

Abstract
Chimeric antigen receptors (CARs) are versatile synthetic receptors that provide T cells with engineered specificity. Clinical success in treating B-cell malignancies has demonstrated the therapeutic potential of CAR-T cells against cancer, and efforts are underway to expand the use of engineered T cells to the treatment of diverse medical conditions, including infections and autoimmune diseases. Here, we review current understanding of the molecular properties of CARs, how this knowledge informs the rational design and characterization of novel receptors, successes and shortcomings of CAR-T cells in the clinic, and emerging solutions for the continued improvement of CAR-T cell therapy.
Keywords: chimeric antigen receptor (CAR), adoptive T-cell therapy, immunotherapy, synthetic biology, protein engineering
The adoptive transfer of T cells expressing chimeric antigen receptors (CARs) have shown remarkable clinical efficacy against advanced B-cell malignancies [1–5]. This clinical success has sparked urgent interest in the development of new CARs and the extension of CAR-T cell therapy to solid tumors as well as applications beyond cancer. As the field accumulates both research and clinical experiences, the potential as well as limitations of this exciting new therapeutic paradigm are coming into clearer focus. In this review, we begin with a discussion of current understanding of the biology behind CAR expression and signaling, which informs the rational design of new CAR molecules. We next survey the clinical outcomes of CAR-T cell therapy trials to date, and discuss challenges highlighted by these bedside experiences. Finally, we discuss emerging solutions that seek to improve upon current CAR designs and describe potential applications of CAR-T cell therapy beyond cancer.
The Biology of CAR Expression and Mechanism of Signaling
CARs are fusion proteins with well-defined functional domains, including an extracellular antigen-binding domain, extracellular spacer, transmembrane domain, and intracellular signaling domain responsible for T-cell activation (Figure 1A) [6]. Second- and third-generation CARs (see Glossary) incorporate one or two additional intracellular costimulatory signaling domains, respectively, to enhance T-cell activation and proliferation (Figure 1B) [6]. A major strength of the CAR platform is its modularity, with interchangeable and structurally distinct options available for each functional domain (Box 1). This modularity has enabled the construction of a variety of functional receptor molecules with diverse antigen specificities and structural properties. Although a full mechanistic understanding of CAR signaling and its effects on T-cell biology remains elusive, recent studies on T-cell receptor (TCR) signaling as well as detailed observations of CAR-T cell behavior in vitro have yielded valuable insights into the mechanism of CAR function and provide guidance for the design and construction of new CARs. Unless otherwise noted, all results discussed in this article refer to human conventional T cells (as opposed to regulatory T cells), and all pre-clinical in vivo results refer to findings obtained from the adoptive transfer of human T cells into immunocompromised mouse models.
Figure 1. Chimeric Antigen Receptor (CAR) Structure and Designs.

(A) CARs are modularly constructed fusion receptors comprising the following protein domains (from N- to C-terminus): extracellular antigen-binding domain, extracellular spacer, transmembrane domain, costimulatory domain(s), and T-cell activation domain. (B) First-generation CARs contain a single intracellular signaling domain, most commonly CD3ζ, that is capable of triggering T-cell activation. Second- and third-generation CARs incorporate one or two costimulatory domains, respectively, and enhance productive T-cell stimulation compared to first-generation CARs. ScFv: single-chain variable fragment; Fc: crystallizable fragment of an antibody; VL: light-chain variable fragment; VH: heavy-chain variable fragment; ITAM, immunoreceptor tyrosine-based activation motif.
Box 1. CAR Parts.
Antigen-binding moiety
The antigen-binding domain in a CAR can consist of any target-binding protein, as long as the molecule remains functional when fused to an N-terminal signal peptide and C-terminal components that constitute the rest of the receptor. Antibody-derived single chain variable fragments (scFvs) are the most commonly used antigen-binding domains, but CARs have also been constructed with other antibody-derived binding components such as nanobodies [151] or natural binding partners of the target antigen [65].
Extracellular spacer
Commonly used extracellular spacers are taken from CD4, CD8, and CD28 extracellular domains as well as the IgG Fc region. Amino acid substitutions are often made to the Fc domain in order to prevent unwanted interactions with Fc gamma receptors (FcRγ) expressed by cells such as monocytes and natural-killer cells [28,152–154].
Transmembrane domain
CAR transmembrane domains typically consist of the membrane-spanning domain of CD4, CD8, CD28, or CD3ζ. Transmembrane domain choice is dictated by whether a molecule remains functional when fused to particular C-terminal signaling domains, and the decision is often based on historical experience. Investigations into CAR signaling mechanisms may shed light on whether the CAR transmembrane domain functions merely as a structural anchor, or plays additional functional roles.
Costimulatory domain
Costimulation augments T-cell activation, leading to increased cytokine production, proliferation, differentiation, and persistence. Costimulatory domains in CARs borrow from a variety of native receptors that shape T-cell activation, with CD28 and 4-1BB intracellular domains being the most common [6]. The relative contributions of CD28 and 4-1BB to CAR-T cell function has been reviewed extensively elsewhere [32,155]. Efforts to combine the strengths of multiple costimulatory domains in third-generation CARs have yielded varying results thus far [32,156–162]. The ability to quantitatively predict the effects of costimulatory signal combinations will likely require a more in-depth mechanistic understanding of CAR signaling than is currently available.
Activation domain
CD3ζ, CD3ε, and FcRγ intracellular domains were regularly used as the activation domain in first-generation CARs, but CD3ζ has emerged as the activation domain of choice in recent years [6]. It remains unclear how the use of different activation domains may alter CAR behavior, but the CD3ζ activation domain in second-generation CARs has been sufficient to mediate clinical efficacy in multiple clinical trials [1–5].
Effect of CAR Expression on T-cell Biology
CAR-encoding transgenes are most commonly introduced into CD4+ and/or CD8+ T cells via viral transduction, resulting in strong constitutive CAR expression [2,7–9]. The gross overexpression of potent signaling domains that constitute the CAR, such as CD3ζ and CD28 or 4-1BB, suggests that CARs have the potential to influence T-cell biology even in the absence of antigen stimulation. Indeed, cases of dramatic tonic signaling have been reported for multiple CAR constructs, with higher basal CAR expression levels correlating with increased tonic signaling and CAR-T cell exhaustion in the absence of antigen exposure (irresponsive cytotoxic T cells) [10–12]. It is worth noting that the specific effects of CAR expression on T-cell biology appear to correlate more strongly with the type of CAR expressed (e.g., CARs containing CD28 vs. 4-1BB) than with the genetic background of the T cells, as illustrated by transcriptional profiling of CD28 and 4-1BB CAR-T cells generated from multiple donors [10]. Furthermore, the number of costimulatory domains incorporated into CAR molecules has been shown to affect the basal phosphorylation levels of signaling proteins important in human T-cell activation (LAT, ZAP-70, SYK, ERK, and LCK) [13], suggesting that the specific composition of CAR molecules can profoundly influence T-cell biology independent of antigen stimulation. A recent study compared the function of T cells expressing a CD19 CAR that was either randomly integrated via retroviral transduction, or site-specifically integrated into the TCR α constant (TRAC) or β2-microglobulin loci, with, or without additional promoter/enhancer elements [12]. The study found that the dynamic pattern of CAR expression regulated by the native TRAC locus contributed to the most sustained anti-tumor efficacy in vivo [12], demonstrating the need to stringently control basal and dynamic CAR expression levels to achieve optimal therapeutic efficacy. Further investigation is needed to determine whether CAR expression from the TRAC locus will serve as a superior strategy across multiple CAR designs and tumor targets.
CAR Signal Initiation and Transduction
Few studies have directly assessed the mechanism by which CARs convert extracellular binding events into intracellular signaling. Nevertheless, research on immunoreceptor triggering and the specific signaling domains utilized in CARs provide a framework for inferring the biophysical and biochemical events in CAR signaling.
As discussed in Box 1, the most common signaling domains in CARs are derived from the cytoplasmic segments of CD3ζ, CD28, and 4-1BB. Both CD3ζ and CD28 contain stretches of positively charged, basic amino-acid residues in their cytoplasmic tails, which are known to closely interact with the negatively charged inner leaflet of the plasma membrane when the receptors are in their resting, “off” state [14–16]. Similarly, 4-1BB contains a basic stretch of amino acids in its cytoplasmic tail, even though its membrane-interacting capabilities have not been investigated in detail. For both CD3ζ, and CD28, receptor triggering leads to dissociation of the intracellular chains from the plasma membrane, suggesting membrane dissociation as a critical step toward signal transduction [15,16]. The exact functional role of receptor-membrane contact remains unclear, but the sequestration of receptor chains inside lipid bilayers could serve as a means to regulate the availability of binding sites that are critical for downstream signaling (Figure 2A) [16,17]. Specifically, the masking and unmasking of immunoreceptor tyrosine-based activation motif (ITAM) phosphorylation sites on CD3ζ chains, or the various signaling-molecule binding sites on CD28, could play a critical role in signal transduction [14,16,17]. Furthermore, the configuration of the receptor chains could influence the clustering of receptors and their relative location to other membrane-tethered signaling molecules, thereby altering the initiation and/or maintenance of signal transduction [15,18].
Figure 2. An Integrated Mechanistic Model of CAR Signaling Initiation.

Research on T-cell receptor (TCR) triggering and the specific signaling domains utilized in CARs suggests the following potential mechanisms working in concert to initiate CAR signaling. (A) Ligand binding could generate mechanical forces that lead to the dissociation of CAR intracellular domains from the plasma membrane, thereby unmasking critical binding sites for downstream signaling molecules. (i) At rest, CAR intracellular domains (e.g. CD28 and CD3ζ) may interact with the plasma membrane, as they do in their native receptor contexts, through basic residue motifs that bind to the negatively charged inner leaflet of the plasma membrane [14–16]. (ii) Upon antigen binding, CAR intracellular domains dissociate from the plasma membrane and adopt a signaling-competent conformation that allows interactions with downstream signaling molecules, including kinases such as ZAP-70 and Lck [15,16]. Phosphorylation of the intracellular domains is thought to lock the domains in the membrane-free state [15]. (B) Extending the receptor deformation model of TCR triggering to CARs suggests that the changes in CAR conformation from (A, i) to (A, ii) may arise from mechanical pulling or pushing between the T cell and the target cell. (i) A pulling force can be transmitted via tension in the CAR extracellular and transmembrane domains to dislodge the intracellular domains from the plasma membrane. (ii) A pushing force may alter the local membrane curvature, thereby reducing the stability of the membrane-associated state of the CAR intracellular domains. (C) In the kinetic segregation model of TCR triggering, bulky phosphatases must be physically segregated from TCRs for T-cell activation domains to transduce signal. Thus, in addition to having accessible (i.e. membrane-free) intracellular signaling domains, CARs may also need to be segregated from phosphatases to initiate signal transduction. (i) Segregation of phosphatases and CARs can occur when CAR/ligand interactions force the T cell and target cell into close apposition and exclude bulky phosphatases from the immunological synapse (IS). (ii) By this logic, CARs with excessively long extracellular spacers that allow phosphatases to comingle with CARs at the IS would not be able to robustly activate T-cell signaling. (D) CARs in resting T cells are localized diffusely together with other surface receptors such as CD45. As the events in (C) take place in response to target-cell engagement, ligated CARs can coalesce into microclusters, which have been confirmed to exclude CD45 and transduce T-cell activation signals [26]. With time, microclusters at the CAR-containing immunological synapse are hypothesized to coalesce and organize with other native surface receptors into the supramolecular activation cluster (SMAC), commonly observed at TCR synapses.
