

ABSTRACT
We give an overview of experimental and computational methods to estimate RNA metabolism rates genome-wide. We then advocate a local definition of RNA metabolism rate at the level of individual phosphodiester bonds. Rates of formation and disappearance of individual bonds are unambiguously defined, in contrast to rates of complete transcripts. We show that over previous approaches, the recently developed transient transcriptome sequencing (TT-seq) protocol allows for estimation of metabolism rates of individual bonds with least positional bias.
KEYWORDS: kinetics, RNA metabolism, RNA sequencing, splicing, transcription
Importance of RNA metabolism rates
All stages of RNA metabolism contribute to the control of gene expression, including RNA synthesis, splicing, and degradation. The ratio between the synthesis and degradation rates determines steady-state levels of mature RNA. Upon a transcriptional trigger, both degradation and splicing rates contribute to the time until which new steady-state levels are reached. Whereas variations in RNA synthesis rates are the major determinants of mRNA levels,1-3 RNA degradation rates further fine-tunes mRNA abundance and can be dynamically changed to shape gene expression.3-9 Combinations of synthesis and degradation rates enable different gene regulatory strategies that can favor turn-over or robustness, and high or low levels of expression.3,10,11 Although the number of genome-wide studies of RNA splicing rates is more limited, it is clear that splicing rates also vary a lot between and within genes,3,11-13 with impact on the composition of the isoform repertoire of a cell. Altogether, precise quantitative measurements of transcription, degradation, and splicing rates are necessary to obtain a deeper understanding of gene expression control and of the underlying mechanisms.
Limitations of steady-state RNA-seq data
At steady-state, production and degradation of every molecular species balance each other. The mature RNA concentration is consequently the ratio of the synthesis rate over the degradation rate (Fig. 1). Hence, steady-state RNA-seq cannot untangle synthesis rate from degradation rates. The same issue affects all proxies for splicing kinetics that are derived from steady-state data. For instance, there is a possible confounding when defining using the ratio of reads spanning exon–exon over exon–intron junctions to assess splicing efficiency.14 Indeed, steady-state levels of exon–exon junction reads are proportional to synthesis over degradation rates and the exon–intron junction reads have steady-state values proportional to synthesis over splicing rates (Fig. 1). Hence, the ratio of exon–exon over exon–intron reads equals to splicing rate over decay rate. Although this ratio allows comparing the splicing rate of different junctions within one gene (under the reasonable assumption that all exon–exon junctions have a similar decay rate) a straightforward comparison of splicing between different genes is biased due to the generally varying decay rates among genes. Similarly, the ratio of exonic over intronic reads15 not only depends on how fast the precursor RNA is processed, but also on both the stability the spliced out introns and on the stability of the mature RNA.16 Altogether, the usage of steady-state RNA-seq to infer rates or variations of rates is intrinsically limited.
Figure 1.
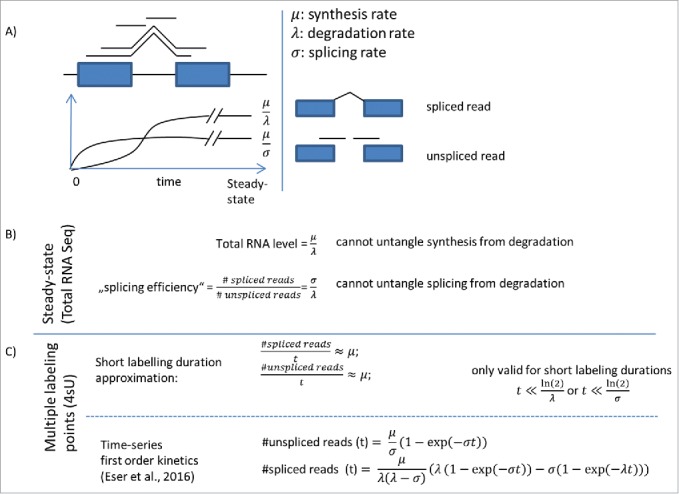
RNA-seq versus metabolically labeling approaches. (A) Sketch of a concentration curve of labeled unspliced and spliced RNA after labeling based on classic first-order kinetics.13 (B) Steady-state data alone is not enough to untangle synthesis, degradation, and splicing rate. In particular, the ratio of spliced over unspliced reads, often taken as a proxy for splicing efficiency depends on degradation rate. (C) For labeling duration much shorter than degradation or splicing time, unspliced reads reflect the RNA synthesis (top). Labeling time series can be analyzed using kinetics model, for instance first-order kinetics (Eser 2016). This time series allows analyzing splice junction formation kinetics (bottom).
