

ABSTRACT
The bacterial small RNA (sRNA) SgrS has been a fruitful model for discovery of novel RNA-based regulatory mechanisms and new facets of bacterial physiology and metabolism. SgrS is one of only a few characterized dual-function sRNAs. SgrS can control gene expression posttranscriptionally via sRNA-mRNA base-pairing interactions. Its second function is coding for the small protein SgrT. Previous work demonstrated that both functions contribute to relief of growth inhibition caused by glucose-phosphate stress, a condition characterized by disrupted glycolytic flux and accumulation of sugar phosphates. The base-pairing activity of SgrS has been the subject of numerous studies, but the activity of SgrT is less well characterized. Here, we provide evidence that SgrT acts to specifically inhibit the transport activity of the major glucose permease PtsG. Superresolution microscopy demonstrated that SgrT localizes to the cell membrane in a PtsG-dependent manner. Mutational analysis determined that residues in the N-terminal domain of PtsG are important for conferring sensitivity to SgrT-mediated inhibition of transport activity. Growth assays support a model in which SgrT-mediated inhibition of PtsG transport activity reduces accumulation of nonmetabolizable sugar phosphates and promotes utilization of alternative carbon sources by modulating carbon catabolite repression. The results of this study expand our understanding of a basic and well-studied biological problem, namely, how cells coordinate carbohydrate transport and metabolism. Further, this work highlights the complex activities that can be carried out by sRNAs and small proteins in bacteria.
IMPORTANCE Sequencing, annotation and investigation of hundreds of bacterial genomes have identified vast numbers of small RNAs and small proteins, the majority of which have no known function. In this study, we explore the function of a small protein that acts in tandem with a well-characterized small RNA during metabolic stress to help bacterial cells maintain balanced metabolism and continue growing. Our results indicate that this protein acts on the glucose transport system, inhibiting its activity under stress conditions in order to allow cells to utilize alternative carbon sources. This work sheds new light on a key biological problem: how cells coordinate carbohydrate transport and metabolism. The study also expands our understanding of the functional capacities of small proteins.
KEYWORDS: Hfq, PTS, RNase E
INTRODUCTION
Uptake and metabolism of carbon sources are regulated by a variety of mechanisms to ensure a steady flow of intermediates through central metabolism. Under most circumstances, glucose is a preferred carbon source for Escherichia coli, yet under some conditions, metabolic flux becomes suboptimal, and glucose transport and metabolism are disfavored (1, 2). One condition of impaired glucose metabolism occurs when cells are exposed to nonmetabolizable glucose analogs (e.g., α-methylglucoside [αMG]) that are taken up and phosphorylated but cannot be metabolized further. This induces the so-called “glucose-phosphate stress response,” which allows cells to reduce sugar phosphate accumulation and recover from stress. If the stress response is inactivated, cells show striking growth defects (3–6) and in some cases even lysis (7, 8). Growth of cells experiencing glucose-phosphate stress is improved by supplementation with glycolytic intermediates downstream of metabolic bottlenecks (8) or if sugar transport is inhibited (4, 9).
Glucose and the analog α-methylglucoside (αMG) are primarily transported into the cell through the major glucose transporter PtsG (EIICBGlc), while another analog, 2-deoxyglucose (2DG), is taken up mainly by the mannose transporter ManXYZ (EIIABCDMan) (10). PtsG and ManXYZ import and concomitantly phosphorylate incoming sugars via the phosphoenolpyruvate phosphotransferase system (PTS) comprised of several proteins that participate in a phosphorelay that begins with the glycolytic intermediate phosphoenolpyruvate (PEP) as the phosphate donor (11). The glucose PTS is especially significant since the EIIAGlc protein (which activates PtsG) has key regulatory roles in catabolite repression, which ensures preferential glucose utilization (12–15).
Our studies characterizing the glucose-phosphate stress response have demonstrated that a small RNA (sRNA), SgrS, whose synthesis is induced by glucose-phosphate stress, is the key regulatory effector of the response (4, 9, 16). Like many other sRNAs in bacteria, SgrS carries out base-pairing-dependent regulation of numerous mRNAs (4, 17–21). Less typical is the second function of SgrS, namely, encoding the 43-amino-acid protein SgrT (9, 22). Unlike any other known dual-function sRNA-small protein pair, SgrS and SgrT act independently in the same pathway, albeit by different mechanisms (9, 22). The base-pairing activity of SgrS ameliorates glucose-phosphate stress by inhibiting translation and promoting degradation of the ptsG and manXYZ mRNA transcripts, thereby preventing synthesis of more sugar transporters during stress. In contrast, SgrT was shown to have no effect on sugar transporter mRNA levels, but it could still reverse growth inhibition caused by stress (9, 22). These and other results suggested that SgrT acted by inhibiting glucose transporter activity (9). SgrT and SgrS base-pairing functions also act at different times during glucose-phosphate stress. The base-pairing function acts immediately, whereas SgrT is not detected until ∼30 min following the onset of stress (22). The goal of the present study was to examine the target specificity and physiological roles of SgrT in the glucose-phosphate stress response. We found that while the SgrS base-pairing activity serves to inhibit further synthesis of both PtsG and ManXYZ, SgrT specifically inhibits the transport activity of PtsG. Consistent with a mechanism of inhibition requiring physical interaction, SgrT localization to the membrane is PtsG dependent. Amino acid residues in the N-terminal region of the IIC (membrane) domain of PtsG confer sensitivity to SgrT-mediated inhibition of sugar transport activity. SgrT-mediated interference with the catabolite repression function of the glucose PTS allows utilization of alternative carbon sources, such as lactose, during glucose-phosphate stress. This provides a clear benefit to cells producing this small protein under glucose-phosphate stress conditions.
RESULTS
Ectopic production of SgrT inhibits cell growth on minimal glucose medium.
The base-pairing activity of SgrS inhibits the synthesis of both the PtsG and ManXYZ transport proteins by inhibiting translation of the corresponding mRNAs (4, 17, 18, 23). Our hypothesis is that SgrT inhibits activity of sugar transport proteins. To test whether SgrT affects PtsG, ManXYZ, or other sugar transporters, strains expressing sgrT from a plasmid were tested for growth on minimal media containing various carbon sources (Fig. 1). Growth of cells producing SgrT showed marked inhibition on glucose compared to cells carrying the vector control. In contrast, growth on all other carbon sources tested appeared similar between vector control and sgrT-expressing cells (Fig. 1). Notably, growth on mannose was unaffected by SgrT, and as ManXYZ is the only mannose transporter, these results suggest that SgrT affects only glucose transporters. Interestingly, the trehalose transporter TreB (EIIBCTre) does not have a cognate EIIA and instead relies on EIIAGlc for activation. If SgrT affected glucose transport by interfering with EIIAGlc activity, we would expect sgrT-expressing cells to demonstrate a growth defect on trehalose. Since cells expressing SgrT grow uninhibited on trehalose (Fig. 1), we postulate that SgrT does not act at the level of EIIAGlc to inhibit glucose transport.
FIG 1.
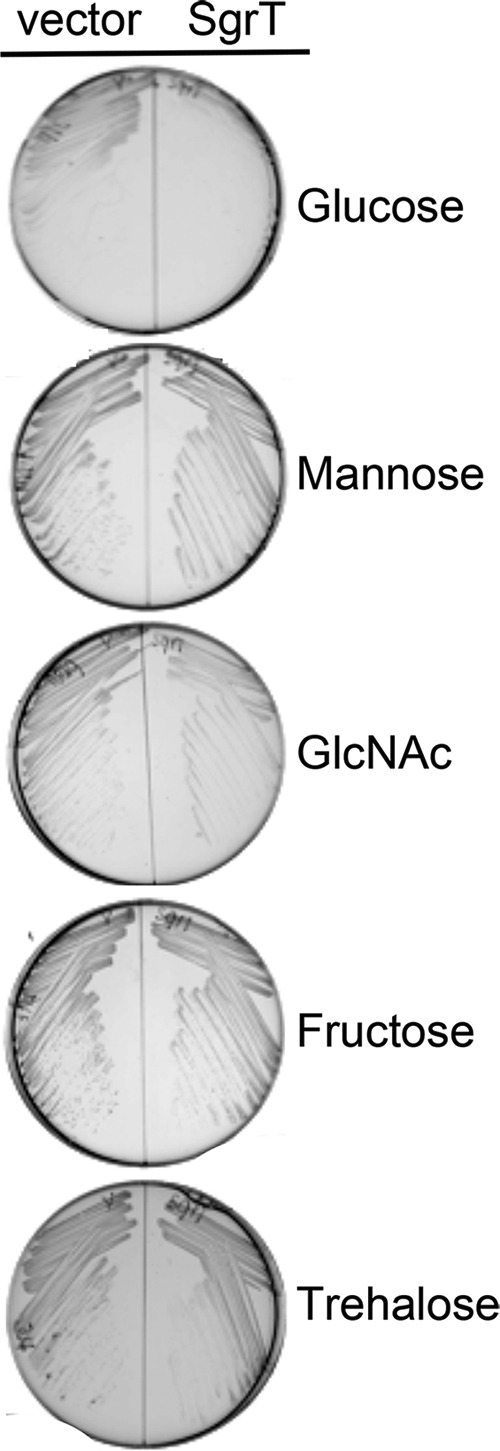
Ectopic production of SgrT inhibits cell growth on minimal glucose medium. ΔsgrS::Kanr lacIq+ cells (CV104) carrying a vector control (pBRCS12 [left]) or Plac-sgrT+ (pBRCV7 [right]) plasmid were grown on minimal medium with ampicillin, IPTG, and one of the following carbon sources (from the top): 0.2% glucose, 0.2% mannose, 0.2% N-acetylglucosamine (GlcNAc), 0.2% fructose, or 0.2% trehalose.
