

SUMMARY
Ultraviolet (UV) radiation is a carcinogen that generates DNA lesions. Here we demonstrate an unexpected role for DGCR8, an RNA binding protein that canonically functions with Drosha to mediate microRNA processing, in the repair of UV-induced DNA lesions. Treatment with UV induced phosphorylation on Serine 153 (S153) of DGCR8 in human and murine cells. S153 phosphorylation was critical for cellular resistance to UV, the removal of UV-induced DNA lesions, and the recovery of RNA synthesis after UV exposure, but not for microRNA expression. The RNA-binding and Drosha-binding activities of DGCR8 were not critical for UV resistance. DGCR8 depletion was epistatic to defects in XPA, CSA and CSB for UV sensitivity. DGCR8 physically interacted with CSB and RNA polymerase II. JNKs were involved in the UV-induced S153 phosphorylation. These findings suggest that UV-induced S153 phosphorylation mediates transcription-coupled nucleotide excision repair of UV-induced DNA lesions in a manner independent of microRNA processing.
Keywords: DGCR8, Drosha, ultraviolet radiation, transcription coupled nucleotide excision repair, JNK, microRNA, DNA repair
eTOC Blurb
Calses et al. find that UV-induced phosphorylation on Serine 153 of DGCR8, an RNA binding protein involved in microRNA processing, is critical for cellular resistance to UV through DNA repair of UV-induced lesions.
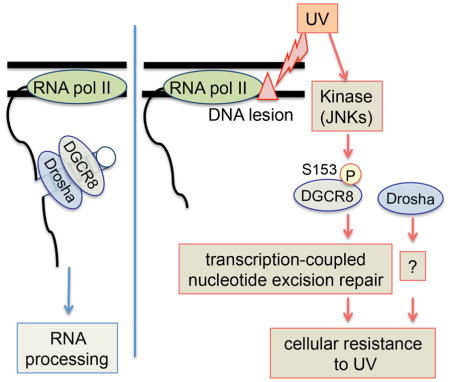
INTRODUCTION
UV irradiation generates DNA photoproducts, mainly cyclobutane pyrimidine dimers (CPDs) and 6-4 photoproducts (6-4PPs), and causes cell cycle arrest, apoptosis, mutagenesis and proliferative transformation (Herrlich et al., 2008). These DNA lesions are repaired by nucleotide excision repair (NER) in mammalian cells (Hanawalt and Spivak, 2008). Deficiencies in NER are associated with human genetic disorders, such as xeroderma pigmentosum (XP), Cockayne syndrome (CS), cerebro-oculo-facio-skeletal syndrome (COFS), UV-sensitive syndrome (UVSS) and trichothiodystrophy (TTD). The NER pathway operates through two subpathways: global genome nucleotide excision repair (GG-NER) and transcription-coupled nucleotide excision repair (TC-NER). In GG-NER, XPC and UV-DDB recognize damage sites throughout the genome, while TC-NER is initiated by RNA polymerase II (RNAPII) that is blocked by DNA lesions during transcription. These subpathways converge on a common downstream pathway to resolve the DNA lesions (Hanawalt and Spivak, 2008).
DGCR8 is a critical protein for microRNA (miRNA) biogenesis (Macias et al., 2013). MiRNAs are small, single-stranded, non-coding RNAs that post-transcriptionally regulate gene expression (Carthew and Sontheimer, 2009) and have been implicated in many biological processes including DNA damage response and repair (Wang and Taniguchi, 2013) and in the pathogenesis of many human diseases (Iorio and Croce, 2012). MiRNAs are initially transcribed as long primary miRNAs (pri-miRNAs) (Carthew and Sontheimer, 2009). Pri-miRNAs are processed by the microprocessor complex, consisting of Drosha (an RNase III-type enzyme), and DGCR8, a double-stranded RNA-binding protein, to generate ~70-nt precursor miRNAs (pre-miRNAs) (Carthew and Sontheimer, 2009). Pre-miRNAs are exported to the cytoplasm where they are cleaved by another RNase III, Dicer, to generate ~22-nt mature miRNA duplexes. Apart from its critical role in miRNA biogenesis, DGCR8 also regulates the stability of some mRNAs and snoRNAs (Macias et al., 2013; Macias et al., 2012). The two dsRNA-binding domains of DGCR8 are required for the recognition of the RNA substrates (Macias et al., 2012; Sohn et al., 2007; Yeom et al., 2006).
DGCR8 is regulated in several ways. MeCP2 binds to the dsRNA-binding domains of DGCR8 and prevents the binding of Drosha to DGCR8 (Cheng et al., 2014). A heme group promotes the dimerization of DGCR8 and facilitates miRNA processing (Weitz et al., 2014). Deacetylation of DGCR8 also enhances miRNA processing (Wada et al., 2012). DGCR8 is phosphorylated at 23 sites and these modifications increase DGCR8 protein stability (Herbert et al., 2013). Some of the 23 phosphorylation sites may be targets of ERK/MAPK (Herbert et al., 2013), but there are likely other kinases involved as well.
miRNAs have been implicated in the cellular response to UV: the expression of a subset of miRNAs is altered in response to UV (Dziunycz et al., 2010; Pothof et al., 2009) and depletion of factors required for miRNA biogenesis leads to cellular sensitivity to UV (Pothof et al., 2009). However, the mechanisms connecting the miRNA biogenesis machinery and UV response remain unclear, and whether UV sensitivity of miRNA biogenesis protein-deficient cells is mediated by the miRNA biogenesis defect is not clear.
Here we show a completely novel and unexpected function of DGCR8 in the repair of UV-induced DNA lesions, which is independent of microRNA processing.
RESULTS
DGCR8 is phosphorylated in response to UV radiation
Initially, we found that DGCR8 was phosphorylated in response to UV by performing western blot analysis of UVC-treated human and murine cells (Figure 1A–B). We subsequently performed mass spectrometry of DGCR8 protein immunopurified from UVC-irradiated human HeLa cells, and identified nine phosphorylation sites (S95, S153, S271, S275, T371, S373, S377, S434 and S619) (Figure 1C and Figure S1A–B). Site-directed mutagenesis experiments for each of these sites revealed that S153 was phosphorylated in a UV-induced manner (Figure 1E). S153 of DGCR8 is conserved among mammals, but not in other vertebrates (Figure 1D and Figure S1C).
Figure 1. S153 of DGCR8 is phosphorylated in response to UV.

(A) Immunoblot. Cell lysates from U2OS cells treated with UVC were treated with λ phosphatase and phosphatase inhibitors, and immunoblotted for DGCR8 and vinculin (loading control). (B) Cell lysates from primary murine embryonic fibroblasts were immunoblotted for Dgcr8. Large gels were used for the western blots shown in (A) and (B) to clearly see the shift of the bands. (C) Schematic illustration of nine phosphorylation sites of human DGCR8 identified by mass spectrometry (not proportional to actual size). The DGCR8 protein contains a WW domain, two dsRNA binding domains (DRBD1 and DRBD2) and a Drosha binding domain (Drosha-BD). (D) Sequence alignment of S153 surrounding amino acids. S153 is conserved among mammals. (E) Immunoblot. U2OS cells depleted of DGCR8 and transfected with shRNA-resistant FLAG-WT DGCR8 or FLAG-S153A DGCR8, +/−UV-irradiated were immunoblotted for DGCR8 or Flag. (F–H) Immunoblot. Indicated cells were irradiated with +/−UV and harvested at the indicated time points, or treated with other agents continuously as indicated. See also Figure S1.
