

Abstract
In the United States of America, prostate cancer is the second most common age-related cancer among men. African-American men have the highest incidence of, and mortality rate from this disease in the United States. According to the American Cancer Society, 29% of all cancer cases and 9% of all cancer deaths are a result of prostate cancer. Individuals who are at highest risk include African-American men, men over 60 years of age, and those with a family history of the disease. African-Americans also have twice the risk of developing prostate cancer as compared to Caucasians. Erythroblastosis virus E26 transformation-specific (ETS) factors play an important role in human cancers. ETS Variant 1 (ETV1), an ETS factor, is notable for its association in prostate cancers, where truncated ETV1 (dETV1) or its full length counterpart is overexpressed in approximately 10% of the prostate cancer patients. Prostate cancer tumorigenesis may be initiated by deregulation of the Wnt/β-catenin pathway. Mutations that stabilize β-catenin were shown to contribute to the loss of cell-growth control in tumorigenesis. We hypothesized that ETV1’s interaction with components of the Wnt/β-catenin pathway may alter β-catenin’s interaction with downstream tumor-suppressor genes, which are critical in regulating apoptosis and cell-growth properties of prostate cells. Our results demonstrate for the first time that ETV1 alters β-catenin activity by activating kinases that regulate Wnt/β-catenin activity through post-translational modification in prostate cancer cells. We further demonstrate that therapeutic agents such as PD98059, that reverse effect of ETV1 on Wnt/β-catenin signaling pathway, can be used to target ETV1-positive prostate cancer cells. These therapeutic agents could have a profound impact on prevention and treatment of prostate cancer which may help to reduce health disparity seen in minority patients. Understanding the role of ETV1 in Wnt/β-catenin pathway will also allow us to develop better diagnostic tools, which can be used as a biomarker for ETV1-positive prostate cancers.
Keywords: ETV1, dETV1, β-Catenin, Phosphorylation, Prostate Cancer, Melanoma, PD98059
INTRODUCTION
The prostate is a walnut-sized gland of the male reproductive system that wraps around the proximal urethra. It can become cancerous by deregulation of certain genes that control cellular growth and apoptosis. In the United States, after lung cancer, prostate cancer is the most common age-related cancer among men (Huang, 2009). By the age of 75, one of nine men will develop prostate cancer (Fortson, 2011). Prostate cancer is more common in African-American men than in Caucasian men over the age of 65 (Bakhsh, 2015). In up to 90% of the patients, prostate cancer metastasizes to bone; in 46% to the lung, and in 25% to the liver (Bakhsh, 2015).
Numerous erythroblastosis virus E26 transformation-specific (ETS) transcription factors contribute to the development of cancers (Oh, 2013; Reddy, 1991; Shin, 2013). ETV1, ETV2, ETV3, ETV4, Fli-1, and ERG are examples of these factors (Fang, 2014; Rao, 1987; Reddy, 1989; Spans, 2013). ETV1, also called ER81, is unique in that it can be both over expressed in prostate cancer as a fusion transcript and as a wild-type, full-length ETV1 (Gasi, 2014; Shin, 2013; Zhang, 2014). ETV1 is notable for its association with prostate cancers, where truncated ETV1 (dETV1), or its full-length counterpart, is overexpressed in approximately 10% of patients (Hermans, 2008; Oh, 2013). When compared to full-length ETV1, truncated ETV1 or dETV1 has 131 fewer amino acids because of gene translocation on its N-terminal end (Oh, 2013). Importantly, induction of invasion and migration are observed in both the full-length and truncated ETV1 expressing prostate cancer cells (Gasi, 2014; Hermans, 2008). Moreover, 40% of all melanoma cases were observed to have ETV1 amplification, where it was shown to promote cellular growth (Chi, 2010; Jané-Valbuena, 2010; Oh, 2013). Prostate-specific antigen (PSA) expression is also enhanced by ETV1 (Gasi, 2014). We currently have a limited understanding of how molecular pathways are affected by elevated ETV1 levels in prostate cancer (Gasi, 2014; Zynda, 2013).
Cancers may be brought on by deregulation of the Wnt/β-catenin pathway (Larue, 2009; Zhang, 2013). Cell regulatory functions throughout embryonic development, as well as homeostasis in adults, are crucially dependent on the Wnt/β-catenin pathway (Larue, 2009). β-catenin is a key element in malignant transformation. However, the mechanism whereby β-catenin participates in malignant transformation is still ambiguous (Larue, 2009; Satyamoorthy, 2002). β-catenin is a unique protein in that it varies in function depending on its location in the cell (Du, 2012; Larue, 2009). Several previous studies have established that in 30% of melanoma cases, cytoplasmic and nuclear β-catenin are overexpressed (Larue, 2009; Webster, 2015). Wnt/β-catenin and MAPK activation are often detected in melanomas; however, it remains unclear how these signaling pathways collaborate during tumorigenesis (Larue, 2009; Satyamoorthy, 2002). The likelihood of a role for β-catenin signaling in cancer is further supported by the observation that the adenoma-tous polyposis coli (APC) tumor-suppressor protein down-regulates excess intracellular β-catenin (Rubinfeld, 1997). Mutations that stabilize β-catenin may contribute to loss of cell-growth control in tumorigenesis (Rubinfeld, 1997). Therefore, it is probable that the β-catenin regulatory properties of ETV1 may also play a role in the tumorigenesis of ETV1-positive prostate cancer.
It is conceivable that ETV1’s interaction with components of this pathway may alter β-catenin interaction with downstream tumor-suppressor genes, which are critical in regulating apoptosis, and cell-growth properties. These alterations can lead to deregulation of growth, differentiation, apoptosis and thus to tumor formation. Understanding the role of ETV1 downstream targets will help to develop better diagnostic tools, potentially using it as a biomarker or exploiting it as a therapeutic target to prevent or treat ETV1-positive prostate cancer. Understanding of prostate cancer biology has increased significantly in recent years. However, additional information is still needed to allow improvements in screening and treatment methods for this disease. Moreover, the discovery of disease-specific target genes will aid in illuminating their involvement in regulatory pathways in ways that can be exploited to develop better treatments for prostate cancer and other human diseases.