SgrT inhibits PtsG but not ManXYZ.
Results of growth experiments suggested that SgrT preferentially affects PtsG but not ManXYZ, and our previous work implicates sugar transport as the most likely step affected by SgrT (9). To further test this, we utilized a transcriptional PsgrS-lacZ fusion to monitor induction of the glucose-phosphate stress response upon exposure of cells to αMG or 2-deoxyglucose (2DG). These two glucose analogs are transported by different PTS proteins: αMG is primarily transported via PtsG (EIICBGlc) (10, 18, 24), whereas 2DG is mainly transported by ManXYZ (EIIABCDMan) (25). We therefore utilized these molecules to probe the activity of SgrT on these two sugar transporters. When cells were exposed to αMG, the β-galactosidase activity of the PsgrS-lacZ strain carrying the vector control was high (Fig. 2A), consistent with uptake of αMG and induction of the glucose-phosphate stress response. In cells expressing SgrT, β-galactosidase activity was reduced by ∼10-fold compared with that of vector control cells (Fig. 2A), supporting the idea that SgrT inhibits uptake of αMG. We noted that sgrT-expressing cells exposed to αMG still had higher levels of β-galactosidase activity than untreated cells and hypothesized that ManXYZ might be responsible for a low level of αMG uptake that was insensitive to SgrT. To test this hypothesis, we repeated the experiments in a manXYZ mutant strain. Compared with the parent strain, the manXYZ mutant showed lower levels of induction of PsgrS-lacZ when exposed to αMG and induction was completely lost when SgrT was produced (Fig. 2A). These data suggest that both PtsG and ManXYZ contribute to uptake of αMG and that SgrT only inhibits the activity of PtsG. To further test whether SgrT could affect transport through ManXYZ, we measured induction of PsgrS-lacZ in response to the ManXYZ-specific substrate 2DG (10). We note that 2DG is a less potent inducer of PsgrS-lacZ than αMG (note the difference in scales between Fig. 2A and B). Nonetheless, 2DG promoted a >10-fold induction of PsgrS-lacZ in vector control cells and a similarly large induction in SgrT-producing cells (Fig. 2B). Together, these data strongly suggest that SgrT specifically affects the transport activity of PtsG but not ManXYZ.
FIG 2.

SgrT inhibits PtsG transport activity but not ManXYZ activity. Three biological replicates of strain CL104 (Δlac mal::lacIq+ sgrS::Tetr) or CL105 (Δlac mal::lacIq+ sgrS::Tetr manXYZ::Kanr) containing a PsgrS-lacZ fusion and harboring either the vector control (pBRCS12) or Plac-sgrT+ (pBRCS1) plasmid were tested for the response to αMG (A) or 2DG (B) as detected by induction of the sgrS promoter. Cells were grown in TB overnight and then subcultured 1:200 and grown to an OD600 of 0.1. IPTG (0.1 mM) was added, and then cultures were split, with half receiving 0.1% 2DG or 0.5% αMG and the other half receiving an equivalent volume of double-distilled water (ddH2O). Samples were taken after 120 min, and β-galactosidase activity was measured.
SgrT localizes to the membrane in a PtsG-dependent manner.
Because SgrT was found to specifically affect uptake of (Fig. 2) and growth on (Fig. 1) substrates of PtsG, we hypothesized that SgrT would interact with PtsG and localize to the cell membrane. In wild-type (WT) or ΔptsG cells, previously characterized and functional SgrT-3×FLAG proteins (9) were labeled by immunofluorescent staining using an anti-FLAG antibody followed by Alexa Fluor 647-labeled secondary antibody and imaged using superresolved single-fluorophore microscopy (26) (Fig. 3). Two-dimensional (2D) projections in the xy plane from a 3D reconstructed image within different z depths are shown in Fig. 3A (1,000 nm) and B (200 nm). Images of individual cells were aligned and projected along the cell's longitudinal axis to the cell cross section (Fig. 3C) to generate heat maps of SgrT distribution, which demonstrates membrane versus cytoplasmic localization of SgrT (Fig. 3D). The intensity of the SgrT-3×FLAG signal is plotted as a function of cell radius (R, where the center of the cell is at 0) (Fig. 3E). In WT cells producing PtsG, SgrT preferentially localized to the cell periphery, indicated by a higher probability of SgrT localization at larger R values in WT cells than in ΔptsG cells (Fig. 3D and E). We note that the observed radial distribution of SgrT in WT cells peaks at ∼200 nm, which is smaller than expected for a live E. coli cell with an average diameter of ∼1 μm. We think two main factors account for this difference. First, we overexpressed SgrT for this imaging experiment, so the cytoplasmic portion of SgrT in excess of what could be bound by PtsG would be expected to shift the histogram toward a lower R value. Additionally, treatment required for the imaging protocol alters cell shape and contributes to a reduction in cell radius. Nevertheless, it is very apparent that deletion of ptsG shifted localization of SgrT away from the periphery (membrane region, higher R values) toward the cytoplasm (lower R values) (Fig. 3D and E). These data are consistent with the model that SgrT localizes to the membrane in a PtsG-dependent manner.
FIG 3.
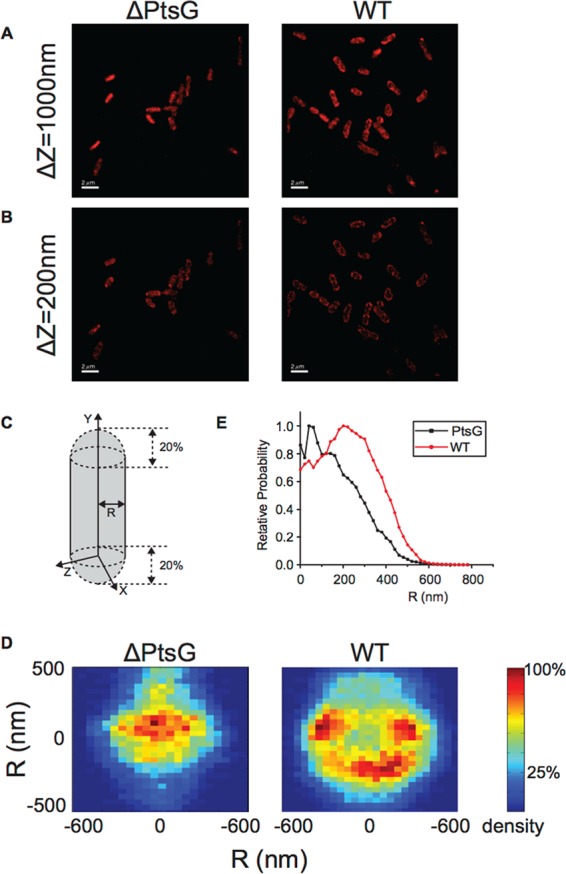
SgrT localizes to the cell membrane in a PtsG-dependent manner. Images in panels A and B are from superresolution microscopy and detection of SgrT-3×FLAG by immunofluorescence. (A) 2D projection images on the xy plane for ΔptsG cells (left) and WT cells (right) for the entire z range of 1,000 nm. (B) 2D projection images on the xy plane for ΔptsG cells (left) and WT cells (right) for the middle z plane (200-nm range). (C) Twenty percent of each end of the poles or septum was cut for 2D projection analysis. (D) 2D projection images on the xz plane for 76 ΔptsG cells (left) and 71 WT cells (right) with the color scale bar. Each pixel is 30 by 30 nm2, and the total number of spots was determined and plotted as heat maps. Cells are combined from two independent experiments. (E) The probability density of finding a spot at R distance from the length axis of cells was plotted by 40-nm binning, with renormalization keeping the peak value of each case as 1.
