

Abstract
Fanconi anemia (FA), dyskeratosis congenita (DC), and Diamond Blackfan anemia (DBA) are 3 of the most common inherited bone marrow failure syndromes (IBMFS), in which the hematologic manifestations can be cured with hematopoietic cell transplantation (HCT). Later in life, these patients face a variety of medical conditions, which may be a manifestation of underlying disease or due to pre-HCT therapy, the HCT, or a combination of all these elements. Very limited long-term follow-up data exist in these populations, with FA the only IBMFS that has specific published data. During the international consensus conference sponsored by the Pediatric Blood and Marrow Transplant Consortium entitled “Late Effects Screening and Recommendations following Allogeneic Hematopoietic Cell Transplant (HCT) for Immune Deficiency and Nonmalignant Hematologic Disease” held in Minneapolis, Minnesota in May of 2016, a half-day session was focused specifically on the unmet needs for these patients with IBMFS. A multidisciplinary group of experts discussed what is currently known, outlined an agenda for future research, and laid out long-term follow-up guidelines based on a combination of evidence in the literature as well as expert opinion. This article addresses the state of science in that area as well as consensus regarding the agenda for future research, with specific screening guidelines to follow in the next article from this group.
Keywords: Late effects, Pediatric allogeneic hematopoietic cell transplantation, Inherited bone marrow failure syndromes, Fanconi anemia, Dyskeratosis congenita, Diamond Blackfan anemia
INTRODUCTION
Fanconi anemia (FA), dyskeratosis congenita (DC), and Diamond Blackfan anemia (DBA) are 3 of the most common inherited bone marrow failure syndromes (IBMFS), for which the hematologic manifestations can be cured with hematopoietic cell transplantation (HCT). With improvements in HCT over time, there are larger numbers of survivors for whom we need to understand long-term health. Many of these patients ultimately face a variety of medical conditions, which may be a manifestation of underlying disease or due to their pre-HCT therapy, their HCT, or a combination of all these elements. This is complicated, as there are limited data regarding late effects in these populations, which impairs our ability to propose approaches that may modify these late effects. An international consensus conference sponsored by the Pediatric Blood and Marrow Transplant Consortium entitled “Late Effects Screening and Recommendations following Allogeneic Hematopoietic Cell Transplant (HCT) for Immune Deficiency and Nonmalignant Hematologic Disease” was held in Minneapolis, Minnesota in May of 2016. During this conference, a half-day session focused specifically on the unmet needs for patients with an IBMFS. The goal was to create better understanding about what is already known, define research gaps, and establish screening recommendations for long-term complications in these patient populations. A multidisciplinary group of experts coordinated multiple phone calls ahead of the conference and met in person at the conference with the focus on the following 6 questions: (1) what about the nature of each disease predisposes patients to late effects, (2) what treatments for each disease predispose patients to late effects, (3) what current HCT methods in each disease predispose patients to late effects, (4) what is known about current HCT outcomes in each disease, (5) what is known about long-term follow-up with or without HCT in each disease, and (6) what should be our agenda for future research based on the most significant gaps in the literature for each disease?
We report here the results of those discussions specifically related to FA, DC, and DBA.
FA
FA is an IBMFS characterized by birth defects, progressive bone marrow failure (BMF), and predisposition to cancer and myelodysplastic syndrome (MDS). The inheritance is primarily autosomal recessive, except FANCB, which is X-linked, and FANCR (RAD51), which is dominant. There at least 21 genes whose gene products combine in the FA/BRCA DNA repair pathway (Figure 1). After DNA damage, the proteins produced by FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL, and FANCM form the core complex, which is required for ubiquitination of the I and D2 proteins, which are in turn required for the downstream complex of FANCD2-ubi, FANCI-ubi, and FANCD1/BRCA2, FANCJ/BACH1/BRIP1, FANCN/PALB2, FANCO/RAD51C, FANCP/SXL4, FANCQ/ERCC4, FANCR/RAD51, FANCS/BRCA1, FANCU/XRCC2, and FANCV/REV7 to form foci for DNA repair [1–5]
Figure 1.
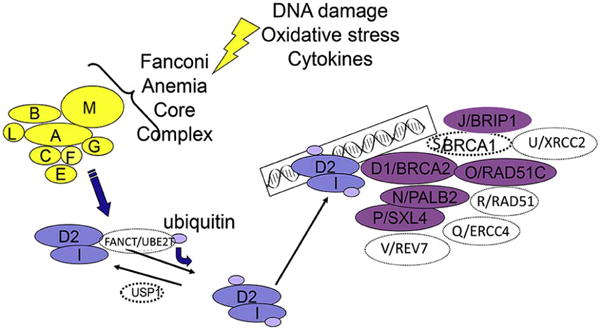
FA/BRCA DNA damage response pathway. This figure is updated from [30].
Acute myeloid leukemia (AML) and solid tumors, including squamous cell carcinoma (SCC) of head and neck and genital region, are the most common malignancies in these patients. The relative risk of AML is increased approximately 500-fold with a cumulative incidence of ~10% by age 50 years, with most individuals diagnosed between the ages of 15 and 35 years [6–9]. Solid tumors may be the first manifestation of FA in individuals who have no birth defects and have not experienced BMF. Head and neck SCC (HNSCCs) are the most common solid tumor in individuals with FA. The risk is 700- to 900-fold that of the general population of comparable age and sex. Compared with the general population, HNSCCs in FA occur at an earlier age (20 to 40 years), present at an advanced stage, and are difficult to treat given limited tolerance to standard chemotherapy and radiation treatments. In contrast with the general population, HNSCC in patients with FA is in the oral cavity, not the oropharynx, and may not be associated with human papillomavirus [10]. In addition, specific subtypes of FA have increased predisposition to a unique set of malignancies. Biallelic pathogenic variants in BRCA2 are associated with early-onset acute leukemia and solid tumors [11–13]. The cumulative probability of any malignancy in patients with biallelic mutations in FANCD1/BRCA2 is 97% by 6 years of age, including AML, medulloblastoma, and Wilms tumor [13]. Similar malignancies are also associated with pathogenic variants in FANCN/PALB2 [14]. This increased risk of primary malignancy adds to the burden of secondary malignancies (leukemia or solid tumors) that may arise after HCT. One study indicated a 4.4-fold higher risk of SCC in patients with FA who had undergone HCT compared with those who had not undergone HCT, with SCC occurring at younger ages in the patients who had undergone HCT [15].
In addition to DNA repair issues [2], there is evidence that FA mutations may lead to increased risk of cancer because of the involvement in other key biological pathways. Studies have implicated excessive reactive oxygen species in FA cells (linked to increased leukemia or SCC) [16,17], mitochondrial defects [18,19] (including a block in mitophagy) [20], and immune function abnormalities in some patients with FA [21,22], suggesting that impaired immune surveillance may contribute to cancer susceptibility in patients with FA.
Chemotherapy and/or radiation used during HCT adds tissue injury and organ damage, which can be particularly severe in patients with a defect in DNA repair and genomic instability. The inherent defective DNA repair in somatic lineages, along with an increased susceptibility to complications of HCT, clearly affect long-term survival of patients with FA.
Patients with FA who progress to severe marrow failure may have been treated in the past with growth factor (granulocyte colony–stimulating factor [G-CSF]) and/or transfusion support. Multiple transfusions lead to iron overload in liver and heart before HCT, potentially increasing the risk of organ damage (eg, sinusoidal obstruction syndrome [SOS]) from chemotherapy and/or radiation used in the conditioning regimen for HCT, which in turn affect overall HCT outcomes.
Androgens remain a treatment modality for BMF in patients with FA. Individuals with FA receiving androgens are at increased risk of side effects, which include virilization and liver toxicity such as elevated liver enzymes, cholestasis, peliosis hepatis (vascular lesion with multiple blood-filled cysts), and hepatic tumors (benign adenomas or malignant carcinomas); these issues have been associated with increased risk for treatment-related mortality during HCT [23,24]. Administration of oral androgens improves blood counts (red cell and platelets) in approximately 50% of individuals with FA and administration of G-CSF improves the neutrophil count in some; however, HCT is currently the only curative therapy for the hematologic manifestations of FA (ie, severe marrow failure, MDS, or leukemia).
