

Abstract
Few kinases have been studied as extensively as protein kinase C (PKC), particularly in the context of cancer. As major cellular targets for the phorbol ester tumor promoters and diacylglycerol (DAG), a second messenger generated by stimulation of membrane receptors, PKC isozymes play major roles in the control of signaling pathways associated with proliferation, migration, invasion, tumorigenesis, and metastasis. However, despite decades of research, fundamental questions remain to be answered or are the subject of intense controversy. Primary among these unresolved issues are the role of PKC isozymes as either tumor promoter or tumor suppressor kinases and the incomplete understanding on isozyme-specific substrates and effectors. The involvement of PKC isozymes in cancer progression needs to be reassessed in the context of specific oncogenic and tumor suppressing alterations. In addition, there are still major hurdles in addressing isozyme-specific function due to the limited specificity of most pharmacological PKC modulators and the lack of validated predictive biomarkers for response, which impacts the translation of these agents to the clinic. In this review we focus on key controversial issues and upcoming challenges, with the expectation that understanding the intricacies of PKC function will help fulfill the yet unsuccessful promise of targeting PKCs for cancer therapeutics.
Keywords: Protein kinase C, phorbol esters, tumor promoter, tumor suppressor, oncogene, cancer therapy
INTRODUCTION
It has been nearly 40 years since the discovery of protein kinase C (PKC), originally identified as a calcium-activated, phospholipid-dependent protein kinase by Nishizuka and co-workers, and established as a cellular receptor for the phorbol ester tumor promoters primarily by Blumberg and colleagues [1–5]. Not surprisingly, PKC represents one of the most extensively studied kinases, with >60,000 citations in PubMed and >10,000 citations associated with cancer. With such an extensive information on PKC regulation and function, it is not unexpected that significant controversies exist in the field. It is not possible to be dogmatic when it comes to defining roles for PKC isozymes in cancer progression, largely due to the fact that these kinases are pleiotropic regulators of cellular function and display complex regulatory mechanisms of activation.
The utmost complexity in the field results from the fact that PKC is a family of highly related isozymes; however, they display obvious (and not so obvious) structural differences that confer unique regulatory and intracellular localization properties. Human PKCs comprise 10 different Ser-Thr kinases, product of nine different genes. Based on their distinctive regulation, they have been classified into “classical/conventional” or calcium-dependent cPKCs (α, βI, βII, and γ), “novel” or calcium-independent nPKCs (δ, ε, η, and θ), and “atypical” aPKCs (ζ and ι) [6, 7] (Figure 1). Phorbol esters, natural compounds that mimic the action of the lipid second messenger diacylglycerol (DAG), bind with high affinity to cPKCs and nPKCs, but not to aPKCs, thus mirroring a scenario of stimulation of receptors (either G-protein-coupled receptors or tyrosine-kinases) coupled to DAG generation [4]. The general assumption is that upon phorbol ester activation, all cPKCs and nPKCs present in a given cell will be allosterically activated and phosphorylate specific substrates. Phorbol esters cause long-lasting activation of PKCs, therefore, cellular responses may be different than those observed by physiologically generated DAG. Of note, the distinctive pattern of substrate phosphorylation, signaling pathway activation and gene expression regulation caused by activation of individual PKCs is responsible for the vast range of cellular responses. The challenge over the years has been to disentangle the intricate network of signaling effectors for each PKC isozyme, which is a massive effort taking into consideration the distinctive patterns of expression and complex regulatory mechanisms governing PKC activation in individual cell types [6, 8].
Figure 1. Protein families with phorbol ester/DAG responsive C1 domains.

Diagram of protein families with C1 domains that bind phorbol esters and diacylglycerol (DAG). DAG is generated upon stimulation of membrane receptors such as tyrosine-kinase and G-protein-coupled receptors. C1, C1 domain; C2, C2 domain; PS, pseudosubstrate domain; PH, pleckstrin homology domain; EF, EF hands; SH2, Src-homology 2 domain; Rac-GAP, Rac GTPase Activating Protein domain; REM, Ras Exchanger Motif; RasGEF, Ras Guanine nucleotide Exchange Factor domain. Note that the figure depicts representative structures for each class, but certain members lack some of these domains or may contain additional domains not depicted here.
Based on the complexities described above, in this review we will focus on the most puzzling questions to be addressed in the field of PKC and cancer. Focusing on these key controversial matters will allow researchers to assess the roles of these kinases in malignant transformation, tumorigenesis and metastasis. Understanding the complexities of PKC regulation and function will accelerate the possibility of targeting discrete members of the PKC family for therapeutic purposes.
IS PKC A TUMOR PROMOTER OR SUPPRESSOR KINASE?
In the context of a cancer cell, activation of PKCs impacts on a number of responses, including proliferation, survival, and motility, ultimately influencing phenotypes associated with tumor progression and metastasis (Figure 2). The first hint that linked PKC with tumor promotion was its identification as a cellular receptor for phorbol esters. These natural compounds, originally isolated as components from croton oil, lack carcinogenic activity, but cause the appearance of skin tumors when applied chronically after a limited dose of an initiating carcinogen agent [9, 10]. Years later, biochemical studies established that phorbol esters bind with high affinity to PKC and that DAG binds to the same site in the protein, a 50 amino acid region known as the C1 domain [1, 4, 11].
Figure 2. PKC isozymes as regulators of cellular function and cancer progression.
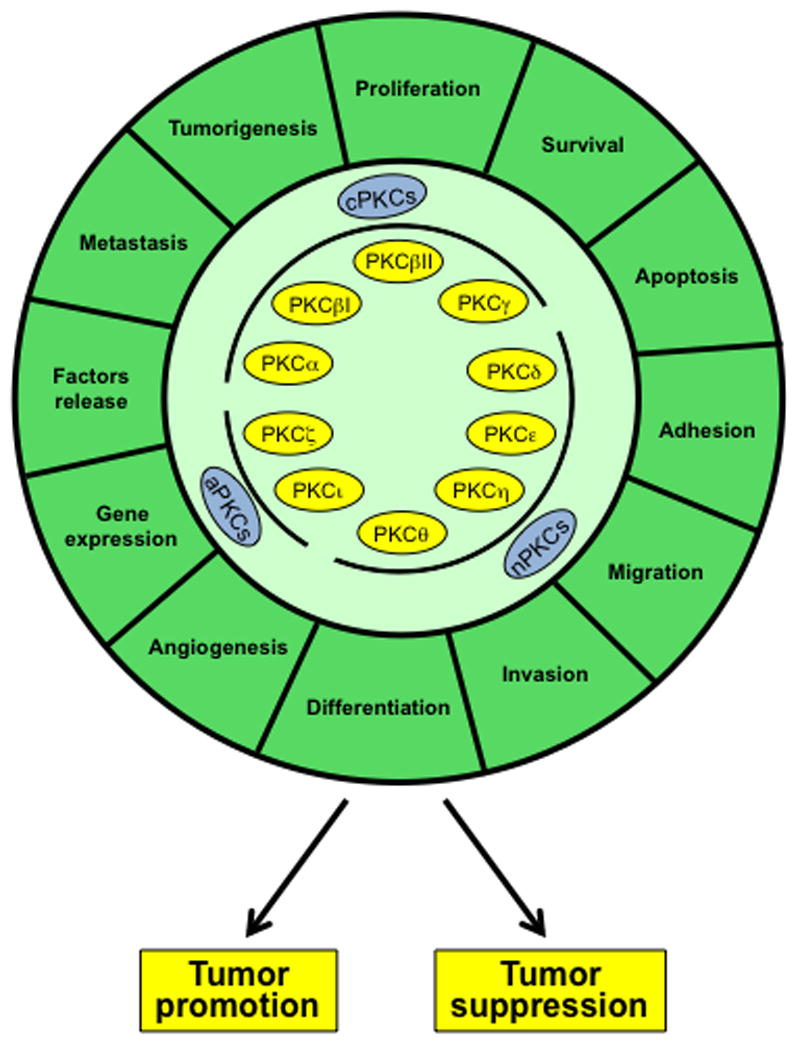
PKCs control multiple functions associated with cancer progression, in many cases in opposite manners. Depending on the context, PKCs can act either as promoters or suppressors of the cancer phenotype.
Whereas initial studies showed that phorbol esters elicit mitogenic responses via PKC activation, contrasting effects were found in a number of cellular models. Actually, phorbol esters could either stimulate or inhibit proliferation, or even trigger an apoptotic response depending on the cellular context [6]. In this regard, it has been established that PKCδ mostly operates as an anti-proliferative kinase that negatively regulates cell cycle progression. Early studies in CHO cells overexpressing PKCδ showed significant accumulation of cells in G2/M and inhibition of cell division in response to phorbol 12-myristate 13-acetate (PMA or TPA) [12]. PKCδ also mediates G1 phase arrest via induction of the cell cycle inhibitor p21 [13]. In addition, PKCδ can be proteolytically activated by caspases and is required for the death effect of apoptotic agents and chemotherapeutic drugs, although this PKC can also mediate apoptotic responses in the absence of proteolytic cleavage [14–16]. Not surprisingly, transgenic overexpression of PKCδ in the mouse skin protects against the tumor promoting effect of phorbol esters [17]. This contrasts with the prominent pro-survival roles for PKCδ and its reported ability to confer growth, survival responses, and a more aggressive phenotype in several cell types. Adding to this controversy, PKCδ has tumor promoting activity in mammary models and pro-migratory/invasive activity in breast cancer, pancreatic cancer, and melanoma models [8, 18, 19]. A similarly complex scenario has been reported for PKCα, which has been shown to promote mitogenic responses in a number of cell types but is also capable of mediating cell cycle arrest programs in others such as the intestinal epithelium, where it strongly represses cyclin D1 translation [8, 20].