The IIC domain of PtsG is required for full sensitivity to SgrT.
Once we established that SgrT inhibits PtsG specifically and its localization to the membrane is dependent on PtsG, we took a genetic approach to assess which region of PtsG makes it susceptible to inhibition by SgrT. PtsG is comprised of three main functional domains: the membrane-bound IICGlc domain, the linker region, and the soluble IIBGlc domain. A previous study demonstrated that the IICGlc domain of PtsG most likely confers substrate specificity for glucose. In that study, a chimeric transporter composed of the PtsG IICGlc domain and the NagE IIBGlcNAc domain (for N-acetylglucosamine transport) was constructed (EIICGlcIIBGlcNAc), and its substrate specificity was assessed (27). The fusion junction was the LKTPGRED motif, which is located in the linker and is identical in PtsG and NagE. It was shown previously that the chimeric EIICGlcIIBGlcNAc (PtsG-NagE) protein could phosphorylate glucose but not GlcNAc, suggesting that the IIC domain dictates the specificity of PtsG for glucose (27). Since our results show that SgrT inhibits growth on glucose but not GlcNAc (Fig. 1), we predicted that SgrT would inhibit growth on glucose of cells expressing the EIICGlcIIBGlcNAc (PtsG-NagE) (Fig. 4A) chimeric protein by targeting the IICGlc domain, which dictates glucose specificity. Using cells producing only one of the transporters chimeric PtsG-NagE (EIICGlcIIBGlcNAc), wild-type PtsG (EIICBGlc), or wild-type NagE (EIICBAGlcNAc), we tested for SgrT-dependent inhibition of growth on glucose minimal medium (Fig. 4B). As expected, cells producing wild-type PtsG grew well in this medium in the absence of SgrT (Fig. 4B, orange line), but when SgrT was produced, these cells were strongly inhibited (Fig. 4B, light blue line). PtsG-NagE cells also grew in glucose minimal medium (Fig. 4B, purple line), consistent with previous data indicating that the IIC domain of PtsG is a primary determinant of glucose specificity. When SgrT was produced in the PtsG-NagE strain, growth was strongly inhibited for ∼16 h, but then cells resumed growth (Fig. 4B, green line). This phenotype and the timing of resumed growth were consistent for many biological replicates. Cells producing NagE were unable to grow on minimal glucose medium, regardless of whether SgrT was produced (Fig. 4B, red and blue lines). Strong growth inhibition of the strain producing the chimeric protein (with only EIIC from PtsG) by SgrT suggests that the key determinants defining SgrT specificity for PtsG reside in the IIC domain. However, the fact that PtsG-NagE cells can escape SgrT-mediated inhibition after prolonged incubation implies that sequences in the IIB domain of PtsG (absent from the chimeric transporter) may also contribute in some way to the PtsG-SgrT interaction.
FIG 4.
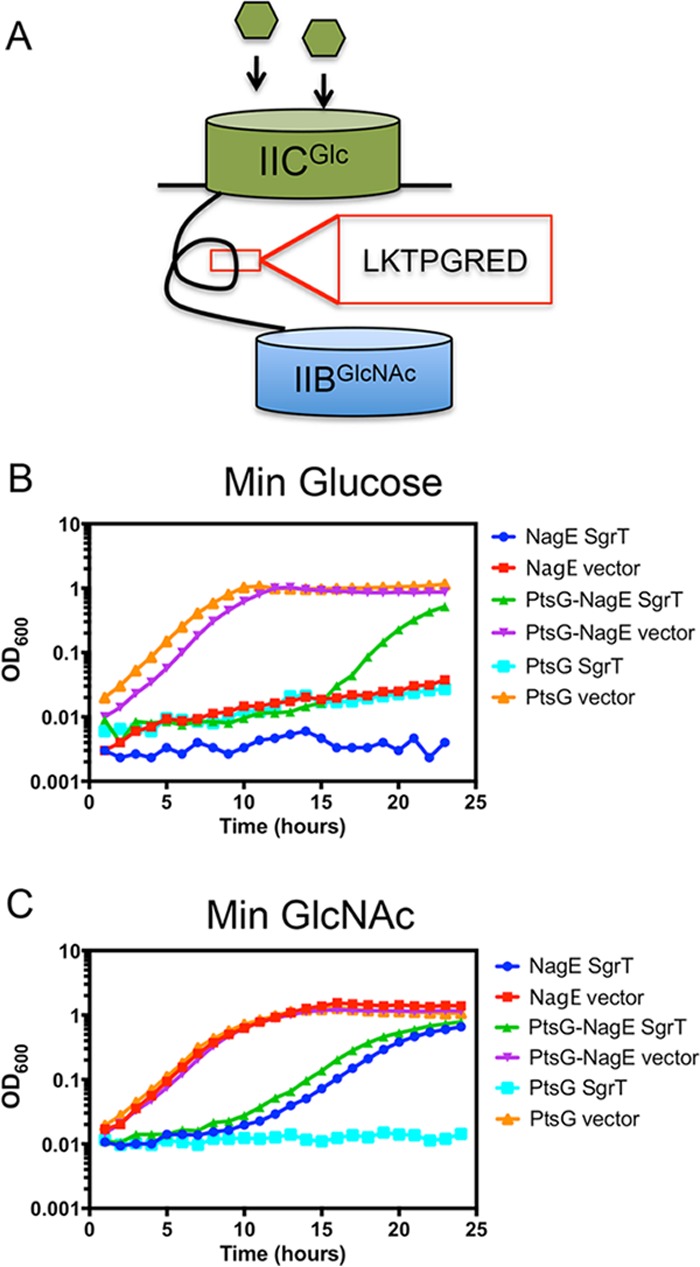
Strong SgrT-mediated growth inhibition requires the IIC domain of PtsG. (A) Schematic depicting the PtsG-NagE (EIICGlc BNag) chimera. The PtsG IIC domain is fused to the NagE IIB domain at their identical linker motif (LKTPGRED). (B) CL174 (NagE) (Δlac mal::lacIq+ sgrS::Tetr manXYZ::cat ΔptsG ΔgalP EIICBANag [NagE+]), CL175 (PtsG-NagE) (Δlac mal::lacIq+ sgrS::Tetr manXYZ::cat ΔgalP EIICGlc EIIBNag+), and CL188 (PtsG) (Δlac mal::lacIq+ sgrS::Tetr manXYZ::cat nagE::Kanr ΔgalP EIICBGlc [PtsG+]) harboring either the vector control (pBRCS12) or Plac-sgrT+ (pBRCS1) plasmid were cultured in M63 glucose medium plus 1 mM IPTG to induce SgrT and monitored for growth inhibition in biological triplicates using a 96-well plate reader. (C) Cultures of the strains used in panel B were grown in M63 GlcNAc medium with 1 mM IPTG to induce SgrT and monitored for growth over time using a 96-well plate reader. Data are the means from three biological replicates.
When cells were grown in GlcNAc, the PtsG strain grew similarly to the NagE strain (Fig. 4C, orange and red lines). The robust growth provided by PtsG on GlcNAc is somewhat surprising given that it has been reported that NagE and ManXYZ transport this substrate (11, 28). Interestingly, a mutation in the IIC domain of PtsG has been reported to allow the use of glucosamine by altering its regulation (29). Perhaps in our strain background, where manXYZ and nagE are deleted, regulation of ptsG is altered in a way that allows GlcNAc utilization. Regardless, we note that SgrT strongly inhibited PtsG-mediated growth on GlcNAc (Fig. 4C, light blue line). This suggests that SgrT can inhibit PtsG activity independent of the transported substrate. Given that both PtsG and NagE promote growth on GlcNAc, it was not surprising to find that chimeric PtsG-NagE also provides for robust growth on this sugar (Fig. 4C, purple line). Interestingly, we found that SgrT could also transiently inhibit growth of both NagE and PtsG-NagE cells on GlcNAc minimal medium (Fig. 4C, blue and green lines). (Note that this transient phenotype is not apparent on plates in Fig. 1 as transient growth inhibition on GlcNAc has already resolved by 24 h.) This result suggests that NagE and PtsG-NagE share determinants conferring partial susceptibility to SgrT. These could be localized to the linker region or other regions of similarity between the PtsG and NagE IIC domains (Fig. 5).
FIG 5.
