

Abstract
The causes of heart failure are diverse. Inherited causes represent an important clinical entity and can be divided into 2 major categories: familial and metabolic cardiomyopathies. The distinct features that might be present in early disease states can become broadly overlapping with other diseases, such as in the case of inherited cardiomyopathies (ie, familial hypertrophic cardiomyopathy or mitochondrial diseases). In this review article, we focus on genetic issues related to advanced heart failure. Because of the emerging importance of this topic and its breadth, we sought to focus our discussion on the known genetic forms of heart failure syndromes, genetic testing, and newer data on pharmacogenetics and therapeutics in the treatment of heart failure, to primarily encourage clinicians to place a priority on the diagnosis and treatment of these potentially treatable conditions.
In the United States, heart failure (HF) with reduced ejection fraction is newly diagnosed in more than 500,000 individuals each year, costs approximately $37 billion, and has an estimated death rate of 50,000 individuals per year.1 The approximate lifetime cost of HF for each individual patient is $100,000 per year.2 Although survival after diagnosis of HF has improved in the past quarter century, the 5-year mortality is still as high as many cancers.3 Based on the Framingham Heart Study, after a new diagnosis of HF, 30-day mortality is approximately 10%, 1-year mortality is 20%–30%, and 5-year mortality is 45%–60%.4
The causes of HF are diverse,5 and the diagnosis and treatment of advanced HF might be challenging for clinicians. The prevalence of inherited cardiomyopathy is underappreciated in advanced HF.6,7 Recent advances in genetic analysis, understanding of the effects of modifier genes, and appreciation for the effect of an individual’s genetic fingerprint on response to therapy provide the basis for greater application of genetics in the diagnosis and treatment of advanced HF. In this review, we seek to provide an up to date framework for understanding the current and future effect of the genetic factors that underpin the etiology and treatment of HF.
Inherited Cardiomyopathies That Might Lead to HF
Inherited forms of cardiomyopathies can be broadly classified into: (1) familial cardiomyopathies (hypertrophic cardiomyopathy [HCM], familial dilated cardiomyopathy [FDCM], arrhythmogenic right ventricular cardiomyopathy [ARVC]), left ventricular (LV) noncompaction (LVNC), and restrictive cardiomyopathy (RCM); and (2) metabolic cardiomyopathies (disorders of fat metabolism (FAOD), mitochondrial function, carbohydrate metabolism, and lysosomes) (Table 1).8
Table 1.
Characteristics of inherited causes of heart failure
| Disease | Prevalence | Inheritance pattern | Investigations |
|---|---|---|---|
| Familial cardiomyopathy | |||
| 1. HCM | Approximately 1 in 5009 | Most: AD10 >50% familial10 |
Unexplained hypertrophy on cardiac imaging, typical ECG abnormalities and genetic testing11 |
| 2. ARVC | Approximately 1 in 200012 | Most: AD with incomplete penetrance13 Rare AR forms13 30–50% familial13 |
Enlargement, aneurysm, and fatty infiltration of the ventricles, ECG changes, and genetic testing12 |
| 3. RCM | 1 in 1000–500014 | Most: AD15 Usually not familial15 |
Generally, atrial arrhythmias, right and left sided filling pressures with clinical signs and symptoms of RVF, and ventricular arrhythmias, with and without genetic testing14 |
| 4. FDCM | Approximately 1 in 250016 | Any pattern: AD (most common), mtDNA, and X-linked16 Variable penetrance16 35% familial16 |
Ventricular dilatation, impaired systolic function, and overall enlargement of ventricular mass16 |
| 5. LVNC | Not well known17 | AD, AR, and X-linked17 | Existing diagnostic criteria might be oversensitive18 Diagnosis is controversial19 |
| Metabolic cardiomyopathy | |||
| 1. FAOD | MCAD: approximately 1 in 10,00020 Others: less common21 |
Most: AR21 | Screening: acylcarnitine profile Confirmation: mutation analysis or fibroblast culture22 |
| 2. Mitochondrial disorders | Adults: approximately 1 in 11,000; children: approximately 1 in 600023 | Any pattern: maternal (through mtDNA) or nuclear DNA in AD, AR or X-linked24 | Muscle biopsy with analysis for defects in mtDNA and/or nuclear DNA24 |
| 3. Glycogen storage diseases | Varies with type25 McArdle disease: approximately 1 in 7650– 42,35525 |
Most: AR26 | Muscle biopsy: increased glycogen stores26 Subtyping: enzyme or DNA analysis26 |
| 3. Disorders of lysosomes | Varies with type27 FD: approximately 1 in 40,000 men27 |
FD is X-linked27 Other patterns reported28 |
Levels of α galactosidase can be tested in WBC and are diagnostic in men but might be normal in clinically affected women Mutational analysis (GLA gene in FD)29 |
AD, autosomal dominant; ARVC, arrhythmogenic right ventricular cardiomyopathy; ECG, electrocardiogram; FAOD, disorders of fat metabolism; FD, Fabry disease; FDCM, familial dilated cardiomyopathy; GLA, α-galactosidase A; HCM, hypertrophic cardiomyopathy; LVNC, left ventricular noncompaction cardiomyopathy; MCAD, medium chain acyl coenzyme A dehydrogenase; mtDNA, mitochondrial DNA; RCM, restrictive cardiomyopathy; RVF, right ventricular failure; WBC, white blood cells.
Familial Cardiomyopathies
The first cardiomyopathy-associated gene was discovered in 1989,30 using linkage analysis, and sparked a rapid expansion in cardiogenetics. Genetic testing has uncovered numerous cardiomyopathy-associated genes (Table 2; refer to Table 3 for nonstandard abbreviations, acronyms, and definitions) that encode proteins underlying the normal structure and function of cardiomyocytes (Fig. 1). The genetic mutation alone cannot yet sufficiently explain the clinical phenotype, and gene-based disease classification of cardiomyopathies remains a hope for the future.8,33 Therefore, the discussion that follows is divided according to clinical phenotype.
Table 2.
Selective genes associated with inherited cardiomyopathies
| Gene (location) | Protein | Disease | Reference(s) |
|---|---|---|---|
| Membrane and or cytoplasm | |||
| HFE (6p21.3) | Hereditary hemochromatosis protein | DCM* | 31 |
| FKTN (9q31.2) | Fukutin | DCM | 16,32 |
| TMEM43 (3p25.1) | Transmembrane protein 43 | ARVC | 33–35 |
| LAMP2 (Xq24) | Lysosome-associated membrane glycoprotein 2 | Danon disease: DCM* HCM* | 10,33,36,37 |
| GLA (Xq22) | α-Galactosidase A | FD* HCM* | 38–41 |
| PRKAG2 (7q36.1) | 5′–AMP-activated protein kinase subunit γ2 | GSD: HCM* | 26 |
| PSEN1 (14q24.3) | Presenilin-1 | DCM | 33–35 |
| TTR (18q12.1) | Transthyretin | Amyloidosis: RCM | 42–44 |
| Ion channels | |||
| SCN5A (3p21) | Cardiac sodium channel | DCM | 33–35 |
| DTNA (18q12.1) | α-Dystrobrevin | LVNC | 17,18 |
| ABCC9 (12p12.1) | SUR2A subunit, KATP channel | DCM | 33–35 |
| ILK (11p15.4) | Integrin-linked kinase | DCM | 33–35 |
| CHRM2 (7q33) | M2-muscarinic acetylcholine receptor | DCM | 33–35 |
| PLN (6q22.1) | Cardiac phospholamban | HCM, ARVC, DCM | 10,33–36 |
| CASQ2 (1p13.1) | Calsequestrin 2 | DCM, LVNC | 33–35 |
| RYR2 (1q43) | Ryanodine receptor 2 | ARVC | 33–35 |
| Cytoskeleton | |||
| CRYAB (11q22.3–q23.1) | α-Crystallin B chain | HCM, DCM | 10,33–36 |
| DMD (Xp21.2) | Dystrophin | DCM | 16,32 |
| DES (2q35) | Desmin | ARVC, RCM, DCM, | 10,33–36,45 |
| LAMA4 (6q21) | Laminin subunit α4 | DCM | 33–35 |
| FKTN (9q31-q33) | Fukutin | DCM | 33–35 |
| FXN (FRDA) (9q21.11) | Frataxin, mitochondrial | HCM | 10,33 |
| Nuclear | |||
| LMNA (1q22) | Lamin A/C | ARVC, DCM | 10,33–36 |
| PRDM16 (1p36.32) | PR domain-containing 16 | DCM | 33–35 |
| NKX2-5 (5q35.1) | Homeodomain transcription factor | DCM, LVNC | 10,33,35,36 |
| EMD (Xq28) | Emerin | DCM | 33–35 |
| Desmosomal | |||
| DSP (6p24) | Desmoplakin | ARVC, DCM, LVNC | 33–36 |
| FHL1 (Xq26) | Four and a half domains protein 1 | HCM | 10,33 |
| FHL2 (2q12.2) | Four and a half domains protein 2 | DCM | 33,35 |
| JUP (17q21) | Junction plakoglobin | ARVC | 33–36 |
| PKP2 (12p11) | Plakophilin-2 | ARVC, DCM | 10,33–36 |
| DSG2 (18q12.1) | Desmoglein-2 | ARVC, DCM | 10,33–36 |
| DSC2 (18q12.1) | Desmocollin-2 | ARVC, DCM | 10,33–36 |
| Sarcomere | |||
| TTN (2q31) | Titin | HCM, ARVC, DCM | 10,33–36 |
| MYH7 (14q12) | Myosin-7 (β-myosin heavy chain) | HCM, RCM, DCM, LVNC | 10,33–36 |
| MYH6 (14q12) | Myosin-6 (α-myosin heavy chain) | HCM, DCM | 10,33,35,36 |
| MYL2 (12q24.11) | Myosin regulatory light chain 2 | HCM, RCM | 10,33,36 |
| MYL3 (3p21.3-p21.2) | Myosin light chain 3 | HCM, RCM | 10,33,36 |
| MYLK2 (20q11.21) | Myosin light chain kinase 2 | HCM | 10,33 |
| MYBPC3 (11p11.2) | Cardiac myosin-binding protein C | HCM, DCM, LVNC | 10,33–36 |
| TNNT2 (1q32) | Cardiac muscle troponin T | HCM, RCM, DCM, LVNC | 10,33–36 |
| TNNI3 (19q13.4) | Cardiac muscle troponin I | HCM, RCM, DCM, LVNC | 10,33–36 |
| TPM1 (15q22.1) | Tropomyosin α1 chain | HCM, RCM, DCM, LVNC | 10,33–36 |
| ACTC1 (15q14) | Cardiac actin | HCM, RCM, DCM, LVNC | 10,33–36 |
| TNNC1 (3p21.1) | Cardiac muscle troponin C | HCM, DCM | 10,33,35,36 |
| Z-disc | |||
| LDB3 (10q22.2-q23.2) | LIM domain-binding protein 3 | HCM, DCM, LVNC | 10,33–36 |
| MURC (9q31.1) | Muscle-restricted coiled-coil | DCM | 35 |
| TCAP (17q12) | Telethonin | HCM, DCM | 10,33,35,36 |
| ANKRD1 (10q23.31) | Ankyrin repeat domain-containing protein 1 | HCM, DCM | 10,33,35,36 |
| MYPN (10q21.3) | Myopalladin | HCM, DCM | 10,33,35,36 |
| ACTN2 (1q42-q43) | α-Actinin-2 | HCM, DCM | 10,33,35,36 |
| Mitochondrial disorder | |||
| MT-TL1 location (mitochondrially located) | Transfer RNA | MELAS, MIDD, CPEO+, MERRF | 46–48 |
| COX15 (10q24.2) | Cytochrome c oxidase assembly protein | Leigh syndrome, complex IV deficiency | 46,49,50 |
| TAZ (Xq28) | Zinc finger and domain-containing protein 18 | Barth syndrome: DCM, LVNC | 46,49,50 |
| SDHA (5p15) | Succinate dehydrogenase | DCM | 46,49,50 |
AMP, adenosine monophosphate; ARVC, arrhythmogenic right ventricular cardiomyopathy; ATP, adenosine triphosphate; CPEO+, chronic progressive external ophthalmoplegia plus; DCM, dilated cardiomyopathy; FD, fabry disease; GSD, glycogen storage diseases; HCM, hypertrophic cardiomyopathy; LVNC, left ventricular noncompaction cardiomyopathy; MELAS, mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episode; MERRF, myoclonic epilepsy with ragged-red fibers; MIDD, maternally inherited diabetes and deafness; RCM, restrictive cardiomyopathy.