MATERIALS AND METHODS
Cell Culture
LNCaP, PNT1A, PC-3, and COS-1: Cells were purchased from American Type Culture Collection (ATCC®). These cells were cultured in RPMI-1640 medium with 10% fetal bovine serum (FBS). Cells were kept at 5% CO2 and 37°C in a Nuaire US AutoFlow CO2 Water-Jacketed Incubator.
Chemicals and Reagents
Cell culture medium RPMI-1640, high glucose with L-Glutamine and HEPES was purchased from American Type Culture Collection (ATCC®). 0.25% Trypsin-EDTA was purchased from Gibco by Life Technologies Corp. PD98059 was purchased from APExBIO. Stock solution of PD98059 was prepared by dissolving it in DMSO. The prepared PD98059 stock solution was stored in −80 °C until it was ready to be used. Bradford’s assay reagent (Bio-Rad Laboratories) with bovine serum albumin (BSA) as protein standard was used to determine protein concentrations for Western blots. ER81 (H-70): sc-28681 rabbit polyclonal antibodies (ETV1), β-catenin (E-5): sc-7963 mouse monoclonal antibodies, and β-actin (C4): sc-47778 mouse monoclonal antibodies, were purchased from Santa Cruz Biotechnology Inc. For Western blot analysis; enhanced chemiluminescence (ECL), secondary anti-mouse and anti-rabbit antibodies were purchased from Amersham, GE Healthcare.
Western Analysis
Cells were seeded in six well plates (Corning Costar) at 2 mL (4.5 × 104 cells/mL) per well. After a 24-hour incubation period, the cells were transfected or treated with drugs. After an additional 24 hours, cells were harvested. All cell culture medium was removed from the six well plates, and washed with ice cold 1× PBS twice. 100 μl lysis buffer (20 mM Hepes (pH 7.5), 100 mM KCl, 0.4 mM EDTA, 0.2% Igepal CA-630, 0.5 mM PMSF, 10 mM β-mercaptoethanol, and protease inhibitor) was used to lyse the cells and collect cell lysate samples into 2 mL microcentrifuge tubes. The samples were then expressed through a 27.5 gauge needle for further lysis of cells. After 30 minute incubation on ice, cells were centrifuged at 12,000× g for 20 minutes. Cell supernatants were collected into clean 2 mL microcentrifuge tubes. Bradford protein assay (Bio-Rad) was used to normalize the amount of protein loaded on the gels. An Ultrospec 3000 UV/Visible Spectrophotometer (Pharmacia Biotech.) set to 595 nm wavelength was used to quantify the protein data. Protein samples were balanced and sample buffer was added to each sample before running them in 4–20% Mini-PROTEIN® TGX™ gels (Bio-Rad) at 100 V. The gel was then transferred onto nitorcellulose blotting membrane (Amersham™ Hybond ECL) at 30 V overnight in a 4 °C refrigerator. Membrane was washed with 1× PBS then incubated for an hour with bovine serum albumin (BSA) solution containing 5% non-fat dry milk. Next, the membrane was incubated with primary antibody (1:200 dilution), then secondary antibody (1:5000 dilution), consecutively for one hour each. After the completion of incubation, the membrane was washed five times with 1× PBS-T solution. Proteins were then detected by enhanced chemiluminescence (ECL) using an LAS3000 imaging system.
Cell Viability Assay
To measure cell viability, LNCaP cells were seeded on 96 well plates at 4 × 104 cells per well. Cells were then allowed to incubate for 24 hours, at which point, vehicle control or the drug PD98059 was added in increasing quantities (10 μM, 20 μM, and 40 μM) for 72 hours. After the 72 hour incubation, cell viability was measured using MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide). A Bio-Rad microplate reader (Bio-Rad Laboratories) was used to quantify spectrophotometric absorbance signal. Inhibitory Concentration (IC50) for the drug PD98059 was calculated using Gnuplot (GNU General Public License).
Luciferase Assay
COS-1 cells were co-transfected with expression vectors β-catenin, TCF-4, pGL3OT (reporter plasmid), and dETV1 (expression plasmid). Transfected cells were incubated for 24 hours and luciferase activity was quantified via the Dual-Glo luciferase assay system protocol as described by the manufacturer (Promega). Luciferase activity measurements were analyzed using Fluoroskan Ascent FL machine and Ascent software (Thermo Electron Corporation). The reporter activity is expressed as relative fluorescence units (RFU).
Calf Intestine Alkaline Phosphatase Assay
1.5 × 105 PNT1A cells grown in six well plates for 24 hours, then transfected with increasing amounts of dETV1 (0.6 μg and 1.4 μg) and a constant amount of β-catenin (0.2 μg) using Lipofectamine 2000 (Invitrogen) as described in the Figure 3. After another 24-hour incubation period, cells were treated with Calf Intestine Alkaline Phosphatase (CIAP).
Figure 3.
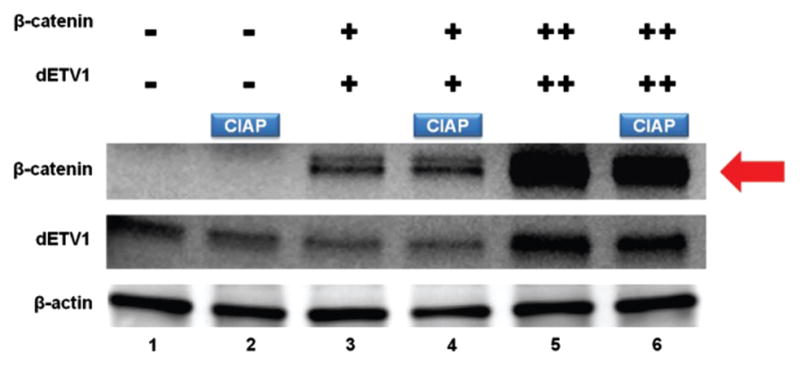
dETV1 induces phosphorylation of β-catenin. PNT1A cells were seeded in six well plates. They were then transfected with empty expression vector or expression plasmid pSG5/HA-dETV1 and pSG5/β-catenin as indicated. Cells were transfected with different concentrations of β-catenin and dETV1 (β-catenin: 0.2 μg, Low dETV1: 0.6 μg, High dETV1: 1.4 μg). After 24 hours of incubation, cells in lane 2, 4, and 6 were treated with Calf Intestine Alkaline Phosphatase (CIAP) and cell lysate was collected and subjected to Western blot analysis.
