

Endocrine therapy is an integral component of systemic therapy for metastatic breast cancer. In addition, targeted therapies are emerging as useful treatments. This article reviews current endocrine treatments of metastatic breast cancer, as well as studies of combination treatments and targeted therapies that interfere with cellular proliferation pathways, as a means of overcoming resistance.
Keywords: Metastatic breast cancer, Endocrine therapy, Aromatase inhibitors, Estrogen receptor‐positive, Selective estrogen‐receptor degrader, Selective estrogen receptor modulators
Abstract
Metastatic breast cancer (MBC) results in substantial morbidity and mortality for women afflicted with this disease. A majority of MBCs are hormone‐responsive and estrogen receptor‐positive, making endocrine therapy (ET) an integral component of systemic therapy. With a primary goal of minimizing the effects of estrogen on hormone‐responsive MBC, ETs are among the first targeted treatments that aim to inhibit the influence of estrogen receptor activation on tumor proliferation. Several biochemical mechanisms have been the focus of drug development for treatment, including selective estrogen‐receptor modulation, aromatase inhibition, and selective estrogen‐receptor degradation. Treatments that exploit these mechanisms have improved survival and quality of life for women with MBC. However, in many cases, resistance to ET limits their effectiveness. Elucidation of the complex cellular signal cascades involved in the development of acquired resistance to ET and the interrelationship of growth factor signaling and estrogen responsiveness have characterized components of these pathways as attractive targets for drug development. Based on these insights and with the aim of overcoming hormone resistance, targeted therapies are emerging as useful treatments for MBC. This article reviews current endocrine treatments of MBC as well as recent and ongoing study of combination treatments and targeted therapies that interfere with cellular proliferation pathways as means of overcoming resistance. The Oncologist 2017;22:507–517
Implications for Practice.
This review provides medical oncologists and other oncology health care providers with a current understanding of the rationale for endocrine therapy in estrogen receptor‐positive metastatic breast cancer and the efficacy and safety profile of available treatment options. Additionally, current concepts regarding the development of treatment resistance and the treatment strategies for overcoming resistance are discussed. Enhancing the current information and the understanding of these topics will assist clinicians in evaluating optimal treatment options for their patients.
摘要
转移性乳腺癌(MBC)在女性人群中的发病率较高, 大量患者因此死亡。MBC以激素反应性和雌激素受体阳性类型居多, 这就使得内分泌治疗(ET)成为全身治疗中不可或缺的一部分。激素反应性MBC的首要治疗目标是尽可能降低雌激素对肿瘤的影响, 在这一背景下, ET成为首批旨在抑制雌激素受体激活对肿瘤增殖影响的靶向治疗方法之一。治疗药物的开发热点集中于数种生化机制, 包括选择性调节雌激素受体、抑制芳香化酶和选择性降解雌激素受体。基于以上机制的治疗方法改善了MBC女性患者的生存期和生活质量。但在诸多病例中, ET耐药限制了治疗的有效性。现已阐明ET获得性耐药形成过程中涉及的复杂细胞信号级联反应以及生长因子信号传导与雌激素反应性之间的相互关系, 由此确定以上通路的组成因素是药物开发的理想靶点。基于以上深入了解, 兼以克服激素耐药为目的, 靶向治疗正逐渐成为MBC的有效治疗方法。本文综述了MBC的现有内分泌治疗, 以及近期完成和正在进行的通过干扰细胞增殖通路来克服耐药性的联合治疗和靶向治疗研究。The Oncologist 2017;22:507‐517
对临床实践的提示:本综述为肿瘤内科医生和其他肿瘤学卫生保健人员提供了以下方面的现有信息:内分泌治疗用于雌激素受体阳性转移性乳腺癌的依据;现有治疗方案的疗效和安全性特征。此外, 还讨论了关于耐药性形成和克服耐药性的治疗策略的现有概念。强化现有信息和对以上主题的理解将有助于临床医生评价患者的最佳治疗选择。
Introduction
Breast cancer (BC) is the most common malignancy and the second leading cause of cancer‐related death among women in the U.S., with approximately 250,000 new cases diagnosed and over 40,000 deaths occurring in 2016 [1]. Based on data from 2006 to 2012, the 5‐year relative survival of individuals with BC of all stages is 89.7% [2]. Although the survival of patients with early BC (defined as cancers that may have spread to nearby lymph nodes but not to distant parts of the body, i.e., stages I, IIA, IIB, and IIIA) is favorable, the survival of patients with advanced metastatic BC (MBC) is poor; the 5‐year relative survival is 100% for stage I, 93% for stage II, 72% for stage III, and 22% for stage IV [3], [4]. Overall, for those with MBC, 5‐year survival is approximately 26% [2]. This review will focus on the use of endocrine therapy (ET) in the management of postmenopausal estrogen receptor (ER)‐positive (ER+) MBC.
Approximately 75% of patients with BC are ER+ [5], [6]. These tumors are associated with better survival than those with low or no ER expression. In two large studies of women at varied stages of BC, 5‐year survival is approximately 10%–15% better for women with ER+ BC than for those with ER‐negative (ER−) BC, ranging between approximately 85%–95% for ER+ and 69%–81% for ER− BC [7], [8]. Estimates suggest that approximately 6% of newly diagnosed BC cases present with metastatic disease and that recurrence from early stage to distant sites occurs in 20%–50% of cases [9], [10]. A population‐based cohort study found that ER+ status was a significant predictor of improved survival in women with MBC [11].
Systemic pharmacotherapy, the mainstay for treating MBC, is aimed at preventing or slowing MBC progression and its related morbidity and maintaining quality of life (QOL) [12], [13]. Retrospective survival analyses suggest that improved systemic treatment options for MBC over the past several decades may be responsible for observed improvement in survival of patients with MBC [11], [14]. Such systemic treatments include chemotherapy, hormonal therapy, biological targeted therapy, and supportive therapy [12]. For patients with advanced MBC, the choice of therapy is based on considerations related to patient characteristics and comorbidities, disease status, prior treatments, and biological characteristics of the tumor [12], [13]. Among the important patient factors are age, menopausal status, performance status (e.g., general well‐being and performance of activities of daily living), and comorbidities, as well as psychological, socioeconomic, and logistical factors. Previous systemic therapy and response, disease‐free interval, potential impact on QOL, and whether the patient has a visceral crisis or is in need of rapid symptom control are factors that are also considered [15]. Recent scientific advances have established hormone‐receptor status and human epidermal growth factor 2 (HER2) status as important predictive markers for disease progression and treatment effectiveness [12], [16].
The main goals of therapy for MBC are palliation with improvement or maintenance of QOL and, potentially, extension of survival [15], [17]. Several population studies document improvement, albeit modest, in mortality rates over the past few decades in patients with MBC [11], [14], [18]. On the other hand, a recent review of data from studies by the Eastern Cooperative Oncology Group found improvement in post‐recurrence survival in the ER+ subgroup but no substantial improvement in overall survival (OS). These differences may be due to the impact of increased availability of effective treatments and improved sequencing of treatments in the population‐based analyses [19].
Cytotoxic chemotherapy and ET have been cornerstones in the management of MBC [16]. Anthracyclines and taxanes are commonly used chemotherapeutic agents in both the early and advanced disease settings [20]. However, they are associated with substantial toxicity, and, with their extensive use, resistance may be observed in individuals with MBC [21]. A variety of ET options are available, including oophorectomy, gonadotropin‐releasing hormone analogues, aromatase inhibitors (AIs), selective ER modulators (SERMS), and selective ER degraders (SERDs) [22], [23].
ET is an effective option for treating pre‐ or postmenopausal women with early‐stage BC or ER+ MBC in the absence of immediate, life‐threatening disease. In the setting of ER+ BC, the efficacy of ET is at least equal to chemotherapy, with a better tolerability profile [24]. Because many women are treated with ET before presenting with MBC, the selection of ETs in postmenopausal ER+ MBC will be influenced by prior exposure and previous outcomes of ET used in adjuvant treatment. With the emphasis on palliation and maintaining QOL for patients with MBC, clinical guidelines generally recommended that cytotoxic chemotherapy be reserved for patients with ER− MBC, those who are refractory to ET, or those who have life‐threatening complications [12].
Materials and Methods
A literature review of PubMed was performed to locate publications on treatment strategies in ER+ MBC related to ET and targeted therapies, particularly those reporting the findings of key clinical trials (i.e., randomized phase 2 and 3) that may influence clinical decision‐making. Manual search strategies were performed to identify relevant conference presentations of interest (i.e., the American Society of Clinical Oncology annual meetings, San Antonio Breast Cancer Symposium, American Association for Cancer Research). Ongoing clinical trials were identified by searching the U.S. National Institutes of Health database (www.clinicaltrials.gov).
Results
Mechanism of Action and Effects of ETs
ETs for ER+ MBC target either estrogen production or the ER system and bind to ER in tumor cells and other human tissues [25], [26]. SERMs, such as tamoxifen and toremifene, are cytostatic agents that competitively bind to ER in tumor cells and breast tissue, producing receptor dimerization and a nuclear complex that decreases DNA synthesis and inhibits estrogenic effects [27]. The downstream effects of the binding of SERMs to ER are tissue‐specific. These effects may also differ among specific agents, acting as antagonists in BC tissue and, alternatively, as partial agonists in some tissues, such as endometrium and bone. SERMs are orally administered, generally well tolerated, and protective for bone mineral density. Common adverse events (AEs) associated with anti‐estrogen therapy include vaginal bleeding (that is more likely attributable to the estrogen agonistic activity of antiestrogen therapy) and hot flashes. Serious AEs such as thromboembolic events have occurred. The incidence of any thromboembolic events in women with early BC receiving adjuvant tamoxifen is approximately 3%–4% [28], [29]. Tissue‐specific effects of tamoxifen on the uterus are associated with a low but significantly increased incidence of endometrial cancer of 2.20 per 1,000 women‐years compared with 0.71 for placebo [30], [31].