Potentially reversible.
Table 3.
Nonstandard abbreviations, acronyms, and definitions
| ADRB: β-adrenergic receptor |
| Alleles: alternative forms of a gene |
| ARVC: arrhythmogenic right ventricular cardiomyopathy |
| CPEO+: chronic progressive external ophthalmoplegia syndrome plus |
| FA: Friedreich ataxia |
| FAOD: disorders of fat metabolism |
| FD: Fabry disease |
| FDCM: familial dilated cardiomyopathy |
| Genes: units of inheritance at specific locations (loci) on a chromosome |
| GSD: glycogen storage diseases |
| HCM: hypertrophic cardiomyopathy |
| Heterozygous: a genotype with 2 different alleles of a gene for a particular trait |
| Homozygous: a genotype with the same allele of a gene for a particular trait |
| IEM: inborn errors of metabolism |
| LCHAD: long chain acyl coenzyme A dehydrogenase deficiency |
| LVNC: left ventricular noncompaction cardiomyopathy |
| MCAD: medium chain acyl coenzyme A dehydrogenase |
| MELAS: mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes syndrome |
| MERRF: myoclonic epilepsy with ragged red fibre syndrome |
| MIDD: maternally inherited diabetes and deafness |
| mtDNA: mitochondrial DNA |
| Mutations: alteration of genetic material producing a new variation |
| Phenotype: detectable expression of a genotype |
| RCM: restrictive cardiomyopathy |
| SCD: sudden cardiac death |
| TRR: familial transthyretin |
| VLCAD: very long chain acyl coenzyme A dehydrogenase deficiency |
| X-linked: a gene that is carried by the X (sex) chromosome |
Figure 1.
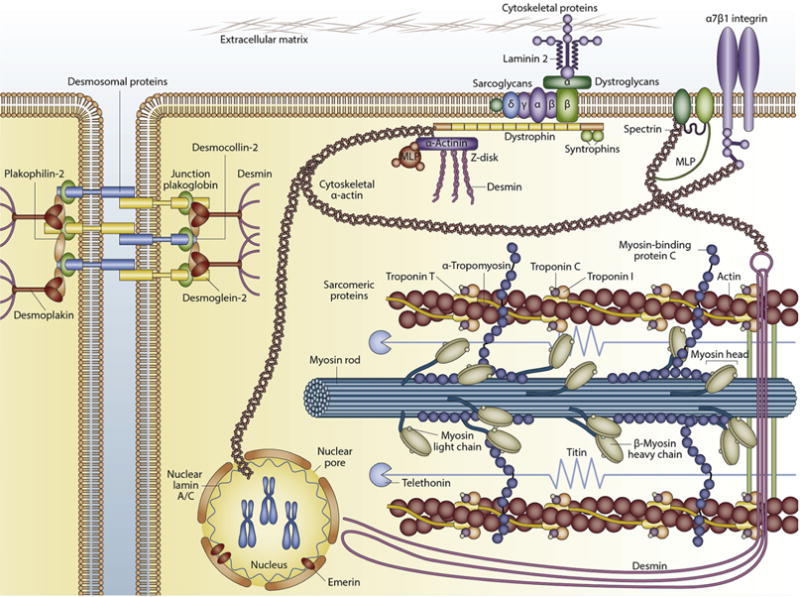
Main types of proteins involved in familial cardiomyopathy. MLP, muscle protein. Reprinted by permission from Macmillan Publishers Ltd: Nature Reviews Cardiology.51 Copyright © 2013.
HCM
HCM is the most common cause of sudden cardiac death (SCD) in otherwise young healthy individuals, and has a prevalence of approximately 1 in 500 (0.2%) worldwide.9 HCM is diagnosed by the observation of unexplained hypertrophy on cardiac imaging (eg, echocardiography) coupled with typical electrocardiographic abnormalities.11 Distribution of hypertrophy is variable with 2 dominant phenotypes characterized by either apical or asymmetric septal hypertrophy.11 Evolution to end-stage HF occurs in a small but significant cohort.52 Disease progression is linked to progressive fibrosis, but the underlying biological pathways are incompletely understood.11 Unfortunately, there is no specific medical therapy that can interrupt this process, and symptom management ultimately followed by heart transplantation is frequently the only option in advanced disease.
HCM is caused by autosomal dominant (AD) mutations in structural genes (Fig. 1, Table 2). Although there are approximately 1400 mutations (largely missense) associated with familial HCM,10,36 no specific genotype has been broadly associated with progression to advanced HF. Approximately 70% of individuals with HCM have mutations in 1 of 2 genes, myosin-7 (β-myosin heavy chain) and cardiac myosin-binding protein C (50% penetrance by age 50 in men; Table 2),33,53 and cardiac muscle troponin T and other genes each account for < 5% of cases (Table 2).54 Approximately 8% of individuals have mutations in more than 1 sarcomere gene, leading to a more severe phenotype and greater likelihood of HF.10
The presence of HF in a patient with unexplained hypertrophy, or HF symptoms out of proportion with degree of hypertrophy, should trigger consideration of inherited phenocopies of HCM like Friedreich ataxia and Fabry disease (FD), and familial transthyretin amyloid cardiomyopathy and senile familial transthyretin cardiomyopathy.55 Genetic testing can be useful to differentiate these entities.56 For example, the FXN gene, which causes Friedreich ataxia, is estimated to affect 1 in 40,000 people and can manifest with adult-onset HCM that begins after age 25 and might present with hypokinetic-dilated cardiomyopathy or LV hypertrophy.56 Overall, the yield of genetic testing in index patients with a family history of HCM is 40%–70%, and in sporadic cases is closer to 34%.33,57–59
The treatment of HCM requires a multidisciplinary approach for control of HF and anginal symptoms, LV outflow tract obstruction, and arrhythmias. Pharmacotherapy with β-blockers (first-line agents), calcium channel blockers, and disopyramide is the backbone of therapy for obstruction.60 Active research on various agents, including statins61 and N-acetylcysteine (ClinicalTrials.gov Identifier: NCT01537926) might expand the therapeutic options in the future. Survival of patients whose disease progresses to require cardiac transplantation is comparable with patients who receive a transplant for non-HCM diseases.62
ARVC
ARVC is characterized by enlargement, aneurysm, and/or dysfunction of the right and/or left ventricle with associated depolarization and repolarization abnormalities and important arrhythmic risk.12 Histological evaluation might reveal characteristic fibrosis, myocyte necrosis, and fatty replacement.12 With an estimated prevalence of 1 in 2000, ARVC is a leading cause of ventricular arrhythmia in young otherwise healthy individuals.12 Asymptomatic patients with ARVC have low rates of disease progression to right ventricular dilation and HF.63 However, when patients have disease that progresses to clinical HF, management can be complicated by right ventricular failure (RVF), which limits treatment options in advanced disease. Aggressive use of ventricular tachycardia ablation might contribute to a decline in ventricular function and progression of disease.64 Frequent intense aerobic exercise might lead to more rapid disease progression,65,66 so aerobic conditioning for ARVC patients should be limited.67
Early challenges in genetic diagnosis were overcome by recognition of severe autosomal recessive (AR) forms associated with palmoplantar keratosis and woolly hair.13 Several genes most commonly transmitted as AD are known to cause ARVC (Table 2), although there are rare AR forms.34 Although nondesmosomal gene mutations (eg, lamin A/C [LMNA] and cardiac sodium channel; Table 2), have been seen, desmosomal proteins account for most cases that meet clinical diagnostic criteria (30%–70% of cases; desmocollin-2, desmoplakin, plakophilin-2, etc; Table 2).33,34 Unfortunately, the signal to noise ratio for ARVC genetic testing is relatively poor, which has created challenges in interpretation of genetic test results.35 Compound or digenic heterozygosity of desmosomal mutations are frequently seen among symptomatic adults,68 implicating multiple genetic factors.