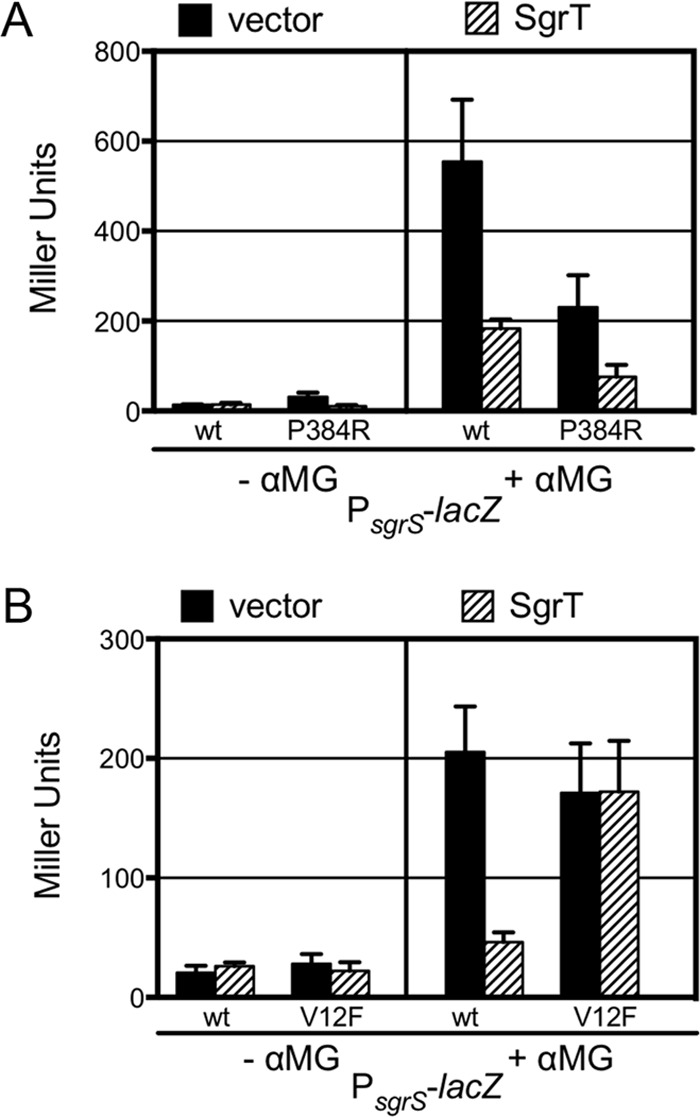
Identification of PtsG residues conferring resistance to SgrT-mediated transport inhibition. (A) CL108 cells (Δlac mal::lacIq+ PsgrS-lacZ [short] sgrS::Tetr ptsG::Kanr) harboring a plasmid containing WT ptsG (pZACL1) or ptsG(P384R) (pZACL2), as well as the vector control (pBRCS12) or Plac-sgrT+ plasmid (pBRCV7), were grown in TB overnight and subcultured 1:100 in TB plus 1 mM IPTG and 50 ng aTc and grown to an OD600 of 0.5. Cultures were then split, and half received treatment with 0.05% αMG. β-Galactoside activity was measured after 45 min. (B) The same procedure was repeated using the ptsG(V12F) mutation (pZACL3).
PtsG(V12F), but not PtsG(P384R), is resistant to SgrT-mediated transport inhibition.
To further define the interactions between the PtsG IIC domain and SgrT, we tested the susceptibilities of various ptsG mutants to SgrT-mediated inhibition of αMG transport as described above, using the PsgrS-lacZ reporter fusion. We began by testing mutants reported to have either increased transport of glucose or αMG or broadened substrate specificities (30, 31). The residues in these mutants reside in the cytoplasmic portion of PtsG. Most of these mutants (S157E, H339Y, K257N, M17T, and D343G) were still inhibited by SgrT. The Jahreis group reported that a PtsG mutant with broadened substrate specificity, PtsG(P384R), was not sensitive to inhibition by SgrT (32). We constructed the same mutant and found that the strain producing PtsG(P384R) was substantially less responsive to induction of PsgrS-lacZ by αMG than the strain producing wild-type PtsG (Fig. 5A). This suggests that this mutation impairs the function of PtsG and reduces transport of αMG. In contrast with the findings of Jahreis and colleagues (33), we found evidence that PtsG(P384R) activity could still be inhibited by SgrT (Fig. 5A). When SgrT was produced in the PtsG(P384R) strain, the fold reduction in PsgrS-lacZ activity was comparable to that observed in the wild-type PtsG strain. Thus, our data do not support the idea that SgrT requires this proline residue (P384) in the PtsG linker region in order to control PtsG activity. Further evidence arguing against this portion of the PtsG linker as a primary determinant of SgrT susceptibility is the conservation of the proline and surrounding residues within the linker between PtsG and NagE; the LKTPGRED motifs are identical in both proteins. If this motif were sufficient to confer susceptibility to SgrT, we would expect ectopic production of SgrT to inhibit NagE activity and growth on N-acetylglucosamine. Instead, SgrT does not strongly inhibit growth on GlcNAc (Fig. 1 and 4C).
Residues at the N terminus of PtsG (within the IIC domain) have previously been implicated in modulating the rate of glucose (and αMG) transport (34, 35). We tested the activity and SgrT sensitivity of another PtsG mutant, PtsG(V12F), which was previously shown to have an enhanced rate of αMG transport (30, 34). This mutant was also tested by Jahreis and coworkers for interaction with SgrT by coimmunoprecipitation, and their results suggested that PtsG(V12F) and SgrT could still interact (32). However, given the discordance of our results for the P384R mutation, we proceeded to test the V12F mutant as described above. In the absence of stress, cells with wild-type and V12F PtsG had similar levels of PsgrS-lacZ activity (Fig. 5B). As expected, when wild-type cells were stressed, PsgrS-lacZ activity increased substantially (Fig. 5B, compare activity in wild-type/vector cells for without αMG versus with αMG). Ectopic production of SgrT reduced this activity (Fig. 5B), consistent with SgrT-mediated inhibition of wild-type PtsG activity. In the PtsG(V12F) strain, exposure to αMG also induced PsgrS-lacZ activity (Fig. 5B, compare activity in V12F/vector cells without αMG versus those with αMG), but production of SgrT had no effect on this activity. These observations suggest that the transport activity of PtsG(V12F) is similar to that of wild-type PtsG but that this mutation renders PtsG(V12F) insensitive to inhibition by SgrT.
SgrT strongly inhibits preexisting PtsG transporters.
Our previous work indicates that mechanistically, the base-pairing activity of SgrS only stops new synthesis of PtsG transporters but has no effect on preexisting transporters (9, 20). In contrast, we hypothesize that SgrT has an immediate inhibitory effect on PtsG activity. To test the relative effects of SgrT and SgrS base pairing on PtsG transport activity, we directly measured the uptake of [14C]αMG by ΔsgrS cells carrying a vector control or ectopically expressing sgrT alone or an sgrS allele possessing only the base-pairing region for 20 min prior to exposure of cells to [14C]αMG. Cells carrying the vector control accumulated [14C]αMG, as evidenced by the increase in cell-associated radioactivity over time (Fig. 6). The negative control, a ΔptsG mutant, failed to accumulate appreciable levels of [14C]αMG. Cells producing SgrT were similar to the ΔptsG mutant and showed very little uptake of [14C]αMG (Fig. 6). In contrast, cells expressing the base-pairing-only SgrS looked very similar to the positive-control cells and accumulated [14C]αMG at a similar rate (Fig. 6). These results support our overall model in which the base-pairing activity of SgrS acts only to inhibit new PtsG synthesis, but the preexisting PtsG (which is extremely stable [20]) remains active and SgrT is required to inhibit this activity.
FIG 6.
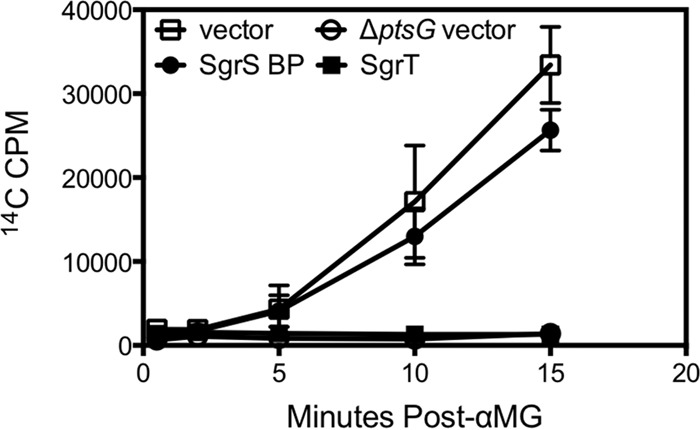
SgrT strongly and immediately inhibits transport activity of PtsG. CL113 cells (Δlac mal::lacIq+ galP::Kanr sgrS::Tetr manXYZ::cat) carrying the vector control (pBRCS12), Plac-sgrT+ (pBRCS1), or sgrS base-pairing-only (pLCV5) plasmid were grown in M63 glycerol medium with 0.1 mM IPTG and then washed with M63 salts medium. Cells were exposed to [14C]αMG and monitored for accumulation of radioactivity over time. The results shown are the average for biological triplicates.
