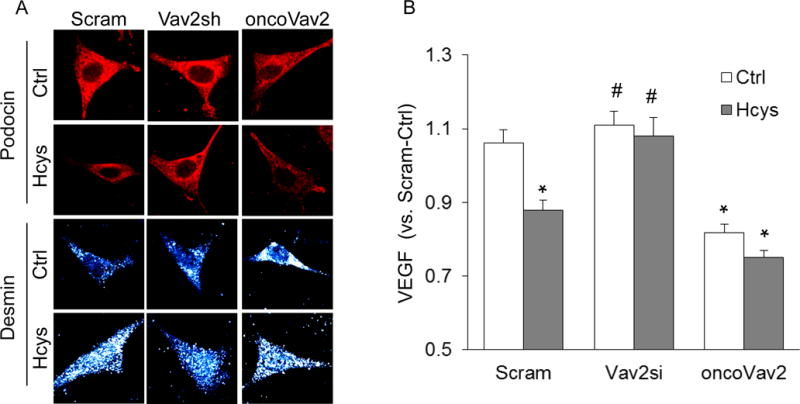
A. Podocin (red) and desmin (gray) staining following Hcys treatment (n = 5). B. Measurement of secreted VEGF in cellular supernatant (n=7). Scram: Scramble; Vav2sh: Vav2 shRNA; Vav2si: Vav2 siRNA, * p<0.05 vs. Ctrl; # p<0.05 vs. Hcys.
Effects of Rac-1 activation on Hcys-induced NOX activity
It is well-defined that activated Rac serves as an anchor protein to regulate NOX activity in many cell types30–32. We have shown that Hcys treatment and oncoVav2 transfection resulted in increased Rac-1 activation in cultured podocytes (Figure 8A). A significant decrease in Rac-1 activity was observed in podocytes with silenced Vav2 gene, suggesting that more Rac-1 remains in the GDP-bound or inactive state (Figure 8A). Coupled with increased GTP-bound Rac-1, we also found that oncoVav2 transfection produced a great increase in O2·−, which was inhibited in the Vav2 shRNA transfected podocytes (Figure 8B).
Figure 8. Vav2 blockade prevented in vitro inhibition of NOX-derived O2·− production.
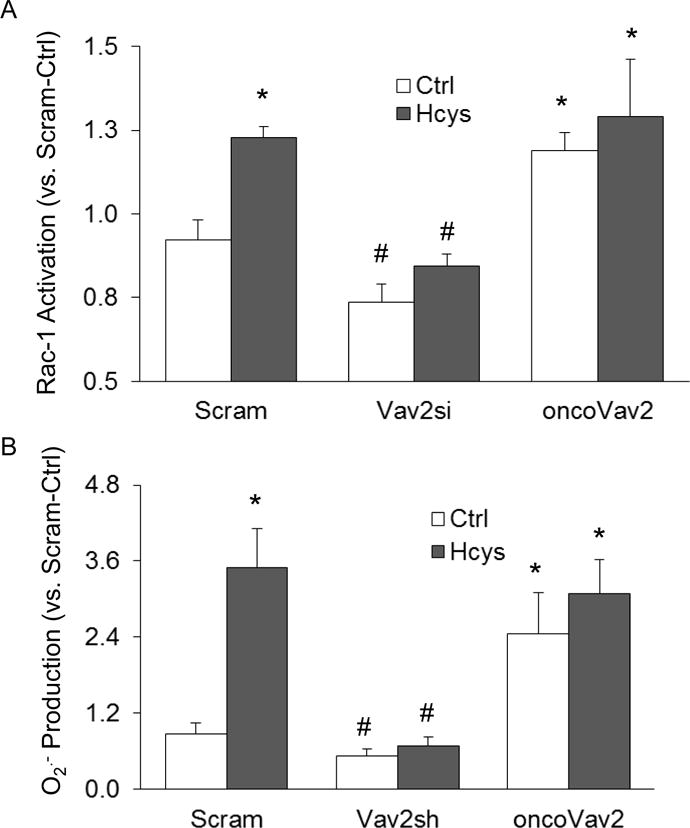
A. Rac-1 activity (n=6) and O2·− production (n=5) (B) in Vav2- and oncoVav2-transfected podocytes in the presence or absence of Hcys. * p<0.05 vs. Ctrl; # p<0.05 vs. Hcys.