Cycloheximide Chase Assay
PC-3 cells (1.5 × 105) were seeded in six well plates and incubated for 24 hours. The next day, the cells were transfected with constant concentrations of β-catenin and increasing concentrations of dETV1 (β-catenin: 0.2 μg, Low dETV1: 0.4 μg, High dETV1: 1.4 μg). After another 24 hours of incubation, Cycloheximide, an inhibitor of protein biosynthesis was added. Cell lysates were then collected at 0, 1, 2, 4, and 6 hour time period consecutively. β-catenin, dETV1, and β-actin protein levels were then measured by Western blot.
Statistical Analysis
Experiment data are presented in mean ± standard deviations (S.D.). Samples were analyzed by t-test whenever applicable.
RESULTS
ETV1 Regulates β-Catenin Activity
Fli-1 belongs to ETS gene family that plays a crucial role in certain cancers (Im, 2000; Paulo, 2012). Previously in our lab, Fli-1 has been shown to diminish retinoic acid receptor (RXR) activity in a dose dependent manner, suggesting that ETS transcription factors function both as transcriptional activators and inhibitors (Kayarthodi, 2014; Ramakrishnan, 2004). Therefore, since ETV1 is also an ETS gene, we reasoned that it may have a similar effect on β-catenin activity. To demonstrate that dETV1 has a regulatory effect on β-catenin, we co-transfected empty vector or increasing amounts of dETV1 expression plasmid along with a reporter gene into cells. Interestingly, dETV1 inhibited β-catenin activity in a dose dependent manner as shown in Figure 1.
Figure 1.
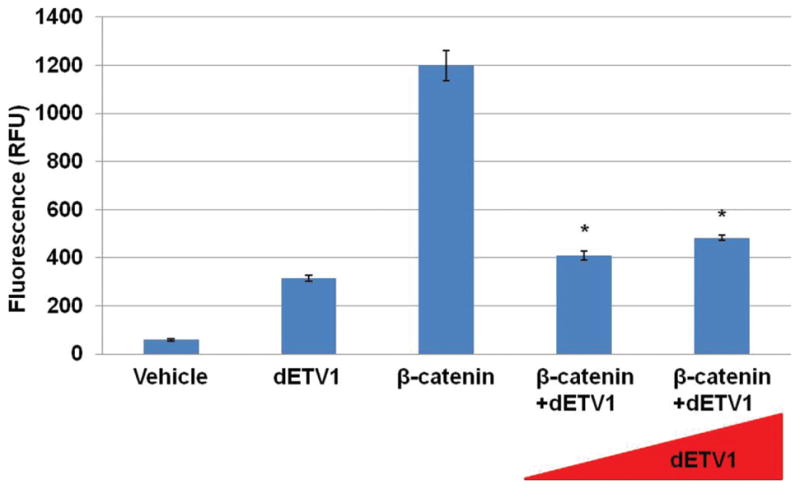
dETV1 regulates β-catenin activity. COS-1 cells were co-transfected with β-catenin, TCF-4, pGL3OT, and dETV1 in increasing concentrations as indicated. After 24 hours of transfection, luciferase activity was examined. The reporter activity is expressed as relative fluorescence units (RFU). Chart values represent mean±S.D. Asterisk represents p < 0.01.
ETV1 Regulates β-Catenin Expression
Since we found β-catenin activity to be diminished with the addition of dETV1, we postulated that ETV1 may regulate β-catenin expression levels. To test this hypothesis, we transfected COS-1 cells with expression plasmids, β-catenin, dETV1 as well as, β-catenin with increasing amount of dETV1, as described in Figure 2(a). Expression of β-catenin and dETV1 was analyzed using Western blot analysis. We observed that increasing dETV1 causes dramatic upregulation of β-catenin expression (Fig. 2(a)). In comparison to control β-catenin expression levels, the concentration of β-catenin with the highest levels of dETV1 is significantly greater as we have shown by Western blot analysis (Fig. 2(a)).
Figure 2.

dETV1 regulates β-catenin expression in different cell types. (a) dETV1 regulates β-catenin expression in COS-1 cells. COS-1 cells were seeded in six well plates. They were transfected with empty expression vector or expression plasmid pSG5/dETV1 and pSG5/β-catenin as indicated. Cells were transfected with different concentrations of β-catenin and dETV1 (β-catenin: 0.2 μg, Low dETV1: 0.4 μg, High dETV1: 1.4 μg). After 48 hours of transfection, cell lysate was collected and subjected to Western blot analysis. (b) dETV1 regulates β-catenin expression in PNT1A cells. Cells were seeded in six well plates. They were trans-fected with empty expression vector or expression plasmid pSG5/dETV1 and pSG5/β-catenin as indicated. Cells were transfected with different concentrations of β-catenin and dETV1 (β-catenin: 0.2 μg, Low dETV1: 0.4 μg, High dETV1: 1.4 μg). After 48 hours of transfection, cell lysate was collected and subjected to Western blot analysis. (c) dETV1 regulates endogenous β-catenin expression in prostate cancer cells (LNCaP). Cells were seeded in six well plates. They were transfected with empty expression vector or expression plasmid pSG5/HA-dETV1 and pSG5/β-catenin as indicated. Cells were transfected with different concentrations of β-catenin and dETV1 (β-catenin: 0.2 μg, Low dETV1: 0.4 μg, High dETV1: 1.4 μg). After 48 hours of transfection, cell lysate was collected and subjected to Western blot analysis.