If the association and dissociation of the intracellular domains from the plasma membrane played a role in CAR signaling, how might extracellular ligand-binding trigger the intracellular dissociation event? It has been shown that T cell/target cell conjugations can generate mechanical forces through spontaneous membrane motions and receptor engagement [19–21]. This observation supports the receptor deformation model of TCR triggering, which posits that the tugging and pulling between a conjugated T cell/target cell pair would ultimately deform a ligated TCR to a signaling-competent conformation [22]. Target-cell ligation may similarly provide a mechanical force to dislodge a CAR’s intracellular domain from the T-cell plasma membrane and trigger receptor signaling (Figure 2B). Such a model, which relies on general mechanical coupling rather than specific binding-induced conformational changes, is consistent with the fact that functional CARs can be constructed from a variety of structurally dissimilar domains. If true, this model would suggest that the extracellular spacer and transmembrane domain of a functional CAR must fall within a certain range of conformational flexibility, and the mechanical properties of these linker domains would likely impact the sensitivity of a CAR.
The receptor deformation model is only one of several leading hypotheses for TCR signaling. Another model, termed the “kinetic segregation” model, posits that TCR signaling is triggered by the exclusion of bulky phosphatases such as CD45 and CD148 from the compact immunological synapse formed between a conjugated pair of T-cell and target-cell membranes [23,24]. It has been shown that cell contacts formed by CAR/cognate ligand conjugation exclude CD45 [25,26]. Furthermore, head-to-head comparisons of CARs with different extracellular spacer lengths revealed that a short-spacer CAR is most effective for targeting membrane-distal epitopes in CD19 and ROR1 [27,28], consistent with the hypothesis that a compact immunological synapse is essential to T-cell signaling by excluding bulky phosphatases (Figure 2C). However, there is a limit to how close T-cell and target-cell membranes can favorably come together due to the thick glycocalyx surrounding each cell and the energetic cost of membrane bending [29]. As such, shorter spacer lengths are not always optimal, particularly when targeting membrane-proximal epitopes [7,28,30], so both the location of target epitopes and the receptor’s extracellular spacer length are important in CAR design. In addition to adjusting cell-cell distance in an immunological synapse, spacer length also affects the mechanical properties of the CAR molecule itself. Therefore, the ability of the CAR to access target epitopes, achieve phosphatase segregation, as well as transduce mechanical forces should be considered in concert, when selecting target epitopes and designing corresponding CAR structures.
CAR stimulation yields many of the same signal transduction events that occur upon TCR stimulation, such as the phosphorylation of CD3ζ ITAMs, Lck, ZAP70, and LAT [13]. CARs also appear to mimic TCRs at the immunological synapse, forming initial microclusters that exclude CD45 and organizing, together with native receptors, into the characteristic bull’s-eye structure of the supramolecular activation cluster (SMAC) (Figure 2D) [25,26]. The importance of clustering in CAR signaling was further highlighted by the observation of a tonic signaling CAR that led to antigen-independent CAR-T cell exhaustion [10]. The propensity of the CAR’s extracellular single-chain variable fragment (scFv) domain to spontaneously aggregate was proposed as the cause of antigen-independent receptor clustering and signaling [10]. These results suggest that care should be taken to avoid domains with innate oligomerization tendencies when designing CARs in order to prevent spontaneous receptor clustering that leads to premature T-cell exhaustion.
The intensity of the costimulatory signal required for proper T-cell activation has been observed to vary inversely with the extent of TCR stimulation [31]. However, CARs provide hard-coded stoichiometry between CD3ζ and costimulatory domains. This fixed dosing of CD3ζ and costimulatory domains potentially underlies the observation that a nonfunctional subpopulation can emerge upon antigen stimulation of second-generation CD19 CARs in both CD4+ and CD8+ T cells [9]. While native costimulatory receptors can still be involved in CAR-T cell activation, exactly how these native receptors interface with CAR-mediated signaling remains to be explored. Nevertheless, overexpression of costimulatory receptor ligands in conjunction with CARs was shown to increase the ability of human CD19 CAR-T cells to reject NALM6 leukemia xenografts in mice [32], suggesting that CAR signaling might benefit from additional costimulation in productively activated T cells.
CARs, TCRs, and BCRs
CARs and TCRs both activate T cells and likely share many of the biophysical and biochemical signaling events discussed above. However, CARs also differ from TCRs in several prominent ways: CARs harbor fewer subunits and fewer ITAMs, do not need co-receptors to support ligand binding, have several-orders higher affinity for their targets, typically interface with much more abundant cognate antigens, and can be activated by a molecularly diverse set of ligands (Figure 3) [33]. In these aspects, CARs are actually more similar to B-cell receptors (BCRs) (Figure 3). The BCR extracellular domain consists of an antibody. Therefore, like CARs, BCRs exhibit high ligand affinity and recognize targets in a major histocompatibility complex (MHC)-unrestricted manner.
Figure 3. Comparison of Stimulatory Immunoreceptors.

Standard CARs combine the antibody-like target-binding properties of B-cell receptors (BCRs) with the T-cell activation abilities of T-cell receptors (TCRs). T-cell costimulatory properties are further incorporated into CARs with the addition of costimulatory domains. KD, dissociation equilibrium constant (with typical range shown for each receptor type); VL: light-chain variable fragment; VH: heavy-chain variable fragment; ITAM, immunoreceptor tyrosine-based activation motif.
BCRs also share many of the signaling characteristics of TCRs, such as the phosphorylation of ITAM motifs and the formation of receptor microclusters that mature into an immunological synapse [34,35]. Notably, BCRs undergo tonic signaling, which is an emerging challenge in CAR engineering efforts [10,11]. These shared themes in natural immunoreceptor signaling may underlie the success of CARs in synthetically combining BCR and TCR properties to activate T cells and trigger productive effector functions.
Predicting CAR Efficacy in the Wet Lab
CAR-T cells are typically assessed for their therapeutic potential first in vitro, then in mouse models, and finally in phase-I clinical trials, with financial and time commitments rising exponentially at each transition. As such, there is a need for in vitro assays that can reliably identify promising CARs at early stages of the bench-to-bedside pipeline. In vitro quantifications of cytokine production, T-cell proliferation, and target-cell lysis are the standard assays by which CARs are evaluated for basic function. However, these assays often fail to predict relative in vivo performance when comparing multiple CARs that demonstrate basic in vitro function [36].
To increase the predictive power of in vitro assays, “stress tests” have become an increasingly common technique to mimic the repetitive stimulations and low effector-to-target ratios characteristic of in vivo environments. For example, it has been shown that repeated challenges with fresh tumor cells can reveal differences among CAR-T cell lines in terms of their long-term survival [36] and their propensities for PD-1–mediated exhaustion [37] in ex vivo cultures, thus providing a means to distinguish high-performing CARs from a pool of merely functional CARs. An in vivo version of this stress test calls for re-challenging a mouse with an additional tumor load once a previous tumor is cleared, and this method was used to demonstrate the superiority of 4-1BB over CD28 in the context of a mesothelin-specific CAR against mesothelioma xenografts [37]. In a similar vein, Sadelain and colleagues developed an in vivo stress test in which mice were treated with serially reduced CAR-T cell doses to determine the lowest dose required for a given CAR construct to achieve anti-tumor efficacy [32]. Using this method, the researchers compared six methods of providing CD28 and/or 4-1BB costimulation and identified a rank order of varying therapeutic efficacies [32]. As these in vitro and in vivo approaches gain credence in unmasking subtle differences in CAR therapeutic efficacy, future work will be able to assess additional fine-tuned adjustments, such as the variation of dosing schedule and mixing of various CAR-T cell products in treatment protocols.
Mouse models of CAR-T cell therapy typically employ immunocompromised mice to allow for the adoptive transfer of human T cells. Such systems differ sharply from the immune environment in the eventual human therapeutic context, and they offer limited insight into how CAR-T cells may interface with a patient’s endogenous immune components. However, the alternative—i.e., evaluating CARs in murine T cells adoptively transferred into immunocompetent mice—comes with the drawback that optimized murine CAR designs do not necessarily translate well to human T-cell performance. As with many other therapeutic modalities, the efficacy of CAR-T cell therapy in mouse models has not always translated to clinical success (e.g. first-generation CARs performed well in mice but consistently underperformed in clinical trials [38–42]). Furthermore, the use of murine models prevents accurate evaluation of CAR cross-reactivity against healthy human tissues that might express the target antigen at low levels [43]. The emerging methods described above are improving our ability to detect subtle functional differences among various CAR-T cell treatments. Nevertheless, definitive validation of these methods will only come when future clinical trials are carried out to evaluate the resulting conclusions.
Clinical Experiences and Challenges
The number of CAR-T cell therapy clinical trials has grown steadily since the turn of the century (Box 2), with 147 clinical trials recruiting as of March 2017 according to www.clinicaltrials.gov. Early clinical trials evaluated first-generation CARs targeting a number of solid-tumor antigens, including CD171 for neuroblastoma [39], folate receptor (FR) for ovarian cancer [40], and carbonic anhydrase IX (CAIX) for renal cell carcinoma [42]. These trials demonstrated the feasibility of generating patient-specific T-cell products, but they also highlighted important challenges. First, no anti-tumor efficacy was observed, with the lack of CAR-T cell persistence after adoptive transfer cited as a major factor [39,40]. Furthermore, although anti-CD171 and anti-FR CAR-T cells were well tolerated by patients, anti-CAIX CAR-T cells led to severe liver toxicities associated with on-target, off-tumor T-cell–mediated attacks on CAIX+ bile-duct human epithelial cells [42]. Subsequent clinical trials incorporated changes in both antigen target and CAR components such as additional costimulatory domains to address the challenges observed in these early clinical experiences.
Box 2. Clinician’s Corner.
CARs are synthetic receptors that can be expressed in T cells to redirect T-cell responses toward specific targets of interest.
CARs that respond to the pan–B-cell marker CD19 have shown robust efficacy in clinical trials for chemotherapy-refractory B-cell malignancies, including lymphocytic leukemia and non-Hodgkin’s lymphoma. In particular, multiple trials have achieved greater than 85% complete response rate in patients with acute lymphocytic leukemia.
Prominent side effects of CAR-T cell therapy observed in the clinic thus far include cytokine release syndrome (CRS, characterized by sudden and dramatic spikes in serum cytokine levels); neurological toxicities (e.g., cerebral edema); and on-target, off-tumor toxicities (e.g., T-cell infiltration into healthy heart or lung tissues). Severe side effects have contributed to cases of patient fatality in CAR-T cell trials targeting CD19 and HER2.
CAR-T cell therapy against non-hematologic malignancies has yielded limited success thus far. Potential obstacles include inefficient T-cell localization to the tumor site, physical barriers to tumor infiltration by T cells, and potent immunosuppressive factors that render T cells dysfunctional in the tumor microenvironment. Active pre-clinical research and clinical trials are attempting to overcome obstacles in the application of CAR-T cells to other cancer types by assessing different target antigens, treatment protocols, and methods to “armor” the CAR-T cell.
In addition to cancer therapy, pre-clinical work has demonstrated the potential efficacy of CAR-engineered T cells in diverse therapeutic applications, such as the control of viral infections and the treatment of autoimmune disorders.
CD19 CAR-T Cell Therapy for B-cell Malignancies
The most prominent CAR-T success story thus far has been the use of second-generation CARs targeting the pan–B-cell marker CD19 [1–5]. In particular, CD19 CAR-T cell therapy has achieved >85% complete response rate in the treatment of acute lymphocytic leukemia (ALL) across multiple trials [44–46]. The success of CD19 CAR-T cell therapy has been attributed to multiple factors. First, CD19 is highly expressed on the vast majority of malignant B cells, and off-tumor toxicity is primarily limited to B-cell aplasia, a condition that can be clinically managed with prophylactic infusions of gamma-globulin [6]. Second, the incorporation of costimulatory domains such as CD28 and 4-1BB has significantly enhanced the potency of T-cell responses and survival upon adoptive transfer (Box 1) [47–51]. Third, liquid tumors such as malignant B cells present a uniquely accessible environment to CAR-T cells, without a number of immunosuppressive factors such as hypoxia and high local transforming growth factor (TGF)-β activity typically associated with solid tumor microenvironments [52,53]. However, despite these important advantages, clinical experiences with CD19 CAR-T cell therapy have also revealed ongoing challenges.
