

SUMMARY
The circadian clock generates biological rhythms of metabolic and physiological processes, including the sleep-wake cycle. We previously identified a missense mutation in the flavin adenine dinucleotide (FAD) binding pocket of CRYPTOCHROME2 (CRY2), a clock protein that causes human advanced sleep phase. This prompted us to examine the role of FAD as a mediator of the clock and metabolism. FAD stabilized CRY proteins leading to increased protein levels. In contrast, knockdown of Riboflavin kinase (Rfk), an FAD biosynthetic enzyme, enhanced CRY degradation. RFK protein levels and FAD concentrations oscillate in the nucleus, suggesting they are subject to circadian control. Knockdown of Rfk combined with a riboflavin-deficient diet altered the CRY levels in mouse liver and the expression profiles of clock and clock-controlled genes (especially those related to metabolism including glucose homeostasis). We conclude that light-independent mechanisms of FAD regulate CRY and contribute to proper circadian oscillation of metabolic genes in mammals.
Keywords: Circadian clock, Circadian rhythms, CRYPTOCHROME, CRY, FAD, metabolism, FBXL3, Riboflavin kinase, protein degradation
eTob blurb
Hirano et al. examine a mechanism in which a co-factor, FAD, regulates CRYPTOCROME proteins and circadian regulation of metabolism. FAD stabilizes CRY proteins by competing with the CRY ubiquitin E3 ligase. Knockdown of Rfk, a biosynthetic enzyme of FAD, alters the expression profiles of CRY and clock-controlled metabolic genes.
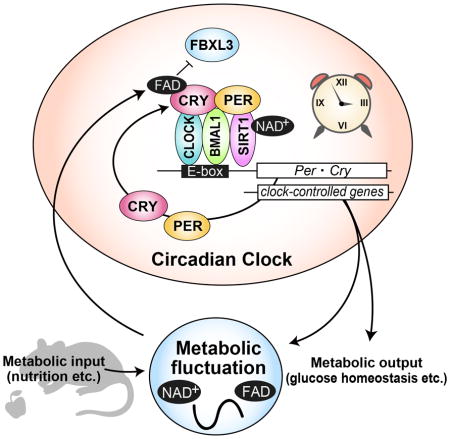
INTRODUCTION
Circadian rhythms are observed in organisms across the phylogenetic spectrum and are recognized in many biological functions from the molecular level through behavior (Schibler and Sassone-Corsi, 2002). Rhythms with approximately 24-hour periodicity are governed by the internal time-keeping system, the circadian clock. At its most basic level, the cell-autonomous circadian clock is constituted by a transcriptional/translational negative feedback loop, in which activators (CLOCK/BMAL1) and repressors (PERIOD1-3 (PER1-3)/CRYPTOCHROME1-2 (CRY1-2)) are coordinately regulated (Hardin and Panda, 2013; Reppert and Weaver, 2002). We have shown that mutations in some of the core clock genes (PER2, PER3, Casein kinase Iδ, CRY2, DEC2) affect the timing of sleep-wake behavior or sleep duration in humans (He et al., 2009; Hirano et al., 2016b; Jones et al., 1999; Toh et al., 2001; Xu et al., 2007; 2005; Zhang et al., 2016). Gene targeting or mutations of the clock genes in rodent models cause circadian phenotypes but also, in some cases, lead to metabolic dysfunction (Doi et al., 2009; Turek et al., 2005; Marcheva et al., 2010; Rudic et al., 2004; Shimba et al., 2011).
The circadian clock is present in many peripheral tissues, playing important roles in local metabolism (Asher and Sassone-Corsi, 2015; Hirota and Fukada, 2004; Panda, 2004; Schibler and Sassone-Corsi, 2002). It has been widely accepted that cellular metabolism is under the control of the circadian clock. Indeed, a majority of metabolites display daily oscillation of tissue and serum concentrations (Dallmann et al., 2012; Eckel-Mahan et al., 2012; Fustin et al., 2012; Hatori et al., 2012). These metabolites then feed back to the circadian clock by modulating enzymatic activity. Recent studies demonstrate that redox homeostasis is interconnected with the circadian clock. Cellular redox state represented by the relative ratio of FAD (oxidized)/NADPH (reduced) oscillates in the rodent SCN where the master clock resides (Wang et al., 2012). In mouse liver, circadian rhythmicity of oxidative function of mitochondria is observed (Kil et al., 2015; Peek et al., 2013). Furthermore, it was demonstrated that even without a functional transcriptional/translational negative feedback loop, the circadian oscillation of the redox state persists in peripheral clocks of various organisms (Edgar et al., 2012; O’Neill and Reddy, 2012; O’Neill et al., 2012). The involvement of NAD+ in circadian regulation has been well characterized (Berger and Sassone-Corsi, 2015; Rutter et al., 2001). The concentration of NAD+ and a rate-limiting enzyme of the biosynthetic pathway of NAD+, nicotinamide phosphoribosyltransferase (NAMPT), cycle in a circadian manner (Nakahata et al., 2009; Ramsey et al., 2009). NAD+ is a classic cofactor critical for enzymatic activity of SIRT1 (NAD+-dependent histone deacetylase) in various species (Blander and Guarente, 2004; Imai et al., 2000; Landry et al., 2000; Rosenberg and Parkhurst, 2002; Tanny et al., 1999). Thus, SIRT1 activity is directed rhythmically by NAD+, resulting in modulation of circadian transcription (Nakahata et al., 2008). In parallel, another group reported that SIRT1-mediated deacetylation of PER2 protein leads to its stabilization (Asher et al., 2008). Compared to NAD+, the physiological importance of FAD has been ignored especially in the mammalian circadian clock. The major function of FAD is as a co-factor of flavoprotein catalyzing metabolic reactions (Powers, 2003). By switching its redox state (FAD⇋FADH2), an electron is transferred to or from the flavoprotein, thus modifying its activity. FAD also acts as a chromophore of photosensing proteins including CRY in plants and Drosophila (Öztürk et al., 2007). In plants and flies, FAD bound to CRY protein detects blue light and contributes to signal transduction pathways generating responses to light signals. In the mammalian molecular clock, CRY1 and CRY2 possess transcriptional repressor activity (Kume et al., 1999). Knockout of Cry1 and Cry2 in mice leads to behavioral rhythms being completely abolished (van der Horst et al., 1999; Vitaterna et al., 1999). Thus, the light-independent function of CRYs has been highlighted. We recently showed that a missense mutation in CRY2 causing amino acid conversion from Ala260 to Thr is responsible for Familial Advanced Sleep Phase (FASP) (Hirano et al., 2016b), which is a sleep trait characterized by early sleep and wake times (Jones et al., 1999). The Ala260 residue resides in the FAD binding pocket of CRY2 (Hitomi et al., 2009). FBXL3 (a CRY2 E3 ubiquitin ligase) binds competitively to the same pocket and promotes ubiquitylation of CRY proteins (Xing et al., 2013). The mouse model harboring the human CRY2 FASP mutation (A260T) recapitulated the human phenotype of advanced activity onset and offset. Although we found that these mutant mice exhibit altered light entrainment, whether this is a result of direct light sensing remains unclear (Hirano et al., 2016b). Thus, our study of the human mutation suggested that FAD may be a key molecule in the circadian clock through CRY regulation in vivo.
Here, we demonstrate that FAD stabilizes CRY proteins. A FAD synthetic enzyme (riboflavin kinase) oscillates in the nucleus, resulting in rhythmic FAD levels. In vivo knockdown of Rfk, Riboflavin (Vitamin B2) kinase essential for FAD synthesis, altered the expression rhythms of CRY1, CRY2 and PER1. Expression levels of some metabolic genes were also affected by Rfk knockdown. These data demonstrate that cycling of cellular FAD ensures the proper oscillation of CRY expression and contributes to the overall regulation of cellular metabolism.
