

Abstract
Background and Objective
Afatinib is an oral irreversible ErbB-Family Blocker indicated for treatment of patients with EGFR mutation positive advanced non-small cell lung cancer. This trial assessed whether renal impairment influences the pharmacokinetics and safety of afatinib.
Methods
This was an open-label, single-dose study. Pharmacokinetic parameters after afatinib 40 mg were investigated in subjects with moderate (n = 8) or severe (n = 8) renal impairment (estimated glomerular filtration rate 30–59 mL/min/1.73 m2 and 15–29 mL/min/1.73 m2, respectively) and healthy matched controls (n = 14). Plasma and urine samples were collected before and up to 14 days after dosing for pharmacokinetic and plasma protein-binding assessment. Primary endpoints were area under the plasma concentration–time curve from time zero to the last quantifiable concentration (AUClast) and maximum plasma concentration (C max) between subjects with renal impairment and healthy matched controls.
Results
Pharmacokinetic profiles and plasma protein binding were similar in all groups. The extent of exposure, as indicated by AUClast and C max, was generally similar between the matched treatment groups, with the exception of the geometric mean ratio of AUClast for subjects with severe renal impairment, which showed a trend towards a higher value compared with matched healthy subjects (150.0 % [90 % CI 105.3–213.7]) Inter-individual variability was moderate (geometric mean coefficient of variation 28–39 % for moderate impairment, 34–42 % for severe impairment). Afatinib was well tolerated and urinary excretion was minimal.
Conclusion
Moderate-to-severe renal impairment had a minor influence on the pharmacokinetics of afatinib that was within the observed inter-individual variability, suggesting that afatinib treatment can be considered in this patient population.
Registered at ClinicalTrials.gov as NCT02096718.
Key Points
| For subjects with moderate renal impairment, there was no clinically relevant change in the pharmacokinetics of afatinib. |
| For subjects with severe renal impairment, the mean exposure [maximum plasma concentration (C max) and area under the plasma concentration–time curve from time zero to time of last quantifiable concentration (AUClast)] values of afatinib were increased by 22 and 50 %, respectively. Inter-individual variability was moderate (geometric mean coefficient of variation 34–42 %). |
| Renal impairment had no effect on the plasma protein binding of afatinib, and urinary excretion of afatinib was minimal. |
Introduction
Afatinib is a potent and selective, irreversible blocker of the ErbB-Family of transmembrane tyrosine kinase receptors, which includes epidermal growth factor receptor (EGFR), and human epidermal growth factor receptors 2, 3, and 4 (HER2, HER3, and HER4) [1, 2]. It has activity in a wide range of tumour cell lines that harbour a hyperactivated ErbB signalling network.
Afatinib has been studied in patients with various solid tumours, including advanced non-small cell lung cancer (NSCLC), squamous cell carcinoma of the lung, and metastatic head and neck cancer [3–6]. It is approved in many countries for treatment of patients with EGFR mutation positive NSCLC [7], and is undergoing regulatory review for treatment of patients with advanced NSCLC of squamous cell histology [8]. In patients with solid tumours, afatinib is absorbed with maximum plasma concentration (C max) achieved at 2–5 h, and is eliminated with an apparent terminal elimination half-life of about 37 h at steady state [9]. Afatinib exposure (C max and area under the plasma concentration–time curve [AUC]) values increase slightly more than proportionally over the dose range 20–50 mg [9]. Following a high-fat/high-caloric meal, afatinib exposure is reduced by ~39–50 % [11], it is, therefore, recommended that patients take afatinib fasted [7]. The major route of elimination of afatinib is via the faeces (85 %) as unchanged drug, with less than 5 % eliminated through the renal pathway [12]. Afatinib shows moderate-to-high binding to plasma proteins (~95 %) in previous studies [13].
Because cancer patients with severe renal impairment (i.e. creatinine clearance [CLCR] of <30 mL/min) were not included in the previous clinical trials, treatment with afatinib in this population has not been recommended to date [7, 14]. The rationale for performing the current trial was to be able to include the treatment of such patients in the dosing recommendations.
Methods
This was a single-centre, open-label, sequential-group, single-dose study (ClinicalTrials.gov identifier: NCT02096718) using a matched-group design. The study protocol was approved by an independent ethics committee (Ethikkommission der Landesärztekammer Schleswig–Holstein, Bad Segeberg, Germany) and competent authority (BfArM, Bonn, Germany), and conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from all subjects before study entry.