ETV1 Regulates β-Catenin Expression in Prostate Cells (PNT1A)
The ETV1 gene plays an instrumental role in prostate cancer (Oh, 2013; Paulo, 2012). For this reason, we examined the effect of dETV1 on the expression of β-catenin in PNT1A cells (human normal prostate cells), allowing us to investigate the possible effect of dETV1 on normal prostate cells. As in the previous COS-1 cell study, we transfected β-catenin, dETV1, and co-transfected increasing amounts of dETV1 with a stable amount of β-catenin into PNT1A cells. As expected, β-catenin expression levels were significantly higher with each increasing addition of dETV1 (Fig. 2(b)). Increased β-catenin protein expression is seen in many cancers (Rubinfeld, 1997). Since dETV1 increased β-catenin levels by such a significant level, it suggests that ETV1 may play a bigger role in prostate cancer than previously realized.
ETV1 Regulates Endogenous β-Catenin Expression in Prostate Cancer Cells (LNCaP)
Since normal prostate cells were so dramatically affected, we wondered if prostate cancer cells (LNCaP) would respond to additional expression of dETV1 since it already harbors the ETS gene, ETV1. To distinguish between the endogenous ETV1 and transfected dETV1, we employed a HA-tagged dETV1, allowing us to examine effects of HA-dETV1 on endogenous β-catenin more closely. As expected, in Figure 2(c), the control shows endogenous β-catenin expression in the LNCaP cells. However, with the expression of increasing concentrations of HA-dETV1, endogenous β-catenin protein expression was increased significantly (Fig. 2(c)). These results suggest that β-catenin expression was increased by more than twofold in the presence of higher amounts of HA-dETV1 (Fig. 2(c)). The data consistently indicates increased expression of β-catenin protein in the presence of higher expression levels of dETV1 both in normal and cancerous prostate cells. Since it is known that β-catenin undergoes post-translational modification through phosphorylation, we hypothesized that the effect seen on β-catenin by dETV1 may be due to post-translational modification of β-catenin such as phosphorylation (Fig. 2(c)).
Calf Intestine Alkaline Phosphatase Assay (CIAP) of β-Catenin in PNT1A Cells
After determining that β-catenin protein levels were distinctly increased in the presence of dETV1 in multiple cell lines (Fig. 2), we pursued further elucidation on this pathway. We show that β-catenin undergoes post-translational modifications as a result of dETV1 co-expression. The effects of ETV1 on phosphorylation levels of β-catenin were analyzed using a calf intestine alkaline phosphatase (CIAP) assay, whereby phosphate groups are enzymatically removed from proteins. PNT1A cells were co-transfected with β-catenin and increasing amount of dETV1 and then treated with CIAP to examine post-translational modification effects on β-catenin. CIAP treatment reduced the size of β-catenin band suggesting that β-catenin undergoes post-translational modification by phosphorylation (Fig. 3). Our data suggest that a considerable amount of β-catenin protein (~23%) was phosphorylated in the presence of high dETV1. Nevertheless, it is important to note that only a minute population of a specific protein may become phosphorylated at any given time. Hence, we are observing CIAP’s effect on only a marginal portion of the available β-catenin protein. Furthermore, densitometry analysis shows a reduction of approximately 23% in β-catenin protein expression, which suggests dETV1 promotes phosphorylation of β-catenin.
Expression of β-Catenin Protein is Stabilized in the Presence of dETV1
Next, we show that the stability of β-catenin is affected when co-transfected with dETV1. We transfected PC-3 cells with a constant amount of β-catenin (0.2 μg) and two increasing concentrations of dETV1 (Low dETV1: 0.4 μg and High ETV1: 1.4 μg) followed by a 24 hour incubation period. Cycloheximide, a protein biosynthesis inhibitor, was then added. Cell lysates were collected at 0, 1, 2, 4, and 6 hour time periods to measure for β-catenin protein stability. The stability of β-catenin in the absence of dETV1 decreased in a time dependent manner from high (100%) to low (~14%) β-catenin protein level over the six hours (Fig. 4). Conversely, when a large amount of dETV1 was expressed along with normal levels of β-catenin, there was an increased stabilization of the β-catenin protein level (~37%) were observed (Fig. 4). Taken together, the data indicate that dETV1 enhances β-catenin protein stabilization.
Figure 4.
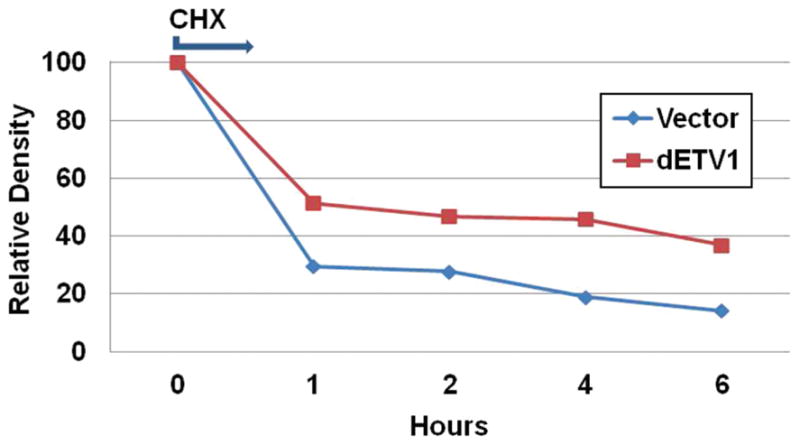
dETV1 stabilizes β-catenin expression. PC-3 cells were seeded and transfected with 0.2 μg of β-catenin (blue line), and with 0.2 μg of β-catenin along with 0.4 μg of dETV1 (red line). After 24 hours of incubation, Cycloheximide, an inhibitor of protein biosynthesis was added. Cell lysates were then collected at 0, 1, 2, 4, and 6 hour time periods and (β-catenin, dETV1, β-actin) protein levels were measured by Western blot followed by densitometry analysis.
