

Abstract
In this article, we report on a Brazilian female patient born to consanguineous parents and presenting with alobar holoprosencephaly, severe eye involvement, and unusual skin hyperpigmented lesions. She was found to have a mutation (c.2240T > C; p.Val751Gly) in exon 15 of the PTCH1 gene. Mutations in this gene are associated with the nevoid basal cell carcinoma syndrome (NBCCS, OMIM 109400) and, in other instances, with holoprosencephaly (holoprosencephaly-7, OMIM 610828). Severe eye involvement ranging from orbital coloboma to microphthalmia has been seldom reported in patients with NBCCS with PTCH1 mutations. To our knowledge, this is the first report of an individual with central nervous system, skin, and eye manifestations due to a PTCH1 mutation. Mechanisms involved in these multisystem manifestations are discussed.
Keywords: PTCH1 gene, eye anomalies, holoprosencephaly, skin, consanguinity
Introduction
Holoprosencephaly (HPE) is a common developmental defect affecting both the forebrain and the face. It is characterized by the incomplete separation of the cerebral hemispheres into distinct right and left halves.1 2 3 Eye anomalies are part of the clinical spectrum of the HPEs, ranging from cyclopia to variable structural eye defects, with the most extreme variant being bilateral anophthalmia.4 Eye anomalies, mainly microphthalmia and orbital cysts, have been reported in patients with nevoid basal cell carcinoma syndrome (NBCCS), also known as Gorlin syndrome (OMIM 109400).5 6 7 Both situations present a common pathogenetic etiology: NBCCS results from deletions or loss-of-function mutations of the PTCH1 gene, whereas central nervous system anomalies such as HPE, microcephaly, and developmental delay have been attributed to gain-of-function mutations or duplications involving this gene.8
Here we report the clinical findings of a patient with HPE who has a previously reported PTCH1 mutation.9 The severe multisystem involvement in this patient led us to consider the possibility of a second recessive condition due to parental consanguinity contributing to the abnormal phenotype.10
Case Report
The female patient (Fig. 1A–C) was born in 1999. She was the first child of a healthy 24-year-old G1P1 white woman and her healthy consanguineous (F = 1/16) 22-year-old husband. The pregnancy was unremarkable and the child was born by normal vaginal delivery at 40 weeks' gestation. The birth weight was 3.25 kg (50th centile), birth length was 45 cm (3rd centile), but the occipitofrontal circumference was not recorded. At birth, eye and craniofacial anomalies were noted. The child had normal vitality and was discharged at day 4. At the age of 12 months, she developed seizures. Clinical examination at the age of 53 months showed microcephaly, hypotelorism, bilateral microphthalmia, large ears, upslanted palpebral fissures, absent anterior nasal spine, agenesis of the nasal septum, diminished frontonasal angle, flat and hypoplastic nose, midline cleft lip/palate, large ears, and delayed neuropsychological development. Results of routine laboratory blood tests were normal. G-banded chromosomes were normal. Skin examination showed a large area of atopic eczema extending from the front of the trunk and several small and irregular nevi mainly observed on the face, neck, dorsum, and limbs. Magnetic resonance imaging of the brain showed alobar HPE and large cysts inside both orbits with small eye remnants. The optic chiasm was present as was the splenium of the corpus callosum. The brainstem was hypoplastic, and cerebellar tonsil and nodule were displaced backward with a large fourth ventricle invading the cisterna magna. There was a small blind meningocele in the occipital region (Fig. 2A–D).
Fig. 1.