RESULTS
FAD stabilizes CRY1 and CRY2
Previously, we demonstrated that FAD increased CRY2 protein levels in cell culture (Hirano et al., 2016b). We predicted that the effect of FAD on protein levels is conserved in CRY1 and 2 since the amino acid sequence of the FAD binding domain is well conserved. Similar to CRY2, treatment of cultured cells with FAD increased CRY1 (Figure 1A). A cycloheximide chase experiment suggested that the protein stability of endogenous CRY1 and CRY2 is also increased by FAD treatment in the nucleus, although they did not reach statistical significance (Figure 1B). CRY2 carrying the A260T mutation in the FAD binding pocket was less stable vs. wild-type (WT) CRY2 in the nucleus (Figure 1C), as previously shown (Hirano et al., 2016b). Of note, mutant CRY2 was not stabilized by FAD treatment, while WT-CRY2 was (Figure 1C). This indicated that proper binding of FAD to CRY is needed for the stabilization.
Figure 1. FAD stabilizes CRY protein.

(A) Effect of FAD on CRY1 and CRY2 protein levels in HEK293 total cell lysates. Forty-two hours after transfection, HEK293 cells were treated with 100 μM FAD for 6 hours. Data are shown as means with SEM (n=3, *: p<0.05 by Student’s t-test). (B) Degradation assay of endogenous CRY1 and CRY2 protein in NIH3T3 cells. The cells were pre-treated with 100 μM FAD (or PBS as a control) for 6 hours, and then treated with 100 μg/ml CHX and/or 100 μM FAD. Cells were fractionated, and nuclear extracts were used for western blotting analysis. CRY protein levels at the starting point (t=0 hours) were normalized to 1. Data are shown as means with SEM. (C) Degradation assay of WT-CRY2 and A260T-CRY2 protein in HEK293 cells. Forty-two hours after transfection, HEK293 cells were pre-treated with FAD for 6 hours and then treated with 100 μg/ml CHX and/or 100 μM FAD. Cells were fractionated. CRY protein levels in the nuclear fraction at t=0 hours were normalized to 1. Data are shown as means with SEM (n=3). Significance (p<0.05) was determined by two-way ANOVA. (D) CRY and RFK expression in NIH3T3 cells transfected with Rfk siRNAs. Over-expressed FLAG-CRY2, endogenous CRY1 and RFK protein levels were analyzed by western blotting (n=3, *: p<0.05 by Dunnett’s test). (E) CRY1 and CRY2 expression in NIH3T3 cells stably expressing Rfk shRNAs (n=3, *: p<0.05 by Dunnett’s test). (F) CRY1 expression in NIH3T3 cells transfected with Flad1 siRNAs (n=3, *: p<0.05 by Student’s t-test). (G) Degradation assay of CRY1 and CRY2 protein in Rfk knockdown cells. NIH3T3 cells were transfected with Rfk siRNA and CRY expression vectors. Forty-two hours after transfection, the cells were treated with 100 μg/ml CHX. Statistical significance (*: p<0.05) was determined by two-way ANOVA followed by Post-hoc test). (H) Degradation assay of endogenous CRY1 and CRY2 protein in NIH3T3 cells stably expressing Rfk shRNAs. Statistical significance (*: p<0.05) was determined by two-way ANOVA followed by post-hoc test). (I) FBXL3 competition assay. FLAG-CRY1-HA-FBXL3 or Myc-CRY2-FLAG-FBXL3 complexes expressed in HEK293 cells were purified using HA or FLAG antibody. FAD was added to CRY-FBXL3 complexes and incubated at 4 °C for 2 hours in vitro.
FAD synthesis is a two-step reaction. Riboflavin (Vitamin B2), a precursor of FAD, is first phosphorylated by Riboflavin kinase (RFK) and converted to flavin mononucleotide (FMN). FMN is then adenylated by FAD synthase (FADS, encoded by Flad1 gene) to form FAD. RFK is thought to be the rate-limiting enzyme of the FAD biosynthetic pathway (Kohlmeier, 2003). We first knocked down the Rfk gene using siRNAs in NIH3T3 cells to examine the effect of reduced FAD levels on CRY proteins. Rfk knockdown decreased RFK protein levels as expected. In addition, CRY2 (Figure 1D, upper) and CRY1 (Figure 1D, bottom) protein levels were also reduced by Rfk knockdown. Cells stably expressing shRNA targeting Rfk showed decreased endogenous CRY1 and CRY2 levels (Figure 1E). Similarly, reduction of CRY1 was observed by knockdown of the Flad1 gene, supporting the effect of FAD deficiency (Figure 1F). Moreover, the degradation of CRY1 and CRY2 was enhanced in NIH3T3 cells either transiently transfected with siRNAs (Figure 1G) or stably expressing shRNA for Rfk (Figure 1H). These results revealed that reduced FAD can promote the degradation of CRY1/2, leading to reduced protein levels.
Structural studies suggested that FAD interferes with the function of FBXL3 by competing with FBXL3 for binding to CRY2 (via the FAD binding pocket) (Nangle et al., 2013; Xing et al., 2013). We and another group previously showed that FAD competitively released CRY2 from CRY2-FBXL3 complex in vitro (Hirano et al., 2016b; Xing et al., 2013). We also found that this competitive function of FAD is significantly reduced by the CRY2 A260T mutation (Hirano et al., 2016b). Similarly, CRY1 is also released from CRY1-FBXL3 complexes by the presence of FAD in vitro in a dose dependent manner (Figure 1I). Together, these results indicated that FAD competes with FBXL3 and increases stability of both CRY1 and CRY2 in cultured cells.
FAD oscillates in the nucleus
We then examined the expression rhythms of genes encoding enzymes critical for FAD biosynthesis. Rfk and Flad1 mRNAs did not show significant expression rhythms (Figure 2A). On the other hand, we observed that nuclear RFK protein levels peaked in the middle of the night (CT18) in constant darkness (Figure 2B). Consistent with this, the nuclear concentration of FAD shows clear daily rhythms (Figure 2C) with the peak time at ZT12. These results suggested that FAD plays a time and location-dependent role to regulate CRY stability by competing with FBXL3, which is supported by the fact that FBXL3 is predominantly localized in the nucleus (Godinho et al., 2007; Hirano et al., 2013; Yoo et al., 2013). Indeed, the peak time of FAD oscillation (around ZT12) coincides with the time when CRY1 and CRY2 protein start accumulating in liver, implying that FAD contributes to this accumulation.
Figure 2. Rhythmic expression of components of the FAD synthetic pathway.

(A) Temporal expression profiles of Per1 (positive control), Rfk, and Flad1 in mouse liver. C57BL/6J mice entrained to LD 12:12 were sacrificed every 6 hours. mRNA levels were normalized to G3pdh. (B) Temporal expression profiles of RFK in mouse liver in DD. For DD, mice were sacrificed every 4 hours on the second day in DD. Liver was fractionated into nuclear and cytosolic fractions. Data are shown as means ± SEM (n=3). Statistical significance (p<0.05) was determined by one-way ANOVA. (C) Relative FAD amounts in the mouse liver nucleus. Data are shown as means ± SEM (n=3).