Next we generated a phospho-specific antibody using a peptide that flanks S153 of human DGCR8 as an immunogen (Figure 1D). Specificity of the antibody was confirmed using S153A mutant cells and phosphatase treatment (Figure 1E and Figure S1D). S153 phosphorylation was detected in untreated cells, and was increased in response to UV in a time- and dose-dependent manner (Figure 1F) in multiple human cell lines, including primary fibroblasts and keratinocytes (Figure 1G). S153 phosphorylation was also induced by treatment with UVB, a chemical UV-mimetic, 4-nitroquinoline-1-oxide (4-NQO), and oxidizers including hydrogen peroxide and potassium bromate, but not with other DNA damaging agents and cellular stresses we have tested (Figure 1H and Figure S1E). We also generated a phospho-specific antibody using a peptide that flanks S153 of murine Dgcr8 as an immunogen (Figure 1D) and confirmed UV-induced phosphorylation of S153 in mouse embryonic fibroblasts (MEFs) (Figure S1F). Subcellular fractionation experiments revealed that S153-phosphorylated DGCR8 was detected in the nuclear soluble and chromatin fractions (Figure S1G). The S153 phosphorylation signal was induced diffusely in the nucleus (Figure S1H), even after localized UV irradiation through a micropore filter (Figure S1I), indicating that the distribution of S153-phosphorylated DGCR8 is not limited to sites of UV-induced DNA lesions. Furthermore, almost all of the UV-irradiated cells showed increased phosphorylation of S153 (Figure S1H–I), suggesting that the UV-induced S153 phosphorylation occurs in a cell cycle-independent manner.
DGCR8 phosphorylation mediates cellular resistance to UV radiation
To test the functional significance of UV-induced phosphorylation of S153, we examined UV sensitivity of DGCR8-deficient cells. Depletion of DGCR8 in a human colorectal cancer cell line (HCT-116) using three independent shRNAs resulted in hypersensitivity to UVC and UVB (Figure 2A and Figure S2A). We also recapitulated the UVC sensitivity phenotype with DGCR8 depletion in human fibroblasts (Figure 4B and Figure S4C). Reintroduction of shRNA-resistant wild-type DGCR8 or a S153D phospho-mimetic mutant into DGCR8-depleted HCT116 cells restored UVC resistance. However, the S153A phospho-mutant failed to restore UVC or UVB resistance (Figure 2B–C and Figure S2A), suggesting that S153 phosphorylation is critical for cellular resistance to UV. Similarly, DGCR8-depleted cells were hypersensitive to hydrogen peroxide, and complementation with wild-type or S153D DGCR8 restored resistance to hydrogen peroxide, while the S153A mutant did not (Figure S2B). Dgcr8 knockout MEFs were sensitive to UVC and UVB compared to Dgcr8 knockout MEFs transduced with wild-type human DGCR8 or wild-type mouse Dgcr8, while Dgcr8 knockout MEFs transduced with S153A mutant human DGCR8 or S153A mutant mouse Dgcr8 were as sensitive to UV as Dgcr8 knockout MEFs (Figures S2C and S2D).
Figure 2. Phosphorylation of S153 on DGCR8 is critical for UV resistance.

(A) UVC sensitivity assay. HCT116 cells were depleted of DGCR8 or XPC and plated for survival after +/− UV irradiation. Immunoblot shows DGCR8 and XPC depletion. (B) Schematic presentation of DGCR8 mutants. Immunoblot shows expression of the indicated shRNA-resistant DGCR8 constructs in DGCR8-depleted HCT116 cells. (C) UVC sensitivity assay. DGCR8-depleted HCT116 cells were transduced with the indicated DGCR8 constructs and plated for survival after +/− UV irradiation. All UVC sensitivity data represent mean values +/− SEM of three independent experiments. (D) IP-western. Cell lysate of UV-treated U2OS cells transduced with the indicated FLAG-tagged DGCR8 constructs were immunoprecipitated with anti-FLAG. Immunoblot was done using the indicated antibodies. (E) Model for two independent functions of DGCR8: 1) S153 phosphorylation-mediated UV resistance and TC-NER, and 2) dsRNA binding- and Drosha binding-domain-mediated RNA processing. See also Figure S2 and Figure S3.
Figure 4. Phosphorylation of S153 on DGCR8 is involved in transcription-coupled nucleotide excision repair.

(A) Removal of CPDs or 6-4PPs assessed by flow cytometry using anti-CPD and anti-6-4PP antibodies. HCT116 cells depleted of indicated genes and complemented with indicated constructs were treated with UVC, and CPD positive cells and 6-4PP positive cells at the indicated time points were measured. Asterisks indicate significant difference (p<0.05) relative to shControl +empty vector transduced cells. (B) UVC sensitivity assay. XPC-, CSA-, CSB-, or XPA-deficient fibroblasts and their corrected counterparts were depleted of DGCR8, and plated for survival after +/−UVC irradiation. (C) Recovery of RNA synthesis (RRS) assay. 5′-Ethynyl uridine (5′ EU) incorporation kinetics after UVC irradiation. U2OS cells transfected with indicated siRNAs and cDNA constructs were subjected to the assay. Asterisks indicate significant difference (p<0.05) relative to siControl +empty vector transduced cells. (D) U2OS cells treated with DRB or transfected with CDK9 siRNA were immunoblotted with the indicated antibodies. All data represent mean values +/− SEM of three independent experiments. See also Figure S4.
Previous studies have shown that mutations introduced into two dsRNA-binding domains of DGCR8 abolish microRNA processing activity (Yeom et al., 2006). Surprisingly, complementation of DGCR8-depleted cells with the dsRNA binding domains mutant (mDRBD1/2) restored UVC and UVB resistance (Figure 2B–C and Figure S2A), indicating that RNA binding activity and microRNA processing function of DGCR8 are not required for UV resistance.
Among the various DGCR8 deletion mutants we tested (Figure 2B and Figure S2E), the Δ692 (Drosha-binding deficient) mutant (Yeom et al., 2006) restored UVC resistance in DGCR8-depleted cells (Figure 2C), while other mutants with large deletions (Δ275, Δ483, and Δ276-773) (Yeom et al., 2006) failed to do so (Figure S2F). In contrast, the Δ692 S153A mutant failed to restore UV resistance in DGCR8-depleted cells (Figure 2C). The Δ692 or the Δ692 S153A mutant proteins were not co-immunoprecipitated with Drosha as expected, while wild-type DGCR8 and the DGCR8 S153A mutant were co-immunoprecipitated with Drosha (Figure 2D). Altogether, these findings indicate that S153 phosphorylation is critical for UV resistance, while the DGCR8-Drosha interaction is not.