Clinically significant chemotherapy and radiation sensitivity related to impaired DNA damage repair have historically made allogeneic HCT for patients with FA extremely challenging [25–27]. In addition, patients with FA and AML who receive cytoreduction before HCT may accumulate additional organ damage. Chronic graft-versus-host disease (GVHD) of mouth and/or genitourinary tract, has been associated with increases in the baseline risk of SCC [28]. HCT also increases the risk of secondary solid tumors and therapy-related MDS or leukemias, as it does the non-FA population but to greater degree [15,29]. Renal insufficiency is rare in patients with FA, despite the fact that about one-quarter of FA patients have structural abnormalities involving kidneys and urinary tract [30]. Renal function may be compromised during and after HCT because of chemotherapy and calcineurin inhibitors. There are little data on long-term kidney outcomes in these high-risk patients.
HCT is ideally performed before onset of MDS or AML and before multiple transfusions [31–33]. HCT approaches and outcomes have improved significantly for patients with FA over the last 3 decades [34–36]. Initial methods for HCT for FA in the late 1970s and early 1980s were associated with excessive toxicity and high mortality rates [37]; since then, better understanding of the biology of the disease led to use of lower doses of total body irradiation (TBI), cyclophosphamide, fludarabine and antithymocyte globulin (ATG) as a common HCT conditioning regimen for patients with FA.
Conditioning regimens containing irradiation are a risk factor for post-transplantation malignancy in patients with FA, but the comparative effects of radiation with other medications is not well understood [38,39]. A number of reports have also shown increased risk of subsequent malignancy associated with chronic GVHD in FA [28,36]. T cell depletion of the donor graft is clearly effective in largely eliminating severe acute and chronic GVHD; however, post-transplantation lymphoproliferative disorder is a well-known complication of HCT conditioning regimens containing ATG or those using ex vivo T cell depletion and has been reported in patients with FA after HCT.
GVHD and graft rejection are 2 major challenges in HCT for patients with FA. Addition of ATG and T cell depletion of donor grafts have decreased GVHD, and the addition of fludarabine has decreased graft rejection. This allowed elimination of TBI from the preparative regimen for FA transplantation from matched sibling donors [40–44]. Five-year survival after a matched sibling transplantation is now approximately 90%. Recent results for unrelated donor transplantation similarly show improvements [34–36,42]. With better HCT outcomes, more patients with FA have survived to adulthood and long-term side effects of HCT, including the increased risk of secondary cancers, have become more apparent.
Reports from Brazil, Germany, and the Netherlands in smaller cohorts have demonstrated that good outcomes can be achieved in unrelated donor setting for patients with FA using non-TBI approaches [45–47]. A more recent prospective multi-institutional US study showed that a radiation-free approach led to overall and disease-free survival comparable to those associated with radiation-containing regimens; however, direct comparison with TBI-based approaches and comparison of late effects with TBI versus non-TBI approaches has not been performed [48].
HCT can restore long-term hematopoiesis and cure the hematologic complications of FA; however, when compared with age-matched controls, these patients do not achieve complete health or normal life expectancy. Patients continue to remain at risk of long-term complications either from transplantation-related factors or secondary to FA-related complications, including those related to congenital anomalies, endocrinopathies, and increased cancer susceptibility (Table 1) [9,49]. Tables 1 and 2 describe overall classes of late effects in patients with FA and discuss findings of specific studies.
Table 1.
Complications in Patients with Fanconi Anemia
| System | Abnormalities |
|---|---|
| Endocrine | Poor growth, growth hormone deficiency, glucose metabolism, thyroid, osteoporosis, hypogonadism, and infertility |
| Liver | Liver enzymes, adenomas, carcinomas, iron overload |
| Renal | Renal function |
| Skin | GVHD abnormalities |
| Hearing | Sensorineural hearing loss |
| Ophthalmology | Cataracts, dry eyes, retinitis |
The etiology of these complications is multifactorial and can be associated with aging alone; however, they can also be exacerbated by HCT preparative regimens, GVHD prophylaxis, GVHD, and antibiotics.
Table 2.
Late Effects after HCT in Patients with FA
| Reference | Number | Main Findings |
|---|---|---|
| Bonfim et al. [36] | 157 | Population was a median 9 years in follow-up mostly conditioned without radiation, with HCT from a matched related donor, and with chronic GVHD in about one-third. Late effects included malignancy in 15, hypothyroidism in 29, gonadal dysfunction in 49, neurologic dysfunction in 25, and hearing loss in 15. |
| Rosenberg et al. [15] | 117 | Population had 508 person-years of follow-up mostly conditioned with radiation and one-half with HCT from a matched related donor and one-half from a matched unrelated donor. Late effects included non-SCC deaths in 48 and SCC in 11. Risk of SCC was 4.4-fold higher after HCT versus FA patients without HCT and SCC occurred at younger ages. GVHD was identified as a significant risk factor. |
| Barnum et al. [50] | 44 | Population was a median 3.1 years in follow-up mostly conditioned with radiation, with HCT from a matched unrelated donor or cord blood, and with chronic GVHD in 5%. Late effects included hypothyroidism in 57%, gonadal dysfunction in 27%, short stature in 50%, vitamin D deficiency in 71%, insulin resistance in 38%, and low bone mineral density in 43%. |
| Rose et al. [51] | 39 | Population details were limited, though most were conditioned with radiation. Identified endocrine issues in those after HCT included hypothyroidism in 65% of children and 43% of adults, growth hormone deficiency in 22% of children and 25% of adults, gonadal dysfunction in 40% of children and 43% of adults, hyperglycemia in 30% of children and 9% of adults, and low bone mineral density in 13% of children and 12% of adults. |
| Anur et al. [39] | 22 | Population was a median 7.4 years in follow-up mostly conditioned with radiation, with HCT from an alternative donor, and without chronic GVHD reported. Late effects included malignancy in 2, hypothyroidism in 10, hyperglycemia in 10, gonadal dysfunction in 12, cataracts in 4, and hearing loss in 5. TBI was a risk factor for late effects. |
| Petryk et al. [52] | 20 | Population was a median 2.8 years in follow-up all conditioned with radiation and with chronic GVHD in 15%. Late effects included lower height and body mass index than controls, lower lean body mass, hypothyroidism in 6, gonadal dysfunction in 3, and lower bone mineral density than controls. |
| Sanders et al. [53] | 15 | Population was a median 20 years in follow-up mostly conditioned without radiation, with HCT from a matched related donor, and with chronic GVHD in about one-third. Late effects included malignancy in 2 with other reported outcomes mixed in with non-FA aplastic anemia patients. |
Recommendations for follow-up of patients with FA after transplantation have been limited in the past; more recently, newer studies are adding to the available data and will further refine current recommendations [36,39,47,50,52,53]. Endocrinopathies, including thyroid dysfunction, growth hormone deficiency, and glucose intolerance, are common in patients with FA independent of HCT. In a series of 120 patients with FA, 48% of patients who had not undergone HCT and 53% of patients who had undergone HCT were found to have hypothyroidism [51]. Glucose intolerance/insulin resistance has been reported in 40% to 80% of patients with FA, independent of HCT. A more recent report showed a similar high incidence of hypothyroidism (45%), with one-half of these patients having evidence of hypothyroidism before HCT [39]. In addition, 9 of 18 patients (50%) who received a TBI-based regimen had insulin resistance/insulin-dependent diabetes mellitus, and 30% of patients developed hypertriglyceridemia. About two-thirds (67%) of postpubertal evaluable males also showed evidence of gonadal dysfunction, similar to prior reports. In this cohort, among 18 patients who received TBI-based regimens, 4 patients also developed cataracts and 5 developed conductive hearing loss.
Female patients with FA, even without transplantation, have shortened reproductive life with late menarche, early menopause, and subfertility [54]. Patients with FA undergoing HCT are at high risk of gonadal dysfunction and infertility after high-dose alkylating agent–based chemotherapy or high-to moderate-dose TBI-based regimens. Pregnancy after HCT for FA is relatively rare. Among 285 female patients 16 years or older with FA who underwent HCT with cyclophosphamide/TBI or cyclophosphamide (n = 3), 10 became pregnant, though these data are limited by lack of information about desire for pregnancy [55].