Kinase Inhibitor PD98059 Interferes with Stabilization of β-Catenin by dETV1
After observing the accumulation, phosphorylation, and stabilization of β-catenin with subsequent addition of dETV1, we considered what effect kinase inhibitors would have since it was recently noted that introduction of ERG or ETV1 expression into normal prostate cells activates a RAS/MAPK gene expression program in the absence of ERK activation (Hollenhorst, 2011). There- fore, dETV1 may be interacting through a similar pathway to affect β-catenin. PNT1A cells were transfected with β-catenin, dETV1, and β-catenin with increasing amounts of dETV1 (β-catenin: 0.2 μg, Low dETV1: 0.6 μg, High dETV1: 1.6 μg). After incubating for 24 hours, the cells were maintained in the presence and in absence of a kinase inhibitor, PD98059. Upon addition of PD98059, the accumulation of β-catenin diminished even with the addition of high concentration dETV1. Densitometry analysis shows β-catenin expression levels were reduced by approximately 40% (Fig. 5).
Figure 5.
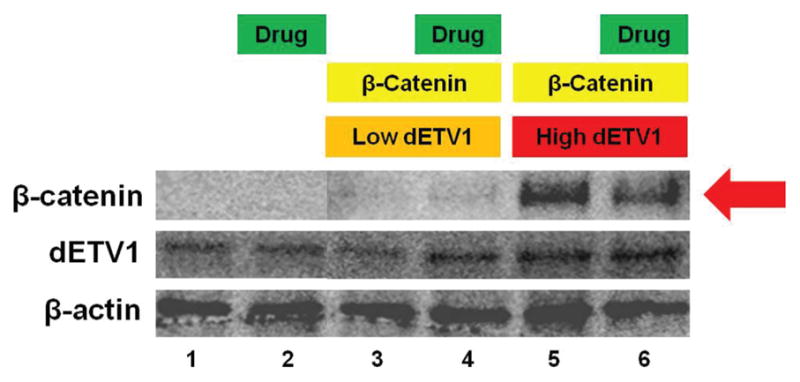
β-catenin expression after PD98059 drug treatment. Prostate cells (PNT1A) were plated in six well plates. They were then transfected with empty expression vector or expression plasmid pSG5/HA-dETV1 and pSG5/β-catenin as indicated. Cells were transfected with different concentrations of β-catenin and dETV1 (β-catenin: 0.2 μg, Low dETV1: 0.6 μg, High dETV1: 1.6 μg). After 24 hours of incubation, cells in lane 2, 4, and 6 were treated with PD98059, a kinase inhibitor and cell lysate was collected and subjected to Western blot analysis. Densitometry analysis was done with Image Gauge software.
PD98059 Cell Viability
Since PD98059 was efficiently able to suppress β-catenin accumulation, we analyzed the effectiveness of this kinase inhibitor on cell viability of prostate cancer cells. We treated LNCaP cells with different concentrations of PD98059 and assessed the drug’s effect via MTT assay after a 72-hour incubation period. The results suggest approximately 60% decreased in cell viability after 72-hour treatment with PD98059 (Fig. 6).
Figure 6.

Kinase inhibitor, PD98059, induces cell death in ETV1-positive prostate cancer (LNCaP) cells. LNCaP cells were seeded in 96 well plates. Cells were then treated with vehicle control and 10 μm, 20 μm, 30 μm, 40 μm. After 72 hours, MTT reagent (tetrazolium bromide salt) and 0.04 M HCL in Isopropanol were added. A plate reader was used at 570 nm to read the plate. Results represent mean ±S.D. Asterisk represents p < 0.01.
DISCUSSION
The conserved Wnt/β-catenin pathway regulates stem-cell pluripotency and cell decisions during embryonic development (Gerlach, 2014). β-catenin mutations in human tumors can prevent GSK-3β phosphorylation and thus lead to its aberrant accumulation (Gerlach, 2014). Up-regulation of β-catenin in the melanoma cell lines (928 and 1335) may have resulted from loss of WT APC, as has been proposed for colon cancer cells (Rubinfeld, 1997). These findings suggest that certain oncogenes are capable of regulating β-catenin levels, which in turn play a crucial role in many cell regulatory activities ranging from embryonic development to adult homeostasis. Another ETS related gene, Fli-1, is able to downregulate RXR activity by sequestering the transcriptional factor CBP through protein–protein interactions (Ramakrishnan, 2004). Thus, the transcriptional inhibitory properties of Fli-1 play an important role in tumorigenesis of Fli-1-positive Ewing sarcomas (Ramakrishnan, 2004). Our observation that ETV1 inhibits β-catenin activity came as a surprise because activation of β-catenin pathway is seen in a variety of human cancers (Nakata, 2015; Reya, 2005). Therefore, it appears that targeting β-catenin activity may also lead to inhibition in tumorigenicity suggesting that homeostasis of β-catenin activity is important in regulating cell growth and development.
After transfecting dETV1 along with β-catenin into different cell lines (COS-1, PNT1A, and LNCaP), we have observed an overall increase of β-catenin expression/accumulation (2–3 fold) as compared to control where we transfected plasmid carrying β-catenin alone into cells. These results suggest that increased β-catenin accumulation within the cells was an oncogenic effect of dETV1. To our knowledge, this is the first demonstration that dETV1 enhances β-catenin levels so dramatically. Recently, ETV1 expression was detected in about 40% of all melanomas examined (Jané-Valbuena, 2010). Upregulation of β-catenin has also been observed in several melanoma cell lines, the reasons for which remain unclear (Rubinfeld, 1997). However, given our results, it is possible that ETV1 may be responsible for the aberrant accumulation of β-catenin in prostate cancer and numerous melanomas. Our results raised the question of how β-catenin activity could be decreasing at the same time expression levels of β-catenin are increasing in ETV1-positive prostate cancers. Others have previously shown that some oncoproteins were capable of inducing post-translational modification of target proteins (Ramakrishnan, 2004). We reasoned that it is possible that ETV1 may induce modification of β-catenin post-translationally, which results in decreased activity. Furthermore, this may lead to accumulation of mature inactive β-catenin molecules which compete with active nascent β-catenin molecules for the formation of TCF-4-mediated transcriptional complex in ETV1-positive prostate cancer cells.