DISCUSSION
The primary goal of this study was to reveal the involvement of the GNEF, Vav2 in activation of the NLRP3 inflammasome during hHcys. Our results showed that Vav2 inhibition by RNA interference attenuated hHcys-induced NLRP3 inflammasome formation and activation in podocytes. Conversely, overexpression of Vav2 resulted in enhanced inflammasome formation and activation in these cells independent of Hcys treatment. Results from the present study also demonstrated that Vav2 inhibition protected podocytes against Hcys-induced glomerular injury. This Vav2-mediated action was associated with NOX activation and increased ROS production in glomeruli.
Distinctive GNEFs facilitate the conversion of GDP for GTP, inciting Rho protein activation33. Among more than 100 GNEFs, the Vav subfamily is highly selective in coordinating intracellular signal transduction pathways associated with NOX activation34,35. Our earlier studies have demonstrated that of the 3 Vav isoforms, both Vav2 and Vav3 are localized in the glomeruli, whereas it is already established in other reports that Vav1 is solely present in hematopoietic cells26. We have shown that by sensing intracellular ROS during hHcys, thioredoxin-interacting protein (TXNIP) associates with NLRP3 to trigger the cascade of downstream events leading to inflammasome activation36. Recently, evidence links Vav2 as a binding partner of TXNIP37. Therefore, we sought to identify whether the detrimental actions of Vav2 is involved in NLRP3 inflammasome assembling, activation and subsequent podocyte dysfunction. It has been reported that internal or external danger signals are capable of initiating the recruitment of ASC and caspase-1, forming the NLRP3 inflammasome complex16,38. This inflammasome complex triggers the production of IL-1β and IL-18, inducing a heightened inflammatory response. Processing of not only the interleukins, but also caspase-1 is considered crucial in the inflammatory process that leads to tissue injury, organ or systemic diseases39. In particular, NLRP3 inflammasome activation has been implicated in the pathogenesis of various renal diseases including acute kidney injury, chronic kidney disease, diabetic nephropathy, crystal related nephropathy and hyperhomocysteinemic nephropathy17,40,41. Renal cells such as podocytes have been documented as the main enriched sites for maturation of glomerular IL-1β production13,15.
To our knowledge, we are the first to report that silencing the Vav2 gene and reduction of its activity using genetic manipulations inhibited the NLRP3 inflammasome by preventing the activation of the small GTPase, Rac-1. Consistent in both our in vivo and in vitro models, our results show that Vav2 shRNA interventions decreased the interactions of inflammasome components NLRP3, ASC and caspase-1 (Figures 1 and 5). Consequently, the absence of inflammasome machinery resulted in decreased IL-1β production in mouse glomeruli (Figure 2) which was associated with diminished caspase-1 activity in podocytes (Figure 7). The most remarkable finding in the present study was that Vav2 overexpression triggered NLRP3 inflammasome formation irrespective of Hcys exposure (Figure 1 and 5), suggesting that Vav2 is crucial in bridging the redox signals derived from NOX activity and NLRP3 inflammasome activation, which may be a critical mechanism responsible for glomerular inflammatory response during hHcys.
There is evidence that abundance of protein leaked in the urine and excessive reabsorption of protein in renal proximal tubular cells are linked to the pathophysiological states of renal disease development42,43. In this regard, Connell et al. demonstrated that in response to albumin overloads, proximal tubule cells encounter direct toxic effects associated with Rac GTPase and NOX activation as well as receptor-mediated endocytosis44. Our results add to this by showing that mice with overexpressed Vav2 had typical features of glomerular damage as evidenced by proteinuria, albuminuria and increased GDI (Figure 3). Downregulation of the Vav2 gene effectively ameliorated these detrimental effects in mouse glomeruli. With respect to the mechanism of glomerular injury during different pathological conditions such as hHcys, podocyte injury is considered to play an important role. It has been reported that disruption of the slit diaphragm, actin rearrangement and foot process effacement are unique indicators of podocyte injury28,45. Our data revealed that Hcys treatment and Vav2 overexpression decreased a podocyte-specific molecule, podocin (Figure 4 and 7), which may result in derangement of the slit diaphragm structure and related cell signaling pathways45. Furthermore, resultant injury to podocytes may lead to the upregulation of the intermediate filament, desmin, which was indeed confirmed in our studies as shown by increased desmin levels in podocytes during hHcys or exposure to high levels of extracellular Hcys. Interestingly, this desmin increase in podocytes during hHcys could be blocked by shRNA of Vav2, but mimicked by oncoVav2 transfection. The results suggest that Vav2 critically contributes to hHcys-induced podocyte injury and glomerular sclerosis.