An Outline for Future Research in FA
Patients with FA who undergo HCT face much brighter prospects today than in years past. We have come a long way in terms of optimizing transplantation outcomes for FA and further improvement in survival may result from a better understanding of late effects resulting from the transplantation itself. This remains challenging as we may not be able to fully separate the progressive problems inherent to the underlying disease from those related to or exaggerated by HCT. FA is the most-studied IBMFS, forming the basis for our understanding of other marrow failure disorders and transplantation strategies for them. Knowledge of late effects in this population will inform future therapies. Now that we have achieved survival rates of 90% or more in patients with FA who have undergone transplantation for marrow failure from sibling donors and over 80% from matched unrelated donors, further optimization of therapy to alter the risk of late effects should become a focus for future study. Rarity of many subtypes of the disease may limit our ability to understand the natural history of different subtypes of FA, but this must be studied, as there will assuredly be differences in long-term outcomes based upon subtype-specific biology. A number of key research priorities moving forward have been identified, including the following:
Large cohorts of patients who had not undergone HCT from FA centers with thorough longitudinal follow-up should be combined to further solidify data regarding the natural history of the disease and yield a robust dataset, against which we will be able to compare the patients with FA who undergo HCT. This, in turn, will lead to a more meaningful comparison in contrast to using HCT cohorts of all nonmalignant conditions or those undergoing HCT for malignant conditions, as the preparative regimens, intensity, and chemotherapy/radiation tolerance are significantly different for patients with FA.
Data from the Center for International Blood and Marrow Transplant Research (CIBMTR) and the European Society for Blood and Marrow Transplantation (EBMT) should be combined to increase the number of patients to quantify late effects after HCT. This effort may be associated with inherent limitations of different approaches utilized for HCT, especially fundamental differences such as T cell depletion or differences in GVHD prophylaxis, which can change late effects significantly related to the altered risk of GVHD, viral infections, and/or post-transplantation lymphoproliferative disorder and, in turn, secondary cancers. The depth of data collection is always an issue with any registry studies compared with prospective studies. It will be important to follow patients treated with the most recent approach of chemotherapy-only preparative regimens to study both short- and long-term effects compared with the historical TBI groups.
Basic research to find the common links or define the disparity between etiology and pathogenesis of drivers of BMF, endocrine problems, leukemogenesis, and carcinogenesis in FA should continue to make progress. This, in turn, will drive the improvements in future HCT and gene therapy efforts to improve global patient outcomes.
DC
DC is a multisystem disorder caused by abnormal telomere biology. There is a phenotypic spectrum that ranges from the classic mucocutaneous triad of oral leukoplakia, reticular skin changes, and abnormal nails to patients with short telomeres and only isolated physical findings [56]. Patients with DC and the related telomere biology disorders (TBD) may present with 1 or a combination of BMF, pulmonary fibrosis, HNSCC, or liver disease. It is important to note that the features of DC change over time and can vary even within the same family. Leukocyte telomeres less than the first percentile for age, measured by flow cytometry with in situ hybridization, are diagnostic of DC [57].
Autosomal dominant, autosomal recessive, and X-linked recessive inheritance patterns have been identified as the causes of DC/TBD and resultant very short telomeres for age. The known genes include ACD, CTC1, DKC1, NAF1, NHP2, NOP10, PARN, POT1, RTEL1, STN1, TERC, TERT, TINF2, and WRAP53 (Figure 2). These 14 genes currently account for about 70% of patients clinically diagnosed with DC [56,58–63]. The genotype/phenotype associations are complex and not fully understood yet [56,58], although there are some correlations between autosomal recessive and X-linked disease, as well as autosomal-dominant pathogenic variants in TINF2 being associated with more severe childhood-onset disease. For example, autosomal-dominant DC/TBD caused by pathogenic variants in TERT, RTEL1, or PARN are associated with adult-onset disease, whereas biallelic or homozygous variants in these genes cause severe DC early in life [59]. A comprehensive diagnostic and clinical care guidelines book has recently been published (www.dcoutreach.org/guidelines).
Figure 2.
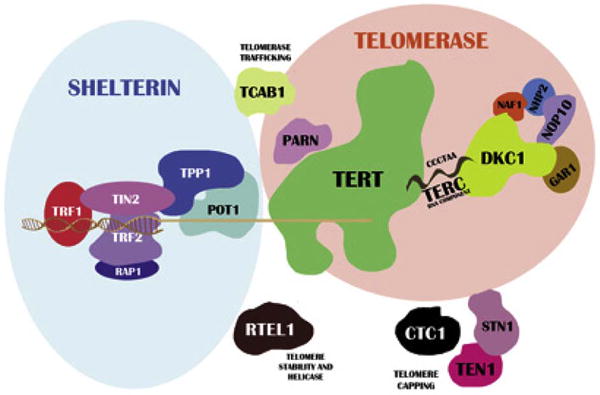
Schematic of telomere biology. The relevant genes are listed in the text, and the figure includes proteins that have not yet been reported to be associated with disease. [Wegman-Ostrosky T, Savage, SA. The genomics of inherited bone marrow failure: from mechanism to the clinic. Br J Haematol. In press.]
Telomeres are a nucleoprotein complex that cap chromosome ends and are essential for chromosomal stability. Telomeres shorten with each cell division because of the inability of DNA polymerase to replicate the 3′ ends of DNA and thus telomere length is a marker of normal aging. The 6 protein shelterin complex serves to protect telomeric ends and telomerase holoenzyme complex extends the nucleotide repeats [57]. When telomeres reach a critically short length, cellular crisis or senescence is triggered. Cancer cells upregulate telomerase, TP53, Rb, and other pathways that allow them to continue to divide despite having critically short telomeres. Patients with DC/TBD have reduced cellular replicative capacity because of the short telomeres caused by their underlying genetic disorder. Pathogenic variants in the telomerase holoenzyme complex limit the ability of cells to extend telomeres, and those in proteins essential in telomere protection result in the loss of telomeres; thus, patient cells lose the ability to replicate sooner than would be expected for their age [59].
The predominant clinical features of DC include BMF, cancer risk, pulmonary fibrosis, and liver cirrhosis [56,58,64,65]. Additional features include enteropathy, enterocolitis, arteriovenous malformations, osteoporosis, avascular necrosis, and immunodeficiency [56,65]. Each of these diagnoses may, in fact, represent the presenting sign of DC in patients without the classic triad. BMF may be seen between 50% and 90% of patients depending on the cohort by the age of 40 [56,58,64]. Cancer has been described in about 15% of all patients. With a median onset of 37 years, approximately 35% of patients will develop cancer by the age of 50. The risk of cancer was reported to be 11-fold compared with the general population and can include MDS (more than 2000-fold increased risk), AML (200-fold), and HNSCC (1154-fold, primarily tongue cancer) [7,56]. The increased risk of primary malignancy is amplified by secondary cancers that can be seen after HCT.
With the development of BMF, patients with DC can be treated with standard supportive care measures, including red blood cell or platelet transfusions, growth factors, and antimicrobial prophylaxis against infections. Generally, these are short-term measures and none represent curative options for BMF. In general, excessive transfusions of red cells or platelets should be avoided as they can complicate future HCT through allogeneic immunization and through iron overload from the red blood cells. A special note regarding the growth factor G-CSF is that it should not be given in combination with androgens because of the risk of splenic rupture [66]. Importantly, immunosuppressive therapy typically used in acquired aplastic anemia is unlikely to be successful with more cases being refractory, relapsing, or progressing to clonal evolution and may only delay or complicate other treatments [58,65].
Androgens, including oxymetholone and more recently danazol, are hormones that have been shown to stabilize and improve peripheral blood counts in DC similar to the efficacy they have shown in FA [56,58,65]. The treatment duration and follow-up have been too short to draw strong conclusions regarding prolonged value and long-term side-effects. Side effects of androgens include impaired liver function, liver tumors, virilization, and behavioral problems. Patients with DC may actually be even more responsive to these hormones than those with FA [65], but they may also develop more side effects because of increased sensitivity [67]. The more recently used androgen danazol may ultimately have fewer side effects and improved efficacy in DC; 1 small study suggested that there may be modulation of TERT gene expression with increase in telomerase [65,68]. It is unknown whether these effects will benefit organ dysfunction aside from BMF.