While the tumor-promoting activity of phorbol esters is well established, is there any significant evidence that PKC isozymes could act as oncogenes? The first reported example of an oncogenic PKC was the novel PKCε isoform. Early studies in fibroblasts and colonic epithelial cells found that PKCε overexpression has transforming activity, as judged by its ability to confer anchorage-independent growth and tumorigenic potential upon inoculation of cells into nude mice [21, 22]. Nevertheless, other than these few examples it became clear that PKCε fails to transform most cell lines in culture. Oncogenic mutations for PKCε have not been reported in cancer, however there is evidence that this kinase is up-regulated in a number of cancers, particularly epithelial tumors. For example, PKCε overexpression has been described in the majority of primary tumors from invasive ductal breast cancer and non-small cell lung cancer (NSCLC) patients [23, 24]. In prostate cancer, PKCε levels are markedly elevated compared to benign prostatic epithelia, and its overexpression has been associated with disease recurrence [25–27]. Studies in mice showed that PKCε overexpression confers a hyperplastic phenotype but is not sufficient to lead to full-blown cancer. For example, prostate-specific PKCε transgenic mice develop prostatic intraepithelial neoplasia (PIN), but these lesions do not progress to cancer [28]. Transgenic overexpression of PKCε in the mouse skin enhances carcinoma formation after tumor promotion and sensitizes skin to the development of squamous cell carcinoma by UVR, which occurs by transducting signals that inhibit apoptosis and enhance proliferation of preneoplastic cells [29]. Thus, whereas existing data precludes us to unambiguously define PKCε as an oncogene, accumulating evidence supports a “pro-oncogenic” role for this kinase. It is possible that PKCε cooperates with other alterations to promote progression to malignancy, a subject of intense research. At a mechanistic level, PKCε-driven phenotypes may be the consequence of the synergistic activation of pathways associated with cell survival and mitogenesis, including PI3K/Akt, Erk, Stat3, and NF-κB [8]. PKCε is also known to cooperate with Ras/Raf-1 [22, 30]. Enhanced DAG production caused by oncogenic stimuli, such as K-Ras [31], may funnel signals primarily through PKCε when it is overexpressed, shifting the balance towards the activation of growth/survival pathways. Whether PKCε is hyperactivated in cancer cells has not been easy to address, mainly due to the lack of reliable tools capable of detecting the activated status of the kinase. One scenario that has yet to be surveyed is the potential contribution of PKCε overexpression to a pro-tumorigenic microenvironment. Since early reports linked PKCε oncogenic activity to the release of TGF-β, and later with the production of a number of cytokines and pro-inflammatory factors [29, 32, 33], overexpressed PKCε could be envisioned as a major component of autonomous oncogenic autocrine loops or a driver of paracrine interactions with cells from the tumor microenvironment. NF-κB, a crucial pathway for regulating the expression of inflammatory cytokines, is a known effector for PKCε, and PKCε stimulates the expression of pro-inflammatory and angiogenic genes, namely COX2, VEGF, and others [8, 34].
In addition to the multiple functional associations between phorbol ester/DAG responsive PKC isozymes and cancer phenotypes, the atypical PKCι isoform has been extensively characterized as an oncogenic kinase. PKCι overexpression is observed in human pancreatic, colon, lung, prostate, breast and ovarian cancer [35]. In lung squamous cell carcinoma, the PKCι gene (PRKCI) co-amplifies with SOX2, resulting in a cooperation that is required for a stem cell-like phenotype [36]. PKCι is an essential mediator of oncogenic K-Ras and is required for the maintenance of the tumor-initiating cell phenotype. Moreover, in ovarian cancer, genetic disruption of PKCι inhibits the clonal expansion, anchorage-independent growth, and tumorigenic properties of tumor initiating cells [37, 38].
As indicated above, PKCs, particularly PKCα and PKCδ, have prominent roles as anti-proliferative and pro-apoptotic kinases in several models [8]. However, can we truly define PKC isozymes as bona-fide tumor suppressors? There have been numerous expression analysis studies using cancer specimens that reported decreased expression of PKCs, including isozymes with growth-inhibitory activity, however causal relationships with disease progression have not been always established. Interestingly, studies using knock-out mice assigned tumor suppressing roles for PKCα, PKCδ, and PKCζ [39–41]. One notable example has been described for PKCα deficient mice, which show elevated tendency to develop spontaneous intestinal tumors. In addition, ApcMin/+ mice show enhanced intestinal tumorigenesis in a PKCα-null background [42].
Recently, a comprehensive study of cancer associated mutations in PKCs has been reported by Newton, Brognard and others [43]. More than 500 mutations within cPKCs, nPKCs and aPKCs, in most cases heterozygous, have been identified in diverse cancers. Functional studies revealed that they are inactivating mutations in most cases, thus highlighting their potential tumor suppressive roles. This was well established for a mutated PKCβ, but still needs to be formally demonstrated for the other mutations present in that PKC and in other PKC isoforms. Interestingly, the PKCβ mutant reduces the expression of PKCα, suggesting that cross-talks between PKCs may also contribute to the tumor suppressing phenotype. Bioinformatics analysis revealed that loss-of-function mutations in PKC isozymes possibly cooperate with co-occurring mutations in defined oncogenes and tumor suppressors known to be regulated by PKC. Other study found a cancer associated loss-of-function mutation in a substrate-specific recruitment motif of PKCι [44], arguing that this oncogenic kinase could also have a tumor suppressing role in some settings. The real contribution of PKC mutations as drivers of tumor formation and their relevance in the context of other oncogenic/tumor suppressor stimuli will be a subject of intense research and debate in the coming years.
WHAT ARE THE PKC SUBSTRATES RELEVANT FOR CELLULAR RESPONSES?
Identifying PKC substrates is of manifest importance, as it will provide a better picture of the signaling effectors and responses by which PKC isozymes relay information upon activation. Potentially, phosphorylated substrates could be used as pharmacological targets, biomarkers for disease progression, or ultimately as tools to monitor therapeutic response for pharmacological PKC modulators. PKC isozymes are very promiscuous kinases that phosphorylate Ser and Thr flanked by basic residues. In addition to the potential overlapping of PKC phosphorylation sites with phosphorylation consensus for other kinases, a major problem is that a vast majority of proteins in the cell contains putative PKC phosphorylation sites, making the identification of physiologically relevant PKC substrates a challenging undertaking. It would be burdensome to identify PKC substrates upon phorbol ester treatment after 32P labeling [45], and most likely these will include substrates of downstream kinases activated by PKC. Optimal substrate phosphorylation motifs for each PKC isozymes have been determined using oriented peptide libraries by Cantley and coworkers [46]. Nonetheless, studies using pseudosubstrate-derived peptides revealed significant overlapping in substrate recognition for individual PKCs [47], arguing that selectivity within the intracellular milieu is dictated by preferential access to substrates upon differential relocalization of each PKC. Given this complexity, it is anticipated that PKC isozymes “talk” to a multitude of signaling pathways and activate multiple gene expression networks. When assessing PKC substrates, another consideration is that long-lasting phorbol ester responses do not necessarily recapitulate the short-term PKC activation caused by DAG physiologically generated upon receptor activation. Therefore, the nature of the PKC phosphorylated substrates and magnitude of phosphorylated responses may differ depending on the stimuli.
Given the fact that PKCs phosphorylate a wide-range of cellular proteins, how can we dissect such complex web of isozyme specific substrates? One potential strategy would entail the use of the chemical genetics approach developed by the Shokat laboratory. This screening method involves engineering a mutant kinase in which a “gatekeeper” large hydrophobic group within the ATP binding pocket is replaced by a small residue, thus allowing the kinase to accept a bulky ATP analogue. Only the mutant kinase, but not the wild-type kinase, can accommodate the bulky analogue, thus it could be used as a traceable kinase to map the phosphorylated products. Despite the usefulness of this method for identifying substrates for kinases such as Src, JNK, and ERK2 [48–50], so far there have been few attempts for PKCs. For example, Rotenberg and colleagues used this approach to identify PKC substrates in mammary cells that co-immunoprecipitate with traceable PKCα, PKCδ, and PKCζ, and established α6-tubulin as an intracellular PKCα substrate [51, 52]. Another example is the identification of PKCδ substrates in neutrophils [53]. As individual PKCs localize to specific intracellular compartments upon activation, the characterization of PKC isozyme specific phosphoproteomes could be done in principle using traceable kinases targeted to discrete intracellular compartments. Once a phosphosubstrate is identified, its phosphorylation state could be used as a read-out for PKC activity in cellular models or tumor specimens by staining with specific phospho-antibodies. Another approach would entail the use of immunoprecipitation followed by detection with an antibody that recognizes the phosphorylated PKC substrate consensus sequence.