The management of advanced HF in ARVC remains a challenge. With predominantly RVF, use of LV assist device therapy, as a bridge to transplantation, remains impossible for most of these patients.69 Caution should be used in applying ablation therapy, which might speed progression to RVF. The 1-year survival after transplantation is 94%, and 88% are alive at an average follow-up of 6.2 ± 4.8 years after transplantation.69
RCM
Inherited RCM is probably more appropriately considered a subtype of HCM.15 Multiple sporadic forms of RCM exist and are associated with infiltrative processes (eg, hemochromatosis31), and environmental exposures (chemotherapy, radiation).14 Phenotypically, the disease presents with atrial arrhythmias, increased right- and left-sided filling pressures with clinical signs and symptoms of RVF, and ventricular arrhythmias.14 RCM is nearly universally associated with substantial and progressive HF, and transplantation outcomes vary significantly among subgroups.70
Hereditary amyloidoses are a heterogeneous group of AD disorders characterized by the extracellular deposition of insoluble protein fibrils.42 Clinical manifestation ranges from polyneuropathy to cardiomyopathy55; the latter is notable with 3 mutations in the transthyretin gene, and is more common among middle-aged black individuals in the United States.43 One mutation (val122-to-ile substitution) associated with late-onset RCM is particularly frequent and under-diagnosed in black individuals, with an estimated allele frequency of 3.9%.44 Because RNA interference (RNAi) against transthyretin shows promise in clinical trials, the importance of this diagnosis cannot be overstated.71
The management of RCM is challenging. RVF leads to congestive hepatopathy72 and cardiorenal syndrome.73 An LV assist device can be applied in only a limited fashion and earlier referral in patients with symptoms of HF is reasonable.74 Only patients with idiopathic RCM have favourable outcomes with cardiac transplantation.70
FDCM
FDCM is characterized by dilatation and impaired LV systolic function with enlargement of ventricular mass and variable penetrance.16 The genetics of FDCM is complex and can follow any inheritance pattern, including AD (most common), mitochondrial DNA (mtDNA), and X-linked.16 The most frequently reported gene mutations are LMNA and TTN (Fig. 1, Table 2), accounting for up to 7% and 25% of patients with FDCM, respectively.16,32 Patients with the LMNA variant are at greater risk of malignant events, especially men or patients with nonmissense mutations (eg, mutations that affect splicing).45 Larger LV size and systolic and diastolic dysfunction predict disease progression in children.75 In adults, asymptomatic FDCM was identified in 4.6% of asymptomatic relatives, and only LV enlargement independently predicted disease progression.76 When FDCM is associated with cardiac conduction disease, increased creatine kinase level, or family history, the yield from genetic testing is highest (15%–25%).33,54 In patients who undergo cardiac transplantation, the 10-year survival rate is 57% in women and 45% in men.77
LVNC
A developmental failure in trabecular compaction causes a 2-layered appearance of the myocardial wall in LVNC.17 Changes in cardiac imaging and oversensitive criteria have led to an epidemic in the diagnosis of LVNC,78 and caution is needed in assignment of this diagnosis. LVNC might be sporadic or familial, with AD, AR, and X-linked inheritance reported.17 Mutations in sarcomere protein genes have been found in up to 50% of patients, with most in the myosin-7 (β-myosin heavy chain) gene, and > 50% that overlap with HCM (Table 2).17 Unfortunately, existing diagnostic criteria lack specificity.18 It might be that LVNC represents a common morphologic trait that overlaps between normal individuals and those with adverse remodelling due to other cardiomyopathic processes.19
Metabolic Cardiomyopathies
There are 4 main groups of inborn errors of metabolism (IEM) with prominent cardiac involvement: FAOD, mitochondrial function, carbohydrate metabolism (muscle glycogenoses), and lysosomes. Cardiac involvement can also occur with other disorders of intermediary metabolism (like organic acidemias, inherited defects of glycosylation, urea cycle defects, etc).79
With the exception of FD (see section on Disorders of Lysosomes), the cardiac features of IEM that cause cardiomyopathy are nonspecific and clinical clues to IEM lie in the pattern of extracardiac organ involvement, including diabetes mellitus, diplopia, hearing loss, skin lesions, neuropathic pain, kidney disease, or a history suggestive of rhabdomyolysis.22,80 Although most IEM that cause cardiomyopathy are very rare (with the exception of medium-chain acyl coenzyme A [CoA] dehydrogenase [MCAD] and FD), there is disease-specific therapy available for most (eg, FD cardiomyopathy).81
FAOD
FAOD are multisystem and clinically heterogeneous FAOD inherited in an AR pattern.21,22 The most common cause of exercise-induced rhabdomyolysis are FAOD80; other clinical features might include neuropathy, hypoglycemia, and cardiomyopathy.22 Cardiomyopathy often results from FAOD that affect the metabolism of long-chain fats like very long-chain acyl-CoA dehydrogenase deficiency and long-chain acyl-CoA dehydrogenase deficiency, but can also occur in more common FAOD.21,82–84 The prevalence of cardiomyopathy in patients with FAOD ranges from 12% to 55% depending on the age of onset.85
MCAD deficiency, an IEM that can present with SCD, was the first FAOD to be included in newborn screening panels.20 Many patients with MCAD are asymptomatic as children and present as adults when their condition decompensates because of intercurrent illness or alcohol use.85 Cardiomyopathy can be the presenting syndrome in adults, with high mortality rates (29%), so the index of suspicion should be high even in previously asymptomatic adults.20 Acylcarnitine profile with tandem-mass spectrometry can identify > 20 different metabolic defects of fatty acid and organic acid metabolism.22 The diagnosis is confirmed with mutation analysis and fibroblast cultures of enzyme activity.
Disorders of mitochondrial function
Mitochondrial disorders are caused by defects in nuclear or mtDNA genes that lead to abnormal cellular bioenergetics.86 Any pattern of inheritance is possible.24,87 Mitochondrial disorders are common (estimated prevalence: approximately 1 in 11,000 adults and approximately 1 in 6000 children).23
There are a number of well described syndromic presentations of mitochondrial disorders. For example, the most common clinical syndrome in adults is chronic progressive external ophthalmoplegia plus, usually associated with multiple mtDNA deletions,49 and manifests with bilateral ptosis and ophthalmoparesis due to myopathy that affects the extraocular muscles.49 The second most common syndrome is the mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episode associated with the m.3243A>G mutation.87 This syndrome causes stroke-like episodes and seizures (each affecting more than 60% of patients), diabetes mellitus (40%), proximal muscle weakness and fatigue (each affecting > 30% of patients).24,46 More common than these syndromic presentations are patients with nonspecific clinical features, in whom gene panels including hundreds or thousands of genes are required to make the diagnosis.88 The cardiac manifestations can be very diverse and include HCM or dilated cardiomyopathy, LVNC, and arrhythmia.47 Patients can even present with HF as adults.48 The diagnosis should be suspected with symptoms (hearing loss, endocrinopathies, exertional myalgia, seizures) and signs (nerve involvement, ataxia, myopathy) of disease involvement outside the cardiovascular system.88
Disorders of carbohydrate metabolism
Muscle glycogen storage diseases (GSD, most AR) are due to defects in glucose metabolism that lead to organomegaly, hypoglycemia and myopathy.26,50 The most common adult form is McArdle disease (due to a PYGM mutation that affects myophosphorylase) with an estimated prevalence of between 1 in 7650 and 1 in 42,355 in Caucasian individuals.25 Severe cardiomyopathy can be a feature of GSD III and IV.89,90 In patients with myopathic GSD, a unique “second wind phenomenon” (an improvement in exercise tolerance 10–30 minutes into exercise, as muscle metabolism switches from carbohydrates to fat) might occur.91 Creatine kinase level is usually increased with myopathic GSD, which can help to distinguish between GSD and FAOD.50
Two disorders classified as either GSD or lysosomal disorders are Pompe disease and Danon disease. Pompe disease cardiomyopathy is much more common and serious in the infantile-onset form.92,93 Danon disease (X-linked dominant condition due to deficiency of lysosome-associated membrane glycoprotein 2) with glycogen accumulation within cardiomyocytes, skeletal myopathy, and cognitive impairment.37 A third disorder, caused by mutations in the 5′ adenosine monophosphate (AMP)-activated protein kinase subunit γ2 (PRKAG2; encoding the enzyme that modulates glucose uptake and glycolysis) is associated with familial HCM,26 with glycogen-associated granule accumulation leading to ventricular hypertrophy, pre-excitation, and conduction system defects (particularly Wolff-Parkinson-White syndrome).94 The glycogen deposition distinguishes familial HCM caused by sarcomere mutations from PRKAG2 cardiomyopathy.95 A PRKAG2 mutation mouse model showed reversibility in cardiac dysfunction and electrophysiological disorders, suggesting that glycogen storage cardiomyopathy can be modulated by a decrease of glycogen content in the heart.96
Disorders of lysosomes
FD is caused by X-linked pattern mutations in the α-galactosidase A gene, which causes a deficiency in α-galactosidase.28 FD is most commonly diagnosed in adults, with average age of death of approximately 60 years in untreated men.97 FD is said to affect an estimated 1 in 40,000 to 60,000 men.27,98 More recent data showed FD mutations in 1 in 1600 men with newborn screening.99 FD screening in patients with HCM show a prevalence from 1% to 10%, therefore FD should be considered in every patient with HCM.38–40
In FD, the buildup of globotriaosylceramide causes acroparesthesias, angiokeratomas (dark red spots on the skin), hypohydrosis, hearing loss, chronic kidney disease, and cardiomyopathy.27 Although this is an X-linked disorder, women can be affected with severe FD manifestations including cardiomyopathy (eg, late enhancement on gadolinium imaging before LV hypertrophy29) and stroke.28
FD can be screened using a simple dried blood spot test.38 Enzyme activity can be normal in affected women, and requires genetic testing for diagnosis.40 FD cardiomyopathy is amenable to enzyme replacement therapy and all patients should be referred to an expert centre for further evaluation.81
Genetic Modifiers of HF
Myocardial stress alters the expression of numerous genes leading to myocardial hypertrophy, dilatation, and interstitial fibrosis.100 These genes, often a re-expression of a fetal gene program, are dynamically regulated in HF.