Interestingly, Drosha depletion also led to hypersensitivity to UVC and UVB, which was rescued by reintroduction of wild-type Drosha, the ΔC114 (miRNA processing-deficient) or the ΔN490 (miRNA processing-deficient and DGCR8 binding-deficient) Drosha mutants (Han et al., 2004) (Figure 3A–B and Figure S2G). These data suggest that the DGCR8 binding or the microRNA processing activities of Drosha are not required for UV resistance. Furthermore, UV-induced phosphorylation of S153 on DGCR8 occurred normally in Drosha-depleted cells (Figure 3C). These findings collectively suggest that DGCR8 and Drosha are independently required for UV resistance and that the microRNA processing activity of the Drosha-DGCR8 microprocessor complex is not required for UV resistance.
Figure 3. Drosha is required for UV resistance independently of DGCR8.

(A) UVC sensitivity assay. HCT116 cells were depleted of Drosha or XPC, and plated for survival after +/−UV irradiation. Immunoblot shows Drosha and XPC depletion. (B) Schematic presentation of Drosha mutants. Immunoblot confirmed expression of the indicated Drosha constructs in Drosha-depleted HCT116 cells. Drosha-depleted HCT116 cells were transduced with the indicated Drosha constructs and subjected to the UVC sensitivity assay. All UVC sensitivity data represent mean values +/− SEM of three independent experiments. (C) U2OS cells depleted of DGCR8 or Drosha were UV irradiated and subjected to immunoblot with the indicated antibodies. See also Figure S2.
DGCR8 phosphorylation does not alter microRNA expression
Next, to determine whether S153 on DGCR8 is critical for miRNA expression, we conducted miRNA profiling using quantitative RT-PCR of 372 human miRNAs in DGCR8-depleted HCT116 cells transduced with either empty vector, wild-type DGCR8, or the S153A mutant. DGCR8-depleted cells transduced with empty vector showed reduction of microRNA expression compared to non-depleted control or wild-type DGCR8-expressing cells, but there was no clear difference of the microRNA expression profiles between the wild-type DGCR8 and S153A DGCR8 cells (Figure S3), suggesting that S153 is not critical for microRNA processing. This is consistent with a recent report that many phosphorylation sites on DGCR8 (including S153) are not critical for its microRNA processing activity (Herbert et al., 2013). These results indicate that the human DGCR8 protein has at least two independent functions: 1) S153 phosphorylation-mediated UV resistance and 2) RNA binding and Drosha binding domain-mediated RNA processing (Figure 2E).
DGCR8 phosphorylation mediates the repair of UV-induced DNA lesions
We next measured the removal of CPDs and 6-4PPs after UV irradiation to test whether DGCR8 and Drosha are involved in the repair of UV-induced DNA lesions. DGCR8 depletion, as well as XPC depletion and CSB depletion, reduced the removal of CPDs and 6-4PPs (Figure 4A and Figure S4A–B). Reintroduction of wild-type DGCR8 into DGCR8-depleted cells restored efficient removal of CPDs and 6-4 PPs, while the S153A-DGCR8 mutant failed to do so, suggesting that UV-induced S153 phosphorylation is required for the efficient repair of UV-induced DNA lesions. In contrast, depletion of Drosha did not reduce the removal (Figure 4A and Figure S4A–B), suggesting that Drosha is not involved in the repair of the UV-induced DNA lesions. The mechanisms by which DGCR8 and Drosha function in UV resistance are therefore distinct.
DGCR8 and factors involved in transcription coupled nucleotide excision repair are epistatic in UV sensitivity
To test whether DGCR8 is involved in NER and to identify which NER subpathway it may be involved in, we performed epistasis analyses (Figure 4B and Figure S4C). We depleted DGCR8 in human patient-derived fibroblasts deficient in XPC, XPA, CSA or CSB and their corrected counterparts. As expected, XPC, XPA, CSA and CSB-deficient cells were hypersensitive to UVC (Yasuda et al., 2007) and DGCR8 depletion sensitized control (corrected) fibroblasts to UVC. DGCR8 depletion further sensitized XPC-deficient cells to UVC, indicating that DGCR8 is not epistatic with XPC, a factor specifically involved in GG-NER. In contrast, DGCR8 depletion did not further sensitize CSB-, CSA- or XPA-deficient fibroblasts to UVC, indicating that DGCR8 is epistatic with CSB, CSA and XPA. CSB and CSA are factors specifically involved in TC-NER, while XPA is involved in both TC-NER and GG-NER. Therefore, these data collectively suggest that DGCR8 functions in TC-NER, but not in GG-NER.
DGCR8 phosphorylation modulates RNA synthesis recovery (RRS) after UV radiation
To confirm the involvement of DGCR8 in TC-NER, we utilized a recovery of RNA synthesis (RRS) assay (Jia et al., 2015) (Figure 4C and Figure S4D). DGCR8-depleted cells showed impaired RRS after UV radiation similarly to CSB-depleted cells. Reintroduction of either the wild-type or S153D DGCR8, but not the S153A DGCR8, into DGCR8-depleted cells rescued RRS after UV, suggesting that S153 phosphorylation is critical for RRS after UV. These findings are consistent with DGCR8 being involved in TC-NER.
Since RNAPII stalling is presumed to be the initial step for TC-NER (Fousteri et al., 2006), we hypothesized that RNAPII stalling triggers S153 phosphorylation. To test this, we treated human cells with a transcription elongation inhibitor, 5,6-Dichlorobenzimidazole 1-β-D-ribofuranoside (DRB). DRB treatment led to increased S153 phosphorylation even in the absence of UV treatment (Figure 4D). Depletion of CDK9, a kinase involved in transcription elongation and a target of DRB, also led to increased phosphorylation of S153 (Figure 4D). These findings suggest that RNAPII stalling caused either by UV-induced DNA lesions or by other mechanisms stimulates S153 phosphorylation.
Furthermore, wild-type DGCR8 was co-immunoprecipitated with RNAPII, CSB and Drosha (Figure 5A–B), but not with XPC (Figure S4E), regardless of UV treatment, consistent with DGCR8’s role in TC-NER. Interestingly, S153A-DGCR8 was also co-immunoprecipitated with RNAPII, CSB and Drosha (Figure 5A–B), indicating that S153 phosphorylation is not critical for the interactions among these factors. CSA or CSB deficiency did not affect UV-induced phosphorylation of S153 (Figure S5A), indicating that S153 phosphorylation is not downstream of CSA or CSB. Furthermore, DGCR8 depletion did not affect the expression of CSA, CSB, or RNAPII (Figure S5B). Therefore, although DGCR8 is involved in TC-NER, its precise mechanism remains to be determined.
Figure 5. DGCR8 interacts with factors involved in transcription-coupled nucleotide excision repair.