Subjects
Male or female non-smoker subjects with renal impairment, aged 18–79 years, and with a body mass index (BMI) between 18.5 and 34.0 kg/m2, were enrolled into two pre-specified groups based on their estimated glomerular filtration rate (eGFR), calculated using the Modification of Diet in Renal Disease (MDRD) formula [15]: (a) moderate renal impairment, eGFR of 30–59 mL/min/1.73 m2, and (b) severe renal impairment, eGFR of 15–29 mL/min/1.73 m2. Each individual was matched with a healthy control (eGFR ≥90 mL/min/1.73 m2) of the same gender, age (±10 %), race, and BMI (±10 %) (Table 1). Healthy subjects were eligible based on the lack of clinically relevant history or physical examination and electrocardiography (ECG) findings, vital signs, and clinical laboratory tests.
Table 1.
Baseline characteristics of the study population included in pharmacokinetic analysis
| Characteristic | Normal renal function (n = 14)a | Moderate renal impairment (n = 8) | Severe renal impairment (n = 8) |
|---|---|---|---|
| Male [no. (%)] | 7 (50.0) | 3 (37.5) | 6 (75.0) |
| Age (years) | 62.1 ± 11.4 | 68.6 ± 11.0 | 61.0 ± 11.9 |
| White race [no. (%)] | 14 (100.0) | 8 (100.0) | 8 (100.0) |
| BMI (kg/m2) | 26.0 ± 3.2 | 26.1 ± 4.6 | 27.6 ± 3.4 |
| eGFR (mL/min/1.73 m2) | 100.9 ± 10.9 | 44.1 ± 11.1 | 20.0 ± 3.9 |
All data are mean ± SD unless stated otherwise. eGFR calculated using the Modification of Diet in Renal Disease Formula [15]
BMI body mass index, eGFR estimated glomerular filtration rate, SD standard deviation
aMatched to subjects with moderate and severe renal impairment. Two healthy subjects were matched both to subjects with severe impairment and to subjects with moderate impairment
The main exclusion criteria in renally impaired subjects were severe cardiovascular disorders in the preceding 6 months; significant or recent acute gastrointestinal disorders with diarrhoea as a major symptom; interstitial lung disease; changes in usual medication in the 4 weeks prior to trial drug administration (other than discontinuation of medications known to interact with P–glycoprotein); history of photosensitivity or recurrent rash (also excluded in healthy subjects); and haemoglobin <9 g/dL indicating severe anaemia. For all study participants, treatment with any medication with a half-life of ≥24 h in the month before or during the trial, or potent P-glycoprotein inhibitors or inducers in the 2 weeks before or during the trial was prohibited. Women who were pregnant, breast-feeding, or those of child-bearing potential not using adequate contraception for 3 months before and during the study were excluded. Males unwilling to use adequate contraception for 3 months after afatinib administration were excluded.
Study Design and Treatments
After an overnight fast (≥10 h), subjects received a single 40 mg dose of afatinib (Giotrif®, Gilotrif®, Boehringer Ingelheim Pharma GmbH & Co. KG, Germany), administered as a film-coated tablet with 240 mL of water in the sitting or standing position. Water was allowed ad libitum except 1 h before and 2 h after dosing, and standardised meals were served at least 4 h after dosing. Subjects were closely observed in the clinic for at least 48 h (healthy subjects and subjects with moderate renal impairment) or at least 72 h (subjects with severe renal impairment) after dosing until discharge, and, subsequently, returned to the clinic for follow-up blood sampling and urine tests. A final safety follow-up visit was arranged 15–17 days post-dosing.
Pharmacokinetic Evaluation
Venous blood samples were collected into potassium-EDTA-anticoagulant tubes before and 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 7, 8, 9, 12, 24, 36, 48, 60, 72, 96, 120, 144, 192, 240, and 312 h after dosing. For pharmacokinetic assessments, approximately 2.7 mL of blood was collected at each time point. All samples were centrifuged within 30 min of collection at ~2000 to 4000 × g (4 °C) for 10 min. Plasma was transferred to two separate tubes and frozen immediately and stored at −20 °C until analysis. A 15 mL sample for determination of plasma protein binding was also collected in 3 × 4.9 mL vials (monovettes coated with EDTA) on day 1 before dosing and centrifuged at 4000 rpm for 10 min at 4 °C. Plasma samples were stored at −20 °C until analysis. Urine was collected in containers before and 0–4, 4–8, 8–12, 12–24, 24–48, and 48–72 h after dosing. To prevent adsorption losses of afatinib, a detergent (Tween 20) was added to the containers before urine collection. For each collection, the urine was weighed and homogenised, and aliquots were stored at −20 °C until analysis.
