

Abstract
Coxiella burnetii, the etiologic agent of acute Q fever and chronic endocarditis, has a unique biphasic life cycle, which includes a metabolically active intracellular form that occupies a large lysosome-derived acidic vacuole. C. burnetii is the only bacterium known to thrive within such an hostile intracellular niche, and this ability is fundamental to its pathogenicity; however, very little is known about genes that facilitate Coxiella's intracellular growth. Recent studies indicate that C. burnetii evolved from a tick-associated ancestor and that the metabolic capabilities of C. burnetii are different from that of Coxiella-like bacteria found in ticks. Horizontally acquired genes that allow C. burnetii to infect and grow within mammalian cells likely facilitated the host shift; however, because of its obligate intracellular replication, C. burnetii would have lost most genes that have been rendered redundant due to the availability of metabolites within the host cell. Based on these observations, we reasoned that horizontally derived biosynthetic genes that have been retained in the reduced genome of C. burnetii are ideal candidates to begin to uncover its intracellular metabolic requirements. Our analyses identified a large number of putative foreign-origin genes in C. burnetii, including tRNAGlu2 that is potentially required for heme biosynthesis, and genes involved in the production of lipopolysaccharide—a virulence factor, and of critical metabolites such as fatty acids and biotin. In comparison to wild-type C. burnetii, a strain that lacks tRNAGlu2 exhibited reduced growth, indicating its importance to Coxiella's physiology. Additionally, by using chemical agents that block heme and biotin biosyntheses, we show that these pathways are promising targets for the development of new anti-Coxiella therapies.
Keywords: Coxiella burnetii, lateral gene transfer, heme biosynthesis, biotin biosynthesis, LPS, glutamic acid, horizontal gene transfer (HGT)
Introduction
Coxiella burnetii is the etiological agent of acute Q fever and a chronic disease commonly manifested as endocarditis (Maurin and Raoult, 1999). Most human infections occur through inhalation of aerosols originating from ruminants that shed C. burnetii during parturition and in milk. The pathogen persists in the environment as a metabolically quiescent small cell variant (SCV), which transforms into a metabolically active large cell variant (LCV) within a lysosome-derived, acidic (pH ~4.5), Coxiella-containing vacuole (CCV) (Voth and Heinzen, 2007). The unique ability of C. burnetii to thrive in this inhospitable vacuole is fundamental to its physiology and pathogenicity; however, the metabolic processes that drive its intracellular growth are unknown.
The evolutionary origin of C. burnetii is also not clearly understood. The closest relatives of C. burnetii are tick-associated bacteria, indicating that C. burnetii evolved from a tick-associated ancestor (Duron et al., 2015; Gottlieb et al., 2015; Smith et al., 2015). Interestingly, Coxiella-like bacteria found in ticks cannot infect mammalian cells and are unable to grow in ACCM-2, a culture medium that supports robust growth of C. burnetii (Omsland et al., 2011; Duron et al., 2015). These observations suggest that despite their close evolutionary relationship, the human pathogen, and the tick-associated strains have different virulence and metabolic capabilities. Concomitantly, when the genomes of C. burnetii were sequenced, it became clear that the pathogen has acquired several virulence and metabolic genes via horizontal gene transfer (HGT). For example, C. burnetii contains a tryptophan biosynthesis operon of Chlamydial origin, and a Type Four Secretion System (TFSS) and eukaryote-like ankyrin repeat sequence-containing effector proteins that are essential for CCV generation (Seshadri et al., 2003; Beare et al., 2009). Horizontal acquisition of foreign DNA occurs in bacteria through transformation, conjugation, or transduction via mobile genetic elements such as plasmids, integrons, bacteriophages, transposons, retrotransposons etc. The novel DNA is stably maintained and spreads through the recipient population if it offers selective advantage (e.g., antibiotic resistance), allowing the bacterium to adapt to the new environment (Eisen, 2000; Frost et al., 2005; Thomas and Nielsen, 2005).
Unlike eukaryotic genomes, which contain large fractions of non-functional DNA (e.g., >80% of human genome), bacterial genomes are tightly packed with functional genes (Moran, 2002; Ochman and Davalos, 2006). In bacteria there is a bias toward deletion over insertion, hence, DNA is retained in a bacterial genome only if selection is acting effectively to preserve it (Mira et al., 2001). For instance, when the tick-associated ancestor of C. burnetii evolved into a mammalian pathogen that replicates only within the CCV, several biosynthetic genes would be rendered redundant if corresponding metabolic intermediates are available within the host cell. In addition, although genes acquired via HGT might have been critical in facilitating the host shift, many of them could become expendable in the new lifestyle (Lo et al., 2015). These superfluous metabolic and HGT-origin genes would subsequently be deleted from the genome due to a lack of selection pressure to maintain them (Ochman and Moran, 2001). Furthermore, the intracellular niche limits C. burnetii's opportunity to gain new genes from the environment via HGT. Thus, intracellular pathogens such as C. burnetii tend to have reduced genomes in comparison to related free-living bacteria (e.g., C. burnetii's genome is ~2 million bp, whereas E. coli's is ~5 million bp). Based on these observations, we reasoned that a significant number of HGT-origin genes that have been retained in C. burnetii would be critical to its intracellular fitness. In this study, we identified a large number of horizontally derived genes, including those for the synthesis of LPS, fatty acids, heme, and biotin that augment the physiological capability of C. burnetii.