It is important to note that in normal Wnt signaling, β-catenin is directed toward degradation upon being properly phosphorylated; however, recent studies demonstrate that deregulated phosphorylation may lead to accumulation. For example, phosphorylated β-catenin released from Axin1 complex may fail to undergo immediate degradation (Garlach, 2014). Garlach’s work also suggests that the degradation of phosphorylated β-catenin is restrained in Wnt-stimulated cells. In certain circumstances, Ser-9 phosphorylation of GSK-3 could stabilize β-catenin (Haq, 2003). The Wnt-1 proto-oncogene also stabilizes β-catenin in mammalian cell culture and promotes tumor formation (Rubinfeld, 1997). Therefore, we tested whether β-catenin undergoes post-translational modification through phosphorylation using alkaline phosphatase treatment. In this phosphatase assay, we have observed that accumulated β-catenin in the presence of ETV1 undergoes dephosphorylation and partial degradation as indicated by thinner and lesser intense band of β-catenin in the Western blot (Fig. 3). These results suggest that ETV1 induces phosphorylation of β-catenin by activation of protein kinases. In support of this conclusion, it was recently shown that kinases were activated in prostate cancers (Faltermeier, 2015).
Kinases, such as AKT, IKKα, and MEK1/2 among many others, are activated in human cancers (Faltermeier, 2015; Yamaguchi, 2012). AKT and IKKα also stabilize β-catenin levels. β-catenin is directly phosphorylated at S552 by AKT, which results in the binding of 14-3-3ζ to β-catenin and stabilization of β-catenin (Fang, 2007; Tian, 2004; Yamaguchi, 2012). Ubiquitination and degradation of β-catenin are prevented when β-catenin is phosphorylated by IKKα (Yamaguchi, 2012). Taken together, these findings suggest that kinase-mediated phosphorylation may be responsible for stabilization of β-catenin. Gene expressions that are regulated by MEK were shown to be activated by ETS proteins (Hollenhorst, 2011; Zhang, 2014). Furthermore, MAP kinase gene is activated in the presence of ETV1 in prostate cells (Hollenhorst, 2011). Therefore, ETV1 may be a critical factor that activates selective kinases that are able to phosphorylate and stabilize β-catenin. Based on this, we set out to determine whether ETV1 is capable of stabilizing β-catenin levels using a cycloheximide chase assay. Indeed, with the introduction of dETV1, there was a delay in β-catenin degradation compared to control. β-catenin levels in control PC-3 cells was degraded as expected in a time-dependent manner after addition of cycloheximide. However, with the addition of dETV1, β-catenin stabilization was observed even at the six-hour period demonstrating that ETV1 can stabilize β-catenin (Fig. 4). Hence, we propose this as a potential mechanism whereby ETV1 plays a role in prostate tumorigenesis.
From the collective data, we hypothesized that normalizing β-catenin activity may restore normal cellular function and enhance growth arrest in prostate cancer cells as shown in Figure 8. β-catenin activity may be modulated by MEK activity, which was shown to be inhibited by PD98059, a kinase inhibitor (Zhang, 2013). Because we postulated β-catenin may be stabilized by a kinase pathway, we used a selective inhibitor of MEK; PD98059 to reverse the effect of ETV1 induced stabilization of β-catenin. Upon treatment with PD98059, β-catenin protein levels decreased markedly even in the presence of dETV1 (Fig. 5). These results suggest that the specific kinase inhibitor, PD98059 interferes with phosphorylation of β-catenin induced by dETV1. In order to test whether PD98059 can be used as a therapeutic drug to target ETV1-positive prostate cancer cells, we tested the effect of PD98059 on the viability of ETV1-positive prostate cancer cells using MTT assay. PD98059 was very effective in targeting the LNCaP cells after 72-hour treatment at micromolar concentrations with an IC50 value of 4.14 μM shown in Figures 6 and 7. The kinase inhibitory function of PD98059 was possibly the reason for lowering the β-catenin expression in prostate cancer cells where there was a high expression of the ETS transcription factor, dETV1. Therefore, we suggest PD98059 or related kinase inhibitors can be used as a therapeutic agent on ETV1-positive prostate and other cancers.
Figure 8.

Molecular mechanism of regulation for β-catenin. ETV1 activates MEK-mediated phosphorylation of β-catenin. This post-translational modification of β-catenin leads to its over-stabilization. MEK inhibitor, PD98059, reverses ETV1-induced phosphorylation of β-catenin leading to normal stabilization of β-catenin, and also induces cell death of ETV1-positive prostate cancer cells.
Figure 7.

PD98059 IC50 required for targeting prostate cancer (LNCaP) cells. Inhibitory Concentration 50 (IC50) of PD98059 on LNCaP cells for 72 hours with different concentrations 10 μM, 20 μM, 30 μM, and 40 μM. Viability of LNCaP cells were assessed by MTT assay. For each concentration of PD98059, viabilities are given as percentages. Gnuplot software was used to calculate IC50 values. For LNCaP cells, IC50 value of PD98059 is 4.14 μM.
Further elucidation on the role of ETV1 in Wnt/β-catenin pathway will allow us to develop better diagnostic tools, which can be used as a biomarker for ETV1-positive prostate cancers. Also, discovery of targeting different pathways will enable us to create novel therapeutic agents such as kinase inhibitors that are highly selective. Our findings in this area will have a profound impact on prevention and treatment of prostate cancer which may help to reduce health disparity seen in minority patients.