FAD affects the circadian period in cell culture
The stability of CRY1 and CRY2 significantly affects the circadian period at molecular and behavioral levels (Godinho et al., 2007; Hirano et al., 2014; 2016a; Ode et al., 2016; Siepka et al., 2007). Additionally, the FASP mouse model expressing A260T-CRY2 protein exhibits shorter circadian period of both behavioral rhythms and cellular rhythms in peripheral tissues (Hirano et al., 2016b). To examine the effect of FAD on the cellular clock, we analyzed PER2::LUC bioluminescence rhythms in the presence of FAD. FAD treatment lengthened the circadian period in a dose-dependent manner (Figure 3A). This is consistent with the previous finding that inhibition of FBXL3 increased CRY protein stability, thus lengthening the circadian period (Godinho et al., 2007; Hirota et al., 2008; Siepka et al., 2007). In agreement with these observations, we found that CRY1 and CRY2 protein accumulated to higher levels when NIH3T3 cells were treated with FAD after been synchronized by Dexamethasone (DEX) (Figure 3B).
Figure 3. Cellular rhythms of PER2::LUC MEFs treated with FAD.

(A) Representative rhythms of PER2::LUC bioluminescence in MEFs from Per2Luc knockin mice. Cellular rhythms of MEFs were synchronized by 100 nM DEX. Media was replaced by a recording media containing 10–100 μM FAD (or PBS) and 100 μM luciferin for recording of bioluminescence. Period lengths of the bioluminescence rhythms are shown as means ± SEM (n=4, *: p<0.05 by Tukey’s test). (B) NIH3T3 cells were synchronized by 100 nM DEX. Media was replaced by fresh media containing 100 μM FAD (or PBS). Total cell lysates were used for western blotting analysis. Data are shown as means ± SEM (n=3, *:p<0.05 by two-way ANOVA followed by Post-hoc test).
Riboflavin deficiency and in vivo knockdown of Rfk led to lower CRY levels
To investigate the in vivo role of FAD, we analyzed the protein expression rhythms in liver of Rfk knockdown mice. We prepared mice by entraining them to LD 12:12 with free access to normal chow or riboflavin (Vitamin B2)-deficient chow. Subsequently, mice underwent tail vein injection with an Rfk siRNA to knockdown Rfk in liver (Figure 4A). We found that 2 weeks of access to a riboflavin-deficient diet did not cause reduction of CRY levels, at least at ZT18 (Figure 4B, control). However, when subjected to a riboflavin-deficient diet, knockdown of Rfk decreased CRY1 and CRY2 at ZT18 (Figure 4B). Knockdown by Rfk siRNA was successful in mice whether they were maintained on a diet of normal or riboflavin-deficient chow. Next, we asked how FAD deficiency affects other clock proteins in constant darkness. Similar to the LD cycle, CRY protein levels were decreased at CT18. The effect of Rfk knockdown seemed time-dependent, and accumulation of nuclear CRY proteins (from CT6 to CT18) was slow in the knockdown mice. Of note, chronic FAD treatment also disrupted the expression rhythms of CRY1 and CRY2 in NIH3T3 cells (Figure 3B). Together, these results suggest a role for FAD in regulating expression rhythms of CRY. Interestingly, PER1 protein was also decreased by Rfk knockdown at CT12 and 18 (Figure 4C, D). Similar alterations were detected in a cell culture experiments, where knockdown of Rfk and Flad1 decreased PER1 and CRY1 protein levels (Figure S1A).
Figure 4. Effect of Rfk knockdown on clock proteins in mouse liver.

(A) Experimental scheme for in vivo knockdown of the Rfk gene. Mice were fed normal or riboflavin-deficient chow for 2 weeks. Rfk siRNA or control siRNA was injected into mice via the tail vein at ZT10-12. Three days or 6 days after the injection, mice were sacrificed to collect liver tissues. (B) Nuclear protein levels of CRY1, CRY2 and RFK at ZT18. TATA-binding protein (TBP) was used as a nuclear marker. (C, D) Nuclear protein levels of PER1, CLOCK, CRY1, CRY2 at indicated CT in DD. Data are shown as means ± SEM (n=3, *:p<0.05 by two-way ANOVA followed by post-hoc test). (E) mRNA levels of indicated clock genes in mouse liver at specified CT in DD. mRNA levels were normalized to G3pdh. Data are shown as means ± SEM (n=3, *:p<0.05 by two-way ANOVA followed by post-hoc test).
Expression of clock genes is altered by Rfk knockdown
In addition to the effects of CRY1 and CRY2 in regulating expression of clock genes, they are also thought to regulate expression of many metabolism-related genes (Oishi et al., 2003). A recent Chromatin Immunoprecipitation Sequencing (ChIP-seq) study indicated that CRY rhythmically occupies not only E-boxes, but also nuclear receptor response elements to control gene transcription (Koike et al., 2012). In Rfk knockdown mice maintained on a riboflavin-deficient diet, gene expression was significantly decreased for Per1 (consistent with reduced PER1 protein levels seen in liver extracts) and other clock genes (Figure 4D and 4E). Decreased accumulation of CRY1/2 proteins around CT12-18 and reduction of clock gene expression were also observed by another siRNA targeting Rfk (Figure S1B and 1C). Among clock components, we found that Per1 mRNA and protein levels were significantly affected by FAD depletion. As Per1 is a target gene of CRY1, we performed a ChIP assay using CRY1 antibody in the knockdown mice. As shown in the previous ChIP-seq study (Koike et al., 2012), the CRY1 ChIP signal at the Per1 promoter E-box is highest at CT0 (Figure S1D). The DNA binding of CRY1 was significantly decreased by Rfk knockdown, suggesting that FAD depletion altered CRY transcriptional activity.
The effect of FAD deficiency on metabolic pathways
We next examined the transcript levels of a series of genes related to metabolism in liver (Figure 5A and S2A). Glucagon promotes gluconeogenesis, and the expression of glucagon-inducible genes is inhibited by CRY protein through suppression of cAMP signaling (Zhang et al., 2010). A small synthetic compound (KL001) that stabilizes CRY was shown to reduce the response to glucagon. Thus, KL001 lowered the induction of major glucagon targets such as Glucose-6-phosphatase (G6pc) and Phosphoenolpryvate carboxykinase 1 (Pck1) genes in mouse hepatocytes (Hirota et al., 2008). We found that G6pc expression was increased by Rfk knockdown at CT6 and 12 in liver extracts of mice on a riboflavin-free diet (Figure 5A), while there was no effect on Pck1. These data suggest that gluconeogenesis is disturbed by depletion of cellular FAD. In addition, we found that blood glucose concentrations showed a tendency to increase and that glucose levels in liver were significantly increased by Rfk knockdown (Figure 5B). In HepG2 cells (derived from a liver hepatocellular carcinoma), FAD treatment decreased while Rfk knockdown increased the oxygen consumption rate (OCR) (Figure 5C), an indicator of cellular metabolism. Enhanced OCR generally represents reduction of glycolysis. Consistent with this, FAD treatment increased the extracellular acidification rate (ECAR), an indicator of glycolysis (Figure 5D). On the other hand, Rfk knockdown reduced the ECAR, indicating that FAD deficiency decreased glycolysis and reversely increased gluconeogenesis, which is promoted by G6pc. Of note, these effects were cancelled by knockdown of CRY1 and CRY2. As shown in Figure 5C, knockdown of Rfk increased OCR in HepG2 cells (Figure 5E left). Knockdown of both CRY1/2 and Rfk decreased OCR rather than further elevating it (Figure 5E, right), suggesting additional regulatory pathway(s) are involved. On the other hand, CRY1/2 knockdown attenuated the effect of Rfk knockdown on ECAR (Figure 5F). A previous study also showed that knockout of Cry1 and Cry2 decreased glycolysis in liver (Peek et al., 2013), supporting the critical role of CRY in glycolysis. Besides glucose homeostasis, some metabolic genes such as Pparg (regulator of hepatic steatosis), Acs1, and Scd1 (regulators of sterol metabolism) were also modulated by the knockdown, suggesting a more global effect of FAD loss on cellular metabolism (Figure 5G and S2B). Taken together, our study emphasizes that cycling FAD feeds back to the circadian clock through its regulation of CRY stability, thereby controlling metabolic pathways (Figure 5H).
