

Abstract
Structural alerts are widely accepted in chemical toxicology and regulatory decision support as a simple and transparent means to flag potential chemical hazards or group compounds into categories for read-across. However, there has been a growing concern that alerts disproportionally flag too many chemicals as toxic, which questions their reliability as toxicity markers. Conversely, the rigorously developed and properly validated statistical QSAR models can accurately and reliably predict the toxicity of a chemical; however, their use in regulatory toxicology has been hampered by the lack of transparency and interpretability. We demonstrate that contrary to the common perception of QSAR models as “black boxes” they can be used to identify statistically significant chemical substructures (QSAR-based alerts) that influence toxicity. We show through several case studies, however, that the mere presence of structural alerts in a chemical, irrespective of the derivation method (expert-based or QSAR-based), should be perceived only as hypotheses of possible toxicological effect. We propose a new approach that synergistically integrates structural alerts and rigorously validated QSAR models for a more transparent and accurate safety assessment of new chemicals.
Keywords: structural alerts, read-across, QSAR, toxicity, green chemistry
Graphical abstract

Introduction
Over the past few decades, environmental chemists have been under increasing political and public pressure to ensure that hazardous chemicals must be identified and replaced by “greener”, i.e., safer alternatives1,2 while avoiding animal testing of every chemical, which is also both financially and experimentally unsustainable.3 In addition, animal models have received much criticism as being unethical, exorbitantly expensive, and unreliable for extrapolating results and findings to humans.4
Computational models have earned recognition as reliable, fast, and inexpensive alternatives for the toxicity evaluation of chemicals during the early stages of drug discovery or environmental safety assessment.5 This assessment often relies on structural alerts, chemical grouping, and read-across. Structural alerts,6 otherwise known as “expert rules”, are molecular substructures that are associated with a particular adverse outcome.7 The popularity of alerts dates back to a series of studies published in late 80s and early 90s on chemical carcinogenicity and mutagenicity.8–12 As the name suggests, expert rules are based on human expertise and are intended to reflect the chemical basis of the mechanism of action or, at least, the molecular initiating event in the case of more complex endpoints.13
Alerts are used to flag potential hazards and to group compounds into categories for read-across (Figure 1).14 Chemical read-across is a data gap filling procedure to assess certain endpoint effect of a chemical by using data for the same endpoint from another chemical (or a group of chemicals), which is (are) considered structurally similar.15 These methods have earned acceptance among toxicologists due to their simplicity, transparency, and ease ofinterpretation.16 However, although chemical read across has been accepted by regulatory agencies, there have been observations that this approach is prone to bias.17–20 On the other hand, though supported by rigorously evaluated statistical significance, Quantitative Structure-Activity Relationships (QSAR) modeling is usually considered as a “black box”21, referencing the perceived lack of interpretability (Figure 1).
Figure 1.

Contrasting alerts- and QSAR-based predictions in chemical safety assessment. Structural alerts are derived on small datasets and used in read-across for flagging unsafe compounds. QSAR models are developed on larger datasets and used to make binary, categorical, or quantitative prediction of compound toxicity.
The use of structural alerts and QSAR models (often collectively referred to as (Q)SAR) have become a major concept in chemical safety assessment and regulatory decision support since the acceptance of Registration, Evaluation, Authorization, and Restriction of Chemicals (REACH) legislation in 2006 by the European Union.22 This law compels manufacturers to provide detailed information on chemicals that are manufactured, marketed, or imported. In the United States, the Environmental Protection Agency (EPA) has been using (Q)SAR methods to support a weight-of-evidence in the hazard assessment of chemicals under the US Toxic Substances Control Act (TSCA).23
Qualitative approaches, such as alerts, chemical grouping, and read-across, have been incorporated into several computerized expert systems, often employing additional layers of secondary modulating effects around each generic expert alert (e.g., OECD QSAR Toolbox24, OCHEM ToxAlerts25, Lhasa’s Derek26, etc.). Also, because analyzing and processing large volumes of experimental data by human experts can be very slow, current expert-based systems often use statistical approaches to discover candidate substructures strongly associated with the target activity. These candidate substructures are then reviewed and curated by experts in the field to provide mechanistic interpretation.
Albeit less commonly appreciated, statistically-based QSAR models can indeed afford mechanistic interpretation.27 However, the difficulty of interpreting QSAR models, especially by non-experts in the field, has led regulatory agencies to prefer the use of simple structural alerts for the prediction of various properties. On the other hand, the ability of QSAR to provide a reliable quantitative assessment of the toxicity potential of a chemical is highly advantageous. Hence, there is a strong need for the development of a method that would combine the predictive power of QSAR models while maintaining transparency for expert evaluation. Recently, integrative approaches of QSAR and read-across have been proposed by our28 and other groups.29–31 Specifically, we have proposed chemical-biological read-across (CBRA), which combines quantitative toxicity prediction with a visualization methodology, ensuring both high predictivity and interpretability of models.28
In this paper, we have compared, contrasted, and attempted to consolidate the two major modern approaches to toxicity assessment. Our objectives were to: (i) investigate the reliability of structural alerts for chemical toxicity and safety assessment; (ii) highlight the advantages and liabilities of both structural alerts and QSAR modeling; (iii) alarm users about the danger of blindly using structural alerts as sole toxicity predictors; and (iv) introduce a new toxicity prediction framework integrating SAR rules and QSAR-based approaches to increase both the transparency and accuracy of predictions.
How reliable are alerts for predicting chemical toxicity?
It is worth noting at the onset of this perspective that terms, such as “toxic/non-toxic”, “sensitizer/non-sensitizer”, and “safe/unsafe” have been used in this paper for simplicity. We fully realize that defining respective biological phenomena in absolute terms is an oversimplification, since the assignment of a chemical to a category, e.g., toxic or non-toxic, depends on the dose or exposure and on the designated threshold of toxicity for a particular endpoint.
As mentioned in the Introduction, alerts have been widely accepted because they can be easily generated and interpreted. These advantages notwithstanding, there has been a growing concern that alerts have limited utility for accurate toxicity assessment. One obvious concern is that most alerts represent functional groups or substructures that can be found in many, both toxic and non-toxic compounds, leading to predictions with overly high sensitivity18 but low specificity. As we discuss below, this phenomenon may be because chemical properties of substructures depend on other groups in the molecule that could influence the reactivity of the substructural alert.