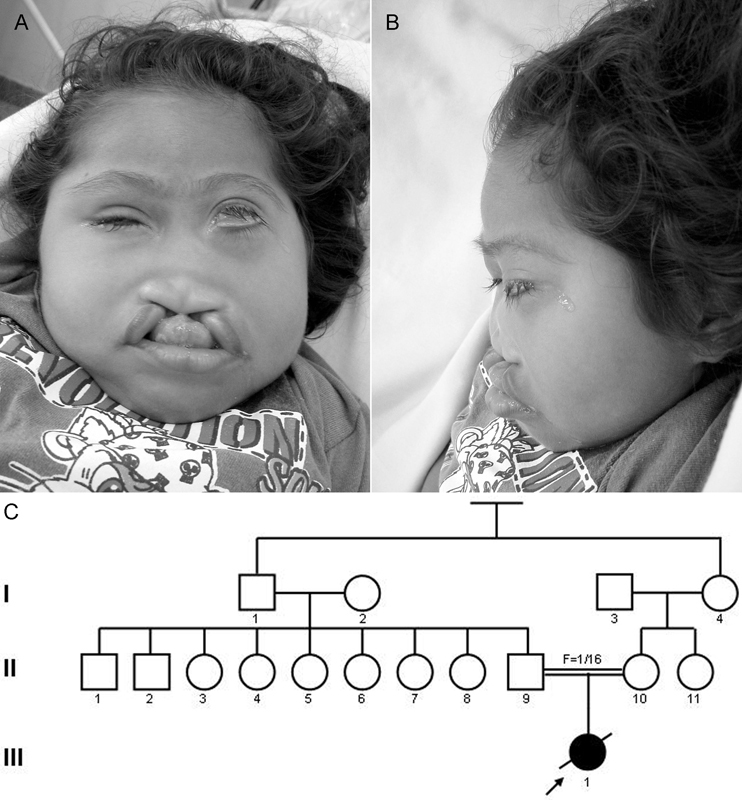
A–C Facial appearance and pedigree of the patient.
Fig. 2.

A–D Magnetic resonance imaging showing alobar holoprosencephaly, microphthalmia, orbital cysts, structural anomalies of the corpus callosum, fourth ventricle, cerebellum, and cisterna magna.
In the present case, we performed mutational analysis of the entire coding region and exon–intron binderies of SHH, TGIF, SIX3, ZIC2, and PTCH1 genes. The primers used to screen these genes have been previously described by Mansilla et al.11 Polymerase chain reaction (PCR) amplifications were performed in a 35-μL reaction mixture, using 20 ng of deoxyribonucleic acid (DNA) template, 1 μL of PCR Amplification Buffer (Invitrogen, Carlsbad, California, United States), 1.5 mM of MgSO4, 200 μM of each dNTP (Amersham Biosciences, Piscataway, New Jersey, United States), 0.25–10 μM of each primer (Invitrogen), and 0.02 U of Taq DNA polymerase/II (Applied Biosystems, Foster City, California, United States). The PCR cycling parameters used for amplification of the all five genes were 95°C for 30 seconds, 60°C for 30 seconds, 72°C for 1 minute, and a final extension of 72°C for 7 minute. Direct sequencing was performed using the Big Dye Terminator v.3.1cycle sequencing Kit (Applied Byosystems, Foster City, California, United States) and ABI3500 DNA Analysis System (Applied Byosystems, 3500 Genetic Analyser- Foster City, California, United States). PolyPhen (http://genetics.bwh.harvard.edu/pph2) was used to predict the effect of the nonsynonymous mutation found in the PTCH1 gene. In exon 15 of the PTCH1 gene, a transversion was identified: c.2240T > C, resulting in p.Val751Gly.9 It was the only variation of note found in any of the genes examined. DNA samples from the parents were not available for analysis.
Discussion
Deletions and loss-of-function mutations of the PTCH1 gene have been reported in patients with NBCCS and other diverse tumors.12 13 14 15
This condition can be traced back to 2,000 BC through studies in ancient paintings from Pompeii and Herculaneum and in Egyptian mummies with related congenital skeletal anomalies.16 17 Variable clinical expression of many Mendelian disorders including NBCCS syndrome and HPE have been credited to multiple genetic hits or environmental exposures.10 18
Mutations in the SHH signaling network genes mainly result in HPE19; however, causal mechanisms that drives at-risk individuals to manifest the phenotype remain unknown to date.20 Relatives presenting with identical mutations can have differing phenotypes including abnormalities of the pituitary gland and corpus callosum, eye colobomas, microphthalmia, choanal aperture stenosis, solitary median maxillary central incisor, “false” upper lip midline cleft, and isolated cleft lip.2 21 22 23 24 Phenotypic variability led some authors to suggest that multiple genetic and environmental agents could regulate the severity of the manifestations and that multiple hits may influence the severity of the phenotype.10
The correlation between the SHH pathway, PTCH1 mutations, and HPE in humans was well established through the report of five unrelated patients with four different missense mutations in PTCH1. Phenotypic variability was remarkable in relatives of these patients who had the same mutation, ranging from normal phenotype (the father of patient 180) to typical HPE-like phenotype (brother of patient 1,335). It was suggested that mutations in the large extracellular loops could affect the ability of PTCH1 to bind SHH and that mutations in the intracellular loops could perturb interactions of the intracellular domains of PTCH1 with Smo or other proteins involved in SHH transduction.21 25 Reports on neural tube defects (NTDs) in patients with NBCCS suggest that PTCH1 could be an etiological factor for NTDs in humans.26
Involvement of eye structures in patients with NBCCS has been reported to be similar to those exhibited in mice and Drosophila melanogaster with patched mutations.27 28 Evidence linking ocular developmental defects with PTCH1 have been previously reported in patients with NBCCS, microphthalmia, and orbital cysts.6 7 24 The association of these findings with HPE in our patient reinforces the clinical heterogeneity observed in patients with PTCH1 mutations; however, the basis for the underlying patterning problems in these mutations is not known and further research is necessary to elucidate the mechanisms involved. In the present case, consanguinity could be responsible for a second recessive condition leading to the extreme severity of the clinical picture observed in the patient.