One important obstacle is the frequent occurrence of cytokine release syndrome (CRS, also known as “cytokine storm”)—i.e., the sudden and dramatic increase in serum levels of various cytokines, including interleukin (IL)-1, IL-2, IL-6, IL-8, IL-10, tumor necrosis factor (TNF)-α, and interferon (IFN)-γ [44,46]. Cytokine production is an important T-cell effector function that appears to correlate with therapeutic efficacy [54,55]. However, severe CRS has also been implicated in the death of multiple patients in CAR-T cell therapy trials [43,56]. Although the administration of tocilizumab (an anti-IL-6 receptor α antibody), etanercept (an anti–TNF-α antibody), or corticosteroids can often provide effective clinical intervention, these agents are not uniformly effective across patients and, in the case of corticosteroids, can be lymphotoxic and directly counter CAR-T cell therapy [57,58]. This in turn, can lead to relapses in patients that had previously achieved complete remission [46]. Another challenge is that severe neurotoxicities have been observed in multiple patients after CD19 CAR-T cell infusion, the cause of which remains unclear [45,46,59]. A phase-II clinical trial conducted by Juno Therapeutics was halted in July 2016 following the death of three patients from cerebral edema. The addition of the chemotherapy drug fludarabine to a lymphodepletion conditioning regimen that already included cyclophosphamide was cited as the source of toxicity, and the U.S. Federal Drug Administration (FDA) lifted the clinical hold after three days. However, the same trial was suspended again in November 2016 following the deaths of two more patients due to cerebral edema. The cause of this complication remains under investigation, and it is not yet certain whether this toxicity is unique to CD19 CAR-T cell therapy. Finally, a sizable fraction of ALL patients who achieved complete remission after CD19 CAR-T cell therapy eventually relapsed with the emergence of CD19− tumor cells [45,60]. It has been shown that antigen escape—i.e., escape of tumor cells from immune surveillance due to the loss of targeted antigens—can be achieved by malignant B cells via frame-shift and missense mutations in CD19, as well as alternative splicing of the cd19 mRNA, each leading to the loss of binding epitopes recognized by CD19 CARs [61].
Despite these challenges, the clinical efficacy of CD19 CAR-T cells has been remarkably robust, and multiple companies are aiming for FDA approval of their CD19 CAR-T cell products in 2017. As such, CD19 CAR-T cells will likely become the first commercially available adoptive T-cell therapy for cancer. Beyond CD19, additional B-cell malignancy antigens such as CD20, CD22, and BCMA are also under active clinical investigation, highlighting exciting advancements in the treatment of refractory B-cell leukemia and lymphoma.
CAR-T Cell Therapy for Non-hematologic Malignancies
In contrast to the success observed in the treatment of B-cell malignancies, clinical evaluations of CAR-T cell therapy for solid tumors have yielded more modest results. The majority of completed clinical trials on solid tumors have utilized first-generation CARs, and their limited therapeutic efficacies are perhaps unsurprising in retrospect [62]. However, trials with second- and third-generation CARs targeting the tumor-associated antigens mesothelin and HER2, respectively, have also been completed, each revealing distinct challenges [43,63].
Specifically, in a phase-I clinical trial, patients with malignant pleural mesothelioma or pancreatic cancer were treated with T cells that had been transfected with mRNA encoding a second-generation mesothelin CAR containing a 4-1BB costimulatory domain [63,64]. Transient, partial response was observed in one patient [63], but the same patient eventually developed anaphylaxis attributed to the generation of IgE antibodies specific to the CAR, which contained a murine scFv [64]. Human anti-mouse antibodies (HAMAs) were detected in a second patient among the group of four patients reported, highlighting the risk of immunogenicity arising from CAR expression.
In a separate phase I trial, a patient with metastatic colon cancer was treated with 1 x 1011 CD8+ T cells expressing a third-generation HER2 CAR containing both CD28 and 4-1BB. The patient experienced severe respiratory distress within 15 minutes of T-cell infusion and died of cardiac arrest 5 days later [43]. Post-mortem analysis confirmed massive T-cell infiltration into the lung, and it was speculated that T-cell activation by low levels of HER2 expression on lung epithelial cells triggered severe CRS and contributed to patient mortality [43].
Although the path to solid tumor treatment with CAR-T cells has been littered with obstacles, several ongoing clinical trials continue to evaluate a number of potential targets, including CEA for colorectal cancers, disialoganglioside GD2 for neuroblastoma and sarcoma, prostate-specific membrane antigen (PSMA) for prostate cancer and melanoma, epidermal growth factor receptor variant three (EGFRvIII) and IL-13 receptor α2 (IL13Rα2) for glioblastoma, as well as new trials targeting mesothelin and HER2 [62]. A recent report presented the case of a patient with recurring, multifocal glioblastoma attaining complete tumor resolution after intraventricular infusion of IL13Rα2 CAR-T cells [65]. Remarkably, the CAR-T cells were able to not only eliminate multiple intracranial tumors but also to resolve spinal metastases. The patient eventually relapsed 228 days after the first CAR-T cell treatment, possibly due to decreased expression of IL13Rα2 on tumor cells [65]. Nevertheless, such results provide evidence that new antigens coupled with novel delivery modalities may lead to improved therapeutic outcomes against solid tumors. Indeed, new methods for local delivery of T cells to the tumor site—such as direct injection into the pleural cavity of the lung for pleural malignancies, or implantation of a T-cell–laden biopolymer at the site of surgical tumor resection—are active areas of research [8,66].
Current Pitfalls and Potential Remedies
Clinical experiences thus far highlight both the promise and the current limitations of CAR-T cell therapy. Several different strategies are under active investigation to address the various challenges that have been identified, with a general trend toward engineering multi-functional T cells that enable greater control after T-cell deployment into the patient.
Increasing Specificity
On-target, off-tumor toxicity has been a major challenge in CART cell therapy, and its potential lethality lends the problem particular urgency. Given a general lack of tumor-exclusive surface antigens, the vast majority of CARs are directed against tumor-associated antigens that are also present on at least a subset of healthy cells. This shared antigen expression profile is responsible for off-tumor toxicities observed in CARs targeting CD19, HER2, and CEA, among others [67]. A number of cancer germline antigens (CGAs, also known as cancer testes antigens) are being evaluated as potential targets, but it remains to be seen whether CGAs that are widely expressed on tumor cells and absent from essential tissues can be identified [68]. An alternative approach has focused on targeting “neoantigens”—i.e., antigenic epitopes that arise from tumor-specific somatic mutations and are thus, unique to diseased (cancerous) cells [68]. Most neoantigen-targeted therapies have focused on the isolation of neoantigen-reactive T cells and/or genes encoding the neoantigen-reactive TCR [69–71]. Nevertheless, mutations in surface-bound receptors (e.g., EGFRvIII) are readily compatible with CAR-mediated targeting [72], and it is also possible to engineer CARs to recognize MHC-presented antigens [73,74].
Given the dearth of “perfect” antigens that are both unique to and uniformly expressed on tumor cells, researchers have actively explored alternative strategies to increase targeting specificity. One approach is to fine-tune the binding affinity and avidity of CARs to identify a “sweet spot,” where the CAR could bind tumor cells harboring high antigen expression while sparing healthy tissues with low antigen expression. By systematically examining mutations to EGFR- and HER2-binding scFv domains, researchers have generated CARs that could specifically target glioma and ovarian carcinoma xenografts, respectively, while avoiding normal cells expressing the same cognate antigen in mice [75,76]. The possibility of tuning the therapeutic window of CAR-T cells by adjusting their ligand-binding affinity and avidity has sustained clinical interest in shared antigens such as HER2 and CEA, despite past observations of severe toxicity [43,77].
Since 2012, a series of studies have explored the idea of increasing targeting specificity by incorporating logical computation capabilities into CAR signaling [78–81]. By using Boolean logic, AND- and NOT-gate CAR-T cells require that the target cells present the correct “combination” of antigens (e.g., “A and B,” or “A but not B”) instead of a single antigen before triggering T-cell activation (Box 3, Figure 4). Such strategies have the potential to significantly increase tumor-targeting specificity, generally at the cost of increasing complexity in the transgenic constructs that must be introduced into the T-cell product. It remains to be seen whether these strategies would support robust therapeutic efficacy in the clinical setting.
Box 3. Boolean-Logic Computation to Increase CAR-T Cell Specificity.
Tumor-targeting specificity of CAR-T cells can be increased by programming T cells to recognize combinations of biomarkers rather than single antigens. Sadelain and colleagues demonstrated the NOT-gate concept by pairing a conventional CAR or TCR with an inhibitory CAR (iCAR) that contains the signaling domains of CTLA-4 or PD-1. The conventional CAR or TCR can trigger T-cell activation upon binding to antigen A, but the presence of antigen B would trigger an inhibitory cascade via the iCAR that overrides any activation signal, yielding “A-but-not-B” signal integration (Figure 4A) [79].
Several AND-gate CAR designs have also been recently described. One design separates the CD3ζ and CD28/4-1BB domains into two receptor chains targeting different antigens, such that full-intensity T-cell activation can only be achieved if both antigens are present to trigger both receptors (Figure 4B) [78]. In another approach, Wang and colleagues designed a “masked CAR” whose antigen-binding domain is blocked by a masking peptide, which can be removed by tumor-associated proteases in the tumor microenvironment (Figure 4C) [81]. As yet another alternative, Lim and colleagues reported a novel “synNotch” receptor [80], which releases a synthetic transcription factor (TF) upon ligand binding to upregulate CAR expression from a cognate synthetic promoter (Figure 4D). The net effect is an AND-gate response in which antigen 1 triggers the synNotch receptor and CAR expression, and a temporally delayed antigen 2 activates the T cell via CAR signaling. In principle, the synNotch platform can be adapted to a wide range of ligand inputs and genetic outputs, but the use of various murine and viral components in this system poses immunogenicity challenges.
It remains to be seen whether these highly engineered systems can achieve the robust T-cell effector function and fine-tuned temporal resolution required to yield clinical efficacy while preventing off-tumor toxicities. Answers to such questions are anticipated in the near future as the field of synthetic immunology moves beyond proof of concept into more physiologically relevant systems.
Figure 4. Increasing Targeting Specificity of CARs by Boolean Logic Calculations.

(A) NOT-gate CAR developed by the Sadelain group paired a conventional CAR or TCR with an inhibitory CAR (iCAR) that contains either PD-1 or CTLA-4. Antigen binding to the iCAR triggers an inhibitory signal that overrides the activation signal from the conventional CAR or TCR [79]. (B-D) Three AND-gate CAR designs. (B) A chimeric costimulatory receptor (CCR) that is equivalent to a third-generation CAR lacking the CD3ζ chain was developed. This CCR is paired with a first-generation CAR, and both receptors must be triggered by their respective cognate antigens to achieve full T-cell activation [78]. (C) The Wang group engineered a “masked CAR” whose antigen-binding domain is blocked by a masking peptide until the peptide is removed via cleavage by a tumor-associated protease [81]. (D) The Lim group developed a synNotch receptor that releases a synthetic transcription factor (TF) upon ligand binding (signal 1). The TF subsequently drives the expression of a CAR from a synthetic, cognate promoter, and the CAR can then responds to its cognate antigen (signal 2) [80]. VL: light-chain variable fragment; VH: heavy-chain variable fragment; scFv: single-chain variable fragment; Ag, antigen; DAP10: DNAX-activating protein 10; FKBP: FK506-binding protein; FRB: FKBP-rapamycin binding domain; tm: transmembrane; ecto: ectoplasmic; cyto: cytoplasmic
Preventing and Mitigating Toxicities
In addition to on-target, off-tumor toxicities, severe side effects such as CRS and neurotoxicity have prompted a search for methods to prevent and manage toxicities. Dose reduction has been considered as a general approach to reduce toxicity. For example, a high dose of T cells (1 x 1011) was used in the original HER2 CAR-T cell trial that led to patient death [43]. A subsequent phase-I/II trial followed a dose-escalation protocol, starting with 1 x 104 cells and ending at a maximum dose of 1 x 108 CAR-T cells [82]. No dose-limiting toxicity was observed within the range tested, but efficacy was also limited to transient partial response in 1 of 17 evaluable patients. T-cell persistence correlated with T-cell dosage, suggesting that T-cell dosing may need to operate within a limited window to support therapeutic efficacy while minimizing toxicity.