Other ETs include the third‐generation, oral AIs. Aromatase is a cytochrome P450 enzyme involved in the synthesis of estrogen. Therefore, AIs function as antiestrogens by decreasing the biosynthesis of estrogen from androgens, the primary estrogen biosynthesis pathway in postmenopausal women [32]. AIs are not beneficial for premenopausal women because the ovaries are the primary site of estrogen biosynthesis prior to menopause. There are two categories of AIs: the steroidal inhibitor exemestane and nonsteroidal inhibitors such as anastrozole and letrozole [32], [33]. Exemestane, a type 1 steroidal AI, binds irreversibly to aromatase, causing permanent inactivation of the enzyme even after the drug is cleared from circulation. Nonsteroidal (type II) AIs such as anastrozole and letrozole bind reversibly to aromatase, thereby inhibiting the synthesis of estrogen [32]. Clinical data pertaining to the effects of AIs on bone mineral density demonstrate that both type I and type II AIs may reversibly increase bone resorption; therefore, these agents may increase the risk of bone fractures [34]. Other potential clinically significant AEs of these agents may include dyslipidemia and joint pain/stiffness [35].
SERDs demonstrate different structure, pharmacologic properties, and molecular activity in comparison with SERMs. In contrast, SERDs are pure ER antagonists, exhibiting exclusively anti‐estrogenic effects [36], by which they block and downregulate ER activity, accelerate degradation of the ER, and inhibit the proliferation of estrogen‐dependent breast tumor cells [36], [37].
Fulvestrant is a SERD approved for treatment of hormone receptor (HR)‐positive (HR+) MBC in postmenopausal women with disease progression following ET. It is similar to tamoxifen in that fulvestrant binds competitively to the ER but with a higher affinity (IC50 0.89 versus .025) [38], [39], [40]. However, in contrast to tamoxifen's partial agonist activity, fulvestrant blocks estrogen‐sensitive gene transcription, resulting in no known agonist activity. In addition, fulvestrant inhibits ER dimerization and translocation to the nucleus and accelerates ER degradation, resulting in complete suppression of estrogenic effects on breast tissue [41], [42]. Common AEs associated with fulvestrant include menopause‐like symptoms such as hot flashes [43].
Another therapeutic option for postmenopausal women with MBC is the semi‐synthetic progestin megestrol. Although its mechanism of action is not yet fully understood, proposed mechanisms include interaction with the steroid (progesterone, glucocorticoid, and androgen) receptors, reduced cellular estrogen uptake, and growth factor interactions, as well as suppression of adrenal steroid production and ovarian secretion of androgens [44]. As described below, the use of megestrol for treatment of ER+ MBC has decreased with the discovery of more effective and tolerable treatments. However, it continues to be a second‐ or third‐line hormonal treatment option for patients who have relapsed on SERM and AI agents [45]. Side effects of megestrol are related to its antiestrogenic and antiandrogenic effects and include weight gain, edema, and breakthrough menstrual bleeding. Potentially serious effects include thromboembolic events [46].
Lastly, other available therapies for postmenopausal women with ER+ MBC include high‐dose estrogen and androgens [12], [47], [48], [49], [50]. Androgenic agents such as nandrolone decanoate have been used as third‐line agents [45], and fluoxymesterone is an option for endocrine‐resistant disease [12], [51], [52]. High‐dose ET (diethylstilbestrol or ethinylestradiol) may represent an option as a salvage treatment for postmenopausal women with late‐stage ER+ MBC after resistance to AI therapy [12], [52].
Clinical Efficacy of ETs
Tamoxifen has been an important ET since seminal studies demonstrated its activity against advanced MBC [53]. Over 3 decades of clinical studies, tamoxifen has demonstrated efficacy and a favorable toxicity profile compared with chemotherapy as first‐line treatment of ER+ MBC. A systematic review of 86 clinical trials found that patients treated with tamoxifen had an objective response rate (ORR = complete response [CR] and partial response [PR]) of 34%; 19% of patients in these studies achieved stable disease (SD) for at least 6 months [54]. Toremifene, another SERM indicated for treatment of postmenopausal women with ER+ MBC, is considered equivalent to tamoxifen in terms of both efficacy and safety [55].
An overview of available ET for ER+ MBC is provided in Table 1 [56–68]. In early studies of second‐line treatment, AIs were superior to megestrol in time to progression (TTP) or other measures of efficacy, with some studies also demonstrating significant improvements in survival. In the last decade, AIs have mostly replaced earlier treatments as first‐ and second‐line treatment in postmenopausal women with advanced MBC [69], [70], [71].
Table 1. Clinical trials of endocrine treatment as monotherapy or in combination with other endocrine therapies for ER+ MBC.
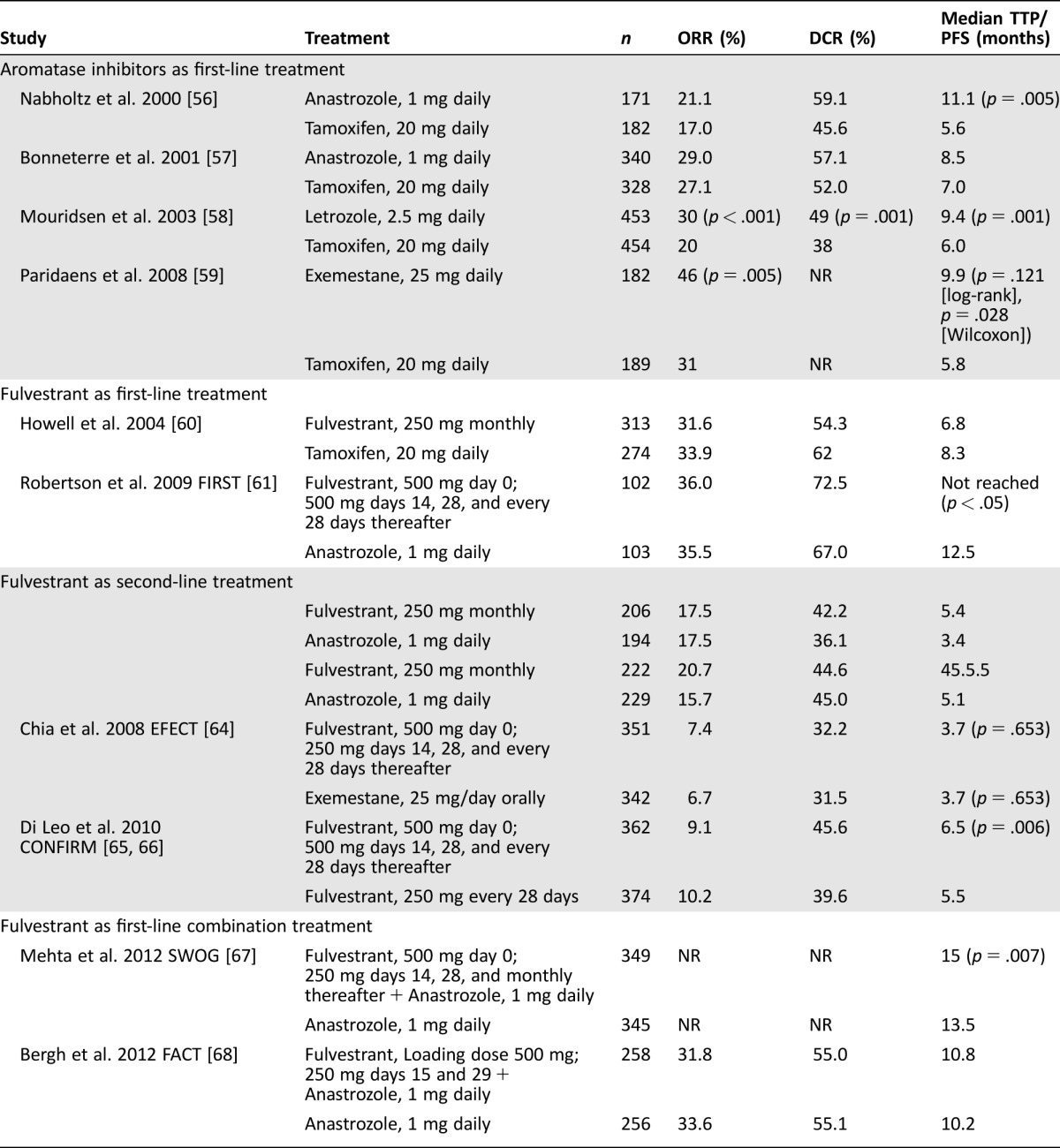
AIs have demonstrated equivalent or superior response compared with tamoxifen as first‐line treatment (Table 1) [56], [57], [58], [59]. Although Bonneterre and colleagues did not report superiority of 1 mg of anastrozole daily compared with 20 mg of tamoxifen daily, fewer than 45% of patients enrolled in TARGET were confirmed to be ER+ [57]. In contrast, Nabholtz and colleagues did demonstrate superiority of anastrozole versus tamoxifen in a cohort that was 85.7% ER+ [56]. Ferretti and colleagues examined 6 phase 3 trials involving 2,787 women treated with AIs versus tamoxifen. They confirmed a significant advantage in ORR, TTP, and disease control rate (DCR = CR + PR + SD), but no difference was found in OS [72]. Tamoxifen was associated with significantly more thromboembolic events and vaginal bleeding than the AIs. Hot flashes, vomiting, and musculoskeletal pain were slightly more frequent with AIs.
Limited data on tamoxifen as second‐line treatment after failure on AIs and progression of MBC suggest clinical benefit in almost 50% of patients, but less than 10% achieved an objective response [73]. Although the implications of the pharmacodynamic differences between type I and type II AIs have not been fully elucidated, study data suggest that sequential administration after initial treatment failure may result in disease control. Among 241 patients who progressed on nonsteroidal AIs, exemestane resulted in clinical CR in 1.2% of patients and PR in 5.4% (ORR 6.6%); SD for at least 6 months was seen in 41.9% of patients, and the median TTP was 14.7 months [74]. In another small exploratory study, exemestane demonstrated clinical efficacy after relapse on nonsteroidal AIs and, likewise, nonsteroidal AIs also exhibited efficacy after relapse on exemestane [75]. In the BOLERO‐2 trial, among 239 patients treated with placebo plus exemestane, ORR was 2.1% by central assessment, and the median progression‐free survival (PFS) was 4.1 months [76].