Figure 5. Rfk knockdown results in altered expression of genes in metabolic pathways.

(A) mRNA levels of G6pc and Pck in mouse liver. mRNA levels were normalized to Tbp. Data are shown as means ± SEM (n=3, *: p<0.05 by two-way ANOVA). (B) Glucose levels in blood serum (left) and liver (right) at CT12. Data are shown as means ± SEM (n=3, *: p<0.05 by Student’s t-test) (C) Oxygen consumption rate assay in HepG2 cells. Cells were transfected with siRNA of Rfk or treated with 100 μM FAD for 24 hours. Data are shown as means ± SEM (n=6). Statistical significance was determined by two-way ANOVA. (D) Extracellular acidification rate assay in HepG2 cells. Cells were transfected with Rfk siRNA or treated with 100 μM FAD for 24 hours before recording fluorescence of a glycolysis probe. Data are shown as means ± SEM (n=5 for FAD treatment, n=9 for Rfk knockdown). Statistical significance was determined by two-way ANOVA. (E) Oxygen consumption rate assay in HepG2 cells transfected with siRNA of Rfk and/or CRY1/2. Data are shown as means ± SEM (n=9) (F) Extracellular acidification rate assay in HepG2 cells transfected with siRNA of Rfk and/or CRY1/2. Data are shown as means ± SEM (n=9) (G) mRNA levels of indicated metabolism-relevant genes in mouse liver. mRNA levels were normalized to Tbp. Data are shown as means ± SEM (n=3, *: p<0.05 by two-way ANOVA). (H) Proposed model of CRY regulation by FAD. The classical clock generates gene expression rhythms including Rfk and Nampt, which rhythmically synthesize FAD and NAD+. FAD binding to CRY prevents FBXL3-mediated CRY degradation, leading to stabilization. Time-dependent FAD synthesis modulates CRY expression rhythms and amplitudes, which regulate the gene expression rhythms of important genes for metabolism.
DISCUSSION
In the present study, we demonstrate that FAD levels are controlled in a circadian manner (Figure 2C). The peak timing of FAD rhythms was around ZT12 when CRY1 and CRY2 proteins start to accumulate in mouse liver (Lee et al., 2001). FAD deficiency affected CRY stability and accumulation in the circadian cycle (Figure 1 and 4C), suggesting that FAD-mediated stabilization contributes to the nuclear accumulation of CRY. We also found that RFK protein levels (the rate-limiting FAD biosynthetic enzyme) in the nucleus show circadian changes (Figure 2B). However, the mechanisms for rhythmic expression of the riboflavin transporters or enzymatic activity of RFK and FADS remain elusive. Moreover, redox homeostasis (ratio of reduced to oxidative forms of FAD) plays a critical role in the proper maintenance of cellular physiology, and recent findings revealed that daily rhythms of redox state is an integral part of circadian regulation (Edgar et al., 2012; O’Neill et al., 2012; Peek et al., 2013; Wang et al., 2012). Thus, in addition to biosynthesis, the cellular redox state can contribute to the oscillation of FAD.
The expression of clock genes and clock-controlled genes was disturbed by Rfk knockdown (Figure 4D, 4E, 5A and 5G). Interestingly, genes displaying significantly altered expression also have high ChIP scores in a comprehensive CRY1 and CRY2 ChIP-seq study (Koike et al., 2012), suggesting that they are direct targets of CRY. For example, ChIP scores in the G6pc promoter were ranked 111th (for CRY1) and 20th (for CRY2) among all genes (>10,000) with specific ChIP signals. On the other hand, Pck1, another target of glucagon, was ranked 852nd (CRY1) and 1658th (CRY2) by ChIP score (Koike et al., 2012) and showed no change with Rfk knockdown (Figure 5A). Furthermore, we found that CRY1/2 knockdown reversed or attenuated the effect of Rfk silencing in OCR and ECAR assays, suggesting the involvement of CRY-FAD regulation in glucose metabolism (Figures 5E and 5F). Congruent with these data, metabolomics analysis revealed altered glucose metabolism under FAD treatment in HepG2 cells (Figure S3). However, to ensure that this is relevant in vivo, further investigation using Cry1/2 knockout mice or mice harboring mutations in the FAD binding domain of CRY1 and CRY2 will be needed. Per1 also scored the highest in the previous ChIP study of CRYs and, in our studies, showed significant alterations in both mRNA expression and protein levels. We observed that Rfk knockdown reduced DNA binding of CRY1 to a target sequence of the Per1 promoter (Figure S1D). However the reduction of Per1 expression by Rfk knockdown argues that its regulation is even more complex. Previously, we and another group reported that mutation at the FAD binding site of CRY altered CRY repressor activity (Hirano et al., 2016b; Hitomi et al., 2009). Therefore, FAD deficiency may affect CRY repressor activity, which cannot be detected by the ChIP assay.
FAD is a co-factor of CRY in various species (Lin and Todo, 2005). In plants and flies, CRY harboring FAD detects blue light and contributes to the biological response to light by changing protein conformation. In plants, Arabidopsis CRY regulates development and growth in response to light. Drosophila CRY is a circadian photosensor contributing to clock resetting in response to light. Furthermore, dCRY acts as a light-dependent magnetosensor and knockout results in loss of magnetosensation (Gegear et al., 2008; 2010). Interestingly, human CRY2 can rescue the light-dependent magnetosensation phenotype of dCry-deficient flies (Foley et al., 2011). Whether CRY2 is actually involved in magnetoreception in mammalian physiology is unknown. On the other hand, mammalian CRYs have acquired repressor activity of transcription and are considered core components in the circadian clock. CRY repressor activity is light-independent (Griffin et al., 1999), and the central clock (SCN) does not detect light directly. Thus, the link between FAD and the mammalian circadian clock in the physiological context was unclear. A recent report revealed that LSD1, a lysine-specific FAD-dependent histone demethylase, regulates the light-resetting of the circadian clock through PKCα-mediated phosphorylation of LSD1 (Nam et al., 2014). Phosphorylated LSD1 (at Ser112) interacts with CLOCK/BMAL1 complexes and modulates E-box-mediated gene expression. Knockout of Lsd1 or knocked-in of a mutation at the phosphorylation site of LSD1 (Ser112 to Ala) led to decreased amplitude of gene expression in mouse embryonic fibroblasts and behavioral rhythms in mice. However, histone demethylation was intact in these Lsd1 mutant mice. Thus, it is likely that the role of LSD1 in circadian phenotypes is independent of FAD. Furthermore, the circadian periods of these Lsd1 mutants remain indistinguishable from normal mice. Here, we found that FAD treatment indeed modulates the circadian period (Figure 3A). The data presented in this study therefore demonstrate that FAD contributes to circadian rhythmicity by binding to CRYs and that a deficiency of FAD causes dysregulations of metabolism.
Riboflavin (Vitamin B2) is an essential nutrient for mammals. Because mammals are unable to synthesize it, we depend on dietary intake of riboflavin (Powers, 2003). Deficiency of riboflavin causes abnormal development, neurodegeneration, cardiovascular disease, alterations of DNA damage repair, and cancer (Powers, 2003). Although the effect of riboflavin deficiency may act through different pathways, some of these effects may be mediated through disturbance of CRY protein levels and altered circadian regulation. Indeed, CRY proteins are highly associated with cancer development and DNA damage (Lee et al., 2013; Ozturk et al., 2009; Papp et al., 2015). In the present study, we found that administration or deletion of FAD altered glucose metabolism through CRYs (Fig. 5), indicating a significant role for the FAD-CRY pathway in normal physiology.