Results and discussion
Identification of horizontally acquired genes in C. burnetii
HGT is a major driver of evolution and adaptation in bacteria (Lerat et al., 2005; Price et al., 2008; Treangen and Rocha, 2011). By examining differences in nucleotide composition (e.g., GC%) HGT-origin genes can be tentatively identified; however, because most transfers occur between closely related bacteria, and because genes from distant organisms will evolve over time to reflect the base composition of the recipient genome (Lawrence and Ochman, 1997), this approach is not always effective. Another common approach is to perform reciprocal-BLAST analysis (e.g., Raghavan et al., 2012), where, if the top hit is from a distantly related organism, the gene could be of putative HGT origin. While this approach provides rapid results, the best BLAST hit may not always be the closest phylogenetic relative of the query gene (Koski and Golding, 2001). This is especially the case for organisms such as C. burnetii that have few close relatives represented in NCBI databases. For instance, C. burnetii is the only defined species within its genus, and complete genomes are available for only three other bacteria within the order Legionellales—Legionella, Rickettsiella, and Diplorickettsia. In order to circumvent this limitation, we utilized a phylogeny informed BLAST-based approach (HGTector, Zhu et al., 2014), but applied very strict criteria to categorize a gene as horizontally acquired. We set Coxiella as the self-group, and Legionellales as the exclusion group, which captured HGT events where only Coxiella has acquired a particular gene from outside of Legionellales and ignored any events where the genes could also have been transferred elsewhere within the order. In addition, because the phylogenetic position of Legionellales within Gammaproteobacteria is not well-resolved (Williams et al., 2010), we also ignored any gene that was potentially gained from another member of the class Gammaproteobacteria. Using this ultra-conservative approach, we were able to identify 172 “high-confidence” horizontally acquired genes on the chromosome of C. burnetii RSA 493 (Figure 1, Dataset S1), whereas, none of the genes located on C. burnetii plasmids (QpH1, QpRS, QpDG) were deemed to be of horizontal origin.
Figure 1.

Horizontally acquired genes in C. burnetii. Two outer rings show ORFs (black bars) on forward and reverse strands, respectively. Third ring shows positions of 172 HGT-origin genes, and the inner ring contains biosynthetic genes examined in this study. Genes are colored according to their putative donors, as shown in the center. Number of genes acquired from each donor is also shown.
Orthologs of all 172 HGT-origin genes are present in C. burnetii Dugway 5J108-111 (NC_009727.1), C. burnetii 3262 (CP013667.1), and C. burnetii Z3055 (NZ_LK937696.1), but we couldn't detect orthologs of CBU_1991 in C. burnetii RSA 331 (CP000890.1), CBU_2021 and CBU_0562 in C. burnetii G_Q212 (NC_011527.1), and CBU_0007a, CBU_0167, CBU_0168, CBU_0768, and CBU_0792 in C. burnetii K_Q154 (NC_011528.1). Of the 172 HGT genes, 18 are located close to transposase genes in C. burnetii RSA 493, suggestive of a role for transposons or insertion sequences in acquiring foreign genes. In addition to the large number of transposases (>30), all C. burnetii strains contain several other selfish genetic elements that are horizontally exchanged between bacteria, including two Group I introns, an intein, and an intervening sequence (Seshadri et al., 2003; Raghavan et al., 2007, 2008; Beare et al., 2009; Warrier et al., 2016). Proliferation of mobile genetic elements and extensive genome rearrangements are hallmarks of bacteria that have recently shifted to host-associated lifestyles, indicating that the obligate intracellular growth of C. burnetii is of recent origin (Ochman and Moran, 2001; McCutcheon and Moran, 2012).
As expected, our analysis excluded several genes that were likely acquired via HGT because they are also present in other members of the order Legionellales e.g., TFSS genes, eukaryote-like sterol reductase genes, plasmid genes with eukaryotic domains etc. (Seshadri et al., 2003; Beare et al., 2009; Gilk et al., 2010; Voth et al., 2013). Without better resolution of C. burnetii's phylogenetic position in the bacterial tree, it is difficult to discern whether these genes were acquired by a common ancestor or were gained independently by each bacterium as they adapted to their respective intracellular niches (Gottlieb et al., 2015). Intriguingly, C. burnetii encodes several intact or pseudogenized genes that encode the components of a Type IV pilus (Seshadri et al., 2003). A related Type IV pilus enables Acinetobacter baumannii to acquire DNA from the environment with high efficiency (Smith et al., 2007), indicating that during an earlier stage during C. burnetii's evolution, a functional Type IV pilus endowed it with the ability to acquire foreign genes proficiently, thereby likely facilitating its transition into a mammalian pathogen from a tick-associated ancestor (Duron et al., 2015; Smith et al., 2015; Gerhart et al., 2016).
HGT contributed to the lipopolysaccharide profile of C. burnetii
HGT-origin genes are strewn all across the C. burnetii genome, indicating, as shown before, that there are no prototypical pathogenicity islands, but, based on their clustered genome locations, several genes appear to have been acquired en bloc (Figure 1). A group of genes of particular interest is CBU_0678 to CBU_0683, which is part of an operon that encodes genes involved in the biosynthesis of LPS (Seshadri et al., 2003; Narasaki and Toman, 2012). We conducted in-depth phylogenetic analyses of CBU_0678, the most upstream gene of this cluster, in order to validate the HGTector results. Based on both Maximum Likelihood and Bayesian phylogenetic analyses (Figure 2, Figure S1), the closest orthologs of this gene are present in members of Alphaproteobacteria. Furthermore, O-polysaccharide biosynthesis genes tend to occur as an operon in most bacteria, and since several genes in this location (CBU_0673, CBU_0676, CBU_0678 to CBU_0682) were also probably acquired from Alphaproteobacteria, it is highly likely that the genes were acquired in a single event. Previous studies have shown that LPS genes are horizontally transferred among bacteria (Nelson and Selander, 1994), probably due to their importance in host-pathogen interactions (Narasaki and Toman, 2012). In fact, full-length LPS is the only C. burnetii virulence factor established in an immunocompetent animal model of infection. It protects the pathogen from innate immune response (Shannon et al., 2005), and avirulent Nine Mile phase II produces a severely truncated LPS due to the loss of 22 LPS biosynthesis genes, including CBU_0678 to CBU_0682 (Beare et al., 2006). The observation that important virulence factors in C. burnetii (e.g., LPS, TFSS, effector proteins) were assembled via HGT illustrates the significance of this process in the evolutionary history of this intracellular pathogen.