Although the implications of the pharmacodynamic differences between type I and type II AIs have not been fully elucidated, study data suggest that sequential administration after initial treatment failure may result in disease control.
As outlined herein, much of the data on the efficacy and safety of fulvestrant is with the initially approved monthly dose of 250 mg as first‐ and second‐line treatment compared with SERMs and AIs (Table 1) [60], [62], [63]. Similar first‐line efficacy with fulvestrant was shown versus tamoxifen and anastrozole [60]. Two clinical trials and a subsequent survival analysis of these studies confirmed similar responses to fulvestrant and anastrozole in second‐line treatment of MBC [62], [63], [77].
In the CONFIRM trial, patients were randomized to receive 500 mg of fulvestrant on days 0, 14, 28, and every 28 days thereafter or 250 mg on days 0, 28, and every 28 days thereafter. The primary endpoint, PFS, was significantly longer for the 500 mg versus 250 mg group (median PFS 6.5 versus 5.5 months, respectively; hazard ratio [HR], 0.80; 95% confidence interval [CI], 0.68–0.94; p = .006). However, ORR and DCR were similar in both arms (ORR, 9.1% versus 10.2%; DCR, 45.6% versus 39.6%) [65]. A final data analysis demonstrated that median OS was 26.4 versus 22.3 months with 500 mg of fulvestrant versus 250 mg, respectively (HR, 0.81; 95% CI, 0.69–0.96; nominal p = .016). These data indicate that 500 mg of fulvestrant is associated with a clinically relevant 4.1‐month difference in median OS and 19% reduction in risk of death compared with250 mg of fulvestrant [66]. The safety profiles of fulvestrant doses were similar, and no new safety concerns were noted.
A dose of 500 mg of fulvestrant was approved in 2010 for treatment of ER+ MBC in postmenopausal women [78]. To assess fulvestrant efficacy in first‐line treatment, the FIRST trial compared 500 mg of fulvestrant with 1 mg of anastrozole as first‐line treatment in ER+ MBC [61]. Fulvestrant was at least as effective as anastrozole in terms of DCR and ORR but was associated with significantly longer TTP. In the OS analysis for FIRST, which was added to the protocol as an endpoint in an amendment after the original protocol was developed, treatment with 500 mg of fulvestrant resulted in a statistically significant OS benefit compared with anastrozole (median OS 54.1 versus 48.4 months, respectively; HR, 0.70; 95% CI, 0.50–0.98; p = .041). This benefit was observed across prespecified subgroups [79]. The phase 3 FALCON trial is being conducted in postmenopausal women with HR‐positive locally advanced BC or MBC who have not previously been treated with hormone therapy. The primary study endpoint is PFS, and the results are expected to be presented in 2016. [80]. The FALCON and FIRST studies differed because FALCON enrollment was limited to de novo patients.
Research continues to examine whether ET with more than one endocrine agent could improve responsiveness over single endocrine agents. Fulvestrant, administered alone or with anastrozole, has also been examined as second‐line treatment versus exemestane after relapse on nonsteroidal AIs. The SoFEA trial in postmenopausal women with HR‐positive BC who relapsed or progressed with locally advanced or metastatic disease on a nonsteroidal AI found no differences between fulvestrant (500 mg on day 1, followed by 250 mg on days 15 and 29, and then every 28 days) plus placebo versus fulvestrant plus anastrozole (1 mg per day), or between fulvestrant plus placebo versus exemestane (25 mg per day) in terms of DCR, ORR, and OS [81]. The FACT trial examined combination treatment of fulvestrant (500 mg on day 1 and 250 mg on days 15 and 29, and thereafter every 28 days) plus 1 mg per day of anastrozole compared with anastrozole alone as first‐line treatment after first relapse on nonsteroidal AIs. It also failed to demonstrate clinical benefit for combination therapy versus monotherapy [68]. The SWOG study used a similar dosing regimen but in patients who had no prior chemotherapy, hormonal therapy, or immunotherapy for metastatic disease. It demonstrated that combination therapy with fulvestrant and anastrozole significantly improved median PFS (15.0 versus 13.5 months; p = .007) and median OS (47.7 versus 41.3 months; p = .049) versus anastrozole alone [67]. Because of the difference in study populations, the percentage of ET‐naïve patients was disproportionately higher in SWOG (59.7%) versus FACT (34.4%), which may have contributed to the difference in the outcomes.
Disease Progression After ET
Evidence exists that ER+ MBC may either be unresponsive to ET (de novo resistance) or lose endocrine responsiveness by upregulating other signaling pathways involved in cell survival and proliferation (i.e., acquired endocrine resistance) [82]. Several mechanisms may be responsible for acquired endocrine resistance, including downregulation or loss of ER expression, ER mutations generating mutant ER isoforms, or altered expression or activity of ER coregulators [83], [84], [85]. Phosphorylation, methylation, ubiquitination, and additional posttranslational modifications of ER and its coregulators have been shown to influence ER activity and sensitivity to ET [84]. Preclinical data suggest that crosstalk between growth factor receptor and ER pathways may mediate the development of resistance to ET in ER+ MBC [86], [87]. Growth factor receptor pathways may act as ER‐independent drivers of tumor growth and survival, leading to the expression of ER‐target genes independently of estrogen binding to ER, therefore conferring resistance to ET [83], [84]. Epidermal growth factor receptor, HER2 epidermal growth factor receptor, and insulin growth factor receptor have been recognized as the most prominent factors contributing to endocrine resistance [84], [87]. As a result, many clinical strategies have focused on co‐targeting these pathways together with ER to overcome endocrine resistance. Upregulation of cell survival signaling, such as the phosphatidylinositide 3‐kinase (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway, or positive regulators of the cell cycle or anti‐apoptotic molecules and downregulation of negative regulators of the cell cycle or pro‐apoptotic molecules can also lead to endocrine resistance [84]. Activation of the PI3K/Akt/mTOR pathway is commonly found in BC [88]. Clinical strategies focusing on co‐targeting the mTOR pathways when added to ET in the setting of prior endocrine resistance result in increased PFS [89].
New Agents for ER+ MBC
Through mechanisms that may include preventing the development of resistance to endocrine treatment, the long‐term efficacy of hormone therapy may be increased when used in combination with novel agents. The mechanistic target of mTOR regulates cell growth, proliferation, and survival. When mTOR inhibitors are combined with AIs, they inhibit cell growth and induce apoptosis [90].
Currently, the only mTOR inhibitor approved for the treatment of postmenopausal women with advanced HR+ BC is everolimus in combination with exemestane after treatment failure with letrozole or anastrozole [89]. Clinical trials have reported efficacy and safety results in studies examining the effects of two mTOR inhibitors (everolimus and temsirolimus) combined with nonsteroidal and steroidal AIs compared with an AI or tamoxifen alone (Table 2) [76], [91], [92], [93], [94], [95], [96]. In the phase 3 HORIZON trial, the addition of temsirolimus to letrozole did not lead to improvement in PFS in AI‐naïve advanced BC. Grade 3 to 4 toxicities were more common in the temsirolimus arm versus the letrozole‐alone arm, including hyperglycemia (4% versus 1%) [96]. Results of the phase 3 BOLERO‐2 study showed that the addition of everolimus to exemestane resulted in significantly improved ORR and PFS [76], [94]. In the final analysis, the median PFS for the combination was 7.8 months versus 3.2 months for exemestane alone (HR, 0.45; 95% CI, 0.38–0.54; p < .001) based on investigator review and 11.0 months versus 4.1 months, respectively, for the combination versus exemestane alone based on central assessment (HR, 0.38; 95% CI, 0.31–0.48; p < .001) [76]. Final investigator‐assessed ORR were 12.6% (95% CI, 9.8–15.9) for the combination versus 1.7% (95% CI, 0.5–4.2) for exemestane alone (p < .001). Corresponding central assessment ORR were 12.6% (95% CI, 9.8–15.9) and 2.1% (95% CI, 0.7–4.8) [76]. Despite a clinically meaningful and statistically significant improvement in PFS, the primary endpoint, adding everolimus to exemestane did not confer a statistically significant improvement in OS [97].
Table 2. Clinical trials of CDK4/6 kinase inhibitors and mTOR inhibitors in combination with AIs for ER+ MBC.