This flaw in the prediction accuracy has caused controversy in the evaluation of the power of alerts by the scientific community. The Organization for Economic Co-operation and Development Organization (OECD) characterizes read-across as a technique used to predict a determined endpoint, but requires that expert judgement is needed and that a justification should be provided.32 The OECD sponsored the development of QSAR Toolbox24, a software application to predict (eco)toxicity based on chemical grouping and read-across that leaves the assessment of the prediction to the end user.33
Another software, Toxtree (http://toxtree.sourceforge.net/), has a skin sensitization module that implements structural alerts.34 Contributing to the confusion about the significance of alerts as toxicity predictors, the developers of Toxtree recently placed a statement on their website35 that they changed the name of the module from “Skin sensitization alerts” to “Skin sensitization reactivity domain” explaining that alerts provide only grouping into a reactivity mode of action and do not predict skin sensitization potential. Although not explicitly reported, this conspicuous change in nomenclature is most likely due to pitfalls and deficiencies in the method. For instance, the use of simple categories led to the misclassification of 25% of compounds evaluated for the respiratory sensitization, including non-sensitizers containing alerts, and sensitizers, that did not contain alerts.36
In another example, Hewitt et al.37 developed new structural alerts for hepatotoxicity but also found that many alerts were likewise present in non-hepatotoxic drugs. Ironically, the authors still declared that they had developed a scheme capable of generating mechanistically-supported structural alerts suitable for identifying chemicals with hepatotoxic potential. Nevertheless, recognizing the inherent contradiction of their findings, they stated that these alerts are not a model to be used for the prediction of hepatotoxicity. In another study,38 the authors could not assign an unambiguous mode of action for most of the identified liver and kidney toxicity alerts. In the paper describing the ToxAlerts tool25, the developers have commented on the limitation of alerts as a helpful technique for flagging potentially toxic compounds but not necessarily predicting toxicity.
Contrasting alerts and QSAR-based predictions
Skin sensitization, being a major environmental and occupational health hazard,39 represents a highly illustrative opportunity to contrast the results of QSAR modeling and alert-based approaches. The aforementioned OECD QSAR Toolbox has a special module for this endpoint, which makes the comparison straightforward.
Previously, we have developed and published skin sensitization QSAR models17 that we compare here with the outcome of the LLNA node of OECD QSAR Toolbox24 and OCHEM ToxAlerts25 web server. To make the comparison fair, we only use compounds (i) not present in databases and (ii) inside the applicability domain of our models. Therefore, 90 compounds (38 sensitizers and 52 non-sensitizers) were used for comparison with QSAR Toolbox, and 246 compounds (160 sensitizers and 86 non-sensitizers) were used for comparison with ToxAlerts. Our models showed a significantly higher prediction accuracy for the same sets of external compounds when compared to both alerts-based tools as evaluated by Positive Predictive Value (PPV), Specificity, Negative Predictive Value (NPV), and Correct Classification Rate (CCR) (Table 1). Although predictions made with QSAR Toolbox and ToxAlerts show higher sensitivity than our models when evaluating the same set of structures, our models featured a much higher PPV. These results indicate that the probability of correctly classifying sensitizers is much higher using QSAR models and that alert-based predictions have a bias towards sensitizers.
Table 1.
Comparison of the prediction accuracies of QSAR models, skin sensitization module of the OECD QSAR Toolbox, and OCHEM ToxAlerts.
| QSAR Toolbox | |||||
| QSAR vs. SAR rule tool | CCR | Sensitivity | PPV | Specificity | NPV |
| QSAR | 0.74 | 0.50 | 0.94 | 0.98 | 0.75 |
| QSAR Toolbox | 0.46 | 0.53 | 0.38 | 0.38 | 0.53 |
| OCHEM ToxAlerts | |||||
| QSAR vs. SAR rule tool | CCR | Sensitivity | PPV | Specificity | NPV |
| QSAR | 0.80 | 0.74 | 0.90 | 0.85 | 0.64 |
| ToxAlerts | 0.60 | 0.84 | 0.71 | 0.36 | 0.55 |
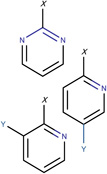
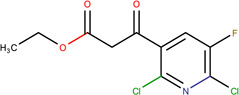
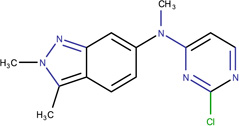
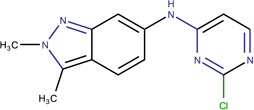
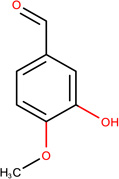
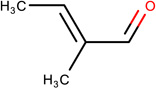
In Table 2, we show three examples of structural alerts flagged by QSAR Toolbox or by ToxAlerts: activated pyridine/pyrimidine, formyl group (aldehydes), and aromatic amines. In addition, we compare experimental toxicity assessment with predictions obtained by our QSAR models for compounds containing the alert substructures obtained by QSAR Toolbox or ToxAlerts. As one can see, all compounds with an activated pyridine/pyrimidine subgroup are non-sensitizers in LLNA assays. These compounds were flagged as sensitizers by QSAR Toolbox, but correctly predicted as non-sensitizers by the QSAR models. Molecules containing two other alerts flagged by ToxAlerts (formyl group and aromatic amines) are not always the sensitizers: examples are provided by 3-hydroxy-4-methoxybenzaldehyde (correctly predicted by QSAR as a non-sensitizer) and (2E)-2-methyl-2-butenal (incorrectly predicted by QSAR as a sensitizer) that contain the formyl group. We also identified the following eleven non-sensitizers containing five or more ToxAlerts each: 3-hydroxy-4-methoxybenzaldehyde, vanillin (ten alerts both), and ethyl vanillin (nine alerts) (see Supplementary Information). These results highlight that the presence of even multiple structural alerts for skin sensitization does not automatically suggest that the respective compounds are toxic and as such should be avoided in commercial products. At the same time, our results show that externally validated QSAR models afford higher accuracy of assessing chemical toxicity than structural alerts.
Table 2.