Plasma and urine concentrations of the free base of afatinib (BIBW 2992 BS) were analysed using validated high-performance liquid chromatography–tandem mass spectrometry methods (Nuvisan GmbH, Neu-Ulm, Germany) [13]. Plasma concentrations within the validated concentration range (0.100–50.0 ng/mL for plasma, 5.00–1000 ng/mL for urine) were used to calculate the pharmacokinetic parameters. Assay performance was assessed by back-calculation of calibration standards, tabulation of the standard curve fit function parameters, and measurement of quality control samples. Quality control samples spiked at three concentrations were analysed for each matrix. Mean assay accuracy [deviation from nominal value (%)] ranged from 3.0 to 3.9 % for plasma and from −0.4 to 1.1 % for urine. Mean precision [coefficient of variation (CV) %] was ≤8.8 % for plasma and ≤5.8 % for urine, respectively. Incurred samples re-analysis confirmed the initial value in 96.3 and 100 % for plasma and urine, thereby demonstrating good assay reproducibility.
In vitro plasma protein binding of afatinib was determined in pre-dose plasma samples after spiking of 150 nmol/L (=72.9 ng/mL) [14C]-radiolabelled afatinib using equilibrium dialysis. Previous in-house studies demonstrated the suitability of this method (Boehringer Ingelheim, Biberach, Germany) [13].
Safety Evaluation
The safety of afatinib was assessed by 12-lead ECG, vital signs (pulse, blood pressure), routine laboratory assessments, physical examination, adverse event (AE) reporting, and assessment of global tolerability by the investigator. AEs were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) Version 3.0.
For safety reasons, individuals with renal impairment were treated sequentially, with moderate renally impaired subjects treated first. Dosing of subjects with renal impairment was performed following a 3 + 5 scheme: with three subjects being initially treated (1 subject dosed per day). Dosing of the remaining 5 subjects was only allowed in the absence of drug-related toxicity of CTCAE grade ≥3 in the first 3 subjects. The decision to proceed with further afatinib administration in the severe renal impairment group was based on a combined assessment of drug-related AEs and interim pharmacokinetic measurements after dosing of subjects in the moderate renal impairment group and their respective controls to more accurately predict exposure and half-life of afatinib in subjects with severe renal impairment. At least 7 days separated dosing of the moderate and severe renal impairment groups. Dosing was continued if no subject had a drug-related AE of CTCAE grade ≥3, or less than 2 subjects had a drug-related AE of CTCAE grade ≥2 within 3 days after dosing. Afatinib 40 mg was used in this study, because it is the daily dose approved for continuous treatment of NSCLC [7].
Statistical and Pharmacokinetic Analysis
The study planned to recruit up to 32 subjects with the aim of entering 8 subjects with moderate renal impairment, 8 subjects with severe renal impairment, and 8–16 healthy subjects matched individually to the subjects with renal impairment. A total of 32 subjects receiving 40 mg afatinib for the primary analysis were judged an adequate sample size, in agreement with regulatory guidance of pharmacokinetic studies in subjects with impaired renal function [16–18].
Standard non-compartmental methods were used to calculate plasma pharmacokinetic parameters. Area under the plasma concentration–time curve was calculated using the log-linear trapezoidal rule from time zero (pre-dose) up to the time of the last quantifiable concentration (AUClast) and extrapolated to infinity (AUC∞). AUC∞ was calculated by the following equation: AUClast + /K e, where is the concentration of afatinib predicted by the regression line for the time of last quantifiable measurement and K e is the terminal rate elimination constant obtained by log-linear regression of the terminal elimination phase data. Terminal half-life was calculated from the terminal rate constant, and renal clearance (CLR,0–72) was calculated as the amount of afatinib excreted in urine/plasma AUC over 72 h.
The aim of the study was to estimate the relative bioavailability of afatinib in subjects with renal impairment compared with subjects with normal renal function. The primary pharmacokinetic endpoints were AUClast and C max; AUC∞ was a secondary endpoint. The log-transformed AUC and C max values for afatinib were compared between groups using an analysis of variance (ANOVA). The model included ‘renal function group’ as fixed effect. For the pairwise comparison between the groups, the difference between the expected means was estimated by the difference in the corresponding least-square means (point estimate); the 90 % two-sided confidence interval (CI, corresponding to a two-sided alpha level of 10 %) based on the t-distribution and P value were also computed. The difference and CI were then back-transformed to the original scale to provide the point estimate (geometric mean) and interval estimates for the ratio of renal impaired subjects and matched healthy subjects. The main focus of the study was to estimate the magnitude of effects; as such, no hypothesis was tested and no equivalence range was specified. CIs and P values were computed, but were interpreted in the context of the exploratory nature of the study; that is, CIs were considered as interval estimates for effects, while P values were considered as an exploratory measure of evidence for effects in the data (though a P value <0.1 indicates formal significance).
Other parameters were presented as descriptive statistics (arithmetic and geometric means [gMean], standard deviation, and gCV). Non-compartmental analysis of plasma concentration–time data was performed using the Phoenix WinNonlin® version 6.3 software (Pharsight Corporation, Cary, NC, USA). Statistical analyses were performed using SAS®, version 9.4 (SAS Institute Inc., Cary, NC, USA).