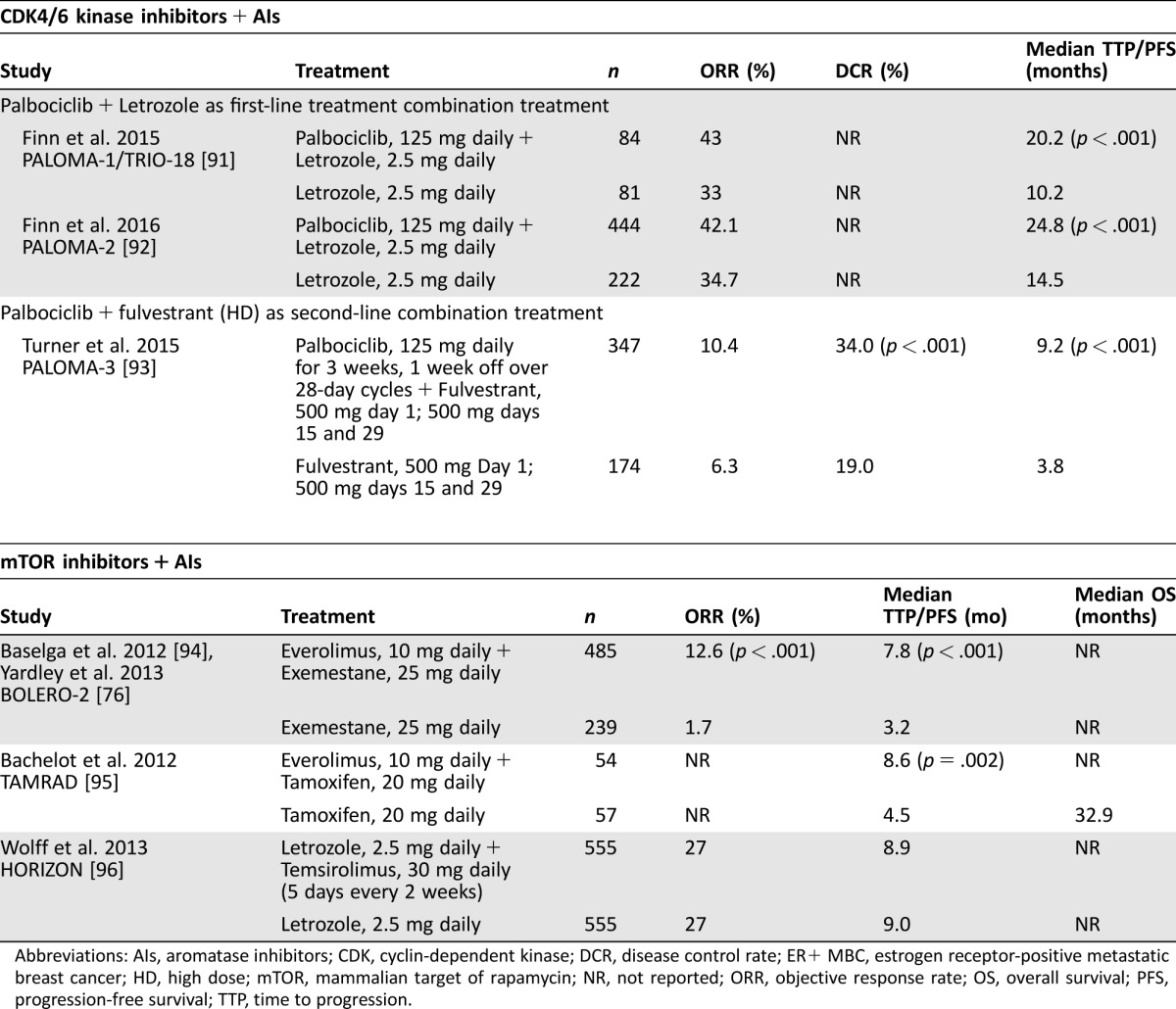
Abbreviations: AIs, aromatase inhibitors; CDK, cyclin‐dependent kinase; DCR, disease control rate; ER+ MBC, estrogen receptor‐positive metastatic breast cancer; HD, high dose; mTOR, mammalian target of rapamycin; NR, not reported; ORR, objective response rate; OS, overall survival; PFS, progression‐free survival; TTP, time to progression.
Approximately half of patients in the combination arm experienced a maximum 1/2 grade toxicity. In the combination arm, AEs of clinical interest included rash, stomatitis, noninfectious pneumonitis, metabolic abnormalities, and infections [76]. Rates of AEs leading to discontinuation that were suspected to be related to at least one study drug were 21.4% in the combination arm versus 3.4% in the exemestane arm. The two most common AEs leading to treatment discontinuation in the combination arm were pneumonitis (5.6%) and stomatitis (2.7%) versus increased gammaglutamyltransferase (1.7%) and increased aspartate aminotransferase (1.3%) in the exemestane arm [76]. The phase 2 TAMRAD trial also found that the addition of everolimus to tamoxifen resulted in improvement in 6‐month DCR (61% [95% CI, 47%–74%] vs 42% [95% CI, 29%–56%]; p = .045) [95]. The median TTP for the combination group was 8.6 months versus 4.5 months for tamoxifen alone, representing a 46% reduction in risk of progression in the combination group (HR, 0.54; 95% CI, 0.36–0.81; p = .002). At the time of analysis, median OS had not been reached by the tamoxifen plus everolimus group and was 32.9 months for tamoxifen alone (HR, 0.45; 95% CI, 0.24–0.81; p = .007) [95].
Cyclin‐dependent kinases (CDKs) are a subgroup of serine/threonine kinases that plays a key role in regulating cell cycle progression [98], [99], [100]. Several studies have identified alterations of cell cycle regulators in human BC and provided a rationale for the potential therapeutic role for CDK4/6 inhibition in breast tumors. A randomized phase 2 study (PALOMA‐1/TRIO‐18) of palbociclib, a highly selective inhibitor of CDK4/6 kinase, administered as 125 mg once daily for 3 weeks followed by 1 week off in 28‐day cycles with or without 2.5 mg of letrozole daily, as first‐line therapy for ER+, HER2‐negative (HER2−) MBC reported a statistically significant improvement in PFS (the primary endpoint) for combination treatment versus letrozole alone (20.2 versus 10.2 months; HR, 0.488; 95% CI, 0.319–0.748; p = .001) [91]. Based on these results, palbociclib in combination with letrozole was approved by the U.S. Food and Drug Administration (FDA) in 2015 for the treatment of postmenopausal women with ER+, HER2− advanced BC as initial endocrine‐based therapy for their metastatic disease [101], [102]. This indication is approved under accelerated approval based on PFS. Continued approval for this indication may be contingent upon verification and description of clinical benefit in a confirmatory trial.
PALOMA‐2 is a randomized double‐blind phase 3 confirmatory study of 666 postmenopausal patients without prior systemic therapy for MBC. As of February 2016, 331 PFS events were reported. Median PFS for the combination of palbociclib and letrozole was 24.8 versus 14.5 months for letrozole alone (HR, 0.58; 95% CI, 0.46–0.72; p < .001). The ORR was 42.1% for palbociclib and letrozole versus 34.7% for letrozole (p = .031; 55.3% versus 44.4% in patients with measurable disease [p = .013]). The clinical benefit rate was 84.9% versus 70.3% (p < .001) [92].
The final analysis of the phase 3 PALOMA‐3 study has recently been reported, although the OS follow‐up is ongoing [103]. PALOMA‐3 investigated the combination of 125 mg of palbociclib and 500 mg of fulvestrant compared with 500 mg of fulvestrant plus placebo in women with HR+, HER2− MBC (n = 521). Eligible patients of any menopausal status could have had one prior line of chemotherapy but could not have extensive symptomatic visceral metastasis or be at risk for life‐threatening complications. Disease relapse or progression had to occur after prior ET. In the palbociclib plus fulvestrant group, PFS was greater versus the fulvestrant plus placebo group (9.5 versus 4.6 months, respectively; HR, 0.46; 95% CI, 0.36–0.59; p < .001) [103]. Treatment response was not significantly impacted by PIC3CA mutation status or hormone‐receptor expression level. Grade 3/4 AEs were more common in the combined group (73% [251/345]) than in the fulvestrant plus placebo group (22% [38/172]). The most common grade 3/4 AEs for the palbociclib plus fulvestrant group and the fulvestrant plus placebo groups, respectively, were neutropenia (65% and 1%), anemia (3% and 2%), and leucopenia (28% and 1%) [103]. Patient‐reported QOL from this study indicated that overall global QOL scores were higher in the palbociclib plus fulvestrant group (66.1) than in the fulvestrant plus placebo group (63.0; p = .0313) [104]. Based on these results, palbociclib in combination with fulvestrant was approved by the U.S. FDA for the treatment of HR+, HER2− advanced BC or MBC in women with disease progression after ET [43], [101].
In regard to ribociclib and abemaciclib, preliminary clinical activity in BC was observed in early phase 1/2 studies. In a dose escalation phase 1 study conducted in 132 patients with advanced solid tumors and lymphomas, preliminary signs of clinical activity of ribociclib were observed, with 2 of 70 evaluable patients experiencing confirmed PRs, one of whom had PIK3CA‐mutation, ER+ BC [105]. The most common drug‐related grade 3/4 AEs were neutropenia (19%), lymphopenia (14%), and leukopenia (12%) [105]. Initial results of the ribociclib plus letrozole combination arm (n = 10) of a phase 1b/2 study in 17 women with ER+, HER2− advanced BC indicated preliminary antitumor activity of this combination: of 6/10 patients with known response, 1 patient had a PR, and 2 patients had SD. The most common drug‐related AEs (all grade/grade 3–4) were neutropenia (90%/50%) and nausea (40%/0%) [105], [106].
In a dose escalation phase 1 study conducted in patients with advanced solid tumors, clinical activity of abemaciclib was observed in MBC patients receiving abemaciclib monotherapy, with 11 of 47 patients (23%) experiencing confirmed PR, all of whom had HR+ BC. Among patients with HR+ MBC, the ORR was 31%, as well as in metastatic HR+ BC patients receiving abemaciclib plus fulvestrant, with 4 of 19 patients (21%) experiencing confirmed PR. The most common (all grade >20%) drug‐related AEs with abemaciclib monotherapy were diarrhea (63%), nausea (45%), fatigue (41%), vomiting (25%), leukopenia (25%), thrombocytopenia (23%), and neutropenia (23%). The most common (all grade >20%) drug‐related AEs with abemaciclib plus fulvestrant were diarrhea (79%), fatigue (68%), nausea (63%), neutropenia (42%), vomiting (42%), anorexia (32%), leukopenia (32%), and abdominal pain (21%) [107].
Numerous PI3K inhibitors are in varying stages of clinical testing in patients with ER+ BC. PIK3CA is mutated in about 35% of ER+ BC, but its prognostic significance is still unclear, as there was no difference in the frequency of PIK3CA mutations between ER+ and ER−, HER2+ tumors [108]. Activation of the PI3K/AKT pathway has been shown to confer resistance to antiestrogens [88], and PI3K inhibition has been shown to overcome endocrine resistance in preclinical testing [109], [110]. Emerging clinical data suggest preliminary clinical activity of PI3K inhibitors in combination with ET in patients with ER+ BC.