The development of cancer in patients with DC can be particularly difficult to manage and requires attention to cancer-specific therapy, which may need tailored. Exposure to chemotherapy and radiation can result in increased risk of prolonged cytopenias as well as pulmonary and hepatic toxicities. MDS or AML would be an indication to proceed to HCT, though with concern for additive toxicities of treating with pre-HCT chemotherapy [56,65]. Non-BMF manifestations, such as pulmonary fibrosis and liver cirrhosis, whether from primary disease or secondary to therapies, are managed with standard supportive care measures specific to the dysfunctional organ. Both lung and liver transplantation have been reported successfully in DC patients and should be given appropriate consideration [69,70]. Subsequent need for immunosuppression may impact already underlying immunodeficiency and risk of cancer development. Attempts at telomere elongation and telomerase activation are being studied in the laboratory, but it is too early to know the potential impact of these therapies [71].
HCT represents the only current curative treatment for BMF, MDS, and leukemia in patients with DC. However, underlying pulmonary and hepatic disease may predispose these patients to increased transplantation-related morbidity and mortality, and HCT may result in increased toxicity in the lungs and liver compared with other transplant recipients [56,65]. Additionally, HCT will not help tissues other than the bone marrow, and the interaction between underlying disease and complications of HCT is not well understood. As mentioned above, exposure to chemotherapy and radiation can result in increased risk of prolonged cytopenias as well as pulmonary and hepatic toxicities. Because of the underlying sensitivity of these patients, early myeloablative conditioning transplantations resulted in significant early and late mortality, with fewer than 30% of patients surviving 10 to 15 years after HCT [64,72,73]. There has been an attempt to modify protocols to have reduced-intensity conditioning (RIC) by lowering or eliminating alkylating agents and radiation.
The development of RIC protocols with fludarabine, lower doses of cyclophosphamide, lower doses of TBI, and serotherapy with ATG or alemtuzumab subsequently showed improved early outcomes, with 9 patients out of 11 surviving [72,74,75]. A nonradiation regimen with fludarabine, lower-dose melphalan, and alemtuzumab resulted in 5 of 7 patients alive in early follow-up [76]. There has also been success in the haploidentical setting with a similar RIC approach and the use of ex vivo T cell depletion [77].
Despite early success with RIC regimens, there is still a significant amount of late mortality after HCT for DC, primarily from pulmonary complications. In a systematic review of survival after both myeloablative and RIC HCT in DC, 109 patients reported in the literature had an overall survival of 57% at 5 years and 23% at 10 years [64]. This report confirmed improvement in early survival after the year 2000, when transplantation was performed before the age of 20 and when the donor was a matched sibling, but with no real change in long-term survival. Although infection, graft failure, and hemorrhage dominate early causes of mortality, pulmonary disease, liver disease, and vascular complications dominate late mortality. These data are affected by a publication bias, a changing definition of telomere disease over the last few years, and the possibility that those with BMF have more severe forms of DC [64]. Conversely, some patients with DC/TBD phenotype and “isolated” BMF have been diagnosed and underwent transplantation as idiopathic severe aplastic anemia, with results different from those with the severe DC-associated phenotype; ie, “classical” DC. Those patients may also have the same poor prognosis as the other DC patients and worsen the overall results of patients who underwent transplantation for apparently acquired severe aplastic anemia.
It is clear, however, that HCT studies in DC need to look beyond traditional 3- to 5-year endpoints and aim to understand and, potentially, prevent late mortality. To further improve outcomes, some investigators are attempting to eliminate radiation and alkylating agents and use only fludarabine with ATG [78] or alemtuzumab (ongoing multi-institution trial based out of Boston Children’s Hospital [www.clinicaltrials.gov, NCT01659606]). Although there is some promise in early results with these approaches, larger numbers and longer follow-up are needed to establish whether these approaches lead to improved outcomes.
There are no dedicated studies examining long-term follow-up in this population to date. The systematic review mentioned above and published in 2016 reviewed all case reports and case series that include late complications, which are primarily pulmonary disease, liver disease, and vascular complications [64]. There is a lag time inherent in survivorship studies. Although further development of studies could examine long-term follow-up in patients treated with earlier conditioning regimens, fully understanding the impact of recent RIC protocols will take more time.
An Outline for Future Research in DC
Reports on both short- and long-term outcomes in DC lag far behind the number of reports that now exist for FA. A number of key research priorities moving forward have been identified, including the following:
Reports on long-term follow-up for those patients where the short-term outcomes have already been reported should be included, as should collaboration on collective retrospective studies with much greater time after HCT. More importantly, prospective trials should include patients with DC who either undergo or do not undergo HCT and require follow-up beyond the traditional 3- to 5-year endpoints previously reported.
Given the rarity of this disease, international collaboration will be required, through mechanisms that already exist such as the CIBMTR and EBMT, as well as through newly formed consortia. An example of this is the Clinical Care Consortium for Telomere Associate Ailments established and formalized within the last few years. There is a need for multiple comparisons over time that can include the comparison between those with DC who undergo HCT with those who do not undergo HCT, as well as with patients who undergo HCT for other reasons, and even the general population. As in other areas, knowledge of long-term outcomes will inform future directions for the treatment of these patients.
Ongoing biological and translational studies that can lead to increased understanding of the genotype-phenotype correlation to better understand natural history and that may alter the role of HCT in the long run are needed. The spectrum of DC-related TBDs is only just being understood and long-term natural history studies of patients with very short telomeres and pathogenic variants in telomere biology genes need to be conducted. Given the growing number of genes associated with DC/TBDs, large, international, collaborative studies are required to better understand the complications before and after HCT.
DBA
DBA is a rare IBMFS frequently responsive to long-term corticosteroid therapy but often needing chronic red cell transfusions. The disorder is characterized by macrocytic anemia, reticulocytopenia, and a selective decrease or absence of erythroid precursors in an otherwise normocellular bone marrow. Congenital physical abnormalities, including thumb abnormalities (classically a triphalangeal thumb), craniofacial defects, and cleft lip/palate as well as short stature, are seen in one-half of the patients with DBA. The inheritance is primarily autosomal dominant, due to mutations in genes for ribosomal proteins; approximately 50% are de novo. Mutations in genes involved in the 40s small ribosomal subunit have been reported in RPS19, RPS10, RPS26, RPS24, RPS17, RPS29, RPS28, RPS27, and RPS20. Those in the 60s large ribosomal subunit include RPL11, RPL5, RPL35A, RPL26, RPL15, RPL31, and RPL27 (Figure 3). Rare patients with clinical DBA had mutations in X-linked GATA1 and TSR2 [79,80]. DBA is also a cancer-predisposition syndrome [81].
Figure 3.
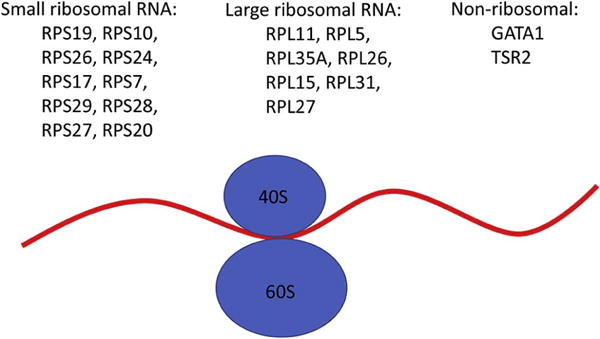
Genes involved in ribosomal biogenesis in Diamond Blackfan anemia.
Generally, patients receive transfusions until the diagnosis is confirmed and remain on transfusion therapy, withholding corticosteroids, for the first year of life if the blood product supply is readily available and safe and reliable venous access is attainable. At around 1 year of age, after vaccinations are completed and the neurodevelopmental risk of corticosteroids is mitigated, a corticosteroid trial is initiated. Corticosteroid doses are weaned to the lowest possible dose to maintain an adequate hemoglobin level. The only known cure for the anemia of DBA is HCT, although approximately 20% of patients will enter a durable remission from either corticosteroid or chronic transfusion therapy [82].