Examples of structural alerts and comparison of experimental and predicted skin sensitization effects for alert-containing compounds.
| Alerts | Compounds containing the alert substructure | ||
|---|---|---|---|
 |
 |
 |
 |
|
Activated pyridine / pyrimidine QSAR Toolbox |
Ethyl 2,6-dichloro-5-fluoro- β-oxo-3-pyridinepropanoate LLNA: non-sensitizer QSAR: non-sensitizer QSAR Toolbox: sensitizer |
N-(2-chloro-4-pyrimidinyl)- N-2,3-trimethyl-2H-indazol- 6-amine LLNA: non-sensitizer QSAR: non-sensitizer QSAR Toolbox: sensitizer |
N-(2-chloro-4-pyrimidinyl)- 2,3-dimethyl-2H-indazol-6- amine LLNA: non-sensitizer QSAR: non-sensitizer QSAR Toolbox: sensitizer |
 |
 |
 |
 |
|
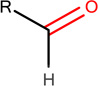
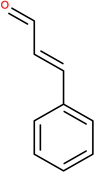
Formyl group OCHEM ID: TA264 |
Cinnamic aldehyde LLNA: Sensitizer ToxAlerts: Sensitizer QSAR: Sensitizer |
3-hydroxy-4- methoxybenzaldehyde LLNA: Non-sensitizer ToxAlerts: Sensitizer QSAR: Non-sensitizer |
(2E)-2-methyl-2-butenal LLNA: Non-sensitizer ToxAlerts: Sensitizer QSAR: Sensitizer |
 |
 |
 |
 |
|
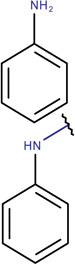
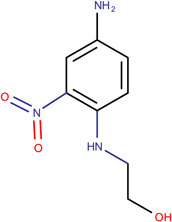
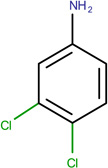
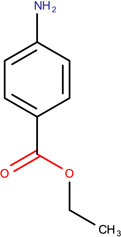
Aromatic amines OCHEM ID: TA311 |
2-(4-Amino-2nitro- phenylamino)-ethanol LLNA: Sensitizer ToxAlerts: Sensitizer QSAR: Sensitizer |
3,4-dichloroaniline LLNA: Sensitizer ToxAlerts: Sensitizer QSAR: Non-sensitizer |
Benzocaine LLNA: Non-sensitizer ToxAlerts: Sensitizer QSAR: Non-sensitizer |
Another example to contrast alerts and QSAR-based predictions is provided by two well-known alerts for the hERG blockage. The hERG K+ channels (or simply hERG) are known to have an essential contribution to heartbeat regulation, and its blockage is closely associated with lethal cardiac arrhythmia.40 This channel has been earmarked as one of the most important anti-targets to be considered in the early stages of drug development due to its high ligand promiscuity.41 Recently, we developed a freely available webserver for the early identification of putative hERG blockers and non-blockers named Pred-hERG (http://labmol.farmacia.ufg.br/predherg/), based on binary and multiclass QSAR models.42,43
The presence of a tertiary amine is one of the most well defined substructure alert for hERG blockage.44,45 Previously,43 we showed sixteen examples of changes in electronic and steric environment of the tertiary amine that could transform a potent hERG blocker to a less potent blocker or even to a non-blocker. In the studied dataset, 5,984 compounds (3,436 blockers and 2,548 non-blockers) contained at least one tertiary amine. Employing this alert only, the positive predictive value (PPV) is 0.57 while QSAR will give a PPV of 0.86. Using tertiary amines to flag hERG blockers would result in a high false positive outcome. Another alert for hERG, arylchloride, follows the same trend. In the studied dataset, 1,277 compounds with at least one arylchloride substructure (854 blockers and 423 non-blockers) were found. Using this alert only, the PPV is 0.67, while QSAR yields a PPV of 0.80. The use of this alert as a predictor results in a CCR of 0.50, whereas the QSAR approach, which correctly predicts non-blockers, results in CCR of 0.78.
In another example, we employed structural alerts to analyze the difference between safe and unsafe drugs.46 This previously described dataset46 included thirteen withdrawn drugs and seven drugs currently on the market. All withdrawn drugs display significant toxicity and side effect. Here, we have used predictions made by previously published QSAR models46 and profiled the compounds with the alerts from the QSAR Toolbox.
The calculated toxicity alerts for the marketed and withdrawn drugs are shown in Table S1 (see Supplementary Information). All withdrawn drugs were predicted as “High, Class III” by toxic hazard models. Only three marketed drugs were expectably predicted as “Low, Class I”. However, four marketed drugs (Valtrex, Microzide, Neurontin and Enoxaparin) were classified as dangerous (Class III). The majority of both the withdrawn and marketed drugs were considered as safe (no alerts) by “Carcinogenicity”, “DNA alerts”, “In vitro mutagenicity”, and both types of “Protein binding alerts”. On the other hand, all marketed drugs (excluding Fenfluramine and Valtrex) were predicted to have the “in vivo mutagenicity” alerts. Thus, all 20 drugs were found to contain at least one alert that categorized them as unsafe. These results show that it is impossible to distinguish the “safe” marketed from “unsafe” withdrawn drugs using the established toxicity alerts, providing yet another illustration of the weakness of structural alerts as reliable drug safety predictors. At the same time, the toxicity of these drugs was reliably assessed by QSAR models (see Table S1).
Complexity of chemical structure questions the significance of single alerts
Structural alerts highlight the importance of specific structural features as determinants of compound toxicity.14 However, biological effects are measured for the entire molecules, raising doubts that a fragment can always adequately define the property of the whole molecule. We examine this interplay between the fragment and the whole molecular structure using a few case studies.
Previously, we have developed QSAR models for predicting binding affinities of ligands to thyroid hormone receptor (THR).47 By interpreting developed models,27 we can gain insight into structural features responsible for the binding affinity of ligands to receptors and guide structural modifications that will modulate binding affinity of ligands. Figure 2 shows that a small change such as replacement of carboxymethyl by carboxyethyl substituent in the same position in a molecule causes changes in many descriptor values. Thus, when a new fragment is added, many descriptors can change their values, reflecting the interconnectivity and mutual influence of all fragments in the molecule.47
Figure 2.

Comparison of the profiles of the most important descriptors for two strong THR binders that differ by only one -CH2- fragment.
The same concept of the mutual influence of substituents in the molecule can be much better illustrated by several studies on the effects of substituents on the toxicity of the nitroaromatics.48–51 In these studies, we have developed QSAR models for rat acute toxicity of nitroaromatics using Simplex Representation of Molecular Structure (SiRMS) descriptors52 and Partial Least Squares modeling approach.53 Interpretation of developed models following the protocol described elsewhere54 demonstrated that although an aromatic ring with a nitro group or groups usually increases toxicity, its contribution varies widely depending on the nature and number of other substituents in the aromatic ring. Thus, our main finding was that although an aromatic nitro group is considered as a toxicophore55, its contribution to toxicity could be significantly modified by other substituents.