One large-scale study of more than 20,000 subjects showed 2 single nucleotide polymorphisms (SNPs) (odds ratio [OR], approximately 1.5) to be associated with HF.101 An SNP in the Ka renal chloride channel gene is associated with HF risk (OR, 1.27 per allele copy),102 which might have a considerable effect at the population level.100,102,103 Other predisposing genetic factors, including epigenetic mechanisms like DNA methylation, histone modifications, adenosine triphosphate-dependent chromatin remodelling, and noncoding RNAs are related to the acquisition of HF, however, their discussion is beyond the scope of this article.103
Genetic Testing
Since the Human Genome Project was completed in 2003,104 sequencing has become faster and less expensive. Whole-exome sequencing (WES) offers a more complete genetic picture with a cost that is equal to or less expensive than gene panels.105 Genetic ascertainment of suspected familial cardiomyopathy using WES54,106 presents several challenges: (1) clinical diagnosis attained in an unclear percentage of index cases; (2) identification of novel and functionally deleterious variants for which significance is unclear; (3) confusion in interpretation of genetic testing results; (4) burden on clinical resources including staff and cost; and (5) effect on relatives, including anxiety and commitment to future screening.
WES can be particularly helpful in determining the genetic factors responsible for familial cardiomyopathy, however, there is a documented lack of genotype-phenotype correlation (Table 2), which makes the use of genetic panels challenging.105 It also offers the potential of new gene discovery.105 Genetic testing should always be performed in a skilled, high-volume setting with expertise in genetic counselling.
Current guidelines recommend genetic testing for any patient with a clinical diagnosis of familial cardiomyopathy, and mutation-specific testing for family members of a genotype-positive proband.33,53 Two of the most compelling reasons for genetic testing in clinical practice are: (1) the identification of family members with no structural abnormalities at potential risk; and (2) clarification of diagnosis in patients with IEM for whom clinical presentation and pattern is similar to familial cardiomyopathies but with differing outcomes (Table 2). Table 4 shows the recommended interval surveillance of adult patients with gene-positive cardiomyopathy. Large studies are needed to assess the effect of genetic diagnosis on response to drug therapies and outcomes.
Table 4.
Evaluation of relatives of patients with cardiomyopathy
| Cardiomyopathy | Interval | Screening |
|---|---|---|
| Gene positive | 12 months | Symptom assessment SCD risk stratification Medication reconciliation Exercise counselling Physical examination ECG and echocardiography |
| 3–5 years (if stable without clinical progression or related complications) | As above | |
| Gene negative | 12–18 months (in those with ventricular enlargement or depressed fractional shortening) | As above |
| 3–5 years (if stable without clinical progression or related complications) | As above |
ECG, electrocardiogram; SCD, sudden cardiac death.
From the Advanced Heart Failure and Cardiomyopathy program, Division of Cardiovascular Medicine, Yale-New Haven Hospital, Yale School of Medicine. Reproduced with permission.
Genetic Approaches to Therapy: Pharmacogenetics and Future Approaches
Pharmacogenomics
The treatment of chronic HF improved markedly with the adoption of 3 drug classes; angiotensin converting enzyme (ACE) inhibitors (ACEi)/angiotensin-II receptor blockers, β-blockers, and aldosterone antagonists, along with implanted cardioverter defibrillators and cardiac resynchronization therapy.
However, there is vast interindividual variation in response to those treatments and a significant amount of the variability is likely due to individual genetic variation.107 Various cytochrome P450, family 2, subfamily D, polypeptide 6 (CYP2D6) alleles in metoprolol users increase the drug’s plasma concentration in patients with *0/*0 genotypes and intermediate genotypes compared with those with 2 fully functional alleles (n = 31; P < 0.01).108 This pronounced effect of the CYP2D6 genotype causes increased blood levels of metoprolol by several fold, which might have an effect on its cardioselectivity. Another study found no association between the various CYP2D6 genotypes and metoprolol-induced adverse effects or efficacy in hypertensive patients109; a subsequent study that examined carvedilol’s pharmacokinetic effects showed similar results.110 In the largest study to examine this association, Bijl et al. showed a lower diastolic blood pressure (approximately 5.4 mm Hg lower) in poor β-blocker metaolizers by CYP2D6 compared with extensive metabolizers who harboured similar polymorphisms.111 However, the results from such studies are rather inconsistent because of small population size and limitations in the study methodology. Therefore, although this pharmacogenetic effect is likely to have theoretical risks on the clinical benefits of β-blocker treatment and adverse drug reactions, investigations of this type might lead to personalized medication regimens and improved clinical outcomes in the future.
Adrenergic receptors variants and β-blockers
In response to decreased cardiac output in chronic HF, several neurohormonal systems, including the sympathetic nervous system, are activated in an effort to maintain adequate circulation. Sympathetic hyperactivation in HF results in five-to sixfold increased circulating levels of norepinephrine compared with healthy control subjects,112 and is increased in direct proportion to the degree of LV dysfunction.113 Chronic stimulation of the adrenergic receptor in HF by the circulating catecholamines norepinephrine and epinephrine result in cardiac dysfunction.
β-Blockers have been the backbone of HF therapy since 1997 when the US Food and Drug Administration approved carvedilol for the treatment of HF.114,115 Nevertheless, there have been several studies that documented a variation in response to β-blocker therapy with some HF patients benefiting more than others.114,116,117 Genetic polymorphisms in the β1-adrenergic receptor (ADRB1) have been examined. One of the most studied polymorphisms in the ADRB1 gene is the Arg389Gly variant, which is highly conserved among species with conflicting results in various studies. The Beta-Blocker Evaluation of Survival Trial (BEST) (ClinicalTrials.gov Identifier: NCT00000560) was a large, randomized clinical trial in which the efficacy of bucindolol in HF was evaluated. The trial was terminated prematurely because of a lack of mortality benefit in the overall trial population; however, mortality benefit was significantly improved in the nonblack subset of patients. In a genetic substudy of BEST, Arg-389 homozygotes treated with bucindolol had a statistically significant age-, sex-, and race-adjusted reduction in mortality (38%) and reduction in mortality or hospitalization (34%) vs placebo.118 In contrast, Gly-389 carriers had no clinical response to bucindolol compared with placebo.118 However, there are conflicting data with regard to other β-blocker therapies. No significant difference in mortality, hospitalization, or drug effect was found in other randomized trials in which the effect of the Arg389Gly variant in patients who took long-acting metoprolol and carvedilol was assessed.117,119
Another variant that has been studied in ADRB1 is a Ser to Gly change at codon 49. Patients with idiopathic dilated cardiomyopathy who were homozygous for Ser49 had poorer prognoses, as measured by occurrence of death or cardiac transplant, compared with Gly49 carriers.120 In a follow-up study, the response to β-blocker therapy was evaluated based on genotype. In patients who took low-dose β-blocker therapy, the 5-year mortality rate was lower among Gly49 carriers than in Ser49 homozygotes.121 In patients who took high-dose β-blockers, there was no significant difference in outcome between genotypes, with better outcomes for Ser49 homozygotes.121
Polymorphisms in the β2-adrenergic receptor (ADRB2) gene have also been extensively studied. In a large prospective cohort study of β-blocker therapy after acute coronary syndrome, Lanfear et al. showed a differential 3-year survival in β-blocker users on the basis of Gly16Arg (46 GA) and Gln27Glu (79 CG) genotypes in ADRB2.122 In another study on the response to carvedilol in HF patients, subjects who were homozygous for Gln27 displayed a lower response rate of improvement of LV function (≥10%) or fractional shortening (≥ 5%) than patients who were homozygous or heterozygous for Glu27 (26% vs 63%; P = 0.003).123 In a further report on the effect of genetic polymorphisms in the sympathetic nervous and the renin-angiotensin-aldosterone system on prognosis in HF, patients carrying 2 copies of the ADRB2 Arg16Gln27 haplotype had increased risk for adverse outcomes (hazard ratio, 1.91; 95% confidence interval, 1.09–3.36).124 Other studies have shown that polymorphisms in ADRB1 and adrenoceptor alpha 2C gene (ADRA2C) synergistically influence the ejection fraction response to β-blocker therapy of HF patients,125,126 and have been associated with the development of hypertension.127 More clinical studies are warranted before genetic testing for predicted response to β-blocker therapy is applied in the clinical setting. Particularly, there is a need to assess multiple adrenergic receptor variants simultaneously to get an accurate picture of the overall effect on pharmacological therapy.
Renin-angiotensin-aldosterone system
Multiple clinical trials have established the survival benefit of ACEi therapy, resulting in their central prominence in modern day HF pharmacotherapy.115,128,129 An insertion/deletion (D) polymorphism in ACE has been shown to explain approximately half of the variance in systemic ACE levels (the D allele associated with increased ACE levels) and is associated with HF incidence and severity.129–132 Because the ACE D allele results in higher ACE levels, it has been theorized that HF patients who carry the D allele might require a higher dose of ACEi to achieve a response similar to individuals who do not carry the D allele. Most studies that assessed intermediate phenotypes of this (ie, serum ACE activity, aldosterone escape, and mean arterial pressure) support this theory.133–136 The relationship between the ACE polymorphism and survival benefit in HF requires further evaluation in larger studies.
In the most extensive pharmacogenetic study of HF patients to assess the ACE polymorphism, a dose-dependent relationship between the ACE D allele and transplant-free survival was observed.137 After a mean follow-up of 33 months, patients who received low-dose ACEi therapy had poorer transplant-free survival associated with the D allele (relative risk of 2.07 for DD homozygotes).137 The relative risk in DD homozygotes was 2.75 for those who did not receive β-blocker therapy.137 As might be expected, there was no significant difference between the effects of the D allele in patients who received high-dose ACEi therapy. So, although the D allele was associated with poorer transplant-free survival, these were the patients who benefited most from ACEi and β-blocker therapy.