Three PI3K inhibitors have reached phase 3 testing in patients with advanced or metastatic HR+ BC. The pan‐PI3K inhibitor buparlisib (BKM120) is being investigated in combination with fulvestrant versus fulvestrant plus placebo in postmenopausal HR+, HER2− advanced BC or MBC refractory to AIs (BELLE‐2, NCT01610284) and those refractory to both AIs and mTOR inhibitors (BELLE‐3, NCT01633060). The findings from the BELLE‐2 study were recently reported. Treatment with buparlisib plus fulvestrant was associated with longer PFS than fulvestrant alone (6.9 versus 5.0 months; p < .001). Patients with ctDNA PIK3CA mutations had much better outcomes when treated with the combination therapy; PFS in the buparlisib plus fulvestrant group was 7 versus 3.2 months in the fulvestrant alone group (p < .001). Serious AEs were experienced by up to 26% of patients who received buparlisib [111]. The other two agents in phase 3 trials are taselisib and alpelisib, both of which selectively target the α‐isoform of class I PI3K. Both of these agents are being investigated in combination with fulvestrant versus fulvestrant plus placebo in postmenopausal ER+, HER2− advanced BC or MBC refractory to AIs (SANDPIPER, NCT02340221 and SOLAR‐1, NCT02437318).
In a phase 1b dose‐finding study of buparlisib in combination with letrozole in 51 postmenopausal women with ER+ MBC refractory to ET, patients were allocated to continuous or intermittent (5 on/2 off days) buparlisib treatment on an every‐4‐week schedule. Two of 20 patients in the continuous arm experienced objective responses (1 CR and 1 PR). None of the 31 patients in the intermittent arm demonstrated a response, but 14 (45%) had SD. Sixteen patients remained free of progression for at least 6 months (6 in the continuous arm; 10 in the intermittent arm). Of these, four were considered to have primary ET resistance [112]. In this study, the most common drug‐related AEs for continuous and intermittent treatment included transaminase elevation (75%/45%), hyperglycemia (70%/48%), fatigue (70%/42%), nausea (65%/26%), alkaline phosphatase increase (60%/19%), anemia (55%/19%), and depression (55%/32%) [112]. The preliminary results of another phase 1 dose escalation trial conducted in 31 patients with ER+ MBC demonstrated that buparlisib combined with fulvestrant has antitumor activity, with 7 of 22 evaluable patients experiencing a PR and 7 patients experiencing SD lasting at least 6 months [113]. In this study, most AEs were grade 1/2 except for grade 3 diarrhea, and the most common (>20%) grade ≥2 AEs were fatigue, alanine/aspartate aminotransferase elevations, and rash.
Initial results of a phase 1b dose escalation study of taselisib combined with letrozole in 28 patients with HR+ advanced BC indicated promising preliminary antitumor activity, with an ORR of 38% in patients with PIK3CA‐mutant tumors [114]. In this study, the most common drug‐related AEs (all grades; ≥10%) included diarrhea, nausea, stomatitis, fatigue, rash, decreased appetite, hyperglycemia, dysgeusia, mucosal inflammation, vomiting, muscle spasms, asthenia, dry mouth, dry skin, pruritus, and increased aspartate aminotransferase. Drug‐related grade 3/4 AEs occurring in more than one patient included diarrhea (14%), hyperglycemia (7%), and mucosal inflammation (7%).
In a dose escalation phase 1 study investigating alpelisib plus fulvestrant in 64 patients with ER+ locally advanced BC or MBC with documented progression on standard therapy, PRs were observed in 2 patients with PIK3CA‐altered tumors evaluable for response (2/33, 6%), but no PRs were observed in the 15 evaluable patients with PIK3CA‐wildtype tumors [115]. In this study, the most common (≥25%) drug‐related AEs (all grades/all doses) were hyperglycemia (41%), diarrhea (34%), nausea (30%), and vomiting (25%). The most common (>10%) drug‐related grade 3/4 AEs (all doses) were maculopapular rash (14%) and hyperglycemia (13%) [114]. Preliminary clinical results indicate that combinations of alpelisib plus an AI (letrozole or exemestane) are well tolerated and have clinical activity [116], [117]. Initial results of the alpelisib plus letrozole combination arm of a phase 1b study in 21 patients with ER+, HER2− advanced BC indicated antitumor activity of this combination, with 3 patients experiencing PR and 6 patients with SD. Among the 18 (56%) patients with PIK3CA mutation, 2 had a PR [116]. The side‐effect profile of this combination was similar to single‐agent alpelisib, with most common AEs being rash, hyperglycemia, nausea, fatigue, and diarrhea.
Other classes of agents that are being tested in ongoing clinical trials for women with ER+ BC include epigenetic modifiers (entinostat) and next‐generation SERDs (GDC‐0810, RAD1901, GDC‐0927, and AZD9496). Preclinical testing of the histone deacetylase inhibitor entinostat have shown that entinostat can restore sensitivity to AI therapy and that the combination of entinostat and letrozole more effectively inhibited tumor growth than either drug alone [118]. The phase 2 ENCORE 301 study investigating entinostat plus exemestane versus placebo plus exemestane in 130 postmenopausal women with locally recurrent or metastatic ER+ BC, progressing on treatment with AIs, reported significantly greater PFS in the combination arm than in the placebo arm (4.3 versus 2.3 months) [119]. A phase 3 trial of entinostat plus exemestane in patients with recurrent ER+ advanced BC or MBC following AI therapy is currently underway (NCT02115282).
The dual‐acting investigational next‐generation oral SERD GDC‐0810 targets the ER as an antagonist and causes degradation of the ER protein. In preclinical studies, GDC‐0810 was shown to induce tumor regressions in both tamoxifen‐sensitive and tamoxifen‐resistant tumor models in vivo [120]. In a phase 1 trial of GDC‐0810 conducted in 41 postmenopausal women with ER+, HER2− advanced BC or MBC, 13 of 31 (42%) patients achieved SD lasting for 6 months or longer [121]. In this study, the most common drug‐related AEs of any grade were diarrhea (63%), fatigue (46%), and nausea (44%). A phase 2 study is currently being conducted with GDC‐0810 in postmenopausal women with advanced BC or MBC previously treated with AIs, including tumors with ESR1 mutations (NCT01823835).
The next‐generation oral SERD/SERM RAD1901 is currently under evaluation in clinical studies [122], [123]. At high doses, RAD1901 acts as a SERD, binding to the ER and inducing degradation of the receptor. At low doses, RAD1901 acts as a SERM with estrogen‐like effects in certain tissues, which can both reduce hot flashes and protect against bone loss. In addition, RAD1901 is able to cross the blood‐brain barrier. Currently, a phase 1 study of RAD1901 is being conducted in ER+, HER2− postmenopausal women with advanced BC (NCT02338349). Two additional SERDs currently in phase 1 testing are GDC‐0927 (NCT02316509) and AZD9496 (NCT02248090) [124] in women with ER+ advanced BC.
Discussion
Treatment of ER+ MBC is palliative, not curative, so consideration of optimal treatment must balance the potential for extending survival with the effects of treatment on the patient's ability to maintain function and QOL. In addition to disease characteristics, improving treatment for ER+ MBC requires considering a wide range of factors, including treatment tolerability, patient preference, and patient QOL.
Treatment of ER+ MBC is palliative, not curative, so consideration of optimal treatment must balance the potential for extending survival with the effects of treatment on the patient's ability to maintain function and QOL.
Postmenopausal women with HR+, HER2− MBC who have received no prior ET may be treated with an AI, a SERM (e.g., tamoxifen, toremifene), or palbociclib plus letrozole as first‐line therapy. Women who have received prior ET may be treated on disease progression without symptomatic visceral disease with an ET not yet administered, preferentially an agent with a different mechanism of action compared with the previous agent, such as a SERD (e.g., fulvestrant), or with novel combinations such as exemestane plus everolimus or palbociclib plus fulvestrant, or with older agents such as megestrol, ethinylestradiol, and fluoxymesterone. Guidance on the sequencing of these therapies is limited, and factors to consider when choosing a therapy for these patients include performance status, comorbidities, and patient preference. Acquired resistance occurs in a substantial proportion of patients. New and emerging targeted therapies aimed at intracellular pathways of proliferation may contribute to sustained responses when combined with ETs.
Acknowledgments
This article was supported by AstraZeneca LP. Writing assistance was provided by Greg Tardie, Ph.D., Paula G. Davis, Ph.D., Elizabeth Goodwin, Ph.D., and the Lockwood Group. Editorial services were provided by Kathleen Major and Joan Hudson (funded by AstraZeneca LP).
Author Contributions
Conception/Design: Virginia G. Kaklamani, William J. Gradishar
Collection and/or assembly of data: Virginia G. Kaklamani, William J. Gradishar
Data analysis and interpretation: Virginia G. Kaklamani, William J. Gradishar
Manuscript writing: Virginia G. Kaklamani, William J. Gradishar
Final approval of manuscript: Virginia G. Kaklamani, William J. Gradishar
Disclosures
Virginia G. Kaklamani: Novartis Pharmaceuticals Corporation (H). The other author indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
References
- 1. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin 2016;66:7–30. [DOI] [PubMed] [Google Scholar]
- 2.National Cancer Institute . SEER Stat Fact Sheets: Breast Cancer. 2016. Available at http://seer.cancer.gov/statfacts/html/breast.html. Accessed March 7, 2016.
- 3.National Cancer Institute . NCI Dictionary of Cancer Terms. 2016. Available at http://www.cancer.gov/publications/dictionaries/cancer-terms?cdrid= 446564. Accessed March 7, 2016.
- 4. American Cancer Society. Breast Cancer Detailed Guide. 2016. Available at http://www.cancer.org/acs/groups/cid/documents/webcontent/003090-pdf.pdf. Accessed March 7, 2016.
