

Abstract
Biotin/(strept)avidin self-assembly is a powerful platform for nanoscale fabrication and capture with many different applications in science, medicine, and nanotechnology. However, biotin/(strept)avidin self-assembly has several well-recognized drawbacks that limit performance in certain technical areas and there is a need for synthetic mimics that can either become superior replacements or operational partners with bio-orthogonal recognition properties. The goal of this tutorial review is to describe the recent progress in making high affinity synthetic association partners that operate in water or biological media. The review starts with a background summary of biotin/(strept)avidin self-assembly and the current design rules for creating synthetic mimics. A series of case studies are presented that describe recent success using synthetic derivatives of cyclodextrins, cucurbiturils, and various organic cyclophanes such as calixarenes, deep cavitands, pillararenes, and tetralactams. In some cases, two complementary partners associate to produce a nanoscale complex and in other cases a ditopic host molecule is used to link two partners. The article concludes with a short discussion of future directions and likely challenges.
1. Background on biotin/(strept)avidin
Biotin/(strept)avidin self-assembly is the basis of a very large number of technologies in science, medicine, nanotechnology and molecular engineering, and the methodology is described in many review articles.1 To briefly summarize, non-covalent association of the small vitamin molecule biotin with the avidin protein (or the structurally similar streptavidin) in water is remarkably strong (Ka ~ 1015 M−1) with a very slow dissociation half-life of days to weeks depending on protein and conditions. The ability to selectively self-assemble two molecular partners in water and at room temperature creates a powerful platform for nanoscale technology development. It is difficult to cleanly divide all the applications that employ biotin/(strept)avidin self-assembly into individual categories, but, the organizational chart in Figure 1 provides some useful distinctions. First it separates biotin/(strept)avidin self-assembly into two operationally discrete groups; (a) Pre-assembly, where the biotin/(strept)avidin association process is used for surface modification or molecular ligation, and produces a functionalized surface or nanoscale object for subsequent use, or (b) In-Situ Capture, where selective assembly occurs within a complex sample and achieves an immediately useful outcome such as affinity purification or localized assembly. These discrete self-assembly processes are the basis for different applications in histology, imaging, diagnosis, catalysis, analysis, and purification.1,2,3,4
Figure 1.
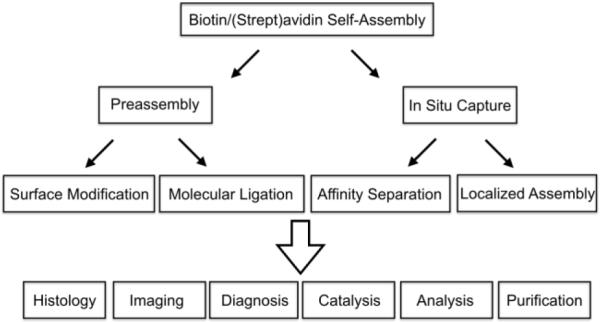
Organizational chart of applications that are based on biotin/(strept)avidin self-assembly.
Although avidin and streptavidin have a very similar quaternary structure comprised of four identical subunits, there are structural distinctions that determine the suitability of each protein for specific applications. In short, avidin is derived from egg-whites and has N-linked apsaragine glycosyl groups and a pI of ~10. The attached sugars and positive charge can induce non-specific association processes which weaken diagnostic performance. Streptavidin, a bacterial-origin protein, has quite a different primary sequence with no attached sugars and a much lower pI of ~5-6. Because of these factors, streptavidin is often the preferred protein for capture or detection methods. While the practical value and broad utility of biotin/(strept)avidin self-assembly is without doubt, there are several well-recognized drawbacks that limit performance in certain technical areas. Some of the major concerns are: (a) The (strept)avidin tetramer has a total molecular weight of ~66KDa, a large size that is a limitation for applications that require rapid diffusion through restricted sites, or applications that require coated surfaces with high coverage densities. (b) Each subunit in the protein tetramer has a biotin binding pocket, and the overall tetravalent affinity for biotin can induce crosslinking and clustering effects which are useful in some cases but undesired in others. (c) The protein is potentially immunogenic, which restricts applications in living subjects. (d) Endogenous biotin can cause background and specificity issues when performing assays with biotin-rich tissues and extracts. (e) Both association partners (biotin and (strept)avidin) are optically transparent and while standard covalent conjugation methods can easily attach a reporter group to biotin, chemical conjugation to the much larger (strept)avidin protein can be problematic due the presence of multiple reactive groups on the protein surface.
Considerable effort has been made to overcome these performance deficiencies by engineering the structure of biotin/(strept)avidin pairs. One well-known example is the commercially available NeutrAvidin which is a deglycosylated derivative of avidin with near neutral pI.5 A major effort has also been made to produce a (strept)avidin derivative with monomeric affinity for biotin. This is a challenging protein design problem because a tryptophan residue from an adjacent subunit is critical for high affinity biotin binding. Thus, it is hard to change the monomer primary sequence in a way that eliminates subunit assembly as a tetramer without reducing the subunit’s affinity for biotin. An early approach produced a mutated streptavidin tetramer with only one subunit having strong monovalent affinity for biotin.1 More recent advances have produced purely monomeric streptavidin-like proteins with high biotin affinity.6,7 These monovalent biotin binding systems have been used to label cell surface proteins without target aggregation and thus facilitate high resolution microscopy of cell surfaces.8 In addition, they have been genetically fused with a second protein of interest to produce an intracellular multifunctional construct that does not oligomerize. Mutagenesis methods have also produced monovalent Strep-Tactin, a modified form of streptavidin that binds specifically to the genetically encodable amino acid sequence, WSHPQFEK. Immobilized Strep-Tactin is predominantly employed for protein purification, but it also can be used for protein tethering in biophysics expriments.9 The alternative approach to modifying biotin/(strept)avidin affinity is to alter the biotin structure and a classic example is 2-iminobiotin whose pH dependent interaction enables easy protein purification.10 Taken together, these various modifications of biotin/(strept)avidin structure and function are impressive advances, but there is still room for additional improvement. In particular, there is a great need for new orthogonal molecular self-assembly systems that can be used simultaneously within a single analytical sample. As described below, these complementary molecular recognition systems can be synthetic or biochemical. But before describing them, it is worth briefly summarizing the key supramolecular features of biotin/(strept)avidin assembly that produce the remarkable thermodynamic and kinetic properties.
A myriad of experimental and computational methods have been used to study biotin/(strept)avidin assembly as a model of high affinity protein–ligand energetics.11 The biotin binding pocket within the folded protein structure has a complementary shape that is lined with a mixture of polar and hydrophobic residues. In the case of streptavidin, the hydrophobic region of the pocket is defined by four tryptophan residues that stack against the hydrocarbon regions of the tetrahydrothiophene ring and valeric acid tail of biotin. The complex is also stabilized by an extensive network of cooperative hydrogen bonds, including five important hydrogen bonds between the ureido ring of biotin and the streptavidin protein (Figure 2). Proximal to the binding pocket is the flexible loop3-4 that closes over the bound biotin and enhances affinity.12 The exceptionally slow rate of biotin dissociation is controlled by the dynamics of loop3-4 and consequently, amino acid point mutations in the loop structure often have large effects on Ka and koff. 13 Significantly faster dissociation rate constants are observed with structural analogues of biotin that bind in the pocket but apparently prevent loop closure.14 Although there is a significant body of knowledge concerning the dynamic and cooperative non-covalent processes that produce the high affinity, recent reports highlight discrepancies that suggest the current binding models are not complete.15 In part, this is due to the large molecular size and structural complexity of the biotin/(strept)avidin system which makes it challenging to conduct accurate molecular dynamics simulations.16
Figure 2.
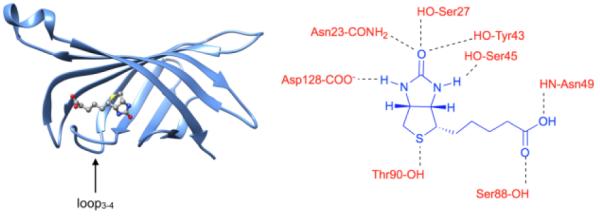
(left) Picture of biotin/streptavidin complex with loop3-4 closed like a lid, (right) important hydrogen bonds within the biotin/streptavidin binding pocket.11
With this background information in mind, the goal of this tutorial review is to describe the most effective synthetic mimics of biotin/(strept)avidin that operate in water or biological media, with a focus on the two generic association systems shown in Figure 3. In the first case, two complementary partners A and B associate to produce a nanoscale complex and in the second case, a ditopic host molecule is used to link two partners C and D. In each case, one or both or the association partners is functionalized by having an attached targeting unit, reporter group, immobilization site, etc. Since the focus is on synthetic mimics of biotin/(strept)avidin that achieve one of the functions or applications listed in Figure 1, the following supramolecular topics are not covered in specific detail; complexation of small molecules inside container molecules, self-assembly of amphiphilic host/guest complexes into larger aggregates, and supramolecular catalysis.
Figure 3.