Eighty percent of DBA patients initially respond to corticosteroid therapy, but one-half of these patients will lose their steroid responsiveness or need to discontinue therapy because of excessive corticosteroid toxicity, such as cataracts, pathologic fractures, poor linear growth, etc [83]. Osteopenia and other corticosteroid side effects may be present at the time the patient resumes chronic transfusion therapy and may be exacerbated by endocrinopathies resulting from iron overload. Thus, specific attention to endocrine function (thyroid, adrenal, gonadal, pituitary, etc.) should be part of any pre-and post-HCT evaluation. In fact, it is recommended that all patients with DBA are followed routinely by an endocrinologist, regardless of treatment. The incidence of osteopenia is not well reported in DBA; nor is the use of bisphosphonate therapy [84]. However, pathologic fractures have been seen in up to 20% of patients [83]. There are normal values for bone densitometry down to age 5 years. Similarly, the use of growth hormone has not been well documented but has been reported to be effective in some DBA patients [85]. Osteosarcoma has also been reported, with an observed to expected ratio of over 30 [86]. It is not clear if the use of growth hormone and the incidence of osteosarcoma are linked; however, 2 patients who developed osteosarcoma had been on growth hormone therapy, 1 of these after HCT.
To date, registries in many countries with granular information have been able to provide outcome data with regards to HCT. The North American Diamond Blackfan Anemia Registry (DBAR) with over 25 years of enrollment has 100 patients who have undergone 102 transplantations; data from these patients are being analyzed currently. The Italian Association of Paediatric Haematology and Oncology-DBA and HCT Registries analyzed 30 allogeneic transplantations from 9 centers [87]. The United Kingdom DBA registry has 10 patients reported who have undergone HCT (de la Fuente J; unpublished data). Registries from France and Germany have data linked to CIBMTR. A current CIBMTR study of 64 patients is underway with 7 years of follow-up.
The DBAR previously reported improved outcomes for patients undergoing HCT at an age younger than 10 years [88,89]. As the majority of the HCT reported were done for transfusion dependence, 1 of the main issues in these patients is hypothesized to be the impact of iron burden on transplantation outcome. Of importance is that cardiac iron burden in DBA patients appears to be greater than in thalassemia patients undergoing chronic transfusion therapy, with increased cardiac deposition disproportionate to total body iron burden [90]. Aside from cardiac iron overload, iron burden resulting in multiple endocrinopathies may create comorbidities in these patients. Therefore, HCT outcomes may be linked to transfusion therapy and the subsequent iron burden rather than directly to the actual age of the patient or the underlying disease. Careful evaluation and management of iron overload should be pursued before transplantation and will likely need to be continued after HCT. Despite a paucity of data to support a direct relationship between transplantation-related morbidity/mortality, aggressive pre-HCT iron chelation can certainly reduce the iron burden to a liver iron concentration of less than 3 mg/g dry weight and may potentially decrease the risk of hepatic SOS and infection. Post-HCT aggressive phlebotomy may be necessary to decrease any remaining iron burden, as routine phlebotomy for follow up laboratory evaluation and time will not significantly reduce the iron burden exacerbated by necessary pre-engraftment red cell transfusions. The post-HCT management of iron burden appears to be vital to long-term survival as deaths due to cardiac iron overload have been reported to the DBAR as late as 5 to 7 years after transplantation (unpublished data), suggesting that the duration of an elevated iron burden exposure during, as well as before, HCT is critical.
As summarized above, DBA is caused by a mutation (or deletion) in 1 of 17 small or large subunit ribosomal protein genes in 65% to 75% of patients [91]; less than 1% of the cases are inherited in an X-linked recessive manner and are due to mutations in the transcription factor GATA1 [80] or the putative RPS26 chaperone TSR2 [79]. Thus, it is vital to document the genotype of the patient and determine that the donor sibling or related donor is not also affected. Unfortunately, there may be incomplete penetrance with variable expression, and genotypically positive donors may be hematologically normal. In the absence of a known genotype, DBA-associated birth defects, macrocytosis, or an elevated fetal hemoglobin or erythrocyte adenosine deaminase activity may reveal that a sibling is likewise affected and should not serve as a donor [92]. As well, there are no genotype/phenotype correlations for cancer risk or of the likelihood of a disproportionately elevated cardiac iron burden. Thus, there is no reliable predictor of these predispositions that may influence transplantation decision-making. There is, however, a possible correlation of immunodeficiency and neutropenia in patients with a mutation/deletion of RPL35A (unpublished data). These patients often have more infections and may require G-CSF and immunoglobulin therapy continuously or intermittently. The complications associated with this genotype may warrant an earlier consideration of HCT.
The deaths reported to the DBAR in past years were mostly due to transplantation-related mortality, such as GVHD and SOS. In more recent years, the deaths of patients who have not undergone HCT have been mainly due to complications from iron overload. As the patients in the DBAR are aging, more cancers are also being reported. Five cancers have now been reported after HCT. Although post-HCT cancers have been reported to the DBAR, data are insufficient to define the risk of cancer after HCT; however it may be higher than the risk without HCT [93].
An Outline for Future Research in DBA
Reports on both short- and long-term outcomes in DBA also lag far behind the number of reports that now exist for FA. A number of key research priorities moving forward have been identified, including the following:
Analysis of all available data is the key to accurate HCT outcome assessment. Data from the CIBMTR and the DBAR may have 70% to 80% of patients with DBA and HCT reported but must be linked to avoid likely duplication of these rare patients. These data sets were not established to discern the nuances of DBA in the context of transplantation. Furthermore, the data collected are complementary, so the data sets must be merged for an effective analysis.
Larger more meaningful studies, which will require appropriate funding for more specific data collection, need new revised forms for DBAR, CIBMTR, and EBMT or other international registry collaborations. In particular, detailed genotypic data may reveal at-risk genotypes and correlations of outcomes with iron burden and prior corticosteroid therapy need to be documented.
Most importantly, increased collaboration between the BMF expert and the stem cell transplantation physician is imperative to appropriately analyze outcomes as well as care for the patients with DBA.
IMPORTANT METHODOLOGIC CHALLENGES IN FUTURE STUDIES OF LATE EFFECTS OF IBMFS
Two questions that one should start with when planning future studies of late effects in IBMFS are (1) what are the major late effects after HCT, and (2) which are related to the transplantation itself or are part of the natural history of the syndrome? The analyses should be stratified according to the specific syndrome, the reason for the HCT (eg, aplastic anemia, MDS, or leukemia), the preparative regimen, source of stem cells, and type and extent of GVHD if it occurred. Ideally, comparison groups should include patients who have and have not undergone transplantation with the same syndrome, 1 syndrome versus another, or the syndrome versus the general population. If cancer risks are studied, comparison of risks with the general population would provide standardized incidence rates. Data could include the cumulative incidence of late effects or mortality, using age rather than interval from transplantation as the time course, to determine the impact of the transplantation on the trajectory to an event for which patients with a specific IBMFS may have an intrinsic risk.
Most important to understanding the role of HCT in late effects is to avoid bias by using the appropriate time scale (age), keeping all data current, minimal patients “lost to follow-up”, common protocols for data collection and analyses, and consideration of time trends (older versus recent treatment modalities) and of birth cohort effects. Analyses can be actuarial, in which the censored subjects are implied to be representative of those who remain in the study. Competing risks, in which experience of an event may change the risk of another event, should be addressed.
Studies can be retrospective, cross-sectional, or prospective, and data may come from large cohorts, consortia of cohorts, or existing registries such as CIBMTR and EBMTR. It is vital to have cohorts of patients who have not undergone transplantation that are as close as possible to those who have, with the caveat that transplantations are done for clinical indications or anticipated indications, and those subjects may therefore be intrinsically different from those who have not had an HCT.