(A–B) IP-western. Cell lysate of UV-treated (or untreated) U2OS cells transduced with the HA-tagged DGCR8 (wild type or S153A mutant) constructs were immunoprecipitated with the indicated antibodies. Immunoblot was done using the indicated antibodies. See also Figure S4 and S5.
Lastly, we sought to identify the kinase that phosphorylates S153 of DGCR8 in response to UV treatment. Since JNKs are known to be activated in response to UV exposure (Johnson and Nakamura, 2007; Lopez-Camarillo et al., 2012), we focused on JNKs. First, treatment with a JNK inhibitor, SP600125, abrogated UV-induced S153 phosphorylation (Figure 6A), while treatment with a JNK activator, anisomycin, induced S153 phosphorylation in the absence of UV treatment (Figure 6B). Second, overexpression of JNK1a1 induced S153 phosphorylation even in untreated cells and the phosphorylation was sustained after UV radiation (Figure 6C). To determine whether JNK directly phosphorylates S153 of DGCR8, we conducted an in vitro kinase assay using GST-tagged wild-type or S153A fragments (aa1-275) of DGCR8 or GST-tagged (aa1-79) C-JUN (positive control) as substrates and immunopurified FLAG-WT-JNK1a1 or kinase dead FLAG-JNK1a1 (APF) from cells treated with and without UV radiation (Figure S6A). As expected, WT-JNK1a1 but not kinase-dead JNK1a1 phosphorylated S63 of C-JUN (Figure S6B). Importantly, WT-JNK1a1 from UV-treated cells phosphorylated S153 of WT-DGCR8 in vitro (Figure 6D, lane 6 and Figure 6E, lane 4), while the kinase-dead JNK1a1 was unable to phosphorylate S153 (Figure 6E, lanes 5 and 6). Taken together, these data suggest that JNKs phosphorylate S153 of DGCR8 in response to UV radiation.
Figure 6. JNK phosphorylates S153 of DGCR8 after UV radiation.

(A) Immunoblot. U2OS cells were pretreated with the JNK inhibitor, SP600125, or DMSO for 2 hours, irradiated with +/−UV, incubated with media containing SP600125 or DMSO, and cell lysates were harvested at the indicated time points. (B) Immunoblot. U2OS cells were continuously treated with the JNK activator, anisomycin, and cell lysates were harvested at the indicated time points. (C) Immunoblot. U2OS cells expressing either vector or FLAG-JNK1a1 were treated with +/−UV, and cell lysates were harvested at the indicated time points. (D) Immunoblot after in vitro kinase assay. Recombinant GST-tagged WT or S153A DGCR8 (aa1-275) fragment was used as a substrate and immunopurified WT JNK1a1 was used as the enzyme. (E) Immunoblot after in vitro kinase assay. Recombinant GST-tagged WT DGCR8 (aa1-275) fragment was used as a substrate and immunopurified WT JNK1a1 or kinase dead JNK1a1 (APF) was used as the enzyme. (F) Schematic presentation of DGCR8 and Drosha functions. The DGCR8-Drosha microprocessor complex processes RNAs. When DNA is damaged by UV, RNAPII stalls and triggers phosphorylation of S153 on DGCR8, which facilitates removal of UV-induced DNA lesions by TC-NER and promotes UV resistance. Drosha is also involved in UV resistance by an unknown mechanism. See also Figure S6.
DISCUSSION
Based on these findings, we propose a novel cellular signaling pathway (the DGCR8-mediated UV response pathway) that connects UV-induced DNA damage, DGCR8 protein and TC-NER (Figure 6F). In this pathway, in response to UV, RNAPII stalls and triggers phosphorylation of S153 of DGCR8 by JNKs, and this phosphorylation facilitates efficient removal of UV-induced DNA lesions through TC-NER in a manner independent of microRNA processing and Drosha. This function is, in turn, important for cellular survival after UV exposure. Drosha also contributes to cellular resistance to UV, but by mechanisms that are independent of DNA repair.
The c-jun N-terminal kinases (JNKs) are stress-activated kinases that are a part of the mitogen-activated protein kinases (MAPK) family. JNKs are stimulated by a plethora of intrinsic and extrinsic stimuli including UV exposure (Johnson and Nakamura, 2007; Lopez-Camarillo et al., 2012). DGCR8 has been shown to be multiply phosphorylated by ERK/MAPK that affects pro-growth miRNA processing (Herbert et al., 2013). Our studies suggest that a single UV-induced phosphorylation site, S153, is critical for UV resistance and that JNKs phosphorylate S153 of DGCR8 in response to UV exposure. These findings suggest that DGCR8 is one of the critical substrates of JNKs in cellular response to UV exposure. Since S153 phosphorylation was not completely inhibited in the presence of a JNK inhibitor (Figure 6A), it is likely that some other kinases also phosphorylate S153 of DGCR8. It has been reported that simple inhibition of JNKs does not affect removal of UV-induced DNA lesions (Rouget et al., 2008), which also suggests involvement of some additional kinases in the phosphorylation of S153. Such kinases remain to be determined. Treatment with cisplatin or mitomycin C did not induce S153 phosphorylation (Figure S1E), while it is reported that such a treatment activates JNKs, suggesting that activation of JNKs is not sufficient to induce S153 phosphorylation and there should be more complex regulatory mechanisms of this process.
We found that DGCR8 is involved in TC-NER and that DGCR8 physically interacts with some known factors involved in TC-NER. However, the precise molecular mechanism by which S153 phosphorylation on DGCR8 regulates TC-NER remains to be determined. Elucidating this mechanism is a focus of our on-going research. S153-phosphorylated DGCR8 may facilitate the recruitment of known or unknown factors involved in TC-NER to stalled RNAPII and mediate the removal of UV-induced DNA lesions. S153-phosphorylated DGCR8 may regulate interaction among TC-NER factors, or some posttranslational modifications of TC-NER factors. Another possibility is that S153 phosphorylated DGCR8 may sequester some negative regulators of TC-NER. The microprocessor complex is recruited to chromatin during transcription and processes pri-miRNA co-transcriptionally (Morlando et al., 2008; Pawlicki and Steitz, 2008). It would be of interest to test whether S153-phosphorylated DGCR8 affects recruitment of any TC-NER factors to RNAPII. However, our functional and epistasis studies, in addition to the physical interactions that we have identified between DGCR8 and TC-NER factors, support a completely novel biological function of DGCR8 in the UV damage response that is independent of miRNA processing. On the other hand, it is still possible that some miRNAs regulate TC-NER, since generation of some miRNAs is not DGCR8-dependent (Chong et al., 2010) (Berezikov et al., 2007).