SgrT-mediated inhibition of PtsG prevents inducer exclusion, promoting growth by allowing utilization of alternative carbon sources.
Inducer exclusion is one of the mechanisms that ensures preferential utilization of glucose by enteric bacteria when glucose is present in a mixture with other carbon sources (36). Inducer exclusion requires the dephosphorylated form of EIIAGlc, which accumulates when cells are actively importing glucose via PtsG. Under these conditions, EIIAGlc interacts with a variety of transport proteins and enzymes to inhibit uptake or utilization of alternative carbon sources (36). In a previous study, we demonstrated robust inducer exclusion when cells were grown in the presence of glucose and lactose, evidenced by very low expression of lac genes, when SgrT was not produced. In contrast, in cells making SgrT, inducer exclusion was relieved and lac genes were highly expressed (9). These data are consistent with the model that SgrT-mediated inhibition of PtsG activity results in accumulation of phospho-EIIAGlc, relieving inducer exclusion and promoting lac expression. Based on these previous results, we hypothesize that SgrT-mediated relief of inducer exclusion may allow cells experiencing glucose-phosphate stress to utilize alternative carbon sources (37). To test this, ΔsgrS cells expressing a vector control or SgrT were grown in the presence of αMG with or without lactose and simultaneously monitored for growth and lac expression (Fig. 7). Without lactose, cells expressing SgrT showed growth improvement compared to vector control and reached a density of 1.0 after 400 min (Fig. 7A). In the presence of lactose, however, SgrT-expressing cells reached the same density in only 200 min and ultimately grew to a higher density. Vector control cell growth was also improved by lactose, but only after a prolonged lag phase (Fig. 7B). SgrT-producing cells also showed ∼2-fold increased endogenous β-galactosidase activity compared with control cells at early time points after stress induction (data not shown), consistent with relief of inducer exclusion in these cells. These data demonstrate that SgrT production not only aids cells in overcoming the stress of accumulating sugar phosphates but also promotes utilization of alternative carbon sources, thereby markedly improving stress recovery and growth.
FIG 7.
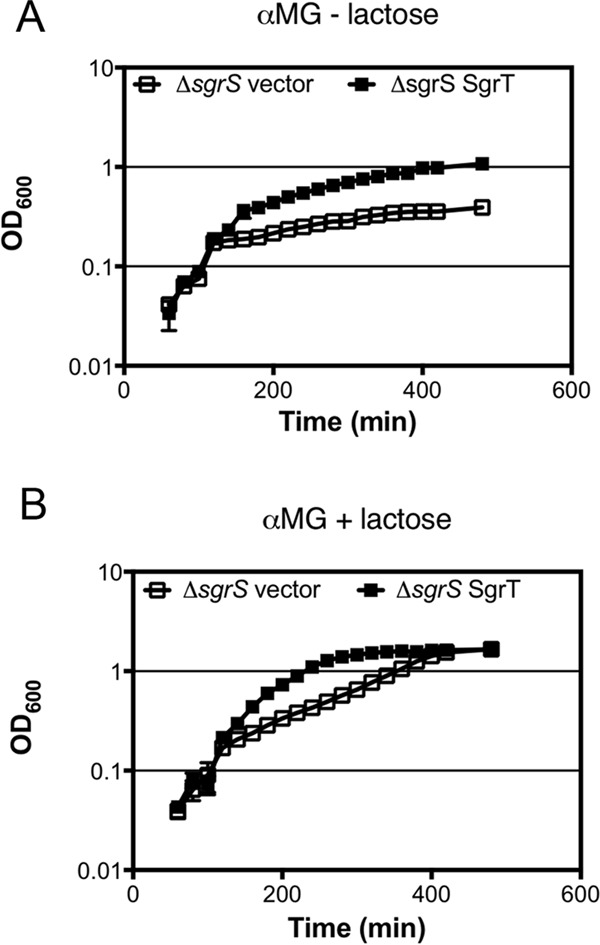
SgrT facilitates use of alternative carbon sources during stress. CS136 cells (MG1655 ΔsgrS::Kanr) carrying the vector control (pHDB3) or Plac-sgrT+ (pBRCV7) plasmid were grown in TB medium with 0.5% αMG plus 1.0 mM IPTG without (A) or with (B) lactose, and growth was measured by recording the optical density at 600 nm over time.
DISCUSSION
SgrS is a versatile sRNA able to produce a functional protein and regulate many targets to alleviate glucose-phosphate stress (23). Among these targets, PtsG and ManXYZ transporters are particularly important, as they are directly responsible for importing stress-inducing molecules (38, 39). SgrT and SgrS base pairing act independently to tackle the problem of sugar transport under stress conditions from two directions. The SgrS base-pairing function is critical and is used first to inhibit further synthesis of sugar transporters (22), after which SgrT is produced to inhibit existing PtsG transporters (22). In this study, we determined that SgrT specifically affects the activity of only one sugar transporter, namely, PtsG (EIICBGlc) (Fig. 1, 2, and 4). This stands in contrast to the base-pairing activity of SgrS, which impacts synthesis of both PtsG (4, 17) and another sugar transporter, ManXYZ (18). We discovered that the IIC domain of PtsG contains determinants that make PtsG but not a highly similar transporter, NagE, susceptible to inhibition by SgrT (Fig. 4). Preferential localization of SgrT to cell membrane regions in ptsG+ but not ΔptsG strains (Fig. 3) suggests that SgrT modulates PtsG activity via physical interactions. Finally, we demonstrated that SgrT-producing cells have a growth advantage during glucose-phosphate stress, particularly when an alternative carbon source is provided (Fig. 7). Cumulatively, our work upholds our model that the protein (SgrT) component of the dual-function sRNA SgrS provides a specific mechanism for inhibiting sugar transport activity and promoting cell growth under glucose-phosphate stress conditions (Fig. 8).
FIG 8.
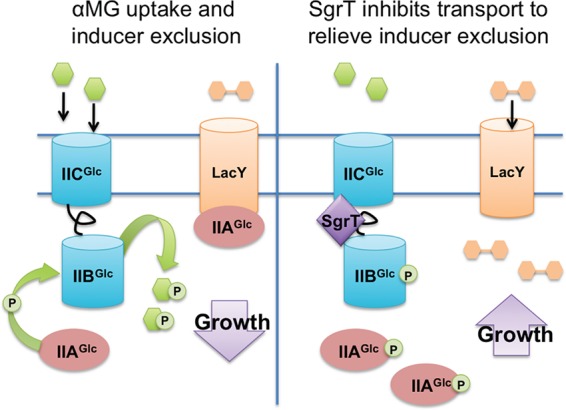
Model for regulation of PtsG activity by SgrT. During active αMG transport (left), PtsG and EIIAGlc are dephosphorylated because phosphate is rapidly transferred to the incoming sugar. EIIAGlc binds the lactose permease protein (among others), inhibiting its activity and enacting inducer exclusion. SgrT binding to PtsG (right) inhibits transport, stopping the flow of phosphate so that phosphorylated EIIAGlc accumulates. P-EIIAGlc no longer inhibits transport or utilization of alternative carbon sources, and thus inducer exclusion is relieved. This allows utilization of other carbon sources (e.g., lactose), leading to promotion of growth.
Under stress, SgrS base-pairing activity inhibits the synthesis of both the PtsG and ManXYZ transporters responsible for importing αMG and 2DG. In contrast, SgrT solely targets PtsG and has no effect on 2DG transport (Fig. 2). Why might SgrT differentiate between these transporters, and what does this tell us about the nature of glucose-phosphate stress? Unlike ManXYZ, the proteins comprising the glucose PTS are critical for metabolic regulation via carbon catabolite repression (CCR). During glucose transport, EIIAGlc (Crr) exists largely in a dephosphorylated state due to rapid phosphate transfer to PtsG. Dephosphorylated EIIAGlc binds and inhibits other transporters, such as LacY, causing inducer exclusion (Fig. 8) (36). Another effect of low phospho-EIIAGlc levels is reduced production of the small molecule cyclic AMP (cAMP), since only phospho-EIIAGlc binds to and activates the adenylate cyclase to stimulate cAMP production (40–42). These two outcomes of CCR—inducer exclusion and reduced cAMP production—together reduce the activity and synthesis, respectively, of transporters for alternative carbon sources, thus favoring glucose utilization. The glucose analog αMG also stimulates CCR, which not only favors further transport of αMG but also prevents uptake and metabolism of other carbon sources. The stress response enacted by SgrS and SgrT counteracts the CCR response, and in this study, we show that this allows the utilization of metabolizable sugars such as lactose, which helps cells thrive during stress (Fig. 8).