Results
Thirty subjects were enrolled and completed the study (14 healthy subjects, 8 subjects with moderate renal impairment, and 8 with severe renal impairment). Two healthy subjects were matched both to subjects with severe impairment and to subjects with moderate impairment. All 30 subjects were included in the pharmacokinetic and safety analyses. All participants were Caucasian. Baseline characteristics of the volunteers are given in Table 1. As the healthy controls were matched to their respective patients, no relevant differences in baseline characteristics existed between the groups.
Pharmacokinetics of Afatinib
There were no notable differences in the plasma concentration–time profiles between subjects with moderate or severe renal impairment and their matched healthy controls (Fig. 1). Afatinib showed a biphasic disposition profile, and a long terminal phase was detected extending out to the last sampling point of 312 h. The extent of exposure, as indicated by AUClast and C max, was generally similar between the matched treatment groups (Table 2), with the exception of the gMean ratio of AUClast for subjects with severe renal impairment, which showed a trend towards a higher value compared with matched healthy subjects (two-sided P value <0.1, Table 3). The secondary endpoint AUC∞ showed similar results to AUClast.
Fig. 1.
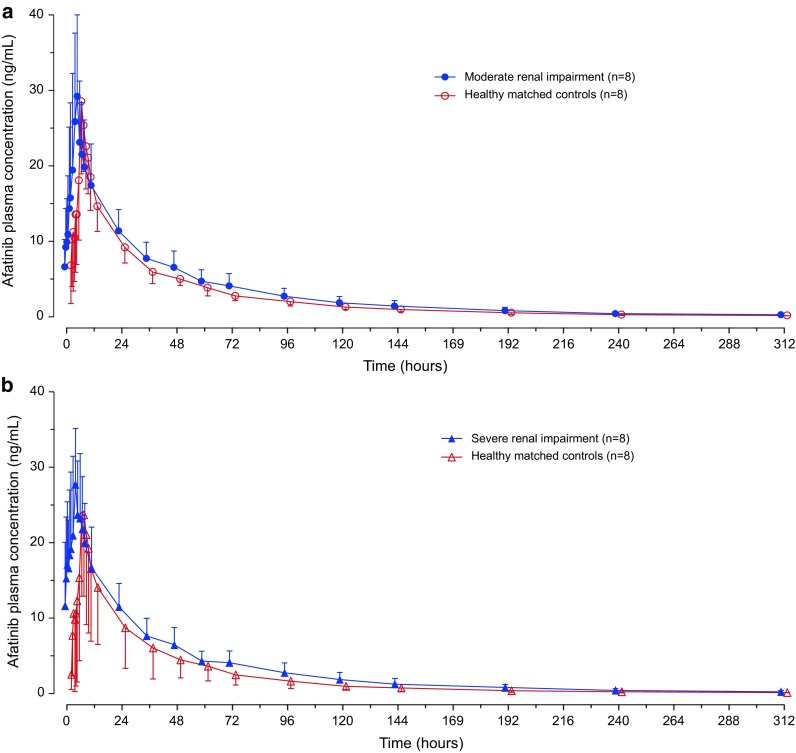
Arithmetic mean (± standard deviation [SD]) plasma concentration–time profiles for afatinib following a single 40 mg dose in subjects with moderate (n = 8) (a) and severe (n = 8) (b) renal impairment compared with matched healthy controls (linear scale). For clarity, data points have been shifted on the x-axis to avoid overlap of error bars and only one whisker of SD is shown per group (renal impairment +SD, healthy subjects −SD)
Table 2.
Geometric mean pharmacokinetic parameters after a single dose of 40 mg afatinib for subjects with moderate or severe renal impairment and matched healthy controls (as grouped by eGFR)
| Parameter and unit | Moderate renal impairment (n = 8) | Matched controls to moderate renal impairment (n = 8) | Severe renal impairment (n = 8) | Matched controls to severe renal impairment (n = 8) |
|---|---|---|---|---|
| Primary endpoints | ||||
| AUClast (ng·h/mL) | 948 (32.9) | 776 (22.9) | 952 (31.3) | 634 (50.8) |
| C max (ng/mL) | 28.7 (44.0) | 28.4 (32.2) | 28.2 (24.5) | 23.2 (42.1) |
| Secondary endpoint | ||||
| AUC∞ (ng·h/mL) | 976 (32.5) | 797 (22.7) | 980 (31.9) | 653 (49.8) |
| Other endpoints | ||||
| t max (h) | 6.0 (5.0 to 8.0) | 5.0 (5.0 to 6.0) | 5.0 (3.0 to 7.0) | 5.5 (5.0 to 7.0) |
| t 1/2 (h) | 73.4 (21.6) | 81.7 (19.4) | 80.2 (15.9) | 75.5 (28.5) |
| CLR,0–72 (mL/min) | 16.1 (26.8) | 25.4 (46.7)a | 8.7 (46.6)a | 29.6 (25.8)b |
| fe0–72 (%) | 1.65 (26.7) | 2.12 (44.2)a | 0.91 (18.5)a | 2.39 (33.5)b |
| Protein binding (%) | 94.7 ± 0.7 | 94.8 ± 0.2 | 94.4 ± 1.0 | 94.7 ± 0.6 |
Data are presented as geometric mean (geometric CV %) or mean ± SD for protein binding or median (range) for t max
AUC last area under the drug plasma concentration–time curve from time 0 to the time of the last quantifiable data point, AUC ∞ area under the drug plasma concentration–time curve from time 0 to infinity, C max maximum drug concentration in plasma, CL R,0-72 renal clearance over 72 h, CV % coefficient of variation (%), fe 0–72 fraction of oral dose observed in urine over 72 h, t 1/2 terminal elimination half-life, t max time to reach C max
a n = 7
b n = 5
Table 3.