Our studies, along with other studies, suggest that factors involved in miRNA processing function in DNA repair and DNA damage response. Deletion of Dicer or Dgcr8 in the developing mouse brain leads to increased DNA damage (Swahari et al., 2016). Furthermore, small non-coding RNAs, termed DNA damage-response RNAs (DDRNAs) or double-strand break (DSB)-induced RNAs (diRNAs), generated at sites of DNA damage are involved in DNA damage response after DSB generation (Francia et al., 2016; Francia et al., 2012) (Wei et al., 2012). These small non-coding RNAs are generated in a manner dependent on Drosha and Dicer. How these small non-coding RNAs interact with the DGCR8-mediated UV response pathway is an interesting issue. Current evidence suggests that they are differently regulated: generation of DDRNAs presumably depends on RNA processing activity of Drosha and Dicer, while the DGCR8-mediated UV response pathway and cellular resistance to UV did not require RNA processing activity of the DGCR8-Drosha complex (Figure 2C and Figure S3). Therefore, we assume that they have distinct functions: DDRNAs are important for DNA damage response after DSB but may not be important for UV resistance or TC-NER, while the DGCR8-mediated UV response pathway is critical for TC-NER.
The mechanism by which Drosha supports UV resistance is unknown. This is a non-canonical function of Drosha, since it did not require DGCR8 binding or microRNA processing activities. It may be related to UV tolerance mechanisms other than DNA repair itself, such as translesion synthesis and suppression of apoptosis.
Our discovery of a novel, miRNA processing-independent function of DGCR8 has several important implications. In humans, DGCR8 is one of the many genes whose heterozygous deletion is associated with DiGeorge syndrome, characterized by developmental abnormalities and behavioral problems (Shiohama et al., 2003). S153-mediated function may or may not contribute to the clinical phenotypes of DiGeorge syndrome and schizophrenia (Zhou et al., 2013), for which single-nucleotide polymorphisms in the microprocessor complex are associated with increased risk. The fact that DiGeorge syndrome is not associated with UV sensitivity suggests that Dgcr8 heterozygosity does not cause a sufficient loss of function to confer this phenotype. Whether mutations in any genes involved in the DGCR8-mediated UV response pathway cause XP, CS, COFS, UVSS and TTD is another important question. Somatic mutations in the catalytic domains of Drosha and DGCR8 occur in Wilms’ tumors (Torrezan et al., 2014). These mutations affect miRNA processing activity of the microprocessor complex, and are unlikely to affect S153 phosphorylation. However, cBioPortal for Cancer Genomics (http://cbioportal.org) (Cerami et al., 2012) revealed several mutations throughout the DGCR8 gene in human cancers. Some of the missense mutations, for example, L152F, S156N (melanoma) and G149C (stomach and lung cancer), flank S153. It remains to be tested whether these mutations alter S153-mediated function.
Multiple Dgcr8-deficient mouse models have been developed (Bezman et al., 2010; Rao et al., 2009; Wang et al., 2007; Yi et al., 2009) with the assumption that DGCR8 functions only in RNA processing. Our results suggest that it is important to determine whether the phenotypes observed in these animal models are mediated by the RNA processing function or the S153-mediated TC-NER function of Dgcr8.
Thus, further elucidation of the non-canonical functions of the microprocessor complex proteins is warranted for our understanding of many diseases.
EXPERIMENTAL PROCEDURES
Cell lines
U2OS, HeLa and HCT116 were obtained from the American Type Culture Collection (ATCC). Mouse embryonic fibroblasts were a gift from Bruce Clurman’s lab (Fred Hutchinson Cancer Research Center (FHCRC)). Immortal human keratinocytes (HaCaT) were a gift from Paul Nghiem’s lab (University of Washington). Primary human foreskin fibroblasts (HFF) were a gift from Denise Galloway’s lab (FHCRC). XPA deficient (GM04312), XPA corrected (GM15876), CSA (GM16094) and CSB (GM16095) deficient human fibroblasts were purchased from Coriell Cell Repositories. XPC deficient cells (XP4PASV) and XPC corrected human fibroblasts (XP4PASV/FLAG-XPC(wt)) were described (Yasuda et al., 2007). Dgcr8 knockout mouse embryonic fibroblasts (MEFs) were purchased from Novus Biologicals and cultured following manufacturers specifications. All cells were maintained at 37°C in a 5% CO2 incubator and grown in DMEM containing 10% FBS, L-glutamine and Pens/Strep. 10 ug/mL of hygromycin B (Life Technologies) was added in the media for XPC cells.
Plasmids, siRNAs, shRNAs and virus production
FLAG or hemagglutinin (HA) tagged- human and mouse DGCR8 and FLAG tagged-human CSA cDNA were cloned into the Nco1 and BamH1 sites of pMMP-IRES-puro retroviral vector (Pulsipher et al., 1998). Phosphomutants (S95A, S153A, S271A, S275A, T371A, S373A, S377A, S434A and S619A) and a phosphomimic mutant (S153D) of DGCR8 were generated using the QuikChange XL Site-Directed Mutagenesis kit (200514-5, Agilent Technologies). The double-stranded RNA binding domain mutant A568K, A569K, A676K, S677K (mDRBD1/2) of DGCR8 was subcloned into pMMP-IRES-puro using the aforementioned sites from pcDNA-mDRBD1/2 DGCR8 (Yeom et al., 2006), a gift from the Narry Kim lab (Seoul National University). FLAG-DGCR8 WT and mutants were mutated using QuikChange XL Site-Directed Mutagenesis kit with primers (1. Forward 5′CTCCCTGCTGAGGACCCTTTTAATTTTTATGGGGCCTCCCTTCTC TCCAAAGGA-3′ 2. Reverse 5′TCCTTTGGAGAGAAGGGAGGCCCCATAAAA ATTAAAAGGGTCCTCAGCAGGGAG -3′) to generate shDGCR8-1 resistant constructs. The Drosha-binding domain mutant (FLAG-Δ692 WT and FLAG-Δ692 S153A) of DGCR8 was also subcloned into pMMP-IRES-puro. FLAG-wild-type-, FLAG-ΔC114- and FLAG-ΔN490-Drosha were subcloned into pBABE puro retroviral vector (a gift from Bruce Clurman’s lab, FHCRC) using BamH1 and Sal1 sites from plasmids (Lee et al., 2006) provided by the Narry Kim lab. CSB construct (pBR-CSB) was a gift from Alan Weiner’s lab (University of Washington) and was used to subclone FLAG-CSB into pBabe-puro retroviral vector using BamH1 and Sal1 sites. pCDNA3 Flag Jnk1a1 (Addgene plasmid # 13798) and pCDNA3 Flag Jnk1a1(apf) (Addgene plasmid # 13846) were a gift from Roger Davis lab (University of Massachusetts). Retroviruses and lentiviruses were produced as described (Wang et al., 2011). All constructs were verified by direct sequencing.
Mass spectrometry
HeLa cells stably infected with pMMPpuroFLAG-DGCR8 were treated with 60 J/m2 of UV-C and 1 hour later, whole-cell extracts were prepared in lysis buffer. IP was performed using monoclonal antibody against FLAG (M2, Santa Cruz) and the IP product was separated by 7.5% SDS-PAGE. Gel pieces corresponding to FLAG-DGCR8 bands were cut from a Coomassie stained gel and subjected to tryptic digestion (Shevchenko et al., 1996). Tryptic peptides were concentrated and desalted using a C18micro ZipTip (Millipore) following the manufacturer’s instructions. The eluted peptides were dried by vacuum centrifugation, resuspended in 7 μL of 2% acetonitrile/0.1% formic acid and 5 μL was analyzed by liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) with a nanoLC 2D (Eksigent) coupled to an OrbiTrap mass spectrometer (ThermoScientific) using an instrument configuration (Licklider et al., 2002).