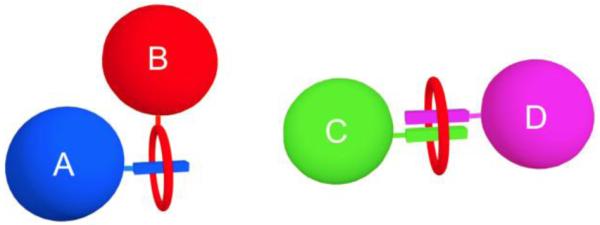
Two synthetic host/guest systems that mimic the self-assembly of biotin/(strept)avidin and achieve the functions and applications listed in Figure 1. (left) the complementary partners A and B associate to produce a nanoscale complex, (right) a ditopic host molecule (red oval) links two partners C and D.
2. Achieving high affinity host•guest complexation in water
In a 2003 review article, Houk and co-workers analyzed the binding thermodynamics of a wide variety of synthetic and biological host•guest systems (Table 1).17 Amongst the synthetic systems, perhaps the most studied class of compounds are α-, β-, and γ-cyclodextrins (CDs) which bind to a variety of hydrophobic small molecules (e.g. derivatives of alkanes, benzene, and polycyclics like adamantane) with an average Ka of 102.5±1.1 M−1 and an average ΔG of −3.5 ± 1.4 kcal mol−1. Accordingly, 95% of CD•guest complexes lie in the range of Ka = 1 to 105 M−1. A similar analysis was performed for a dataset of 973 organic host•guest complexes that included various cyclophanes and cucurbit[6]uril and delivered an average Ka of 103.4±1.6 M−1 which corresponds to a ΔG of −4.6±2.1 kcal mol−1. For these synthetic receptors 95% of the Ka values are in the 1 – 107 M−1 range which is only slightly wider than for cyclodextrins. Compared to biotin/(strept)avidin these monovalent binding affinities are modest and need to be increased for use in many applications.
Table 1.
Binding constants of synthetic hosts, antibodies, proteins and enzymes with neutral organic compounds.
| Host type | Guest type | Mean log Ka |
Standard deviation[a] |
−ΔG [kcal mol−1] |
|---|---|---|---|---|
| cyclodextrin | organic molecule |
2.5 | 1.1 | 3.5 ± 1.4 |
| organic host[b] |
organic molecule |
3.4 | 1.6 | 4.6 ± 2.1 |
| catalytic antibody |
substrate | 3.5 | 1.0 | 4.8 ± 1.3 |
| enzyme | substrate | 3.7 | 1.3 | 5.1 ± 1.7 |
| albumin | organic molecule |
4.6 | 0.9 | 6.3 ± 1.3 |
| catalytic antibody |
transition state |
6.6 | 2.0 | 9.0 ± 2.7 |
| receptor | drug | 7.3 | 1.5 | 9.5 ± 2.1 |
| antibody | antigen | 8.1 | 2.0 | 11.1 ± 2.7 |
| enzyme | inhibitor | 8.6 | 2.1 | 11.7 ± 2.8 |
| enzyme | transition state |
16.0 | 4.0 | 21.9 ± 5.4 |
In units of log Ka; 68% of cases fall within one standard deviation of the mean value, 95% within two standard deviations for a normal distribution.
In aqueous solvent.
Summarized in Figure 4 are four molecular design strategies to increase host/guest affinity in water. First, and foremost, is Cram’s principle of preorganization (Figure 4a) which states that “the more highly hosts and guests are organized for binding and low solvation prior to their complexation, the more stable will be their complexes.” When considered in the context of aqueous solution, this principle dictates that the host cavity should not contain polar groups (e.g. H-bond acceptors or donors) that are strongly solvated by water.18 The second molecular design strategy is to introduce electrostatic (e.g. ion-ion or ion-dipole) interactions between host and guest.19 For example, Sugammadex is an anionic β-cyclodextrin derivative that strongly binds to the positively charged steroidal neuromuscular blocking agent rocuronium with Ka = 1.05 × 107 M−1 in water and is used to reverse the residual side effects of rocuronium post-surgically (Figure 4b).20 The third design strategy (Figure 4c) is to exploit the hydrophobic effect. Empirically, it is known that removal of hydrocarbon surface area from water contributes approximately −0.04 kcal−1 mol−1 Å−2 to the free energy of binding. Therefore, increasing the host and guest size and maximizing buried surface area provides an avenue to increase affinity.17 A molecular understanding of the hydrophobic effect is a major ongoing research topic. Studies of protein systems have concluded that the presence of enthalpically high energy waters of solvation in the binding pocket is a key factor that promotes high affinity protein•ligand binding and may be one reason why protein•ligand affinities generally exceed host•guest binding.21 Recent insight provided by the groups of Nau, Scherman, and DeSimone have made a connection between high-affinity protein•ligand binding and high affinity host•guest binding.22,23 For example, they find that the number of H2O molecules inside cucurbit[n]urils (CB[n]) changes from 2 (CB[5]) to 22 (CB[10]) as the volume of the cavity increases. These H2O molecules are high energy because: 1) the geometrical constraints of the cavity preclude a full complement of H-bonds, and 2) the CB[n] cavity has extremely low polarizability and thus poor dispersion interactions exist between the CB[n] cavity and the encapsulated H2O molecules. The authors performed MD simulations of the potential energies of the encapsulated waters as a function of CB[n] and found that CB[7] represents a sweet spot that provides the largest overall enthalpic driving force which is consistent with the experiment. The water molecules inside CB[6] are high energy but few (3-4) whereas those inside CB[8] are many (10) but of overall lower potential energy. A corollary of this insight with clear connections to Cram’s principle of pre-organization is to design the host to maximize both the number and energy of encapsulated water molecules.22,23 Finally, the fourth design strategy (Figure 4d) is the concept of multivalency24 which involves the covalent linking of n hosts and n guests to create systems that depending on the efficiency of linker design can achieve binding affinities on the order of Ka,mono2. A full description of these approaches is beyond the scope of this tutorial review and the reader is referred to authoritative reviews.17–19,24
Figure 4.
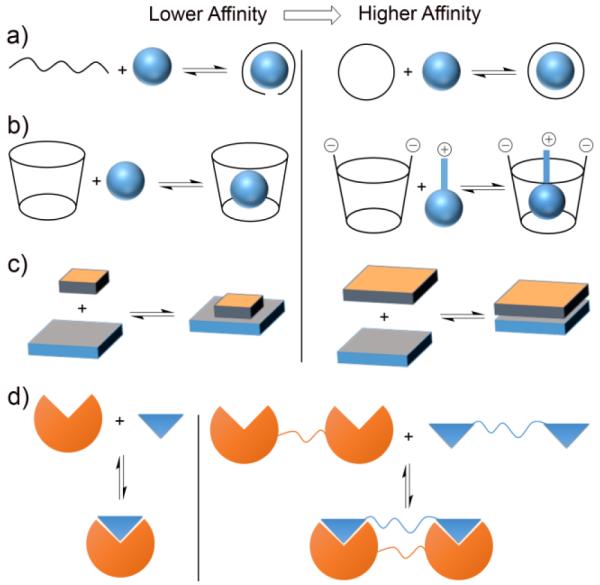
Four general design strategies to increase host•guest binding affinity in water: a) preorganization, b) electrostatics, c) hydrophobic effect, and d) multivalency.
3. Host•guest mimics of biotin/(strept)avidin
The last decade has witnessed a resurgence in the design and development of container molecules for organic guests. These new synthetic systems have become valuable as experimentally tractable models of high affinity association and detailed experimental and computational studies have produced valuable new molecular level insight. The recent advances in conjugation chemistry using high yielding covalent click reactions has greatly facilitated emerging efforts to structurally modify these new synthetic association systems for many of the applications described in Figure 1 above. The following sections describe case studies from the recent literature that illustrate the practical potential of these synthetic mimics of biotin/(strept)avidin. The collection is representative and not an exhaustive summary.
3a. Cyclodextrins
The affinity of unfunctionalized CD toward its guests rarely exceeds 104 M−1. Accordingly, the use of CD as a component of a synthetic affinity pair requires the use of multiple non-covalent interactions in the form of multivalent systems comprising either: a) two or more CDs and two or more guests or b) one CD, one guest, and additional non-covalent interactions (e.g. protein•protein or DNA hybridization). In this section we present examples of both approaches.