Diuretics, Aldosterone Antagonists, and Digoxin Therapy
The clinical implication of pharmacogenetics on other cardiovascular-related drugs have been reported albeit the data are limited. One study that examined individuals receiving a thiazide diuretic who were carriers of 1 or 2 copies of the Trp460 variant allele showed greater protection from the combined outcome of myocardial infarction and stroke (OR, 0.49; 95% confidence interval, 0.32–0.77) than patients with Gly460.138 Verstuyft et al. examined the multidrug resistance gene (MDR1) C3435T SNP on digoxin plasma concentration and showed that individuals who were homozygous TT had the highest digoxin concentrations.139 In another study in African Americans with resistant hypertension who were randomized to treatment with placebo, spironolactone, amiloride, or combination, the spironolactone arm with rs3890011 GG and GC (variants of cytochrome P450, family 4, subfamily A, polypeptide 11 [CYP4A11]) but not in CC homozygotes (P = 0.002), had reduced blood pressure when compared with the other drugs.140 Collectively, there is currently no role for pharmacogenetic testing in the clinical use of these medications and further studies are needed to replicate these results, which would then have important therapeutic implications in the management of cardiovascular disease.140
Future Approaches to Genetic Therapies
It has been the goal of the Clinical Pharmacogenetics Implementation Consortium of the National Institutes of Health’s Pharmacogenomics Research Network (http://www.pgrn.org) and the Pharmacogenomics Knowledge Base (PharmGKB; http://www.pharmgkb.org) to provide guidelines for gene therapy to facilitate the translation and clinical use of pharmacogenomics.141 One of these approaches includes promotion of high volume laboratories to construct a genomic profile that can preemptively be tested in patients who take certain drugs and aid with individualized therapy.
There has been some progress in gene therapy as a futuristic option to treat HF. For example, one study that involved the transfer of the sarcoplasmic reticulum Ca2+ ATPase gene, with the help of a vector, into the cardiac myocardium to rescue HF,142 showed potential beneficial effects on the cardiomyocyte.143 However, there are unprecedented challenges in translating gene therapy to human patients and clinical practice, such as inefficient delivery methods and host immune responses, to technological limitations in health care systems and provider awareness about genomics.
Several microRNA experiments have provided some hope for the therapeutic application of microRNA in heart disease. Carè et al. showed that in vivo inhibition of microRNA 133 by a single infusion of an antagomir caused marked and sustained cardiac hypertrophy.144 A recent study in a mouse model revealed that increased expression of endogenous microRNA 25 contributed to declining cardiac function during HF and can be halted by injection of an antisense oligonucleotide against microRNA 25, and thereby improve cardiac function and survival relative to control.145
Collectively, more studies are warranted to help determine the individual’s genomic profile, which might be of value in the tailoring of therapy in patients with HF and the understanding of precision medicine.
Conclusions
The causes of advanced HF are diverse. Inherited etiologies represent an important and potentially treatable group of disorders with which clinicians should become very familiar. Genetic technology has revolutionized cardiovascular genetics, and as we continue to discover novel disease-causing genes on an unprecedented scale, new methods and technologies to rapidly assess the functional significance of variants singly, or in combination, will evolve. In this review, we focused on the various inherited phenotypes of cardiomyopathy, treatment challenges, and future genetic therapies.
In summary, we recommend a careful 3-generation pedigree search for history of acquired or inherited cardiometabolic diseases, including SCD, and a thorough review of systems to help ascertain the etiology; a careful assessment for potentially reversible causes of cardiomyopathy (eg, FD, hemochromatosis, HCM etc); a tailored approach to early referral for consideration of various therapies, including those for advanced HF, however, we would not recommend genetic testing for SNPs in patients at this point in time; and judicious collaboration with a centre of expertise in cardiovascular genetics for counselling and testing in all patients with possible advanced HF not due to typical ischemic etiologies.
Acknowledgments
Sandra Sirrs has received funding for travel support and speaker fees from Genzyme Canada and Shire Human Genetics Therapies.
Footnotes
Disclosures
The remaining authors have no conflicts of interest to disclose.
References
- 1.Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statisticse2013 update: a report from the American Heart Association. Circulation. 2013;127:e6–245. doi: 10.1161/CIR.0b013e31828124ad. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dunlay SM, Shah ND, Shi Q, et al. Lifetime costs of medical care after heart failure diagnosis. Circ Cardiovasc Qual Outcomes. 2011;4:68–75. doi: 10.1161/CIRCOUTCOMES.110.957225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braunwald E. Heart failure. JACC Heart Fail. 2013;1:1–20. doi: 10.1016/j.jchf.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 4.Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol. 2011;8:30–41. doi: 10.1038/nrcardio.2010.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McMurray JJ, Petrie MC, Murdoch DR, Davie AP. Clinical epidemiology of heart failure: public and private health burden. Eur Heart J. 1998;19(suppl):9–16. [PubMed] [Google Scholar]
- 6.Monserrat L, Hermida M, Bouzas B, et al. Familial dilated cardiomyopathy in patients transplanted for idiopathic dilated cardiomyopathy [in Spanish] Rev Esp Cardiol. 2002;55:725–32. doi: 10.1016/s0300-8932(02)76691-8. [DOI] [PubMed] [Google Scholar]
- 7.Rowin EJ, Maron BJ, Kiernan MS, et al. Advanced heart failure with preserved systolic function in nonobstructive hypertrophic cardiomyopathy: under-recognized subset of candidates for heart transplant. Circ Heart Fail. 2014;7:967–75. doi: 10.1161/CIRCHEARTFAILURE.114.001435. [DOI] [PubMed] [Google Scholar]
- 8.Maron BJ, Towbin JA, Thiene G, et al. Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006;113:1807–16. doi: 10.1161/CIRCULATIONAHA.106.174287. [DOI] [PubMed] [Google Scholar]
- 9.Maron BJ, Gardin JM, Flack JM, et al. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92:785–9. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 10.Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. 2003;107:2227–32. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 11.Klues HG, Schiffers A, Maron BJ. Phenotypic spectrum and patterns of left ventricular hypertrophy in hypertrophic cardiomyopathy: morphologic observations and significance as assessed by two-dimensional echocardiography in 600 patients. J Am Coll Cardiol. 1995;26:1699–708. doi: 10.1016/0735-1097(95)00390-8. [DOI] [PubMed] [Google Scholar]
- 12.Basso C, Corrado D, Bauce B, Thiene G. Arrhythmogenic right ventricular cardiomyopathy. Circ Arrhythm Electrophysiol. 2012;5:1233–46. doi: 10.1161/CIRCEP.111.962035. [DOI] [PubMed] [Google Scholar]
- 13.Coonar AS, Protonotarios N, Tsatsopoulou A, et al. Gene for arrhythmogenic right ventricular cardiomyopathy with diffuse non-epidermolytic palmoplantar keratoderma and woolly hair (Naxos disease) maps to 17q21. Circulation. 1998;97:2049–58. doi: 10.1161/01.cir.97.20.2049. [DOI] [PubMed] [Google Scholar]
- 14.Kushwaha SS, Fallon JT, Fuster V. Restrictive cardiomyopathy. N Engl J Med. 1997;336:267–76. doi: 10.1056/NEJM199701233360407. [DOI] [PubMed] [Google Scholar]
- 15.Sen-Chowdhry S, Syrris P, McKenna WJ. Genetics of restrictive cardiomyopathy. Heart Fail Clin. 2010;6:179–86. doi: 10.1016/j.hfc.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 16.McNally EM, Golbus JR, Puckelwartz MJ. Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin Invest. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pantazis AA, Elliott PM. Left ventricular noncompaction. Curr Opin Cardiol. 2009;24:209–13. doi: 10.1097/HCO.0b013e32832a11e7. [DOI] [PubMed] [Google Scholar]
- 18.Kohli SK, Pantazis AA, Shah JS, et al. Diagnosis of left-ventricular noncompaction in patients with left-ventricular systolic dysfunction: time for a reappraisal of diagnostic criteria? Eur Heart J. 2008;29:89–95. doi: 10.1093/eurheartj/ehm481. [DOI] [PubMed] [Google Scholar]
- 19.Arbustini E, Weidemann F, Hall JL. Left ventricular noncompaction: a distinct cardiomyopathy or a trait shared by different cardiac diseases? J Am Coll Cardiol. 2014;64:1840–50. doi: 10.1016/j.jacc.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lang TF. Adult presentations of medium-chain acyl-CoA dehydrogenase deficiency (MCADD) J Inherit Metab Dis. 2009;32:675–83. doi: 10.1007/s10545-009-1202-0. [DOI] [PubMed] [Google Scholar]
- 21.Tein I. Disorders of fatty acid oxidation. Handb Clin Neurol. 2013;113:1675–88. doi: 10.1016/B978-0-444-59565-2.00035-6. [DOI] [PubMed] [Google Scholar]
- 22.Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2010;33:527–32. doi: 10.1007/s10545-010-9090-x. [DOI] [PubMed] [Google Scholar]