CONCLUSION
This review discusses the current state of knowledge regarding long-term follow-up of patients with FA, DC, and DBA after HCT. In addition, we outline an agenda for future research to address key missing data needed to further define long-term effects and possibly lead to trials that could either prevent or mitigate specific effects. In a subsequent publication, this working group will provide specific screening guidelines for these 3 diseases. Because of time constraints, it was not possible to address many other forms of IBMFS through this consensus conference. There was recognition of the need to also work on continued understanding and consensus recommendations for other IBMFS as we move forward.
Acknowledgments
Pediatric Blood and Marrow Transplant Consortium, St. Baldrick’s Foundation to M.A.P. B.P.A. and S.A.S. are supported by the Intramural Research Program of the National Cancer Institute of the National Institutes of Health. The views expressed in this paper do not reflect the official policies of the Department of Health and Human Services, nor does mention of trade names, commercial practices, or organizations imply endorsement by the US Government. The content is solely the responsibility of the authors and does not necessarily represent the official views of those that provided funding.
Financial disclosure: This work was supported in part by grants from the National Institutes of Health (1R13CA159788-01 [to M.A.P., K.S.B.], U01HL069254 [to M.A.P.], R01 CA078938 [to K.S.B.]).
Footnotes
Conflict of interest statement: The authors have no relevant conflicts of interest to disclose.
References
- 1.Bogliolo M, Surrallés J. Fanconi anemia: a model disease for studies on human genetics and advanced therapeutics. Curr Opin Genet Dev. 2015;33:32–40. doi: 10.1016/j.gde.2015.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Mamrak NE, Shimamura A, Howlett NG. Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome Fanconi anemia. Blood Rev. 2016 doi: 10.1016/j.blre.2016.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ameziane N, May P, Haitjema A, et al. A novel Fanconi anaemia subtype associated with a dominant-negative mutation in RAD51. Nat Commun. 2015;6:8829. doi: 10.1038/ncomms9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bluteau D, Masliah-Planchon J, Clairmont C, et al. Biallelic inactivation of REV7 is associated with Fanconi anemia. J Clin Invest. 2016;126:3580–3584. doi: 10.1172/JCI88010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang AT, Kim T, Wagner JE, et al. A dominant mutation in human RAD51 reveals its function in DNA interstrand crosslink repair independent of homologous recombination. Mol Cell. 2015;59:478–490. doi: 10.1016/j.molcel.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberg PS, Alter BP, Ebell W. Cancer risks in Fanconi anemia: findings from the German Fanconi Anemia Registry. Haematologica. 2008;93:511–517. doi: 10.3324/haematol.12234. [DOI] [PubMed] [Google Scholar]
- 7.Alter BP, Giri N, Savage SA, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol. 2010;150:179–188. doi: 10.1111/j.1365-2141.2010.08212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tamary H, Nishri D, Yacobovich J, et al. Frequency and natural history of inherited bone marrow failure syndromes: the Israeli Inherited Bone Marrow Failure Registry. Haematologica. 2010;95:1300–1307. doi: 10.3324/haematol.2009.018119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003;101:822–826. doi: 10.1182/blood-2002-05-1498. [DOI] [PubMed] [Google Scholar]
- 10.Alter BP, Giri N, Savage SA, Quint WG, de Koning MN, Schiffman M. Squamous cell carcinomas in patients with Fanconi anemia and dyskeratosis congenita: a search for human papillomavirus. Int J Cancer. 2013;133:1513–1515. doi: 10.1002/ijc.28157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wagner JE, Tolar J, Levran O, et al. Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia, and Fanconi anemia. Blood. 2004;103:3226–3229. doi: 10.1182/blood-2003-09-3138. [DOI] [PubMed] [Google Scholar]
- 12.Myers K, Davies SM, Harris RE, et al. The clinical phenotype of children with Fanconi anemia caused by biallelic FANCD1/BRCA2 mutations. Pediatr Blood Cancer. 2012;58:462–465. doi: 10.1002/pbc.23168. [DOI] [PubMed] [Google Scholar]
- 13.Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007;44:1–9. doi: 10.1136/jmg.2006.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reid S, Schindler D, Hanenberg H, et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA-N and predispose to childhood cancer. Nat Genet. 2007;39:162–164. doi: 10.1038/ng1947. [DOI] [PubMed] [Google Scholar]
- 15.Rosenberg PS, Socié G, Alter BP, Gluckman E. Risk of head and neck squamous cell cancer and death in patients with Fanconi anemia who did and did not receive transplants. Blood. 2005;105:67–73. doi: 10.1182/blood-2004-04-1652. [DOI] [PubMed] [Google Scholar]
- 16.Du W, Adam Z, Rani R, Zhang X, Pang Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid Redox Signal. 2008;10:1909–1921. doi: 10.1089/ars.2008.2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Du W, Rani R, Sipple J, et al. The FA pathway counteracts oxidative stress through selective protection of antioxidant defense gene promoters. Blood. 2012;119:4142–4151. doi: 10.1182/blood-2011-09-381970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kumari U, Ya Jun W, Huat Bay B, Lyakhovich A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene. 2014;33:165–172. doi: 10.1038/onc.2012.583. [DOI] [PubMed] [Google Scholar]
- 19.Ponte F, Sousa R, Fernandes AP, et al. Improvement of genetic stability in lymphocytes from Fanconi anemia patients through the combined effect of α-lipoic acid and N-acetylcysteine. Orphanet J Rare Dis. 2012;7:28. doi: 10.1186/1750-1172-7-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shyamsunder P, Esner M, Barvalia M, et al. Impaired mitophagy in Fanconi anemia is dependent on mitochondrial fission. Oncotarget. 2016 doi: 10.18632/oncotarget.11161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Myers KC, Bleesing JJ, Davies SM, et al. Impaired immune function in children with Fanconi anaemia. Br J Haematol. 2011;154:234–240. doi: 10.1111/j.1365-2141.2011.08721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giri N, Alter BP, Penrose K, et al. Immune status of patients with inherited bone marrow failure syndromes. Am J Hematol. 2015;90:702–708. doi: 10.1002/ajh.24046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scheckenbach K, Morgan M, Filger-Brillinger J, et al. Treatment of the bone marrow failure in Fanconi anemia patients with danazol. Blood Cells Mol Dis. 2012;48:128–131. doi: 10.1016/j.bcmd.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 24.Fanconi Anemia: Guidelines for Diagnosis and Management. Fourth. Fanconi Anemia Research Fund, Inc; Eugene, Oregon, USA: 2014. [Google Scholar]
- 25.Gluckman E, Devergie A, Schaison G, et al. Bone marrow transplantation in Fanconi anaemia. Br J Haematol. 1980;45:557–564. doi: 10.1111/j.1365-2141.1980.tb07178.x. [DOI] [PubMed] [Google Scholar]
- 26.Gluckman E, Devergie A, Dutreix J. Radiosensitivity in Fanconi anaemia: application to the conditioning regimen for bone marrow transplantation. Br J Haematol. 1983;54:431–440. doi: 10.1111/j.1365-2141.1983.tb02117.x. [DOI] [PubMed] [Google Scholar]
- 27.Gluckman E. Bone marrow transplantation in Fanconi’s anemia. Stem Cells. 1993;11(suppl 2):180–183. doi: 10.1002/stem.5530110829. [DOI] [PubMed] [Google Scholar]
- 28.Guardiola P, Socié G, Li X, et al. Acute graft-versus-host disease in patients with Fanconi anemia or acquired aplastic anemia undergoing bone marrow transplantation from HLA-identical sibling donors: risk factors and influence on outcome. Blood. 2004;103:73–77. doi: 10.1182/blood-2003-06-2146. [DOI] [PubMed] [Google Scholar]