Protein-binding molecular switches via cyclodextrin•guest interactions
A very nice example of a system that uses β-CD•guest complexation to control protein binding was published by Jayawickramarajah in 2011.25 Figure 5a shows the structures of three DNA-small molecule chimeras (DC) based on a core 19-mer oligonucleotide that bears adamantane and β-CD groups at its 5′ and 3′ termini, respectively. DC1 – DC1” assume a stem loop conformation that is further stabilized by intramolecular β-CD-adamantane complexation. UV/Vis thermal denaturation experiments demonstrate that DC1 – DC1” are substantially stabilized relative to analogous oligonucleotides lacking the adamantane and β-CD units. Addition of a complementary oligonucleotide strand (ODN3) resulted in duplex formation, unfolding of the hairpin, and exposure of the adamantane headgroup as monitored by the fluorescence changes of the pyrrolo dC unit of DC1’. Finally, the ability of ODN to simultaneously unfold fluorescein labelled DC1” and promote binding of DC1”•ODN3 to β-lactoglobulin which is a promiscuous binder of hydrophobes in its β-barrel calyx was demonstrated. Fluorescence anisotropy measurements established that DC1”•ODN3 binds as well to β-lactoglobulin as a positive control oligonucleotide with a fully exposed headgroup. As β-CDs are known to bind a wide range of drugs and drug like molecules, this strategy can be used to target a wide range of proteins in an oligonucleotide responsive manner.
Figure 5.

a) Oligonucleotide triggered unfolding and protein binding of β-CD•adamantane stabilized hairpins. b) Self-assembled heterosequenced porphyrin nanowires based on tetravalent β-CD•adamantane complexation. (Reprinted by permission from refs 25 and 26. Copyright 2010 and 2011, American Chemical Society)
Porphyrin nanowires
As described above, β-CD•guest complexes do not typically display association constants above 104 M−1. Therefore, to incorporate CD within a high affinity supramolecular pair requires a design based on multivalency. A classic example of such an approach was reported by Jayawickramarajah, who decorated octapropargyloxy Zn-porphyrins with eight β-CD groups or eight adamantane groups and achieved disc-like architectures with four CDs or adamantanes pointing up and four pointing down (Figure 5b).26 Self-assembly of the components was evidenced by UV/Vis spectroscopy suggesting the formation of porphyrin nanowires. TEM images at low concentration revealed the presence of 3.5 nm wide rods hundreds of nanometers long which was confirmed by STM measurements. The supramolecular architecture separated alternating donor and acceptor porphyrins by 2 nm and enabled fast and efficient energy transfer (3.13 × 109 s−1, 81%) as measured by time-correlated single photon counting. The tetravalent binding of β-CD to adamantane was so robust that nanowire assembly persisted even at 80 °C or in the presence of excess AdCO2−.
Macroscopic self-assembly controlled by β-CD•guest molecular recognition events
The self-assembly of molecules into well defined and functional structures by non-covalent interactions has been studied intensively in the past 25 years. In contrast, the self assembly of macroscopic objects by molecular recognition events is rare, but would be an extremely useful assembling tool in materials science. To demonstrate such a concept, Harada and co-workers prepared cross linked polyacrylamide gels that were derivatized with α-CD or β-CD and complementary gels that were derivatized with adamantane, n-butyl, and t-butyl groups and observed their self-assembly processes upon shaking in water (Figure 6)27 For example, mixing β-CD-gel and Ad-gel gives the β-CD-gel/Ad-gel assembly which is so robust that it does not dissociate below 90 °C (Figure 6b). Pairs of gels that do not contain tight binding affinity pairs (e.g. α-CD-gel and Ad-gel) adhere more weakly to each other. The molecular recognition specific nature of the adhesion was proved by adding excess of free β-CD or adamantaneamine which competed for CD•guest complexation and decreased adhesion of the macroscopic objects. To test the limits of this type of assembly the authors color coded (dye labelled) different gels and monitored their assembly in three and four component mixtures. Self-assembly of square pieces of α-CD-gel, t-Bu-gel, and n-Bu gel gives selectively a network of α-CD-gel/n-Bu-gel along with free t-Bu gel which can be understood on the basis of the molecular recognition preferences of α-CD. Conversely, a mixture of β-CD-gel, t-Bu-gel, and n-Bu gel gives selectively a network of β-CD-gel/t-Bu-gel along with free n-Bu gel (Figure 6c). Finally, all four components (α-CD-gel, β-CD-gel, t-Bu-gel, and n-Bu-gel) undergo self-sorting assembly without evidence of cross-talk (Figure 6d). This beautiful work points a way to harness molecular recognition events to control the self-assembly of functional macroscopic objects.
Figure 6.
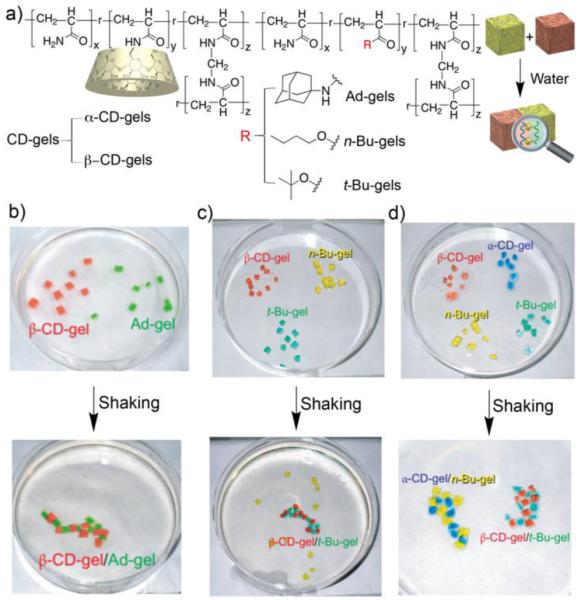
a) Structures of host and guest functionalized cross linked polyacrylamides for macroscopic self-assembly, b) β-CD-gel and Ad-gel system, c) β-CD-gel, t-Bu-gel, n-Bu-gel system, d) self-sorting α-CD-gel, β-CD-gel, n-Bu-gel, t-Bu-gel system. (Reprinted by permission from ref 27. Copyright 2011, Nature Publishing Group).
3b. Cucurbit[n]urils
The last decade has witnessed the discovery, application, and elucidation of the molecular level reasons for ultratight binding of CB[n] receptors.28 For example, in 2005, the Isaacs lab discovered that a larger CB[n] homologue (CB[7]) exhibited remarkably tight binding toward adamantaneammonium (Ka = 4.2 × 1012 M−1) and trimethylammoniomethyl ferrocene (Ka = 3.3 × 1011 M−1) in water by NMR competition experiments which was subsequently confirmed by Kaifer, Inoue, and Kim by ITC competition experiments.29,30 Subsequent work identified bis(trimethylammoniomethyl) ferrocene (Ka = 3 × 1015 M−1) and culminated with measurement of Ka = 7 × 1017 M−1 for CB[7]•Diamantane(NMe3)22+.31,32
These extremely high affinities mean that CB[n] hosts are well suited for creation of synthetic mimics of biotin/(strept)avidin technology. For smaller CB[n] (e.g CB[7]), 1:1 host:guest complexes are typically formed which dictates that the two components (e.g. A and B) to be non-covalently linked must be functionalized with CB[n] host and a guest for CB[n], respectively, as shown in Figure 3 (left). Conversely, the larger CB[n] (e.g. CB[8]) possess the ability to bind two or more guests simultaneously to form homo- or heteroternary complexes (e.g. CB[8]•first guest•second guest) as illustrated in Figure 3 (right). In this section we provide examples of both types of systems.
Supramolecular fishing for plasma membrane proteins
Plasma membrane proteins have a plethora of biological functions, but their isolation is challenging due to their inherent hydrophobicity and currently relies on biotin/(strept)avidin technology. In 2011, Kim and co-workers reported the use of CB[7]•guest chemistry as an alternative for the isolation of plasma membrane proteins (Figure 7).33 For this purpose, they partially oxidized CB[7] to yield (HO)nCB[7] (n ≈ 6) and then performed the reaction with N-hydroxsuccinimidyl sepharose beads (Et3N, DMSO) to yield the HO-CB[7]-bead conjugate at a loading level of 0.88 nmol CB[7] / mg Sepharose. Separately, bovine serum albumin (BSA) as model protein was reacted with a ferrocene derivative to yield Fc-BSA. Next, affinity pull down assays were performed using the Sepharose-CB[7] beads to capture Fc-BSA on its own or from a more complex mixture comprising Fc-BSA in HEK293 cell lysate. The captured proteins were released from the resin by heating (95 °C) and the denatured proteins analyzed by SDS-PAGE to establish that only Fc-BSA was affinity captured based on CB[7]•Fc complexation. For comparison, related experiments were performed using commercially available streptavidin beads which established that the Sepharose-CB[7] beads capture 75% of Fc-Hemoglobin whereas only 33% Biotin-Hemoglobin was captured by the streptavidin beads. Importantly, the Fc-proteins could also be released from Sepharose-CB[7] by incubating with excess DiaminoFC which binds much tighter (≈ 1 × 1015 M−1) toward CB[7] that AminoFc does. Recently, Urbach and Isaacs extended this system by immobilizing a monofunctionalized CB[7]-azide on Sepharose beads by click chemistry and showed that it could even be used to capture native proteins that contain an N-terminal phenylalanine residue (e.g. insulin and human growth hormone).34
Figure 7.