To explore the mechanism by which Vav2 activates the NLRP3 inflammasome and thereby lead to podocyte injury and glomerular sclerosis, we examined the effects of Vav2 interventions on Rac activity and consequent NOX activation. It is well known that instigation of NOX as a functional signaling complex is dependent on the translocation of its cytosolic subunits (p47phox and p67phox) to the membrane in which this platform initiates the generation of superoxide (O2·−)22,46. The Rac protein is crucial in these migratory events, serving as a binary switch that cycles between active GTP-bound and inactive GDP-bound states. However, the molecular cues directly responsible for Rac protein activation and subsequent NOX activity following prolonged exposure to Hcys remained unclear. From our findings, we confirmed that expression of constitutively active Vav2, can itself drive the activation of the Rac-1 protein (Figure 8A) and the aggregation of a functional NOX complex, which produces O2·− even without Hcys stimulus (Figure 8B). Moreover, blockade of Vav2 attenuated enhancement of Rac activity and O2·− generation, even in the presence of Hcys, a known activator of ROS. Results from these experiments illustrate that Vav2 can promote injurious consequences in podocytes and that inhibition of Vav2 function may terminate the development and progression of Hcys-induced podocyte injury and consequent glomerular sclerosis.
Altogether, the results from the present study signify that Vav2 is critically involved in Hcys-induced NLRP3 inflammasome formation and activation, which may critically contribute to podocyte dysfunction and glomerular injury. These results also demonstrate that Rac-1-NOX activation and subsequent production of O2·− are major triggering mechanisms of the NLRP3 inflammasome in podocytes, which is consistent with previous reports14,36. Vav2 may be a therapeutic target for termination of early events in hyperhomocysteinemic nephropathy and ESRD.
Highlights.
Guanine nucleotide exchange factor Vav2 is essential for hHcys-induced Rac-1 activation, NADPH oxidase activity and activation of the NLRP3 inflammasome.
Overexpression of Vav2 enhanced NLRP3 inflammasome formation and activation in glomeruli of hHcys mice.
NADPH oxidase activation by enhanced Vav2 alone is sufficient to trigger inflammasome activation and contribute to glomerular injury.
Acknowledgments
SOURCES OF FUNDING
This work was supported by grants DK54927, HL075316, HL57244 (to P.L) and DK104031 (to K.B) from National Institutes of Health.