Another successful approach to identify PKC substrates was Stable Isotope Labeling with Amino acid in Cell culture (SILAC)-based quantitative phosphoproteomics. A study designed to investigate PKCδ substrates implicated in cell death in systematic and dynamic manners revealed a series of candidate effectors of a cleaved kinase mutant. This analysis also revealed a network of phosphorylations by PKCδ downstream kinases [54]. Phosphoproteomics profiling has been also used to identify nuclear PKCβI substrates in embryonic stem cells [55]. Combining these laborious approaches with appropriate functional analysis may provide highly valuable information on isozyme-specific signaling networks and clues for potential effectors and biomarkers. Furthermore, it is also important to consider that the nature of substrates will differ depending on the cell type, thus the expectation is that prominent differences in phosphorylation would be expected between normal and transformed cells.
WHY IS GENERATING ISOZYME SPECIFIC PKC MODULATORS SO CHALLENGING?
At a structural level, PKC isozymes have two well defined regions, an N-terminal regulatory region comprising the C1 and C2 domains, and the C-terminal catalytic region responsible for ATP binding and phosphotransferase activity. The C1 domain, site for phorbol ester/DAG binding, is duplicated in tandem (C1a and C1b) in cPKCs and nPKCs [56]. DAG (or phorbol ester) binding to the C1 domain is a crucial step in the allosteric activation of PKCs. The PKCδ C1b domain structure, solved by X-ray crystalography in 1994 [57], showed that phorbol esters insert into a narrow groove between two pulled-apart β strands at one tip of the domain, which creates a contiguous hydrophobic surface that is key for the association with the plasma membrane. The single C1 domain present in aPKCs retains the overall conformation of phorbol ester-responsive C1 domains, however it possesses residues that interfere with the formation of the C1 domain-ligand-membrane complex [58, 59]. The C2 domain in cPKCs binds calcium, whereas this domain in nPKCs is primarily a calcium-unresponsive phospholipid-binding module [6, 7]. Active PKCs form a complex with lipid, ligand and proteins through the regulatory domain, thus providing substantial opportunities for pharmacological targeting.
As with most kinases, generation of highly selective inhibitors for PKC isozymes became a challenge. The greatest degree of sequence homology and structural resemblance among PKC isozymes is in the catalytic domain, thus designing inhibitors capable of discriminating among individual members of the PKC family has been exceedingly problematic. Still, relatively potent PKC inhibitors directed against the ATP-binding pocket in the catalytic domain have been identified. However, despite some exceptions, most catalytic inhibitors display little or no selectivity among the different members of the PKC family. Most common inhibitors are summarized in Table 1, and also described elsewhere [7, 60].
Table 1.
Most common classes of PKC inhibitors
| Class | Examples | PKC isozyme selectivity | Other targets | Potential as anticancer agents |
|---|---|---|---|---|
| ATP-competitive inhibitors | Bisindolylmaleimide I | Non-selective | Other kinases | Yes |
| Gö6976 | cPKCs | |||
| Enzastaurin | Some selectivity for PKCβ | |||
| C1 domain ligands | Bryostatin 1 | cPKC/nPKC activator in vitro, functional inhibitor in cells/in vivo | “Non-PKC” phorbol ester receptors (PKDs, RasGRPs, chimaerins, others) | Yes |
| Protein-protein interaction inhibitors | Aurothiomalate | PKCι (inhibits interaction with Par6) | Unknown | Yes |
| εV1-2 | PKCε (inhibits interaction with RACK) | Yes | ||
| Substrate-competitive inhibitors | Pseudosubstrate-derived peptides | Limited | Unknown | No |
| PIF-pocket inhibitors | PS315 | Unknown | Unknown | Unknown |
Targeting autoinhibitory domains is a promising strategy to develop selective PKC inhibitors. Peptides designed to target the PKC pseudosubstrate domain implicated in autoinhibition have been widely used; however, the prediction is that based on the limited recognition selectivity of pseudosubstrate derived peptides by individual PKCs [7, 47], these agents might lack specificity. Studies using pseudosubstrates inhibitors may have to be reinterpreted with specific tools or molecular/genetic approaches. It is important to highlight a well documented example of PKCζ allosteric inhibition by targeting a motif known as the PIF-pocket, known to interact intramolecularly with the C1 domain [61, 62]. It seems that, at least for this aPKC, autoinhibition cannot be solely explained by a pseudosubstrate mechanism, thus one may envision targeting other autoinhibitory interactions as means for the development of selective PKC modulatory agents.
A successful approach to target PKC in an isozyme-specific manner has been developed by the laboratory of Daria Mochly-Rosen. Peptide modulators have been generated that inhibit protein-protein interactions crucial for directing PKC translocation to intracellular compartments. The rationale behind this methodology is that PKC isozymes interact with anchoring proteins, such as RACKs, that selectively bind to PKCs in an active state. Peptide derived from various regions in PKC, such as the C2 domain, can prevent the interaction with RACKs and inhibit PKCs in an isozyme-selective manner [60]. Notably, one of these peptides that targets the pro-oncogenic PKCε (εV1-2), has anti-tumorigenic activity in xenograft models of non-small lung cancer and impairs lung cancer cell motility signaling [63, 64]. Through a completely different mechanism, aurothiomalate, a compound that disrupts the interaction between PKCι and Par6, blocks transformed growth in models where this aPKC has prominent roles [65].
When it comes to selectivity for activation of PKC isozymes, there are also major challenges ahead. The first comprehensive characterization of C1 domain ligands showed that DAG has essentially the same binding affinity for all cPKCs and nPKCs. However, significant differences could be observed for non-physiological C1 domain ligands. Most notably, under limiting calcium conditions, phorbol esters, 12-deoxyphorbol esters, and most prominently the ligands mezerein and thymeleatoxin, display significantly lower affinities for nPKCs relative to cPKCs [47]. Some of these ligands have significant potential as isozyme-selective tools in cellular models; however, this has not been fully exploited despite the vast heterogeneity in their responses, including the perplexing fact that some display anti-tumor promoter activity in mouse models [66, 67].
One of the known complexities in C1 domain pharmacology is the non-equivalency of C1a and C1b domains. There have been a number of studies underscoring different ligand recognition properties of individual C1 domains and selective relocalization to distinctive intracellular compartments [56, 68–70]. Whereas the mechanistic basis for the non-equivalency is not well understood, one hypothesis is that the unique lipid composition of internal cellular membranes contributes to the differential C1 domain interactions. Notably, C1 domains can act as protein interacting modules, and protein-protein interactions via the C1 domain can be critical drivers of localization and function. C1 domain interacting proteins include the ER-Golgi protein Tmp21-I, the cell matrix protein fascin, the centrosome protein pericentrin, and the small G-protein RhoA [56, 69–73].
The most comprehensive effort to rationally design C1 domain ligands comes from the combined efforts of the Blumberg, Marquez, and Lee laboratories. Their approach involves the generation of five-membered ring DAG lactones obtained by constraining the glycerol backbone. The rationale behind this approach is that the low binding affinity of DAG relative to phorbol esters was attributable to the flexibility of the glycerol backbone, thus cyclization of the structure would reduce the entropic penalty associated with DAG binding. By modifying DAG lactones through incorporation of different branched hydrophobic chains it has been possible to generate ligands with preferential affinity for discrete PKCs or “non-PKC” phorbol ester receptors (see below) [74–77]. Although DAG lactones have not yet been fully characterized in a physiological setting, the expectation is that they could be rationally designed to dissect isozyme-specific responses.
DO “NON-PKC” PHORBOL ESTER/DAG RECEPTORS CONTRIBUTE TO CANCER PROGRESSION?
An important concept that emerged more than two decades ago is that proteins with C1 domains but distinctive from PKCs are also capable of binding DAG and phorbol esters with high affinity (Table 2). One of these families of phorbol ester/DAG receptors, protein kinase D (PKD), is structurally related to PKC, with duplicated C1 domains, whereas others are totally unrelated to PKC. This last group includes lipid kinases (diacylglycerol kinases or DGKs), guanine nucleotide exchange factors for small GTPases (RasGRPs), GTPase activating proteins (chimaerin Rac-GAPs), and components of the synaptic vesicle fusion protein complex (Munc-13 isoforms) (see Figure 1). In vitro binding assays determined that they bind phorbol esters with high affinity in a phospholipid-dependent manner, and respond to phorbol esters in cellular models [6, 78–82]. Understanding the relative contribution of “non-PKC” phorbol ester receptors to cell physiology (i.e., whether they can be activated by receptors coupled to DAG generation) and disease has been a major undertaking. Understandably, it is reasonable to question whether phorbol ester responses could be mediated, at least in part, by proteins other than PKCs.
Table 2.