- 23.Schaefer AM, McFarland R, Blakely EL, et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008;63:35–9. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 24.Koopman WJ, Willems PH, Smeitink JA. Monogenic mitochondrial disorders. N Engl J Med. 2012;366:1132–41. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 25.De Castro M, Johnston J, Biesecker L. Determining the prevalence of McArdle disease from gene frequency by analysis of next-generation sequencing data [e-pub ahead of print] Genet Med. doi: 10.1038/gim.2015.9. http://dx.doi.org/10.1038/gim.2015.9, accessed March 5 2015. [DOI] [PMC free article] [PubMed]
- 26.Arad M, Maron BJ, Gorham JM, et al. Glycogen storage diseases presenting as hypertrophic cardiomyopathy. N Engl J Med. 2005;352:362–72. doi: 10.1056/NEJMoa033349. [DOI] [PubMed] [Google Scholar]
- 27.Sachdev B, Takenaka T, Teraguchi H, et al. Prevalence of Anderson-Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105:1407–11. doi: 10.1161/01.cir.0000012626.81324.38. [DOI] [PubMed] [Google Scholar]
- 28.Weidemann F, Niemann M, Warnock DG, Ertl G, Wanner C. The Fabry cardiomyopathy: models for the cardiologist. Annu Rev Med. 2011;62:59–67. doi: 10.1146/annurev-med-090910-085119. [DOI] [PubMed] [Google Scholar]
- 29.Niemann M, Herrmann S, Hu K, et al. Differences in Fabry cardiomyopathy between female and male patients: consequences for diagnostic assessment. JACC Cardiovasc Imaging. 2011;4:592–601. doi: 10.1016/j.jcmg.2011.01.020. [DOI] [PubMed] [Google Scholar]
- 30.Jarcho JA, McKenna W, Pare JA, et al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med. 1989;321:1372–8. doi: 10.1056/NEJM198911163212005. [DOI] [PubMed] [Google Scholar]
- 31.Alexander J, Kowdley KV. HFE-associated hereditary hemochromatosis. Genet Med. 2009;11:307–13. doi: 10.1097/GIM.0b013e31819d30f2. [DOI] [PubMed] [Google Scholar]
- 32.Herman DS, Lam L, Taylor MR, et al. Truncations of titin causing dilated cardiomyopathy. N Engl J Med. 2012;366:619–28. doi: 10.1056/NEJMoa1110186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ackerman MJ, Priori SG, Willems S, et al. HRS/EHRA expert consensus statement on the state of genetic testing for the channelopathies and cardiomyopathies: this document was developed as a partnership between the Heart Rhythm Society (HRS) and the European Heart Rhythm Association (EHRA) Europace. 2011;13:1077–109. doi: 10.1093/europace/eur245. [DOI] [PubMed] [Google Scholar]
- 34.Milting H, Klauke B. Molecular genetics of arrhythmogenic right ventricular dysplasia/cardiomyopathy. Nat Clin Pract Cardiovasc Med. 2008;5:E1. doi: 10.1038/ncpcardio1350. [author reply E2] [DOI] [PubMed] [Google Scholar]
- 35.Tester DJ, Ackerman MJ. Genetic testing for potentially lethal, highly treatable inherited cardiomyopathies/channelopathies in clinical practice. Circulation. 2011;123:1021–37. doi: 10.1161/CIRCULATIONAHA.109.914838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niimura H, Patton KK, McKenna WJ, et al. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation. 2002;105:446–51. doi: 10.1161/hc0402.102990. [DOI] [PubMed] [Google Scholar]
- 37.Cheng Z, Fang Q. Danon disease: focusing on heart. J Hum Genet. 2012;57:407–10. doi: 10.1038/jhg.2012.72. [DOI] [PubMed] [Google Scholar]
- 38.Hagege AA, Caudron E, Damy T, et al. Screening patients with hypertrophic cardiomyopathy for Fabry disease using a filter-paper test: the FOCUS study. Heart. 2011;97:131–6. doi: 10.1136/hrt.2010.200188. [DOI] [PubMed] [Google Scholar]
- 39.Palecek T, Honzikova J, Poupetova H, et al. Prevalence of Fabry disease in male patients with unexplained left ventricular hypertrophy in primary cardiology practice: prospective Fabry cardiomyopathy screening study (FACSS) J Inherit Metab Dis. 2014;37:455–60. doi: 10.1007/s10545-013-9659-2. [DOI] [PubMed] [Google Scholar]
- 40.Havndrup O, Christiansen M, Stoevring B, et al. Fabry disease mimicking hypertrophic cardiomyopathy: genetic screening needed for establishing the diagnosis in women. Eur J Heart Fail. 2010;12:535–40. doi: 10.1093/eurjhf/hfq073. [DOI] [PubMed] [Google Scholar]
- 41.Terryn W, Deschoenmakere G, De Keyser J, et al. Prevalence of Fabry disease in a predominantly hypertensive population with left ventricular hypertrophy. Int J Cardiol. 2013;167:2555–60. doi: 10.1016/j.ijcard.2012.06.069. [DOI] [PubMed] [Google Scholar]
- 42.Hund E, Linke RP, Willig F, Grau A. Transthyretin-associated neuropathic amyloidosis. Pathogenesis and treatment. Neurology. 2001;56:431–5. doi: 10.1212/wnl.56.4.431. [DOI] [PubMed] [Google Scholar]
- 43.Benson MD. Inherited amyloidosis. J Med Genet. 1991;28:73–8. doi: 10.1136/jmg.28.2.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jacobson DR, Pastore RD, Yaghoubian R, et al. Variant-sequence transthyretin (isoleucine 122) in late-onset cardiac amyloidosis in black Americans. N Engl J Med. 1997;336:466–73. doi: 10.1056/NEJM199702133360703. [DOI] [PubMed] [Google Scholar]
- 45.van Rijsingen IA, Arbustini E, Elliott PM, et al. Risk factors for malignant ventricular arrhythmias in lamin a/c mutation carriers a European cohort study. J Am Coll Cardiol. 2012;59:493–500. doi: 10.1016/j.jacc.2011.08.078. [DOI] [PubMed] [Google Scholar]
- 46.Silvestri G, Ciafaloni E, Santorelli FM, et al. Clinical features associated with the A–>G transition at nucleotide 8344 of mtDNA (“MERRF mutation”) Neurology. 1993;43:1200–6. doi: 10.1212/wnl.43.6.1200. [DOI] [PubMed] [Google Scholar]
- 47.Yaplito-Lee J, Weintraub R, Jamsen K, et al. Cardiac manifestations in oxidative phosphorylation disorders of childhood. J Pediatr. 2007;150:407–11. doi: 10.1016/j.jpeds.2006.12.047. [DOI] [PubMed] [Google Scholar]
- 48.Nakagawa H, Okayama S, Kamon D, et al. Refractory high output heart failure in a patient with primary mitochondrial respiratory chain disease. Intern Med. 2014;53:315–9. doi: 10.2169/internalmedicine.53.1386. [DOI] [PubMed] [Google Scholar]
- 49.Pfeffer G, Sirrs S, Wade NK, Mezei MM. Multisystem disorder in late-onset chronic progressive external ophthalmoplegia. Can J Neurol Sci. 2011;38:119–23. doi: 10.1017/s031716710001115x. [DOI] [PubMed] [Google Scholar]
- 50.Talente GM, Coleman RA, Alter C, et al. Glycogen storage disease in adults. Ann Intern Med. 1994;120:218–26. doi: 10.7326/0003-4819-120-3-199402010-00008. [DOI] [PubMed] [Google Scholar]
- 51.Wilde AA, Behr ER. Genetic testing for inherited cardiac disease. Nat Rev Cardiol. 2013;10:571–83. doi: 10.1038/nrcardio.2013.108. [DOI] [PubMed] [Google Scholar]
- 52.Melacini P, Basso C, Angelini A, et al. Clinicopathological profiles of progressive heart failure in hypertrophic cardiomyopathy. Eur Heart J. 2010;31:2111–23. doi: 10.1093/eurheartj/ehq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gersh BJ, Maron BJ, Bonow RO, et al. 2011 ACCF/AHA Guideline for the Diagnosis and Treatment of Hypertrophic Cardiomyopathy: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Developed in collaboration with the American Association for Thoracic Surgery, American Society of Echocardiography, American Society of Nuclear Cardiology, Heart Failure Society of America, Heart Rhythm Society, Society for Cardiovascular Angiography and Interventions, and Society of Thoracic Surgeons. J Am Coll Cardiol. 2011;58:e212–60. doi: 10.1016/j.jacc.2011.06.011. [DOI] [PubMed] [Google Scholar]
- 54.Teekakirikul P, Kelly MA, Rehm HL, Lakdawala NK, Funke BH. Inherited cardiomyopathies: molecular genetics and clinical genetic testing in the postgenomic era. J Mol Diagn. 2013;15:158–70. doi: 10.1016/j.jmoldx.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 55.Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31. doi: 10.1186/1750-1172-8-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Casazza F, Morpurgo M. The varying evolution of Friedreich’s ataxia cardiomyopathy. Am J Cardiol. 1996;77:895–8. doi: 10.1016/S0002-9149(97)89194-1. [DOI] [PubMed] [Google Scholar]
- 57.Bos JM, Will ML, Gersh BJ, et al. Characterization of a phenotype-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2014;89:727–37. doi: 10.1016/j.mayocp.2014.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ. Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc. 2005;80:739–44. doi: 10.1016/S0025-6196(11)61527-9. [DOI] [PubMed] [Google Scholar]
- 59.Andersen PS, Havndrup O, Hougs L, et al. Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat. 2009;30:363–70. doi: 10.1002/humu.20862. [DOI] [PubMed] [Google Scholar]
- 60.Spirito P, Seidman CE, McKenna WJ, Maron BJ. The management of hypertrophic cardiomyopathy. N Engl J Med. 1997;336:775–85. doi: 10.1056/NEJM199703133361107. [DOI] [PubMed] [Google Scholar]
- 61.Nagueh SF, Lombardi R, Tan Y, et al. Atorvastatin and cardiac hypertrophy and function in hypertrophic cardiomyopathy: a pilot study. Eur J Clin Invest. 2010;40:976–83. doi: 10.1111/j.1365-2362.2010.02349.x. [DOI] [PubMed] [Google Scholar]
- 62.Maron MS, Kalsmith BM, Udelson JE, Li W, DeNofrio D. Survival after cardiac transplantation in patients with hypertrophic cardiomyopathy. Circ Heart Fail. 2010;3:574–9. doi: 10.1161/CIRCHEARTFAILURE.109.922872. [DOI] [PubMed] [Google Scholar]
- 63.Riley MP, Zado E, Bala R, et al. Lack of uniform progression of endocardial scar in patients with arrhythmogenic right ventricular dysplasia/cardiomyopathy and ventricular tachycardia. Circ Arrhythm Electrophysiol. 2010;3:332–8. doi: 10.1161/CIRCEP.109.919530. [DOI] [PubMed] [Google Scholar]
- 64.Philips B, Madhavan S, James C, et al. Outcomes of catheter ablation of ventricular tachycardia in arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Arrhythm Electrophysiol. 2012;5:499–505. doi: 10.1161/CIRCEP.111.968677. [DOI] [PubMed] [Google Scholar]