EXPERIMENTAL PROCEDURES
Mice
All experimental protocols were conducted according to US National Institutes of Health guidelines for animal research and were approved by the Institutional Animal Care and Use Committee at the University of California, San Francisco. Mice were kept in cages with free access to food and water. Riboflavin-deficient chow was purchased from Research Diets Inc.(New Brunswick, NJ).
Cell culture and constructs
HEK293 cells, NIH3T3 cells and HepG2 cells were purchased from ATTC. Authentication of the cell lines was performed by ATCC according to their protocol. Cultures were tested for mycoplasma contamination and all cell lines used for experiments were mycoplasma-free. HEK293 cells and NIH3T3 cells were cultured in DMEM (Corning Inc.) containing 10% FBS and 100 U/ml Penicillin-Streptomycin (Thermo Fisher Scientific) and maintained by standard methods. HepG2 cells were maintained in MEM containing 10% FBS, non-essential amino acids and 1 mM Sodium Pyruvate. Mouse embryonic fibroblasts (MEFs) were prepared from E12.5 embryos of Per2luc knockin mice (Yoo et al., 2004). After removing the head, paws and internal organs, embryos were chopped and incubated in 0.25% trypsin in PBS for 24 hr at 4°C. After incubation for 30 min at 37°C in 0.25% trypsin in PBS, cells were dissociated by pipetting in DMEM. Supernatant was cultured in a cell culture dish with DMEM and maintained by standard methods. Cells were transfected with Lipofectamine 3000 transfection reagent (Thermo Fisher Scientific) according to manufacturer’s protocol. DNA constructs used for transfections are listed in supplemental experimental procedures (Hirano et al., 2016b).
Bioluminescence rhythms in cell culture
Cellular rhythms were synchronized by treatment with 100 nM dexamethasone (DEX) for 2 hours. Media was changed to the recording media: phenol-red free DMEM (Sigma Aldrich) containing 10mM HEPES-pH7.0, 3.5g/L D-glucose, 0.1 mM luciferin potassium salt (Thermo Fisher Scientific). Bioluminescence was continuously recorded in a LumiCycle 32 instrument (Actimetrics, Wilmette, IL). Bioluminescence was detrended by subtracting 24-hr average of bioluminescence using the LumiCycle analysis software to calculated periods.
Expression profiles of proteins and genes
C57BL/6J male mice (~10 weeks old) were entrained to LD 12:12 for at least 10 days. For expression rhythms of RFK in DD, mice were sacrificed every 4 hours in dim red light on the 2nd day of DD. For LD, mice were sacrificed every 6 hours. Liver tissues were collected, followed by nuclear extraction (Yoshitane et al., 2009) and mRNA extraction.
Nuclear and cytosolic fractionation of cells
Cells were resuspended in the lysis buffer [10mM HEPES-KOH (pH 7.8 at 4°C), 10mM KCl. 0.1mM EDTA, 0.2% NP-40] containing 1mM DTT and protease inhibitor (Thermo Fisher Scientific) and incubated for 10 min on ice. Cell membranes were disrupted by vortexing twice for 15 sec, followed by centrifugation for 10 min at 20,000×g. The pellet (nuclear fraction) was washed with the lysis buffer without NP-40 and dissolved in SDS sample buffer [62.5mM Tris-HCl (pH 6.8), 50mM DTT, 2% SDS, 10% glycerol].
Western Blotting
For whole-cell extracts, HEK293 cells were lysed in SDS sample buffer [62.5mM Tris-HCl (pH 6.8), 50mM DTT, 2% SDS, 10% glycerol]. Preparation of the cytosolic and the nuclear fractions of mouse liver was performed as previously described (Yoshitane et al., 2009). Protein samples were separated by SDS-PAGE. Tissues were transfered to PVDF membranes (Millipore) with blocking in T-TBS [50mM Tris-HCl (pH 7.4), 137mM NaCl, 0.1% Tween 20] containing 1% Skim milk. Primary antibodies were reacted in the blocking solution at 4°C overnight. Then, secondary antibodies were reacted in the blocking solution at RT for 2 hr. Proteins were detected with the Western Lightning Plus ECL (PerkinElmer). Band intensities were determined using Image J software. β-actin was used as a loading control for total cell lysates, and TBP was used as the nuclear marker. Proteins were detected with the following antibodies: anti-cMyc 9E10 (Santa Cruz Biotechnology, sc-40), anti-FLAG M2 (Sigma Aldrich, F1804), anti-HA Y11 (Santa Cruz Biotechnology, sc-805-G), anti-β-actin (Abcam, AC-15), anti-LMNB1 (Abcam, ab16048), anti-TBP (Santa Cruz Inc.) anti-hPER1 (Thermo Fisher Scientific, PA1-524), anti-mPER2 (Alpha Diagnostic International, PER-21A), anti-CLOCK (MBL, D333-3), anti-CRY1 (MBL, PM081) and anti-CRY2 (MBL, PM082). Secondary antibodies used were goat anti-mouse IgG-HRP (Santa Cruz Biotechnology, sc-2005), goat anti-rabbit IgG-HRP (Santa Cruz Biotechnology, sc-2006) and goat anti-guinea pig IgG-HRP (Santa Cruz Biotechnology, sc2438).
Real-time qPCR
Total RNA was extracted by TRIzol reagent (Thermo Fisher Scientific) from liver samples and purified by RNeasy Mini kit (QIAGEN). cDNA was synthesized from 1.5 μg RNA by GoScript (Promega) using dT15 primer. Quantification of mRNA was performed with GoTaq Real-Time qPCR Kits (Promega) using gene specific primers (Supplemental Table1). mRNA levels were normalized by mouse Gapdh or Tbp levels.
FAD concentration
FAD was measured with an FAD assay kit (Sigma Aldrich) following the manufacture’s protocol. Briefly, the nuclear extract was added to 2 volumes of 8%(w/w) Perchloric Acid (PCA), followed by vortexing the mixture for 30 seconds. The lysate was centrifuged for 10 min at 1,500×g and the supernatant was used for FAD measurements.
FAD competition assay
Flavin adenine dinucleotide disodium salt hydrate (FAD, Sigma Aldrich) was diluted in PBS to make a 100 mM stock solution. HEK293 cells were transfected with plasmid vectors for CRY (FLAG-CRY1 or CRY2-Myc-His) and FBXL3 (HA-FBXL3 or FLAG-FBXL3). Forty-two hours after transfection, the cells were treated with 10 μM MG132 (Calbiochem) for 6 hours. CRY-FBXL3 complex was purified with anti-FLAG M2 affinity gel (Sigma Aldrich) or HA antibody. FAD was incubated with CRY1-FBXL3 complex binding to anti-FLAG M2 affinity gel or protein G sepharose (GE Healthcare) in 40 μl PBS for 2 hours on ice. After centrifugation, the supernatant was collected as the ‘released CRY’ sample. CRY still binding to FBXL3 was eluted by adding SDS sample buffer.
Tail vein injection of siRNA
siRNA for Rfk was purchased from Thermo Fisher Scientific (siRNA#1:cat# S79629, siRNA#2:cat# S79628). siRNA solution for the injection was prepared according to the manual of Invivofectamine 3.0 (Thermo Fisher Scientific). BALB/c male mice (8 weeks old) were fed a normal diet or riboflavin-free diet for 2 weeks. Mice (10 weeks old at the injection) were anesthetized with Isoflurane during the procedure, and siRNA targeting Rfk or equivalent amounts of control siRNA (3 mol/mice each siRNA) were injected through the tail vein using a 28G syringe. Injections were performed at ZT10-12.