- 5. Nadji M, Gomez‐Fernandez C, Ganjei‐Azar P et al. Immunohistochemistry of estrogen and progesterone receptors reconsidered: Experience with 5,993 breast cancers. Am J Clin Pathol 2005;123:21–27. [DOI] [PubMed] [Google Scholar]
- 6. Rugo HS. The breast cancer continuum in hormone‐receptor‐positive breast cancer in postmenopausal women: Evolving management options focusing on aromatase inhibitors. Ann Oncol 2008;19:16–27. [DOI] [PubMed] [Google Scholar]
- 7. Bentzon N, Düring M, Rasmussen BB et al. Prognostic effect of estrogen receptor status across age in primary breast cancer. Int J Cancer 2008;122:1089–1094. [DOI] [PubMed] [Google Scholar]
- 8. Yu KD, Wu J, Shen ZZ et al. Hazard of breast cancer‐specific mortality among women with estrogen receptor‐positive breast cancer after five years from diagnosis: Implication for extended endocrine therapy. J Clin Endocrinol Metab 2012;97:E2201–E2209. [DOI] [PubMed] [Google Scholar]
- 9. Brewster AM, Hortobagyi GN, Broglio KR et al. Residual risk of breast cancer recurrence 5 years after adjuvant therapy. J Natl Cancer Inst 2008;100:1179–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lu J, Steeg PS, Price JE et al. Breast cancer metastasis: Challenges and opportunities. Cancer Res 2009;69:4951–4953. [DOI] [PubMed] [Google Scholar]
- 11. Chia SK, Speers CH, D'yachkova Y et al. The impact of new chemotherapeutic and hormone agents on survival in a population‐based cohort of women with metastatic breast cancer. Cancer 2007;110:973–979. [DOI] [PubMed] [Google Scholar]
- 12.National Comprehensive Cancer Network . Clinical Practice Guidelines in Oncology. Breast Cancer. V1‐2016. 2016. Available at http://www.nccn.org/professionals/physician_gls/pdf/breast.pdf. Accessed March 7, 2016.
- 13. Bernard‐Marty C, Cardoso F, Piccart MJ. Facts and controversies in systemic treatment of metastatic breast cancer. The Oncologist 2004;9:617–632. [DOI] [PubMed] [Google Scholar]
- 14. Giordano SH, Buzdar AU, Smith TL et al. Is breast cancer survival improving? Cancer 2004;100:44–52. [DOI] [PubMed] [Google Scholar]
- 15. Chung CT, Carlson RW. Goals and objectives in the management of metastatic breast cancer. The Oncologist 2003;8:514–520. [DOI] [PubMed] [Google Scholar]
- 16. Glück S, Arteaga CL, Osborne CK. Optimizing chemotherapy‐free survival for the ER/HER2‐positive metastatic breast cancer patient. Clin Cancer Res 2011;17:5559–5561. [DOI] [PubMed] [Google Scholar]
- 17. Aranda S, Schofield P, Weih L et al. Meeting the support and information needs of women with advanced breast cancer: A randomised controlled trial. Br J Cancer 2006;95:667–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dawood S, Broglio K, Gonzalez‐Angulo AM et al. Trends in survival over the past two decades among white and black patients with newly diagnosed stage IV breast cancer. J Clin Oncol 2008;26:4891–4898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tevaarwerk AJ, Gray RJ, Schneider BP et al. Survival in patients with metastatic recurrent breast cancer after adjuvant chemotherapy: Little evidence of improvement over the past 30 years. Cancer 2013;119:1140–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pronzato P, Rondini M. First line chemotherapy of metastatic breast cancer. Ann Oncol 2006;17(suppl 5):v165–v168. [DOI] [PubMed] [Google Scholar]
- 21. von Minckwitz G, Martin M, Wilson G et al. Optimizing taxane use in MBC in the emerging era of targeted chemotherapy. Crit Rev Oncol Hematol 2013;85:315–331. [DOI] [PubMed] [Google Scholar]
- 22. Buzdar AU, Hortobagyi G. Update on endocrine therapy for breast cancer. Clin Cancer Res 1998;4:527–534. [PubMed] [Google Scholar]
- 23. Lumachi F, Santeufemia DA, Basso SM. Current medical treatment of estrogen receptor‐positive breast cancer. World J Biol Chem 2015;6:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Colleoni M, Gelber S, Coates AS et al. Influence of endocrine‐related factors on response to perioperative chemotherapy for patients with node‐negative breast cancer. J Clin Oncol 2001;19:4141–4149. [DOI] [PubMed] [Google Scholar]
- 25.GTx Inc. Fareston [package insert]. Memphis, TN, 2012.
- 26.AstraZeneca Pharmaceuticals LP . Nolvadex [package insert]. Wilmington, DE, 2012.
- 27. Baumann CK, Castiglione‐Gertsch M. Estrogen receptor modulators and down regulators: Optimal use in postmenopausal women with breast cancer. Drugs 2007;67:2335–2353. [DOI] [PubMed] [Google Scholar]
- 28. Baum M, Budzar AU, Cuzick J et al. Anastrozole alone or in combination with tamoxifen versus tamoxifen alone for adjuvant treatment of postmenopausal women with early breast cancer: First results of the ATAC randomised trial. Lancet 2002;359:2131–2139. [DOI] [PubMed] [Google Scholar]
- 29. Fisher B, Costantino JP, Wickerham DL et al. Tamoxifen for prevention of breast cancer: Report of the National Surgical Adjuvant Breast and Bowel Project P‐1 Study. J Natl Cancer Inst 1998;90:1371–1388. [DOI] [PubMed] [Google Scholar]
- 30. Jordan VC. Tamoxifen: Toxicities and drug resistance during the treatment and prevention of breast cancer. Annu Rev Pharmacol Toxicol 1995;35:195–211. [DOI] [PubMed] [Google Scholar]
- 31.Teva Pharmaceuticals USA . Tamoxifen Citrate [package insert]. Sellersville, PA, 2012.
- 32. Buzdar AU, Robertson JF, Eiermann W et al. An overview of the pharmacology and pharmacokinetics of the newer generation aromatase inhibitors anastrozole, letrozole, and exemestane. Cancer 2002;95:2006–2016. [DOI] [PubMed] [Google Scholar]
- 33. Harwood KV. Advances in endocrine therapy for breast cancer: Considering efficacy, safety, and quality of life. Clin J Oncol Nurs 2004;8:629–637. [DOI] [PubMed] [Google Scholar]
- 34. Perez EA, Weilbaecher K. Aromatase inhibitors and bone loss. Oncology (Williston Park) 2006;20:1029–1039; discussion 1039–1040, 1042, 1048. [PMC free article] [PubMed] [Google Scholar]
- 35. Lønning PE, Eikesdal HP. Aromatase inhibition 2013: Clinical state of the art and questions that remain to be solved. Endocr Relat Cancer 2013;20:R183–R201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lumachi F, Luisetto G, Basso SM et al. Endocrine therapy of breast cancer. Curr Med Chem 2011;18:513–522. [DOI] [PubMed] [Google Scholar]
- 37. Yeh WL, Shioda K, Coser KR et al. Fulvestrant‐induced cell death and proteasomal degradation of estrogen receptor α protein in MCF‐7 cells require the CSK c‐Src tyrosine kinase. PLoS One 2013;8:e60889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wakeling AE. Similarities and distinctions in the mode of action of different classes of antioestrogens. Endocr Relat Cancer 2000;7:17–28. [DOI] [PubMed] [Google Scholar]
- 39. Wakeling AE, Bowler J. Steroidal pure antioestrogens. J Endocrinol 1987;112:R7–R10. [DOI] [PubMed] [Google Scholar]
- 40. Wakeling AE, Dukes M, Bowler J. A potent specific pure antiestrogen with clinical potential. Cancer Res 1991;51:3867–3873. [PubMed] [Google Scholar]
- 41. Buzdar AU. Fulvestrant–A novel estrogen receptor antagonist for the treatment of advanced breast cancer. Drugs Today (Barc) 2008;44:679–692. [DOI] [PubMed] [Google Scholar]
- 42. Morris C, Wakeling A. Fulvestrant (‘Faslodex')–A new treatment option for patients progressing on prior endocrine therapy. Endocr Relat Cancer 2002;9:267–276. [DOI] [PubMed] [Google Scholar]
- 43.AstraZeneca Pharmaceuticals LP . Faslodex [package insert]. Wilmington, DE, 2016.
- 44. Bines J, Dienstmann R, Obadia RM et al. Activity of megestrol acetate in postmenopausal women with advanced breast cancer after nonsteroidal aromatase inhibitor failure: A phase II trial. Ann Oncol 2014;25:831–836. [DOI] [PubMed] [Google Scholar]
- 45. Cardoso F, Harbeck N, Fallowfield L et al. Locally recurrent or metastatic breast cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow‐up. Ann Oncol 2012;23(suppl 7):vii11–vii19. [DOI] [PubMed] [Google Scholar]
- 46.Par Pharmaceutical Inc . Megestrol acetate tablets, USP [package insert]. Spring Valley, NY, 2013.