Figure 2.
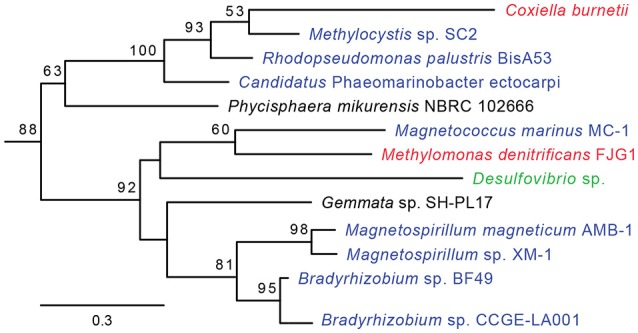
CBU_0678 was gained via HGT. A Maximum Likelihood phylogeny for CBU_0678, a LPS biosynthesis gene, is shown. Gammaproteobacteria are in red, Alphaproteobacteria in blue, Delta/Epsilonproteobacteria in green, and Planctomycetes in black. Bootstrap values of > 50 are indicated at the nodes.
HGT enhanced C. burnetii's fatty acid and biotin metabolism
Another set of genes in C. burnetii that is of putative foreign origin is CBU_0034 to CBU_0038 (Figure 1). This gene cluster appears to have originated in either a Deltaproteobacteria or a Spirochete. We validated the HGT origin of this presumptive operon through phylogenetic analysis of its first gene, CBU_0038 (Figure 3A, Figure S2). Furthermore, this set of genes has a similar arrangement in both C. burnetii and in Spirochaeta africana DSM 8902, its best BLAST hit (Dataset S1), indicating that the genes were transferred en bloc. In addition, an IS1111A transposase (CBU_0040) is proximally located to the operon, illustrating a probable role for this mobile genetic element in HGT (Figure 3B). Based on homology, CBU_0034 to CBU_0038 encode ACP, FabB, FabZ, FabA, and FabH, respectively, which are involved in the synthesis of unsaturated fatty acids (Zhang and Rock, 2008; Feng and Cronan, 2009). In contrast, vertically inherited genes (CBU_0493 to CBU_0497) are responsible for the synthesis of saturated fatty acids (Gilk, 2012), denoting that C. burnetii's ability to produce unsaturated fatty acids was enhanced through HGT. C. burnetii also contains a putative fatty acid desaturase (CBU_0920) that introduces double bonds into existing fatty acids (Gilk, 2012). This gene also appears to be of HGT origin, but was not included in our analysis because it is present in other Legionellales. In addition to fatty acid biosynthesis genes, C. burnetii has also gained three pyruvate dehydrogenases (CBU_0686, CBU_0692, and CBU_0693) (Figure 1). Pyruvate dehydrogenase complex converts pyruvate into acetyl-CoA, which is a critical metabolite for both fatty acid biosynthesis and ATP generation (de Kok et al., 1998). Thus, HGT has played a significant role in shaping C. burnetii's fatty acid metabolism. Furthermore, a recent study showed that transposon-mediated disruption of CBU_0035 and CBU_0038 resulted in reduced C. burnetii growth within Vero cells, suggesting that this process is a critical constituent of the pathogen's physiology (Martinez et al., 2014).
Figure 3.

A horizontally acquired fatty acid biosynthesis operon in C. burnetii. (A) Maximum Likelihood phylogenetic tree for CBU_0038, a fatty acid biosynthesis gene. Gammaproteobacteria is colored red, Delta/Epsilonproteobacteria in green, Spirochaetes in brown, Bacteriodetes in purple, and Firmicutes in blue. Bootstrap values of > 50 are indicated at the nodes. (B) Fatty acid biosynthesis genes have similar arrangement in C. burnetii and Spirochaeta africana, and an IS1111A transposase is located next to the operon in C. burnetii.
Fatty acid biosynthesis enzymes require biotin as a cofactor, and biotin in turn is synthesized by utilizing a portion of the fatty acid biosynthesis pathway (Lin et al., 2010). Unlike most other bacteria, C. burnetii contains two copies of the gene bioC, the first committed step in biotin production. The two bioC genes are only 45% similar at the nucleotide level, indicating that they were not formed by a recent duplication event in C. burnetii. Further, bioC.2 (CBU_1004) has an 11 bp overlap with bioH and is part of the bioA-bioBFHCD-birA regulon (CBU_1008 to CBU_1002), whereas bioC.1 (CBU_0467) is a single gene located at a different part of the genome, indicating that bioC1 is of horizontal origin. To understand the functional relevance of these genes, we examined their expression levels in C. burnetii grown in ACCM-2 and within Vero cells (Warrier et al., 2014). The RNA-seq data revealed that all biotin biosynthesis genes (bioA, bioB, bioC.1, bioC.2, bioD, bioF, and bioH) were expressed under both conditions, but their expression was significantly higher in Vero cells than in ACCM-2 (Table S1). In contrast, the expression of birA, the transcriptional repressor of biotin operon showed the opposite pattern, suggesting that biotin is likely synthesized under both conditions, with possible upregulation within the host cell. Furthermore, a small molecule (MAC13772) that blocks biotin biosynthesis in E. coli (Zlitni et al., 2013) inhibited the growth of C. burnetii in ACCM-2, indicating that the production of biotin is a critical process in this pathogen (Figure 4). Biotin synthesis has been shown to be critical to the virulence of other human pathogens such as Francisella tularensis and Mycobacterium tuberculosis (Woong Park et al., 2011; Feng et al., 2014). Thus, novel pharmaceutical agents that block this process hold promise as a potential broad-spectrum agent to treat intracellular pathogens.