gMean ratios (%) for AUC and C max of afatinib 40 mg (subjects with renal impairment versus controls), with 90 % confidence intervals (CIs) in each renal impairment group based on estimated glomerular filtration rate (eGFR) according to the Modification of Diet in Renal Disease formula [15]
| Parameter | Renal impairment group | gMean ratio % (90 % CI)a | Inter-individual gCV [%]b | P value |
|---|---|---|---|---|
| AUClast | Moderate | 122.2 (95.7–156.0) | 28.3 | 0.17 |
| Severe | 150.0 (105.3–213.7) | 41.9 | 0.06c | |
| C max | Moderate | 101.2 (72.9–140.3) | 38.5 | 0.95 |
| Severe | 121.7 (90.8–163.2) | 34.2 | 0.26 | |
| AUC∞ | Moderate | 122.4 (96.1–155.9) | 28.0 | 0.16 |
| Severe | 150.1 (105.6–213.3) | 41.5 | 0.06c |
AUC last area under the drug plasma concentration–time curve from time 0 to the time of the last quantifiable data point, AUC ∞ area under the drug plasma concentration–time curve from time 0 to infinity, CI confidence interval, C max maximum drug concentration in plasma, gCV geometric coefficient of variation (%), gMean geometric mean
aRatio of gMeans (renal impairment subjects to healthy subjects). Statistical assessment of differences in pharmacokinetic parameters between subjects with moderate and severe renal impairment and healthy subjects was performed using separate ANOVA models
bSee Table 2 for the individual group means for each treatment group
cTwo-sided P values <0.1 indicate formal significance
In subjects with moderate renal impairment, the gMean AUClast of 948 ng·h/mL was slightly increased compared with the value of 776 ng·h/mL in matched healthy controls; gMean C max values were similar at 28.7 and 28.4 ng/mL, respectively. Inter-individual variability in exposure (AUClast and C max) was moderate (gCV 33–44 %) in subjects with moderate renal impairment and slightly less (gCV 23–32 %) in matched healthy controls. In subjects with severe renal impairment, the gMean AUClast of 952 ng·h/mL was moderately increased compared with the value of 634 ng·h/mL in healthy matched controls. Geometric mean C max was also moderately higher in subjects with severe renal impairment, at 28.2 ng/mL compared with 23.2 ng/mL in healthy controls. Inter-individual variability in exposure (AUClast and C max) was low-to-moderate in subjects with severe renal impairment (gCV 25–31 %) and moderate in matched healthy controls (gCV 42–51 %; Table 2).
Graphical representations of the individual and gMean values for the primary endpoints for subjects with moderate renal impairment demonstrated a wider spread of values for both AUClast and C max compared with matched healthy controls, but displaying a large amount of overlap between the two groups (Fig. 2). Compared with subjects with severe renal impairment, matched healthy controls had slightly lower values overall for AUClast and C max, although the spread of values was of relatively equal magnitude. There was also considerable overlap between values of subjects with severe renal impairment and matched healthy controls (Fig. 2).
Fig. 2.

Comparison of individual and gMean AUClast (a) and C max (b) in subjects with moderate and severe renal impairment and matched healthy controls. AUC last area under the drug plasma concentration-time curve from time 0 to the time of the last quantifiable data point, C max maximum drug concentration in plasma, gMean geometric mean
The median time-to-peak plasma concentration (t max) (6 h in subjects with moderate renal impairment, 5 h in their healthy matched controls, 5 h in subjects with severe renal impairment, and 5.5 h in their healthy matched controls) and t max range were similar between all subject groups. The gMean terminal half-life ranged from 73 to 82 h and was comparable for subjects with renal impairment and normal renal function (Table 2).