Acknowledgments
We thank all the members of Drs. Reddy and Rao laboratories. This research is supported by DOD W81XWH-09-1-0236, DOD W81XWH-08-1-0628, DOD W81XWH-10-1-0418 grants, NIH 2U54CA118948, 3U54CA118638-05S1 to Dr. Reddy, GCC Distinguished Cancer Scholar Awards to Dr. Reddy and Dr. Rao, RCMI, U54 RR026137 and U54 MD007588. Sharif Morsalin is supported by Research Initiative for Scientific Enrichment (RISE) program.
List of Abbreviations
- CBP
CREB-binding protein
- ERG
ETS Related Gene
- ERK
Extracellular signal-regulated kinases
- Fli-1
Friend leukemia virus integration 1
- GSK-3β
Glycogen synthase kinase 3 beta
- HA
Hemagglutinin
- IKKα
IκB kinase alpha
- MAPK
Mitogen-activated protein kinases
- MEK
MAPK/Erk kinase
References
- Bakhsh MU, Lee S, Ahmad S, Takher J, Pareek A, Syed U, Seashore J, Szemraj E. Should prostate cancer be considered as a differential diagnosis in patients with osteolytic bone lesions? Eur Rev Med Pharmacol Sci. 2015;19:4791–94. [PubMed] [Google Scholar]
- Chi P, Chen Y, Zhang L, Guo X, Wongvipat J, Shamu T, Fletcher JA, Dewell S, Maki RG, Zheng D, Antonescu CR, Allis CD, Sawyers CL. ETV1 is a lineage survival factor that cooperates with KIT in gastrointestinal stromal tumours. Nature. 2010;467:849–53. doi: 10.1038/nature09409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du C, Zhang C, Li Z, Biswas MH, Balaji KC. Beta-catenin phosphorylated at threonine 120 antagonizes generation of active beta-catenin by spatial localization in trans-Golgi network. PLoS One. 2012;7:e33830. doi: 10.1371/journal.pone.0033830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faltermeier CM, Drake JM, Clark PM, Smith BA, Zong Y, Volpe C, Mathis C, Morrissey C, Castor B, Huang J, Witte ON. Functional screen identifies kinases driving prostate cancer visceral and bone metastasis. Proc Natl Acad Sci USA. 2016;113:E172–E181. doi: 10.1073/pnas.1521674112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang D, Hawke D, Zheng Y, Xia Y, Meisenhelder J, Nika H, Mills GB, Kobayashi R, Hunter T, Lu Z. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J Biol Chem. 2007;282:11221–29. doi: 10.1074/jbc.M611871200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Xu H, Yang C, Kayarthodi S, Matthews R, Rao VN, Reddy ES. Molecular mechanism of activation of transforming growth factor beta/smads signaling pathway in Ets related gene-positive prostate cancers. J Pharm Sci Pharmacol. 2014;1:82–85. doi: 10.1166/jpsp.2014.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Xu H, Yang C, Morsalin S, Kayarthodi S, Rungsrisuriyachai K, Gunnal U, Mckenzie B, Rao VN, Reddy ES. Ets related gene and Smad3 proteins collaborate to activate transforming growth factor-beta mediated signaling pathway in ETS related gene-positive prostate cancer cells. J Pharm Sci Pharmacol. 2014;1:175–81. doi: 10.1166/jpsp.2014.1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortson WS, Kayarthodi S, Fujimura Y, Xu H, Matthews R, Grizzle WE, Rao VN, Bhat GK, Reddy ES. Histone deacetylase inhibitors, valproic acid and trichostatin-A induce apoptosis and affect acetylation status of p53 in ERG-positive prostate cancer cells. Int J Oncol. 2011;39:111–19. doi: 10.3892/ijo.2011.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasi TD, Boormans J, Hermans K, Trapman J. ETS fusion genes in prostate cancer. Endocr Relat Cancer. 2014;21:R143–R152. doi: 10.1530/ERC-13-0390. [DOI] [PubMed] [Google Scholar]
- Gerlach JP, Emmink BL, Nojima H, Kranenburg O, Maurice MM. Wnt signalling induces accumulation of phosphorylated beta-catenin in two distinct cytosolic complexes. Open Biol. 2014;4:140120. doi: 10.1098/rsob.140120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq S, Michael A, Andreucci M, Bhattacharya K, Dotto P, Walters B, Woodgett J, Kilter H, Force T. Stabilization of beta-catenin by a Wnt-independent mechanism regulates cardiomyocyte growth. Proc Natl Acad Sci USA. 2003;100:4610–15. doi: 10.1073/pnas.0835895100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermans KG, van der Korput HA, van Marion R, van de Wijngaart DJ, Zielvan der Made A, Dits NF, Boormans JL, van der Kwast TH, van Dekken H, Bangma CH, Korsten H, Kraaij R, Jenster G, Trapman J. Truncated ETV1, fused to novel tissue-specific genes, and full-length ETV1 in prostate cancer. Cancer Res. 2008;68:7541–49. doi: 10.1158/0008-5472.CAN-07-5930. [DOI] [PubMed] [Google Scholar]
- Hollenhorst PC, Ferris MW, Hull MA, Chae H, Kim S, Graves BJ. Oncogenic ETS proteins mimic activated RAS/MAPK signaling in prostate cells. Genes Dev. 2011;25:2147–57. doi: 10.1101/gad.17546311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Waknitz M. ETS gene fusions and prostate cancer. Am J Transl Res. 2009;1:341–51. [PMC free article] [PubMed] [Google Scholar]
- Im YH, Kim HT, Lee C, Poulin D, Welford S, Sorensen PH, Denny CT, Kim SJ. EWS-FLI1, EWS-ERG, and EWS-ETV1 oncoproteins of Ewing tumor family all suppress transcription of transforming growth factor beta type II receptor gene. Cancer Res. 2000;60:1536–40. [PubMed] [Google Scholar]