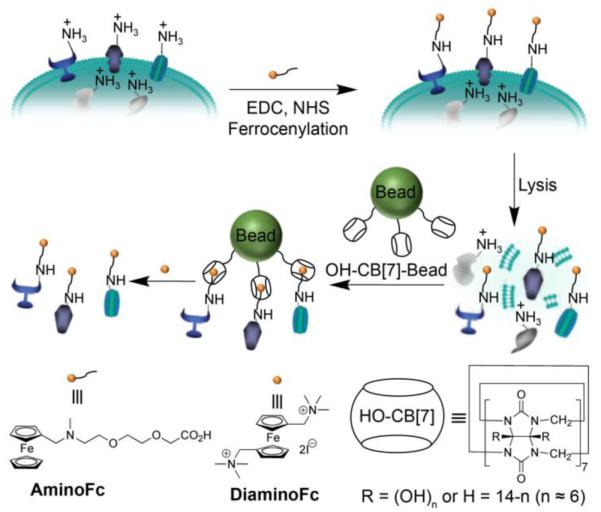
Supramolecular fishing for ferrocenylated membrane proteins. (Reprinted by permission from ref 33. Copyright 2011, Nature Publishing Group).
Supramolecular PEGylation of biopharmaceuticals
The rise of biopharmaceuticals as alternates to small molecule drugs has brought with it challenges due to their generally lower chemical and/or structural stability. Accordingly, great efforts have been devoted to: 1) finding excipients to improve the stability of biopharmaceuticals and 2) developing covalent modification methods (e.g. PEGylation) to confer enhanced stability. As an alternative to the covalent PEGylation, a team comprising the Langer, Anderson, and Isaacs groups have introduced the concept of supramolecular PEGylation.35 For this purpose, the clickable monofunctionalized CB[7]-azide compound was reacted with dibenzocyclooctyne derivatized PEGMW (MW = 5, 10, 30 kD) to yield CB[7]-PEGMW conjugates (Figure 8a). By virtue of their CB[7] cavity, the CB[7]-PEGMW conjugates have the ability to selectively bind to proteins that contain N-terminal hydrophobic amino acids (e.g. phenylalanine of insulin)36 with high affinity (Kd = 2.1 μM, Figure 8b). The results of UV/Vis, fluorescence, GPC, and MALDI-MS indicate that insulin and CB[7]-PEGMW combine to non-covalently PEGylate the N-terminus of insulin. The stability of the insulin•CB[7]-PEGMW complexes against aggregation was assayed by UV/Vis transmittance at 540 nm which revealed that insulin•CB[7]-PEGMW are stable over 100 days whereas insulin alone was found to aggregate within 14 hours. The in vitro biological activity of the 100 day aged insulin•CB[7]-PEGMW samples were measured by dose-response studies of AKT phosphorylation and were superior to that of insulin. Finally, Insulin•CB[7]-PEG30000 maintains normal blood glucose levels of STZ diabetic mice much longer than insulin alone (Figure 8c). The excellent selectivity and affinity of the CB[7]•N-terminal phenylalanine recognition serves as a synthetic equivalent of biotin/(strept)avidin that permits selective non-covalent functionalization of proteins.
Figure 8.
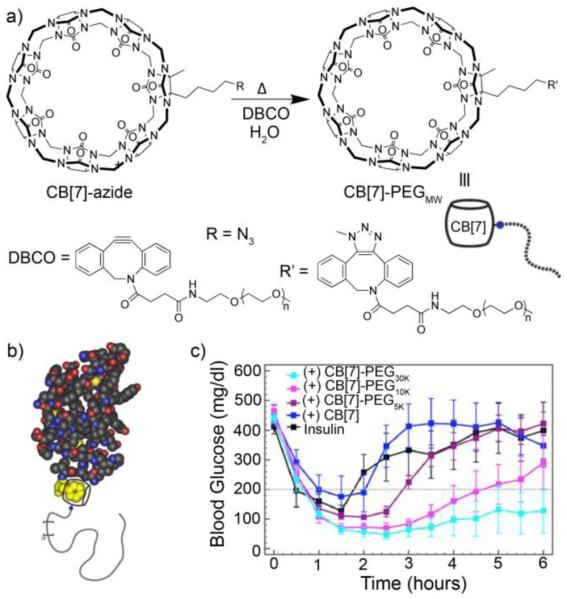
a) Preparation of CB[7]-PEGMW conjugates by click chemistry, b) illustration of the non-covalent Insulin•CB[7]-PEGMW complexes, and c) plot of blood glucose levels of STZ diabetic mice upon treatment with insulin, or CB[7] or CB[7]-PEGMW versus time. (Reprinted by permission from ref 36. Copyright 2016, National Academy of Science).
Vesicle fusion event detected by CB[n]•guest complexation
In the body, the fusion of vesicles is a trigger that leads to cellular processes like vesicle trafficking, exocytosis, and the release of neurotransmitters. In biology, the vesicle fusion process involves SNAREs which act as molecular linkers, but details of the process are still unresolved. Recently, Kim and co-workers demonstrated how a CB[7]•guest affinity pair could be used as a new tool to detect vesicle fusion events by Fluorescence Resonance Energy Transfer (FRET).37 For this purpose, monoallyloxy-CB[7] was reacted with cysteamine by thiol-ene reaction and then reacted with N-hydroxysuccinimidyl-Cy3 to afford Cy3-CB[7] and separately an adamantane derivative was reacted with N-hydroxysuccinimidyl-Cy5 to give Ad-Cy5. As expected, the tight binding between CB[7] and adamantane ammonium ions (Ka ≈ 1012 M−1) results in formation of the Cy3-CB[7]•Ad-Cy5 complex which displays a strong FRET emission at ≈ 670 nm. Next, the Cy3-CB[7] and Ad-Cy5 components were separately incorporated inside v-vesicles which contain embedded v-SNARE VAMP-2 and t-vesicles which contain embedded t-SNARE proteins of approximately 80 nm diameter. Upon vesicle fusion, the Cy3-CB[7]•Ad-Cy5 affinity FRET pair is reconstituted and detected. Interestingly, the authors were able to observe the presence and kinetics of fusion pore flickering events that occur during membrane fusion (Figure 9b). The work suggests approaches to perform multicolor FRET to monitor short lived events in complex biological processes.
Figure 9.
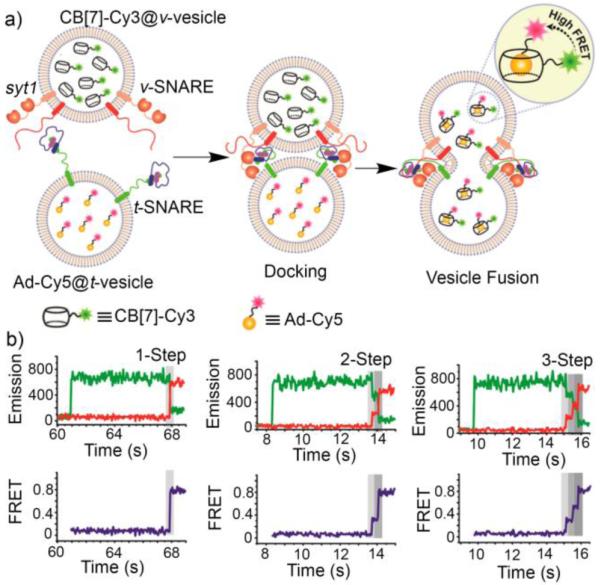
a) Illustration of the v-SNARE / t-SNARE vesicle fusion process monitored by formation of the Cy3-CB[7]•Ad-Cy5 FRET affinity pair, and b) fluorescence intensity time traces for three individual fusion events with different levels of multistep flickering as reported by the Cy3-CB[7]•Ad-Cy5 FRET affinity pair. (Reprinted by permission from ref 37. Copyright 2015, American Chemical Society).
Microfluidic preparation of supramolecular microcapsules
Microcapsules have a variety of important applications including cell encapsulation, drug delivery, and electronic displays. In 2012, Scherman and co-workers showed how a strategic combination of microfluidics, polymers, gold nanoparticles, and CB[8] heteroternary complexation allows the facile preparation of supramolecular microcapsules (Figure 10).38 For this purpose, gold nanoparticles were decorated with methyl viologen ligands and a polyacrylate was appended with 2-naphthol units which jointly enables CB[8] heteroternary complexation. Aqueous solutions of each of the three components were mixed in a microfluidic system in the presence of fluorous oil which resulted in the formation of monodisperse supramolecular nanoparticles (60 μm diameter) held together by CB[8] ternary complexation (Figure 10a,b). Fluorescence microscopy established that the polymer-CB[8]-gold nanoparticle composite resides on the surface of the microspheres (Figure 10c). The internal aqueous cavity can be filled with E. coli that express green fluorescent protein or by FITC labelled dextran (Figure 10c). Reduction of the viologen units by addition of sodium dithionite (Na2SO4) results in one electron reduction of MV2+ to MV+• and dissociation of the heteroternary complex which results in the release of encapsulated cargo (Figure 10d) over 12 hours.
Figure 10.