Glucose concentration
Four days after the injection of siRNAs, blood was collected from mouse tail veins. Blood was incubated for 60 min at 37°C and centrifuged at 3000 rpm for 20 min. Supernatant was collected and used for a glucose oxidation assay (Sigma Aldrich). For glucose concentration in liver lysate, proteins were removed by PCA precipitation. One third volume of 4 M PCA was added to total liver lysate, followed by incubation on ice for 5 min. After centrifuge at 13,000xg for 2 min, supernatant was collected. Ice-cold 2M KOH was added to titrate pH to ~7.0. Samples were centrifuged at 13,000xg for 15 min at 4°C, and supernatant was used for the glucose oxidation assay (Sigma Aldrich).
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR)
HepG2 cells were cultured in 96-well plates and transfected with indicated siRNAs. Twenty-four hours after the transfection, cell culture media were replaced by media containing the OCR fluorescence probe (Abcam, OCR kit) and cultured for 24 hours. Media was changed to recording media; phenol-red free DMEM (Sigma Aldrich) containing 10mM HEPES-pH7.0, 3.5g/L D-glucose and 10% FBS. Florescence was continuously measured every 2.5 min or 5 min by with a SynergyTM H4 Hybrid Multi-Mode Microplate Reader (BioTek). For ECAR, transfected cells were cultured for 48 hours, and culture media was changed to glycolysis buffer containing probe (Abcam, ECAR kit). Florescence was measured every 1.5 min using a SynergyTM H4 Hybrid Multi-Mode Microplate Reader (BioTek).
Statistical analysis
All error bars in the figures represent SEM. No statistical analysis was used to predetermine the sample sizes. Experiments were not randomized and were not analyzed blindly. Data was statistically analyzed using R software and Prism7. To assess statistical significance, data were obtained from ≥3 independent experiments. Two-tailed paired Student’s t-test, Tukey’s test, one-way ANOVA or two-way ANOVA followed by Sidak test (as post-hoc test) was used. P values <0.05 were considered to represent a statistically significant difference.
Supplementary Material
HIGHLIGHTS.
FAD stabilizes CRYPTOCHROME (CRY) proteins by competing with FBXL3.
FAD concentration in the nucleus has a daily rhythm.
FAD lengthens the circadian period.
In vivo knockdown of Riboflavin kinase (Rfk) alters CRY and PER1 expression rhythms.
Acknowledgments
This work was funded by NIH grant GM079180 and HL059596 to L.J.P. and Y-H.F and by the William Bowes Neurogenetics Fund. This work was also supported by NIH grant P30 DK063720 to the UCSF Diabetes Research Center. L.J.P. is an investigator of the Howard Hughes Medical Institute. A.H. was supported by the Japanese Society for the Promotion of Science (JSPS), the Kanae Foundation for the promotion of medical science, and the Uehara Memorial Life science foundation. We thank Dr. Masako Asahina (UCSF) for providing the HepG2 cell line.
Footnotes
AUTHORS CONTRIBUTIONS: A.H. performed experiments. D. B. performed metabolomics analysis. A.H, Y.-H.F, and L.J.P designed experiments, analyzed data and wrote the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Asher G, Sassone-Corsi P. Time for food: the intimate interplay between nutrition, metabolism, and the circadian clock. Cell. 2015;161:84–92. doi: 10.1016/j.cell.2015.03.015. [DOI] [PubMed] [Google Scholar]
- Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. SIRT1 Regulates Circadian Clock Gene Expression through PER2 Deacetylation. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- Berger SL, Sassone-Corsi P. Cold Spring Harb Perspect Biol. 2015. Metabolic Signaling to Chromatin. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander G, Guarente L. The Sir2 Family of Protein Deacetylases. Annu Rev Biochem. 2004;73:417–435. doi: 10.1146/annurev.biochem.73.011303.073651. [DOI] [PubMed] [Google Scholar]
- Dallmann R, Viola AU, Tarokh L, Cajochen C, Brown SA. The human circadian metabolome. Proc Natl Acad Sci U S a. 2012;109:2625–2629. doi: 10.1073/pnas.1114410109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi M, Takahashi Y, Komatsu R, Yamazaki F, Yamada H, Haraguchi S, Emoto N, Okuno Y, Tsujimoto G, Kanematsu A, et al. Salt-sensitive hypertension in circadian clock–deficient Cry-null mice involves dysregulated adrenal Hsd3b6. Nature Medicine. 2009;16:67–74. doi: 10.1038/nm.2061. [DOI] [PubMed] [Google Scholar]
- Eckel-Mahan KL, Patel VR, Mohney RP, Vignola KS, Baldi P, Sassone-Corsi P. Coordination of the transcriptome and metabolome by the circadian clock. Proc Natl Acad Sci U S a. 2012;109:5541–5546. doi: 10.1073/pnas.1118726109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RS, Green EW, Zhao Y, van Ooijen G, Olmedo M, Qin X, Xu Y, Pan M, Valekunja UK, Feeney KA, et al. Peroxiredoxins are conserved markers of circadian rhythms. Nature. 2012;485:459–464. doi: 10.1038/nature11088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley LE, Gegear RJ, Reppert SM. Human cryptochrome exhibits light-dependent magnetosensitivity. Nature Communications. 2011;2:356. doi: 10.1038/ncomms1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turek Fred W, Joshu Corinne, Kohsaka Akira, Lin Emily, Ivanova Ganka, McDearmon Erin, Laposky A, Losee-Olson S, Easton A, Jensen DR, et al. Obesity and Metabolic Syndrome in Circadian Clock Mutant Mice. Science. 2005;108:1043–1045. doi: 10.1126/science.1108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fustin JM, Doi M, Yamada H, Komatsu R, Shimba S, Okamura H. Rhythmic nucleotide synthesis in the liver: temporal segregation of metabolites. CellReports. 2012;1:341–349. doi: 10.1016/j.celrep.2012.03.001. [DOI] [PubMed] [Google Scholar]
- Gegear RJ, Casselman A, Waddell S, Reppert SM. Cryptochrome mediates light-dependent magnetosensitivity in Drosophila. Nature. 2008;454:1014–1018. doi: 10.1038/nature07183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegear RJ, Foley LE, Casselman A, Reppert SM. Animal cryptochromes mediate magnetoreception by an unconventional photochemical mechanism. Nature. 2010;463:804–807. doi: 10.1038/nature08719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godinho SIH, Maywood ES, Shaw L, Tucci V, Barnard AR, Busino L, Pagano M, Kendall R, Quwailid MM, Romero MR, et al. The after-hours mutant reveals a role for Fbxl3 in determining mammalian circadian period. Science. 2007;316:897–900. doi: 10.1126/science.1141138. [DOI] [PubMed] [Google Scholar]
- Griffin EA, Staknis D, Weitz CJ. Light-independent role of CRY1 and CRY2 in the mammalian circadian clock. Science. 1999;286:768–771. doi: 10.1126/science.286.5440.768. [DOI] [PubMed] [Google Scholar]