Figure 4.
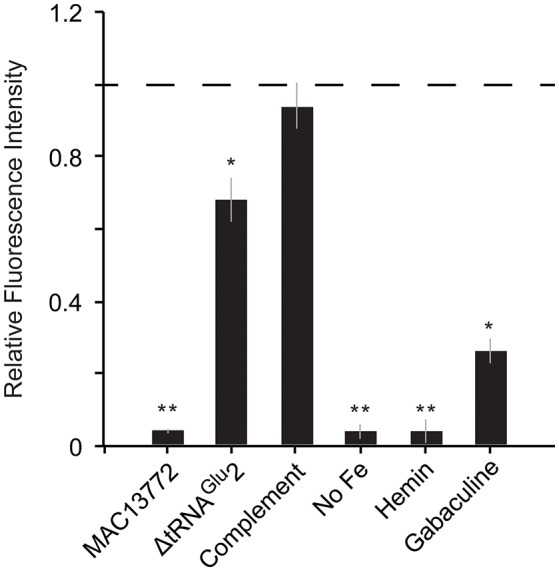
Heme and biotin syntheses are critical to C. burnetii's growth. Growth of C. burnetii in ACCM-2 after 7 days was measured using PicoGreen. Fluorescence of each strain relative to that of control (wild-type grown in ACCM-2; dashed line) is shown. MAC13772: ACCM-2 supplemented with 300 μg/ml MAC13772, a biotin biosynthesis inhibitor; ΔtRNAGlu2: tRNAGlu2-deletion strain; Complement: tRNAGlu2-deletion complemented with intact tRNAGlu2 on pAM100; No Fe: ACCM-2 without FeSO4; Hemin: ACCM-2 with hemin in place of FeSO4; Gabaculine: ACCM-2 supplemented with 100 μM gabaculine, a heme biosynthesis inhibitor. Statistically significant differences in growth from control are indicated by (**) p < 0.001 and (*) p < 0.01 (unpaired t-test).
Heme biosynthesis is an essential metabolic process in C. burnetii
C. burnetii's genome is small compared to that of free-living bacteria such as E. coli (~2 and 5 Mb, respectively). Correspondingly, it contains less than half the number of tRNAs than in E. coli (42 and 89, respectively). This reduction has occurred due to loss of redundant tRNAs; for instance, while E. coli has five copies of tRNAIle, C. burnetii has only one copy. In contrast to other tRNAs, C. burnetii contains an additional tRNAGlu isoacceptor (tRNAGlu2, anticodon CUC) that is not found in any other Gammaproteobacteria, denoting that it was gained via HGT (Figure 5A). Additionally, a toxin-antitoxin system (CBU_0284, CBU0285) that was probably horizontally acquired is located adjacent to tRNAGlu2, further strengthening the evidence for its putative HGT origin (Figure 5B). Because tRNAGlu2 is retained in such a streamlined genome, it most likely has a critical function; in fact, a tRNAGlu2-deletion strain had significantly slower growth than the wild-type strain in ACCM-2, which was restored in a complementation strain, signifying the importance of the HGT-derived tRNA to optimum bacterial fitness (Figure 4). However, the major function of tRNAGlu2 is unlikely to be protein synthesis because tRNAGlu2 (anticodon CUC) cannot decode the major glutamate codon GAA (76%), whereas tRNAGlu1 (anticodon UUC) can decode both GAA and GAG codons efficiently. Additionally, Coxiella proteins are not enriched for glutamate in comparison to E. coli (~6% of all codons is for Glu in both bacteria). Taken together, these data suggest that protein biosynthesis is not tRNAGlu2's major function in C. burnetii.
Figure 5.
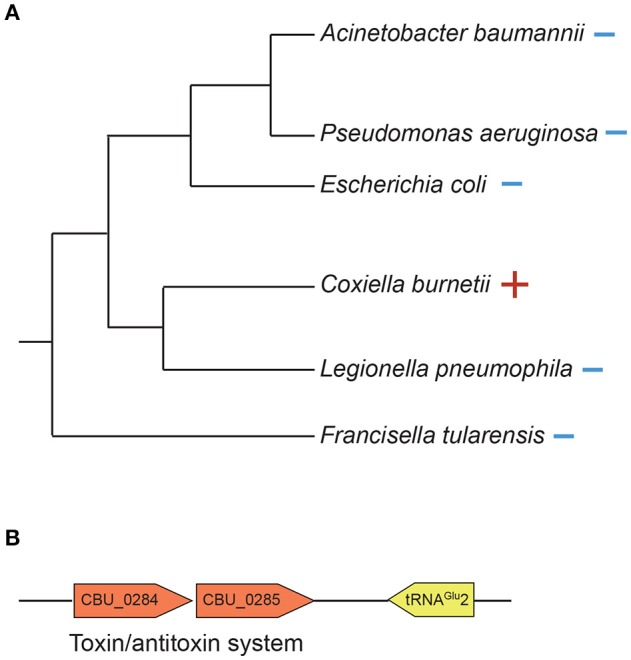
tRNAGlu2 was horizontally acquired by C. burnetii. (A) 16S rDNA phylogenetic tree of Gammaproteobacteria showing the presence (+) or absence (–) of tRNAGlu(anticodon CUC). (B) A putative toxin/antitoxin system is located adjacent to tRNAGlu2.