Western blot analysis
Whole-cell lysates were prepared using sample buffer (0.05 mol/L Tris-HCl (pH 6.8), 2% SDS, 6% β-mercaptoethanol), resolved by polyacrylamide gel (NuPage, Life Technologies) electrophoresis and transferred onto nitrocellulose membranes. Chemiluminescence was used for detection, and membranes were digitally scanned by Imagequant LAS 4000 (GE Biosciences).
Immunofluorescent microscopy
To detect pS153 DGCR8 by immunofluorescence, cells were treated with 60 J/m2 UVC. After one hour, cells were fixed with 4% paraformaldehyde (Santa Cruz Biotechnology) for 10 minutes at room temperature and subsequently permeabilized with PBS containing 0.5% Tween-20 (Fisher Scientific) for 10 minutes at room temperature. After several washes with PBS containing 0.5% Tween-20 (washing buffer), cells were incubated over night at 4 °C with anti-phosphoS153 (1:1000). After several washes with PBS containing 0.5% Tween-20, cells were incubated with Alexa-fluor 488 goat anti-rabbit (A11034, Life Technologies) secondary antibody for one hour. Nuclei were counterstained with 4,6-diami- dino-2-phenylindole (DAPI, 1 μg/mL). Coverslips were mounted on slides in Vectashield (Vector Laboratories). Image acquisitions were made with a TE2000 Nikon microscope equipped with a 20x immersion objective and a CCD camera (CoolSNAP ES, Photometrics).
Flow cytometry
Cells were treated with UVC (12 J/m2 for detection of CPDs, or 60 J/m2 for detection of 6-4PPs) and fixed with 2% paraformaldehyde at 37 °C for 10 minutes. Then, cells were treated with cold 90% methanol for 30 minutes at −20 °C and treated with DNase (Promega) for 1 hour at 37 °C. After blocking with PBS containing 1% BSA for 10 minutes at room temperature, cells were incubated with anti-CPD (1:1000) or anti-6-4PPs (1:1000) diluted in PBS containing 1% BSA and 0.25% Tween 20 overnight at 4 °C. After washing with PBS containing 1% BSA and 0.05% Tween 20, cells were incubated with fluorescein-conjugated anti-mouse antibody (1:1000) (715-095-151, Jackson ImmunoResearch) for 1 hour in the dark at room temperature. Nuclei were counterstained with propidium iodide (2 μg/mL) (P1470, Sigma-Aldrich) and treated with RNase A (100 μg/mL) (Invitrogen) for 20 minutes at 37°C. Flow cytometric analyses were conducted with a BD FACS Canto II analyzer and the FlowJo software.
Recovery of RNA synthesis (RRS) assay
RRS assays were done as described with some modifications (Jia et al., 2015; Nakazawa et al., 2010). U2OS cells stably expressing siRNA resistant FLAG-WT DGCR8, FLAG-S153A DGCR8 or FLAG-S153D-DGCR8 were transfected with siRNA against endogenous DGCR8. Forty eight hours after transfection of siRNAs, cells were irradiated with UVC (16 J/m2) and immediately incubated with 5′-ethynyuridine (EU) (50 μM, Life Technologies) for 2 hours (Jia et al., 2015; Nakazawa et al., 2010). Then, cells were fixed and permeabilized at the indicated time points. After blocking with PBS containing 10% FBS, cells were incubated for 1 hour at room temperature with Alexa-fluor 488 goat anti-rabbit (A11034, Life Technologies) secondary antibody. Nuclei were counterstained with DAPI (1 μg/mL).
Purification of recombinant GST-DGCR8 protein and in vitro kinase assay
DNA fragment corresponding to aa 1-275 FLAG-tagged WT or S153A DGCR8 was cloned into pGEX-4T-1 vector (GE Healthcare). The recombinant plasmids were transformed into E. coli BL21 (DE3) cells (EMD Millipore). GST-DGCR8-WT and GST-DGCR8-S153A proteins were expressed and purified using GST-Bind resin (EMD Millipore) according to the manufacturer’s instructions. Human JUN partial-length recombinant protein (aa 1-79) with a GST-tag (Novus Biologicals) was used a positive control. The in vitro kinase assay was performed as described (Chadee and Kyriakis, 2010).
Cell survival assay
Cells were seeded into 12-well plates at 8 × 103/well (HCT-116), 1 × 104/well (U2OS), 1 × 104/well (XP and CS fibroblasts) and treated with UVC (254nm) from a UV Stratalinker 1800 (Stratagene) or UVB (output range 280–320nm) at indicated doses using the method described (Wallace et al., 2012). After incubation for 5–7 days, cells were stained with crystal violet as described (Wang et al., 2011).
Statistics
All statistical analyses were done using Student’s t-test (2-tail). P value < 0.05 was considered significant.
MicroRNA profiling
RNA from stable isogenic cell lines (HCT-116) depleted of endogenous DGCR8 by shDGCR8-1, transduced with shRNA-resistant FLAG-WT, FLAG-S153A DGCR8 or empty vector, were extracted using RNeasy mini kit (Qiagen). RNA from four biological replicates were submitted to Exiqon for miRNA real-time PCR using miRCURY LNA™ Universal RT microRNA PCR human panel 1 which contains 372 human miRNAs.
Supplementary Material
HIGHLIGHTS.
Ultraviolet Radiation (UV) induces phosphorylation on S153 of DGCR8.
Phosphorylation on S153 of DGCR8 mediates cellular resistance to UV.
S153 phosphorylation mediates transcription coupled nucleotide excision repair.
JNKs are involved in UV-induced phosphorylation of S153 of DGCR8.
Acknowledgments
We thank Drs. Javier F Cáceres, Alan Weiner, Ronald Cheung and Muneesh Tewari for critical reading of the manuscript, Kanan Lathia for technical assistance. We thank Drs. V. Narry Kim and Alan Weiner, the Galloway lab (FHCRC), and the Clurman lab (FHCRC) for reagents. This work was supported by Howard Hughes Medical Institute, the National Institutes of Health (NIH)/NHLBI (R21 HL092978 to T.T.), NIH/NCI (R01 CA125636 to T.T.), NIH/NIEHS (R21 ES026326 to T.T.), NIH/NIAMS (R01-AR049832 and R01-AR067722 to P.N.), Fanconi Anemia Research Fund (to T.T.), and Obliteride Pilot Grant (to T.T.). Y.W. was a research fellow supported by Canadian Institute of Health Research. J.W. and P.C. were supported by PHS NRSA 2T32 GM007270 from NIGMS. K.D. is a Thomsen Breast Cancer Fellow. This work was supported by Grants-in-Aid to K.S. (JSPS KAKENHI Grant Numbers JP16H06307 and JP16H01311). The Fred Hutch Proteomics Facility is partially funded by Cancer Center Support Grant P30 CA015704 from the NIH. There is no conflict of interest.