Another strategy for preventing or lessening toxicity is to reduce the longevity of CAR expression. Instead of virally integrating CAR-encoding genes into patient-derived T cells, transient transfection of CAR-encoding mRNA can enable temporary CAR expression [63,83]. A major trade-off associated with transient CAR expression is the need to infuse multiple doses of T cells into each patient; indeed, repeated infusion is accompanied by an increased risk of immune rejection of CAR-T cells, as illustrated by the previously discussed clinical trial targeting mesothelin [64].
Taking a different approach, Lim and colleagues proposed a system in which a second-generation CAR is split into two chains that can be reconstituted into a functional receptor by the dimerization-inducing molecule rapamycin or its analog [84]. Such a system enables temporal control over the availability of functional CAR molecules via the use of the trigger molecule, but the inability to spatially constrain the trigger molecule could limit its utility in preventing off-tumor toxicity [84].
In lieu of toxicity prevention, strategies to rapidly eliminate CAR-T cells if and when patients experience severe toxicity have also been proposed. Incorporation of herpes simplex virus thymidine kinase (HSV-TK) into engineered cells allows their specific removal with the administration of ganciclovir [85,86]. However, immunogenicity associated with HSV-TK can lead to rapid depletion of T cells expressing this transgene [87]. Another suicide strategy with clinical promise is the inducible caspase 9 (iCasp9) protein, which dimerizes into the functional, pro-apoptotic form upon the addition of AP1903, a chemical inducer of dimerization [88]. A third strategy is to express a truncated, non-ligand-binding, non-signaling copy of human EGFR (huEGFRt), which allows the selective depletion of huEGFRt+ T cells with the anti-EGFR monoclonal antibody cetuximab [89]. Furthermore, the huEGFRt protein can simultaneously serve as a marker for engineered T cells in cell sorting as well as in immunochemistry [4,5], rendering it a multipurpose addition to therapeutic CAR-T cells.
Increasing T-cell Persistence and Effector Function
Although toxicity management has been a major focus in CAR-T cell development, an equally important challenge is generating T cells with sufficient anti-tumor activities to achieve therapeutic efficacy. Which starting T-cell population is optimal for the generation of therapeutic CAR-T cells remains an unresolved question [90]. Empirical evidence generated in non-CAR adoptive T-cell therapy models has variously supported the use of naïve [91], central memory [92], and stem-cell memory T cells [93]. Despite the lack of definitive proof for any particular T-cell subtype as the optimal therapeutic candidate, an emerging consensus is that less differentiated phenotypes are more likely to provide the proliferative potential required for long-term engraftment, which is strongly correlated with superior therapeutic efficacy (Figure 5A) [68]. Recent work by Riddell and colleagues in a CD19 CAR mouse model identified CD8+ central memory and CD4+ naïve T cell populations as being superior among naïve, central memory, and effector memory compartments in eradicating lymphoma xenografts in mice [94]. Moreover, the CD4+ and CD8+ T cells subsets presented synergistic antitumor efficacy when combined [94]. These results informed the design of an ongoing clinical trial that uses CD19 CAR-T cells derived from defined starting T-cell subsets (NCT01865617). The trial’s initial report described 30 patients who received bulk CD4+ CAR-T cells mixed with CD8+ CAR-T cells that were derived from either central-memory or bulk CD8+ T cells [95]. No statistically significant inferences could be made when contrasting the two CD8+ CAR-T cell groups, in part due to large patient-to-patient variability in cell doses and lymphodepletion regimens, as well as to the high overall remission rate regardless of the type of CD8+ CAR-T cells used (27 out of 29 evaluable patients achieved bone-marrow remission) [95]. However, the trial is ongoing and may eventually provide sufficient statistical power to identify superior T-cell phenotypes in CAR-T cell therapy.
Figure 5. Strategies to Enhance T-cell Persistence and Effector Function.
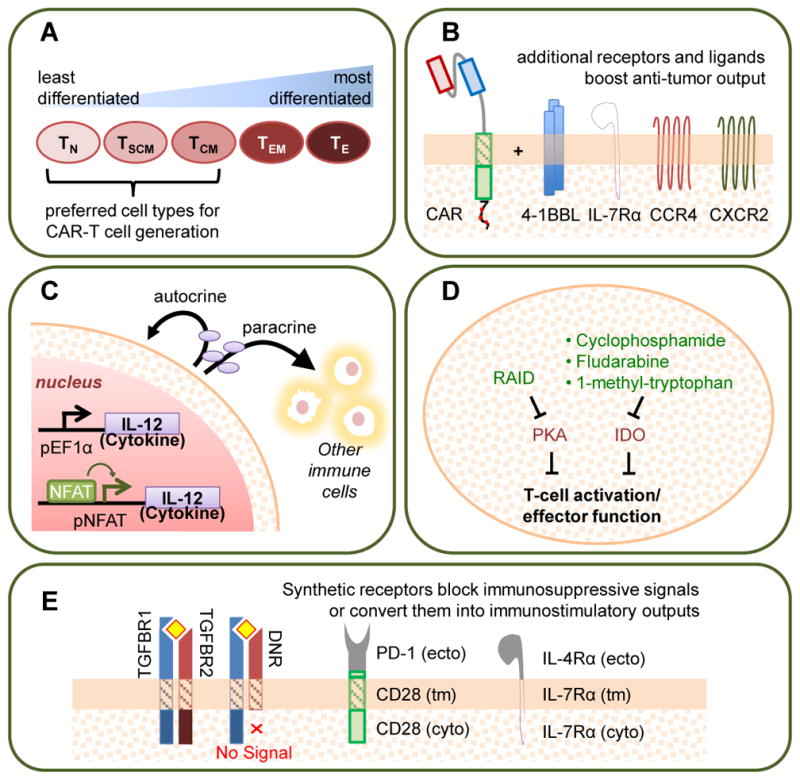
Several engineering approaches have been shown to increase T-cell proliferation, persistence, and anti-tumor effector functions such as cytotoxicity and cytokine production. These strategies include: (A) Using less differentiated cell types as starting material for CAR-T cell manufacturing. TN: naïve T cell; TSCM: stem-cell memory T cell; TCM: central memory T cell; TEM: effector memory T cell; TE: effector T cell. (B) “Armoring” CAR-T cells with additional transgenic receptors or receptor ligands provide costimulation, enhance cytokine signaling, and/or promote migration [32,96–98]. (C) Equipping T cells with stimulatory cytokines expressed from constitutive or inducible promoters (pNFAT, pEF1alpha) [99,163]. (D) Blocking inhibitory signaling pathways through either the expression of transgenic peptides or the administration of pharmaceutical drugs. RAID: regulatory subunit I anchoring disruptor. PKA: protein kinase A. IDO: indoleamine 2,3-dioxygenase [102,103]. (E) Generating chimeric receptors that either abolish endogenous signaling pathways or convert inhibitory ligand inputs into stimulatory signal outputs [104,105,107–109]. TGFBR: TGF-β receptor; DNR: dominant-negative TGF-β receptor. PD-1: programmed-death 1; IL-12: interleukin 12; IL-4Rα and IL-7Rα: interleukin receptors 4 alpha and 7 alpha; CCR4: C-C Motif Chemokine Receptor 4; CXCR2: C-X-C chemokine receptor type 2; tm: transmembrane; ecto: ectoplasmic; cyto: cytoplasmic
Beyond choosing an appropriate starting population, the addition of transgenic features may also enhance T-cell persistence and effector function. The incorporation of costimulatory molecules such as the 4-1BB ligand (4-1BBL) [32], cytokine receptors such as IL-7 receptor α [96], and chemokine receptors such as CCR4 and CXCR2 [97,98] has been shown to enhance the persistence, reduce the exhaustion, and increase the anti-tumor efficacy of human CD4+ and CD8+ T cells (Figure 5B). Another way to generate “armored” CAR-T cells with enhanced function involves the transgenic overexpression of cytokines such as IL-2, IL-12, and IL-15, which promote T-cell proliferation and effector functions (Figure 5C) [99]. For example, it has been reported that human CD4+ and CD8+ T cells programmed to inducibly express IL-12 can enhance tumor rejection by resisting regulatory T cell (Treg)-mediated inhibition [100]. However, the pleitropic effects of potent cytokines makes their transgenic overexpression a delicate balancing act between therapeutic immune activation and pathologic overstimulation. For example, the administration of T cells engineered to inducibly express IL-12 resulted in transient clinical responses in multiple patients with metastatic melanoma, but at the cost of severe liver toxicities [101].
The maintenance of T-cell activation and effector function is regulated by an intricate balance between stimulatory and inhibitory signals. Therefore, the negation of inhibitory signals can be as effective as the promotion of activating signals in optimizing T-cell function. For example, both genetic and chemical approaches have been used to introduce inhibitors into human CAR-T cells to counter the activity of tumor-associated immunosuppressive factors such as adenosine, prostaglandin E2, and indoleamine 2,3 dioxygenase (IDO) metabolic products (Figure 5D) [102,103]. In addition, studies of the adoptive transfer of both murine and human tumor-targeting T cells, have demonstrated that the expression of a dominant-negative TGF-β receptor (DNR)—i.e., a truncated TGF-β receptor 2 chain that lacks the cytoplasmic signaling domain—can enhance the eradication of established tumors in both immunocompetent and immunodeficient mice (Figure 5E) [104,105]. The combination of the DNR with a HER2 CAR is currently under clinical evaluation (NCT00889954). Furthermore, inhibitory signals can also be removed by genetic knockout of inhibitory receptors such as PD-1 [106], and this strategy is now being evaluated in the clinic (NCT02793856).
Going one step further, fusion receptors that actively convert inhibitory ligand inputs to stimulatory functional outputs have also been reported. Specifically, fusion proteins that combine the extracellular ligand-binding domain of an inhibitory receptor (e.g., PD-1 or IL-4 receptor) with the cytoplasmic signaling domain of an immunostimulatory receptor (e.g., CD28, IL-7, or IL-2/15 βc receptor) have been shown to effectively rewire T-cell responses to otherwise inhibitory input signals (Figure 5E) [107–109]. The ability to maintain T-cell persistence, sustain anti-tumor effector functions, and prevent pre-mature T-cell exhaustion will play a critical role in the development of robust therapeutic T cells for cancer immunotherapy.
Preventing Tumor Escape
Loss of antigen expression has been cited as the cause or potential cause of tumor relapse in CAR-T cell therapy targeting CD19 and IL13Rα [45,60,65]. Furthermore, tumor populations are known to be highly heterogeneous and characterized by considerable intra-tumor variations in mutational profiles and gene-expression signatures [110]; thus, a single CAR may not be sufficient to recognize all tumor clones in a given patient. To address the challenge of antigen escape, we and others have developed bispecific CAR-T cells with a broadened range of antigen recognition [30,111–114]. Three general strategies have been explored for the generation of bispecific CAR-T cells: dual CAR (expressing two full-length CARs in each T cell), pooled CAR (combining two T-cell products, each expressing one CAR), and single-chain bispecific CAR (expressing a single CAR molecule that can recognize two different antigens).
The dual-CAR and pooled-CAR strategies can readily make use of conventional, single-input CAR constructs. However, the dual-CAR strategy must contend with either (i) the packaging limit of viral vectors used to transduce T cells, with increasing payload size being strongly correlated with decreasing transduction efficiency [115,116], or (ii) the toxicities and inefficiencies associated with multiple transductions steps required to introduce each CAR separately. Although the pooled-CAR strategy avoids these drawbacks, it requires the generation of two cell products for each patient, and only a portion of the infused cells would be able to recognize a particular antigen. In a head-to-head comparison, the dual-CAR strategy has been shown to be superior to the pooled-CAR approach [114].