This finding can be illustrated using an example of chlorosubstituted nitrobenzenes (Figure 3). Although one could expect that an increase of the number of chlorine substituents would result in an increase of toxicity, the influence of chlorine is ambiguous and strongly depends on the structural environment. For instance, a chlorine atom in the ortho-position to the nitro group is present in both the most toxic compounds (2,6-dichloronitrobenzene) and the least toxic compounds (2,3,5-trichloronitrobenzene). Overall, the insertion of a chlorine substituent in nitrobenzene increases its toxicity; the ortho-isomer is the most toxic. Introduction of the second chlorine results in the large toxicity changes, which are observed for dichloronitrobenzenes.48 Addition of chlorine substituents decreases the difference in toxicity between the isomers. Moreover, the accumulation of chlorine atoms in the benzene ring decreases their influence on toxicity, i.e., the increase in toxicity is not proportional to the number of chlorine atoms or, even more, the addition of chlorine decreases the toxicity.48
Figure 3.

Evolution of toxicity of chloronitrobenzenes. The digits in the circles correspond to the positions of chlorines in aromatic ring.
We also analyzed the effect of sequential insertion of chlorine substituents into the benzene ring.48 In Figure 4, the toxicity of each molecule is represented as six separate contributions of the corresponding carbon of the aromatic ring and its substituent. Insertion of a chlorine atom in the ortho-position to the nitro group leads to toxicity increase in comparison with nitrobenzene. This effect is not limited to the chlorine atom only. In fact, the contributions to toxicity of all other atoms are augmented (except C–H bond in ortho-position to the C–Cl bond, Figure 4). Insertion of an additional chlorine adjacent to the previous ortho-chlorine has only a small effect on toxicity. Although the new C–Cl bond (position 3) increases the toxicity of the molecule, the contributions of the nitro group and other C–Cl fragment (position 2) have been diminished. Thus, in spite of the redistribution of influence on toxicity between different fragments of 2,3-dichloronitrobenzene, the toxicity of the whole compound hardly changes compared to 2-chloronitrobenzene. A dramatic change in toxicity was predicted for 2,3,5-trichloronitrobenzene. Substitution of hydrogen by chlorine in position 5 results in substantial lowering of toxicity. This resulted in the diminishing toxicity of all analyzed fragments, especially chlorine in position 2.48
Figure 4.

Relative influence of structural fragments on toxicity of chlorosubstituted nitrobenzenes.
Several examples discussed in this section emphasize the important conclusion that compound toxicity can be substantially affected by the mutual interference between its structural components. Moreover, individual substructures are not acting directly and independently, as is saliently presumed by the structural alerts concept. Instead, various substructures, even including distant neighbors, mutually influence their contributions. Interpretation of QSAR models could help not only to find such substructures, but also identify their preferred neighbors and their relative position in the molecule to increase or decrease the desired and undesired properties of the molecule, respectively, which provides an avenue toward computationally-driven design of green chemicals. Examples of molecular design driven by structural hypotheses generated by the interpretation of QSAR models are discussed in the next section.
Interpretation of QSAR models: Pulling the rabbit out of the black box
An important benefit of cheminformatics analysis is the use of structural alerts established from the interpretation of QSAR models and/or cluster analysis to design new compounds with improved characteristics. Although QSAR modeling is usually referred to as a “black box” approach, many studies conducted both by our group alone,27,56 or in collaboration with other groups,57,58 and by other groups59–61 have shown that the models could be interpreted in terms of structural features responsible for activity or toxicity.
Interpretation of properly validated models could result in statistically-significant structural alerts that could be very useful in the molecular design of novel compounds with desired properties. We can illustrate this point by several QSAR studies of antiviral activity, in which we successfully designed novel potent compounds using structural alerts derived from QSAR models.52,62–67 The workflow used in these studies is shown in Figure 5 using the design of agents against rhinovirus 2 as an example. First, we collected, curated, and integrated all information related to the target of interest and compounds hitting this target (Figure 5A). In the next step, QSAR models were developed and rigorously validated (Figure 5B). Then, inverse task solution was performed using one of the approaches for the interpretation of QSAR models developed by our group in previous years.56,58,66 For illustration, we color-coded atoms and structural fragments of the modeling set compounds according to their partial contributions to the overall activity of the molecule identified by QSAR models. Examples of such color-coded structures are shown in Figure 5C. Atoms and structural fragments enhancing or reducing antiviral activity are colored in red or green, respectively, whilst indifferent atoms are colored in gray. As can be seen from Figure 5C, the contribution of the same structural moiety in different molecules can vary dramatically, which demonstrates again the strong influence of structural surroundings of a fragment on the properties of molecules. Analysis of such color-coded structures and the contributions of surrounding-dependent fragments throughout the whole training set is very useful for targeted design of new compounds. Next, the average contributions of considered fragments are summarized by all properties of interest (Figure 5D). This allows the obtained trends to be transformed into structural alerts covering not only presence or absence of a certain substituent, but also its various physical-chemical properties. For instance, in the study of activity of [(biphenyloxy)propyl]isoxazole derivatives against rhinovirus, we have established the requirements for the length and charge of the terminal substituent in the molecule (Figure 5E). In the last step of our workflow (Figure 5F), these alerts were successfully used in the design of novel antiviral agents against influenza62, herpes63,64, rhinovirus65, and coxsackievirus67.
Figure 5.

A workflow for generating QSAR-based structural alerts. As an example, QSAR-based structural alerts were used to guide molecular design of antiviral agents against human rhinovirus serotype 2.
In another example of structural optimization, we used QSAR models of skin sensitization17 and skin permeability68 to delineate a putative pathway of structural optimization to improve chemical properties using both imputed data from QSAR models and experimental data. We illustrated this approach using an example of putative stepwise structural optimization of the permeability of pentanoic acid, considering experimental data and predictions using developed models (Figure 6). Starting from this compound of relatively low permeability (logKp = −2.7), several transformation steps can increase its permeability more than 10-fold and convert it to n-heptanol (logKp = −1.50). N-heptanol is predicted as a sensitizer, which is confirmed by its Material Safety Data Sheet entry,69 but it can be transformed to octanoic acid, which has similar permeability (logKp = −1.60) as n-heptanol while lacking its sensitization potential. Similar schemes concerning various environmental toxicity endpoints could be extremely useful in green chemistry to design safe schemes of synthesis of new compounds of interest.68
Figure 6.

Example of a structural transformation of pentanoic acid to octanoic acid to improve skin permeability (modified from Alves et al.68).