Estimation of rates using RNA metabolic labeling
To circumvent these limitations, alternative protocols are used that directly probe the kinetics. One class of approach is based on transcriptional arrest.17-19 However, great care should be taken when using transcriptional arrest, because arresting transcription is a major stress on cells and because transcription and degradation are globally coupled.8 Alternatively, Dölken et al.20 developed a technique based on metabolic labeling which has been successfully applied to many eukaryotes including yeast, fly, mouse, and human.3,12,21 The key idea is to use a modified nucleotide, usually 4-thiouridine (4sU), to tag newly synthesized RNA starting from one point in time. Labeling durations as short as 90 sec have been applied allowing the investigation of very rapid events such as the degradation of short-lived non-coding RNAs and splicing.22 The approach is often applied with a single labeling duration. However, a whole labeling time course of a steady-state cell population can be also used giving more time points to investigate the kinetics and fit the rates.13,22 Studying time-dependence of the rates upon a stress11 or during the cell cycle9 require mathematical modeling of the time dependency of the kinetic parameters.
To accurately determine the rates, one further has to model the underlying read generating process in great detail. Even though the absolute amount of labeled RNA of any transcript increases with labeling duration, the number of sequenced reads of short-lived transcripts decreases, as short-lived RNAs represent a decreasing fraction of all purified RNAs. Hence, to fit a kinetic model to read counts and to obtain absolute measures of half-life, a normalization factor that correspond to the overall amount of labeled RNA in the samples prior to purification must be estimated. This has been either done using spike-ins23 or by fitting a global model jointly across all genes.3,13,24 Moreover, it is important to control for cross-contamination with unlabeled RNA,3,13 especially for short durations where the proportion of unlabeled RNA in the sample can be so large that small cross-contamination can lead to a large fraction of reads in the purified samples. Also, inaccurate determination of the feature length (exon, intron, junction) as well as GC-content can introduce artificial correlations between the kinetic rates and, for example, length of a gene. Hence, correlations between length (transcript, 5′-UTR, 3′-UTR) and any kinetic parameter should be considered with great care. Estimations of the synthesis rate are particularly sensitive to these biases compared with splicing and decay. We observed great bias for short genes in fission yeast (mostly non-coding).13 In general, long observation periods are desirable. However, 4sU induced inhibition of RNA translation for long labeling periods give an upper limit of 1 h–2 h of labeling time.25
Defining rates of individual phosphodiester bonds
The notion of RNA synthesis rates, degradation rates, and the splicing rates of introns lead to practical and conceptual difficulties due to the interleaved nature of transcription. A single gene locus can give rise to many splice isoforms simultaneously as well as many overlapping non-coding RNAs. It is not possible to unambiguously allocate each read to either of these transcripts. Statistical models that try to untangle the concentration of overlapping isoforms exist, but lead to highly coupled estimates. Hence, delineating the RNA metabolism rates of overlapping transcripts is extremely difficult. The issue is not only technical but also biological. Adding to splicing variation, the widespread variations in 5′ and 3′ end imply that there is an extremely large number of unique RNA sequences that are transcribed from one gene.26 Variations in the exact transcription start sites may affect synthesis rates, and variations in the 3′ end can affect RNA stability by adding or removing cis-regulatory motifs with role in RNA degradation. Therefore, similar isoforms may have significantly different RNA metabolism rates.