The generation of single-chain bispecific CARs requires structural optimization and, therefore, a potential increase in up-front cost at the receptor development stage compared to the other two strategies [30]. However, once a bispecific CAR has been developed, the generation of CAR-T cell products would benefit from increased transduction efficiency or reduced number of distinct cell products required compared with the dual-CAR or pooled-CAR strategy, respectively. Accumulating knowledge on the structure-function correlation in CAR designs will continue to facilitate the development of novel CARs with increased functional capabilities.
Moving Beyond Artisanal T-cell Production
To date, CAR-T cell therapy has been driven largely by academic centers that have developed in-house expertise on the design, production, and administration of CAR-T cell products. Although several companies have emerged in the wake of promising clinical reports, the prospect of widespread application of CAR-T cell therapy hinges on the ability to move T-cell manufacturing toward robust, standardizable production processes. In its current form, CAR-T cells are primarily a personalized medicine that must be tailor-made for each patient. This individualization increases treatment costs and imposes a treatment-time delay while new cell products are generated. Furthermore, standardization of product quality is extremely difficult as the starting material necessarily varies with individual patients.
Several ongoing clinical trials are evaluating the use of donor-derived T cells, with graft-versus-host disease (GVHD) as the primary safety consideration [117]. Early clinical reports indicate promising outcomes for the use of allogeneic CD19 CAR-T cells in the treatment of patients who have received prior allogeneic hematopoietic stem-cell transplants [118]. Efforts toward the generation of “universal” CAR-T cells have focused on eliminating the endogenous TCR using gene-editing techniques such as CRISPR/Cas9, transcription activator-like effector nucleases (TALENs), and zinc-finger nucleases (ZFNs) [106,119–122]. Notably, two recent studies introduced CAR transgenes into the TRAC locus to simultaneously yield CAR expression and eliminate native TCR activity [12,123]. Interestingly, expressing a CAR in the TRAC locus yielded particularly potent CAR-T cells with reduced tonic signaling, terminal differentiation, and exhaustion [12]. In a first-in-human clinical application, TALEN-edited CD19 CAR-T cells lacking the endogenous TCR α chain and CD52 (whose absence renders engineered T cells insensitive to the lymphodepleting agent alemtuzumab) demonstrated clinical efficacy in the treatment of two infants with progressing ALL [124]. However, both patients also exhibited symptoms of skin GVHD that were attributed to the presence of residual TCR+ donor T cells, highlighting room for further improvement [124]. Instead of eliminating the endogenous TCR, an alternative strategy is to integrate the CAR into a starting T-cell population with known TCR specificity, particularly toward Epstein-Barr virus (EBV), which can be found in the majority of the human population in the form of latent infection [125–128]. The use of T cells with a known TCR specificity reduces the risk of unexpected off-tumor toxicities. Furthermore, it has been proposed that generating CART cells from EBV-specific T cells can enhance productive T-cell activation through costimulatory signals provided by latently infected antigen-presenting cells in vivo [125–127]. GD2 CARs integrated into EBV-specific T cells are currently being evaluated in the clinic for the treatment of GD2+ sarcomas (NCT01953900).
Better Together: Prospects of Combination Therapy
CAR-T cell therapy is one of several new advancements in immunotherapy in recent years, and the prospect of combining different treatment modalities has been under active evaluation. Current protocols for adoptive T-cell transfer typically include conditioning chemotherapy, which enhances anti-tumor efficacy by depleting endogenous lymphocytes (including regulatory T cells) and maximizing homeostatic proliferative support for transferred T cells [1,90,129]. In the case of patients with B-cell malignancies, conditioning chemotherapy may also reduce disease burden, which can further enhance T-cell proliferation and persistence [1]. More recently, synergistic possibilities between CAR-T cell therapy and checkpoint blockade have become a topic of great interest [37,130,131]. Because T-cell exhaustion has been cited as a primary cause of failures in T-cell–mediated anti-tumor immunity [132–135], combination therapies that support sustained T-cell effector function have the potential to significantly improve treatment outcomes. Indeed, a recent clinical report suggests that PD-1 blockade can enhance the anti-tumor efficacy of CD19 CAR-T cells in patients who fail to respond to CD19 CAR-T cells alone, although additional studies will be required to elucidate detailed mechanisms behind the observed therapeutic synergy [136].
Diverse Therapeutic Applications
Beyond cancer therapy, efforts have also been underway to use CARs to combat infectious and autoimmune diseases in pre-clinical models. Antiviral approaches have targeted Hepatitis B [137], Hepatitis C [138], and HIV [139,140] infections, where virus-infected cells can be distinguished by surface presentation of specific viral proteins. An antifungal CAR uses the extracellular domain of the pattern-recognition receptor Dectin-1 as its antigen-binding domain and effectively targets carbohydrate epitopes on the opportunistic fungus Apergillus, disrupting its germination [141]. The Dectin-1 CAR demonstrates that pattern-recognition receptors from the innate immune system may serve as a source of antigen-binding domains for CARs that target pathogens.
A recent study demonstrated the novel use of CAR-T cells against the antibody-mediated autoimmune disease pemphigus vulgaris (PV) [26]. This strategy takes advantage of the fact that PV’s pathology is largely attributed to a single cellular source—i.e., pathogenic, anti-Dsg3 antibody–expressing memory B cells—whose surface marker can be recognized by CARs [26]. CAR-T cells have also been engineered to attack autoreactive T cells, including both CD4+ and CD8+ targets. Specifically, these CARs utilize an MHC-I or MHC-II/autoantigenic peptide complex as their extracellular domain, which enables ligation with autoreactive TCRs on target T cells [142,143]. Intriguingly, such redirected T cells were reported to ameliorate experimental autoimmune encephalomyelitis (EAE, a murine model of multiple sclerosis) by inhibiting the activity of multiple autoreactive T cell clones, including ones whose cognate epitopes were different from that targeted by CAR-T cells [142]. Furthermore, the pathologic Th1-dominant responses to autoantigens were replaced by protective Th2 responses after CAR-T cell treatment, suggesting a true reprogramming of the immune response mediated by adoptively transferred T cells [142].
The potent immunosuppressive capabilities of Tregs have also been harnessed with CAR engineering to address therapeutic approaches for autoimmune diseases. For instance, Tregs engineered with the aforementioned CARs can recognize autoreactive TCRs and both prevent and treat EAE in mice [144]. In cases where autoreactive immune cells cannot be identified or do not exist, CARs can direct Treg activity to the site of inflammation—such as the brain (in the case of EAE) [145] or the colon (in the case of murine colitis) [146]—to attenuate local inflammatory activity. Furthermore, human CAR-Tregs with engineered specificity to human major histocompatibility HLA-A2 have been shown to both attenuate GVHD caused by adoptively transferred HLA-A2+ peripheral blood mononuclear cells (PBMCs) and minimize rejection of HLA-A2+ human PBMCs and skin tissues in mice [147–149]. In addition, Tregs engineered with CARs that bind factor VIII (FVIII, a blood-clotting protein used as replacement therapy in hemophilia A patients) were found to suppress the production of anti-FVIII antibodies and reduce the proliferation of FVIII-reactive T cells in vitro [150], thus returning tolerance for the FVIII clotting factor. Taken together, these results suggest that CAR Tregs might be used to promote tolerance in the host to autoimmunogenic stimuli, allogeneic tissues, and allergens.
Concluding Remarks
The clinical success of CD19 CAR-T cells has inspired tremendous interest in adoptive T-cell therapy in recent years. Accumulating knowledge on T-cell biology, CAR signaling, and rational protein engineering promises to support the continuing improvement of CAR-T cell therapy for applications in cancer and beyond. Novel engineering strategies that produce T cells with increasingly complex functionalities have been developed to address issues such as tumor-targeting specificity and longevity of T-cell response in vivo, and a major task facing the field is the transition from proof-of-concept studies to the development of clinically implementable technologies. Although imperfect, pre-clinical mouse models have been used to successfully identify an arsenal of strategies to improve CAR-T cell efficacy against immunosuppressed solid tumors; future work must identify which strategies or combinations of strategies will hold true in the human clinical context. As CAR-T therapy advances through clinical trials toward widespread application, a significant number of important questions remain to be answered (see Outstanding Questions). Nevertheless, as a highly programmable, living immunotherapeutic strategy, CAR-T cells are poised to provide an alternative treatment paradigm for a variety of diseases still awaiting effective treatment options.
Outstanding Questions Box.
Which antigens can serve as effective and non-toxic targets for non–B-cell malignancies? Antigens that facilitate effective tumor-elimination while avoiding off-tumor toxicities remain elusive. The ability to identify better antigens for safe and efficacious tumor targeting will widen the applicability of adoptive T-cell therapy for cancer.
How can we overcome challenges associated with solid tumors—including antigen choice, immune suppression, access to “cold” tumors that lack inflammatory signatures, and infiltration into solid tumor masses? Multiple factors surrounding tumor access and maintenance of T-cell functionality within tumor microenvironments will need to be addressed to increase therapeutic efficacy against solid tumors.
How can we achieve high-throughput optimization of new CAR constructs with predictable in vivo functionality? CAR engineering has relied on labor-intensive, time-consuming in vitro characterizations that often fail to predict in vivo functionality. The identification of in vitro assays with higher predictive value and the development of higher-throughput T-cell isolation, expansion, and characterization methods will facilitate the generation of novel CARs for new disease targets.
How can we develop widely accessible T-cell manufacturing processes that can generate high-quality T-cell products? Current knowledge on T-cell manufacturing is concentrated in a few academic centers and companies affiliated with such centers. Widespread application of T-cell therapy will necessitate the development of robust T-cell manufacturing processes that can be consistently replicated in non-specialist medical centers.
Could we generate donor-derived “universal CAR-T cells” that can be pre-manufactured for off-the-shelf use? The personalized nature of current CAR-T cell therapy increases treatment cost and limits its availability. Off-the-shelf CAR-T cells would significantly broaden the accessibility of this treatment paradigm.
Trends Box.
CAR T-cell therapy has shown remarkable clinical efficacy against B-cell leukemias, but there is significant room for improvement in treatment approaches against non-hematologic malignancies.
CAR design has relied heavily on historical experience and trial-and-error. Accumulating knowledge of T-cell biology, CAR signaling, and rational protein engineering is facilitating increasingly sophisticated CAR designs.
Novel in vitro and in vivo characterization methods for CAR function enable detection of subtle differences across CAR designs and may better predict CAR performance in the clinic.
Various strategies have been employed to generate “armored” T cells against immunosuppression and exhaustion, and to improve T-cell persistence.
Promising pre-clinical data support the extension of CAR-T cell therapy to the treatment of infections and autoimmune diseases.
Acknowledgments
We acknowledge all the works that could not be cited due to space constraints. This work was supported by the National Institutes of Health (F30CA183528 to ZC and DP5OD012133 to YYC).