- Hardin PE, Panda S. Circadian timekeeping and output mechanisms in animals. Current Opinion in Neurobiology. 2013;23:724–731. doi: 10.1016/j.conb.2013.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatori M, Vollmers C, Zarrinpar A, DiTacchio L, Bushong EA, Gill S, Leblanc M, Chaix A, Joens M, Fitzpatrick JAJ, et al. Time-restricted feeding without reducing caloric intake prevents metabolic diseases in mice fed a high-fat diet. Cell Metabolism. 2012;15:848–860. doi: 10.1016/j.cmet.2012.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Y, Jones CR, Fujiki N, Xu Y, Guo B, Holder JL, Rossner MJ, Nishino S, Fu YH. The Transcriptional Repressor DEC2 Regulates Sleep Length in Mammals. Science. 2009;325:866–870. doi: 10.1126/science.1174443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Kurabayashi N, Nakagawa T, Shioi G, Todo T, Hirota T, Fukada Y. In Vivo Role of Phosphorylation of Cryptochrome 2 in the Mouse Circadian Clock. Molecular and Cellular Biology. 2014;34:4464–4473. doi: 10.1128/MCB.00711-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Nakagawa T, Yoshitane H, Oyama M, Kozuka-Hata H, Lanjakornsiripan D, Fukada Y. USP7 and TDP-43: Pleiotropic Regulation of Cryptochrome Protein Stability Paces the Oscillation of the Mammalian Circadian Clock. PLoS ONE. 2016a;11:e0154263. doi: 10.1371/journal.pone.0154263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Shi G, Jones CR, Lipzen A, Pennacchio LA, Xu Y, Hallows WC, McMahon T, Yamazaki M, Ptáček LJ, et al. A Cryptochrome 2 mutation yields advanced sleep phase in humans. eLife. 2016b;5:e16695. doi: 10.7554/eLife.16695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Yumimoto K, Tsunematsu R, Matsumoto M, Oyama M, Kozuka-Hata H, Nakagawa T, Lanjakornsiripan D, Nakayama KI, Fukada Y. FBXL21 Regulates Oscillation of the Circadian Clock through Ubiquitination and Stabilization of Cryptochromes. Cell. 2013;152:1106–1118. doi: 10.1016/j.cell.2013.01.054. [DOI] [PubMed] [Google Scholar]
- Hirota T, Fukada Y. Resetting mechanism of central and peripheral circadian clocks in mammals. Zool Sci. 2004;21:359–368. doi: 10.2108/zsj.21.359. [DOI] [PubMed] [Google Scholar]
- Hirota T, Lewis WG, Liu AC, Lee JW, Schultz PG, Kay SA. A chemical biology approach reveals period shortening of the mammalian circadian clock by specific inhibition of GSK-3β. Proc Natl Acad Sci USa. 2008;105:20746–20751. doi: 10.1073/pnas.0811410106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hitomi K, DiTacchio L, Arvai AS, Yamamoto J, Kim ST, Todo T, Tainer JA, Iwai S, Panda S, Getzoff ED. Functional motifs in the (6-4) photolyase crystal structure make a comparative framework for DNA repair photolyases and clock cryptochromes. Proc Natl Acad Sci U S a. 2009;106:6962–6967. doi: 10.1073/pnas.0809180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- Jones CR, Campbell SS, Zone SE, Cooper F, DeSano A, Murphy PJ, Jones B, Czajkowski L, Ptáček LJ. Familial advanced sleep-phase syndrome: A short-period circadian rhythm variant in humans. Nature Medicine. 1999;5:1062–1065. doi: 10.1038/12502. [DOI] [PubMed] [Google Scholar]
- Kil IS, Ryu KW, Lee SK, Kim JY, Chu SY, Kim JH, Park S, Rhee SG. Circadian Oscillation of Sulfiredoxin in the Mitochondria. Molecular Cell. 2015;59:651–663. doi: 10.1016/j.molcel.2015.06.031. [DOI] [PubMed] [Google Scholar]
- Kohlmeier M. Nutrient Metabolism: Structures, Functions, and Genes. Cambridge, MA: Academic Press; 2003. [Google Scholar]
- Koike N, Yoo SH, Huang HC, Kumar V, Lee C, Kim TK, Takahashi JS. Transcriptional Architecture and Chromatin Landscape of the Core Circadian Clock in Mammals. Science. 2012;338:349–354. doi: 10.1126/science.1226339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume K, Zylka MJ, Sriram S, Shearman LP, Weaver DR, Jin X, Maywood ES, Hastings MH, Reppert SM. mCRY1 and mCRY2 Are Essential Components of the Negative Limbof the Circadian Clock Feedback Loop. Cell. 1999;98:193–205. doi: 10.1016/s0092-8674(00)81014-4. [DOI] [PubMed] [Google Scholar]
- Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L, Sternglanz R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc Natl Acad Sci USa. 2000;97:5807–5811. doi: 10.1073/pnas.110148297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Etchegaray JP, Cagampang FR, Loudon AS, Reppert SM. Posttranslational mechanisms regulate the mammalian circadian clock. Cell. 2001;107:855–867. doi: 10.1016/s0092-8674(01)00610-9. [DOI] [PubMed] [Google Scholar]
- Lee JH, Gaddameedhi S, Öztürk N, Ye R, Sancar A. DNA Damage-Specific Control of Cell Death by Cryptochrome in p53-Mutant Ras-Transformed Cells. Cancer Research. 2013;73:785–791. doi: 10.1158/0008-5472.CAN-12-1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C, Todo T. The cryptochromes. Genome Biol. 2005;6:220. doi: 10.1186/gb-2005-6-5-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcheva B, Ramsey KM, Buhr ED, Kobayashi Y, Su H, Ko CH, Ivanova G, Omura C, Mo S, Vitaterna MH, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466:627–631. doi: 10.1038/nature09253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science. 2009;324:654–657. doi: 10.1126/science.1170803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahata Y, Yoshida M, Takano A, Soma H, Yamamoto T, Yasuda A, Nakatsu T, Takumi T. A direct repeat of E-box-like elements is required for cell-autonomous circadian rhythm of clock genes. BMC Mol Biol. 2008;9:1. doi: 10.1186/1471-2199-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HJ, Boo K, Kim D, Han DH, Choe HK, Kim CR, Sun W, Kim H, Kim K, Lee H, et al. Phosphorylation of LSD1 by PKCα is crucial for circadian rhythmicity and phase resetting. Molecular Cell. 2014;53:791–805. doi: 10.1016/j.molcel.2014.01.028. [DOI] [PubMed] [Google Scholar]
- Nangle S, Xing W, Zheng N. Crystal structure of mammalian cryptochrome in complex with a small molecule competitor of its ubiquitin ligase. Cell Research. 2013;23:1417–1419. doi: 10.1038/cr.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ode KL, Ukai H, Susaki EA, Narumi R, Matsumoto K, Hara J, Koide N, Abe T, Kanemaki MT, Kiyonari H, et al. Knockout-Rescue Embryonic Stem Cell-Derived Mouse Reveals Circadian-Period Control by Quality and Quantity of CRY1. Molecular Cell. 2016:1–16. doi: 10.1016/j.molcel.2016.11.022. [DOI] [PubMed] [Google Scholar]
- Oishi K, Miyazaki K, Kadota K, Kikuno R, Nagase T, Atsumi GI, Ohkura N, Azama T, Mesaki M, Yukimasa S, et al. Genome-wide expression analysis of mouse liver reveals CLOCK-regulated circadian output genes. J Biol Chem. 2003;278:41519–41527. doi: 10.1074/jbc.M304564200. [DOI] [PubMed] [Google Scholar]
- Ozturk N, Lee JH, Gaddameedhi S, Sancar A. Loss of cryptochrome reduces cancer risk in p53 mutant mice. Proc Natl Acad Sci U S a. 2009;106:2841–2846. doi: 10.1073/pnas.0813028106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öztürk N, Song SH, Özgür S, Selby CP, Morrison L, Partch C, Zhong D, Sancar A. Structure and Function of Animal Cryptochromes. Cold Spring Harbor Symposia on Quantitative Biology. 2007;72:119–131. doi: 10.1101/sqb.2007.72.015. [DOI] [PubMed] [Google Scholar]