Hence, although summary statistics at the level of a whole gene are certainly useful simplifications, we argue that a definition of rates for individual bonds helps clarifying the notions and devising clear mathematical models. We distinguish five types of phosphodiester bonds: exonic, exon–intron, intronic, intron–exon, and exon–exon bonds (Fig. 2). The production rate of the four first types is equal to the synthesis rate or transcription rate of these bonds. In steady-state culture conditions, the junction formation rate of the exon–exon bonds also equals the transcription rate, assuming no loss during RNA processing. However, tracking exon–exon bonds during a labeling time course will allow studying splicing kinetics and, in particular, the delay between transcription and junction formation. The degradation rates of exon–intron bonds are the cleavage rate of the donor sites, and the degradation rates of the intron–exon bonds are the cleavage rate of the acceptor sites. For one single intron, those cleavage rates do not need to be equal to each other because of a likely longer half-life of the donor site bonds, since they are transcribed before the acceptor sites, and also because alternative splicing imply that one donor site can correspond to multiple acceptor sites. The synthesis rates of these bonds are the sum of the synthesis rates of all transcripts (including all alternatively spliced isoforms) containing them. In contrast, the degradation rates of single bonds do not trivially relate to the degradation rate of the RNA species (including all alternatively spliced isoforms) that contain them. They are some combination of those, in a way that depend on degradation kinetics and amount of each RNA species. Nonetheless, both the synthesis rates and the degradation rates of individual bonds at steady state are well-defined quantities.
Figure 2.
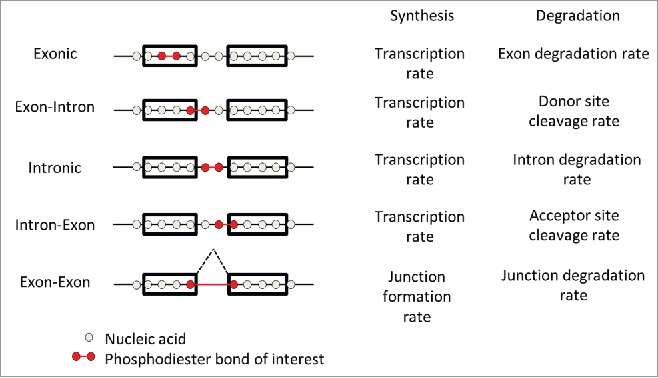
Synthesis and degradation rates of individual phosphodiester bonds. To simplify the figure, we only consider a gene without overlapping transcripts. However, the definitions apply to configurations with overlapping transcripts and can also be defined for non-canonical splicing (cryptic-, circular-, and trans-splicing). The interpretation of the synthesis and degradation rate of each bond is given to the right.
One should note that hand in hand with the definition of rates described above, a careful annotation of exon and intron boundaries is required. To this end, it is important to not rely on annotations but to adopt a data-driven approach with read mapping algorithms allowing de novo identification of splice sites. It would also be interesting to address non-canonical types of splicing (circular-, trans-splicing) which could be revealed by such de novo identifications, possibly with adapted protocol. Also for these non-canonical cases, the definition of metabolism rates of phosphodiester bonds applies and will be a useful concept.
Transient-transcriptome profiling
We recently contributed to the development of a protocol, transient-transcriptome profiling,23 which will be instrumental for estimating metabolism rates of individual bonds as it addresses an important positional bias that the standard 4sU-seq protocol has. Standard 4sU-seq leads to an overrepresentation of reads from the 5′ ends of genes due to labeling of already on-going transcription products. This effect is particular important in higher eukaryotes, where the polymerase takes a significantly longer time to transcribe a gene in its full extent (typically 20 min in human), than the labeling durations required for studying rapid events such as splicing or degradation of short lived RNAs. One consequence is that parts of the reads sequenced with 4sU-seq protocols are actually not labeled, and that these tend to be more present in the 5′ end of genes (Fig. 3). Hence, synthesis and degradation rates estimates based on standard 4sU-seq protocols are biased by gene length.23 Another effect is due to co-transcriptional splicing. In the standard 4sU-seq protocol, the pulled down RNAs may have introns in their 5′ end already spliced out prior to the labeling. Consequently, there is a relative higher amount of exon–exon reads in the 5′ end of genes and thus the introns toward the 5′ end of genes appear to be spliced faster. Whether the first introns are generally spliced faster than other introns, as single-gene microscopy has indicated, is difficult to assess form standard 4sU-seq data. In contrast, we expect data obtained by TT-seq23 to show a more uniform and less biased coverage of exon–exon reads.