Glossary
- 4-1BB (CD137)
a member of the tumor necrosis factor receptor family that is typically upregulated in activated T cells and provides costimulatory signals upon binding to 4-1BB ligand (4-1BBL); its cytoplasmic domain can be incorporated into CAR molecules
- Affinity
the strength of a given non-covalent binding interaction between two molecules, such as the binding strength of one antibody binding site to its cognate antigen’s epitope
- Allogeneic
adjective describing cells or tissues from a genetically dissimilar individual of the same species
- Anaphylaxis
an acute, severe allergic reaction that can be life-threatening in the absence of immediate medical treatment
- Anti-Dsg3 antibody
antibody targeting desmogelin 3 (Dsg3), a protein that is essential to cell-cell adhesion in the epidermis
- Antigen escape
escape of tumor cells from immune surveillance due to loss of targeted antigen
- Avidity
the overall, accumulated binding strength of multiple non-covalent binding interactions between two molecules or macromolecules; influenced by binding affinity, the number of binding interactions that exist between the two molecules, as well as additional factors such as the proximity of the separate binding sites
- B-cell aplasia
depletion of B cells
- Cas9
a family of prokaryotic nucleases that, when complexed with the appropriate guide RNA sequences, can be directed to specific DNA sequences and generate double-stranded breaks in the DNA, leading to DNA repair processes that may or may not introduce insertions, deletions, and/or mutations in the targeted DNA sequence
- CD28
a surface receptor that naturally binds to CD80 (B7.) and CD86 (B7.2) to provide costimulatory signals required for proper T-cell activation. Its cytoplasmic domain can be incorporated into CAR molecules
- CD3ζ (CD247)
a subunit of the T-cell receptor (TCR) complex that is phosphorylated to initiate T-cell activation signaling networks
- CD45
a receptor-type protein phosphatase with a long rigid extracellular domain; has been shown to play a role in T-cell activation by calibrating the phosphorylation state of kinases and phosphoproteins associated with TCR signaling
- CD148
a receptor-type protein phosphatase with a long rigid extracellular domain; has been shown to negatively regulate TCR signaling
- Central memory T (TCM) cells
a subset of memory T cells characterized by the expression of CD45RO, CD62L, and CCR7; TCM cells can achieve long-term persistence and readily proliferate and differentiate into effector T cells in response to antigen stimulation
- Checkpoint blockade
a family of therapeutic strategies that aim to promote immune responses against diseases (primarily cancers) by blocking checkpoints, such as PD-1 and CTLA-4 signaling, that would otherwise inhibit immune-cell function
- Conventional T cells
T cells that are not of the regulatory phenotype; could be naïve, stem-cell memory, memory, or effector T cells
- CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9
a prokaryotic system that enables the introduction of insertions, deletions, and/or mutations at specific genomic locations
- Dectin-1
a pattern-recognition receptor that specifically binds to β-glucans, which are glucose polymers expressed on fungi cell walls
- Epstein-Barr virus (EBV)
a common virus in the herpes family; most humans have coexisting latent infection and anti-EBV adaptive immunity
- Fc (fragment crystallizable) region
the base region of an antibody that interacts with Fc receptors and the complement system
- First-generation CARs
CARs that do not contain any costimulatory domains
- Graft-versus-host disease (GVHD)
a medical condition in which transplanted tissue from a genetically dissimilar person attacks host cells, leading to tissue damage
- Immunological synapse
the interface formed between a lymphocyte (e.g. a T cell) and an antigen-presenting cell or target cell
- Immunoreceptor tyrosine-based activation motif (ITAM)
tyrosine-containing motifs that are found in stimulatory receptors in immune cells and are phosphorylated to initiate signal transduction
- Kinetic segregation model of TCR triggering
a proposed model for TCR signaling which posits that TCR signaling is triggered by the exclusion of bulky phosphatases such as CD45 and CD148 from the compact immunological synapse formed between a T cell and its target cell or an antigen-presenting cell
- Lymphodepletion conditioning
chemotherapeutic treatment administered to cancer patients in order to eliminate existing lymphocytes prior to the adoptive transfer of therapeutic cells
- Naïve T (TN) cells
a subset of T cells that have never encountered antigen stimulation and are characterized by the expression of CD45RA, CD62L, and CCR7
- Nanobodies
antibodies that contain a single, monomeric variable domain; initially derived from camelid and fish heavy-chain antibodies
- Pemphigus vulgaris (PV)
an autoimmune disorder of the skin caused by antibodies that target desmogelin (Dsg) proteins present in epithelial cells, ultimately leading to the loss of cell-cell adhesion in the epidermis and blisters in the skin
- Peripheral blood mononuclear cells (PBMCs)
all blood cells found within circulating blood that possesses a round nucleus, including lymphocytes and monocytes
- Receptor deformation model of TCR triggering
a proposed model for TCR signaling which posits that the tugging and pulling between a conjugated T cell/target cell pair deforms the ligated TCR to a conformation that can initiate signaling
- Regulatory T cells
a CD4+/CD25hi/FOXP3+ T-cell subtype whose primary function is to maintain tolerance toward self-antigens and prevent autoimmunity by suppressing the proliferation and activity of immune cells, particularly effector T cells
- Second-generation CARs
CARs that contain one costimulatory domain
- Single-chain variable fragment (scFv)
a fusion protein comprising the variable regions of the heavy and light chains of an antibody connected via a linker peptide sequence
- Stem-cell memory T (TSCM) cells
a subset of T cells that exhibit properties associated with both naïve and memory T cells; TSCM cells have been suggested to have superior self-renewal capability compared to memory T cells and to possess the ability to differentiate into both memory and effector T cells
- Stress tests
in vitro or in vivo assays designed to test the limits of T-cell effector functions, typically by repeatedly challenging T cells with antigen stimulations or by reducing the T-cell dosage relative to tumor burden
- Supramolecular activation cluster (SMAC)
an assembly of receptors arranged in concentric circles at the interface between a lymphocyte (e.g. a T cell) and an antigen-presenting cell or target cell
- T-cell exhaustion
a state of dysfunction in which T cells exhibit reduced functional activities, including proliferation, cytokine production, and cytotoxicity
- Th1 response
Type-1 helper T-cell response—characterized by the production of interferon-γ, interleukin (IL)-2, and tumor necrosis factor-α, among other cytokines—that is particularly effective against intracellular pathogens
- Th2 response
Type-2 helper T-cell response—characterized by the production of IL-4, IL-5, IL-6, IL-10, among other cytokines—that is particularly effective against helminthes and other extracellular pathogens
- Third-generation CARs
CARs that contain two costimulatory domains
- Tonic signaling
receptor signaling in the absence of ligand engagement
- T-cell receptor α constant (TRAC) locus
genetic location within the TCRα gene that encodes the constant region of the TCR α subunit of the TCR complex
- Tumor-associated antigens
antigens that are expressed by tumor cells but can also be present on normal tissues
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Brentjens RJ, et al. Safety and persistence of adoptively transferred autologous CD19-targeted T cells in patients with relapsed or chemotherapy refractory B-cell leukemias. Blood. 2011;118:4817–28. doi: 10.1182/blood-2011-04-348540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kochenderfer JN, et al. Chemotherapy-Refractory Diffuse Large B-Cell Lymphoma and Indolent B-Cell Malignancies Can Be Effectively Treated With Autologous T Cells Expressing an Anti-CD19 Chimeric Antigen Receptor. J Clin Oncol. 2015;33:540–549. doi: 10.1200/JCO.2014.56.2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garfall AL, et al. Chimeric Antigen Receptor T Cells against CD19 for Multiple Myeloma. N Engl J Med. 2015;373:1040–7. doi: 10.1056/NEJMoa1504542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turtle CJ, et al. Immunotherapy of non-Hodgkins lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016;8:355ra116–355ra116. doi: 10.1126/scitranslmed.aaf8621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang X, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016;127:2980–90. doi: 10.1182/blood-2015-12-686725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sadelain M, et al. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov. 2013;3:388–398. doi: 10.1158/2159-8290.CD-12-0548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu L, et al. Inclusion of Strep-tag II in design of antigen receptors for T-cell immunotherapy. Nat Biotechnol. 2016;34:430–434. doi: 10.1038/nbt.3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adusumilli PS, et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci Transl Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang ZL, et al. Identification and selective expansion of functionally superior T cells expressing chimeric antigen receptors. J Transl Med. 2015;13:161. doi: 10.1186/s12967-015-0519-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long AH, et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nat Med. 2015;21:581–590. doi: 10.1038/nm.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frigault MJ, et al. Identification of Chimeric Antigen Receptors That Mediate Constitutive or Inducible Proliferation of T Cells. Cancer Immunol Res. 2015;3:356–367. doi: 10.1158/2326-6066.CIR-14-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eyquem J, et al. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature. 2017;543:113–117. doi: 10.1038/nature21405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karlsson H, et al. Evaluation of Intracellular Signaling Downstream Chimeric Antigen Receptors. PLoS One. 2015;10:e0144787. doi: 10.1371/journal.pone.0144787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aivazian D, Stern LJ. Phosphorylation of T cell receptor zeta is regulated by a lipid dependent folding transition. Nat Struct Biol. 2000;7:1023–6. doi: 10.1038/80930. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, et al. Basic residues in the T-cell receptor ζ cytoplasmic domain mediate membrane association and modulate signaling. Proc Natl Acad Sci U S A. 2011;108:19323–19328. doi: 10.1073/pnas.1108052108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dobbins J, et al. Binding of the cytoplasmic domain of CD28 to the plasma membrane inhibits Lck recruitment and signaling. Sci Signal. 2016;9:ra75. doi: 10.1126/scisignal.aaf0626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu C, et al. Regulation of T Cell Receptor Activation by Dynamic Membrane Binding of the CD3ε Cytoplasmic Tyrosine-Based Motif. Cell. 2008;135:702–713. doi: 10.1016/j.cell.2008.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Merwe PA, et al. Why Do Some T Cell Receptor Cytoplasmic Domains Associate with the Plasma Membrane? Front Immunol. 2012;3:29. doi: 10.3389/fimmu.2012.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li YC, et al. Cutting Edge: mechanical forces acting on T cells immobilized via the TCR complex can trigger TCR signaling. J Immunol. 2010;184:5959–63. doi: 10.4049/jimmunol.0900775. [DOI] [PubMed] [Google Scholar]
- 20.Husson J, et al. Force Generation upon T Cell Receptor Engagement. PLoS One. 2011;6:e19680. doi: 10.1371/journal.pone.0019680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu KH, Butte MJ. T cell activation requires force generation. J Cell Biol. 2016;213:535–542. doi: 10.1083/jcb.201511053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma Z, et al. Surface-anchored monomeric agonist pMHCs alone trigger TCR with high sensitivity. PLoS Biol. 2008;6:e43. doi: 10.1371/journal.pbio.0060043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cordoba SP, et al. The large ectodomains of CD45 and CD148 regulate their segregation from and inhibition of ligated T-cell receptor. Blood. 2013;121:4295–302. doi: 10.1182/blood-2012-07-442251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang VT, et al. Initiation of T cell signaling by CD45 segregation at “close contacts. Nat Immunol. 2016;17:574–582. doi: 10.1038/ni.3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.James JR, Vale RD. Biophysical mechanism of T-cell receptor triggering in a reconstituted system. Nature. 2012;487:64–9. doi: 10.1038/nature11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ellebrecht CT, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353:179–184. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hudecek M, et al. Receptor Affinity and Extracellular Domain Modifications Affect Tumor Recognition by ROR1-Specific Chimeric Antigen Receptor T Cells. Clin Cancer Res. 2013;19:3153–3164. doi: 10.1158/1078-0432.CCR-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hudecek M, et al. The Nonsignaling Extracellular Spacer Domain of Chimeric Antigen Receptors Is Decisive for In Vivo Antitumor Activity. Cancer Immunol Res. 2015;3:125–135. doi: 10.1158/2326-6066.CIR-14-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dustin ML, Depoil D. New insights into the T cell synapse from single molecule techniques. Nat Rev Immunol. 2011;11:672–84. doi: 10.1038/nri3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zah E, et al. T Cells Expressing CD19/CD20 Bispecific Chimeric Antigen Receptors Prevent Antigen Escape by Malignant B Cells. Cancer Immunol Res. 2016;4:498–508. doi: 10.1158/2326-6066.CIR-15-0231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3:939–51. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 32.Zhao Z, et al. Structural Design of Engineered Costimulation Determines Tumor Rejection Kinetics and Persistence of CAR T Cells. Cancer Cell. 2015;28:415–428. doi: 10.1016/j.ccell.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Harris DT, Kranz DM. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends Pharmacol Sci. 2016;37:220–230. doi: 10.1016/j.tips.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Harwood NE, Batista FD. Early Events in B Cell Activation. Annu Rev Immunol. 2010;28:185–210. doi: 10.1146/annurev-immunol-030409-101216. [DOI] [PubMed] [Google Scholar]