a) Microfluidic self-assembly of supramolecular microcapsules with high monodispersity by CB[8] heteroternary complexation between naphthol polyacrylates and viologen derived gold nanoparticles. b) proposed microcapsule structure, c) fluorescence imaging of the rhodamine labelled microcapsules loaded with FITC-dextran, and d) fluorescent images of the microcapsule decomposition in Na2S2O4 solution. (Reprinted by permission from ref 38. Copyright 2015, American Association for the Advancement of Science).
Supramolecular control over split luciferase complementation
In pioneering work Urbach and coworkers have demonstrated that peptides with N-terminal FGG sequences undergo self-assembly with CB[8] to form homoternary complexes with high affinity (Ka = 1.5 × 1011 M−2, Figure 11a).39 Very recently, Brunsveld and co-workers used this homoternary complexation inside CB[8] to reconstitute a split luciferase.40 Figure 11b shows schematic representations of two inactive fragments of firefly luciferase that had been appended with N-terminal FGG units (NFluc437 and CFluc398) which bind to each other with Kd = 112 μM (Ka = 8,900 M−1). At concentrations 100-fold below Kd, the NFluc437•CFluc398 assembly does not form significantly, and it was incompetent in the Promega luciferase assay. However, the addition of CB[8] (2.5 μM) to a solution of NFluc437 and CFluc 398 (each 0.5 μM) promotes the assembly of NFluc437•CB[8]•CFluc398 resulting in 20-fold enhancement of luciferase activity. Addition of 3,5-dimethyladamantaneamine (memantine, mem) which is a tight binding partner of CB[8] (Ka = 4.3 × 1011 M−1) to NFluc437•CB[8]•CFluc398 completely shuts down luciferase activity due to competitive sequestration of CB[8] as CB[8]•memantine (Figure 11c). Subsequent addition of CB[8] reactivates luciferase activity. This work points a path toward the use of CB[n] to control the biological function of enzymes and drugs in vitro and even in vivo.
Figure 11.
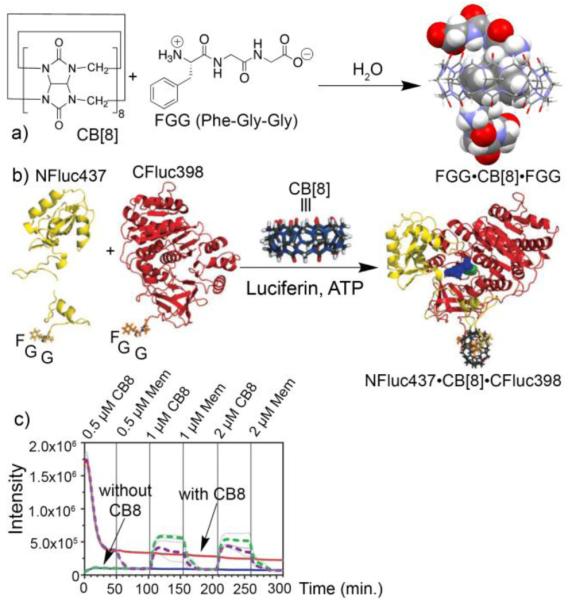
a) Formation of homoternary complex CB[8]•(FGG)2, b) illustration of the reconstitution of NFluc437•CB[8]•CFluc398, and c) kinetic data showing the CB[8] responsive enzymatic catalysis. (Reprinted by permission from refs 39 and 40. Copyright 2006 and 2016, American Chemical Society and Wiley-VCH).
Viologen studded metal organic polyhedron for drug delivery
Isaacs and co-workers recently self-assembled a Fujita-type metal organic polyhedron (MOP1) studded with 24 methylviologen (MV) units and demonstrated that it can bind non-covalently with 24 CB[8] molecules (Figure 12a). This CB[8] capped MOP was further able to form heteroternary complexes with a naphthol derivatized doxorubicin prodrug (dox prodrug) to give the drug loaded MOP2.41 By virtue of its acid labile hydrazone linkage dox prodrug was expected to exhibit pH responsive behavior at the acidic pH of cancer cells. Metabolic assays show that MOP2 is 10-fold more cytotoxic toward HeLa cells than equimolar quantities of doxorubicin prodrug. This enhanced cytotoxicity is due to a combination of enhanced cellular uptake of drug-loaded MOP2 and enhanced dox release as demonstrated by flow cytometry and confocal fluorescence microscopy. These results demonstrate that the strategic merger of the structural features of MOPs with the recognition properties of CB[n] enable interesting biological applications like drug delivery and also suggests their utility for targeted delivery and bioimaging.
Figure 12.
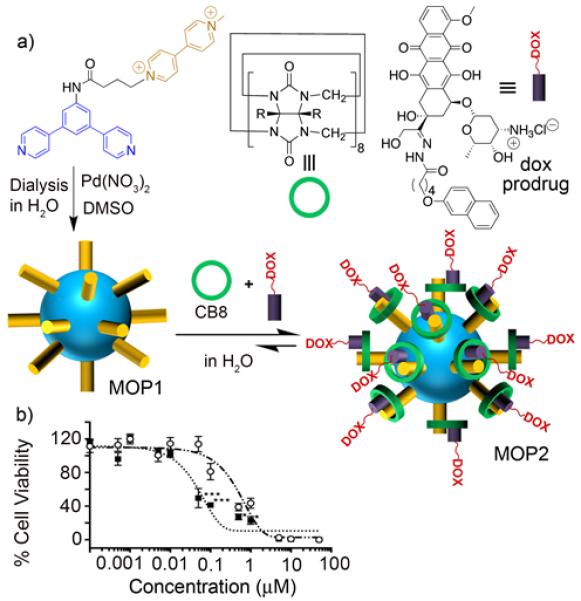
a) Self-assembly of MOP1 and subsequent formation of MOP2 by heteroternary complexation with dox prodrug inside CB[8]. b) Metabolic assay showing the 10-fold enhanced cytotoxicity of MOP2 (⑤) compared to dox prodrug (o). (Reprinted by permission from ref 41. Copyright 2016, American Chemical Society).
3c. Cyclophanes
The molecular recognition properties of synthetic, water-soluble cyclophanes have been investigated for 30 years. Early studies assessed their ability to encapsulate small hydrophobic guests inside the cyclophane cavity and form discrete solution-state complexes. Building on this knowledge, researchers have moved to more sophisticated molecular designs that enable self-assembly of functional nanoscale objects. The following sections describe four different cyclophane systems that have sufficient guest affinity in water to be useful as functional mimics of biotin/(streptavidin). The first two structures (calix[n]arenes and deep cavitands) have bowl-shaped cavities that accept a guest from one side, whereas the other two structures (pillar[n]arenes, tetralactam) have symmetrical cylindrical-shapes that can be threaded by a guest from either end.
Functionalizing liposomes using embedded cyclophanes
The ability to independently alter the upper and lower rims of the calix[n]arene bowl makes it relatively easy to create amphiphilic structures. The group of Liu and coworkers nicely showed that an amphiphilic p-sulfonatocalix[4]arene with long alkyl chains on lower rim that can embed within liposome bilayer membranes (Figure 13). The multiple negatively charged sulfonate groups located on the upper rim promote strong association of a hydrophobic cation within the calixarene cavity. This means that a calix[4]arene modified liposome surface can be readily functionalized with multiple guests without disrupting the bilayer integrity. In one case, the liposome surface was functionalized with a mixture of a biotin targeting ligand (BtPy) and a fluorescent probe (FITCPy). The bifunctionalized liposomes were used for targeted fluorescence imaging of MCF7 human breast cancer cells that express a high number of biotin receptors on the cell surface.42
In a conceptually similar way, Cheng, Hooley and coworkers reported that attachment of anionic groups to the upper rim of a self-folding deep cavitand produces an amphiphilic host structure that can embed within a bilayer membrane (Figure 14) and achieve selective recognition of guests with an attached trimethylammonium (R-NMe3+) group. Guest affinity is promoted by cation-π interactions with the electron rich aromatic surface of the cavitand cavity and binding selectivity is governed by host/guest shape complementarity. The association constants are generally modest but strong enough to promote multivalent association of protein-choline conjugates with a surface-supported bilayer containing the functionalized cavitand. Membrane affinities of choline-tagged proteins (cyt c, myoglobin, hemoglobin) were measured using surface plasmon resonance (SPR) and similar Ka values ~ 105 M−1 were observed regardless of protein size.43 Subsequent studies showed that the membrane-embedded deep cavitand can also anchor choline-conjugated polymerization initiators to the membrane surface and permit in situ synthesis of amine-containing polymethacrylate patches at the water: membrane interface. The micrometer sized polymer patches were subsequently modified in situ to allow display of fluorescent reporters or epitopes for protein immobilization.44
Figure 14.