An alternate function for tRNAGlu2 is in heme biosynthesis. Heme is an iron-containing tetrapyrrole that serves multiple cellular functions, including in respiration, energy generation, oxidative reactions, and signal transduction (Almiron et al., 2001). The universal precursor of tetrapyrrole biosynthesis is 5-aminolevulinic acid (ALA) (Anzaldi and Skaar, 2010). There are two alternate pathways in nature for the synthesis of ALA: C5 pathway found in most bacteria, and Shemin pathway present in most eukaryotes, including humans (Frankenberg et al., 2003). The starting point of the C5 pathway is Glutamyl-tRNAGlu, which is converted into ALA using two consecutive enzymes HemA and HemL. In Shemin pathway, ALA is synthesized from Succinyl-CoA and glycine by the enzyme ALAS (Frankenberg et al., 2003). The remaining steps are shared between the C5 and Shemin pathways. We examined the C. burnetii genome and discovered that it encodes an intact C5 heme biosynthesis pathway, and RNA-seq data confirmed that these genes are expressed during growth (Table S1). Additionally, heme biosynthesis pathway is conserved in all C. burnetii strains but is absent in non-pathogenic Coxiella present in ticks, and a C. burnetii strain with a transposon insertion in the hemL gene had reduced growth in Vero cells (Martinez et al., 2014), implying the importance of heme biosynthesis to the human pathogen.
Several bacteria augment heme biosynthesis with heme transported from the outside (Mike et al., 2013). C. burnetii encodes a transporter (feoAB) for ferrous iron, which is required for heme biosynthesis, but has no known transporters for ferric ions or heme. Consequently, we reasoned that the pathogen might not be able to utilize extracellular heme as its sole source of iron. To test this, we inoculated equal amounts of Coxiella into ACCM-2 medium with FeSO4 (standard recipe) or into ACCM-2 in which FeSO4 was replaced with equimolar amount of hemin. Growth was measured after 7 days using PicoGreen as described previously (Martinez et al., 2015). As shown in Figure 4, Coxiella growth in FeSO4-containing ACCM-2 was significantly higher than in hemin-containing ACCM-2 (p < 0.001, unpaired t-test), whereas the fluorescence measurements between the control (ACCM-2 without FeSO4) and the 1hemin-containing ACCM-2 samples were not significantly different (p > 0.05, unpaired t-test), showing that C. burnetii cannot utilize external heme. To test C. burnetii's requirement for heme biosynthesis, we treated C. burnetii with increasing concentrations of gabaculine, an inhibitor of HemL (Wang et al., 1997), and found that 100 uM gabaculine significantly inhibited bacterial growth (p < 0.01, unpaired t-test) (Figure 4). These data show the importance of heme biosynthesis to C. burnetii's normal physiology, and indicates that heme biosynthesis genes are potential targets for the development of new anti-Coxiella therapies.
In conclusion, by examining a few critical biosynthetic pathways we show that HGT has played an important role in shaping the metabolic capability of C. burnetii. Due to the uncertainty in C. burnetii's phylogenetic relationship with other Gammaproteobacteria, we focused on a subset of genes that were likely acquired from distantly related bacteria. However, horizontal exchange usually occurs at higher frequency between closely related bacteria (Ochman et al., 2000), and hence, it is highly likely that an even larger proportion of genes in C. burnetii are of HGT origin. Availability of more genomes of bacteria in the phylogenetic neighborhood of C. burnetii along with more detailed evolutionary, genetic and functional analyses are required to identify all HGT-origin genes in C. burnetii and to fully understand their impact on the pathogen's physiology and pathogenicity.
Materials and methods
C. burnetii growth assay
C. burnetii Nine Mile phase II RSA 439 was grown in ACCM-2 medium as described previously (Omsland et al., 2011; Warrier et al., 2014), and incubated at 37°C, 2.5% O2, and 5.0% CO2 using a Galaxy 170 R incubator (New Brunswick Scientific, NJ). Chloramphenicol (8 μg/ml) and/or Kanamycin (375 μg/ml) were added as necessary. Growth was measured using PicoGreen as described previously (Martinez et al., 2015). Briefly, 50 μl of culture was mixed with 5 μl of Triton X-100 Surfact-Amps 10% detergent solution (Thermo Scientific) in 96-well black-bottom Cliniplates (Thermo Scientific), and allowed to incubate at room temperature for 10 min with shaking. PicoGreen (Life Technologies) was diluted 1:200 in TE buffer and 55 μl was added to the wells, and incubated at room temperature with shaking for 5 min. Wells were excited at 495 nm and emission was read at 519 nm using a Victor X5 2030 Multiplate Reader (Perkin Elmer). To determine whether Coxiella can utilize external heme as its sole iron source, ferrous sulfate was either omitted from ACCM-2 preparations or substituted with hemin (Alfa Aesar), and to assay the importance of heme and biotin biosyntheses, gabaculine (Enzo Life Sciences) or MAC13772 (Maybridge) was added to ACCM-2. Briefly, a 10 mM stock of hemin was made in 1.5 M NaOH, a 300 mg/ml stock of MAC13772 was made in dimethyl sulfoxide (DMSO), and a 100 mM stock of gabaculine was made in distilled water. One microliter of solution from these stocks was added to 1 ml of ACCM-2 to attain final concentrations of 10 μM hemin, 300 μg/ml MAC13772, or 100 μM gabaculine. ACCM-2 containing the same amount of solvent was used as control, and growth was measured after 7 days using PicoGreen as described above.