Integration of structural alerts and QSAR
Using the example of aquatic toxicity of nitroaromatic compounds against Tetrahymena pyriformis, we demonstrated earlier50 that, in addition to producing statistically-sound structural alerts that influence activity, it is beneficial to combine mechanism-based alerts with QSAR predictions. Structural alerts were used to classify the investigated compounds into two putative mechanisms of action (redox cyclers and nucleophilic attack). Compounds lacking these alerts could not be assigned any mechanism and formed the third subset. We succeeded in developing two robust and predictive mechanism-based (local) QSAR models for Tetrahymena pyriformis. These models, however, had a limited applicability domain. No acceptable models were built for the third subset. Next, we used all compounds to build a mechanism-free (global) consensus model, which had much better coverage, but was less predictive. Expectedly, mechanism-based local models for redox cyclers were unable to predict the toxicity of compounds acting through nucleophilic attack and vice versa. Thus, structural alerts could be useful in combination with local QSAR model to enable the correct prognosis of toxicity.
The best results were achieved by choosing the most appropriate of the two local models for predicting a new compound possessing structural alerts. If a new compound lacked the alerts, global model should be used. Compared to traditional local and global models, this approach affords the highest external predictive accuracy and the largest coverage.
Similar results were obtained for the skin sensitization dataset. Although protein binding is an initiating event for skin sensitization and protein-binding alerts are based on well-established organic chemistry principles, existing alerts alone cannot predict skin sensitizers efficiently. However, if there are enough data, alerts could be used for assigning a mechanism of action to investigate compounds and to develop local QSAR models (Table S2). The latter could be united with mechanism-ignorant global model in a consensus ensemble that will have comparable or higher predictive power and coverage than separate models (Table S3).
Another approach to overcome the drawbacks of individual structural alerts is the Chemical-Biological Read-Across (CBRA),28 which infers the activity of a compound from those of its chemical and biological analogs. CBRA assesses the similarity between chemical substances based on their (i) computed structural properties classically regarded as chemical descriptors or (ii) experimentally-obtained or predicted results of biological measurements regarded as biological descriptors. The methodology of CBRA is described elsewhere in great details.28 We conceived CBRA as a next-generation read-across to better comprehend the complexity of toxicity prediction.28
CBRA can be visually represented as a radial plot (see Figure 7). The compound of interest is represented by a large central node surrounded by additional nodes representing the k-nearest neighbors in biological (left side) and in the chemical (right side) space. Each neighbor-node is colored based on its observed activity (e.g., red = toxic, green = non-toxic). The relative position of each neighbor-node from the central compound-node is based on the Tanimoto similarity between these two compounds: the more similar two compounds are, the closer to the central node the neighbor-node is. The nearest neighbors in both chemical and biological space (i.e., shortest edges) are placed closest to the 12 o'clock position.
Figure 7.

Radial CBRA plot for eugenol. The central node representing the target compound eugenol is surrounded by biological (left side) and chemical (right side) neighbors. Jaccard distance is used to position the neighbors towards the target compound. Edges and nodes are colored according to the known activity classification (red = toxic, green = non-toxic).
CBRA plots provide a visual means of studying the structure-activity relationships for a given compound or a cluster of similar compounds. This is especially useful for understanding: (i) the neighborhood (whether there are common neighbors between the chemical space and the biological space) and (ii) the activity landscape in which the compound resides (whether neighbors have common activities). Thus, CBRA can identify activity and/or similarity cliffs, which can be leveraged to guide the design of greener chemicals.
In utilizing a similarity-weighted aggregation, CBRA maximizes the complementarities between chemical and biological data. In particular, conflicting predictions from chemical and biological models are resolved, resulting in overall accuracy gains. CBRA was compared with other hybrid approaches such as pooling the chemical and biological space into a hybrid space and straightforward ensemble modeling, which equally weigh predictions from a chemical model and a biological model. On four datasets, CBRA was equivalent to or better than the other two hybrid approaches.28
CBRA combines the simplicity and transparency of read-across methods with the benefits afforded by more sophisticated techniques such as ensemble modeling and instance-based learning.70 Another advantage is the use of not only chemical but also biological data to increase the reliability of chemical hazard assessment. The performance of CBRA may be further optimized by setting a similarity threshold or by optimizing the number of chemical or biological neighbors separately. The drawback of this method is that it requires biological data to make new predictions. However, this deficiency could be overcome by developing conventional QSAR models of all contributing biological assays as will be presented by our group in a separate publication elsewhere.
Summary
In this paper we have discussed the potential caveats of using structural alerts alone for risk assessment as well as the more reliable and yet less interpretable predictive performance of QSAR models. The simple use of alerts for chemical read-across has been pointed out as an erroneous application. Therefore, read-across should be used to help an expert to make a final decision only after analyzing all available information sources, e.g., QSAR predictions, in vitro and in vivo outcomes, etc.71–73 Obviously, only rigorously validated approaches could be used as such sources of information.
As we have shown, structural alerts are extremely promiscuous if used alone to predict biological activity and their use may be harmful in the drug discovery pipeline as well as for the safety assessment. Thus, there is a need to develop novel methods that would combine the transparency and interpretability of structural alerts with the predictive power of QSAR models.
Multiple examples of computational toxicity studies considered in this paper suggest a synergism of structural alerts and QSAR models to predict activity or toxicity. Structural alerts can affect the design of safer compounds. However, the global effect of such modification can only be predicted by a QSAR model and not by isolated alerts alone. These considerations advance the integration of structural alerts and QSAR models for designing novel compounds and predicting their toxicity.
In summary, in place of the status quo shown in Figure 1, we propose an integrative approach for safety assessment of new chemicals using both structural alerts and QSAR models (Figure 8). In this approach, the alerts serve as transparent hypotheses (which could be derived by traditional SAR analysis or model interpretation) that can be either reinforced or refuted by statistically significant and externally validated QSAR models. Any alert should be viewed as a structural hypothesis of chemical action, but its predictive power should be buttressed by QSAR predictions and, if possible, by experimental validation.
Figure 8.

Integrative approach for chemical safety assessment of new chemicals by combining structural alerts and QSAR models.
Conclusions
We position this paper as a programmatic statement that could potentially transform the mindsets of both researchers and regulators. We especially would like to stress that we did not only wish to alarm the scientific community about the limitations of structural alerts, which could be very useful for understanding of the underlying toxicity mechanisms. Our main goals were to show how the toxicity prediction should not be done by blindly relying on structural alerts as well as how to boost safety assessment by combining the strongest parts of the alerts and QSAR models.