“Non-PKC” phorbol ester/DAG receptors in cancer
| Class | Isotypes | Phorbol ester binding* | Activity | Roles in cancer |
|---|---|---|---|---|
|
| ||||
| Protein kinase D | PKD1 | + | Protein kinases | Diverse roles in promotion or suppression of tumorigenesis and metastasis, primarily in solid tumors |
| PKD2 | + | |||
| PKD3 | + | |||
|
| ||||
| RasGRP | RasGRP1 | + | Ras/Rap1 Guanine nucleotide Exchange Factors (GEFs) | Diverse roles in oncogenesis of blood cancers and solid tumors |
| RasGRP2 | − | |||
| RasGRP3 | + | |||
| RasGRP4 | + | |||
|
| ||||
| Chimaerin | α1-chimaerin | + | Rac GTPase Activating Proteins (GAPs) | Tumor suppression in glioma and breast cancer |
| α2-chimaerin | + | |||
| β1-chimaerin | + | |||
| β2-chimaerin | + | |||
| β3-chimaerin | + | |||
|
| ||||
| Munc-13 | Munc-13-1 | + | Coordination of vesicle docking, priming and fusion | Unknown |
| Munc-13-2 | ? | |||
| Munc-13-3 | ? | |||
| Munc-13-4 | ? | |||
|
| ||||
| Diacylglycerol-kinase | DGKα | − | Lipid kinases | Limited knowledge for phorbol ester-responsive DGKs |
| DGKβ | + | |||
| DGKγ | + | |||
| DGKδ | − | |||
| DGKε | − | |||
| DGKη | − | |||
| DGKκ | − | |||
| DGKθ | − | |||
| DGKι | − | |||
| DGKζ | − | |||
Not all DGKs have been examined for phorbol ester binding, but structural predictions on C1 domains suggest that other tan DGKβ and DGKγ all other DGKs should be insensitive to phorbol esters.
Ras-GRPs are multidomain proteins with a single C1 domain that catalyze GDP to GTP exchange in Ras. Among the four members of the family, only RasGRP2 (which is also a Rap nucleotide exchange factor) fails to bind phorbol esters, but all other members (RasGRP1, RasGRP3, and RasGRP4) are high affinity phorbol ester receptors [82–84]. DAG exerts a dual role in RasGRP activation, directly via the RasGRP C1 domain and indirectly through PKC-mediated RasGRP phosphorylation [85]. RasGRPs express in different subsets of hematopoietic lineages, with RasGRP1 primarily expressing in T cells, RasGRP3 in B cells, T cells and macrophages, and RasGRP4 expressed in mast cells, neutrophils and thymocytes. RasGRPs are prominent effectors of T cell (RasGRP1) and B cell (RasGRP3) receptor stimulation and mediate Ras signaling activation [86]. Notably, RasGRP1 plays important roles in blood cancers, most notably RasGRP1 in T cell acute lymphoblastic leukemia/lymphoma (T-ALL) [86, 87]. Elevated levels of RasGRP1 as well as oncogenic K-Ras mutations seem to be mutually exclusive events that both contribute, albeit mechanistic differences, to T-ALL development [86, 88]. In addition, there is growing evidence that RasGRPs contribute to the pathogenesis of B cell lymphoma and acute myeloid leukemia (AML) [86]. Given the emerging role of immune cells in cancer development and targeted therapy, it would be important to examine the potential involvement of RasGRPs in this context. With regard to epithelial cancers, Patricia Lorenzo’s group established a role for RasGRP1 in non-melanoma skin cancer. Elevated RasGRP1 levels in keratinocytes results in Ras activation, an effect that is further augmented by phorbol ester treatment. Moreover, using mouse models, this group found that RasGRP1 contributes to skin tumor progression [89, 90]. RasGRP3 is up-regulated in prostate tumors, and experiments using cell lines suggest that it contributes to growth and survival [91]. Functions of RasGRP3 in the progression of other cancers, including melanoma, glioma, and breast cancer, have been suggested [86, 92, 93], but causal relationships with disease progression need to be investigated.
Chimaerins are a family of Rac-GAP proteins, thereby they accelerate GTP hydrolysis from the small G-protein Rac, a major player in cancer cell motility. The chimaerin family comprises at least five members: α1-, α2-, β1-, β2-, and β3- chimaerin [6, 94, 95]. These Rac-GAPs bind phorbol esters in vitro with high affinity, and they can be activated by receptors coupled to DAG generation, such as tyrosine-kinase receptors, therefore redistributing to the plasma membrane where they bind to and inactivate its partner Rac. For example, EGF promotes translocation of β2-chimaerin to the plasma membrane in a DAG-dependent manner via binding to its C1 domain, which acts as a “brake” that limits Rac activation [96, 97]. Mechanistically, redistribution of β2-chimaerin to membranes differs from that of PKCs in that it also requires the adaptor protein Nck1 for anchoring purposes. Nck binding is mediated by an atypical Pro-rich domain adjacent to the C1 domain [98]. Early studies identified β2-chimaerin as a tumor suppressor gene that is down-regulated in high-grade glioma [99]. Later, this Rac-GAP was found to be down-regulated in breast cancer cell lines and in a few number of human breast cancer samples. Restoring β2-chimaerin expression in breast cancer cells leads to significant phenotypic changes, including reduced proliferation, motility, and tumorigenic potential [100–102]. A very recent study suggested a more complex role for β2-chimaerin in breast cancer, as down-regulation of its expression in vivo in MMTV-Neu/Her2 mice accelerates tumor onset, although it delays tumor progression. The significant increase in the number of preneoplastic lesions in mammary glands from MMTV-Neu/Her2 mice in a β2-chimaerin null-background supports a role for this DAG-responsive Rac-GAP as a tumor suppressor. However, despite the reduction in latency, tumors in a β2-chimaerin null background exhibit a less aggressive behavior. It has been also noted that reduced β2-chimaerin expression in human breast cancer inversely correlates with E-cadherin expression and associates with reduced relapse-free survival [103].
PKD isozymes are another family of “non-PKC” phorbol ester/DAG receptors that belong to the calcium/calmodulin-dependent kinase (CaMK) superfamily. The family comprises 3 related members (PKD1, PKD2 and PKD3) [104, 105]. Emerging evidence links PKD kinases to pathways implicated in cancer progression, such as ERK, JNK and NF-κB, and additionally were found to drive processes such as motility and invasion that are crucial to the metastatic dissemination of cancer cells [104, 106–108]. There is compelling evidence for a differential expression of PKD isozymes and non-overlapping functions in cancer. Notably, several reports showed up-regulation of PKDs in cancer, such as PKD1 in pancreatic and skin cancer, or PKD3 in prostate cancer. Nevertheless, down-regulation has been also observed in several cancer types, for example PKD1 in breast, colon, and gastric cancer. Interestingly, PKD1 negatively-regulates cell migration and epithelial-to-mesenchymal transition (EMT) in breast cancer cells, and loss of PKD1 in invasive breast cancer is associated with reduced expression of E-cadherin. On the other hand, PKD2 and PKD3 support cancer cell migration, as well as increase the expression of metalloproteases and urokinase-type plasminogen activator, thus augmenting ECM remodeling, and are possible inducers of EMT. PKD3 has been largely associated with disease progression in breast and prostate cancer [105]. The reported requirement of PKD1 for driving the reprogramming process of pancreatic acinar cells to a ductal phenotype as well as progression to pancreatic intraepithelial neoplasia (PanIN), together with its role as a downstream K-Ras effector [109], highlight the diversity of effects of these kinases depending on the cancer type. It is worth mentioning that a pharmacological inhibitor of PKD blocks prostate cancer cell proliferation and tumorigenesis, demonstrating the promising therapeutic potential of targeted inhibition of these kinases for cancer treatment [107]. Nonetheless, potent and selective isozyme-specific PKD inhibitors are still needed. For a comprehensive description on PKD regulation and their roles in cancer, readers should refer to excellent published reviews [105, 110, 111].
ARE PKC ISOZYMES SUITABLE TARGETS FOR CANCER THERAPEUTICS?
PKC isozymes sit at the crossroads of multiple signaling pathways and hence are obvious therapeutic targets for multiple diseases, including cancer, diabetes, heart failure and Alzheimer’s disease. The observed alterations in PKC expression in many cancer types, together with the wide-ranging effects mediated by PKC isozymes in crucial processes involved in malignant transformation and metastasis, strongly justifies efforts to develop PKC targeting drugs for cancer treatment. It is fair to say, however, that success has been limited. This is not unexpected due to the complex biological roles of PKC isozymes, both redundant and opposite, and their significant functional heterogeneity in different cancer types.
Most efforts in the search for small molecule PKC activity modulators have been focused on targeting the catalytic domain. This has proven to be difficult largely because of the high homology with other protein kinases and especially among the members of the PKC family. As mentioned above, the ATP binding site in the catalytic domain is well conserved among the protein kinase superfamily and certainly within the members of the PKC family. Several PKC inhibitors used in clinical trials, such as UCN-01 (7-hydroxystaurosporine) or midostaurin (N-benzoyl staurosporine, PKC412), turned out to target other kinases in addition to PKC [60, 112–114]. A well studied PKC inhibitor is enzastaurin, a small molecule originally described as selective for PKCβ, although it can also inhibit other PKCs via targeting the ATP-binding site [60]. Cellular-based studies demonstrated enhanced cytotoxicity by combination of enzastaurin with other chemotherapeutic agents. Nonetheless, this did not proove to be highly beneficial in a clinical setting. At the time of submission of this review, there have been 48 clinical trials using enzastaurin listed in ClinicalTrial.gov, both as a single agent or in combination standard of care therapy.