Generation of tRNAGlu2 deletion and complementation strains
To delete tRNAGlu2, ~1200 bp on each side of the gene along with Chloramphenicol acetyltransferase (CAT) gene was cloned into the vector pJC-Kan, and the mutant was generated as described previously (Beare et al., 2012). Insertion of the CAT gene in place of tRNAGlu2 was confirmed using PCR and DNA sequencing. To generate a complementation strain we used pKM244, a low-copy plasmid that is maintained stably in C. burnetii (Chen et al., 2010). Because the tRNAGlu2-deletion strain already contained a CAT gene, we amplified a kanamycin resistance gene driven by 1169P from pJB-Kan (Beare et al., 2012) and cloned it into pKM244 using NheI and AatII to generate pAM100. We inserted tRNAGlu2 along with its flanking intergenic regions into pAM100 using BamHI. C. burnetii was transformed (400 ohms, 2.5 kV, 25 mF) with either empty pAM100 or pAM100 with cloned tRNAGlu2, as described previously (Omsland et al., 2011; Beare et al., 2012). Growth in ACCM-2 of wild-type (with empty pAM100), deletion (with empty pAM100) and complementation strains were measured at day-7 using PicoGreen as described above.
Detection of horizontal gene transfer
Horizontally acquired genes were identified using HGTector (Zhu et al., 2014). Coxiella was set as self-group, and Legionellales was set as exclusion group. BLAST parameter thresholds were set at 70% identity and an E-value of at least 1e-5. To validate the HGT data, phylogenetic analyses were conducted for several putative HGT genes. To this end, TBLASTN search was conducted against NCBI nr database and the top 100 matches with at least 30% identity, 70% coverage, and E-value of 1e-10 were chosen. Sequence alignment was performed using Clustal Omega (Sievers et al., 2011), and ambiguously aligned regions were removed using Gblocks (Talavera and Castresana, 2007). The evolution model GTR+I+G (General Time Reversible plus Invariant sites plus Gamma distribution) was selected using jModelTest2 (Darriba et al., 2012). Bayesian trees were constructed using MrBayes as implemented in Geneious (Huelsenbeck and Ronquist, 2001; Kearse et al., 2012). A chain length of 1,000,000 was used with a burn-in fraction of 25% and sampling every 100 trees. Maximum Likelihood trees were constructed using RAxML (Stamatakis et al., 2008) as implemented in Geneious with 1,000 bootstrap replicates.
Author contributions
AM, JM, MB, PB, and RR conducted the experiments, analyzed the data, and prepared the manuscript.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported in part by NIH grants AI123464 and AI126385 (to RR). JM was supported by a Sigma Xi Grant-in-Aid of Research award (G201510151633590). MB was supported by grants from the Agence Nationale de la Recherche (ANR-14-CE14-0012-01; AttaQ), ERA-NET Infect-ERA (ANR-13-IFEC-0003; EUGENPATH), and the ATIP-AVENIR programme). PB was supported by funding from the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases.
Supplementary material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fcimb.2017.00174/full#supplementary-material
References
- Almiron M., Martinez M., Sanjuan N., Ugalde R. A. (2001). Ferrochelatase is present in Brucella abortus and is critical for its intracellular survival and virulence. Infect. Immun. 69, 6225–6230. 10.1128/IAI.69.10.6225-6230.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anzaldi L. L., Skaar E. P. (2010). Overcoming the heme paradox: heme toxicity and tolerance in bacterial pathogens. Infect. Immun. 78, 4977–4989. 10.1128/IAI.00613-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare P. A., Larson C. L., Gilk S. D., Heinzen R. A. (2012). Two systems for targeted gene deletion in Coxiella burnetii. Appl. Environ. Microbiol. 78, 4580–4589. 10.1128/AEM.00881-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare P. A., Unsworth N., Andoh M., Voth D. E., Omsland A., Gilk S. D., et al. (2009). Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella. Infect. Immun. 77, 642–656. 10.1128/IAI.01141-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beare P. A., Samuel J. E., Howe D., Virtaneva K., Porcella S. F., Heinzen R. A., et al. (2006). Genetic diversity of the Q fever agent, Coxiella burnetii, assessed by microarray-based whole-genome comparisons. J. Bacteriol. 188, 2309–2324. 10.1128/JB.188.7.2309-2324.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C., Banga S., Mertens K., Weber M. M., Gorbaslieva I., Tan Y., et al. (2010). Large-scale identification and translocation of type IV secretion substrates by Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 107, 21755–21760. 10.1073/pnas.1010485107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D., Taboada G. L., Doallo R., Posada D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Methods 9, 772. 10.1038/nmeth.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kok A., Hengeveld A. F., Martin A., Westphal A. H. (1998). The pyruvate dehydrogenase multi-enzyme complex from Gram-negative bacteria. Biochim. Biophys. Acta 1385, 353–366. 10.1016/S0167-4838(98)00079-X [DOI] [PubMed] [Google Scholar]
- Duron O., Noël V., McCoy K. D., Bonazzi M., Sidi-Boumedine K., Morel O., et al. (2015). The recent evolution of a maternally-inherited endosymbiont of ticks led to the emergence of the Q fever pathogen, Coxiella burnetii. PLoS Pathog. 11:e1004892. 10.1371/journal.ppat.1004892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen J. A. (2000). Horizontal gene transfer among microbial genomes: new insights from complete genome analysis. Curr. Opin. Genet. Dev. 10, 606–611. 10.1016/S0959-437X(00)00143-X [DOI] [PubMed] [Google Scholar]