We have demonstrated that blind reliance on structural alerts could lead researchers astray. We are not suggesting to use QSAR models instead of structural alerts. Although in this study we compared both approaches and demonstrated that “black box” QSAR predictions usually provide the user with statistically more accurate predictions, we also showed how alerts could serve as actionable structural hypotheses that could be validated by QSAR predictions. We have proposed an integrative approach for designing new green chemicals by the structural modification of existing functional but toxic compounds using a combination of structural alerts and QSAR models.
Another important point is that the influence of any part of compound on its biological effect is not constant and strongly depends on its structural environment. Thus, any alert, even derived by mechanistic interpretation of statistically significant QSAR models does not have automatic predictive power. Alerts should be viewed as a structural hypothesis of chemical action only and their true predictive power should be confirmed by QSAR predictions and, if possible, by experimental validation.
The major recommendations discussed in our paper are as follows:
Toxicity prediction solely based on structural alerts is unreliable in most cases and should be avoided;
Regardless of how alerts are identified, they act within the whole chemical structure and therefore their effect on chemical toxicity is dependent on their structural environment; the extent of interdependency should be evaluated on large datasets;
Once the alert is identified by any method, its significance should be confirmed by QSAR models or, even better, by experiment;
Structural alerts should be combined with QSAR and/or CBRA models to improve the accuracy of toxicity prediction;
Albeit structural alerts often fail as toxicity predictors they may be useful to split a large dataset into mechanism-based subsets for developing local QSAR models;
An intelligent combination of alerts and QSAR models can be used for rationally designing functional compounds devoid of toxicity, i.e., for green chemistry applications.
Supplementary Material
Acknowledgments
This study was supported in part by NIH (GM096967). VA thanks FAPEG (grant 201310267001095), CNPq (grant 400760/2014-2), and CAPES.
Footnotes
Authors’ contributions
This manuscript combined a series of separately written, invited contributions from the various coauthors. All sections were written and compiled from contributions of multiple coauthors. Primary attributions for the various contributed sections are as follows: Introduction – V. Alves and A. Tropsha; How reliable are alerts for predicting chemical toxicity? – V. Alves and S. Capuzzi; Contrasting alerts and QSAR-based predictions – V. Alves, E. Muratov, R. Braga, C. Andrade, and A. Zakharov; Complexity of chemical structure questions the significance of single alerts – V. Alves, E. Muratov, R. Politi, S. Farag, and V. Kuz’min; Interpretation of QSAR models: Pulling the rabbit out of the black box – E. Muratov, V. Kuz’min, V. Alves; Integration of structural alerts and QSAR – E. Muratov, V. Kuz’min, E. Mokshyna, V. Alves, A. Sedykh, Y. Low, and D. Fourches; Summary and Conclusions – V. Alves, E. Muratov, and A. Tropsha. Figures were done by V. Alves and E. Muratov. Substantial assistance in editing of all contributions was provided by V. Alves, S. Capuzzi, and E. Muratov; and final editing was accomplished by A. Tropsha, who also takes primary responsibility for the final content.
Conflict of interests
The authors declare no actual or potential conflict of interests.
References
- 1.Schulte PA, McKernan LT, Heidel DS, Okun AH, Dotson GS, Lentz TJ, Geraci CL, Heckel PE, Branche CM. Environ. Heal. 2013;12:31. doi: 10.1186/1476-069X-12-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collins T. Green Chem. 2003;5:G51–G52. [Google Scholar]
- 3.Burden N, Sewell F, Chapman K. PLOS Biol. 2015;13:1–8. doi: 10.1371/journal.pbio.1002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Greek R, Menache A. Int. J. Med. Sci. 2013;10:206–221. doi: 10.7150/ijms.5529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Naven R, Louise-May S. Hum. Exp. Toxicol. 2015;34:1304–1309. doi: 10.1177/0960327115605440. [DOI] [PubMed] [Google Scholar]
- 6.OECD. Report of the workshop on structural alerts for the OECD (Q)SAR application toolbox. [accessed 23 June 2016];2009 http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?cote=env/jm/mono(2009)4&doclanguage=en. [Google Scholar]
- 7.Blagg J. Burger’s Medicinal Chemistry and Drug Discovery. Hoboken, NJ, USA: John Wiley & Sons, Inc.; 2010. pp. 301–334. [Google Scholar]
- 8.Ashby J, Tennant RW. Mutat. Res. 1991;257:229–306. doi: 10.1016/0165-1110(91)90003-e. [DOI] [PubMed] [Google Scholar]
- 9.Ashby J, Tennant RW. Mutat. Res. 1988;204:17–115. doi: 10.1016/0165-1218(88)90114-0. [DOI] [PubMed] [Google Scholar]
- 10.Ashby J. Environ. Mutagen. 1985;7:919–921. doi: 10.1002/em.2860070613. [DOI] [PubMed] [Google Scholar]
- 11.Rosenkranz HS, Klopman G. Mutagenesis. 1990;5:333–361. doi: 10.1093/mutage/5.4.333. [DOI] [PubMed] [Google Scholar]
- 12.McGregor D. Mutat. Res. 1989;222:300–306. [PubMed] [Google Scholar]
- 13.Allen TEH, Goodman JM, Gutsell S, Russell PJ. Chem. Res. Toxicol. 2014;27:2100–2112. doi: 10.1021/tx500345j. [DOI] [PubMed] [Google Scholar]