A second class of PKC modulators with anti-cancer activity are the bryostatins, macrocyclic lactones derived from the marine bryozoan Bugula neritina. Bryostatins bind and activate PKCs via the C1 domain [115]. Early studies revealed that, paradoxically, bryostatin 1 failed to induce many typical phorbol ester responses, including tumor promotion in skin. Furthermore, this agent inhibits tumor formation in the classical DMBA/TPA paradigm [66]. The differential pharmacological properties of bryostatin 1 may be linked to unique patterns of PKC isozyme relocalization [116]. Bryostatin 1 has marked anti-proliferative, apoptotic, and cytotoxic effects in cancer cells. In addition, this agent induces differentiation of leukemic cells, setting the basis for its clinical use in differentiation therapy for leukemia patients. Bryostatin 1 has been examined both as single agent and in combination therapies for hematopoietic cancers as well as for a range of solid tumors, including pancreatic, lung, renal, esophageal and ovarian cancer. The most common adverse effect of bryostatin 1 is myalgia, although this agent is well tolerated in combination with other therapeutic regimes [112, 117, 118]. Bryostatin 1 is scarce in nature and is in limited supply, therefore attempts are being made to simplify its structure and develop highly potent surrogates that could be clinically efficacious. Picolog, a synthetically accessible bryostatin analogue with picomolar affinity for PKC, induces apoptosis in mantle cell lymphoma cell lines and inhibits tumor growth in a mouse model of MYC-induced lymphoma [119, 120]. However, this and other bryostatin derivatives, such as Merle 23, and neristatin 1, have not yet been tested in patients. Other group of C1 domain ligands with clinical use are the ingenols, such as ingenol mebutate (Picato®). Ingenol derivatives bind to PKC isozymes with similar in vitro affinity, although differential activation of PKCs in cellular models has been described [121]. This drug entered the pharmaceutical market in 2012 for the treatment of actinic keratosis, and seems to be effective for the treatment of basal cell carcinomas [122].
A third approach to target PKC involves modulating its expression. Aprinocarsen (ISIS 3521) is a 20-base antisense oligonucleotide that targets PKCα. The rationale behind the development of this agent was the growth inhibitory effect of PKCα depletion on cancer cell or tumor growth. Aprinocarsen markedly reduced proliferation and invasion of cancer cells, as well as xenograft tumor growth. Clinical trials have been established for patients with prostate, breast, ovarian, colon, lung, glioblastoma, melanoma, and other cancers [112]. However, this drug has been discontinued due to limited clinical benefit, which is not surprising considering that PKCα is not necessarily a driver of proliferation and tumorigenesis, and the lack of a validated predictive biomarker for response.
Lastly, with regard to PKC modulators acting through disruption of protein-protein interactions, a promising candidate is aurothiomalate. This drug is related to the antiarthritic gold compound auranofilin, and it has been repurposed for cancer treatment due to its inhibitory effect on the PKCι-Par6 interaction. Aurothiomalate blocks PKCι signaling and inhibits transformed growth in a number of cancer cell lines in culture as well as in mouse models [123]. A phase I dose escalation study with this inhibitor has been reported for patients with lung, ovarian and pancreatic cancer [124]. There are other candidate PKC inhibitors in this category with anti-cancer potential, such as the PKCε inhibitor εV1-2 (KAI-1678), which was tested for safety and efficacy to treat neuropathic pain [125]. Based on the reported anti-tumorigenic activity of this inhibitor [63], it could be conceivably used as an anti-cancer agent.
FINAL REMARKS
While here we chose to discuss five of the most pressing issues in the field of PKC and cancer, other open questions remain. For example, can PKCs be used as biomarkers of disease progression? Can PKCs predict responders vs. non-responders to therapy? PKCs regulate many pathways involved in cellular transformation and metastatic dissemination of cancer cells. This is done in strict isozyme- and context-dependent manners, and often in opposite directions. The interplay between PKC isozymes and their effectors is complex, making it hard to predict the impact that inhibiting specific isozymes will have in the clinic. It is imperative to better understand the complex relationship of individual PKCs with their effectors, and a better appreciation of their interactions with oncogenic stimuli characteristic of every tumor type is warranted, as predictably discrete PKC isozymes may contribute to the progression of subsets of cancers. This would be particularly important to assess the effect of PKC modulators in patients in a personalized manner. The poor understanding of PKC substrates and limited availability of tools to determine activated status of PKCs represent major drawbacks for objectively assessing the effect of PKC targeting agents in patients, and validated predictive biomarkers for response are urgently needed. Addressing fundamental aspects of PKC biology and pharmacology would be required to enlighten the potential clinical benefits of PKC targeting agents.
Acknowledgments
M.G.K.’s laboratory is supported by grants R01-CA189765, R01-CA196232, and R01 ES026023 from NIH, and PC130641 from the Department of Defense.
References
- 1.Castagna M, Takai Y, Kaibuchi K, et al. Direct activation of calcium-activated, phospholipid-dependent protein kinase by tumor-promoting phorbol esters. J Biol Chem. 1982;257:7847–7851. [PubMed] [Google Scholar]
- 2.Driedger PE, Blumberg PM. Specific binding of phorbol ester tumor promoters. Proc Natl Acad Sci U S A. 1980;77:567–571. doi: 10.1073/pnas.77.1.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konig B, Di Nitto PA, Blumberg PM. Phospholipid and Ca++ dependency of phorbol ester receptors. J Cell Biochem. 1985;27:255–265. doi: 10.1002/jcb.240270307. [DOI] [PubMed] [Google Scholar]
- 4.Sharkey NA, Leach KL, Blumberg PM. Competitive inhibition by diacylglycerol of specific phorbol ester binding. Proc Natl Acad Sci U S A. 1984;81:607–610. doi: 10.1073/pnas.81.2.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takai Y, Kishimoto A, Iwasa Y, et al. Calcium-dependent activation of a multifunctional protein kinase by membrane phospholipids. J Biol Chem. 1979;254:3692–3695. [PubMed] [Google Scholar]
- 6.Griner EM, Kazanietz MG. Protein kinase C and other diacylglycerol effectors in cancer. Nat Rev Cancer. 2007;7:281–294. doi: 10.1038/nrc2110. [DOI] [PubMed] [Google Scholar]
- 7.Wu-Zhang AX, Newton AC. Protein kinase C pharmacology: refining the toolbox. Biochem J. 2013;452:195–209. doi: 10.1042/BJ20130220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garg R, Benedetti LG, Abera MB, et al. Protein kinase C and cancer: what we know and what we do not. Oncogene. 2014;33:5225–5237. doi: 10.1038/onc.2013.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ewing MW, Conti CJ, Kruszewski FH, et al. Tumor progression in Sencar mouse skin as a function of initiator dose and promoter dose, duration, and type. Cancer Res. 1988;48:7048–7054. [PubMed] [Google Scholar]
- 10.Yuspa SH. Mechanisms of initiation and promotion in mouse epidermis. IARC Sci Publ. 1984:191–204. [PubMed] [Google Scholar]
- 11.Ono Y, Fujii T, Igarashi K, et al. Phorbol ester binding to protein kinase C requires a cysteine-rich zinc-finger-like sequence. Proc Natl Acad Sci U S A. 1989;86:4868–4871. doi: 10.1073/pnas.86.13.4868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Watanabe T, Ono Y, Taniyama Y, et al. Cell division arrest induced by phorbol ester in CHO cells overexpressing protein kinase C-delta subspecies. Proc Natl Acad Sci U S A. 1992;89:10159–10163. doi: 10.1073/pnas.89.21.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nakagawa M, Oliva JL, Kothapalli D, et al. Phorbol ester-induced G1 phase arrest selectively mediated by protein kinase Cdelta-dependent induction of p21. J Biol Chem. 2005;280:33926–33934. doi: 10.1074/jbc.M505748200. [DOI] [PubMed] [Google Scholar]
- 14.Fujii T, Garcia-Bermejo ML, Bernabo JL, et al. Involvement of protein kinase C delta (PKCdelta) in phorbol ester-induced apoptosis in LNCaP prostate cancer cells. Lack of proteolytic cleavage of PKCdelta. J Biol Chem. 2000;275:7574–7582. doi: 10.1074/jbc.275.11.7574. [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Guerrico AM, Meshki J, Xiao L, et al. Molecular mechanisms of protein kinase C-induced apoptosis in prostate cancer cells. J Biochem Mol Biol. 2005;38:639–645. doi: 10.5483/bmbrep.2005.38.6.639. [DOI] [PubMed] [Google Scholar]