- Feng Y., Cronan J. E. (2009). Escherichia coli unsaturated fatty acid synthesis: complex transcription of the fabA gene and in vivo identification of the essential reaction catalyzed by FabB. J. Biol. Chem. 284, 29526–29535. 10.1074/jbc.M109.023440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y., Napier B. A., Manandhar M., Henke S. K., Weiss D. S., Cronan J. E. (2014). A Francisella virulence factor catalyses an essential reaction of biotin synthesis. Mol. Microbiol. 91, 300–314. 10.1111/mmi.12460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankenberg N., Moser J., Jahn D. (2003). Bacterial heme biosynthesis and its biotechnological application. Appl. Microbiol. Biotechnol. 63, 115–127. 10.1007/s00253-003-1432-2 [DOI] [PubMed] [Google Scholar]
- Frost L. S., Leplae R., Summers A. O., Toussaint A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Microbiol. 3, 722–732. 10.1038/nrmicro1235 [DOI] [PubMed] [Google Scholar]
- Gerhart J. G., Moses A. S., Raghavan R. (2016). A Francisella-like endosymbiont in the Gulf Coast tick evolved from a mammalian pathogen. Sci. Rep. 6:33670. 10.1038/srep33670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilk S. D. (2012). Role of lipids in Coxiella burnetii infection. Adv. Exp. Med. Biol. 984, 199–213. 10.1007/978-94-007-4315-1_10 [DOI] [PubMed] [Google Scholar]
- Gilk S. D., Beare P. A., Heinzen R. A. (2010). Coxiella burnetii expresses a functional Δ24 sterol reductase. J. Bacteriol. 192, 6154–6159. 10.1128/JB.00818-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb Y., Lalzar I., Klasson L. (2015). Distinctive genome reduction rates revealed by genomic analyses of two Coxiella-like endosymbionts in ticks. Genome Biol. Evol. 7, 1779–1796. 10.1093/gbe/evv108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck J. P., Ronquist F. (2001). MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17, 754–755. 10.1093/bioinformatics/17.8.754 [DOI] [PubMed] [Google Scholar]
- Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., et al. (2012). Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. 10.1093/bioinformatics/bts199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koski L. B., Golding G. B. (2001). The closest BLAST hit is often not the nearest neighbor. J. Mol. Evol. 52, 540–542. 10.1007/s002390010184 [DOI] [PubMed] [Google Scholar]
- Lawrence J. G., Ochman H. (1997). Amelioration of bacterial genomes: rates of change and exchange. J. Mol. Evol. 44, 383–397. 10.1007/PL00006158 [DOI] [PubMed] [Google Scholar]
- Lerat E., Daubin V., Ochman H., Moran N. A. (2005). Evolutionary origins of genomic repertoires in bacteria. PLoS Biol. 3:e130. 10.1371/journal.pbio.0030130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S., Hanson R. E., Cronan J. E. (2010). Biotin synthesis begins by hijacking the fatty acid synthetic pathway. Nat. Chem. Biol. 6, 682–688. 10.1038/nchembio.420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo W. S., Gasparich G. E., Kuo C. H. (2015). Found and lost: the fates of horizontally acquired genes in arthropod-symbiotic Spiroplasma. Genome Biol. Evol. 7, 2458–2472. 10.1093/gbe/evv160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E., Cantet F., Bonazzi M. (2015). Generation and multi-phenoty1ning of Coxiella burnetii transposon mutants. J. Vis. Exp. e52851. 10.3791/52851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez E., Cantet F., Fava L., Norville I., Bonazzi M. (2014). Identification of OmpA, a Coxiella burnetii protein involved in host cell invasion, by multi-phenotypic high-content screening. PLoS Pathog. 10:e1004013. 10.1371/journal.ppat.1004013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin M., Raoult D. (1999). Q fever. Clin. Microbiol. Rev. 12, 518–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCutcheon J. P., Moran N. A. (2012). Extreme genome reduction in symbiotic bacteria. Nat. Rev. Microbiol. 10, 13–26. 10.1038/nrmicro2670 [DOI] [PubMed] [Google Scholar]
- Mike L. A., Dutter B. F., Stauff D. L., Moore J. L., Vitko N. P., Aranmolate O., et al. (2013). Activation of heme biosynthesis by a small molecule that is toxic to fermenting Staphylococcus aureus. Proc. Natl. Acad. Sci. U.S.A. 110, 8206–8211. 10.1073/pnas.1303674110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mira A., Ochman H., Moran N. A. (2001). Deletional bias and the evolution of bacterial genomes. Trends Genet. 17, 589–596. 10.1016/S0168-9525(01)02447-7 [DOI] [PubMed] [Google Scholar]
- Moran N. A. (2002). Microbial minimalism: genome reduction in bacterial pathogens. Cell 108, 583–586. 10.1016/S0092-8674(02)00665-7 [DOI] [PubMed] [Google Scholar]
- Narasaki C. T., Toman R. (2012). Lipopolysaccharide of Coxiella burnetii. Adv. Exp. Med. Biol. 984, 65–90. 10.1007/978-94-007-4315-1_4 [DOI] [PubMed] [Google Scholar]
- Nelson K., Selander R. K. (1994). Intergeneric transfer and recombination of the 6-phosphogluconate dehydrogenase gene (gnd) in enteric bacteria. Proc. Natl. Acad. Sci. U.S.A. 91, 10227–10231. 10.1073/pnas.91.21.10227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochman H., Davalos L. M. (2006). The nature and dynamics of bacterial genomes. Science 311, 1730–1733. 10.1126/science.1119966 [DOI] [PubMed] [Google Scholar]
- Ochman H., Moran N. A. (2001). Genes lost and genes found: evolution of bacterial pathogenesis and symbiosis. Science 292, 1096–1099. 10.1126/science.1058543 [DOI] [PubMed] [Google Scholar]
- Ochman H., Lawrence J. G., Groisman E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. 10.1038/35012500 [DOI] [PubMed] [Google Scholar]