Figure 3.
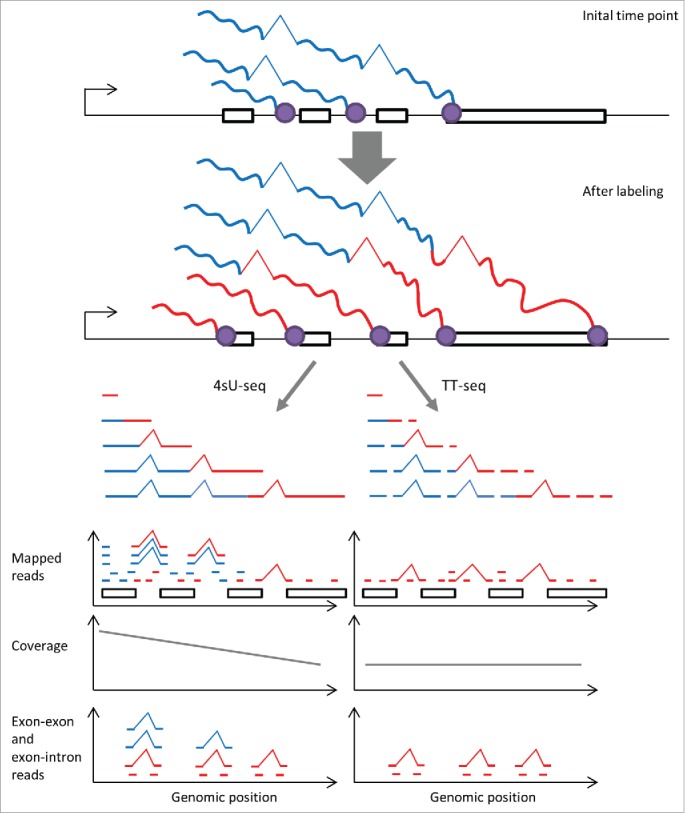
TT-seq enables uniform mapping of the human transient transcriptome. When labeling with 4sU for a shorter duration than the time required for polymerase to complete transcription, only short part (red) of a nascent transcript gets labeled (top). In standard 4sU-seq, the complete nascent transcript is purified and sequenced. This leads to higher coverage in the 5′ end of genes, and due to co-transcriptional splicing, to high ratio of exon–exon reads over exon–intron reads toward the genes 5′ end (bottom left). In contrast TT-seq, whose fragmentation steps allows sequencing only the labeled part of nascent transcripts, leads to a more uniform coverage along transcripts and a more uniform distribution of the ratio of exon–exon over exon–intron reads (bottom right). Having a uniform coverage is important to study the metabolism of individual bonds in an unbiased fashion.
In conclusion, we predict that TT-seq will become an important protocol to study splicing kinetics genome-wide, as it alleviates positional biases that former labeling protocols entail. To analyze RNA metabolism with TT-seq data, we suggest a switch from a gene-level to single-bond level focus.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Livia Caizzi, Björn Schwalb, and Patrick Cramer for feedback on the manuscript.
Funding
LW was supported by the Juniorverbund in der Systemmedizin “mitOmics” grant FKZ 01ZX1405A.