Footnotes
AUTHOR CONTRIBUTIONS
P.C.C. designed and performed experiments, analyzed data, and wrote the manuscript. K.K.D. analyzed data, and wrote the manuscript. N.T. performed experiments and analyzed data. Y.C. and B.E.C. provided technical advice and generated reagents. M.K. and P.N. provided technical advice and input for interpretation, and edited the manuscript. J.H., Y.W and C. J. provided technical advice and input for interpretation. P.R.G. performed mass spectrometry, analyzed data, and wrote the manuscript. K.S. and M.S. provided technical advice and input for interpretation. T.T. conceptualized and directed the study, analyzed data, and wrote the manuscript.
Supplemental information is available for this article.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berezikov E, Chung WJ, Willis J, Cuppen E, Lai EC. Mammalian mirtron genes. Mol Cell. 2007;28:328–336. doi: 10.1016/j.molcel.2007.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezman NA, Cedars E, Steiner DF, Blelloch R, Hesslein DG, Lanier LL. Distinct Requirements of MicroRNAs in NK Cell Activation, Survival, and Function. J Immunol. 2010;185:3835–3846. doi: 10.4049/jimmunol.1000980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136:642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chadee DN, Kyriakis JM. Activation of SAPK/JNKs in vitro. Methods Mol Biol. 2010;661:59–73. doi: 10.1007/978-1-60761-795-2_3. [DOI] [PubMed] [Google Scholar]
- Cheng TL, Wang Z, Liao Q, Zhu Y, Zhou WH, Xu W, Qiu Z. MeCP2 suppresses nuclear microRNA processing and dendritic growth by regulating the DGCR8/Drosha complex. Dev Cell. 2014;28:547–560. doi: 10.1016/j.devcel.2014.01.032. [DOI] [PubMed] [Google Scholar]
- Chong MM, Zhang G, Cheloufi S, Neubert TA, Hannon GJ, Littman DR. Canonical and alternate functions of the microRNA biogenesis machinery. Genes Dev. 2010 doi: 10.1101/gad.1953310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Dziunycz P, Iotzova-Weiss G, Eloranta JJ, Lauchli S, Hafner J, French LE, Hofbauer GF. Squamous cell carcinoma of the skin shows a distinct microRNA profile modulated by UV radiation. J Invest Dermatol. 2010;130:2686–2689. doi: 10.1038/jid.2010.169. [DOI] [PubMed] [Google Scholar]
- Fousteri M, Vermeulen W, van Zeeland AA, Mullenders LH. Cockayne syndrome A and B proteins differentially regulate recruitment of chromatin remodeling and repair factors to stalled RNA polymerase II in vivo. Mol Cell. 2006;23:471–482. doi: 10.1016/j.molcel.2006.06.029. [DOI] [PubMed] [Google Scholar]
- Francia S, Cabrini M, Matti V, Oldani A, d’Adda di Fagagna F. DICER, DROSHA and DNA damage response RNAs are necessary for the secondary recruitment of DNA damage response factors. J Cell Sci. 2016;129:1468–1476. doi: 10.1242/jcs.182188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia S, Michelini F, Saxena A, Tang D, de Hoon M, Anelli V, Mione M, Carninci P, d’Adda di Fagagna F. Site-specific DICER and DROSHA RNA products control the DNA-damage response. Nature. 2012;488:231–235. doi: 10.1038/nature11179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Kim YK, Jin H, Kim VN. The Drosha-DGCR8 complex in primary microRNA processing. Genes Dev. 2004;18:3016–3027. doi: 10.1101/gad.1262504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol. 2008;9:958–970. doi: 10.1038/nrm2549. [DOI] [PubMed] [Google Scholar]
- Herbert KM, Pimienta G, DeGregorio SJ, Alexandrov A, Steitz JA. Phosphorylation of DGCR8 increases its intracellular stability and induces a progrowth miRNA profile. Cell Rep. 2013;5:1070–1081. doi: 10.1016/j.celrep.2013.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrlich P, Karin M, Weiss C. Supreme EnLIGHTenment: damage recognition and signaling in the mammalian UV response. Mol Cell. 2008;29:279–290. doi: 10.1016/j.molcel.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio MV, Croce CM. MicroRNA dysregulation in cancer: diagnostics, monitoring and therapeutics. A comprehensive review. EMBO Mol Med. 2012;4:143–159. doi: 10.1002/emmm.201100209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia N, Nakazawa Y, Guo C, Shimada M, Sethi M, Takahashi Y, Ueda H, Nagayama Y, Ogi T. A rapid, comprehensive system for assaying DNA repair activity and cytotoxic effects of DNA-damaging reagents. Nat Protoc. 2015;10:12–24. doi: 10.1038/nprot.2014.194. [DOI] [PubMed] [Google Scholar]
- Johnson GL, Nakamura K. The c-jun kinase/stress-activated pathway: regulation, function and role in human disease. Biochim Biophys Acta. 2007;1773:1341–1348. doi: 10.1016/j.bbamcr.2006.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Analytical chemistry. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Lee Y, Han J, Yeom KH, Jin H, Kim VN. Drosha in Primary MicroRNA Processing. Cold Spring Harb Symp Quant Biol. 2006;71:51–57. doi: 10.1101/sqb.2006.71.041. [DOI] [PubMed] [Google Scholar]
- Licklider LJ, Thoreen CC, Peng J, Gygi SP. Automation of nanoscale microcapillary liquid chromatography-tandem mass spectrometry with a vented column. Analytical chemistry. 2002;74:3076–3083. doi: 10.1021/ac025529o. [DOI] [PubMed] [Google Scholar]
- Lopez-Camarillo C, Ocampo EA, Casamichana ML, Perez-Plasencia C, Alvarez-Sanchez E, Marchat LA. Protein kinases and transcription factors activation in response to UV-radiation of skin: implications for carcinogenesis. Int J Mol Sci. 2012;13:142–172. doi: 10.3390/ijms13010142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macias S, Cordiner RA, Caceres JF. Cellular functions of the microprocessor. Biochem Soc Trans. 2013;41:838–843. doi: 10.1042/BST20130011. [DOI] [PubMed] [Google Scholar]
- Macias S, Plass M, Stajuda A, Michlewski G, Eyras E, Caceres JF. DGCR8 HITS-CLIP reveals novel functions for the Microprocessor. Nat Struct Mol Biol. 2012;19:760–766. doi: 10.1038/nsmb.2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I, Proudfoot NJ. Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol. 2008;15:902–909. doi: 10.1038/nsmb.1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa Y, Yamashita S, Lehmann AR, Ogi T. A semi-automated non-radioactive system for measuring recovery of RNA synthesis and unscheduled DNA synthesis using ethynyluracil derivatives. DNA Repair (Amst) 2010;9:506–516. doi: 10.1016/j.dnarep.2010.01.015. [DOI] [PubMed] [Google Scholar]