Acknowledgments
This study was supported in part by a grant from the CNPq (grant nos. 470996/2006–4 and 301926/2007–7) to author A. R.-C. The authors are indebted to Dr. Jeff Murray and the laboratory staff.
References
- 1.Cohen M M Jr. The hedgehog signaling network. Am J Med Genet A. 2003;123A(1):5–28. doi: 10.1002/ajmg.a.20495. [DOI] [PubMed] [Google Scholar]
- 2.Marini M, Cusano R, De Biasio P. et al. Previously undescribed nonsense mutation in SHH caused autosomal dominant holoprosencephaly with wide intrafamilial variability. Am J Med Genet A. 2003;117A(2):112–115. doi: 10.1002/ajmg.a.10163. [DOI] [PubMed] [Google Scholar]
- 3.Hahn J S, Plawner L L. Evaluation and management of children with holoprosencephaly. Pediatr Neurol. 2004;31(2):79–88. doi: 10.1016/j.pediatrneurol.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Rahimov F, Ribeiro L A, de Miranda E, Richieri-Costa A, Murray J C. GLI2 mutations: phenotypical manifestations in four Brazilian patients. How wide is the phenotype? Am J Med Genet A. 2006;140:2571–2576. doi: 10.1002/ajmg.a.31370. [DOI] [PubMed] [Google Scholar]
- 5.OMIM – Online Mendelian Inheritance in Man An Online Catalog of Human Genes and Genetic Disorders Available at http://www.omim.org/entry/109400. Accessed January 21, 2016
- 6.Ragge N K, Salt A, Collin J R, Michalski A, Farndon P A. Gorlin syndrome: the PTCH gene links ocular developmental defects and tumour formation. Br J Ophthalmol. 2005;89(8):988–991. doi: 10.1136/bjo.2004.061390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rodrigues A L, Carvalho A, Cabral R. et al. Multiple nevoid basal cell carcinoma syndrome associated with congenital orbital teratoma, caused by a PTCH1 frameshift mutation. Genet Mol Res. 2014;13(3):5654–5663. doi: 10.4238/2014.July.25.21. [DOI] [PubMed] [Google Scholar]
- 8.Derwińska K, Smyk M, Cooper M L, Bader P, Cheung S W, Stankiewicz P. PTCH1 duplication in a family with microcephaly and mild developmental delay. Eur J Hum Genet. 2009;17(2):267–271. doi: 10.1038/ejhg.2008.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ribeiro L A, Murray J C, Richieri-Costa A. PTCH mutations in four Brazilian patients with holoprosencephaly and in one with holoprosencephaly-like features and normal MRI. Am J Med Genet A. 2006;140(23):2584–2586. doi: 10.1002/ajmg.a.31369. [DOI] [PubMed] [Google Scholar]
- 10.Ming J E, Muenke M. Multiple hits during early embryonic development: digenic diseases and holoprosencephaly. Am J Hum Genet. 2002;71(5):1017–1032. doi: 10.1086/344412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mansilla M A, Cooper M E, Goldstein T. et al. Contributions of PTCH gene variants to isolated cleft lip and palate. Cleft Palate Craniofac J. 2006;43(1):21–29. doi: 10.1597/04-169R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindström E, Shimokawa T, Toftgård R, Zaphiropoulos P G. PTCH mutations: distribution and analyses. Hum Mutat. 2006;27(3):215–219. doi: 10.1002/humu.20296. [DOI] [PubMed] [Google Scholar]
- 13.Tostar U, Malm C J, Meis-Kindblom J M, Kindblom L G, Toftgård R, Undén A B. Deregulation of the hedgehog signalling pathway: a possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J Pathol. 2006;208(1):17–25. doi: 10.1002/path.1882. [DOI] [PubMed] [Google Scholar]
- 14.Larsen A K, Mikkelsen D B, Hertz J M, Bygum A. Manifestations of Gorlin-Goltz syndrome. Dan Med J. 2014;61(5):A4829. [PubMed] [Google Scholar]