- 47. Haddow A, Watkinson JM, Paterson E et al. Influence of synthetic oestrogens on advanced malignant disease. Br Med J 1944;2:393–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ingle JN. Estrogen as therapy for breast cancer. Breast Cancer Res 2002;4:133–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Iwase H, Yamamoto Y, Yamamoto‐Ibusuki M et al. Ethinylestradiol is beneficial for postmenopausal patients with heavily pre‐treated metastatic breast cancer after prior aromatase inhibitor treatment: A prospective study. Br J Cancer 2013;109:1537–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lønning PE, Taylor PD, Anker G et al. High‐dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat 2001;67:111–116. [DOI] [PubMed] [Google Scholar]
- 51. Manni A, Arafah BM, Pearson OH. Androgen‐induced remissions after antiestrogen and hypophysectomy in stage IV breast cancer. Cancer 1981;48:2507–2509. [DOI] [PubMed] [Google Scholar]
- 52. Schifeling DJ, Jackson DV, Zekan PJ et al. Fluoxymesterone as third line endocrine therapy for advanced breast cancer. A phase II trial of the Piedmont Oncology Association. Am J Clin Oncol 1992;15:233–235. [DOI] [PubMed] [Google Scholar]
- 53. Tormey DC and Jordan VC. Long‐term tamoxifen adjuvant therapy in node‐positive breast cancer: A metabolic and pilot clinical study. Breast Cancer Res Treat 1984;4:297–302. [DOI] [PubMed] [Google Scholar]
- 54. Litherland S, Jackson IM. Antioestrogens in the management of hormone‐dependent cancer. Cancer Treat Rev 1988;15:183–194. [DOI] [PubMed] [Google Scholar]
- 55. Mao C, Yang ZY, He BF et al. Toremifene versus tamoxifen for advanced breast cancer. Cochrane Database Syst Rev: 2012;7:CD008926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Nabholtz JM, Buzdar A, Pollak M et al. Anastrozole is superior to tamoxifen as first‐line therapy for advanced breast cancer in postmenopausal women: Results of a North American multicenter randomized trial. Arimidex Study Group. J Clin Oncol 2000;18:3758–3767. [DOI] [PubMed] [Google Scholar]
- 57. Bonneterre J, Buzdar A, Nabholtz JM et al. Anastrozole is superior to tamoxifen as first‐line therapy in hormone receptor positive advanced breast carcinoma. Cancer 2001;92:2247–2258. [DOI] [PubMed] [Google Scholar]
- 58. Mouridsen H, Gershanovich M, Sun Y et al. Superior efficacy of letrozole versus tamoxifen as first‐line therapy for postmenopausal women with advanced breast cancer: Results of a phase III study of the International Letrozole Breast Cancer Group. J Clin Oncol 2001;19:2596–2606. [DOI] [PubMed] [Google Scholar]
- 59. Paridaens RJ, Dirix LY, Beex LV et al. Phase III study comparing exemestane with tamoxifen as first‐line hormonal treatment of metastatic breast cancer in postmenopausal women: The European Organisation for Research and Treatment of Cancer Breast Cancer Cooperative Group. J Clin Oncol 2008;26:4883–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Howell A, Robertson JF, Abram P et al. Comparison of fulvestrant versus tamoxifen for the treatment of advanced breast cancer in postmenopausal women previously untreated with endocrine therapy: A multinational, double‐blind, randomized trial. J Clin Oncol 2004;22:1605–1613. [DOI] [PubMed] [Google Scholar]
- 61. Robertson JF, Llombart‐Cussac A, Rolski J et al. Activity of fulvestrant 500 mg versus anastrozole 1 mg as first‐line treatment for advanced breast cancer: Results from the FIRST study. J Clin Oncol 2009;27:4530–4535. [DOI] [PubMed] [Google Scholar]
- 62. Osborne CK, Pippen J, Jones SE et al. Double‐blind, randomized trial comparing the efficacy and tolerability of fulvestrant versus anastrozole in postmenopausal women with advanced breast cancer progressing on prior endocrine therapy: Results of a North American trial. J Clin Oncol 2002;20:3386–3395. [DOI] [PubMed] [Google Scholar]
- 63. Howell A, Robertson JF, Quaresma Albano J et al. Fulvestrant, formerly ICI 182,780, is as effective as anastrozole in postmenopausal women with advanced breast cancer progressing after prior endocrine treatment. J Clin Oncol 2002;20:3396–3403. [DOI] [PubMed] [Google Scholar]
- 64. Chia S, Gradishar W, Mauriac L et al. Double‐blind, randomized placebo controlled trial of fulvestrant compared with exemestane after prior nonsteroidal aromatase inhibitor therapy in postmenopausal women with hormone receptor‐positive, advanced breast cancer: Results from EFECT. J Clin Oncol 2008;26:1664–1670. [DOI] [PubMed] [Google Scholar]
- 65. Di Leo A, Jerusalem G, Petruzelka L et al. Results of the CONFIRM phase III trial comparing fulvestrant 250 mg with fulvestrant 500 mg in postmenopausal women with estrogen receptor‐positive advanced breast cancer. J Clin Oncol 2010;28:4594–4600. [DOI] [PubMed] [Google Scholar]
- 66. Di Leo A, Jerusalem G, Petruzelka L et al. Final analysis of overall survival for the Phase III CONFIRM trial: Fulvestrant 500 mg versus 250 mg. Cancer Res 2012;72(suppl 24):Abstract S1–S4. [Google Scholar]
- 67. Mehta RS, Barlow WE, Albain KS et al. Combination anastrozole and fulvestrant in metastatic breast cancer. N Engl J Med 2012;367:435–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bergh J, Jönsson PE, Lidbrink EK et al. FACT: An open‐label randomized phase III study of fulvestrant and anastrozole in combination compared with anastrozole alone as first‐line therapy for patients with receptor‐positive postmenopausal breast cancer. J Clin Oncol 2012;30:1919–1925. [DOI] [PubMed] [Google Scholar]
- 69. Buzdar A, Douma J, Davidson N et al. Phase III, multicenter, double‐blind, randomized study of letrozole, an aromatase inhibitor, for advanced breast cancer versus megestrol acetate. J Clin Oncol 2001;19:3357–3366. [DOI] [PubMed] [Google Scholar]
- 70. Buzdar AU, Jonat W, Howell A et al. Anastrozole versus megestrol acetate in the treatment of postmenopausal women with advanced breast carcinoma: Results of a survival update based on a combined analysis of data from two mature phase III trials. Arimidex Study Group. Cancer 1998;83:1142–1152. [PubMed] [Google Scholar]
- 71. Kaufmann M, Bajetta E, Dirix LY et al. Exemestane is superior to megestrol acetate after tamoxifen failure in postmenopausal women with advanced breast cancer: Results of a phase III randomized double‐blind trial. The Exemestane Study Group. J Clin Oncol 2000;18:1399–1411. [DOI] [PubMed] [Google Scholar]
- 72. Ferretti G, Bria E, Giannarelli D et al. Second‐ and third‐generation aromatase inhibitors as first‐line endocrine therapy in postmenopausal metastatic breast cancer patients: A pooled analysis of the randomised trials. Br J Cancer 2006;94:1789–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Thürlimann B, Robertson JF, Nabholtz JM et al. Efficacy of tamoxifen following anastrozole ('Arimidex') compared with anastrozole following tamoxifen as first‐line treatment for advanced breast cancer in postmenopausal women. Eur J Cancer 2003;39:2310–2317. [DOI] [PubMed] [Google Scholar]
- 74. Lønning PE, Bajetta E, Murray R et al. Activity of exemestane in metastatic breast cancer after failure of nonsteroidal aromatase inhibitors: A phase II trial. J Clin Oncol 2000;18:2234–2244. [DOI] [PubMed] [Google Scholar]
- 75. Bertelli G, Garrone O, Merlano M et al. Sequential treatment with exemestane and non‐steroidal aromatase inhibitors in advanced breast cancer. Oncology 2005;69:471–477. [DOI] [PubMed] [Google Scholar]
- 76. Yardley DA, Noguchi S, Pritchard KI et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO‐2 final progression‐free survival analysis. Adv Ther 2013;30:870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Howell A, Pippen J, Elledge RM et al. Fulvestrant versus anastrozole for the treatment of advanced breast carcinoma: A prospectively planned combined survival analysis of two multicenter trials. Cancer 2005;104:236–239. [DOI] [PubMed] [Google Scholar]
- 78.United States Food and Drug Administration. Faslodex label and approval history. 2010. Available at http://www.accessdata.fda.gov/drugsatfda_docs/appletter/2010/021344s007,s012ltr.pdf. Accessed August 9, 2016.
- 79. Robertson JF, Llombart‐Cussac A, Feltl D et al. Fulvestrant 500 mg versus anastrozole as first‐line treatment for advanced breast cancer: Overall survival from the Phase II ‘FIRST’ study. Cancer Res 2015;75(suppl 9):Abstract S6–S04. [Google Scholar]
- 80.AstraZeneca Pharmaceuticals LP. ClinicalTrials.gov web site. A global study to compare the effects of fulvestrant and arimidex in a subset of patients with breast cancer (FALCON). Available at www.clinicaltrials.gov/ct2/show/NCT01602380. Accessed March 7, 2016.
- 81. Johnston SR, Kilburn LS, Ellis P et al. Fulvestrant plus anastrozole or placebo versus exemestane alone after progression on non‐steroidal aromatase inhibitors in postmenopausal patients with hormone‐receptor‐positive locally advanced or metastatic breast cancer (SoFEA): A composite, multicentre, phase 3 randomised trial. Lancet Oncol 2013;14:989–998. [DOI] [PubMed] [Google Scholar]
- 82. Hayes E, Nicholson RI, Hiscox S. Acquired endocrine resistance in breast cancer: Implications for tumour metastasis. Front Biosci (Landmark Ed) 2011;16:838–848. [DOI] [PubMed] [Google Scholar]
- 83. Glück S. Extending the clinical benefit of endocrine therapy for women with hormone receptor‐positive metastatic breast cancer: Differentiating mechanisms of action. Clin Breast Cancer 2014;14:75–84. [DOI] [PubMed] [Google Scholar]
- 84. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med 2011;62:233–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Suba Z. The pitfall of the transient, inconsistent anticancer capacity of antiestrogens and the mechanism of apparent antiestrogen resistance. Drug Des Devel Ther 2015;9:4341–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Aguilar H, Solé X, Bonifaci N et al. Biological reprogramming in acquired resistance to endocrine therapy of breast cancer. Oncogene 2010;29:6071–6083. [DOI] [PubMed] [Google Scholar]
- 87. Arpino G, Wiechmann L, Osborne CK et al. Crosstalk between the estrogen receptor and the HER tyrosine kinase receptor family: Molecular mechanism and clinical implications for endocrine therapy resistance. Endocr Rev 2008;29:217–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Miller TW, Balko JM, Arteaga CL. Phosphatidylinositol 3‐kinase and antiestrogen resistance in breast cancer. J Clin Oncol 2011;29:4452–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Novartis Pharmaceuticals Corporation. Afinitor [package insert]. East Hanover, NJ, 2016.