- 35.Malissen B, Bongrand P. Early T cell activation: integrating biochemical, structural, and biophysical cues. Annu Rev Immunol. 2015;33:539–61. doi: 10.1146/annurev-immunol-032414-112158. [DOI] [PubMed] [Google Scholar]
- 36.Kunkele A, et al. Functional Tuning of CARs Reveals Signaling Threshold above Which CD8+ CTL Antitumor Potency Is Attenuated due to Cell Fas-FasL-Dependent AICD. Cancer Immunol Res. 2015;3:368–379. doi: 10.1158/2326-6066.CIR-14-0200. [DOI] [PubMed] [Google Scholar]
- 37.Cherkassky L, et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130–3144. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jensen MC, et al. Antitransgene Rejection Responses Contribute to Attenuated Persistence of Adoptively Transferred CD20/CD19-Specific Chimeric Antigen Receptor Redirected T Cells in Humans. Biol Blood Marrow Transplant. 2010;16:1245–1256. doi: 10.1016/j.bbmt.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park JR, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 40.Kershaw MH, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Till BG, et al. Adoptive immunotherapy for indolent non-Hodgkin lymphoma and mantle cell lymphoma using genetically modified autologous CD20-specific T cells. Blood. 2008;112:2261–71. doi: 10.1182/blood-2007-12-128843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamers CHJ, et al. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J Clin Oncol. 2006;24:e20–2. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 43.Morgan RA, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther. 2010;18:843–51. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brentjens RJ, et al. CD19-Targeted T Cells Rapidly Induce Molecular Remissions in Adults with Chemotherapy-Refractory Acute Lymphoblastic Leukemia. Sci Transl Med. 2013;5:177ra38. doi: 10.1126/scitranslmed.3005930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Maude SL, et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Davila ML, et al. Efficacy and Toxicity Management of 19-28z CAR T Cell Therapy in B Cell Acute Lymphoblastic Leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Finney HM, et al. Activation of resting human primary T cells with chimeric receptors: costimulation from CD28, inducible costimulator, CD134, and CD137 in series with signals from the TCR zeta chain. J Immunol. 2004;172:104–13. doi: 10.4049/jimmunol.172.1.104. [DOI] [PubMed] [Google Scholar]
- 48.Imai C, et al. Chimeric receptors with 4-1BB signaling capacity provoke potent cytotoxicity against acute lymphoblastic leukemia. Leukemia. 2004;18:676–684. doi: 10.1038/sj.leu.2403302. [DOI] [PubMed] [Google Scholar]
- 49.Milone MC, et al. Chimeric Receptors Containing CD137 Signal Transduction Domains Mediate Enhanced Survival of T Cells and Increased Antileukemic Efficacy In Vivo. Mol Ther. 2009;17:1453–1464. doi: 10.1038/mt.2009.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Savoldo B, et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. J Clin Invest. 2011;121:1822–6. doi: 10.1172/JCI46110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kowolik CM, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 52.Hatfield SM, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. 2015;7:277ra30. doi: 10.1126/scitranslmed.aaa1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Torre-Amione G, et al. A highly immunogenic tumor transfected with a murine transforming growth factor type beta 1 cDNA escapes immune surveillance. Proc Natl Acad Sci U S A. 1990;87:1486–90. doi: 10.1073/pnas.87.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kalos M, et al. T cells with chimeric antigen receptors have potent antitumor effects and can establish memory in patients with advanced leukemia. Sci Transl Med. 2011;3:95ra73. doi: 10.1126/scitranslmed.3002842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Porter DL, et al. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med. 2011;365:725–33. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brentjens R, et al. Treatment of chronic lymphocytic leukemia with genetically targeted autologous T cells: case report of an unforeseen adverse event in a phase I clinical trial. Mol Ther. 2010;18:666–8. doi: 10.1038/mt.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maude SL, et al. Managing Cytokine Release Syndrome Associated With Novel T Cell-Engaging Therapies. Cancer J. 2014;20:119–122. doi: 10.1097/PPO.0000000000000035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee DW, et al. Current concepts in the diagnosis and management of cytokine release syndrome. Blood. 2014;124:188–95. doi: 10.1182/blood-2014-05-552729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee DW, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet. 2015;385:517–28. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grupp SA, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013;368:1509–18. doi: 10.1056/NEJMoa1215134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sotillo E, et al. Convergence of Acquired Mutations and Alternative Splicing of CD19 Enables Resistance to CART-19 Immunotherapy. Cancer Discov. 2015;5:1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Johnson LA, June CH. Driving gene-engineered T cell immunotherapy of cancer. Cell Res. 2017;27:38–58. doi: 10.1038/cr.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beatty GL, et al. Mesothelin-Specific Chimeric Antigen Receptor mRNA-Engineered T Cells Induce Antitumor Activity in Solid Malignancies. Cancer Immunol Res. 2014;2:112–120. doi: 10.1158/2326-6066.CIR-13-0170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maus MV, et al. T Cells Expressing Chimeric Antigen Receptors Can Cause Anaphylaxis in Humans. Cancer Immunol Res. 2013;1:26–31. doi: 10.1158/2326-6066.CIR-13-0006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown CE, et al. Regression of Glioblastoma after Chimeric Antigen Receptor T-Cell Therapy. N Engl J Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stephan SB, et al. Biopolymer implants enhance the efficacy of adoptive T-cell therapy. Nat Biotechnol. 2015;33:97–101. doi: 10.1038/nbt.3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol. 2013;31:999–1008. doi: 10.1038/nbt.2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Klebanoff CA, et al. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med. 2016;22:26–36. doi: 10.1038/nm.4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tran E, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer. N Engl J Med. 2016;375:2255–2262. doi: 10.1056/NEJMoa1609279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Linnemann C, et al. High-throughput epitope discovery reveals frequent recognition of neo-antigens by CD4+ T cells in human melanoma. Nat Med. 2014;21:81–85. doi: 10.1038/nm.3773. [DOI] [PubMed] [Google Scholar]
- 71.Tran E, et al. Cancer Immunotherapy Based on Mutation-Specific CD4+ T Cells in a Patient with Epithelial Cancer. Science. 2014;344:641–645. doi: 10.1126/science.1251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Johnson LA, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med. 2015;7:275ra22. doi: 10.1126/scitranslmed.aaa4963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Willemsen RA, et al. T cell retargeting with MHC class I-restricted antibodies: the CD28 costimulatory domain enhances antigen-specific cytotoxicity and cytokine production. J Immunol. 2005;174:7853–8. doi: 10.4049/jimmunol.174.12.7853. [DOI] [PubMed] [Google Scholar]
- 74.Liu H, et al. Targeting Alpha-Fetoprotein (AFP)-MHC Complex with CAR T-Cell Therapy for Liver Cancer. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1203. [DOI] [PubMed] [Google Scholar]
- 75.Liu X, et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer Res. 2015;75:3596–607. doi: 10.1158/0008-5472.CAN-15-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Caruso HG, et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer Res. 2015;75:3505–3518. doi: 10.1158/0008-5472.CAN-15-0139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Parkhurst MR, et al. T Cells Targeting Carcinoembryonic Antigen Can Mediate Regression of Metastatic Colorectal Cancer but Induce Severe Transient Colitis. Mol Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kloss CC, et al. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol. 2013;31:71–75. doi: 10.1038/nbt.2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fedorov VD, et al. PD-1- and CTLA-4-Based Inhibitory Chimeric Antigen Receptors (iCARs) Divert Off-Target Immunotherapy Responses. Sci Transl Med. 2013;5:215ra172–215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Roybal KT, et al. Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell. 2016;164:770–779. doi: 10.1016/j.cell.2016.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Han X, et al. Masked Chimeric Antigen Receptor for Tumor-Specific Activation. Mol Ther. 2017;25:274–284. doi: 10.1016/j.ymthe.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ahmed N, et al. Human Epidermal Growth Factor Receptor 2 (HER2) -Specific Chimeric Antigen Receptor-Modified T Cells for the Immunotherapy of HER2-Positive Sarcoma. J Clin Oncol. 2015;33:1688–1696. doi: 10.1200/JCO.2014.58.0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barrett DM, et al. Treatment of advanced leukemia in mice with mRNA engineered T cells. Hum Gene Ther. 2011;22:1575–86. doi: 10.1089/hum.2011.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wu C-Y, et al. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science. 2015;350:aab4077. doi: 10.1126/science.aab4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bonini C, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science. 1997;276:1719–24. doi: 10.1126/science.276.5319.1719. [DOI] [PubMed] [Google Scholar]
- 86.Tiberghien P, et al. Administration of herpes simplex-thymidine kinase-expressing donor T cells with a T-cell-depleted allogeneic marrow graft. Blood. 2001;97:63–72. doi: 10.1182/blood.v97.1.63. [DOI] [PubMed] [Google Scholar]
- 87.Berger C, et al. Analysis of transgene-specific immune responses that limit the in vivo persistence of adoptively transferred HSV-TK-modified donor T cells after allogeneic hematopoietic cell transplantation. Blood. 2006;107:2294–302. doi: 10.1182/blood-2005-08-3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Di Stasi A, et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang X, et al. A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood. 2011;118:1255–1263. doi: 10.1182/blood-2011-02-337360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer. Immunol Rev. 2014;257:56–71. doi: 10.1111/imr.12132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hinrichs CS, et al. Adoptively transferred effector cells derived from naive rather than central memory CD8+ T cells mediate superior antitumor immunity. Proc Natl Acad Sci U S A. 2009;106:17469–74. doi: 10.1073/pnas.0907448106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Berger C, et al. Adoptive transfer of effector CD8+ T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Klebanoff CA, et al. Determinants of Successful CD8+ T-Cell Adoptive Immunotherapy for Large Established Tumors in Mice. Clin Cancer Res. 2011;17:5343–5352. doi: 10.1158/1078-0432.CCR-11-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sommermeyer D, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo. Leukemia. 2016;30:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Turtle CJ, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients. J Clin Invest. 2016;126:2123–38. doi: 10.1172/JCI85309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Perna SK, et al. Interleukin-7 mediates selective expansion of tumor-redirected cytotoxic T lymphocytes (CTLs) without enhancement of regulatory T-cell inhibition. Clin Cancer Res. 2014;20:131–9. doi: 10.1158/1078-0432.CCR-13-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Di Stasi A, et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392–402. doi: 10.1182/blood-2009-03-209650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Peng W, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res. 2010;16:5458–68. doi: 10.1158/1078-0432.CCR-10-0712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Pegram HJ, et al. CD28z CARs and Armored CARs. Cancer J. 2014;20:127–133. doi: 10.1097/PPO.0000000000000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhang L, et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Mol Ther. 2011;19:751–9. doi: 10.1038/mt.2010.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang L, et al. Tumor-infiltrating lymphocytes genetically engineered with an inducible gene encoding interleukin-12 for the immunotherapy of metastatic melanoma. Clin Cancer Res. 2015;21:2278–88. doi: 10.1158/1078-0432.CCR-14-2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ninomiya S, et al. Tumor indoleamine 2,3-dioxygenase (IDO) inhibits CD19-CAR T cells and is downregulated by lymphodepleting drugs. Blood. 2015;125:3905–16. doi: 10.1182/blood-2015-01-621474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Newick K, et al. Augmentation of CAR T-cell Trafficking and Antitumor Efficacy by Blocking Protein Kinase A Localization. Cancer Immunol Res. 2016;4:541–51. doi: 10.1158/2326-6066.CIR-15-0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7:1118–22. doi: 10.1038/nm1001-1118. [DOI] [PubMed] [Google Scholar]