The use of alternative non-PTS carbon sources may be especially beneficial for glucose-phosphate stress recovery since the PTS-dependent transport of αMG depletes PEP, which subsequently cannot be replenished by metabolism. In fact, the depletion of such critical metabolic intermediates is thought to be the cause of stress as opposed to any inherent toxicity associated with sugar phosphate accumulation, as the addition of sugar phosphates downstream of the metabolic block allows cells to recover even while αMG is actively transported (8). Although the exact cause of stress in natural environments is still unknown, it is clear that conditions resulting in accumulation of nonmetabolizable phosphorylated intermediates (or their analogs) of the early steps of glycolysis promote rapid induction of the stress response. There is evidence that αMG and 2DG exist in nature. Klebsiella pneumoniae can metabolize αMG as a carbon source (43), and αMG has been found in environmental water samples (44). Saccharomyces cerevisiae can detoxify 2DG (45). Gammaproteobacteria, mainly enteric bacteria, are the only organisms known to possess this particular stress response (46, 47). Whether these sugar analogs or similar compounds are present in the gut or other environmental niches where these organisms reside is undetermined.
It is worth noting that our experiments demonstrated a clear role for the IIC domain of PtsG in conferring susceptibility to SgrT, whereas others (32, 33) have suggested that the linker region connecting the IIC and IIB domains is involved in SgrT binding. Bimolecular fluorescence complementation experiments performed by the Jahreis group (32) tested the interactions between SgrT and various lengths of PtsG by fusing them to different domains of green fluorescent protein (GFP) and monitoring GFP complementation by fluorescence. They showed that complementation occurred most strongly when both the IIC domain and linker were present but found no interaction between SgrT and the IIC domain alone (32). We note that these experiments utilized protein fusions between PtsG fragments or SgrT and different domains of GFP. Our experience with SgrT protein fusions has been that any but small epitopes strongly impair SgrT function and prevent it from controlling PtsG (C. R. Lloyd and C. K. Vanderpool, unpublished data). Thus, it is possible that bimolecular fluorescence complementation assays with protein fusions did not fully capture relevant SgrT-PtsG interactions. These results, along with those we present here, suggest that determinants in both the linker and IIC domain are important for SgrT interaction. Interestingly, in a cross-linking and copurification experiment (using SgrT tagged with the small hemagglutinin [HA] epitope), Kosfeld and Jahreis found evidence that SgrT preferentially interacts with dephosphorylated PtsG (32), which would be the predominant form present during active glucose transport as the phosphate is rapidly transferred to the incoming sugar. This observation suggests that SgrT might only associate with PtsG during active transport. We envision a model in which SgrT interacts with PtsG and blocks sugar passage directly or causes a conformational change that prevents phosphorylation of PtsG. This would leave the EIIAGlc protein in a phosphorylated state—unable to bind and inhibit the lactose permease and other carbon utilization proteins, thereby enabling use of any other available carbon sources (Fig. 8). More structure-function studies are necessary to determine if the interaction between SgrT and EIICBGlc is direct or requires an intermediate and to elucidate the molecular mechanism of SgrT activity.
MATERIALS AND METHODS
Bacterial strain construction.
All strains and plasmids used in this study are described in Table 1. The pBR322 derivative plasmids harboring SgrT used in this study were previously described (9). To create the pZA plasmid derivatives of pZA31-R (48), wild-type E. coli MG1655 ptsG was cloned into the BamHI and NdeI sites of the plasmid, and mutants were created using QuikChange mutagenesis (Agilent Technologies). All Δlac strains are derivatives of DJ480 (D. Jin, National Cancer Institute). CL104 was created by P1 transduction of an sgrS::Tetr mutation into a strain containing a PsgrS-lacZ transcriptional fusion (CV5202). CL108 was subsequently created by transducing a ptsG::Kanr mutation from the Keio collection (49) into CL104. Strain CL113 was constructed by P1 transduction of galP::Kanr (from the Keio collection) and manXYZ::cat cassettes into strain CS216 (sgrS::Tetr mal::lacIq+). CL174, CL175, and CL188 are all derivatives of CL113 from which kanamycin cassettes were removed using the FLP-mediated site-specific recombination methods described in reference 50. The PtsG-NagE hybrid created in CL175 was generated by PCR amplification of the first 1,050 nucleotides of PtsG linked to the upstream kanamycin cassette from the ycfH::Kanr mutant (Keio collection) and recombining this linear PCR product into NM300-1 (which carries a mini-λ encoding λ Red functions) (51) via transformation at the nagE locus and then P1 transducing the hybrid into CL174.
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Descriptiona | Reference or source |
|---|---|---|
| Strains | ||
| CV104 | ΔlacX74 mal::lacIq ΔsgrS::Kanr | 4 |
| CS136 | Δlac ΔsgrS::Kanr | 54 |
| CL104 | Δlac mal::lacIq PsgrS-lacZ (short) sgrS::Tetr | This study |
| CL105 | Δlac mal::lacIq PsgrS-lacZ (short) sgrS::Tetr manXYZ::Kanr | This study |
| CL108 | Δlac mal::lacIq PsgrS-lacZ (short) sgrS::Tetr ptsG::Kanr | This study |
| CL113 | Δlac mal::lacIq sgrS::Tetr galP::Kanr manXYZ::cat | This study |
| CL143 | Δlac mal::lacIq sgrS::Tetr galP::FRT ptsG::Kanr manXYZ::cat | This study |
| CL174 | Δlac mal::lacIq sgrS::Tetr galP::FRT ptsG::FRT manXYZ::cat | This study |
| CL175 | Δlac mal::lacIq sgrS::Tetr galP::FRT ptsG::FRT manXYZ::cat EIICGlcNAc::EIICGlc (at nagE locus linked to Kanr) | This study |
| CL188 | Δlac mal::lacIq sgrS::Tetr galP::FRT ptsG::FRT nagE::Kanr | This study |
| Plasmids | ||
| pBRCS12 | pBRPlac vector control | 54 |
| pBRCV7 | pBRPlac with sgrT coding sequence under control of Plac promoter | 9 |
| pBRCS1 | pBRCV7 with 3×FLAG tag inserted at C terminus of SgrT | 9 |
| pLCV5 | pLCV1 with point mutation that changes 5th codon of sgrT to UAA | 9 |
| pHDB3 | pBR322 derivative, vector control | 55 |
| pZA31R | Vector | 48 |
| pZACL1 | ptsG WT | This study |
| pZACL2 | ptsG(P384R) | This study |
| pZACL3 | ptsG(V12F) | This study |
FRT, FLP recombination target.
Media and growth conditions.
Bacteria were cultured in Luria-Bertani (LB) medium at 37°C unless otherwise specified in the figure legends. For experiments investigating the overexpression of SgrT on various carbon sources, CV104 cells harboring pBRCS12 or pBRCV7 plasmids were plated on minimal M63 agar plates containing 0.2% glucose, N-acetylglucosamine (GlcNAc), fructose, trehalose, or mannose plus ampicillin and 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) to induce SgrT. For growth experiments in minimal media, CL174, CL175, and CL188 cells containing pBRCS12 or pBRCS1 plasmids were grown overnight in M63 with 0.4% glycerol plus ampicillin and then subcultured 1:200 in M63 with 0.2% glucose plus ampicillin and 1 mM IPTG to induce SgrT. For growth curves examining growth with or without addition of lactose under glucose-phosphate stress, CS136 cells harboring pHDB3 or pBRCV7 were grown as previously described (9). Growth curve experiments conducted past 12 h were performed in a plate reader. Independent triplicate cultures were grown overnight in M63 glycerol (0.4%) and then subcultured 1:200 in either 0.2% glucose or 0.2% GlcNAc and grown for 1 h, and then 0.2 ml was transferred into the wells of a 96-well plate and optical density (OD) was monitored for 24 h. Experiments investigating the sensitivity of ptsG mutants to SgrT regulation were performed using MacConkey agar plates containing 1 mM IPTG to induce sgrT, 50 ng anhydrous tetracycline (aTc) to induce ptsG, 0.5% αMG to induce the stress response, 100 mg/ml ampicillin and 25 mg/ml chloramphenicol to select for the SgrT- and PtsG-harboring plasmids, respectively, and 1% lactose.
β-Galactosidase assays.
CL104 and CL105 strains harboring pBRCS12 or pBRCS1 plasmids were grown overnight in tryptone broth (TB) with ampicillin and subcultured 1:100 into TB medium with 1 mM IPTG to induce SgrT. Cells were grown to an OD600 of 0.5, split, and either stressed with 0.5% αMG or 0.2% 2DG or left unstressed. Samples were taken after 120 min, and Miller assays were performed (52). For CL108 strains harboring the pBRCS12 or pBRCV7 and pZACL1, pZACL2, or pZACL3 plasmids, the same protocol was used and PtsG synthesis was induced with 50 ng/ml aTc.