- 90. Boulay A, Rudloff J, Ye J et al. Dual inhibition of mTOR and estrogen receptor signaling in vitro induces cell death in models of breast cancer. Clin Cancer Res 2005;11:5319–5328. [DOI] [PubMed] [Google Scholar]
- 91. Finn RS, Crown JP, Lang I et al. The cyclin‐dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first‐line treatment of oestrogen receptor‐positive, HER2‐negative, advanced breast cancer (PALOMA‐1/TRIO‐18): A randomised phase 2 study. Lancet Oncol 2015;16:25–35. [DOI] [PubMed] [Google Scholar]
- 92. Finn RS, Martin M, Rugo HS et al. PALOMA‐2: Primary results from a phase III trial of palbociclib (P) with letrozole (L) compared with letrozole alone in postmenopausal women with ER+/HER2– advanced breast cancer (ABC). J Clin Oncol 2016;34:507a. [Google Scholar]
- 93. Turner NC, Ro J, André F et al. Palbociclib in hormone‐receptor‐positive advanced breast cancer. N Engl J Med 2015;373:209–219. [DOI] [PubMed] [Google Scholar]
- 94. Baselga J, Campone M, Piccart M et al. Everolimus in postmenopausal hormone‐receptor‐positive advanced breast cancer. N Engl J Med 2012;366:520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Bachelot T, Bourgier C, Cropet C et al. Randomized phase II trial of everolimus in combination with tamoxifen in patients with hormone receptor‐positive, human epidermal growth factor receptor 2‐negative metastatic breast cancer with prior exposure to aromatase inhibitors: A GINECO study. J Clin Oncol 2012;30:2718–2724. [DOI] [PubMed] [Google Scholar]
- 96. Wolff AC, Lazar AA, Bondarenko I et al. Randomized phase III placebo‐controlled trial of letrozole plus oral temsirolimus as first‐line endocrine therapy in postmenopausal women with locally advanced or metastatic breast cancer. J Clin Oncol 2013;31:195–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Piccart M, Hortobagyi GN, Campone M et al. Everolimus plus exemestane for hormone‐receptor‐positive, human epidermal growth factor receptor‐2‐negative advanced breast cancer: Overall survival results from BOLERO‐2. Ann Oncol 2014;25:2357–2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Caldon CE, Daly RJ, Sutherland RL et al. Cell cycle control in breast cancer cells. J Cell Biochem 2006;97:261–274. [DOI] [PubMed] [Google Scholar]
- 99. Lundberg AS, Weinberg RA. Control of the cell cycle and apoptosis. Eur J Cancer 1999;35:1886–1894. [DOI] [PubMed] [Google Scholar]
- 100. Sutherland RL, Musgrove EA. Cyclins and breast cancer. J Mammary Gland Biol Neoplasia 2004;9:95–104. [DOI] [PubMed] [Google Scholar]
- 101.Pfizer Inc. Ibrance [package insert]. New York, NY, 2015.
- 102.United States Food and Drug Administration. FDA approves Ibrance for postmenopausal women with advanced breast cancer. 2015. Available at http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm432871.htm. Accessed March 7, 2016.
- 103. Cristofanilli M, Turner NC, Bondarenko I et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone‐receptor‐positive, HER2‐negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA‐3): Final analysis of the multicentre, double‐blind, phase 3 randomised controlled trial. Lancet Oncol 2016;17:425–439. [DOI] [PubMed] [Google Scholar]
- 104. Harbeck N, Iyer S, Turner N et al. Quality of life with palbociclib plus fulvestrant in previously treated hormone receptor‐positive, HER2‐negative metastatic breast cancer: Patient‐reported outcomes from the PALOMA‐3 trial. Ann Oncol 2016;27:1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Infante JR, Shapiro G, Witteveen P et al. A phase I study of the single‐agent CDK4/6 inhibitor LEE011 in pts with advanced solid tumors and lymphomas J Clin Oncol 2014;32(suppl 5s):2528a. [Google Scholar]
- 106. Juric D, Hamilton E, Garcia Estévez L et al. Phase Ib/II study of LEE011 and BYL719 and letrozole in ER+, HER2– breast cancer: Safety, preliminary efficacy and molecular analysis. Cancer Res 2015;75(suppl 9):P5–19‐24a. [Google Scholar]
- 107. Patnaik A, Rosen LS, Tolaney SM et al. Efficacy and safety of abemaciclib, an inhibitor of CDK4 and CDK6, for patients with breast cancer, non‐small cell lung cancer, and other solid tumors. Cancer Discov 2016;6:740–753. [DOI] [PubMed] [Google Scholar]
- 108. Stemke‐Hale K, Gonzalez‐Angulo AM, Lluch A et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 2008;68:6084–6091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Creighton CJ, Fu X, Hennessy BT et al. Proteomic and transcriptomic profiling reveals a link between the PI3K pathway and lower estrogen‐receptor (ER) levels and activity in ER+ breast cancer. Breast Cancer Res 2010;12:R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Miller TW, Balko JM, Fox EM et al. ERα‐dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov 2011;1:338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Baselga J, Im SA, Iwata H et al. PIK3CA status in circulating tumor DNA (ctDNA) predicts efficacy of buparlisib (BUP) plus fulvestrant (FULV) in postmenopausal women with endocrine‐resistant HR+/HER2– advanced breast cancer (BC): First results from the randomized, phase III BELLE‐2 trial. Cancer Res 2016;76 (suppl 4):S6–01a. [Google Scholar]
- 112. Mayer IA, Abramson VG, Isakoff SJ et al. Stand up to cancer phase Ib study of pan‐phosphoinositide‐3‐kinase inhibitor buparlisib with letrozole in estrogen receptor‐positive/human epidermal growth factor receptor 2‐negative metastatic breast cancer. J Clin Oncol 2014;32:1202–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ma CX, Luo J, Naughton M et al. A phase I study of BKM120 and fulvestrant in postmenopausal women with estrogen receptor positive metastatic breast cancer. Cancer Res 2015;75(suppl 9):PD5–6a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Saura C, Sachdev J, Patel MR et al. Ph1b study of the PI3K inhibitor taselisib (GDC‐0032) in combination with letrozole in patients with hormone receptor‐positive advanced breast cancer. Cancer Res 2015;75(suppl 9):PD5–2a. [Google Scholar]
- 115. Janku F, Juric D, Cortes J et al. Phase I study of the PI3Kα inhibitor BYL719 plus fulvestrant in patients with PIK3CA‐altered and wild type ER+/HER2‐ locally advanced or metastatic breast cancer. Cancer Res 2015;75(suppl 9):PD5–5a. [Google Scholar]
- 116. Mayer IA, Gupta AV, Balko J et al. SU2C phase Ib study of the PI3Kα inhibitor BYL719 with letrozole in ER+/HER2– metastatic breast cancer (MBC). J Clin Oncol 2014;32(suppl 5s):516a. [Google Scholar]
- 117. Shah PD, Moynahan ME, Modi S et al. Phase I trial: PI3Kα inhibitor BYL719 plus aromatase inhibitor (AI) for patients with hormone receptor‐positive (HR+) metastatic breast cancer (MBC). Cancer Res 2015;75(suppl 9):PD5‐3a. [Google Scholar]
- 118. Sabnis GJ, Goloubeva O, Chumsri S et al. Functional activation of the estrogen receptor‐α and aromatase by the HDAC inhibitor entinostat sensitizes ER‐negative tumors to letrozole. Cancer Res 2011;71:1893–1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Yardley DA, Ismail‐Khan RR, Melichar B et al. Randomized phase II, double‐blind, placebo‐controlled study of exemestane with or without entinostat in postmenopausal women with locally recurrent or metastatic estrogen receptor‐positive breast cancer progressing on treatment with a nonsteroidal aromatase inhibitor. J Clin Oncol 2013;31:2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Lai A, Kahraman M, Govek S et al. Identification of GDC‐0810 (ARN‐810), an orally bioavailable selective estrogen receptor degrader (SERD) that demonstrates robust activity in tamoxifen‐resistant breast cancer xenografts. J Med Chem 2015;58:4888–4904. [DOI] [PubMed] [Google Scholar]
- 121. Dickler M, Bardia A, Mayer I et al. A first‐in‐human phase I study to evaluate the oral selective estrogen receptor degrader GDC‐0810 (ARN‐810) in postmenopausal women with estrogen receptor+ HER2‐, advanced/metastatic breast cancer. Cancer Res 2015;75(suppl 15):CT231a. [Google Scholar]
- 122. Wardell SE, Nelson ER, Chao CA et al. Evaluation of the pharmacological activities of RAD1901, a selective estrogen receptor degrader. Endocr Relat Cancer 2015;22:713–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Garner F, Shomali M, Paquin D et al. RAD1901: A novel, orally bioavailable selective estrogen receptor degrader that demonstrates antitumor activity in breast cancer xenograft models. Anticancer Drugs 2015;26:948–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. De Savi C, Bradbury RH, Rabow AA et al. Optimization of a novel binding motif to (E)‐3‐(3,5‐Difluoro‐4‐((1R,3R)‐2‐(2‐fluoro‐2‐methylpropyl)‐3‐methyl‐2,3,4,9‐tetra hydro‐1H‐pyrido[3,4‐b]indol‐1‐yl)phenyl)acrylic acid (AZD9496), a potent and orally bioavailable selective estrogen receptor downregulator and antagonist. J Med Chem 2015;58:8128–8140. [DOI] [PubMed] [Google Scholar]