- Jané-Valbuena J, Widlund HR, Perner S, Johnson LA, Dibner AC, Lin WM, Baker AC, Nazarian RM, Vijayendran KG, Sellers WR, Hahn WC, Duncan LM, Rubin MA, Fisher DE, Garraway LA. An oncogenic role for ETV1 in melanoma. Cancer Res. 2010;70:2075–84. doi: 10.1158/0008-5472.CAN-09-3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayarthodi S, Fujimura Y, Fang J, Morsalin S, Rao VN, Reddy ES. Anti-epileptic drug targets ewing sarcoma. J Pharm Sci Pharmacol. 2014;1:87–100. doi: 10.1166/jpsp.2014.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larue L, Luciani F, Kumasaka M, Champeval D, Demirkan N, Bonaventure J, Delmas V. Bypassing melanocyte senescence by beta-catenin: A novel way to promote melanoma. Pathol Biol (Paris) 2009;57:543–47. doi: 10.1016/j.patbio.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Nakata A, Yoshida R, Yamaguchi R, Yamauchi M, Tamada Y, Fujita A, Shimamura T, Imoto S, Higuchi T, Nomura M, Kimura T, Nokihara H, Higashiyama M, Kondoh K, Nishihara H, Tojo A, Yano S, Miyano S, Gotoh N. Elevated beta-catenin pathway as a novel target for patients with resistance to EGF receptor targeting drugs. Sci Rep. 2015;5:13076. doi: 10.1038/srep13076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S, Shin S, Lightfoot SA, Janknecht R. 14-3-3 proteins modulate the ETS transcription factor ETV1 in prostate cancer. Cancer Res. 2013;73:5110–19. doi: 10.1158/0008-5472.CAN-13-0578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulo P, Barros-Silva JD, Ribeiro FR, Ramalho-Carvalho J, Jeronimo C, Henrique R, Lind GE, Skotheim RI, Lothe RA, Teixeira MR. FLI1 is a novel ETS transcription factor involved in gene fusions in prostate cancer. Genes Chromosomes Cancer. 2012;51:240–49. doi: 10.1002/gcc.20948. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R, Fujimura Y, Zou JP, Liu F, Lee L, Rao VN, Reddy ES. Role of protein–protein interactions in the anti-apoptotic function of EWS-Fli-1. Oncogene. 2004;23:7087–94. doi: 10.1038/sj.onc.1207927. [DOI] [PubMed] [Google Scholar]
- Rao VN, Papas TS, Reddy ES. ERG, a human ETS- related gene on chromosome 21: Alternative splicing, polyadenylation, and translation. Science. 1987;237:635–39. doi: 10.1126/science.3299708. [DOI] [PubMed] [Google Scholar]
- Reddy ES, Rao VN. ERG, an ETS-related gene, codes for sequence-specific transcriptional activators. Oncogene. 1991;6:2285–89. [PubMed] [Google Scholar]
- Reddy ES, Rao VN, Papas TS. The ERG gene: A human gene related to the ETS oncogene. Proc Natl Acad Sci USA. 1987;84:6131–35. doi: 10.1073/pnas.84.17.6131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Robbins P, El-Gamil M, Albert I, Porfiri E, Polakis P. Stabilization of beta-catenin by genetic defects in melanoma cell lines. Science. 1997;275:1790–92. doi: 10.1126/science.275.5307.1790. [DOI] [PubMed] [Google Scholar]
- Satyamoorthy K, Herlyn M. Cellular and molecular biology of human melanoma. Cancer Biol Ther. 2002;1:14–17. doi: 10.4161/cbt.1.1.32. [DOI] [PubMed] [Google Scholar]
- Shin S, Oh S, An S, Janknecht R. ETS variant 1 regulates matrix metalloproteinase-7 transcription in LNCaP prostate cancer cells. Oncol Rep. 2013;29:306–14. doi: 10.3892/or.2012.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spans L, Clinckemalie L, Helsen C, Vanderschueren D, Boonen S, Lerut E, Joniau S, Claessens F. The genomic landscape of prostate cancer. Int J Mol Sci. 2013;14:10822–51. doi: 10.3390/ijms140610822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian Q, Feetham MC, Tao WA, He XC, Li L, Aebersold R, Hood L. Proteomic analysis identifies that 14-3-3zeta interacts with beta-catenin and facilitates its activation by Akt. Proc Natl Acad Sci USA. 2004;101:15370–75. doi: 10.1073/pnas.0406499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster MR, Kugel CH, III, Weeraratna AT. The Wnts of change: How Wnts regulate phenotype switching in melanoma. Biochim Biophys Acta. 2015;1856:244–51. doi: 10.1016/j.bbcan.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Hsu JL, Hung MC. Regulation of ubiquitination-mediated protein degradation by survival kinases in cancer. Front Oncol. 2012;2:15. doi: 10.3389/fonc.2012.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Zhang H, Wang N, Zhao C, Zhang H, Deng F, Wu N, He Y, Chen X, Zhang J, Wen S, Liao Z, Zhang Q, Zhang Z, Liu W, Yan Z, Luu HH, Haydon RC, Zhou L, He TC. Modulation of beta-catenin signaling by the inhibitors of MAP kinase, tyrosine kinase, and PI3-kinase pathways. Int J Med Sci. 2013;10:1888–98. doi: 10.7150/ijms.6019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Gu ML, Zhou XX, Ma H, Yao HP, Ji F. Altered expression of ETV1 and its contribution to tumorigenic phenotypes in gastrointestinal stromal tumors. Oncol Rep. 2014;32:927–34. doi: 10.3892/or.2014.3281. [DOI] [PubMed] [Google Scholar]
- Zynda E, Jackson MW, Bhattacharya P, Kandel ES. ETV1 positively regulates transcription of tumor suppressor ARF. Cancer Biol Ther. 2013;14:1167–73. doi: 10.4161/cbt.26883. [DOI] [PMC free article] [PubMed] [Google Scholar]