- 16.Reyland ME, Jones DN. Multifunctional roles of PKCdelta: Opportunities for targeted therapy in human disease. Pharmacol Ther. 2016;165:1–13. doi: 10.1016/j.pharmthera.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reddig PJ, Dreckschmidt NE, Ahrens H, et al. Transgenic mice overexpressing protein kinase Cdelta in the epidermis are resistant to skin tumor promotion by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1999;59:5710–5718. [PubMed] [Google Scholar]
- 18.Allen-Petersen BL, Carter CJ, Ohm AM, Reyland ME. Protein kinase Cdelta is required for ErbB2-driven mammary gland tumorigenesis and negatively correlates with prognosis in human breast cancer. Oncogene. 2014;33:1306–1315. doi: 10.1038/onc.2013.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mauro LV, Grossoni VC, Urtreger AJ, et al. PKC Delta (PKCdelta) promotes tumoral progression of human ductal pancreatic cancer. Pancreas. 2010;39:e31–41. doi: 10.1097/MPA.0b013e3181bce796. [DOI] [PubMed] [Google Scholar]
- 20.Hizli AA, Black AR, Pysz MA, Black JD. Protein kinase C alpha signaling inhibits cyclin D1 translation in intestinal epithelial cells. J Biol Chem. 2006;281:14596–14603. doi: 10.1074/jbc.M601959200. [DOI] [PubMed] [Google Scholar]
- 21.Mischak H, Goodnight JA, Kolch W, et al. Overexpression of protein kinase C-delta and -epsilon in NIH 3T3 cells induces opposite effects on growth, morphology, anchorage dependence, and tumorigenicity. J Biol Chem. 1993;268:6090–6096. [PubMed] [Google Scholar]
- 22.Perletti GP, Concari P, Brusaferri S, et al. Protein kinase Cepsilon is oncogenic in colon epithelial cells by interaction with the ras signal transduction pathway. Oncogene. 1998;16:3345–3348. doi: 10.1038/sj.onc.1201871. [DOI] [PubMed] [Google Scholar]
- 23.Bae KM, Wang H, Jiang G, et al. Protein kinase C epsilon is overexpressed in primary human non-small cell lung cancers and functionally required for proliferation of non-small cell lung cancer cells in a p21/Cip1-dependent manner. Cancer Res. 2007;67:6053–6063. doi: 10.1158/0008-5472.CAN-06-4037. [DOI] [PubMed] [Google Scholar]
- 24.Pan Q, Bao LW, Kleer CG, et al. Protein kinase C epsilon is a predictive biomarker of aggressive breast cancer and a validated target for RNA interference anticancer therapy. Cancer Res. 2005;65:8366–8371. doi: 10.1158/0008-5472.CAN-05-0553. [DOI] [PubMed] [Google Scholar]
- 25.Aziz MH, Manoharan HT, Church DR, et al. Protein kinase Cepsilon interacts with signal transducers and activators of transcription 3 (Stat3), phosphorylates Stat3Ser727, and regulates its constitutive activation in prostate cancer. Cancer Res. 2007;67:8828–8838. doi: 10.1158/0008-5472.CAN-07-1604. [DOI] [PubMed] [Google Scholar]
- 26.Cornford P, Evans J, Dodson A, et al. Protein kinase C isoenzyme patterns characteristically modulated in early prostate cancer. Am J Pathol. 1999;154:137–144. doi: 10.1016/S0002-9440(10)65260-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu D, Foreman TL, Gregory CW, et al. Protein kinase cepsilon has the potential to advance the recurrence of human prostate cancer. Cancer Res. 2002;62:2423–2429. [PubMed] [Google Scholar]
- 28.Benavides F, Blando J, Perez CJ, et al. Transgenic overexpression of PKCepsilon in the mouse prostate induces preneoplastic lesions. Cell Cycle. 2011;10:268–277. doi: 10.4161/cc.10.2.14469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verma AK, Wheeler DL, Aziz MH, Manoharan H. Protein kinase Cepsilon and development of squamous cell carcinoma, the nonmelanoma human skin cancer. Mol Carcinog. 2006;45:381–388. doi: 10.1002/mc.20230. [DOI] [PubMed] [Google Scholar]
- 30.Dann SG, Golas J, Miranda M, et al. p120 catenin is a key effector of a Ras-PKCvarepsilon oncogenic signaling axis. Oncogene. 2014;33:1385–1394. doi: 10.1038/onc.2013.91. [DOI] [PubMed] [Google Scholar]
- 31.Wolfman A, Macara IG. Elevated levels of diacylglycerol and decreased phorbol ester sensitivity in ras-transformed fibroblasts. Nature. 1987;325:359–361. doi: 10.1038/325359a0. [DOI] [PubMed] [Google Scholar]
- 32.Cacace AM, Ueffing M, Han EK, et al. Overexpression of PKCepsilon in R6 fibroblasts causes increased production of active TGFbeta. J Cell Physiol. 1998;175:314–322. doi: 10.1002/(SICI)1097-4652(199806)175:3<314::AID-JCP9>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 33.Gutierrez-Uzquiza A, Lopez-Haber C, Jernigan DL, et al. PKCepsilon Is an Essential Mediator of Prostate Cancer Bone Metastasis. Mol Cancer Res. 2015;13:1336–1346. doi: 10.1158/1541-7786.MCR-15-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garg R, Blando J, Perez CJ, et al. Activation of nuclear factor kappaB (NF-kappaB) in prostate cancer is mediated by protein kinase C epsilon (PKCepsilon) J Biol Chem. 2012;287:37570–37582. doi: 10.1074/jbc.M112.398925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parker PJ, Justilien V, Riou P, et al. Atypical protein kinase Ciota as a human oncogene and therapeutic target. Biochem Pharmacol. 2014;88:1–11. doi: 10.1016/j.bcp.2013.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Justilien V, Walsh MP, Ali SA, et al. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. 2014;25:139–151. doi: 10.1016/j.ccr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scotti ML, Smith KE, Butler AM, et al. Protein kinase C iota regulates pancreatic acinar-to-ductal metaplasia. PLoS One. 2012;7:e30509. doi: 10.1371/journal.pone.0030509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Y, Hill KS, Fields AP. PKCiota maintains a tumor-initiating cell phenotype that is required for ovarian tumorigenesis. Mol Cancer Res. 2013;11:1624–1635. doi: 10.1158/1541-7786.MCR-13-0371-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galvez AS, Duran A, Linares JF, et al. Protein kinase Czeta represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol Cell Biol. 2009;29:104–115. doi: 10.1128/MCB.01294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hill KS, Erdogan E, Khoor A, et al. Protein kinase Calpha suppresses Kras-mediated lung tumor formation through activation of a p38 MAPK-TGFbeta signaling axis. Oncogene. 2014;33:2134–2144. doi: 10.1038/onc.2013.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim JY, Valencia T, Abu-Baker S, et al. c-Myc phosphorylation by PKCzeta represses prostate tumorigenesis. Proc Natl Acad Sci U S A. 2013;110:6418–6423. doi: 10.1073/pnas.1221799110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oster H, Leitges M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 2006;66:6955–6963. doi: 10.1158/0008-5472.CAN-06-0268. [DOI] [PubMed] [Google Scholar]
- 43.Antal CE, Hudson AM, Kang E, et al. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Linch M, Sanz-Garcia M, Soriano E, et al. A cancer-associated mutation in atypical protein kinase Ciota occurs in a substrate-specific recruitment motif. Sci Signal. 2013;6:ra82. doi: 10.1126/scisignal.2004068. [DOI] [PubMed] [Google Scholar]
- 45.Kiss Z, Steinberg RA. Phorbol ester-mediated protein phosphorylations in S49 mouse lymphoma cells. Cancer Res. 1985;45:2732–2740. [PubMed] [Google Scholar]
- 46.Nishikawa K, Toker A, Johannes FJ, et al. Determination of the specific substrate sequence motifs of protein kinase C isozymes. J Biol Chem. 1997;272:952–960. doi: 10.1074/jbc.272.2.952. [DOI] [PubMed] [Google Scholar]
- 47.Kazanietz MG, Areces LB, Bahador A, et al. Characterization of ligand and substrate specificity for the calcium-dependent and calcium-independent protein kinase C isozymes. Mol Pharmacol. 1993;44:298–307. [PubMed] [Google Scholar]
- 48.Eblen ST, Kumar NV, Shah K, et al. Identification of novel ERK2 substrates through use of an engineered kinase and ATP analogs. J Biol Chem. 2003;278:14926–14935. doi: 10.1074/jbc.M300485200. [DOI] [PubMed] [Google Scholar]
- 49.Habelhah H, Shah K, Huang L, et al. Identification of new JNK substrate using ATP pocket mutant JNK and a corresponding ATP analogue. J Biol Chem. 2001;276:18090–18095. doi: 10.1074/jbc.M011396200. [DOI] [PubMed] [Google Scholar]
- 50.Shah K, Shokat KM. A chemical genetic screen for direct v-Src substrates reveals ordered assembly of a retrograde signaling pathway. Chem Biol. 2002;9:35–47. doi: 10.1016/s1074-5521(02)00086-8. [DOI] [PubMed] [Google Scholar]
- 51.Abeyweera TP, Chen X, Rotenberg SA. Phosphorylation of alpha6-tubulin by protein kinase Calpha activates motility of human breast cells. J Biol Chem. 2009;284:17648–17656. doi: 10.1074/jbc.M902005200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen X, Zhao X, Abeyweera TP, Rotenberg SA. Analysis of substrates of protein kinase C isoforms in human breast cells by the traceable kinase method. Biochemistry. 2012;51:7087–7097. doi: 10.1021/bi300999c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weng YC, Wang G, Messing RO, Chou WH. Identification of lipocalin-2 as a PKCdelta phosphorylation substrate in neutrophils. J Biomed Sci. 2015;22:21. doi: 10.1186/s12929-015-0129-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xia L, Wang TD, Shen SM, et al. Phosphoproteomics study on the activated PKCdelta-induced cell death. J Proteome Res. 2013;12:4280–4301. doi: 10.1021/pr400089v. [DOI] [PubMed] [Google Scholar]