Footnotes
DISCLOSURES
None
References
- 1.Wu Chia-Chao, Zheng Cai-Mei, Lin Yuh-Feng, Lo Lan, et al. Role of homocysteine in end-stage renal disease. Clinical biochemistry. 2012;45(16–17):1286–94. doi: 10.1016/j.clinbiochem.2012.05.031. [DOI] [PubMed] [Google Scholar]
- 2.Norlund L, Grubb A, Fex G, Leksell H, et al. The increase of plasma homocysteine concentrations with age is partly due to the deterioration of renal function as determined by plasma cystatin C. Clinical chemistry and laboratory medicine. 1998;36(3):175–8. doi: 10.1515/CCLM.1998.032. [DOI] [PubMed] [Google Scholar]
- 3.Bostom AG, Lathrop L. Hyperhomocysteinemia in end-stage renal disease: prevalence, etiology, and potential relationship to arteriosclerotic outcomes. Kidney international. 1997;52(1):10–20. doi: 10.1038/ki.1997.298. [DOI] [PubMed] [Google Scholar]
- 4.Moustapha A, Gupta A, Robinson K, Arheart K, et al. Prevalence and determinants of hyperhomocysteinemia in hemodialysis and peritoneal dialysis. Kidney international. 1999;55(4):1470–5. doi: 10.1046/j.1523-1755.1999.00378.x. [DOI] [PubMed] [Google Scholar]
- 5.McCully KS. Vascular pathology of homocysteinemia: implications for the pathogenesis of arteriosclerosis. The American journal of pathology. 1969;56(1):111–28. [PMC free article] [PubMed] [Google Scholar]
- 6.Miller A, Mujumdar V, Shek E, Guillot J, et al. Hyperhomocyst(e)inemia induces multiorgan damage. Heart and vessels. 2000;15(3):135–43. doi: 10.1007/s003800070030. [DOI] [PubMed] [Google Scholar]
- 7.Tsakiris DA, Tschöpl M, Jäger K, Haefeli WE, et al. Circulating cell adhesion molecules and endothelial markers before and after transluminal angioplasty in peripheral arterial occlusive disease. Atherosclerosis. 1999;142(1):193–200. doi: 10.1016/s0021-9150(98)00175-0. [DOI] [PubMed] [Google Scholar]
- 8.Zhang Chun, Yi Fan, Xia Min, Boini Krishna M, et al. ‘NMDA Receptor-Mediated Activation of NADPH Oxidase and Glomerulosclerosis in Hyperhomocysteinemic Rats’. Antioxidants & Redox Signaling. 2010;13(7):975–986. doi: 10.1089/ars.2010.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yi Fan, Li Pin-Lan. Mechanisms of homocysteine-induced glomerular injury and sclerosis. American journal of nephrology. 2008;28(2):254–64. doi: 10.1159/000110876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sibrian-Vazquez Martha, Escobedo Jorge O, Lim Soojin, Samoei George K, Strongin Robert M. Homocystamides promote free-radical and oxidative damage to proteins. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(2):551–4. doi: 10.1073/pnas.0909737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Zhi-Zhang, Zou Ai-Ping. Homocysteine enhances TIMP-1 expression and cell proliferation associated with NADH oxidase in rat mesangial cells. Kidney international. 2003;63(3):1012–20. doi: 10.1046/j.1523-1755.2003.00825.x. [DOI] [PubMed] [Google Scholar]
- 12.Yi F, Zhang AY, Li N, Muh RW, et al. Inhibition of ceramide-redox signaling pathway blocks glomerular injury in hyperhomocysteinemic rats. Kidney international. 2006;70(1):88–96. doi: 10.1038/sj.ki.5001517. [DOI] [PubMed] [Google Scholar]
- 13.Abais Justine M, Zhang Chun, Xia Min, Liu Qinglian, et al. NADPH oxidase-mediated triggering of inflammasome activation in mouse podocytes and glomeruli during hyperhomocysteinemia. Antioxidants & redox signaling. 2013;18(13):1537–48. doi: 10.1089/ars.2012.4666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abais Justine M, Xia Min, Li Guangbi, Gehr Todd WB, et al. Contribution of endogenously produced reactive oxygen species to the activation of podocyte NLRP3 inflammasomes in hyperhomocysteinemia. Free radical biology & medicine. 2014;67:211–20. doi: 10.1016/j.freeradbiomed.2013.