There were quantifiable urinary concentrations of afatinib over the entire sampling interval (up to 72 h post-dose) in all measured subjects. In subjects with moderate renal impairment, renal clearance over 72 h was 63 % of that of matched healthy controls (CLR,0–72 of 16.1 and 25.4 mL/min, respectively). In subjects with severe renal impairment, renal clearance over 72 h was 29 % of that of matched healthy controls (CLR,0–72 of 8.7 and 29.6 mL/min, respectively). The total cumulative fraction of afatinib excreted in the urine (fe0–72) in subjects with renal impairment was generally low and comparable with matched controls (gMean values between 0.91 and 2.39 %; Table 2).
Effect of Renal Impairment on Protein Binding
The arithmetic mean ± SD fraction of [14C] afatinib bound to plasma proteins in pre-dose plasma samples spiked ex vivo (target concentration 72.9 ng/mL) was high (>94 %) and comparable across all groups (Table 2). The overall mean percentage protein binding was 94.6 ± 0.7 %. As such, renal impairment did not appear to influence plasma protein binding of afatinib.
Safety and Tolerability
Single-dose afatinib 40 mg was well tolerated, with few AEs. None of the subjects experienced serious AEs or discontinued the study due to an AE. AEs were reported in two (13 %) subjects with renal impairment (two moderate) and four (29 %) healthy control subjects. All AEs were of Grade 1 severity on the CTCAE v 3.0 scale. Two subjects had AEs that were considered treatment-related. A headache occurred in a healthy subject and diarrhoea in one subject with moderate renal impairment. All AEs had resolved by the end of the trial. There were no other clinically relevant changes in laboratory parameters, vital signs or ECG.
Discussion
Following administration of a single dose of 40 mg afatinib, exposure levels were similar in subjects with moderate or severe renal impairment compared with matched healthy controls. The pharmacokinetic characteristics of afatinib either in healthy subjects or in subjects with moderate/severe renal impairment within this trial were consistent with the pharmacokinetic characteristics of afatinib in the previous trials of cancer patients with various advanced solid tumours [9]. Although afatinib exposure was increased in subjects with severe renal impairment, this change was well within the observed moderate inter-individual variability (mean gCV ~42 % for AUC). Thus, moderate-to-severe renal impairment has only a minor influence on the pharmacokinetics of afatinib. This result is important, since afatinib is often administered to elderly people who may have impaired renal function.
Absolute afatinib exposure values (C max and AUC) were comparable in moderate and severely renally impaired subjects. Subjects with moderate renal impairment had approximately 20 % higher total exposure to afatinib when compared with matched healthy controls. Peak exposure between the two groups was similar. In subjects with severe renal impairment, total afatinib exposure was approximately 50 % higher than that for healthy matched controls, whereas peak exposure was increased by approximately 20 %. The higher relative total exposure in subjects with severe renal impairment was largely due to the lower exposure in the matched healthy controls for this analysis (i.e. AUClast 634 ng·h/mL in severe renal impairment controls versus 776 ng·h/mL in moderate impairment controls), since exposure in the two renal impairment groups was essentially the same (AUClast 952 and 948 ng·h/mL, respectively). A similar trend was observed for C max. A possible explanation for the lower exposure in the severe renal impairment controls was the higher proportion of male subjects in this group compared with moderate renal impairment controls (75 versus 38 %, respectively). In a population pharmacokinetic analysis of patients with advanced solid tumours receiving continuous doses of afatinib [19], it was found that male patients had a 15 % lower exposure compared with females after accounting for all other covariate effects (in particular weight).
As expected, renal clearance rates of afatinib and fractional renal excretion appeared to be inversely related to renal function; in comparison with matched healthy control subjects, subjects with moderate renal impairment had a moderate decrease in renal clearance and fraction renally excreted, while subjects with severe renal impairment had marked decreases. However, because renal excretion of afatinib plays only a small role in elimination (that is, less than 5 % of afatinib is eliminated by the kidneys), these decreases resulted in only minor changes in total and peak afatinib exposure.
Renal impairment had a relatively minor effect on the absorption, distribution, and/or elimination of afatinib. The terminal half-life of afatinib in all treatment groups was similar, although the gMean value was about twice that previously reported (73–82 h in the current study versus a mean of 27 h for a single 40 mg dose in previous trials) [9]. This was most likely due to the longer sampling period (312 h) in this trial compared with that in other trials (24 or 72 h) [9]. In addition, the lower limit of quantification for afatinib in this study (0.1 ng/mL) was lower than that in the previous studies (0.5 ng/mL) [9]. Similar findings were observed in another study in subjects with hepatic impairment [13], where sampling up to 312 h after dosing extended the terminal elimination phase and was considered to be negligible. In addition, renal impairment had no effect on the plasma protein binding of afatinib, which was moderate-to-high in all treatment groups. The results were consistent with the previous ex vivo and in vitro plasma protein-binding results of afatinib [13].