- 14.Enoch SJ, Roberts DW. In: Chemical Toxicity Prediction: Category Formation and Read-Across. Cronin M, Madden J, Enoch S, Roberts D, editors. Royal Society of Chemistry; 2013. pp. 30–43. [Google Scholar]
- 15.Cronin MTD. Chemical Toxicity Prediction: Category Formation and Read-Across. 2013:1–29. [Google Scholar]
- 16.Cronin MTD. In: Chemical Toxicity Prediction: Category Formation and Read-Across. Cronin M, Madden J, Enoch S, Roberts D, editors. Royal Society of Chemistry; 2013. pp. 155–167. [Google Scholar]
- 17.Alves VM, Muratov EN, Fourches D, Strickland J, Kleinstreuer N, Andrade CH, Tropsha A. Toxicol. Appl. Pharmacol. 2015;284:262–272. doi: 10.1016/j.taap.2014.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stepan AF, Walker DP, Bauman J, Price DA, Baillie TA, Kalgutkar AS, Aleo MD. Chem. Res. Toxicol. 2011;24:1345–1410. doi: 10.1021/tx200168d. [DOI] [PubMed] [Google Scholar]
- 19.Liu R, Yu X, Wallqvist A. J. Cheminform. 2015;7:4. doi: 10.1186/s13321-015-0053-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Low YS, Caster O, Bergvall T, Fourches D, Zang X, Norén GN, Rusyn I, Edwards R, Tropsha A. J. Am. Med. Informatics Assoc. 2015 doi: 10.1093/jamia/ocv127. ocv127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raunio H. Front. Pharmacol. 2011;2:33. doi: 10.3389/fphar.2011.00033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.European Union. Off. J. Eur. Union. 2007:3–280. [Google Scholar]
- 23.ECHA. The Use of Alternatives to Testing on Animals for the REACH Regulation: Second Report under Article 117(3) of the REACH Regulation. [accessed 17 January 2016];2014 http://echa.europa.eu/documents/10162/13639/alternatives_test_animals_2014_en.pdf. [Google Scholar]
- 24.OECD. QSAR Toolbox v. 3.2. [accessed 6 October 2014];2014 http://www.qsartoolbox.org/ [Google Scholar]
- 25.Sushko I, Salmina E, Potemkin Va, Poda G, Tetko IV. J. Chem. Inf. Model. 2012;52:2310–2316. doi: 10.1021/ci300245q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridings JE, Barratt MD, Cary R, Earnshaw CG, Eggington CE, Ellis MK, Judson PN, Langowski JJ, Marchant CA, Payne MP, Watson WP, Yih TD. Toxicology. 1996;106:267–279. doi: 10.1016/0300-483x(95)03190-q. [DOI] [PubMed] [Google Scholar]
- 27.Polishchuk P, Kuz’min V, Artemenko A, Muratov E. Mol. Inform. 2013;32:843–853. doi: 10.1002/minf.201300029. [DOI] [PubMed] [Google Scholar]
- 28.Low Y, Sedykh A, Fourches D, Golbraikh A, Whelan M, Rusyn I, Tropsha A. Chem. Res. Toxicol. 2013;26:1199–1208. doi: 10.1021/tx400110f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Benfenati E, Roncaglioni A, Petoumenou MII, Cappelli CII, Gini G. SAR QSAR Environ. Res. 2015;26:605–618. doi: 10.1080/1062936X.2015.1078408. [DOI] [PubMed] [Google Scholar]
- 30.Price N, Chaudhry Q. Food Chem. Toxicol. 2014;71:136–141. doi: 10.1016/j.fct.2014.05.022. [DOI] [PubMed] [Google Scholar]
- 31.Lozano S, Poezevara G, Halm-Lemeille MP, Lescot-Fontaine E, Lepailleur A, Bissell-Siders R, Crémilleux B, Rault S, Cuissart B, Bureau R. J. Chem. Inf. Model. 2010;50:1330–1339. doi: 10.1021/ci100092x. [DOI] [PubMed] [Google Scholar]
- 32.OECD. Grouping of Chemicals: Chemical Categories and Read-Across. [accessed 10 February 2016]; http://www.oecd.org/env/ehs/risk-assessment/groupingofchemicalschemicalcategoriesandread-across.htm. [Google Scholar]
- 33.OECD Environment Health and Safety Publications. Guidance document for using the OECD (Q)SAR application Toolbox to develop chemical categories according to the OECD guidance on grouping of chemicals, 2009. [accessed 10 February 2016];2009 5 http://www.oecd.org/officialdocuments/publicdisplaydocumentpdf/?doclanguage=en&cote=env/jm/mono. [Google Scholar]
- 34.Enoch SJ, Cronin MTD, Schultz TW, Madden JC. Chem. Res. Toxicol. 2008;21:513–520. doi: 10.1021/tx700322g. [DOI] [PubMed] [Google Scholar]
- 35.Toxtree, Skin sensitisation reactivity domains. [accessed 2 August 2016]; http://toxtree.sourceforge.net/skinsensitisation.html. [Google Scholar]
- 36.Enoch SJ, Roberts DW, Cronin MTD. Chem. Res. Toxicol. 2010;23:1547–1555. doi: 10.1021/tx100218h. [DOI] [PubMed] [Google Scholar]
- 37.Hewitt M, Enoch SJ, Madden JC, Przybylak KR, Cronin MTD. Crit. Rev. Toxicol. 2013;43:537–558. doi: 10.3109/10408444.2013.811215. [DOI] [PubMed] [Google Scholar]
- 38.Pizzo F, Gadaleta D, Lombardo A, Nicolotti O, Benfenati E. Chem. Cent. J. 2015;9:62. doi: 10.1186/s13065-015-0139-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diepgen TL, Coenraads PJ. Kanerva’s Occupational Dermatology. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012. pp. 51–58. [Google Scholar]
- 40.Picard S, Goineau S, Guillaume P, Henry J, Hanouz J-L, Rouet R. Cardiovasc. Toxicol. 2011;11:285–307. doi: 10.1007/s12012-011-9133-z. [DOI] [PubMed] [Google Scholar]
- 41.Mitcheson JS, Chen J, Lin M, Culberson C, Sanguinetti MC. Proc. Natl. Acad. Sci. U. S. A. 2000;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Braga RC, Alves VM, Silva MFB, Muratov E, Fourches D, Lião LM, Tropsha A, Andrade CH. Mol. Inform. 2015;34:698–701. doi: 10.1002/minf.201500040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Braga RC, Alves VM, Silva MFB, Muratov E, Fourches D, Tropsha A, Andrade CH. Curr. Top. Med. Chem. 2014;14:1399–1415. doi: 10.2174/1568026614666140506124442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Springer C, Sokolnicki KL. Chem. Cent. J. 2013;7:167. doi: 10.1186/1752-153X-7-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li Q, Jørgensen FS, Oprea T, Brunak S, Taboureau O. Mol. Pharm. 2008;5:117–127. doi: 10.1021/mp700124e. [DOI] [PubMed] [Google Scholar]