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Chun, Boini Krishna M, Xia Min, Abais Justine M, et al. Activation of Nod-like receptor protein 3 inflammasomes turns on podocyte injury and glomerular sclerosis in hyperhomocysteinemia. Hypertension (Dallas, Tex : 1979) 2012;60(1):154–62. doi: 10.1161/HYPERTENSIONAHA.111.189688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinon Fabio, Mayor Annick, Tschopp Jürg. The inflammasomes: guardians of the body. Annual review of immunology. 2009;27:229–65. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- 17.Lamkanfi Mohamed, Dixit Vishva M. Inflammasomes and their roles in health and disease. Annual review of cell and developmental biology. 2012;28:137–61. doi: 10.1146/annurev-cellbio-101011-155745. [DOI] [PubMed] [Google Scholar]
- 18.Dostert Catherine, Pétrilli Virginie, Van Bruggen Robin, Steele Chad, et al. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science (New York, NY) 2008;320(5876):674–7. doi: 10.1126/science.1156995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imaeda AvlinB, Watanabe Azuma, Sohail Muhammad A, Mahmood Shamail, et al. Acetaminophen-induced hepatotoxicity in mice is dependent on Tlr9 and the Nalp3 inflammasome. The Journal of clinical investigation. 2009;119(2):305–14. doi: 10.1172/JCI35958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinon Fabio, Pétrilli Virginie, Mayor Annick, Tardivel Aubry, Tschopp Jürg. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440(7081):237–41. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 21.Lee Hye-Mi, Kim Jwa-Jin, Kim Hyun Jin, Shong Minho, et al. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes. 2013;62(1):194–204. doi: 10.2337/db12-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gill Pritmohinder S, Wilcox Christopher S. NADPH Oxidases in the Kidney’. Antioxidants & Redox Signaling. 2006;8(9–10):1597–1607. doi: 10.1089/ars.2006.8.1597. [DOI] [PubMed] [Google Scholar]
- 23.Mouawad Flaviana, Tsui Harmony, Takano Tomoko. Role of Rho-GTPases and their regulatory proteins in glomerular podocyte function. Canadian journal of physiology and pharmacology. 2013;91(10):773–82. doi: 10.1139/cjpp-2013-0135. [DOI] [PubMed] [Google Scholar]
- 24.Schmidt Anja, Hall Alan. Guanine nucleotide exchange factors for Rho GTPases: turning on the switch. Genes & development. 2002;16(13):1587–609. doi: 10.1101/gad.1003302. [DOI] [PubMed] [Google Scholar]
- 25.Yi Fan, Chen Qi-Zheng, Jin Si, Li Pin-Lan. Mechanism of homocysteine-induced Rac1/NADPH oxidase activation in mesangial cells: role of guanine nucleotide exchange factor Vav2. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2007;20(6):909–18. doi: 10.1159/000110451. [DOI] [PubMed] [Google Scholar]
- 26.Yi Fan, Xia Min, Li Ningjun, Zhang Chun, et al. Contribution of guanine nucleotide exchange factor Vav2 to hyperhomocysteinemic glomerulosclerosis in rats. Hypertension (Dallas, Tex : 1979) 2009;53(1):90–6. doi: 10.1161/HYPERTENSIONAHA.108.115675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yi Fan, Zhang Andrew Y, Janscha Jennifer L, Li Pin-Lan, Zou Ai-Ping. Homocysteine activates NADH/NADPH oxidase through ceramide-stimulated Rac GTPase activity in rat mesangial cells. Kidney international. 2004;66(5):1977–87. doi: 10.1111/j.1523-1755.2004.00968.x. [DOI] [PubMed] [Google Scholar]
- 28.Nagata Michio. Podocyte injury and its consequences. Kidney international. 2016;89(6):1221–30. doi: 10.1016/j.kint.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 29.Raij Leopoldo, Tian Runxia, Wong Jenny S, He John Cijiang, Campbell Kirk N. Podocyte Injury: The Role of Proteinuria, Urinary Plasminogen and Oxidative Stress. American journal of physiology. Renal physiology. 2016 doi: 10.1152/ajprenal.00162.2016. p. ajprenal.00162.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moldovan L, Irani K, Moldovan I, Finkel T, Goldschmidt-Clermont PJ. The actin cytoskeleton reorganization induced by Rac1 requires the production of superoxide. Antioxidants & redox signaling. 1999;1(1):29–43. doi: 10.1089/ars.1999.1.1-29. [DOI] [PubMed] [Google Scholar]