- 15.Chung J H, Bunz F. A loss-of-function mutation in PTCH1 suggests a role for autocrine hedgehog signaling in colorectal tumorigenesis. Oncotarget. 2013;4(12):2208–2211. doi: 10.18632/oncotarget.1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ponti G, Manfredini M, Ruini C. Wall paintings facies and their possible genetic correlates in the ancient Pompeii: a bio-anthropologic message from the past? Gene. 2016;589(2):151–156. doi: 10.1016/j.gene.2016.04.038. [DOI] [PubMed] [Google Scholar]
- 17.Ponti G, Pellacani G, Tomasi A, Sammaria G, Manfredini M. Skeletal stigmata as keys to access to the composite and ancient Gorlin-Goltz syndrome history: the Egypt, Pompeii and Herculaneum lessons. Gene. 2016;589(2):104–111. doi: 10.1016/j.gene.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 18.Levanat S, Gorlin R J, Fallet S, Johnson D R, Fantasia J E, Bale A E. A two-hit model for developmental defects in Gorlin syndrome. Nat Genet. 1996;12(1):85–87. doi: 10.1038/ng0196-85. [DOI] [PubMed] [Google Scholar]
- 19.Dubourg C, Lazaro L, Pasquier L. et al. Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: mutation review and genotype-phenotype correlations. Hum Mutat. 2004;24(1):43–51. doi: 10.1002/humu.20056. [DOI] [PubMed] [Google Scholar]
- 20.Petryk A, Graf D, Marcucio R. Holoprosencephaly: signaling interactions between the brain and the face, the environment and the genes, and the phenotypic variability in animal models and humans. Wiley Interdiscip Rev Dev Biol. 2015;4(1):17–32. doi: 10.1002/wdev.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ming J E, Roessler E, Muenke M. Human developmental disorders and the Sonic hedgehog pathway. Mol Med Today. 1998;4(8):343–349. doi: 10.1016/s1357-4310(98)01299-4. [DOI] [PubMed] [Google Scholar]
- 22.Hehr U, Gross C, Diebold U. et al. Wide phenotypic variability in families with holoprosencephaly and a sonic hedgehog mutation. Eur J Pediatr. 2004;163(7):347–352. doi: 10.1007/s00431-004-1459-0. [DOI] [PubMed] [Google Scholar]
- 23.Lazaro L, Dubourg C, Pasquier L. et al. Phenotypic and molecular variability of the holoprosencephalic spectrum. Am J Med Genet A. 2004;129A(1):21–24. doi: 10.1002/ajmg.a.30110. [DOI] [PubMed] [Google Scholar]
- 24.Chassaing N, Davis E E, McKnight K L. et al. Targeted resequencing identifies PTCH1 as a major contributor to ocular developmental anomalies and extends the SOX2 regulatory network. Genome Res. 2016;26(4):474–485. doi: 10.1101/gr.196048.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ming J E, Kaupas M E, Roessler E. et al. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. 2002;110(4):297–301. doi: 10.1007/s00439-002-0695-5. [DOI] [PubMed] [Google Scholar]
- 26.Roudgari H, Farndon P A, Murray A D, Hardy C, Miedzybrodzka Z. Is PATCHED an important candidate gene for neural tube defects? Cranial and thoracic neural tube defects in a family with Gorlin syndrome: a case report. Clin Genet. 2012;82(1):71–76. doi: 10.1111/j.1399-0004.2011.01725.x. [DOI] [PubMed] [Google Scholar]
- 27.Thomas C, Ingham P W. Hedgehog signaling in the Drosophila eye and head: an analysis of the effects of different patched trans-heterozygotes. Genetics. 2003;165(4):1915–1928. doi: 10.1093/genetics/165.4.1915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moshiri A, Reh T A. Persistent progenitors at the retinal margin of ptc+/- mice. J Neurosci. 2004;24(1):229–237. doi: 10.1523/JNEUROSCI.2980-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]