- 105.Bendle GM, et al. Blockade of TGF-β Signaling Greatly Enhances the Efficacy of TCR Gene Therapy of Cancer. J Immunol. 2013;191:3232–3239. doi: 10.4049/jimmunol.1301270. [DOI] [PubMed] [Google Scholar]
- 106.Ren J, et al. Multiplex genome editing to generate universal CAR T cells resistant to PD1 inhibition. Clin Cancer Res. 2016 doi: 10.1158/1078-0432.CCR-16-1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Liu X, et al. A Chimeric Switch-Receptor Targeting PD1 Augments the Efficacy of Second-Generation CAR T Cells in Advanced Solid Tumors. Cancer Res. 2016;76:1578–1590. doi: 10.1158/0008-5472.CAN-15-2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Mohammed S, et al. Improving Chimeric Antigen Receptor-Modified T Cell Function by Reversing the Immunosuppressive Tumor Microenvironment of Pancreatic Cancer. Mol Ther. 2017;25:249–258. doi: 10.1016/j.ymthe.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wilkie S, et al. Selective Expansion of Chimeric Antigen Receptor-targeted T-cells with Potent Effector Function using Interleukin-4. J Biol Chem. 2010;285:25538–25544. doi: 10.1074/jbc.M110.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Gerlinger M, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Grada Z, et al. TanCAR: A Novel Bispecific Chimeric Antigen Receptor for Cancer Immunotherapy. Mol Ther Nucleic Acids. 2013;2:e105. doi: 10.1038/mtna.2013.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hegde M, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther. 2013;21:2087–101. doi: 10.1038/mt.2013.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hegde M, et al. Tandem CAR T cells targeting HER2 and IL13Rα2 mitigate tumor antigen escape. J Clin Invest. 2016;126:3036–52. doi: 10.1172/JCI83416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ruella M, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest. 2016;126:3814–3826. doi: 10.1172/JCI87366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bos TJ, et al. Large double copy vectors are functional but show a size-dependent decline in transduction efficiency. J Biotechnol. 2010;150:37–40. doi: 10.1016/j.jbiotec.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 116.Kumar M, et al. Systematic determination of the packaging limit of lentiviral vectors. Hum Gene Ther. 2001;12:1893–905. doi: 10.1089/104303401753153947. [DOI] [PubMed] [Google Scholar]
- 117.Yang Y, et al. Challenges and opportunities of allogeneic donor-derived CAR T cells. Curr Opin Hematol. 2015;22:509–15. doi: 10.1097/MOH.0000000000000181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brudno JN, et al. Allogeneic T Cells That Express an Anti-CD19 Chimeric Antigen Receptor Induce Remissions of B-Cell Malignancies That Progress After Allogeneic Hematopoietic Stem-Cell Transplantation Without Causing Graft-Versus-Host Disease. J Clin Oncol. 2016;34:1112–1121. doi: 10.1200/JCO.2015.64.5929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Provasi E, et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat Med. 2012;18:807–15. doi: 10.1038/nm.2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Torikai H, et al. A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood. 2012;119:5697–5705. doi: 10.1182/blood-2012-01-405365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Poirot L, et al. Multiplex Genome-Edited T-cell Manufacturing Platform for “Off-the-Shelf” Adoptive T-cell Immunotherapies. Cancer Res. 2015;75:3853–3864. doi: 10.1158/0008-5472.CAN-14-3321. [DOI] [PubMed] [Google Scholar]
- 122.Liu X, et al. CRISPR-Cas9-mediated multiplex gene editing in CAR-T cells. Cell Res. 2017;27:154–157. doi: 10.1038/cr.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.MacLeod DT, et al. Integration of a CD19 CAR into the TCR Alpha Chain Locus Streamlines Production of Allogeneic Gene-Edited CAR T Cells. Mol Ther. 2017 doi: 10.1016/j.ymthe.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Qasim W, et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci Transl Med. 2017;9:eaaj2013. doi: 10.1126/scitranslmed.aaj2013. [DOI] [PubMed] [Google Scholar]
- 125.Rossig C, et al. Epstein-Barr virus-specific human T lymphocytes expressing antitumor chimeric T-cell receptors: potential for improved immunotherapy. Blood. 2002;99:2009–16. doi: 10.1182/blood.v99.6.2009. [DOI] [PubMed] [Google Scholar]
- 126.Savoldo B, et al. Epstein Barr virus specific cytotoxic T lymphocytes expressing the anti-CD30zeta artificial chimeric T-cell receptor for immunotherapy of Hodgkin disease. Blood. 2007;110:2620–30. doi: 10.1182/blood-2006-11-059139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Pule MA, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nat Med. 2008;14:1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Barker JN, et al. Successful treatment of EBV-associated posttransplantation lymphoma after cord blood transplantation using third-party EBV-specific cytotoxic T lymphocytes. Blood. 2010;116:5045–5049. doi: 10.1182/blood-2010-04-281873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Gattinoni L, et al. Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med. 2005;202:907–12. doi: 10.1084/jem.20050732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.John LB, et al. Anti-PD-1 Antibody Therapy Potently Enhances the Eradication of Established Tumors By Gene-Modified T Cells. Clin Cancer Res. 2013;19:5636–5646. doi: 10.1158/1078-0432.CCR-13-0458. [DOI] [PubMed] [Google Scholar]
- 131.Gargett T, et al. GD2-specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but can be Protected From Activation-induced Cell Death by PD-1 Blockade. Mol Ther. 2016;24:1135–1149. doi: 10.1038/mt.2016.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ahmadzadeh M, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood. 2009;114:1537–44. doi: 10.1182/blood-2008-12-195792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Sakuishi K, et al. Targeting Tim-3 and PD-1 pathways to reverse T cell exhaustion and restore anti-tumor immunity. J Exp Med. 2010;207:2187–94. doi: 10.1084/jem.20100643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Baitsch L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–60. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Burns WR, et al. Lack of specific gamma-retroviral vector long terminal repeat promoter silencing in patients receiving genetically engineered lymphocytes and activation upon lymphocyte restimulation. Blood. 2009;114:2888–99. doi: 10.1182/blood-2009-01-199216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chong EA, et al. PD-1 Blockade Modulates Chimeric Antigen Receptor (CAR) Modified T Cells and Induces Tumor Regression: Refueling the CAR. Blood. 2016 doi: 10.1182/blood-2016-09-738245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Krebs K, Böttinger N, Huang LR, Chmielewski M, Arzberger S, Gasteiger G, Jäger C, Schmitt E, Bohne F, Aichler M, Uckert W, Abken H, Heikenwalder M, Knolle PPU. T cells expressing a chimeric antigen receptor that binds hepatitis B virus envelope proteins control virus replication in mice. Gastroenterology. 2013;145:456–465. doi: 10.1053/j.gastro.2013.04.047. [DOI] [PubMed] [Google Scholar]
- 138.Sautto GA, et al. Chimeric antigen receptor (CAR)-engineered T cells redirected against hepatitis C virus (HCV) E2 glycoprotein. Gut. 2016;65:512–23. doi: 10.1136/gutjnl-2014-308316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Romeo C, Seed B. Cellular immunity to HIV activated by CD4 fused to T cell or Fc receptor polypeptides. Cell. 1991;64:1037–46. doi: 10.1016/0092-8674(91)90327-u. [DOI] [PubMed] [Google Scholar]
- 140.Deeks SG, et al. A Phase II Randomized Study of HIV-Specific T-Cell Gene Therapy in Subjects with Undetectable Plasma Viremia on Combination Antiretroviral Therapy. Mol Ther. 2002;5:788–797. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- 141.Kumaresan PR, et al. Bioengineering T cells to target carbohydrate to treat opportunistic fungal infection. Proc Natl Acad Sci U S A. 2014;111:10660–10665. doi: 10.1073/pnas.1312789111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jyothi MD, et al. Targeting autoantigen-specific T cells and suppression of autoimmune encephalomyelitis with receptor-modified T lymphocytes. Nat Biotechnol. 2002;20:1215–20. doi: 10.1038/nbt758. [DOI] [PubMed] [Google Scholar]
- 143.Margalit A, et al. Chimeric beta2 microglobulin/CD3zeta polypeptides expressed in T cells convert MHC class I peptide ligands into T cell activation receptors: a potential tool for specific targeting of pathogenic CD8(+) T cells. Int Immunol. 2003;15:1379–87. doi: 10.1093/intimm/dxg136. [DOI] [PubMed] [Google Scholar]
- 144.Mekala DJ, Geiger TL. Immunotherapy of autoimmune encephalomyelitis with redirected CD4+CD25+ T lymphocytes. Blood. 2005;105:2090–2092. doi: 10.1182/blood-2004-09-3579. [DOI] [PubMed] [Google Scholar]
- 145.Fransson M, et al. CAR/FoxP3-engineered T regulatory cells target the CNS and suppress EAE upon intranasal delivery. J Neuroinflammation. 2012;9:112. doi: 10.1186/1742-2094-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Blat D, et al. Suppression of murine colitis and its associated cancer by carcinoembryonic antigen-specific regulatory T cells. Mol Ther. 2014;22:1018–28. doi: 10.1038/mt.2014.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.MacDonald KG, et al. Alloantigen-specific regulatory T cells generated with a chimeric antigen receptor. J Clin Invest. 2016;126:1413–1424. doi: 10.1172/JCI82771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Boardman DA, et al. Expression of a chimeric antigen receptor specific for donor HLA class I enhances the potency of human regulatory T cells in preventing human skin transplant rejection. Am J Transplant. 2016 doi: 10.1111/ajt.14185. [DOI] [PubMed] [Google Scholar]
- 149.Noyan F, et al. Prevention of allograft rejection by use of regulatory T cells with a MHC-specific chimeric antigen receptor. Am J Transplant. 2016 doi: 10.1111/ajt.14175. [DOI] [PubMed] [Google Scholar]
- 150.Yoon J, et al. FVIII-specific human chimeric antigen receptor (CAR) T-regulatory cells suppress T-and B-cell responses to FVIII. Blood. 2016 doi: 10.1182/blood-2016-07-727834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Jamnani FR, et al. T cells expressing VHH-directed oligoclonal chimeric HER2 antigen receptors: Towards tumor-directed oligoclonal T cell therapy. Biochim Biophys Acta - Gen Subj. 2014;1840:378–386. doi: 10.1016/j.bbagen.2013.09.029. [DOI] [PubMed] [Google Scholar]
- 152.Hombach A, et al. Adoptive immunotherapy with genetically engineered T cells: modification of the IgG1 Fc “spacer” domain in the extracellular moiety of chimeric antigen receptors avoids “off-target” activation and unintended initiation of an innate immune response. Gene Ther. 2010;17:1206–1213. doi: 10.1038/gt.2010.91. [DOI] [PubMed] [Google Scholar]
- 153.Almåsbak H, et al. Inclusion of an IgG1-Fc spacer abrogates efficacy of CD19 CAR T cells in a xenograft mouse model. Gene Ther. 2015;22:391–403. doi: 10.1038/gt.2015.4. [DOI] [PubMed] [Google Scholar]
- 154.Jonnalagadda M, et al. Chimeric Antigen Receptors With Mutated IgG4 Fc Spacer Avoid Fc Receptor Binding and Improve T Cell Persistence and Antitumor Efficacy. Mol Ther. 2015;23:757–768. doi: 10.1038/mt.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.van der Stegen SJC, et al. The pharmacology of second-generation chimeric antigen receptors. Nat Rev Drug Discov. 2015;14:499–509. doi: 10.1038/nrd4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Pulè MA, et al. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther. 2005;12:933–941. doi: 10.1016/j.ymthe.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 157.Wang J, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther. 2007;18:712–25. doi: 10.1089/hum.2007.028. [DOI] [PubMed] [Google Scholar]
- 158.Wilkie S, et al. Retargeting of human T cells to tumor-associated MUC1: the evolution of a chimeric antigen receptor. J Immunol. 2008;180:4901–9. doi: 10.4049/jimmunol.180.7.4901. [DOI] [PubMed] [Google Scholar]
- 159.Tammana S, et al. 4-1BB and CD28 Signaling Plays a Synergistic Role in Redirecting Umbilical Cord Blood T Cells Against B-Cell Malignancies. Hum Gene Ther. 2010;21:75–86. doi: 10.1089/hum.2009.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhong XS, et al. Chimeric antigen receptors combining 4-1BB and CD28 signaling domains augment PI3kinase/AKT/Bcl-XL activation and CD8+ T cell-mediated tumor eradication. Mol Ther. 2010;18:413–20. doi: 10.1038/mt.2009.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Carpenito C, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci U S A. 2009;106:3360–5. doi: 10.1073/pnas.0813101106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Haso W, et al. Anti-CD22-chimeric antigen receptors targeting B-cell precursor acute lymphoblastic leukemia. Blood. 2013;121:1165–1174. doi: 10.1182/blood-2012-06-438002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Pegram HJ, et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–41. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]