- 31.Li Su-min, Zeng Ling-wen, Feng Lin, Chen Dong-bao. Rac1-dependent intracellular superoxide formation mediates vascular endothelial growth factor-induced placental angiogenesis in vitro. Endocrinology. 2010;151(11):5315–25. doi: 10.1210/en.2010-0178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yi Fan, Chen Qi-Zheng, Jin Si, Li Pin-Lan. Mechanism of homocysteine-induced Rac1/NADPH oxidase activation in mesangial cells: role of guanine nucleotide exchange factor Vav2. Cellular physiology and biochemistry : international journal of experimental cellular physiology, biochemistry, and pharmacology. 2007;20(6):909–18. doi: 10.1159/000110451. [DOI] [PubMed] [Google Scholar]
- 33.Overbeck AF, Brtva TR, Cox AD, Graham SM, et al. Guanine nucleotide exchange factors: activators of Ras superfamily proteins. Molecular reproduction and development. 1995;42(4):468–76. doi: 10.1002/mrd.1080420415. [DOI] [PubMed] [Google Scholar]
- 34.Liu BP, Burridge K. Vav2 activates Rac1, Cdc42, and RhoA downstream from growth factor receptors but not beta1 integrins. Molecular and cellular biology. 2000;20(19):7160–9. doi: 10.1128/mcb.20.19.7160-7169.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu Yunhao, Collins Caitlin, Kiosses William B, Murray Ann M, et al. A novel pathway spatiotemporally activates Rac1 and redox signaling in response to fluid shear stress. The Journal of cell biology. 2013;201(6):863–73. doi: 10.1083/jcb.201207115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abais Justine M, Xia Min, Li Guangbi, Chen Yang, et al. Nod-like receptor protein 3 Lduring hyperhomocysteinemia. The Journal of biological chemistry. 2014;289(39):27159–68. doi: 10.1074/jbc.M114.567537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu Shasha, Wu Xue, Zong Minru, Tempel Wolfram, et al. Structural basis for a novel interaction between TXNIP and Vav2. FEBS letters. 2016;590(6):857–65. doi: 10.1002/1873-3468.12110. [DOI] [PubMed] [Google Scholar]
- 38.Schroder Kate, Zhou Rongbin, Tschopp Jurg. The LRP3 inflammasome: a sensor for metabolic danger? Science (New York, NY) 2010;327(5963):296–300. doi: 10.1126/science.1184003. [DOI] [PubMed] [Google Scholar]
- 39.Denes A, Lopez-Castejon G, Brough D. Caspase-1: is IL-1 just the tip of the ICEberg? Cell death & disease. 2012;3:e338. doi: 10.1038/cddis.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hutton Holly L, Ooi Joshua D, Holdsworth Stephen R, Kitching A Richard. The LRP3 inflammasome in kidney disease and autoimmunity. Nephrology (Carlton, Vic) 2016;21(9):736–44. doi: 10.1111/nep.12785. [DOI] [PubMed] [Google Scholar]
- 41.Darisipudi Murthy N, Knauf Felix. An update on the role of the inflammasomes in the pathogenesis of kidney diseases. Pediatric nephrology (Berlin, Germany) 2016;31(4):535–44. doi: 10.1007/s00467-015-3153-z. [DOI] [PubMed] [Google Scholar]
- 42.Gekle Michael. Renal tubule albumin transport. Annual review of physiology. 2005;67:573–94. doi: 10.1146/annurev.physiol.67.031103.154845. [DOI] [PubMed] [Google Scholar]
- 43.Hsu Raymond K, Hsu Chi-Yuan. The Role of Acute Kidney Injury in Chronic Kidney Disease. Seminars in nephrology. 2016;36(4):283–92. doi: 10.1016/j.semnephrol.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Whaley-Connell Adam T, Morris E Matthew, Rehmer Nathan, Yaghoubian J Cipporah, et al. Albumin activation of NAD(P)H oxidase activity is mediated via Rac1 in proximal tubule cells. American journal of nephrology. 2007;27(1):15–23. doi: 10.1159/000098432. [DOI] [PubMed] [Google Scholar]
- 45.Shankland SJ. The podocyte’s response to injury: Role in proteinuria and glomerulosclerosis’. Kidney International. 2006;69(12):2131–2147. doi: 10.1038/sj.ki.5000410. [DOI] [PubMed] [Google Scholar]
- 46.Diebold BA, Bokoch GM. Molecular basis for Rac2 regulation of phagocyte NADPH oxidase. Nature immunology. 2001;2(3):211–5. doi: 10.1038/85259. [DOI] [PubMed] [Google Scholar]