The single-dose trial design was chosen, because previous trials have demonstrated that single-dose data accurately describe afatinib pharmacokinetics and, thus, can be extrapolated to steady-state conditions [9], and thus, similar effects on exposure can be expected after multiple dosing, as observed in this trial. As a multiple-dosing trial would not be ethically justifiable in subjects with renal impairment, a single-dose design was planned, which is also in accordance with the regulatory guidelines for the conduct of such studies [16–18]. The matched groups were considered appropriate to perform comparisons between exposure and degree of renal impairment, while considering factors known to impact renal function (sex, race, age, and BMI). The open-label design was considered appropriate, because evaluation of exposure was based on pharmacokinetic assessments, which were not expected to be influenced by the investigator’s or subject’s knowledge of the treatment group. In addition, the same dose of afatinib was administered to all subjects. The sequential dosing of subjects was important to minimise risk to subjects with renal impairment; time effects resulting from sequential treatment were expected to be minimal. Inclusion of 8 subjects with renal impairment per renal function group was considered to be sufficient for the exploratory evaluation of afatinib pharmacokinetics.
The current analysis showing that moderate and severe renal impairment had a minimal effect on the pharmacokinetics of a single-afatinib dose is consistent with data from a population pharmacokinetic analysis of patients with advanced solid tumours receiving continuous doses of afatinib [19]. In this analysis, renal impairment, as measured by a decrease in CLCR levels, was a statistically significant covariate influencing afatinib exposure. However, predicted increases in total exposure were only mild-to-moderate: it was estimated that compared with a population with CLCR of 79 mL/min (mild renal impairment), exposure at steady-state (AUC) would be increased to 113 % in a population with CLCR of 60 mL/min (moderate renal impairment), and to 142 % in a population with CLCR of 30 mL/min (severe renal impairment), consistent with the results seen in this study.
The single 40 mg dose of afatinib was well tolerated by most subjects. All AEs were of mild intensity. The overall frequency of AEs was lower in subjects with renal impairment compared with healthy subjects, and no events were observed in the severe renal impairment group. The overall proportion of subjects reporting AEs was in keeping with the previous healthy volunteer trials [12, 19].
Clinical studies show that the inter-individual variability in afatinib total exposure is high (gCV ~67–85 %) [3]. The high inter-individual variability in exposure (AUC; gCV 60–92 %) was also demonstrated in a meta-analysis of patients with advanced solid tumours who received afatinib doses ranging between 20 and 50 mg [9]. Similarly, a population pharmacokinetic analysis of patients with solid tumours treated with afatinib 40 mg estimated a 90 % prediction interval for AUC in a typical patient of 45–220 % [19]. Thus, the mean exposure increase of 50 % in subjects with severe renal impairment is well within the range of previously reported inter-individual variability of afatinib and is not likely to be clinically relevant, as this patient population should have a similar risk of experiencing AEs as patients with normal renal function receiving afatinib treatment. Based on this, treatment with afatinib can be considered in patients with severe renal impairment. However, patients should be closely monitored for AEs and, if required, managed according to the well-established dose adaptation schedule [3].
Conclusion
A single dose of 40 mg of afatinib was well tolerated, and had a minor influence on total and peak exposure for subjects with moderate or severe renal impairment compared with matched healthy controls. The pharmacokinetic and tolerability findings suggest that afatinib treatment can be considered in subjects with moderate or severe renal impairment.
Acknowledgments
The study was conducted at CRS Clinical Research Services Kiel GmbH, Kiel, Germany (principal investigator, Atef Halabi). Trial programming and statistical support were provided by Datamap GmbH, Freiburg im Breisgau, Germany. Boehringer Ingelheim was responsible for the design and conduct of the study, and the collection and management of the data. The authors were responsible for the analysis and interpretation of the data and the preparation of the manuscript.
Compliance with Ethical Standards
Funding This study was funded by Boehringer Ingelheim Pharma GmbH & Co KG, Germany.