- 55.Costa-Junior HM, Garavello NM, Duarte ML, et al. Phosphoproteomics profiling suggests a role for nuclear betaIotaPKC in transcription processes of undifferentiated murine embryonic stem cells. J Proteome Res. 2010;9:6191–6206. doi: 10.1021/pr100355k. [DOI] [PubMed] [Google Scholar]
- 56.Colon-Gonzalez F, Kazanietz MG. C1 domains exposed: from diacylglycerol binding to protein-protein interactions. Biochim Biophys Acta. 2006;1761:827–837. doi: 10.1016/j.bbalip.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 57.Zhang G, Kazanietz MG, Blumberg PM, Hurley JH. Crystal structure of the cys2 activator-binding domain of protein kinase C delta in complex with phorbol ester. Cell. 1995;81:917–924. doi: 10.1016/0092-8674(95)90011-x. [DOI] [PubMed] [Google Scholar]
- 58.Kazanietz MG, Bustelo XR, Barbacid M, et al. Zinc finger domains and phorbol ester pharmacophore. Analysis of binding to mutated form of protein kinase C zeta and the vav and c-raf proto-oncogene products. J Biol Chem. 1994;269:11590–11594. [PubMed] [Google Scholar]
- 59.Pu Y, Kang JH, Sigano DM, et al. Diacylglycerol lactones targeting the structural features that distinguish the atypical C1 domains of protein kinase C zeta and iota from typical C1 domains. J Med Chem. 2014;57:3835–3844. doi: 10.1021/jm500165n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mochly-Rosen D, Das K, Grimes KV. Protein kinase C, an elusive therapeutic target? Nat Rev Drug Discov. 2012;11:937–957. doi: 10.1038/nrd3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lopez-Garcia LA, Schulze JO, Frohner W, et al. Allosteric regulation of protein kinase PKCzeta by the N-terminal C1 domain and small compounds to the PIF-pocket. Chem Biol. 2011;18:1463–1473. doi: 10.1016/j.chembiol.2011.08.010. [DOI] [PubMed] [Google Scholar]
- 62.Zhang H, Neimanis S, Lopez-Garcia LA, et al. Molecular mechanism of regulation of the atypical protein kinase C by N-terminal domains and an allosteric small compound. Chem Biol. 2014;21:754–765. doi: 10.1016/j.chembiol.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 63.Caino MC, Lopez-Haber C, Kim J, et al. Proteins kinase Cvarepsilon is required for non-small cell lung carcinoma growth and regulates the expression of apoptotic genes. Oncogene. 2012;31:2593–2600. doi: 10.1038/onc.2011.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Caino MC, Lopez-Haber C, Kissil JL, Kazanietz MG. Non-small cell lung carcinoma cell motility, rac activation and metastatic dissemination are mediated by protein kinase C epsilon. PLoS One. 2012;7:e31714. doi: 10.1371/journal.pone.0031714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Erdogan E, Lamark T, Stallings-Mann M, et al. Aurothiomalate inhibits transformed growth by targeting the PB1 domain of protein kinase Ciota. J Biol Chem. 2006;281:28450–28459. doi: 10.1074/jbc.M606054200. [DOI] [PubMed] [Google Scholar]
- 66.Hennings H, Blumberg PM, Pettit GR, et al. Bryostatin 1, an activator of protein kinase C, inhibits tumor promotion by phorbol esters in SENCAR mouse skin. Carcinogenesis. 1987;8:1343–1346. doi: 10.1093/carcin/8.9.1343. [DOI] [PubMed] [Google Scholar]
- 67.Szallasi Z, Krsmanovic L, Blumberg PM. Nonpromoting 12-deoxyphorbol 13-esters inhibit phorbol 12-myristate 13-acetate induced tumor promotion in CD-1 mouse skin. Cancer Res. 1993;53:2507–2512. [PubMed] [Google Scholar]
- 68.Bogi K, Lorenzo PS, Szallasi Z, et al. Differential selectivity of ligands for the C1a and C1b phorbol ester binding domains of protein kinase Cdelta: possible correlation with tumor-promoting activity. Cancer Res. 1998;58:1423–1428. [PubMed] [Google Scholar]
- 69.Wang H, Kazanietz MG. p23/Tmp21 differentially targets the Rac-GAP beta2-chimaerin and protein kinase C via their C1 domains. Mol Biol Cell. 2010;21:1398–1408. doi: 10.1091/mbc.E09-08-0735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang H, Xiao L, Kazanietz MG. p23/Tmp21 associates with protein kinase Cdelta (PKCdelta) and modulates its apoptotic function. J Biol Chem. 2011;286:15821–15831. doi: 10.1074/jbc.M111.227991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Anilkumar N, Parsons M, Monk R, et al. Interaction of fascin and protein kinase Calpha: a novel intersection in cell adhesion and motility. EMBO J. 2003;22:5390–5402. doi: 10.1093/emboj/cdg521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chen D, Purohit A, Halilovic E, et al. Centrosomal anchoring of protein kinase C betaII by pericentrin controls microtubule organization, spindle function, and cytokinesis. J Biol Chem. 2004;279:4829–4839. doi: 10.1074/jbc.M311196200. [DOI] [PubMed] [Google Scholar]
- 73.Pang H, Guo Z, Su W, et al. RhoA-Rho kinase pathway mediates thrombin- and U-46619-induced phosphorylation of a myosin phosphatase inhibitor, CPI-17, in vascular smooth muscle cells. Am J Physiol Cell Physiol. 2005;289:C352–360. doi: 10.1152/ajpcell.00111.2005. [DOI] [PubMed] [Google Scholar]
- 74.Ann J, Yoon S, Baek J, et al. Design and synthesis of protein kinase C epsilon selective diacylglycerol lactones (DAG-lactones) Eur J Med Chem. 2015;90:332–341. doi: 10.1016/j.ejmech.2014.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Blumberg PM, Kedei N, Lewin NE, et al. Wealth of opportunity - the C1 domain as a target for drug development. Curr Drug Targets. 2008;9:641–652. doi: 10.2174/138945008785132376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Garcia LC, Donadio LG, Mann E, et al. Synthesis, biological, and biophysical studies of DAG-indololactones designed as selective activators of RasGRP. Bioorg Med Chem. 2014;22:3123–3140. doi: 10.1016/j.bmc.2014.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pu Y, Perry NA, Yang D, et al. A novel diacylglycerol-lactone shows marked selectivity in vitro among C1 domains of protein kinase C (PKC) isoforms alpha and delta as well as selectivity for RasGRP compared with PKCalpha. J Biol Chem. 2005;280:27329–27338. doi: 10.1074/jbc.M414132200. [DOI] [PubMed] [Google Scholar]
- 78.Areces LB, Kazanietz MG, Blumberg PM. Close similarity of baculovirus-expressed n-chimaerin and protein kinase C alpha as phorbol ester receptors. J Biol Chem. 1994;269:19553–19558. [PubMed] [Google Scholar]
- 79.Caloca MJ, Fernandez N, Lewin NE, et al. Beta2-chimaerin is a high affinity receptor for the phorbol ester tumor promoters. J Biol Chem. 1997;272:26488–26496. doi: 10.1074/jbc.272.42.26488. [DOI] [PubMed] [Google Scholar]
- 80.Kazanietz MG. Eyes wide shut: protein kinase C isozymes are not the only receptors for the phorbol ester tumor promoters. Mol Carcinog. 2000;28:5–11. doi: 10.1002/(sici)1098-2744(200005)28:1<5::aid-mc2>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 81.Kazanietz MG, Lewin NE, Bruns JD, Blumberg PM. Characterization of the cysteine-rich region of the Caenorhabditis elegans protein Unc-13 as a high affinity phorbol ester receptor. Analysis of ligand-binding interactions, lipid cofactor requirements, and inhibitor sensitivity. J Biol Chem. 1995;270:10777–10783. doi: 10.1074/jbc.270.18.10777. [DOI] [PubMed] [Google Scholar]
- 82.Lorenzo PS, Beheshti M, Pettit GR, et al. The guanine nucleotide exchange factor RasGRP is a high -affinity target for diacylglycerol and phorbol esters. Mol Pharmacol. 2000;57:840–846. [PubMed] [Google Scholar]
- 83.Czikora A, Lundberg DJ, Abramovitz A, et al. Structural Basis for the Failure of the C1 Domain of Ras Guanine Nucleotide Releasing Protein 2 (RasGRP2) to Bind Phorbol Ester with High Affinity. J Biol Chem. 2016;291:11133–11147. doi: 10.1074/jbc.M116.725333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Johnson JE, Goulding RE, Ding Z, et al. Differential membrane binding and diacylglycerol recognition by C1 domains of RasGRPs. Biochem J. 2007;406:223–236. doi: 10.1042/BJ20070294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zheng Y, Liu H, Coughlin J, et al. Phosphorylation of RasGRP3 on threonine 133 provides a mechanistic link between PKC and Ras signaling systems in B cells. Blood. 2005;105:3648–3654. doi: 10.1182/blood-2004-10-3916. [DOI] [PubMed] [Google Scholar]