[14C]αMG transport assays.
CL113 cells with pBRCS12 or pBRCS1 were grown overnight in 0.4% M63 glycerol and then subcultured 1:200 in fresh M63 glycerol to an OD600 of 0.2 and induced with 0.1 mM IPTG for 5 min. Cells were then pelleted on ice and prepared exactly as described in reference 8, except for the addition of glucose-6-phosphate.
Immunostaining and superresolved single-fluorophore microscopic imaging.
CL108 cells carrying 3×FLAG-tagged SgrT(pBRCS1) in the presence and absence of ptsG were grown in LB overnight and then diluted 1:100 into fresh LB and grown to OD600 of ∼0.3. Cells were then induced for SgrT and PtsG expression by 50 μM IPTG and 50 ng/ml anhydrous tetracycline, respectively, for 30 min. Cells were collected and fixed with 4% formaldehyde and immobilized on the Lab-Tek chambered coverglass coated with poly-l-lysine (Sigma-Aldrich P8920) at room temperature. Then cells were washed with 1× phosphate-buffered saline (PBS) three times and then permeabilized with 10 mg/ml lysozyme (Sigma-Aldrich L6876) dissolved in 25 mM Tris-Cl (pH 8), 10 mM EDTA, and 50 mM glucose for 30 min at room temperature. Cells were then washed with 1× PBS three times and were treated with 3% bovine serum albumin (BSA) in 1× PBS for 1 h at room temperature. Then cells were incubated with the primary anti-FLAG antibody (from mouse) in 0.3% BSA–1× PBS for 1 h at room temperature. Cells were washed three times with 0.3% BSA in 1× PBS and then incubated with Alexa Fluor 647-labeled secondary antibody in 0.3% BSA–1× PBS for 1 h at room temperature. Then cells were washed three additional times with 0.3% BSA in 1× PBS. Superresolution imaging was performed as previously described (53) on an inverted microscope excited with a 647-nm laser and a 405-nm laser. The emission was separated and collected by a dichroic mirror and notch filters for the 647-nm laser, and data were recorded by an EMCCD camera. A cylindrical lens was inserted in the emission path for 3D imaging. Each STORM image was reconstructed by about 2 to ∼30,000 frames with 30 or 60 ms of exposure. Samples were all imaged in buffer with 10 mM NaCl, 50 mM Tris (pH 8), 10% glucose, pyranose oxidase (final concentration, 1.11 U/ml [Sigma-Aldrich P4234]) and catalase (final concentration, 10 KU/ml [EMD Millipore 219001]). Image reconstruction was performed as previously described (53).
ACKNOWLEDGMENTS
We are grateful to James Slauch and to members of the Vanderpool and Slauch laboratories for stimulating discussions and suggestions.
This work was supported by National Institutes of Health grant GM092830.
Footnotes
For a commentary on this article, see https://doi.org/10.1128/JB.00130-17.
REFERENCES
- 1.Richards GR, Vanderpool CK. 2011. Molecular call and response: the physiology of bacterial small RNAs. Biochim Biophys Acta 1809:525–531. doi: 10.1016/j.bbagrm.2011.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bren A, Park JO, Towbin BD, Dekel E, Rabinowitz JD, Alon U. 2016. Glucose becomes one of the worst carbon sources for E. coli on poor nitrogen sources due to suboptimal levels of cAMP. Sci Rep 6:24834. doi: 10.1038/srep24834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kimata K, Tanaka Y, Inada T, Aiba H. 2001. Expression of the glucose transporter gene, ptsG, is regulated at the mRNA degradation step in response to glycolytic flux in Escherichia coli. EMBO J 20:3587–3595. doi: 10.1093/emboj/20.13.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vanderpool CK, Gottesman S. 2004. Involvement of a novel transcriptional activator and small RNA in post-transcriptional regulation of the glucose phosphoenolpyruvate phosphotransferase system. Mol Microbiol 54:1076–1089. doi: 10.1111/j.1365-2958.2004.04348.x. [DOI] [PubMed] [Google Scholar]
- 5.Morita T, El-Kazzaz W, Tanaka Y, Inada T, Aiba H. 2003. Accumulation of glucose 6-phosphate or fructose 6-phosphate is responsible for destabilization of glucose transporter mRNA in Escherichia coli. J Biol Chem 278:15608–15614. doi: 10.1074/jbc.M300177200. [DOI] [PubMed] [Google Scholar]
- 6.Sun Y, Vanderpool CK. 2013. Physiological consequences of multiple-target regulation by the small RNA SgrS in Escherichia coli. J Bacteriol 195:4804–4815. doi: 10.1128/JB.00722-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Irani MH, Maitra PK. 1977. Properties of Escherichia coli mutants deficient in enzymes of glycolysis. J Bacteriol 132:398–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Richards GR, Patel MV, Lloyd CR, Vanderpool CK. 2013. Depletion of glycolytic intermediates plays a key role in glucose-phosphate stress in Escherichia coli. J Bacteriol 195:4816–4825. doi: 10.1128/JB.00705-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wadler CS, Vanderpool CK. 2007. A dual function for a bacterial small RNA: SgrS performs base pairing-dependent regulation and encodes a functional polypeptide. Proc Natl Acad Sci U S A 104:20454–20459. doi: 10.1073/pnas.0708102104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henderson PJ, Giddens RA, Jones-Mortimer MC. 1977. Transport of galactose, glucose and their molecular analogues by Escherichia coli K12. Biochem J 162:309–320. doi: 10.1042/bj1620309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Postma PW, Lengeler JW, Jacobson GR. 1993. Phosphoenolpyruvate:carbohydrate phosphotransferase systems of bacteria. Microbiol Rev 57:543–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hogema BM, Arents JC, Bader R, Eijkemans K, Yoshida H, Takahashi H, Aiba H, Postma PW. 1998. Inducer exclusion in Escherichia coli by non-PTS substrates: the role of the PEP to pyruvate ratio in determining the phosphorylation state of enzyme IIAGlc. Mol Microbiol 30:487–498. doi: 10.1046/j.1365-2958.1998.01053.x. [DOI] [PubMed] [Google Scholar]
- 13.Kimata K, Takahashi H, Inada T, Postma P, Aiba H. 1997. cAMP receptor protein-cAMP plays a crucial role in glucose-lactose diauxie by activating the major glucose transporter gene in Escherichia coli. Proc Natl Acad Sci U S A 94:12914–12919. doi: 10.1073/pnas.94.24.12914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gutierrez-Rios RM, Freyre-Gonzalez JA, Resendis O, Collado-Vides J, Saier M, Gosset G. 2007. Identification of regulatory network topological units coordinating the genome-wide transcriptional response to glucose in Escherichia coli. BMC Microbiol 7:53. doi: 10.1186/1471-2180-7-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inada T, Kimata K, Aiba H. 1996. Mechanism responsible for glucose-lactose diauxie in Escherichia coli: challenge to the cAMP model. Genes Cells 1:293–301. doi: 10.1046/j.1365-2443.1996.24025.x. [DOI] [PubMed] [Google Scholar]
- 16.Vanderpool CK, Gottesman S. 2007. The novel transcription factor SgrR coordinates the response to glucose-phosphate stress. J Bacteriol 189:2238–2248. doi: 10.1128/JB.01689-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kawamoto H, Koide Y, Morita T, Aiba H. 2006. Base-pairing requirement for RNA silencing by a bacterial small RNA and acceleration of duplex formation by Hfq. Mol Microbiol 61:1013–1022. doi: 10.1111/j.1365-2958.2006.05288.x. [DOI] [PubMed] [Google Scholar]
- 18.Rice JB, Vanderpool CK. 2011. The small RNA SgrS controls sugar-phosphate accumulation by regulating multiple PTS genes. Nucleic Acids Res 39:3806–3819. doi: 10.1093/nar/gkq1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Papenfort K, Podkaminski D, Hinton JC, Vogel J. 2012. The ancestral SgrS RNA discriminates horizontally acquired Salmonella mRNAs through a single G-U wobble pair. Proc Natl Acad Sci U S A 109:E757–E764. doi: 10.1073/pnas.1119414109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papenfort K, Sun Y, Miyakoshi M, Vanderpool CK, Vogel J. 2013. Small RNA-mediated activation of sugar phosphatase mRNA regulates glucose homeostasis. Cell 153:426–437. doi: 10.1016/j.cell.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bobrovskyy M, Vanderpool CK. 2016. Diverse mechanisms of post-transcriptional repression by the small RNA regulator of glucose-phosphate stress. Mol Microbiol 99:254–273. doi: 10.1111/mmi.13230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balasubramanian D, Vanderpool CK. 2013. Deciphering the interplay between two independent functions of the small RNA regulator SgrS in Salmonella. J Bacteriol 195:4620–4630. doi: 10.1128/JB.00586-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bobrovskyy M, Vanderpool CK. 2013. Regulation of bacterial metabolism by small RNAs using diverse mechanisms. Annu Rev Genet 47:209–232. doi: 10.1146/annurev-genet-111212-133445. [DOI] [PubMed] [Google Scholar]