- Omsland A., Beare P. A., Hill J., Cockrell D. C., Howe D., Hansen B., et al. (2011). Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl. Environ. Microbiol. 77, 3720–3725. 10.1128/AEM.02826-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price M. N., Dehal P. S., Arkin A. P. (2008). Horizontal gene transfer and the evolution of transcriptional regulation in Escherichia coli. Genome Biol. 9:R4. 10.1186/gb-2008-9-1-r4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan R., Hicks L. D., Minnick M. F. (2008). Toxic introns and parasitic intein in Coxiella burnetii: legacies of a promiscuous past. J. Bacteriol. 190, 5934–5943. 10.1128/JB.00602-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan R., Miller S. R., Hicks L. D., Minnick M. F. (2007). The unusual 23S rRNA gene of Coxiella burnetii: two self-splicing group I introns flank a 34-base-pair exon, and one element lacks the canonical omegaG. J. Bacteriol. 189, 6572–6579. 10.1128/JB.00812-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raghavan R., Sloan D. B., Ochman H. (2012). Antisense transcription is pervasive but rarely conserved in enteric bacteria. MBio 3:e00156-12. 10.1128/mBio.00156-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seshadri R., Paulsen I. T., Eisen J. A., Read T. D., Nelson K. E., Nelson W. C., et al. (2003). Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc. Natl. Acad. Sci. U.S.A. 100, 5455–5460. 10.1073/pnas.0931379100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon J. G., Howe D., Heinzen R. A. (2005). Virulent Coxiella burnetii does not activate human dendritic cells: role of lipopolysaccharide as a shielding molecule. Proc. Natl. Acad. Sci. U.S.A. 102, 8722–8727. 10.1073/pnas.0501863102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith M. G., Gianoulis T. A., Pukatzki S., Mekalanos J. J., Ornston L. N., Gerstein M., et al. (2007). New insights into Acinetobacter baumannii pathogenesis revealed by high-density pyrosequencing and transposon mutagenesis. Genes Dev. 21, 601–614. 10.1101/gad.1510307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. A., Driscoll T., Gillespie J. J., Raghavan R. (2015). A Coxiella-like endosymbiont is a potential vitamin source for the Lone Star Tick. Genome Biol. Evol. 7, 831–838. 10.1093/gbe/evv016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A., Hoover P., Rougemont J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. 10.1080/10635150802429642 [DOI] [PubMed] [Google Scholar]
- Talavera G., Castresana J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. 10.1080/10635150701472164 [DOI] [PubMed] [Google Scholar]
- Thomas C. M., Nielsen K. M. (2005). Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721. 10.1038/nrmicro1234 [DOI] [PubMed] [Google Scholar]
- Treangen T. J., Rocha E. P. (2011). Horizontal transfer, not duplication, drives the expansion of protein families in prokaryotes. PLoS Genet. 7:e1001284 10.1371/journal.pgen.1001284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voth D. E., Heinzen R. A. (2007). Lounging in a lysosome: the intracellular lifestyle of Coxiella burnetii. Cell. Microbiol. 9, 829–840. 10.1111/j.1462-5822.2007.00901.x [DOI] [PubMed] [Google Scholar]
- Voth D. E., Beare P. A., Howe D., Sharma U. M., Samoilis G., Cockrell D. C., et al. (2013). The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV secretion system substrates. J. Bacteriol. 193, 1493–1503. 10.1128/JB.01359-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. Y., Brown L., Elliott M., Elliott T. (1997). Regulation of heme biosynthesis in Salmonella typhimurium: activity of glutamyl-tRNA reductase (HemA) is greatly elevated during heme limitation by a mechanism which increases abundance of the protein. J. Bacteriol. 179, 2907–2914. 10.1128/jb.179.9.2907-2914.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier I., Hicks L. D., Battisti J. M., Raghavan R., Minnick M. F. (2014). Identification of novel small RNAs and characterization of the 6S RNA of Coxiella burnetii. PLoS ONE 9:e100147. 10.1371/journal.pone.0100147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrier I., Walter M. C., Frangoulidis D., Raghavan R., Hicks L. D., Minnick M. F. (2016). The intervening sequence of Coxiella burnetii: characterization and evolution. Front. Cell. Infect. Microbiol. 6:83. 10.3389/fcimb.2016.00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams K. P., Gillespie J. J., Sobral B. W., Nordberg E. K., Snyder E. E., Shallom J. M., et al. (2010). Phylogeny of gammaproteobacteria. J. Bacteriol. 192, 2305–2314. 10.1128/JB.01480-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woong Park S., Klotzsche M., Wilson D. J., Boshoff H. I., Eoh H., Manjunatha U., et al. (2011). Evaluating the sensitivity of Mycobacterium tuberculosis to biotin deprivation using regulated gene expression. PLoS Pathog. 7:e1002264. 10.1371/journal.ppat.1002264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. M., Rock C. O. (2008). Membrane lipid homeostasis in bacteria. Nat. Rev. Microbiol. 6, 222–233. 10.1038/nrmicro1839 [DOI] [PubMed] [Google Scholar]
- Zhu Q., Kosoy M., Dittmar K. (2014). HGTector: an automated method facilitating genome-wide discovery of putative horizontal gene transfers. BMC Genomics 15:717. 10.1186/1471-2164-15-717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlitni S., Ferruccio L. F., Brown E. D. (2013). Metabolic suppression identifies new antibacterial inhibitors under nutrient limitation. Nat. Chem. Biol. 9, 796–804. 10.1038/nchembio.1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.