- 65.Kirchhof P, Fabritz L, Zwiener M, et al. Age- and training-dependent development of arrhythmogenic right ventricular cardiomyopathy in heterozygous plakoglobin-deficient mice. Circulation. 2006;114:1799–806. doi: 10.1161/CIRCULATIONAHA.106.624502. [DOI] [PubMed] [Google Scholar]
- 66.James CA, Bhonsale A, Tichnell C, et al. Exercise increases age-related penetrance and arrhythmic risk in arrhythmogenic right ventricular dysplasia/cardiomyopathy-associated desmosomal mutation carriers. J Am Coll Cardiol. 2013;62:1290–7. doi: 10.1016/j.jacc.2013.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Maron BJ, Chaitman BR, Ackerman MJ, et al. Recommendations for physical activity and recreational sports participation for young patients with genetic cardiovascular diseases. Circulation. 2004;109:2807–16. doi: 10.1161/01.CIR.0000128363.85581.E1. [DOI] [PubMed] [Google Scholar]
- 68.Xu T, Yang Z, Vatta M, et al. Compound and digenic heterozygosity contributes to arrhythmogenic right ventricular cardiomyopathy. J Am Coll Cardiol. 2010;55:587–97. doi: 10.1016/j.jacc.2009.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tedford RJ, James C, Judge DP, et al. Cardiac transplantation in arrhythmogenic right ventricular dysplasia/cardiomyopathy. J Am Coll Cardiol. 2012;59:289–90. doi: 10.1016/j.jacc.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.DePasquale EC, Nasir K, Jacoby DL. Outcomes of adults with restrictive cardiomyopathy after heart transplantation. J Heart Lung Transplant. 2012;31:1269–75. doi: 10.1016/j.healun.2012.09.018. [DOI] [PubMed] [Google Scholar]
- 71.Coelho T, Adams D, Silva A, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369:819–29. doi: 10.1056/NEJMoa1208760. [DOI] [PubMed] [Google Scholar]
- 72.Megalla S, Holtzman D, Aronow WS, et al. Predictors of cardiac hepatopathy in patients with right heart failure. Med Sci Monit. 2011;17:CR537–41. doi: 10.12659/MSM.881977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hillege HL, Nitsch D, Pfeffer MA, et al. Renal function as a predictor of outcome in a broad spectrum of patients with heart failure. Circulation. 2006;113:671–8. doi: 10.1161/CIRCULATIONAHA.105.580506. [DOI] [PubMed] [Google Scholar]
- 74.Topilsky Y, Pereira NL, Shah DK, et al. Left ventricular assist device therapy in patients with restrictive and hypertrophic cardiomyopathy. Circ Heart Fail. 2011;4:266–75. doi: 10.1161/CIRCHEARTFAILURE.110.959288. [DOI] [PubMed] [Google Scholar]
- 75.Molina KM, Shrader P, Colan SD, et al. Predictors of disease progression in pediatric dilated cardiomyopathy. Circ Heart Fail. 2013;6:1214–22. doi: 10.1161/CIRCHEARTFAILURE.113.000125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mahon NG, Murphy RT, MacRae CA, et al. Echocardiographic evaluation in asymptomatic relatives of patients with dilated cardiomyopathy reveals preclinical disease. Ann Intern Med. 2005;143:108–15. doi: 10.7326/0003-4819-143-2-200507190-00009. [DOI] [PubMed] [Google Scholar]
- 77.Regitz-Zagrosek V, Petrov G, Lehmkuhl E, et al. Heart transplantation in women with dilated cardiomyopathy. Transplantation. 2010;89:236–44. doi: 10.1097/TP.0b013e3181c35255. [DOI] [PubMed] [Google Scholar]
- 78.Niemann M, Stork S, Weidemann F. Left ventricular noncompaction cardiomyopathy: an overdiagnosed disease. Circulation. 2012;126:e240–3. doi: 10.1161/CIRCULATIONAHA.112.095059. [DOI] [PubMed] [Google Scholar]
- 79.Brunetti-Pierri N, Lamance KM, Lewis RA, Craigen WJ. 30-year follow-up of a patient with classic citrullinemia. Mol Genet Metab. 2012;106:248–50. doi: 10.1016/j.ymgme.2012.03.011. [DOI] [PubMed] [Google Scholar]
- 80.Hannah-Shmouni F, McLeod K, Sirrs S. Recurrent exercise-induced rhabdomyolysis. CMAJ. 2012;184:426–30. doi: 10.1503/cmaj.110518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Weidemann F, Niemann M, Breunig F, et al. Long-term effects of enzyme replacement therapy on fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. 2009;119:524–9. doi: 10.1161/CIRCULATIONAHA.108.794529. [DOI] [PubMed] [Google Scholar]
- 82.Antunes AP, Nogueira C, Rocha H, Vilarinho L, Evangelista T. Intermittent rhabdomyolysis with adult onset associated with a mutation in the ACADVL gene. J Clin Neuromusc Dis. 2013;15:69–72. doi: 10.1097/CND.0000000000000012. [DOI] [PubMed] [Google Scholar]
- 83.Xiong D, He H, James J, et al. Cardiac-specific VLCAD deficiency induces dilated cardiomyopathy and cold intolerance. Am J Physiol Heart Circ Physiol. 2014;306:H326–38. doi: 10.1152/ajpheart.00931.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Behrend AM, Harding CO, Shoemaker JD, et al. Substrate oxidation and cardiac performance during exercise in disorders of long chain fatty acid oxidation. Mol Genet Metab. 2012;105:110–5. doi: 10.1016/j.ymgme.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Baruteau J, Sachs P, Broue P, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study from 187 patients. Complementary data. J Inherit Metab Dis. 2014;37:137–9. doi: 10.1007/s10545-013-9628-9. [DOI] [PubMed] [Google Scholar]
- 86.Arbustini E, Diegoli M, Fasani R, et al. Mitochondrial DNA mutations and mitochondrial abnormalities in dilated cardiomyopathy. Am J Pathol. 1998;153:1501–10. doi: 10.1016/S0002-9440(10)65738-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Thorburn DR. Mitochondrial disorders: prevalence, myths and advances. J Inherit Metab Dis. 2004;27:349–62. doi: 10.1023/B:BOLI.0000031098.41409.55. [DOI] [PubMed] [Google Scholar]
- 88.Parikh S, Goldstein A, Koenig MK, et al. Diagnosis and management of mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2015;17:689–701. doi: 10.1038/gim.2014.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moses SW, Parvari R. The variable presentations of glycogen storage disease type IV: a review of clinical, enzymatic and molecular studies. Curr Mol Med. 2002;2:177–88. doi: 10.2174/1566524024605815. [DOI] [PubMed] [Google Scholar]
- 90.Crushell E, Treacy EP, Dawe J, Durkie M, Beauchamp NJ. Glycogen storage disease type III in the Irish population. J Inherit Metab Dis. 2010;33(suppl 3):S215–8. doi: 10.1007/s10545-010-9096-4. [DOI] [PubMed] [Google Scholar]
- 91.Haller RG, Vissing J. Spontaneous “second wind” and glucose-induced second “second wind” in McArdle disease: oxidative mechanisms. Arch Neurol. 2002;59:1395–402. doi: 10.1001/archneur.59.9.1395. [DOI] [PubMed] [Google Scholar]
- 92.Ben-Ami R, Puglisi J, Haider T, Mehta D. The Mount Sinai Hospital clinicalpathological conference: a 45-year-old man with Pompe’s disease and dilated cardiomyopathy. Mount Sinai J Med. 2001;68:205–12. [PubMed] [Google Scholar]
- 93.Herzog A, Hartung R, Reuser AJ, et al. A cross-sectional single-centre study on the spectrum of Pompe disease, German patients: molecular analysis of the GAA gene, manifestation and genotype-phenotype correlations. Orphanet J Rare Dis. 2012;7:35. doi: 10.1186/1750-1172-7-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gollob MH, Green MS, Tang AS, et al. Identification of a gene responsible for familial Wolff-Parkinson-White syndrome. N Engl J Med. 2001;344:1823–31. doi: 10.1056/NEJM200106143442403. [DOI] [PubMed] [Google Scholar]
- 95.Kelly BP, Russell MW, Hennessy JR, Ensing GJ. Severe hypertrophic cardiomyopathy in an infant with a novel PRKAG2 gene mutation: potential differences between infantile and adult onset presentation. Pediatr Cardiol. 2009;30:1176–9. doi: 10.1007/s00246-009-9521-3. [DOI] [PubMed] [Google Scholar]
- 96.Wolf CM, Arad M, Ahmad F, et al. Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations. Circulation. 2008;117:144–54. doi: 10.1161/CIRCULATIONAHA.107.726752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet Med. 2009;11:790–6. doi: 10.1097/GIM.0b013e3181bb05bb. [DOI] [PubMed] [Google Scholar]
- 98.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281:249–54. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 99.Lin HY, Chong KW, Hsu JH, et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet. 2009;2:450–6. doi: 10.1161/CIRCGENETICS.109.862920. [DOI] [PubMed] [Google Scholar]
- 100.Creemers EE, Pinto YM. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res. 2011;89:265–72. doi: 10.1093/cvr/cvq308. [DOI] [PubMed] [Google Scholar]
- 101.Smith NL, Felix JF, Morrison AC, et al. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circ Cardiovasc Genet. 2010;3:256–66. doi: 10.1161/CIRCGENETICS.109.895763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cappola TP, Matkovich SJ, Wang W, et al. Loss-of-function DNA sequence variant in the CLCNKA chloride channel implicates the cardio-renal axis in interindividual heart failure risk variation. Proc Natl Acad Sci U S A. 2011;108:2456–61. doi: 10.1073/pnas.1017494108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Creemers EE, Wilde AA, Pinto YM. Heart failure: advances through genomics. Nat Rev Genet. 2011;12:357–62. doi: 10.1038/nrg2983. [DOI] [PubMed] [Google Scholar]
- 104.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–45. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 105.Johansen Taber KA, Dickinson BD, Wilson M. The promise and challenges of next-generation genome sequencing for clinical care. JAMA Intern Med. 2014;174:275–80. doi: 10.1001/jamainternmed.2013.12048. [DOI] [PubMed] [Google Scholar]
- 106.Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745–55. doi: 10.1038/nrg3031. [DOI] [PubMed] [Google Scholar]