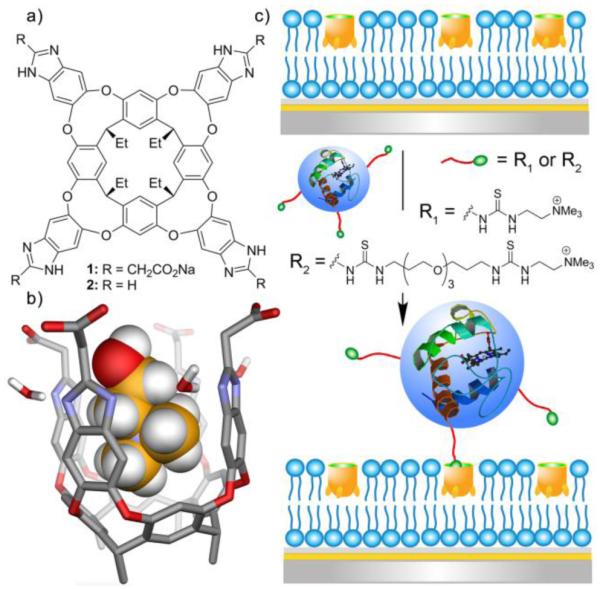
a) Deep cavitand hosts; b) minimized structure of the cavitand:choline complex (SPARTAN, AM1 force field); c) association of choline-labelled cyt c protein to the surface of a membrane bilayer with embedded deep cavitand receptor. (Reprinted with permission from ref 43. Copyright 2014, American Chemical Society).
Pillararene-based drug delivery vesicle
In the last decade pillar[n]arenes have emerged as a new series of versatile cyclophanes. The electron rich side-walls produce strong affinity for electron deficient guest such as pyridinium cations. Not only can pillararenes form discrete host/guest pairs, they can also form amphiphilic complexes that further self-aggregate to create larger aggregates such as micelles and vesicles. A number of research reports have shown that hierarchical pillar[n]arene-based self-assembly is a promising way to fabricate multicomponent nanomaterials with targeting and stimuli-responsive features. An impressive example, reported by Yang and coworkers, is a drug delivery vesicle created by spontaneous self-assembly of an amphiphilic pillar[5]arene complex (Figure 15).45 Each end of the pillar[5]arene host was modified with five cationic tryptophan units and the encapsulated pyridinium guest was linked to a galactose targeting ligand that has affinity for the asialoglycoprotein receptor on cancer cells. Upon sonication, the doubly functionalized and amphiphilic host/guest complex self-assembled to form vesicles. The cancer drug doxorubicin was loaded inside the vesicles and leakage experiments indicated reasonable stability at physiological pH. Cell studies showed that the appended galactose ligands on the vesicle surface promoted asialoglycoprotein mediated endocytosis by HepG2 cells. Furthermore, there was evidence for enhanced vesicle accumulation at the cell nucleus, apparently due to the tryptophan groups acting as multivalent ligands for the nuclear DNA. There was also an enhanced cell killing effect indicating the potential value of this self-assembled drug delivery capsule with dual biomarker targeting properties.
Figure 15.
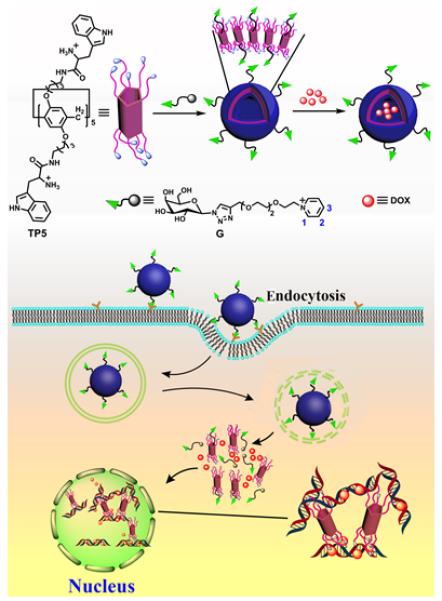
Self-assembly of the the doubly functionalized and amphiphilic pillar[5]arene complex to form vesicles which are used for targeted drug delivery into cancer cells. (Reprinted with permission from ref 45. Copyright 2016, American Chemical Society).
Pre-assembled fluorescent probe for molecular imaging
The tetralactam macrocycle in Figure 16, developed by the Smith group, is a recent addition to the portfolio of high affinity water-soluble cyclophanes.46 The empty macrocycle has a highly preorganized structure and the binding cavity is lined with a synergistic mixture of convergent polar hydrogen bonding sites and hydrophobic surfaces. Most biological association systems, including biotin/(strep)avidin, use similar types of amphiphilic binding pockets to recognize complementary guests with high affinity and shape selectivity. Association studies in water have shown that the tetralactam host binds two structurally similar dyes, squaraine and croconaine, with nanomolar dissociation constants (Figure 16)46,47. The remarkable high affinity is driven by strong hydrogen binding between the dye oxygen atoms and the four macrocycle NH residues, with simultaneous coplanar stacking of the dye aromatic surfaces against the anthracene sidewalls of the macrocycle. Squaraines are highly fluorescent dyes with intense and narrow deep-red absorption and emission bands that make them very attractive for many types of optical imaging, sensing and light harvesting applications. Croconaine dyes are weakly fluorescent, and their sharp and intense absorption bands at ~800 nm are ideal for near-infrared absorption based imaging or laser heating applications. Tetralactam encapsulation of both dye systems leads to favorable changes in dye photophysical properties such as red shifted absorption and emission maxima bands and turn on squaraine fluorescence. Systematic structural studies have evaluated the change in Ka and association kinetics as a function of dye structure and uncovered two interesting trends.48 One is that attachment of long polyethylene glycol chains to the nitrogen atoms at each end of the dye structures has virtually no effect on Ka or kon. But kon is greatly affected by the steric size of the second N-substituent attached to each dye termini. This means that the association kinetics can be fine-tuned by subtle changes in dye structure without altering affinity. The conclusion of these studies is that dyes with N-propyl groups have excellent macrocycle threading properties that permit rapid pre-assembly of targeted fluorescent probes for molecular imaging. The first reported example of this approach to probe pre-assembly is shown in Figure 16.49 A tetralactam macrocycle was threaded onto a fluorescent PEGylated squaraine scaffold containing one or two squaraine docking stations. Appended to the macrocycle periphery were six iminodiacetate units that have weak affinity for bone surfaces. Mouse imaging experiments showed that the pre-assembled probe with twelve bone-targeting iminodiacetate ligands produced more bone accumulation than an analogous pre-assembled probe with six iminodiacetate ligands. The mechanical stability of the pre-assembled fluorescent probes was very high with no probe unthreading after 24 hours inside a living mouse. The results suggest that this versatile pre-assembly method can be used to produce libraries of multivalent near-infrared fluorescent probes for many types of imaging and diagnostic applications. Furthermore, it is likely that this high affinity tetralactam/dye association process can also be exploited for many of the in-situ capture applications described in Figure 1 above.
Figure 16.
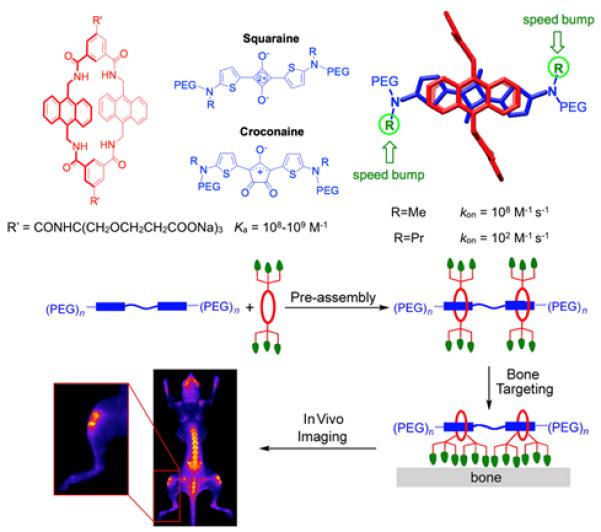
(top) Water-soluble tetralactam host with high affinity for squaraine and croconaine dyes. (bottom) Pre-assembly of near-infrared fluorescent molecular probes with multivalent affinity for bone surfaces. (Adapted with permission from ref 48 and 49. Copyright 2016, American Chemical Society).
3d. Biological based association systems
Biotin/(strept)avidin is not the only naturally occurring biomolecular association system to be exploited for self-assembly applications. There are several quite useful association systems with a biological basis but space constraints only allow a short synopsis of each. One of the best known involves the reversible coordination of chelated transition metal cations such as Ni(II) by proteins or peptides containing multiple histidine residues with imidazole side chains.50 A peptide or protein containing a sequence of six consecutive histidines (His-tagged) forms a moderately stable complex with Ni(II)-nitrilotriacetate (Ni-NTA) in water (Kd ~ 10 μM) that can be reversed by addition of competing free imidazole. Immobilization of the Ni-NTA on a solid surface enables a multivalent binding of the peptide or protein to the surface. This reversible interaction is the basis of many protein affinity chromatography and surface immobilization procedures that are very useful in molecular biology and biotechnology. This multivalent approach to enhanced affinity has also been utilized by researchers who have developed solution-state molecular probes with multiple Ni-NTA units for protein targeting to bilayer membranes, site-specific protein labelling with spectroscopic probes, and labelling of proteins inside live cells or on the cell surface.