- 46.Zakharov AV, Lagunin AA, Filimonov DA, Poroikov VV. Chem. Res. Toxicol. 2012;25:2378–2385. doi: 10.1021/tx300247r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Politi R, Rusyn I, Tropsha A. Toxicol. Appl. Pharmacol. 2014;280:177–189. doi: 10.1016/j.taap.2014.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kuz’min VE, Muratov EN, Artemenko AG, Gorb L, Qasim M, Leszczynski J. J. Comput. Aided. Mol. Des. 2008;22:747–759. doi: 10.1007/s10822-008-9211-x. [DOI] [PubMed] [Google Scholar]
- 49.Kuz’min VE, Muratov EN, Artemenko AG, Gorb L, Qasim M, Leszczynski J. Chemosphere. 2008;72:1373–1380. doi: 10.1016/j.chemosphere.2008.04.045. [DOI] [PubMed] [Google Scholar]
- 50.Artemenko AG, Muratov EN, Kuz’min VE, Muratov NN, Varlamova EV, Kuz’mina AV, Gorb LG, Golius A, Hill FC, Leszczynski J, Tropsha A. SAR QSAR Environ. Res. 2011;22:575–601. doi: 10.1080/1062936X.2011.569950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tinkov OV, Ognichenko LN, Kuz’min VE, Gorb LG, Kosinskaya AP, Muratov NN, Muratov EN, Hill FC, Leszczynski J. Struct. Chem. 2016;27:191–198. [Google Scholar]
- 52.Kuz’min VE, Artemenko AG, Muratov EN. J. Comput. Aided. Mol. Des. 2008;22:403–421. doi: 10.1007/s10822-008-9179-6. [DOI] [PubMed] [Google Scholar]
- 53.Rännar S, Lindgren F, Geladi P, Wold S. J. Chemom. 1994;8:111–125. [Google Scholar]
- 54.Kuz’min VE, Muratov EN, Artemenko AG, Varlamova EV, Gorb L, Wang J, Leszczynski J. QSAR Comb. Sci. 2009;28:664–677. [Google Scholar]
- 55.Benigni R, Bossa C. Curr. Comput. Aided-Drug Des. 2006;2:169–176. [Google Scholar]
- 56.Kuz’min VE, Polishchuk PG, Artemenko AG, Andronati SA. Mol. Inform. 2011;30:593–603. doi: 10.1002/minf.201000173. [DOI] [PubMed] [Google Scholar]
- 57.Tropsha A, Gramatica P, Gombar VK. QSAR Comb. Sci. 2003;22:69–77. [Google Scholar]
- 58.Cherkasov A, Muratov EN, Fourches D, Varnek A, Baskin II, Cronin M, Dearden J, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz’min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A. J. Med. Chem. 2014;57:4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stanton DT. Curr. Comput. Aided. Drug Des. 2012;8:107–127. doi: 10.2174/157340912800492357. [DOI] [PubMed] [Google Scholar]
- 60.Ajmani S, Jadhav K, Kulkarni SA. J. Chem. Inf. Model. 2006;46:24–31. doi: 10.1021/ci0501286. [DOI] [PubMed] [Google Scholar]
- 61.Baskin II, Ait AO, Halberstam NM, Palyulin VA, Zefirov NS. SAR QSAR Environ. Res. 2002;13:35–41. doi: 10.1080/10629360290002073. [DOI] [PubMed] [Google Scholar]
- 62.Kuz’min VE, Artemenko AG, Lozitsky VP, Muratov EN, Fedtchouk AS, Dyachenko NS, Nosach LN, Gridina TL, Shitikova LI, Mudrik LM, Mescheriakov AK, Chelombitko VA, Zheltvay AI, Vanden Eynde J-J. Acta Biochim. Pol. 2002;49:157–168. [PubMed] [Google Scholar]
- 63.Kuz’min VE, Artemenko AG, Lozytska RN, Fedtchouk AS, Lozitsky VP, Muratov EN, Mescheriakov AK. SAR QSAR Environ. Res. 2005;16:219–230. doi: 10.1080/10659360500037206. [DOI] [PubMed] [Google Scholar]
- 64.Artemenko AG, Muratov EN, Kuz’min VE, Kovdienko NA, Hromov AI, Makarov VA, Riabova OB, Wutzler P, Schmidtke M. J. Antimicrob. Chemother. 2007;60:68–77. doi: 10.1093/jac/dkm172. [DOI] [PubMed] [Google Scholar]
- 65.Kuz’min VE, Artemenko AG, Muratov EN, Volineckaya IL, Makarov VA, Riabova OB, Wutzler P, Schmidtke M. J. Med. Chem. 2007;50:4205–4213. doi: 10.1021/jm0704806. [DOI] [PubMed] [Google Scholar]
- 66.Muratov EN, Artemenko AG, Varlamova EV, Polischuk PG, Lozitsky VP, Fedchuk AS, Lozitska RL, Gridina TL, Koroleva LS, Sil’nikov VN, Galabov AS, Makarov VA, Riabova OB, Wutzler P, Schmidtke M, Kuz’min VE. Future Med. Chem. 2010;2:1205–1226. doi: 10.4155/fmc.10.194. [DOI] [PubMed] [Google Scholar]
- 67.Muratov EN, Varlamova EV, Artemenko AG, Khristova T, Kuz’min VE, Makarov VA, Riabova OB, Wutzler P, Schmidtke M. Future Med. Chem. 2011;3:15–27. doi: 10.4155/fmc.10.278. [DOI] [PubMed] [Google Scholar]
- 68.Alves VM, Muratov EN, Fourches D, Strickland J, Kleinstreuer N, Andrade CH, Tropsha A. Toxicol. Appl. Pharmacol. 2015;284:273–280. doi: 10.1016/j.taap.2014.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.OXEA. n-Heptanol. [accessed 18 November 2014];2013 http://www.oxea-chemicals.com/download/wercs/MTA5MDAjZW4jcHMjYXVzIzEzNjg2MTA1NzIwMDAjb3hlYSMxI0Q=/10900-en-ps-us.pdf. [Google Scholar]
- 70.Gonzalez C, Lerch JF, Lebiere C. Cogn. Sci. 2003;27:591–635. [Google Scholar]
- 71.Patlewicz G, Ball N, Becker RA, Booth ED, Cronin MTD, Kroese D, Steup D, van Ravenzwaay B, Hartung T. ALTEX. 2014;31:387–396. doi: 10.14573/altex.1410071. [DOI] [PubMed] [Google Scholar]
- 72.Patlewicz G, Ball N, Boogaard PJ, Becker RA, Hubesch B. Regul. Toxicol. Pharmacol. 2015;72:117–133. doi: 10.1016/j.yrtph.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 73.Ball N, Cronin MTDMTD, Shen J, Blackburn K, Booth EDED, Bouhifd M, Donley E, Egnash L, Hastings C, Juberg DRDR, Kleensang A, Kleinstreuer N, Kroese EDD, Lee ACAC, Luechtefeld T, Maertens A, Marty S, Naciff JMJM, Palmer J, Pamies D, Penman M, Richarz A-NA-N, Russo DPDP, Stuard SBSB, Patlewicz G, Van Ravenzwaay B, Wu S, Zhu H, Hartung T. ALTEX. 2016;33:149–166. doi: 10.14573/altex.1601251. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.