- 107.Talameh JA, McLeod HL, Adams KF, Jr, Patterson JH. Genetic tailoring of pharmacotherapy in heart failure: optimize the old, while we wait for something new. J Card Fail. 2012;18:338–49. doi: 10.1016/j.cardfail.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 108.Rau T, Heide R, Bergmann K, et al. Effect of the CYP2D6 genotype on metoprolol metabolism persists during long-term treatment. Pharmacogenetics. 2002;12:465–72. doi: 10.1097/00008571-200208000-00007. [DOI] [PubMed] [Google Scholar]
- 109.Zineh I, Beitelshees AL, Gaedigk A, et al. Pharmacokinetics and CYP2D6 genotypes do not predict metoprolol adverse events or efficacy in hypertension. Clin Pharmacol Ther. 2004;76:536–44. doi: 10.1016/j.clpt.2004.08.020. [DOI] [PubMed] [Google Scholar]
- 110.Sehrt D, Meineke I, Tzvetkov M, Gultepe S, Brockmoller J. Carvedilol pharmacokinetics and pharmacodynamics in relation to CYP2D6 and ADRB pharmacogenetics. Pharmacogenomics. 2011;12:783–95. doi: 10.2217/pgs.11.20. [DOI] [PubMed] [Google Scholar]
- 111.Bijl MJ, Visser LE, van Schaik RH, et al. Genetic variation in the CYP2D6 gene is associated with a lower heart rate and blood pressure in beta-blocker users. Clin Pharmacol Ther. 2009;85:45–50. doi: 10.1038/clpt.2008.172. [DOI] [PubMed] [Google Scholar]
- 112.Hasking GJ, Esler MD, Jennings GL, et al. Norepinephrine spillover to plasma in patients with congestive heart failure: evidence of increased overall and cardiorenal sympathetic nervous activity. Circulation. 1986;73:615–21. doi: 10.1161/01.cir.73.4.615. [DOI] [PubMed] [Google Scholar]
- 113.Thomas JA, Marks BH. Plasma norepinephrine in congestive heart failure. Am J Cardiol. 1978;41:233–43. doi: 10.1016/0002-9149(78)90162-5. [DOI] [PubMed] [Google Scholar]
- 114.Eichhorn EJ, Bristow MR. The Carvedilol Prospective Randomized Cumulative Survival (COPERNICUS) trial. Curr Control Trials Cardiovasc Med. 2001;2:20–3. doi: 10.1186/cvm-2-1-020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147–239. doi: 10.1016/j.jacc.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 116.Kotecha D, Manzano L, Altman DG, et al. Individual patient data meta-analysis of beta-blockers in heart failure: rationale and design. Syst Rev. 2013;2:7. doi: 10.1186/2046-4053-2-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Effect of metoprolol CR/XL in chronic heart failure: Metoprolol CR/XL Randomised Intervention Trial in Congestive Heart Failure (MERIT-HF) Lancet. 1999;353:2001–7. [PubMed] [Google Scholar]
- 118.Liggett SB, Mialet-Perez J, Thaneemit-Chen S, et al. A polymorphism within a conserved beta(1)-adrenergic receptor motif alters cardiac function and beta-blocker response in human heart failure. Proc Natl Acad Sci U S A. 2006;103:11288–93. doi: 10.1073/pnas.0509937103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Mialet Perez J, Rathz DA, Petrashevskaya NN, et al. Beta 1-adrenergic receptor polymorphisms confer differential function and predisposition to heart failure. Nat Med. 2003;9:1300–5. doi: 10.1038/nm930. [DOI] [PubMed] [Google Scholar]
- 120.Borjesson M, Magnusson Y, Hjalmarson A, Andersson B. A novel polymorphism in the gene coding for the beta(1)-adrenergic receptor associated with survival in patients with heart failure. Eur Heart J. 2000;21:1853–8. doi: 10.1053/euhj.1999.1994. [DOI] [PubMed] [Google Scholar]
- 121.Magnusson Y, Levin MC, Eggertsen R, et al. Ser49Gly of beta1-adrenergic receptor is associated with effective beta-blocker dose in dilated cardiomyopathy. Clin Pharmacol Ther. 2005;78:221–31. doi: 10.1016/j.clpt.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 122.Lanfear DE, Jones PG, Marsh S, et al. Beta2-adrenergic receptor genotype and survival among patients receiving beta-blocker therapy after an acute coronary syndrome. JAMA. 2005;294:1526–33. doi: 10.1001/jama.294.12.1526. [DOI] [PubMed] [Google Scholar]
- 123.Kaye DM, Smirk B, Williams C, et al. Beta-adrenoceptor genotype influences the response to carvedilol in patients with congestive heart failure. Pharmacogenetics. 2003;13:379–82. doi: 10.1097/00008571-200307000-00002. [DOI] [PubMed] [Google Scholar]
- 124.Shin J, Lobmeyer MT, Gong Y, et al. Relation of beta(2)-adrenoceptor haplotype to risk of death and heart transplantation in patients with heart failure. Am J Cardiol. 2007;99:250–5. doi: 10.1016/j.amjcard.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 125.Lobmeyer MT, Gong Y, Terra SG, et al. Synergistic polymorphisms of beta1 and alpha2C-adrenergic receptors and the influence on left ventricular ejection fraction response to beta-blocker therapy in heart failure. Pharmacogenet Genomics. 2007;17:277–82. doi: 10.1097/FPC.0b013e3280105245. [DOI] [PubMed] [Google Scholar]
- 126.Chen L, Meyers D, Javorsky G, et al. Arg389Gly-beta1-adrenergic receptors determine improvement in left ventricular systolic function in nonischemic cardiomyopathy patients with heart failure after chronic treatment with carvedilol. Pharmacogenet Genomics. 2007;17:941–9. doi: 10.1097/FPC.0b013e3282ef7354. [DOI] [PubMed] [Google Scholar]
- 127.Johnson AD, Newton-Cheh C, Chasman DI, et al. Association of hypertension drug target genes with blood pressure and hypertension in 86,588 individuals. Hypertension. 2011;57:903–10. doi: 10.1161/HYPERTENSIONAHA.110.158667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). The CONSENSUS Trial Study Group. N Engl J Med. 1987;316:1429–35. doi: 10.1056/NEJM198706043162301. [DOI] [PubMed] [Google Scholar]
- 129.Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. The SOLVD Investigators. N Engl J Med. 1991;325:293–302. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 130.Rigat B, Hubert C, Alhenc-Gelas F, et al. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–6. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Schut AF, Bleumink GS, Stricker BH, et al. Angiotensin converting enzyme insertion/deletion polymorphism and the risk of heart failure in hypertensive subjects. Eur Heart J. 2004;25:2143–8. doi: 10.1016/j.ehj.2004.08.026. [DOI] [PubMed] [Google Scholar]
- 132.Fatini C, Sticchi E, Marcucci R, et al. ACE insertion/deletion, but not-240A>T polymorphism, modulates the severity in heart failure. J Investig Med. 2008;56:1004–10. doi: 10.2310/JIM.0b013e31818e8028. [DOI] [PubMed] [Google Scholar]
- 133.Struthers AD. Aldosterone escape during angiotensin-converting enzyme inhibitor therapy in chronic heart failure. J Card Fail. 1996;2:47–54. doi: 10.1016/s1071-9164(96)80009-1. [DOI] [PubMed] [Google Scholar]
- 134.Andersson B, Sylven C. The DD genotype of the angiotensin-converting enzyme gene is associated with increased mortality in idiopathic heart failure. J Am Coll Cardiol. 1996;28:162–7. doi: 10.1016/0735-1097(96)00098-8. [DOI] [PubMed] [Google Scholar]
- 135.Myerson SG, Montgomery HE, Whittingham M, et al. Left ventricular hypertrophy with exercise and ACE gene insertion/deletion polymorphism: a randomized controlled trial with losartan. Circulation. 2001;103:226–30. doi: 10.1161/01.cir.103.2.226. [DOI] [PubMed] [Google Scholar]
- 136.Cicoira M, Zanolla L, Rossi A, et al. Failure of aldosterone suppression despite angiotensin-converting enzyme (ACE) inhibitor administration in chronic heart failure is associated with ACE DD genotype. J Am Coll Cardiol. 2001;37:1808–12. doi: 10.1016/s0735-1097(01)01237-2. [DOI] [PubMed] [Google Scholar]
- 137.McNamara DM, Holubkov R, Postava L, et al. Pharmacogenetic interactions between angiotensin-converting enzyme inhibitor therapy and the angiotensin-converting enzyme deletion polymorphism in patients with congestive heart failure. J Am Coll Cardiol. 2004;44:2019–26. doi: 10.1016/j.jacc.2004.08.048. [DOI] [PubMed] [Google Scholar]
- 138.Psaty BM, Smith NL, Heckbert SR, et al. Diuretic therapy, the alpha-adducin gene variant, and the risk of myocardial infarction or stroke in persons with treated hypertension. JAMA. 2002;287:1680–9. doi: 10.1001/jama.287.13.1680. [DOI] [PubMed] [Google Scholar]
- 139.Verstuyft C, Schwab M, Schaeffeler E, et al. Digoxin pharmacokinetics and MDR1 genetic polymorphisms. Eur J Clin Pharmacol. 2003;58:809–12. doi: 10.1007/s00228-003-0567-5. [DOI] [PubMed] [Google Scholar]
- 140.Laffer CL, Elijovich F, Eckert GJ, et al. Genetic variation in CYP4A11 and blood pressure response to mineralocorticoid receptor antagonism or ENaC inhibition: an exploratory pilot study in African Americans. J Am Soc Hypertens. 2014;8:475–80. doi: 10.1016/j.jash.2014.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Relling MV, Klein TE. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin Pharmacol Ther. 2011;89:464–7. doi: 10.1038/clpt.2010.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Niwano K, Arai M, Koitabashi N, et al. Lentiviral vector-mediated SERCA2 gene transfer protects against heart failure and left ventricular remodeling after myocardial infarction in rats. Mol Ther. 2008;16:1026–32. doi: 10.1038/mt.2008.61. [DOI] [PubMed] [Google Scholar]
- 143.del Monte F, Hajjar RJ, Harding SE. Overwhelming evidence of the beneficial effects of SERCA gene transfer in heart failure. Circ Res. 2001;88:E66–7. doi: 10.1161/hh1101.092004. [DOI] [PubMed] [Google Scholar]
- 144.Carè A, Catalucci D, Felicetti F, et al. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–8. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 145.Wahlquist C, Jeong D, Rojas-Munoz A, et al. Inhibition of miR-25 improves cardiac contractility in the failing heart. Nature. 2014;508:531–5. doi: 10.1038/nature13073. [DOI] [PMC free article] [PubMed] [Google Scholar]