References
- [1].Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature 2011; 473:337-342; PMID: 21593866; http://dx.doi.org/25497548 10.1038/nature10098 [DOI] [PubMed] [Google Scholar]
- [2].Marguerat S, Lawler K, Brazma A, Bähler J. Contributions of transcription and mRNA decay to gene expression dynamics of fission yeast in response to oxidative stress. RNA Biol 2014; 11:12; PMID: 25007214; http://dx.doi.org/25497548 10.4161/rna.29196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Rabani M, Raychowdhury R, Jovanovic M, Rooney M, Stumpo DJ, Pauli A, Hacohen N, Schier AF, Blackshear PJ, Friedman N et al.. High-resolution sequencing and modeling identifies distinct dynamic RNA regulatory strategies. Cell 2014; 159:1698-1710; PMID:25497548; http://dx.doi.org/ 10.1016/j.cell.2014.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Raghavan A, Ogilvie RL, Reilly C, Abelson ML, Vasdewani J, Krathwohl M, Bohjanen PR. Genome-wide analysis of mRNA decay in resting and activated primary human T lymphocytes. Nucleic Acids Res 2002; 30:5529-5538; PMID:12490721; http://dx.doi.org/ 10.1093/nar/gkf682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Braun KA, Young ET. Coupling mRNA synthesis and decay. Mol Cell Biol 2014; 34:4078-4087; PMID:25154419; http://dx.doi.org/ 10.1128/MCB.00535-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Munchel SE, Shultzaberger RK, Takizawa N, Weis K. Dynamic profiling of mRNA turnover reveals gene-specific and system-wide regulation of mRNA decay. Mol Biol Cell 2011; 22:2787-2795; PMID:21680716; http://dx.doi.org/ 10.1091/mbc.E11-01-0028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pai AA, Cain CE, Mizrahi-Man O, De Leon S, Lewellen N, Veyrieras J-B, Degner JF, Gaffney DJ, Pickrell JK, Stephens M et al.. The contribution of RNA decay quantitative trait loci to inter-individual variation in steady-state gene expression levels. PLoS Genet 2012; 8:e1003000; PMID:23071454; http://dx.doi.org/ 10.1371/journal.pgen.1003000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sun M, Schwalb B, Schulz D, Pirkl N, Etzold S, Larivière L, Maier KC, Seizl M, Tresch A, Cramer P. Comparative dynamic transcriptome analysis (cDTA) reveals mutual feedback between mRNA synthesis and degradation. Genome Res 2012; 22:1350-1359; PMID:22466169; http://dx.doi.org/ 10.1101/gr.130161.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Eser P, Demel C, Maier KC, Schwalb B, Pirkl N, Martin DE, Cramer P, Tresch A. Periodic mRNA synthesis and degradation co-operate during cell cycle gene expression. Mol Syst Biol 2014; 10:717; PMID:24489117; http://dx.doi.org/ 10.1002/msb.134886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zeisel A, Köstler WJ, Molotski N, Tsai JM, Krauthgamer R, Jacob-Hirsch J, Rechavi G, Soen Y, Jung S, Yarden Y et al.. Coupled pre-mRNA and mRNA dynamics unveil operational strategies underlying transcriptional responses to stimuli. Mol Syst Biol 2011; 7:529; PMID:21915116; http://dx.doi.org/ 10.1038/msb.2011.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N et al.. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol 2011; 29:436-442; PMID:21516085; http://dx.doi.org/ 10.1038/nbt.1861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Windhager L, Bonfert T, Burger K, Ruzsics Z, Krebs S, Kaufmann S, Malterer G, L'hernault A, Schilhabel M, Schreiber S et al.. Ultrashort and progressive 4sU-tagging reveals key characteristics of RNA processing at nucleotide resolution. Genome Res 2012; 22:2031-2042; PMID:22539649; http://dx.doi.org/ 10.1101/gr.131847.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Eser P, Wachutka L, Maier KC, Demel C, Boroni M, Iyer S, Cramer P, Gagneur J. Determinants of RNA metabolism in the Schizosaccharomyces pombe genome. Mol Syst Biol 2016; 12:857; PMID:26883383; http://dx.doi.org/ 10.15252/msb.20156526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bähler J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 2008; 453:1239-1243; PMID:18488015; http://dx.doi.org/ 10.1038/nature07002 [DOI] [PubMed] [Google Scholar]