Conflict of interest
SW, DS, RK, DG, GW, AC, and SW are full time employees of Boehringer Ingelheim. AW is an employee of Datamap GmbH, which has been contracted by Boehringer Ingelheim for trial programming and statistical support for the conduct of this study and has received financial support for its services. AH is an employee of Clinical Research Services, which has been contracted by Boehringer Ingelheim as CRO for the conduct of this study and has received financial support for its services.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the independent ethics committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
References
- 1.Li D, Ambrogio L, Shimamura T, et al. BIBW2992, an irreversible EGFR/HER2 inhibitor highly effective in preclinical lung cancer models. Oncogene. 2008;27:4702–4711. doi: 10.1038/onc.2008.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Solca F, Dahl G, Zoephel A, et al. Target binding properties and cellular activity of afatinib (BIBW 2992), an irreversible ErbB family blocker. J Pharmacol Exp Ther. 2012;343:342–350. doi: 10.1124/jpet.112.197756. [DOI] [PubMed] [Google Scholar]
- 3.Sequist LV, Yang JC, Yamamoto N, et al. Phase III study of afatinib or cisplatin plus pemetrexed in patients with metastatic lung adenocarcinoma with EGFR mutations. J Clin Oncol. 2013;31:3327–3334. doi: 10.1200/JCO.2012.44.2806. [DOI] [PubMed] [Google Scholar]
- 4.Wu YL, Zhou C, Hu CP, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. Lancet Oncol. 2014;15:213–222. doi: 10.1016/S1470-2045(13)70604-1. [DOI] [PubMed] [Google Scholar]
- 5.Seiwert TY, Fayette J, Cupissol D, et al. A randomized, phase II study of afatinib versus cetuximab in metastatic or recurrent squamous cell carcinoma of the head and neck. Ann Oncol. 2014;25:1813–1820. doi: 10.1093/annonc/mdu216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Soria JC, Felip E, Cobo M, et al. Afatinib versus erlotinib as second-line treatment of patients with advanced squamous cell carcinoma of the lung (LUX-Lung 8): an open-label randomised controlled phase 3 trial. Lancet Oncol. 2015;16:897–907. doi: 10.1016/S1470-2045(15)00006-6. [DOI] [PubMed] [Google Scholar]
- 7.Boehringer Ingelheim. Gilotrif [prescribing information]. Ridgefield: Boehringer Ingelheim Pharmaceuticals, Inc.; 2016. http://www.gilotrif.com/. Accessed 4 June 2016.
- 8.European Medicines Agency. Committee for Medicinal Products for Human Use (CHMP). Gilotrif, summary of opinion. 2016. http://www.ema.europa.eu/docs/en_GB/document_library/Summary_of_opinion/human/002280/WC500202381.pdf. Accessed 23 March 2016.
- 9.Wind S, Schmid M, Erhardt J, et al. Pharmacokinetics of afatinib, a selective irreversible ErbB family blocker, in patients with advanced solid tumours. Clin Pharmacokinet. 2013;52:1101–1109. doi: 10.1007/s40262-013-0091-4. [DOI] [PubMed] [Google Scholar]
- 10.Yap TA, Vidal L, Adam J, et al. Phase I trial of the irreversible EGFR and HER2 kinase inhibitor BIBW 2992 in patients with advanced solid tumors. J Clin Oncol. 2010;28:3965–72. doi: 10.1200/JCO.2009.26.7278. [DOI] [PubMed] [Google Scholar]
- 11.Stopfer P, Marzin K, Narjes H, et al. Afatinib pharmacokinetics and metabolism after oral administration to healthy male volunteers. Cancer Chemother Pharmacol. 2012;69:1051–1061. doi: 10.1007/s00280-011-1803-9. [DOI] [PubMed] [Google Scholar]
- 12.Schnell D, Buschke S, Fuchs H, et al. Pharmacokinetics of afatinib in subjects with mild or moderate hepatic impairment. Cancer Chemother Pharmacol. 2014;74:267–275. doi: 10.1007/s00280-014-2484-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.European Medicines Agency. Giotrif. Summary of Product Characteristics, version 24 May 2016. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002280/WC500152392.pdf. Accessed 7 June 2016.
- 14.Levey AS, Coresh J, Greene T, et al. Expressing the Modification of Diet in Renal Disease Study equation for estimating glomerular filtration rate with standardized serum creatinine values. Clin Chem. 2007;53:766–772. doi: 10.1373/clinchem.2006.077180. [DOI] [PubMed] [Google Scholar]
- 15.US FDA Center for Drug Evaluation and Research. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER). Guidance for industry: pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. 1998. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm072127.pdf. Accessed 5 Jan 2016.
- 16.US FDA Center for Drug Evaluation and Research. US Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER). Guidance for industry: pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. 2010. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM204959.pdf. Accessed 2 Jan 2016.
- 17.European Medicines Agency. Committee for Human Medicinal Products (CHMP). Note for guidance on the evaluation of the pharmacokinetics of medicinal products in patients with impaired renal function. 2004. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003123.pdf. Accessed 5 Jan 2016. Accessed 2 Jan 2016.
- 18.Freiwald M, Schmid U, Fleury A, et al. Population pharmacokinetics of afatinib, an irreversible ErbB family blocker, in patients with various solid tumors. Cancer Chemother Pharmacol. 2014;73:759–770. doi: 10.1007/s00280-014-2403-2. [DOI] [PubMed] [Google Scholar]
- 19.Wind S, Giessmann T, Jungnik A, et al. Pharmacokinetic drug interactions of afatinib with rifampicin and ritonavir. Clin Drug Investig. 2014;34:173–182. doi: 10.1007/s40261-013-0161-2. [DOI] [PubMed] [Google Scholar]