- O’Neill JS, Reddy AB. Circadian clocks in human red blood cells. Nature. 2012;469:498–503. doi: 10.1038/nature09702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill JS, van Ooijen G, Dixon LE, Troein C, Corellou F, Bouget FY, Reddy AB, Millar AJ. Circadian rhythms persist without transcription in a eukaryote. Nature. 2012;469:554–558. doi: 10.1038/nature09654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda S. It’s All in the Timing: Many Clocks, Many Outputs. Journal of Biological Rhythms. 2004;19:374–387. doi: 10.1177/0748730404269008. [DOI] [PubMed] [Google Scholar]
- Papp SJ, Huber AL, Jordan SD, Kriebs A, Nguyen M. DNA damage shifts circadian clock time via Hausp-dependent Cry1 stabilization. eLife. 2015 doi: 10.7554/eLife.04883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, Ilkayeva O, Marcheva B, Kobayashi Y, Omura C, et al. Circadian Clock NAD+ Cycle Drives Mitochondrial Oxidative Metabolism in Mice. Science. 2013;342:1243417–1243417. doi: 10.1126/science.1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powers HJ. Riboflavin (vitamin B-2) and health. Am J Clin Nutr. 2003;77:1352–1360. doi: 10.1093/ajcn/77.6.1352. [DOI] [PubMed] [Google Scholar]
- Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, Hong HK, Chong JL, Buhr ED, Lee C, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science. 2009;324:651–654. doi: 10.1126/science.1171641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR. Coordination of circadian timing in mammals. Nature. 2002;418:935–941. doi: 10.1038/nature00965. [DOI] [PubMed] [Google Scholar]
- Rosenberg MI, Parkhurst SM. Drosophila Sir2 is required for heterochromatic silencing and by euchromatic Hairy/E(Spl) bHLH repressors in segmentation and sex determination. Cell. 2002;109:447–458. doi: 10.1016/s0092-8674(02)00732-8. [DOI] [PubMed] [Google Scholar]
- Rudic RD, McNamara P, Curtis AM, Boston RC, Panda S, Hogenesch JB, FitzGerald GA. BMAL1 and CLOCK, Two Essential Components of the Circadian Clock, Are Involved in Glucose Homeostasis. PLoS Biol. 2004;2:e377. doi: 10.1371/journal.pbio.0020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter J, Reick M, Wu LC, McKnight SL. Regulation of clock and NPAS2 DNA binding by the redox state of NAD cofactors. Science. 2001;293:510–514. doi: 10.1126/science.1060698. [DOI] [PubMed] [Google Scholar]
- Schibler U, Sassone-Corsi P. A web of circadian pacemakers. Cell. 2002;111:919–922. doi: 10.1016/s0092-8674(02)01225-4. [DOI] [PubMed] [Google Scholar]
- Shimba S, Ogawa T, Hitosugi S, Ichihashi Y, Nakadaira Y, Kobayashi M, Tezuka M, Kosuge Y, Ishige K, Ito Y, et al. Deficient of a Clock Gene, Brain and Muscle Arnt-Like Protein-1 (BMAL1), Induces Dyslipidemia and Ectopic Fat Formation. PLoS ONE. 2011;6:e25231. doi: 10.1371/journal.pone.0025231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepka SM, Yoo SH, Park J, Song W, Kumar V, Hu Y, Lee C, Takahashi JS. Circadian mutant Overtime reveals F-box protein FBXL3 regulation of cryptochrome and period gene expression. Cell. 2007;129:1011–1023. doi: 10.1016/j.cell.2007.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanny JC, Dowd GJ, Huang J, Hilz H, Moazed D. An enzymatic activity in the yeast Sir2 protein that is essential for gene silencing. Cell. 1999;99:735–745. doi: 10.1016/s0092-8674(00)81671-2. [DOI] [PubMed] [Google Scholar]
- Toh KL, Jones CR, He Y, Eide EJ, Hinz WA, Virshup DM, Ptáček LJ, Fu YH. An hPer2 phosphorylation site mutation in familial advanced sleep phase syndrome. Science. 2001;291:1040–1043. doi: 10.1126/science.1057499. [DOI] [PubMed] [Google Scholar]
- van der Horst GTJ, Muijtjens M, Kobayashi K, Takano R, Kanno SI, Takao M, de Wit J, Verkerk A, Eker APM, van Leenen D, et al. Mammalian Cry1 and Cry2 are essential for maintenance of circadian rhythms. Nature. 1999;398:627–630. doi: 10.1038/19323. [DOI] [PubMed] [Google Scholar]
- Vitaterna MH, Selby CP, Todo T, Niwa H, Thompson C, Fruechte EM, Hitomi K, Thresher RJ, Ishikawa T, Miyazaki J, et al. Differential regulation of mammalian period genes and circadian rhythmicity by cryptochromes 1 and 2. Proc Natl Acad Sci USa. 1999;96:12114–12119. doi: 10.1073/pnas.96.21.12114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TA, Yu YV, Govindaiah G, Ye X, Artinian L, Coleman TP, Sweedler JV, Cox CL, Gillette MU. Circadian rhythm of redox state regulates excitability in suprachiasmatic nucleus neurons. Science. 2012;337:839–842. doi: 10.1126/science.1222826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing W, Busino L, Hinds TR, Marionni ST, Saifee NH, Bush MF, Pagano M, Zheng N. SCFFBXL3 ubiquitin ligase targets cryptochromes at their cofactor pocket. Nature. 2013;496:64–68. doi: 10.1038/nature11964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Toh KL, Jones CR, Shin JY, Fu YH, Ptáček LJ. Modeling of a Human Circadian Mutation Yields Insights into Clock Regulation by PER2. Cell. 2007;128:59–70. doi: 10.1016/j.cell.2006.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Padiath QS, Shapiro RE, Jones CR, Wu SC, Saigoh N, Saigoh K, Ptáček LJ, Fu YH. Functional consequences of a CKIdelta mutation causing familial advanced sleep phase syndrome. Nature. 2005;434:640–644. doi: 10.1038/nature03453. [DOI] [PubMed] [Google Scholar]
- Yoo SH, Mohawk JA, Siepka SM, Shan Y, Huh SK, Hong HK, Kornblum I, Kumar V, Koike N, Xu M, et al. Competing E3 ubiquitin ligases govern circadian periodicity by degradation of CRY in nucleus and cytoplasm. Cell. 2013;152:1091–1105. doi: 10.1016/j.cell.2013.01.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SH, Yamazaki S, Lowrey PL, Shimomura K, Ko CH, Buhr ED, Siepka SM, Hong HK, Oh WJ, Yoo OJ, et al. PERIOD2::LUCIFERASE real-time reporting of circadian dynamics reveals persistent circadian oscillations in mouse peripheral tissues. Proc Natl Acad Sci USa. 2004;101:5339–5346. doi: 10.1073/pnas.0308709101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitane H, Takao T, Satomi Y, Du NH, Okano T, Fukada Y. Roles of CLOCK Phosphorylation in Suppression of E-Box-Dependent Transcription. Mo Cell Biol. 2009;29:3675–3686. doi: 10.1128/MCB.01864-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang EE, Liu Y, Dentin R, Pongsawakul PY, Liu AC, Hirota T, Nusinow DA, Sun X, Landais S, Kodama Y, et al. Cryptochrome mediates circadian regulation of cAMP signaling and hepatic gluconeogenesis. Nature Medicine. 2010;16:1152–1156. doi: 10.1038/nm.2214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Hirano A, Hsu PK, Jones CR, Sakai N, Okuro M, McMahon T, Yamazaki M, Xu Y, Saigoh N, et al. A PERIOD3 variant causes a circadian phenotype and is associated with a seasonal mood trait. Proc Natl Acad Sci USA. 2016;113:E1536–E1544. doi: 10.1073/pnas.1600039113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.