Within the proteome there is a myriad of protein dimerization systems where the partners have a complementary hot spot motif on each surface that becomes the association interface.51 The de novo design of peptide dimerization partners is a challenging task and at present there are only a few structural motifs that can be customized for high affinity intermolecular self-assembly. The best known are α-helical peptide systems that coil together like strands in a rope to form a coiled-coil structure. In nature, coiled-coils are formed as key intermediates during important biological functions such as intracellular regulation and membrane fusion. Coiled-coil dimerization forms an interface with a hydrophobic core and periphery of attractive charge interactions. Using this self-assembly paradigm, numerous synthetic coiled-coils pairs have been designed and investigated as building blocks for assembly, targeting, and drug delivery.
Finally, it is worth noting that the selective pairing of encoded DNA or RNA strands is widely used as fabrication and targeting platform in nanomedicine and nanobiotechnology.52 In vivo applications are sometimes limited by susceptibility to fragmentation catalysed by nucleases so unnatural analogies such as peptide nucleic acids (PNA) have been developed.
4. Future directions and challenges
As stated in the introduction, there is a great need for new orthogonal molecular self-assembly systems that can be employed simultaneously in the same analysis sample. This would enable next generation applications such as hierarchical assembly that builds alternating layers or shells, pull down assays that separate different sets of labelled protein targets, pretargeting technologies that deliver independent payloads to different sites, etc. The crucial first step is to develop robust synthetic chemistry that produces suitably functionalized host and guest conjugates. While methods to monofunctionalize CD and CB structures have been reported, additional progress will have an immediate and major positive impact. In some cases, it is reasonably straightforward to monofunctionalize cyclophanes, but it is harder to create cyclophanes that do not self-aggregate in water. Another important near-future goal for the field is to achieve bio-orthogonality. That is, no cross-reactivity between different affinity pairs and no strong competition by the endogenous proteins in biological media. Most likely this will be first demonstrated by employing mixtures of CB systems in conjunction with biotin/(strept)avidin. Another likely strategy to be soon demonstrated is a combination of covalent and non-covalent click chemistry. The number of spontaneous covalent reactions that work well in biological environments is growing. But a limitation with most click reactions is a relatively slow bimolecular rate constant. One possible solution is to create bifunctional probes that use non-covalent recognition to initiate the association event and then consummate the union with irreversible covalent bond formation. For success in vivo there is the added demand that the association pairs are metabolically stable and also non-toxic. For example, a possible concern using pillararenes for in vivo applications is biological oxidation of the electron rich rings within the structures. Looking to the longer term, it is likely that synthetic mimics of biotin/(strept)avidin can be created with triggered binding ability. Using light or related stimuli it may be possible to activate binding events with high spatiotemporal control. This will lead to new applications that presently have not yet been contemplated.
Figure 13.
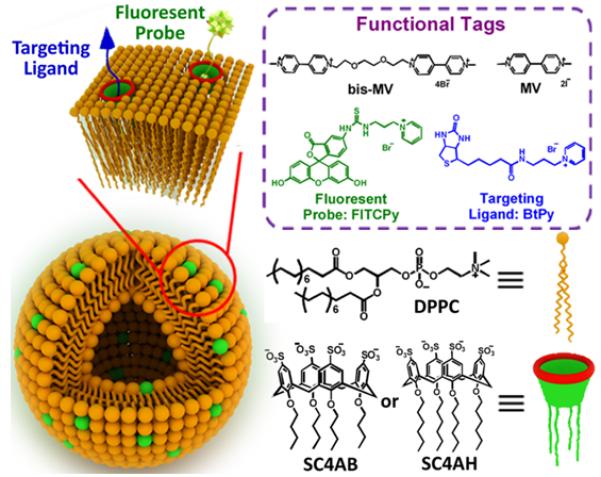
Schematic illustration of multifunctional liposome and noncovalent surface modification via calixarene host guest interaction. (Reprinted with permission from ref 42. Copyright 2016, American Chemical Society).
Key learning points.
Applications of biotin/(strept)avidin self-assembly can be divided into two operationally discrete groups, pre-assembly and in-situ capture.
Most synthetic container molecules have relatively low guest affinities in water.
Molecular design strategies to increase host affinity include preorganization, electrostatics, hydrophobic effect, and multivalency.
Most of the newer self-assembly applications rely on high affinity host•guest complexes of cucurbit[n]urils (CB[n]).
The synthetic flexibility of organic cyclophanes provides new opportunities for increased functionality
Acknowledgements
We are grateful for funding support from the NSF (CHE-1404911 to L.I. and CHE-1401783 to B.S.) and NIH (CA168365 to L.I. and GM059078 to B.S.).
Author Photos Bios

Wenqi Liu (left) was born in Shandong, China. He received his BS in chemistry from Shandong University in 2013, where he worked with Prof. Aiyou Hao. He is currently a PhD student in Prof. Bradley D. Smith’s research group at the University of Notre Dame, USA. His ongoing research focuses on molecular recognition using a tetralactam macrocycle in water and development of preassembly and in situ capture technology.
Bradley D. Smith (right) was born in Melbourne, Australia. He joined the Department of Chemistry and Biochemistry at the University of Notre Dame in 1991 and currently he is Director of the Notre Dame Integrated Imaging Facility. He also serves as Associate Editor of the ACS journal Bioconjugate Chemistry. He has published around 230 research papers in the fields of bioorganic and supramolecular chemistry applied to biological systems. A current research topic is the creation of molecular imaging probes for detecting cancer and microbial infections.

Soumen K. Samanta (left) was born in West Bengal, India and graduated with a MSc degree from IIT Kanpur (2007) under the direction of Prof. P. K. Bharadwaj. Subsequently, he was received a PhD degree from Universität Siegen (2013) studying molecular machines with Prof. Michael Schmittel. Currently, Soumen is a postdoctoral fellow at the University of Maryland, USA in the Isaacs lab studying drug delivery systems incorporating metal organic cages and cucurbit[n]uril.
Lyle Isaacs (right) was born in New York City, New York, where he attended the Bronx High School of Science. After obtaining a BS degree from the University of Chicago in 1991, he performed doctoral research with Professor Francois Diederich (MS 1992, UCLA; PhD 1995, ETH-Zürich). After an NIH postdoctoral fellowship with Professor George Whitesides at Harvard, he joined the faculty at the Department of Chemistry and Biochemistry at the University of Maryland, College Park, in 1998 where he is currently Professor of Chemistry. Lyle was elected Fellow of the American Association for the Advancement of Science in 2013 for his work on the development of cucurbit[n]uril molecular containers and self-sorting systems.