- 86.Ksionda O, Limnander A, Roose JP. RasGRP Ras guanine nucleotide exchange factors in cancer. Front Biol (Beijing) 2013;8:508–532. doi: 10.1007/s11515-013-1276-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ksionda O, Melton AA, Bache J, et al. RasGRP1 overexpression in T-ALL increases basal nucleotide exchange on Ras rendering the Ras/PI3K/Akt pathway responsive to protumorigenic cytokines. Oncogene. 2016;35:3658–3668. doi: 10.1038/onc.2015.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hartzell C, Ksionda O, Lemmens E, et al. Dysregulated RasGRP1 responds to cytokine receptor input in T cell leukemogenesis. Sci Signal. 2013;6:ra21. doi: 10.1126/scisignal.2003848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Luke CT, Oki-Idouchi CE, Cline JM, Lorenzo PS. RasGRP1 overexpression in the epidermis of transgenic mice contributes to tumor progression during multistage skin carcinogenesis. Cancer Res. 2007;67:10190–10197. doi: 10.1158/0008-5472.CAN-07-2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sharma A, Fonseca LL, Rajani C, et al. Targeted deletion of RasGRP1 impairs skin tumorigenesis. Carcinogenesis. 2014;35:1084–1091. doi: 10.1093/carcin/bgu016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yang D, Kedei N, Li L, et al. RasGRP3 contributes to formation and maintenance of the prostate cancer phenotype. Cancer Res. 2010;70:7905–7917. doi: 10.1158/0008-5472.CAN-09-4729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee HK, Finniss S, Cazacu S, et al. RasGRP3 regulates the migration of glioma cells via interaction with Arp3. Oncotarget. 2015;6:1850–1864. doi: 10.18632/oncotarget.2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nagy Z, Kovacs I, Torok M, et al. Function of RasGRP3 in the formation and progression of human breast cancer. Mol Cancer. 2014;13:96. doi: 10.1186/1476-4598-13-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kazanietz MG. Novel “nonkinase” phorbol ester receptors: the C1 domain connection. Mol Pharmacol. 2002;61:759–767. doi: 10.1124/mol.61.4.759. [DOI] [PubMed] [Google Scholar]
- 95.Zubeldia-Brenner L, Gutierrez-Uzquiza A, Barrio-Real L, et al. beta3-chimaerin, a novel member of the chimaerin Rac-GAP family. Mol Biol Rep. 2014;41:2067–2076. doi: 10.1007/s11033-014-3055-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Colon-Gonzalez F, Leskow FC, Kazanietz MG. Identification of an autoinhibitory mechanism that restricts C1 domain-mediated activation of the Rac-GAP alpha2-chimaerin. J Biol Chem. 2008;283:35247–35257. doi: 10.1074/jbc.M806264200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang H, Yang C, Leskow FC, et al. Phospholipase Cgamma/diacylglycerol-dependent activation of beta2-chimaerin restricts EGF-induced Rac signaling. EMBO J. 2006;25:2062–2074. doi: 10.1038/sj.emboj.7601098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gutierrez-Uzquiza A, Colon-Gonzalez F, Leonard TA, et al. Coordinated activation of the Rac-GAP beta2-chimaerin by an atypical proline-rich domain and diacylglycerol. Nat Commun. 2013;4:1849. doi: 10.1038/ncomms2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yuan S, Miller DW, Barnett GH, et al. Identification and characterization of human beta 2-chimaerin: association with malignant transformation in astrocytoma. Cancer Res. 1995;55:3456–3461. [PubMed] [Google Scholar]
- 100.Menna PL, Skilton G, Leskow FC, et al. Inhibition of aggressiveness of metastatic mouse mammary carcinoma cells by the beta2-chimaerin GAP domain. Cancer Res. 2003;63:2284–2291. [PubMed] [Google Scholar]
- 101.Yang C, Liu Y, Lemmon MA, Kazanietz MG. Essential role for Rac in heregulin beta1 mitogenic signaling: a mechanism that involves epidermal growth factor receptor and is independent of ErbB4. Mol Cell Biol. 2006;26:831–842. doi: 10.1128/MCB.26.3.831-842.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang C, Liu Y, Leskow FC, et al. Rac-GAP-dependent inhibition of breast cancer cell proliferation by {beta}2-chimerin. J Biol Chem. 2005;280:24363–24370. doi: 10.1074/jbc.M411629200. [DOI] [PubMed] [Google Scholar]
- 103.Casado-Medrano V, Barrio-Real L, Garcia-Rostan G, et al. A new role of the Rac-GAP beta2-chimaerin in cell adhesion reveals opposite functions in breast cancer initiation and tumor progression. Oncotarget. 2016;7:28301–28319. doi: 10.18632/oncotarget.8597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Durand N, Borges S, Storz P. Functional and therapeutic significance of protein kinase D enzymes in invasive breast cancer. Cell Mol Life Sci. 2015;72:4369–4382. doi: 10.1007/s00018-015-2011-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Durand N, Borges S, Storz P. Protein Kinase D Enzymes as Regulators of EMT and Cancer Cell Invasion. J Clin Med. 2016:5. doi: 10.3390/jcm5020020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bernhart E, Damm S, Wintersperger A, et al. Protein kinase D2 regulates migration and invasion of U87MG glioblastoma cells in vitro. Exp Cell Res. 2013;319:2037–2048. doi: 10.1016/j.yexcr.2013.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Tandon M, Salamoun JM, Carder EJ, et al. SD-208, a novel protein kinase D inhibitor, blocks prostate cancer cell proliferation and tumor growth in vivo by inducing G2/M cell cycle arrest. PLoS One. 2015;10:e0119346. doi: 10.1371/journal.pone.0119346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wei N, Chu E, Wipf P, Schmitz JC. Protein kinase d as a potential chemotherapeutic target for colorectal cancer. Mol Cancer Ther. 2014;13:1130–1141. doi: 10.1158/1535-7163.MCT-13-0880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liou GY, Doppler H, Braun UB, et al. Protein kinase D1 drives pancreatic acinar cell reprogramming and progression to intraepithelial neoplasia. Nat Commun. 2015;6:6200. doi: 10.1038/ncomms7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.LaValle CR, George KM, Sharlow ER, et al. Protein kinase D as a potential new target for cancer therapy. Biochim Biophys Acta. 2010;1806:183–192. doi: 10.1016/j.bbcan.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rozengurt E. Protein kinase D signaling: multiple biological functions in health and disease. Physiology (Bethesda) 2011;26:23–33. doi: 10.1152/physiol.00037.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Martiny-Baron G, Fabbro D. Classical PKC isoforms in cancer. Pharmacol Res. 2007;55:477–486. doi: 10.1016/j.phrs.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 113.Senderowicz AM. Cell cycle modulators for the treatment of lung malignancies. Clin Lung Cancer. 2003;5:158–168. doi: 10.3816/CLC.2003.n.028. [DOI] [PubMed] [Google Scholar]
- 114.Seynaeve CM, Kazanietz MG, Blumberg PM, et al. Differential inhibition of protein kinase C isozymes by UCN-01, a staurosporine analogue. Mol Pharmacol. 1994;45:1207–1214. [PubMed] [Google Scholar]
- 115.Kazanietz MG, Lewin NE, Gao F, et al. Binding of [26-3H]bryostatin 1 and analogs to calcium-dependent and calcium-independent protein kinase C isozymes. Mol Pharmacol. 1994;46:374–379. [PubMed] [Google Scholar]
- 116.von Burstin VA, Xiao L, Kazanietz MG. Bryostatin 1 inhibits phorbol ester-induced apoptosis in prostate cancer cells by differentially modulating protein kinase C (PKC) delta translocation and preventing PKCdelta-mediated release of tumor necrosis factor-alpha. Mol Pharmacol. 2010;78:325–332. doi: 10.1124/mol.110.064741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kollar P, Rajchard J, Balounova Z, Pazourek J. Marine natural products: bryostatins in preclinical and clinical studies. Pharm Biol. 2014;52:237–242. doi: 10.3109/13880209.2013.804100. [DOI] [PubMed] [Google Scholar]
- 118.Ruan BF, Zhu HL. The chemistry and biology of the bryostatins: potential PKC inhibitors in clinical development. Curr Med Chem. 2012;19:2652–2664. doi: 10.2174/092986712800493020. [DOI] [PubMed] [Google Scholar]
- 119.DeChristopher BA, Fan AC, Felsher DW, Wender PA. “Picolog,” a synthetically-available bryostatin analog, inhibits growth of MYC-induced lymphoma in vivo. Oncotarget. 2012;3:58–66. doi: 10.18632/oncotarget.438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lopez-Campistrous A, Song X, Schrier AJ, et al. Bryostatin analogue-induced apoptosis in mantle cell lymphoma cell lines. Exp Hematol. 2012;40:646–656. e642. doi: 10.1016/j.exphem.2012.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kedei N, Lundberg DJ, Toth A, et al. Characterization of the interaction of ingenol 3-angelate with protein kinase C. Cancer Res. 2004;64:3243–3255. doi: 10.1158/0008-5472.can-03-3403. [DOI] [PubMed] [Google Scholar]
- 122.Bettencourt MS. Treatment of superficial basal cell carcinoma with ingenol mebutate gel, 0. 05% Clin Cosmet Investig Dermatol. 2016;9:205–209. doi: 10.2147/CCID.S109531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Murray NR, Kalari KR, Fields AP. Protein kinase Ciota expression and oncogenic signaling mechanisms in cancer. J Cell Physiol. 2011;226:879–887. doi: 10.1002/jcp.22463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mansfield AS, Fields AP, Jatoi A, et al. Phase I dose escalation study of the PKCiota inhibitor aurothiomalate for advanced non-small-cell lung cancer, ovarian cancer, and pancreatic cancer. Anticancer Drugs. 2013;24:1079–1083. doi: 10.1097/CAD.0000000000000009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cousins MJ, Pickthorn K, Huang S, et al. The safety and efficacy of KAI-1678- an inhibitor of epsilon protein kinase C (epsilonPKC)-versus lidocaine and placebo for the treatment of postherpetic neuralgia: a crossover study design. Pain Med. 2013;14:533–540. doi: 10.1111/pme.12058. [DOI] [PubMed] [Google Scholar]