- 29.Armenian SH, Sun CL, Kawashima T, et al. Long-term health-related outcomes in survivors of childhood cancer treated with HSCT versus conventional therapy: a report from the Bone Marrow Transplant Survivor Study (BMTSS) and Childhood Cancer Survivor Study (CCSS) Blood. 2011;118:1413–1420. doi: 10.1182/blood-2011-01-331835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010;24:101–122. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.MacMillan ML, Wagner JE. Haematopoeitic cell transplantation for Fanconi anaemia - when and how? Br J Haematol. 2010;149:14–21. doi: 10.1111/j.1365-2141.2010.08078.x. [DOI] [PubMed] [Google Scholar]
- 32.Mehta P, Locatelli F, Stary J, Smith FO. Bone marrow transplantation for inherited bone marrow failure syndromes. Pediatr Clin North Am. 2010;57:147–170. doi: 10.1016/j.pcl.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 33.Khan NE, Rosenberg PS, Alter BP. Preemptive bone marrow transplantation and event-free survival in Fanconi anemia. Biol Blood Marrow Transplant. 2016;22:1888–1892. doi: 10.1016/j.bbmt.2016.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peffault de Latour R, Porcher R, Dalle JH, et al. Allogeneic hematopoietic stem cell transplantation in Fanconi anemia: the European Group for Blood and Marrow Transplantation experience. Blood. 2013;122:4279–4286. doi: 10.1182/blood-2013-01-479733. [DOI] [PubMed] [Google Scholar]
- 35.MacMillan ML, DeFor TE, Young JA, et al. Alternative donor hematopoietic cell transplantation for Fanconi anemia. Blood. 2015;125:3798–3804. doi: 10.1182/blood-2015-02-626002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bonfim C, Ribeiro L, Nichele S, et al. Long-term survival, organ function, and malignancy after hematopoietic stem cell transplantation for Fanconi anemia. Biol Blood Marrow Transplant. 2016;22:1257–1263. doi: 10.1016/j.bbmt.2016.03.007. [DOI] [PubMed] [Google Scholar]
- 37.Gluckman E, Auerbach AD, Horowitz MM, et al. Bone marrow transplantation for Fanconi anemia. Blood. 1995;86:2856–2862. [PubMed] [Google Scholar]
- 38.Deeg HJ, Socie G, Schoch G, et al. Malignancies after marrow transplantation for aplastic anemia and Fanconi anemia: a joint Seattle and Paris analysis of results in 700 patients. Blood. 1996;87:386–392. [PubMed] [Google Scholar]
- 39.Anur P, Friedman DN, Sklar C, et al. Late effects in patients with Fanconi anemia following allogeneic hematopoietic stem cell transplantation from alternative donors. Bone Marrow Transplant. 2016;51:938–944. doi: 10.1038/bmt.2016.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boulad F, Gillio A, Small TN, et al. Stem cell transplantation for the treatment of Fanconi anaemia using a fludarabine-based cytoreductive regimen and T-cell-depleted related HLA-mismatched peripheral blood stem cell grafts. Br J Haematol. 2000;111:1153–1157. doi: 10.1046/j.1365-2141.2000.02443.x. [DOI] [PubMed] [Google Scholar]
- 41.Locatelli F, Zecca M, Pession A, et al. The outcome of children with Fanconi anemia given hematopoietic stem cell transplantation and the influence of fludarabine in the conditioning regimen: a report from the Italian pediatric group. Haematologica. 2007;92:1381–1388. doi: 10.3324/haematol.11436. [DOI] [PubMed] [Google Scholar]
- 42.Wagner JE, Eapen M, MacMillan ML, et al. Unrelated donor bone marrow transplantation for the treatment of Fanconi anemia. Blood. 2007;109:2256–2262. doi: 10.1182/blood-2006-07-036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ayas M, Al-Jefri A, Al-Mahr M, et al. Stem cell transplantation for patients with Fanconi anemia with low-dose cyclophosphamide and antithymocyte globulins without the use of radiation therapy. Bone Marrow Transplant. 2005;35:463–466. doi: 10.1038/sj.bmt.1704787. [DOI] [PubMed] [Google Scholar]
- 44.Pasquini R, Carreras J, Pasquini MC, et al. HLA-matched sibling hematopoietic stem cell transplantation for Fanconi anemia: comparison of irradiation and nonirradiation containing conditioning regimens. Biol Blood Marrow Transplant. 2008;14:1141–1147. doi: 10.1016/j.bbmt.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonfim C, Ribeiro L, Nichele S, et al. Excellent outcome for Fanconi anemia patients undergoing Hematopoietic Stem Cell Transplantation (HSCT) without radiation: a single center experience on 103 patients. Biol Blood Marrow Transplant. 2015;21:S94. [Google Scholar]
- 46.Chao MM, Kuehl JS, Strauss G, et al. Outcomes of mismatched and unrelated donor hematopoietic stem cell transplantation in Fanconi anemia conditioned with chemotherapy only. Ann Hematol. 2015;94:1311–1318. doi: 10.1007/s00277-015-2370-7. [DOI] [PubMed] [Google Scholar]
- 47.Smetsers SE, Smiers FJ, Bresters D, Sonnevelt MC, Bierings MB. Four decades of stem cell transplantation for Fanconi anaemia in the Netherlands. Br J Haematol. 2016;174:952–961. doi: 10.1111/bjh.14165. [DOI] [PubMed] [Google Scholar]
- 48.Mehta PA, Davies SM, Myers K, et al. Chemotherapy-only preparative regimen for alternative donor hematopoietic cell transplantation for patients with Fanconi Anemia(FA): results of a Multi-Institutional Study. Biol Blood Marrow Transplant. 2015:S104a. [Google Scholar]
- 49.Giri N, Batista DL, Alter BP, Stratakis CA. Endocrine abnormalities in patients with Fanconi anemia. J Clin Endocrinol Metab. 2007;92:2624–2631. doi: 10.1210/jc.2007-0135. [DOI] [PubMed] [Google Scholar]
- 50.Barnum JL, Petryk A, Zhang L, et al. Endocrinopathies, bone health, and insulin resistance in patients with Fanconi anemia after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2016;22:1487–1492. doi: 10.1016/j.bbmt.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rose SR, Myers KC, Rutter MM, et al. Endocrine phenotype of children and adults with Fanconi anemia. Pediatr Blood Cancer. 2012;59:690–696. doi: 10.1002/pbc.24095. [DOI] [PubMed] [Google Scholar]
- 52.Petryk A, Polgreen LE, Barnum JL, et al. Bone mineral density in children with Fanconi anemia after hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2015;21:894–899. doi: 10.1016/j.bbmt.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sanders JE, Woolfrey AE, Carpenter PA, et al. Late effects among pediatric patients followed for nearly 4 decades after transplantation for severe aplastic anemia. Blood. 2011;118:1421–1428. doi: 10.1182/blood-2011-02-334953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sklavos MM, Stratton P, Giri N, Alter BP, Savage SA, Pinto LA. Reduced serum levels of anti-Müllerian hormone in females with inherited bone marrow failure syndromes. J Clin Endocrinol Metab. 2015;100:E197–E203. doi: 10.1210/jc.2014-2838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nabhan SK, Bitencourt MA, Duval M, et al. Fertility recovery and pregnancy after allogeneic hematopoietic stem cell transplantation in Fanconi anemia patients. Haematologica. 2010;95:1783–1787. doi: 10.3324/haematol.2010.023929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Savage S. Dyskeratosis congenita. In: Pagon RAAM, Ardinger HH, Wallace SE, et al., editors. GeneReviews ® [Internet] Seattle: Seattle (WA): University of Washington; 1993–2016. updated 2016 May 26. [Google Scholar]
- 57.Alter BP, Rosenberg PS, Giri N, Baerlocher GM, Lansdorp PM, Savage SA. Telomere length is associated with disease severity and declines with age in dyskeratosis congenita. Haematologica. 2012;97:353–359. doi: 10.3324/haematol.2011.055269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Alter BP. Diagnosis, genetics, and management of inherited bone marrow failure syndromes. Hematology Am Soc Hematol Educ Program. 2007:29–39. doi: 10.1182/asheducation-2007.1.29. [DOI] [PubMed] [Google Scholar]
- 59.Bertuch AA. The molecular genetics of the telomere biology disorders. RNA Biol. 2016;13:696–706. doi: 10.1080/15476286.2015.1094596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ballew BJ, Savage SA. Updates on the biology and management of dyskeratosis congenita and related telomere biology disorders. Expert Rev Hematol. 2013;6:327–337. doi: 10.1586/ehm.13.23. [DOI] [PubMed] [Google Scholar]