- [15].Khodor YL, Rodriguez J, Abruzzi KC, Tang C-HA, Marr MT, Rosbash M. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev 2011; 25:2502-2512; PMID:22156210; http://dx.doi.org/ 10.1101/gad.178962.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gaidatzis D, Burger L, Florescu M, Stadler MB. Analysis of intronic and exonic reads in RNA-seq data characterizes transcriptional and post-transcriptional regulation. Nat Biotech 2015; 33:722-729; PMID: 26098447; http://dx.doi.org/9845373 10.1038/nbt.3269 [DOI] [PubMed] [Google Scholar]
- [17].Holstege FC, Jennings EG, Wyrick JJ, Lee TI, Hengartner CJ, Green MR, Golub TR, Lander ES, Young RA. Dissecting the regulatory circuitry of a eukaryotic genome. Cell 1998; 95:717-728; PMID:9845373; http://dx.doi.org/ 10.1016/S0092-8674(00)81641-4 [DOI] [PubMed] [Google Scholar]
- [18].Wang Y, Liu CL, Storey JD, Tibshirani RJ, Herschlag D, Brown PO. Precision and functional specificity in mRNA decay. Proc Natl Acad Sci U S A 2002; 99:5860-5865; PMID:11972065; http://dx.doi.org/ 10.1073/pnas.092538799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Grigull J, Mnaimneh S, Pootoolal J, Robinson MD, Hughes TR. Genome-wide analysis of mRNA stability using transcription inhibitors and microarrays reveals posttranscriptional control of ribosome biogenesis factors. Mol Cell Biol 2004; 24:5534-5547; PMID:15169913; http://dx.doi.org/ 10.1128/MCB.24.12.5534-5547.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Dölken L, Ruzsics Z, Rädle B, Mages RG, Hoffmann R, Dickinson P, Forster T. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA 2008; 14:1959-1972; PMID:18658122; http://dx.doi.org/ 10.1261/rna.1136108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Miller C, Schwalb B, Maier K, Schulz D, Dümcke S, Zacher B, Mayer A, Sydow J, Marcinowski L, Dölken L et al.. Dynamic transcriptome analysis measures rates of mRNA synthesis and decay in yeast. Mol Syst Biol 2011; 7:458; PMID:21206491; http://dx.doi.org/ 10.1038/msb.2010.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Barrass JD, Reid JEA, Huang Y, Hector RD, Sanguinetti G, Beggs JD, Granneman S. Transcriptome-wide RNA processing kinetics revealed using extremely short 4tU labeling. Genome Biol 2015; 16:282; PMID:26679539; http://dx.doi.org/ 10.1186/s13059-015-0848-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schwalb B, Michel M, Zacher B, Frühauf K, Demel C, Tresch A, Gagneur J, Cramer P. TT-seq maps the human transient transcriptome. Science 2016; 352:1225-1228; PMID:27257258; http://dx.doi.org/ 10.1126/science.aad9841 [DOI] [PubMed] [Google Scholar]
- [24].de Pretis S, Kress T, Morelli MJ, Melloni GEM, Riva L, Amati B, Pelizzola M, De Pretis S, Kress T, Morelli MJ et al.. INSPEcT: a computational tool to infer mRNA synthesis, processing and degradation dynamics from RNA- and 4sU-seq time course experiments. Bioinformatics 2015; 31:1-7; PMID:25189783; http://dx.doi.org/ 10.1093/bioinformatics/btv288 [DOI] [PubMed] [Google Scholar]
- [25].Burger K, Mühl B, Kellner M, Rohrmoser M, Gruber-Eber A, Windhager L, Friedel CC, Dölken L, Eick D. 4-thiouridine inhibits rRNA synthesis and causes a nucleolar stress response. RNA Biol 2013; 10:1623-1630; PMID:24025460; http://dx.doi.org/ 10.4161/rna.26214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pelechano V, Wei W, Steinmetz LM. Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 2013; 497:127-131; PMID:23615609; http://dx.doi.org/ 10.1038/nature12121 [DOI] [PMC free article] [PubMed] [Google Scholar]