- 24.Stock JB, Waygood EB, Meadow ND, Postma PW, Roseman S. 1982. Sugar transport by the bacterial phosphotransferase system. The glucose receptors of the Salmonella typhimurium phosphotransferase system. J Biol Chem 257:14543–14552. [PubMed] [Google Scholar]
- 25.Rephaeli AW, Saier MH Jr. 1980. Substrate specificity and kinetic characterization of sugar uptake and phosphorylation, catalyzed by the mannose enzyme II of the phosphotransferase system in Salmonella typhimurium. J Biol Chem 255:8585–8591. [PubMed] [Google Scholar]
- 26.Huang B, Jones SA, Brandenburg B, Zhuang X. 2008. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat Methods 5:1047–1052. doi: 10.1038/nmeth.1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hummel U, Nuoffer C, Zanolari B, Erni B. 1992. A functional protein hybrid between the glucose transporter and the N-acetylglucosamine transporter of Escherichia coli. Protein Sci 1:356–362. doi: 10.1002/pro.5560010307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones-Mortimer MC, Kornberg HL. 1980. Amino-sugar transport systems of Escherichia coli K12. J Gen Microbiol 117:369–376. [DOI] [PubMed] [Google Scholar]
- 29.Plumbridge J. 2000. A mutation which affects both the specificity of PtsG sugar transport and the regulation of ptsG expression by Mlc in Escherichia coli. Microbiology 146:2655–2663. doi: 10.1099/00221287-146-10-2655. [DOI] [PubMed] [Google Scholar]
- 30.Notley-McRobb L, Ferenci T. 2000. Substrate specificity and signal transduction pathways in the glucose-specific enzyme II (EIIGlc) component of the Escherichia coli phosphotransferase system. J Bacteriol 182:4437–4442. doi: 10.1128/JB.182.16.4437-4442.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Erni B. 2003. Glucose transport by the bacterial phosphotransferase system (PTS): an interface between energy- and signal transduction, p 115–138. In Winkelmann G. (ed), Microbial transport systems. Wiley-VCH Verlag GmbH, Weinheim, Germany. [Google Scholar]
- 32.Kosfeld A, Jahreis K. 2012. Characterization of the interaction between the small regulatory peptide SgrT and the EIICBGlc of the glucose-phosphotransferase system of E. coli K-12. Metabolites 2:756–774. doi: 10.3390/metabo2040756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gabor E, Gohler AK, Kosfeld A, Staab A, Kremling A, Jahreis K. 2011. The phosphoenolpyruvate-dependent glucose-phosphotransferase system from Escherichia coli K-12 as the center of a network regulating carbohydrate flux in the cell. Eur J Cell Biol 90:711–720. doi: 10.1016/j.ejcb.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 34.Aboulwafa M, Chung YJ, Wai HH, Saier MH Jr. 2003. Studies on the Escherichia coli glucose-specific permease, PtsG, with a point mutation in its N-terminal amphipathic leader sequence. Microbiology 149:763–771. doi: 10.1099/mic.0.25731-0. [DOI] [PubMed] [Google Scholar]
- 35.Buhr A, Daniels GA, Erni B. 1992. The glucose transporter of Escherichia coli. Mutants with impaired translocation activity that retain phosphorylation activity. J Biol Chem 267:3847–3851. [PubMed] [Google Scholar]
- 36.Deutscher J, Francke C, Postma PW. 2006. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol Mol Biol Rev 70:939–1031. doi: 10.1128/MMBR.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Richards GR, Vanderpool CK. 2012. Induction of the Pho regulon suppresses the growth defect of an Escherichia coli sgrS mutant, connecting phosphate metabolism to the glucose-phosphate stress response. J Bacteriol 194:2520–2530. doi: 10.1128/JB.00009-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plumbridge J. 1998. Expression of ptsG, the gene for the major glucose PTS transporter in Escherichia coli, is repressed by Mlc and induced by growth on glucose. Mol Microbiol 29:1053–1063. doi: 10.1046/j.1365-2958.1998.00991.x. [DOI] [PubMed] [Google Scholar]
- 39.Kimata K, Inada T, Tagami H, Aiba H. 1998. A global repressor (Mlc) is involved in glucose induction of the ptsG gene encoding major glucose transporter in Escherichia coli. Mol Microbiol 29:1509–1519. doi: 10.1046/j.1365-2958.1998.01035.x. [DOI] [PubMed] [Google Scholar]
- 40.Park YH, Lee BR, Seok YJ, Peterkofsky A. 2006. In vitro reconstitution of catabolite repression in Escherichia coli. J Biol Chem 281:6448–6454. doi: 10.1074/jbc.M512672200. [DOI] [PubMed] [Google Scholar]
- 41.Feucht BU, Saier MH Jr. 1980. Fine control of adenylate cyclase by the phosphoenolpyruvate:sugar phosphotransferase systems in Escherichia coli and Salmonella typhimurium. J Bacteriol 141:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Harwood JP, Gazdar C, Prasad C, Peterkofsky A, Curtis SJ, Epstein W. 1976. Involvement of the glucose enzymes II of the sugar phosphotransferase system in the regulation of adenylate cyclase by glucose in Escherichia coli. J Biol Chem 251:2462–2468. [PubMed] [Google Scholar]
- 43.Pikis A, Hess S, Arnold I, Erni B, Thompson J. 2006. Genetic requirements for growth of Escherichia coli K12 on methyl-alpha-d-glucopyranoside and the five alpha-d-glucosyl-d-fructose isomers of sucrose. J Biol Chem 281:17900–17908. doi: 10.1074/jbc.M601183200. [DOI] [PubMed] [Google Scholar]
- 44.Pitt WW Jr, Jolley RL, Scott CD. 1975. Determination of trace organics in municipal sewage effluents and natural waters by high-resolution ion-exchange chromatography. Environ Sci Technol 9:1068–1073. doi: 10.1021/es60110a013. [DOI] [Google Scholar]
- 45.Randez-Gil F, Blasco A, Prieto JA, Sanz P. 1995. DOGR1 and DOGR2: two genes from Saccharomyces cerevisiae that confer 2-deoxyglucose resistance when overexpressed. Yeast 11:1233–1240. doi: 10.1002/yea.320111303. [DOI] [PubMed] [Google Scholar]
- 46.Horler RS, Vanderpool CK. 2009. Homologs of the small RNA SgrS are broadly distributed in enteric bacteria but have diverged in size and sequence. Nucleic Acids Res 37:5465–5476. doi: 10.1093/nar/gkp501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Peer A, Margalit H. 2014. Evolutionary patterns of Escherichia coli small RNAs and their regulatory interactions. RNA 20:994–1003. doi: 10.1261/rna.043133.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levine E, Zhang Z, Kuhlman T, Hwa T. 2007. Quantitative characteristics of gene regulation by small RNA. PLoS Biol 5:e229. doi: 10.1371/journal.pbio.0050229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H. 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol Syst Biol 2:2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellermeier CD, Janakiraman A, Slauch JM. 2002. Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290:153–161. doi: 10.1016/S0378-1119(02)00551-6. [DOI] [PubMed] [Google Scholar]
- 51.Court DL, Swaminathan S, Yu D, Wilson H, Baker T, Bubunenko M, Sawitzke J, Sharan SK. 2003. Mini-lambda: a tractable system for chromosome and BAC engineering. Gene 315:63–69. doi: 10.1016/S0378-1119(03)00728-5. [DOI] [PubMed] [Google Scholar]
- 52.Miller JH. 1972. Experiments in molecular genetics. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY. [Google Scholar]
- 53.Fei J, Singh D, Zhang Q, Park S, Balasubramanian D, Golding I, Vanderpool CK, Ha T. 2015. RNA biochemistry. Determination of in vivo target search kinetics of regulatory noncoding RNA. Science 347:1371–1374. doi: 10.1126/science.1258849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wadler CS, Vanderpool CK. 2009. Characterization of homologs of the small RNA SgrS reveals diversity in function. Nucleic Acids Res 37:5477–5485. doi: 10.1093/nar/gkp591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ulbrandt ND, Newitt JA, Bernstein HD. 1997. The E. coli signal recognition particle is required for the insertion of a subset of inner membrane proteins. Cell 88:187–196. doi: 10.1016/S0092-8674(00)81839-5. [DOI] [PubMed] [Google Scholar]