- 61.Stanley SE, Gable DL, Wagner CL, et al. Loss-of-function mutations in the RNA biogenesis factor NAF1 predispose to pulmonary fibrosis-emphysema. Sci Transl Med. 2016;8:351ra107. doi: 10.1126/scitranslmed.aaf7837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Takai H, Jenkinson E, Kabir S, et al. A POT1 mutation implicates defective telomere end fill-in and telomere truncations in Coats plus. Genes Dev. 2016;30:812–826. doi: 10.1101/gad.276873.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Simon AJ, Lev A, Zhang Y, et al. Mutations in STN1 cause coats plus syndrome and are associated with genomic and telomere defects. J Exp Med. 2016;213:1429–1440. doi: 10.1084/jem.20151618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Barbaro P, Vedi A. Survival after hematopoietic stem cell transplant in patients with dyskeratosis congenita: systematic review of the literature. Biol Blood Marrow Transplant. 2016;22:1152–1158. doi: 10.1016/j.bbmt.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 65.Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124:2775–2783. doi: 10.1182/blood-2014-05-526285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Giri N, Pitel PA, Green D, Alter BP. Splenic peliosis and rupture in patients with dyskeratosis congenita on androgens and granulocyte colony-stimulating factor. Br J Haematol. 2007;138:815–817. doi: 10.1111/j.1365-2141.2007.06718.x. [DOI] [PubMed] [Google Scholar]
- 67.Khincha PP, Wentzensen IM, Giri N, Alter BP, Savage SA. Response to androgen therapy in patients with dyskeratosis congenita. Br J Haematol. 2014;165:349–357. doi: 10.1111/bjh.12748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Townsley DM, Dumitriu B, Liu D, et al. Danazol treatment for telomere diseases. N Engl J Med. 2016;374:1922–1931. doi: 10.1056/NEJMoa1515319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mahansaria SS, Kumar S, Bharathy KG, Pamecha V. Liver transplantation after bone marrow transplantation for end stage liver disease with severe hepatopulmonary syndrome in dyskeratosis congenita: a literature first. J Clin Exp Hepatol. 2015;5:344–347. doi: 10.1016/j.jceh.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Giri N, Lee R, Faro A, et al. Lung transplantation for pulmonary fibrosis in dyskeratosis congenita: case report and systematic literature review. BMC Blood Disord. 2011;11:3. doi: 10.1186/1471-2326-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Agarwal S, Loh YH, McLoughlin EM, et al. Telomere elongation in induced pluripotent stem cells from dyskeratosis congenita patients. Nature. 2010;464:292–296. doi: 10.1038/nature08792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dietz AC, Orchard PJ, Baker KS, et al. Disease-specific hematopoietic cell transplantation: nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant. 2011;46:98–104. doi: 10.1038/bmt.2010.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gadalla SM, Sales-Bonfim C, Carreras J, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant. 2013;19:1238–1243. doi: 10.1016/j.bbmt.2013.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Brown M, Myers D, Shreve N, Rahmetullah R, Radhi M. Reduced intensity conditioning regimen with fludarabine, cyclophosphamide, low dose TBI and alemtuzumab leading to successful unrelated umbilical cord stem cell engraftment and survival in two children with dyskeratosis congenita. Bone Marrow Transplant. 2016;51:744–746. doi: 10.1038/bmt.2015.333. [DOI] [PubMed] [Google Scholar]
- 75.Nishio N, Takahashi Y, Ohashi H, et al. Reduced-intensity conditioning for alternative donor hematopoietic stem cell transplantation in patients with dyskeratosis congenita. Pediatr Transplant. 2011;15:161–166. doi: 10.1111/j.1399-3046.2010.01431.x. [DOI] [PubMed] [Google Scholar]
- 76.Nelson AS, Marsh RA, Myers KC, et al. A reduced-intensity conditioning regimen for patients with dyskeratosis congenita undergoing hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2016;22:884–888. doi: 10.1016/j.bbmt.2016.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Algeri M, Comoli P, Strocchio L, et al. Successful T-cell-depleted haploidentical hematopoietic stem cell transplantation in a child with dyskeratosis congenita after a fludarabine-based conditioning regimen. J Pediatr Hematol Oncol. 2015;37:322–326. doi: 10.1097/MPH.0000000000000283. [DOI] [PubMed] [Google Scholar]
- 78.Vuong LG, Hemmati PG, Neuburger S, et al. Reduced-intensity conditioning using fludarabine and antithymocyte globulin alone allows stable engraftment in a patient with dyskeratosis congenita. Acta Haematol. 2010;124:200–203. doi: 10.1159/000318721. [DOI] [PubMed] [Google Scholar]
- 79.Gripp KW, Curry C, Olney AH, et al. Diamond-Blackfan anemia with mandibulofacial dystostosis is heterogeneous, including the novel DBA genes TSR2 and RPS28. Am J Med Genet A. 2014;164A:2240–2249. doi: 10.1002/ajmg.a.36633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sankaran VG, Ghazvinian R, Do R, et al. Exome sequencing identifies GATA1 mutations resulting in Diamond-Blackfan anemia. J Clin Invest. 2012;122:2439–2443. doi: 10.1172/JCI63597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Vlachos A, Blanc L, Lipton JM. Diamond Blackfan anemia: a model for the translational approach to understanding human disease. Expert Rev Hematol. 2014;7:359–372. doi: 10.1586/17474086.2014.897923. [DOI] [PubMed] [Google Scholar]
- 82.Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008;142:859–876. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lipton JM, Atsidaftos E, Zyskind I, Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer. 2006;46:558–564. doi: 10.1002/pbc.20642. [DOI] [PubMed] [Google Scholar]
- 84.Lahoti A, Harris YT, Speiser PW, Atsidaftos E, Lipton JM, Vlachos A. Endocrine dysfunction in Diamond-Blackfan Anemia (DBA): a report from the DBA Registry (DBAR) Pediatr Blood Cancer. 2016;63:306–312. doi: 10.1002/pbc.25780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Howell JC, Joshi SA, Hornung L, Khoury J, Harris RE, Rose SR. Growth hormone improves short stature in children with Diamond-Blackfan anemia. Pediatr Blood Cancer. 2015;62:402–408. doi: 10.1002/pbc.25341. [DOI] [PubMed] [Google Scholar]
- 86.Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood. 2012;119:3815–3819. doi: 10.1182/blood-2011-08-375972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Fagioli F, Quarello P, Zecca M, et al. Haematopoietic stem cell transplantation for Diamond Blackfan anaemia: a report from the Italian Association of Paediatric Haematology and Oncology Registry. Br J Haematol. 2014;165:673–681. doi: 10.1111/bjh.12787. [DOI] [PubMed] [Google Scholar]
- 88.Vlachos A, Federman N, Reyes-Haley C, Abramson J, Lipton JM. Hematopoietic stem cell transplantation for Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Bone Marrow Transplant. 2001;27:381–386. doi: 10.1038/sj.bmt.1702784. [DOI] [PubMed] [Google Scholar]
- 89.Aghalar JAE, Lipton JM, Vlachos A. Improved outcomes in Diamond Blackfan anemia treated via stem cell transplant since the year 2000. Blood. 2009;114:3202. [Google Scholar]
- 90.Roggero S, Quarello P, Vinciguerra T, Longo F, Piga A, Ramenghi U. Severe iron overload in Blackfan-Diamond anemia: a case-control study. Am J Hematol. 2009;84:729–732. doi: 10.1002/ajh.21541. [DOI] [PubMed] [Google Scholar]
- 91.Vlachos A, Dahl N, Dianzani I, Lipton JM. Clinical utility gene card for: Diamond-Blackfan anemia–update 2013. Eur J Hum Genet. 2013;21 doi: 10.1038/ejhg.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood. 2010;116:3715–3723. doi: 10.1182/blood-2010-02-251090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vlachos ARP, Kang J, Atsidaftos E, Alter BP, Lipton JM. Myelodysplastic Syndrome and Gastrointestinal Carcinomas Characterize the Cancer Risk in Diamond Blackfan Anemia: A Report from the Diamond Blackfan Anemia Registry. American Society of Hematology; 2016. [Google Scholar]