Notes and references
- 1.Dundas CM, Demonte D, Park S. Appl. Microbiol. Biotechnol. 2013;97:9343–9353. doi: 10.1007/s00253-013-5232-z. [DOI] [PubMed] [Google Scholar]
- 2.Patra M, Zarschler K, Pietzsch H-J, Stephan H, Gasser G. Chem. Soc. Rev. 2016;45:6415–6431. doi: 10.1039/c5cs00784d. [DOI] [PubMed] [Google Scholar]
- 3.Lesch HP, Kaikkonen MU, Pikkarainen JT, Ylä-Herttuala S. Expert Opin. Drug Deliv. 2010;7:551–564. doi: 10.1517/17425241003677749. [DOI] [PubMed] [Google Scholar]
- 4.Heinisch T, Ward TR. Acc. Chem. Res. 2016;49:1711–1721. doi: 10.1021/acs.accounts.6b00235. [DOI] [PubMed] [Google Scholar]
- 5.Marttila AT, Laitinen OH, Airenne KJ, Kulik T, Bayer EA, Wilchek M, Kulomaa MS. FEBS Lett. 2000;467:31–36. doi: 10.1016/s0014-5793(00)01119-4. [DOI] [PubMed] [Google Scholar]
- 6.Lee JM, Kim JA, Yen TC, Lee IH, Ahn B, Lee Y, Hsieh CL, Kim HM, Jung Y. Angew. Chem., Int. Ed. 2016;55:3393–3397. doi: 10.1002/anie.201510885. [DOI] [PubMed] [Google Scholar]
- 7.Lim KH, Huang H, Pralle A, Park S. Biotechnol. Bioeng. 2013;110:57–67. doi: 10.1002/bit.24605. [DOI] [PubMed] [Google Scholar]
- 8.Chamma I, Letellier M, Butler C, Tessier B, Lim K-H, Gauthereau I, Choquet D, Sibarita J-B, Park S, Sainlos M, Thoumine O. Nat. Commun. 2016;7:10773. doi: 10.1038/ncomms10773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baumann F, Bauer MS, Milles LF, Alexandrovich A, Gaub HE, Pippig DA. Nat. Nanotechnol. 2016;11:89–94. doi: 10.1038/nnano.2015.231. [DOI] [PubMed] [Google Scholar]
- 10.Orr GA. J. Biol. Chem. 1981;256:761–766. [PubMed] [Google Scholar]
- 11.DeChancie J, Houk KN. J. Am. Chem. Soc. 2007;129:5419–5429. doi: 10.1021/ja066950n. and referecnes therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song J, Li Y, Ji C, Zhang JZH. Sci. Rep. 2015;5:7906. doi: 10.1038/srep07906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chu V, Stayton PS, Freitag S, Le Trong I, Stenkamp RE. Protein Sci. 2008;7:848–859. doi: 10.1002/pro.5560070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Terai T, Kohno M, Boncompain G, Sugiyama S, Saito N, Fujikake R, Ueno T, Komatsu T, Hanaoka K, Okabe T, Urano Y, Perez F, Nagano T. J. Am. Chem. Soc. 2015;137:10464–10467. doi: 10.1021/jacs.5b05672. [DOI] [PubMed] [Google Scholar]
- 15.Baugh L, Le Trong I, Stayton PS, Stenkamp RE, Lybrand TP. Biochemistry. 2016;55:5201–5203. doi: 10.1021/acs.biochem.6b00698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu F, Zhang J, Mei Y. Sci. Rep. 2016;6:27190. doi: 10.1038/srep27190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Houk KN, Leach AG, Kim SP, Zhang X. Angew. Chem., Int. Ed. 2003;42:4872–4897. doi: 10.1002/anie.200200565. and references therein. [DOI] [PubMed] [Google Scholar]
- 18.C DJ, Cram JM. Container Molecules and Their Guests. The Royal Society of Chemistry; 1997. [Google Scholar]
- 19.Honig B, Nicholls A. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- 20.Bom A, Bradley M, Cameron K, Clark JK, Van Egmond J, Feilden H, MacLean EJ, Muir AW, Palin R, Rees DC, Zhang M-Q. Angew. Chem., Int. Ed. 2002;41:265–270. doi: 10.1002/1521-3773(20020118)41:2<265::aid-anie265>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 21.Snyder PW, Mecinovic J, Moustakas DT, Thomas SW, Harder M, Mack ET, Lockett MR, Héroux A, Sherman W, Whitesides GM. Proc. Natl. Acad. Sci. U. S. A. 2011;108:17889–17894. doi: 10.1073/pnas.1114107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Biedermann F, Nau WM, Schneider HJ. Angew. Chem., Int. Ed. 2014;53:11158–11171. doi: 10.1002/anie.201310958. [DOI] [PubMed] [Google Scholar]
- 23.Biedermann F, Uzunova VD, Scherman OA, Nau WM, De Simone A. J. Am. Chem. Soc. 2012;134:15318–15323. doi: 10.1021/ja303309e. [DOI] [PubMed] [Google Scholar]
- 24.Mammen M, Choi SK, Whitesides GM. Angew. Chem., Int. Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Harris DC, Saks BR, Jayawickramarajah J. J. Am. Chem. Soc. 2011;133:7676–7679. doi: 10.1021/ja2017366. [DOI] [PubMed] [Google Scholar]
- 26.Fathalla M, Neuberger A, Li SC, Schmehl R, Diebold U, Jayawickramarajah J. J. Am. Chem. Soc. 2010;132:9966–9967. doi: 10.1021/ja1030722. [DOI] [PubMed] [Google Scholar]
- 27.Harada A, Kobayashi R, Takashima Y, Hashidzume A, Yamaguchi H. Nat. Chem. 2011;3:34–7. doi: 10.1038/nchem.893. [DOI] [PubMed] [Google Scholar]
- 28.Shetty D, Khedkar JK, Park KM, Kim K. Chem. Soc. Rev. 2015;44:8747–8761. doi: 10.1039/c5cs00631g. [DOI] [PubMed] [Google Scholar]
- 29.Liu S, Ruspic C, Mukhopadhyay P, Chakrabarti S, Zavalij PY, Isaacs L. J. Am. Chem. Soc. 2005;127:15959–15967. doi: 10.1021/ja055013x. [DOI] [PubMed] [Google Scholar]
- 30.Jeon WS, Moon K, Park SH, Chun H, Ko YH, Lee JY, Lee ES, Samal S, Selvapalam N, Rekharsky MV, Sindelar V, Sobransingh D, Inoue Y, Kaifer AE, Kim K. J. Am. Chem. Soc. 2005;127:12984–12989. doi: 10.1021/ja052912c. [DOI] [PubMed] [Google Scholar]
- 31.Rekharsky MV, Mori T, Yang C, Ko YH, Selvapalam N, Kim H, Sobransingh D, Kaifer AE, Liu S, Isaacs L, Chen W, Moghaddam S, Gilson MK, Kim K, Inoue Y. Proc. Natl. Acad. Sci. U. S. A. 2007;104:20737–20742. doi: 10.1073/pnas.0706407105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao L, Sekutor M, Zavalij PY, Mlinaric-Majerski K, Glaser R, Isaacs L. Angew. Chem., Int. Ed. 2014;53:988–993. doi: 10.1002/anie.201309635. [DOI] [PubMed] [Google Scholar]
- 33.Lee D-W, Park K, Banerjee M, Ha S, Lee T, Suh K, Paul S, Jung H, Kim J, Selvapalam N, Ryu S, Kim K. Nat. Chem. 2011;3:154–159. doi: 10.1038/nchem.928. [DOI] [PubMed] [Google Scholar]
- 34.Li W, Bockus AT, Vinciguerra B, Isaacs L, Urbach AR. Chem. Commun. 2016;52:8537–8540. doi: 10.1039/c6cc03193e. [DOI] [PubMed] [Google Scholar]
- 35.Webber MJ, Appel EA, Vinciguerra B, Cortinas AB, Thapa LS, Jhunjhunwala S, Isaacs L, Langer R, Anderson DG. Proc. Natl. Acad. Sci. U. S. A. 2016;113:14189–14194. doi: 10.1073/pnas.1616639113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Urbach AR, Ramalingam V. Isr. J. Chem. 2011;51:664–678. [Google Scholar]
- 37.Gong B, Choi B-K, Kim J-Y, Shetty D, Ko YH, Selvapalam N, Lee NK, Kim K. J. Am. Chem. Soc. 2015;137:8908–8911. doi: 10.1021/jacs.5b05385. [DOI] [PubMed] [Google Scholar]
- 38.Zhang J, Coulston RJ, Jones ST, Geng J, Scherman OA, Abell C. Science. 2012;335:690. doi: 10.1126/science.1215416. [DOI] [PubMed] [Google Scholar]
- 39.Heitmann LM, Taylor AB, Hart PJ, Urbach AR. J. Am. Chem. Soc. 2006;128:12574–12581. doi: 10.1021/ja064323s. [DOI] [PubMed] [Google Scholar]
- 40.Bosmans RPG, Briels JM, Milroy LG, de Greef TFA, Merkx M, Brunsveld L. Angew. Chem., Int. Ed. 2016;55:8899–8903. doi: 10.1002/anie.201602807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Samanta SK, Moncelet D, Briken V, Isaacs L. J. Am. Chem. Soc. 2016;138:14488–14496. doi: 10.1021/jacs.6b09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang YX, Zhang YM, Wang YL, Liu Y. Chem. Mater. 2015;27:2848–2854. [Google Scholar]
- 43.Ghang YJ, Lloyd JJ, Moehlig MP, Arguelles JK, Mettry M, Zhang X, Julian RR, Cheng Q, Hooley RJ. Langmuir. 2014;30:10161–10166. doi: 10.1021/la502629d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perez L, Ghang YJ, Williams PB, Wang Y, Cheng Q, Hooley RJ. Langmuir. 2015;31:11152–11157. doi: 10.1021/acs.langmuir.5b03124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang K, Chang Y, Wen J, Lu Y, Pei Y, Cao S, Wang F, Pei Z. Chem. Mater. 2016;28:1990–1993. [Google Scholar]
- 46.Peck EM, Liu W, Spence GT, Shaw SK, Davis AP, Destecroix H, Smith BD. J. Am. Chem. Soc. 2015;137:8668–8672. doi: 10.1021/jacs.5b03573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu W, Peck EM, Smith BD. J. Phys. Chem. B. 2016;120:995–1001. doi: 10.1021/acs.jpcb.5b11961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu W, Peck EM, Hendzel KD, Smith BD. Org. Lett. 2015;17:5268–5271. doi: 10.1021/acs.orglett.5b02633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Peck EM, Battles PM, Rice DR, Roland FM, Norquest KA, Smith BD. Bioconjug. Chem. 2016;27:1400–1410. doi: 10.1021/acs.bioconjchem.6b00173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.You C, Piehler J. Anal. Bioanal. Chem. 2014;406:3345–3357. doi: 10.1007/s00216-014-7803-y. [DOI] [PubMed] [Google Scholar]
- 51.Apostolovic B, Danial M, Klok H-A. Chem. Soc. Rev. 2010;39:3541. doi: 10.1039/b914339b. [DOI] [PubMed] [Google Scholar]
- 52.Patwa A, Gissot A, Bestel I, Barthélémy P. Chem. Soc. Rev. 2011;40:5844–5854. doi: 10.1039/c1cs15038c. [DOI] [PubMed] [Google Scholar]