- Pawlicki JM, Steitz JA. Primary microRNA transcript retention at sites of transcription leads to enhanced microRNA production. J Cell Biol. 2008;182:61–76. doi: 10.1083/jcb.200803111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pothof J, Verkaik NS, van IW, Wiemer EA, Ta VT, van der Horst GT, Jaspers NG, van Gent DC, Hoeijmakers JH, Persengiev SP. MicroRNA-mediated gene silencing modulates the UV-induced DNA-damage response. EMBO J. 2009;28:2090–2099. doi: 10.1038/emboj.2009.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulsipher M, Kupfer GM, Naf D, Suliman A, Lee JS, Jakobs P, Grompe M, Joenje H, Sieff C, Guinan E, et al. Subtyping analysis of Fanconi anemia by immunoblotting and retroviral gene transfer. Mol Med. 1998;4:468–479. [PMC free article] [PubMed] [Google Scholar]
- Rao PK, Toyama Y, Chiang HR, Gupta S, Bauer M, Medvid R, Reinhardt F, Liao R, Krieger M, Jaenisch R, et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ Res. 2009;105:585–594. doi: 10.1161/CIRCRESAHA.109.200451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch A, Bellew M, Eng J, Fitzgibbon M, Holzman T, Hussey P, Igra M, Maclean B, Lin CW, Detter A, et al. Computational Proteomics Analysis System (CPAS): an extensible, open-source analytic system for evaluating and publishing proteomic data and high throughput biological experiments. Journal of proteome research. 2006;5:112–121. doi: 10.1021/pr0503533. [DOI] [PubMed] [Google Scholar]
- Roberts WC. Safety of fenofibrate--US and worldwide experience. Cardiology. 1989;76:169–179. doi: 10.1159/000174488. [DOI] [PubMed] [Google Scholar]
- Rockstroh M, Müller SA, Jende C, Kerzhner A, von Bergen MM, Tomm JM. Cell fractionation - an important tool for compartment proteomics. Journal of Integrated OMICS. 2011;1:135–143. [Google Scholar]
- Rouget R, Auclair Y, Loignon M, Affar el B, Drobetsky EA. A sensitive flow cytometry-based nucleotide excision repair assay unexpectedly reveals that mitogen-activated protein kinase signaling does not regulate the removal of UV-induced DNA damage in human cells. J Biol Chem. 2008;283:5533–5541. doi: 10.1074/jbc.M706257200. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Wilm M, Vorm O, Mann M. Mass spectrometric sequencing of proteins silver-stained polyacrylamide gels. Analytical chemistry. 1996;68:850–858. doi: 10.1021/ac950914h. [DOI] [PubMed] [Google Scholar]
- Shiohama A, Sasaki T, Noda S, Minoshima S, Shimizu N. Molecular cloning and expression analysis of a novel gene DGCR8 located in the DiGeorge syndrome chromosomal region. Biochemical and biophysical research communications. 2003;304:184–190. doi: 10.1016/s0006-291x(03)00554-0. [DOI] [PubMed] [Google Scholar]
- Sohn SY, Bae WJ, Kim JJ, Yeom KH, Kim VN, Cho Y. Crystal structure of human DGCR8 core. Nat Struct Mol Biol. 2007;14:847–853. doi: 10.1038/nsmb1294. [DOI] [PubMed] [Google Scholar]
- Swahari V, Nakamura A, Baran-Gale J, Garcia I, Crowther AJ, Sons R, Gershon TR, Hammond S, Sethupathy P, Deshmukh M. Essential Function of Dicer in Resolving DNA Damage in the Rapidly Dividing Cells of the Developing and Malignant Cerebellum. Cell Rep. 2016;14:216–224. doi: 10.1016/j.celrep.2015.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torrezan GT, Ferreira EN, Nakahata AM, Barros BD, Castro MT, Correa BR, Krepischi AC, Olivieri EH, Cunha IW, Tabori U, et al. Recurrent somatic mutation in DROSHA induces microRNA profile changes in Wilms tumour. Nat Commun. 2014;5:4039. doi: 10.1038/ncomms5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Kikuchi J, Furukawa Y. Histone deacetylase 1 enhances microRNA processing via deacetylation of DGCR8. EMBO Rep. 2012;13:142–149. doi: 10.1038/embor.2011.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace NA, Robinson K, Howie HL, Galloway DA. HPV 5 and 8 E6 abrogate ATR activity resulting in increased persistence of UVB induced DNA damage. PLoS pathogens. 2012;8:e1002807. doi: 10.1371/journal.ppat.1002807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Huang JW, Li M, Cavenee WK, Mitchell PS, Zhou X, Tewari M, Furnari FB, Taniguchi T. MicroRNA-138 Modulates DNA Damage Response by Repressing Histone H2AX Expression. Mol Cancer Res. 2011;9:1100–1111. doi: 10.1158/1541-7786.MCR-11-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Taniguchi T. MicroRNAs and DNA damage response: Implications for cancer therapy. Cell Cycle. 2013;12:32–42. doi: 10.4161/cc.23051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei W, Ba Z, Gao M, Wu Y, Ma Y, Amiard S, White CI, Rendtlew Danielsen JM, Yang YG, Qi Y. A role for small RNAs in DNA double-strand break repair. Cell. 2012;149:101–112. doi: 10.1016/j.cell.2012.03.002. [DOI] [PubMed] [Google Scholar]
- Weitz SH, Gong M, Barr I, Weiss S, Guo F. Processing of microRNA primary transcripts requires heme in mammalian cells. Proc Natl Acad Sci U S A. 2014;111:1861–1866. doi: 10.1073/pnas.1309915111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda G, Nishi R, Watanabe E, Mori T, Iwai S, Orioli D, Stefanini M, Hanaoka F, Sugasawa K. In vivo destabilization and functional defects of the xeroderma pigmentosum C protein caused by a pathogenic missense mutation. Mol Cell Biol. 2007;27:6606–6614. doi: 10.1128/MCB.02166-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeom KH, Lee Y, Han J, Suh MR, Kim VN. Characterization of DGCR8/Pasha, the essential cofactor for Drosha in primary miRNA processing. Nucleic Acids Res. 2006;34:4622–4629. doi: 10.1093/nar/gkl458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi R, Pasolli HA, Landthaler M, Hafner M, Ojo T, Sheridan R, Sander C, O’Carroll D, Stoffel M, Tuschl T, et al. DGCR8-dependent microRNA biogenesis is essential for skin development. Proc Natl Acad Sci U S A. 2009;106:498–502. doi: 10.1073/pnas.0810766105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang J, Lu X, Song X, Ye Y, Zhou J, Ying B, Wang L. Evaluation of six SNPs of MicroRNA machinery genes and risk of schizophrenia. J Mol Neurosci. 2013;49:594–599. doi: 10.1007/s12031-012-9887-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.